Myasthenia gravis
Myasthenia gravis is a comparatively rare autoimmune disease in which there is an autoantibody to the acetylcholine nicotinic N2 receptor system. The autoantibody can impair the responsiveness of the neuromuscular junction to acetylcholine (ACh; Ch. 27) by three distinct mechanisms:
 increased receptor destruction by complement binding,
increased receptor destruction by complement binding,
crosslinking of receptors, which causes increased receptor internalisation,
The result is that fewer functional receptors are available for ACh, which is therefore less likely to depolarise the muscle cell sufficiently to reach its threshold firing potential. Consequently, in myasthenia gravis there is skeletal muscle weakness. In healthy skeletal muscle, repetitive nerve stimulation leads to a reduction in the numbers of sensitive receptors, but this causes no physiological reduction in muscle activity due to the large number of spare receptors. However, in myasthenia gravis, with repetitive stimulation the smaller receptor pool reduces receptor availability to a level at which increasing numbers of muscle fibres fail to fire. This produces the characteristic rapid muscle fatigue on exertion. The earliest symptoms of myasthenia gravis are often diplopia, which arises from weakness of the extraocular muscles, or ptosis. In 85% of cases the symptoms progress to involve many other muscle groups, particularly producing bulbar, facial and proximal limb weakness.
The thymus gland plays a part in the genesis of the immune response in myasthenia gravis, although its precise role remains uncertain. About 80% of people with myasthenia gravis have an abnormality in the thymus, which is usually lymphoreticular hyperplasia if the onset of the condition is at an early age or a thymoma if the onset is over the age of 40 years.
Symptomatic treatment of the weakness in myasthenia gravis is achieved by prolongation of the action of ACh through inhibiting acetylcholinesterase, the enzyme responsible for its hydrolysis. However, immunosuppression is also important for disease control.
Acetylcholinesterase inhibitors
Mechanism of action and effects
Acetylcholinesterase (AChE) inhibitors block the breakdown of ACh released from presynaptic neurons. Details of their mechanisms of action are found in Chapter 4. They enhance the effect of ACh at all synaptic connections at which it is the neurotransmitter, but their therapeutic actions in myasthenia gravis are by increasing the longevity of ACh at nicotinic N2 receptors (Ch. 4). Unwanted effects arise from the excessive actions of ACh at nicotinic N1 receptors in autonomic ganglia and at muscarinic receptors in postganglionic nerve endings in the parasympathetic nervous system and in sweat glands in the sympathetic nervous system.
Pharmacokinetics and clinical uses
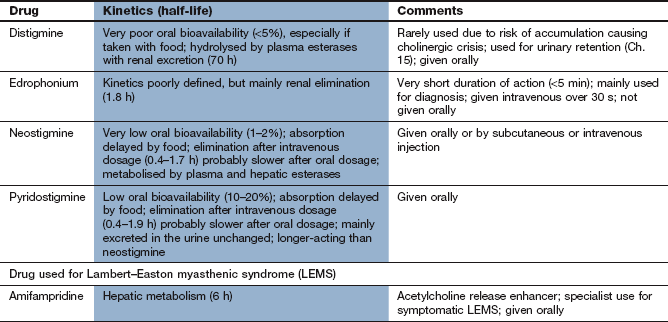
Neostigmine and pyridostigmine are quaternary amines that are slowly and incompletely absorbed from the gut. As a result, oral doses need to be approximately 10 times greater than parenteral doses to be effective. They have short elimination half-lives (1–2 h) due to a combination of renal tubular secretion and some hepatic metabolism. They do not readily cross the blood–brain barrier (see Ch. 9 for AChE inhibitors that cross the blood–brain barrier and are used in Alzheimer's disease). Both neostigmine and pyridostigmine can be used to treat myasthenia gravis, but pyridostigmine is preferred because of its longer duration of action. Neostigmine has a faster onset of action, and is also used by intravenous injection to reverse the effect of competitive neuromuscular blockers (Ch. 27).
Edrophonium has a very short duration of action (2–5 min), owing largely to rapid tissue distribution. It is given as an intravenous bolus to test the therapeutic response to AChE inhibitors in myasthenia gravis (see below) but is of no value in treatment.
Unwanted effects
Unwanted effects arise because the AChE inhibitors are effective at all sites where ACh is released. They are experienced by up to one-third of people treated and are more troublesome with neostigmine than with pyridostigmine. Peripheral muscarinic receptor agonist effects, which can be blocked by co-administration of a muscarinic receptor antagonist such as propantheline, include:
diarrhoea, abdominal cramps, excessive salivation,
bradycardia, hypotension (uncommon),
bronchoconstriction (see Ch. 12),
Excessive dosage of AChE inhibitors will lead to a depolarising neuromuscular blockade by ACh. Initially there may be muscle twitching and cramps, followed by weakness through the build-up of excess ACh (see below).
Management of myasthenia gravis
When the diagnosis of myasthenia gravis is suspected, useful diagnostic information can be obtained rapidly by pharmacological testing. In untreated people with myasthenia an intravenous injection of the short-acting AChE inhibitor edrophonium will produce clinical improvement within 1 min that lasts for about 5 min. Detection of circulating N2 receptor antibodies, and electromyographic tests that demonstrate abnormal fatigability in multiple muscle fibres, are used to confirm the diagnosis.
Treatment
Symptomatic treatment of myasthenia gravis is with an AChE inhibitor, which inhibits the normal rapid breakdown of ACh and thereby enhances the activity of ACh released by nerve stimulation. Pyridostigmine is commonly used since its action is more consistent than that of neostigmine, the dosing frequency is less, and there are fewer muscarinic unwanted effects. The onset of action of pyridostigmine is after about 30–45 min with a duration of action of 3–6 h. An antimuscarinic agent (such as propantheline) may be necessary to block any parasympathomimetic actions of pyridostigmine, especially if large doses are given. The type of interaction between the autoantibody and the receptor probably determines the effectiveness of treatment in an individual. Some individuals do not respond well to AChE inhibitors, while unwanted effects may preclude the use of adequate doses in others. In addition, muscle groups do not all respond equally well to AChE inhibitors; ptosis and diplopia with are the most resistant to treatment.
Excessive dosage of an AChE inhibitor can lead to prolonged stimulation of the N2 receptors by ACh, resulting in a depolarising blockade of the neuromuscular junction similar to that produced by suxamethonium (succinylcholine; Ch. 27). Therefore, muscle weakness in myasthenia gravis can be the result of either inadequate dosage (‘myasthenic crisis’) or excessive dosage (‘cholinergic crisis’) with an AChE inhibitor. The safest way to distinguish these problems is to use assisted ventilation and temporarily withdraw the AChE inhibitor.
Generalised myasthenia is usually treated by immunosuppression with a corticosteroid such as prednisolone (Ch. 44). Corticosteroids are also used for those who are severely ill. They probably act by suppressing T-cell proliferation and reducing antibody synthesis. Initial high-dose corticosteroid therapy can make the weakness worse, particularly in the first few hours, possibly due to a direct effect on neuromuscular transmission. A clinical response is usually apparent after one month, but maximum benefit is delayed for up to 9 months. Ciclosporin (alone or with a corticosteroid) or cyclophosphamide with a corticosteroid (Ch. 38) are used if there is a poor response to a corticosteroid alone, but the dosage of ciclosporin is usually limited by nephrotoxicity. Long-term immunosuppression is usually necessary, since relapse frequently occurs on withdrawal of therapy.
Plasma exchange to remove circulating ACh receptor antibodies can produce a short-term response in severe disease. With repeated plasma exchanges improvement is seen after one day, with a maximum response after 1–2 weeks that is sustained for 2–8 weeks. An alternative is the use of intravenous immunoglobulin, of which IgG is the active component. It produces improvement after about 4 days, and an optimal response after 1–2 weeks that is sustained for 6–15 weeks. Immunoglobulin treatment is usually better tolerated than plasma exchange.
Thymectomy can induce remission in myasthenia gravis, although this can be delayed for up to five years. It is most effective for early onset disease with lymphoreticular hyperplasia and positive receptor antibodies, when it produces complete remission within five years in about 40% and significant improvement in a further 35%. Thymectomy is also used to remove a thymoma, although the clinical benefit is often less clear-cut.
Some drugs can interfere with neuromuscular transmission and exacerbate the symptoms of myasthenia gravis. Those most often implicated include aminoglycoside antibiotics (Ch. 51), β-adrenoceptor antagonists (Ch. 5), phenytoin (Ch. 23), chloroquine and penicillamine (Ch. 30). There is also altered sensitivity to neuromuscular junction blockers, with increased response to competitive (non-depolarising) neuromuscular junction blockers but resistance to depolarising neuromuscular junction blockers (Ch. 27).
Lambert–Eaton myasthenic syndrome
Lambert–Eaton myasthenic syndrome (LEMS) is a clinical syndrome that resembles myasthenia gravis. It is often a paraneoplastic syndrome, with about 60% associated with malignancy (often small-cell lung cancer). LEMS is caused by antibodies to P/Q-type voltage-gated Ca2+ channels on the presynaptic membrane of motor nerves at the neuromuscular junction. These channels are responsible for Ca2+ influx into the neuron that initiates ACh release. LEMS presents with leg and arm weakness and autonomic disturbance such as postural hypotension. While it can involve bulbar muscles, eye muscles are less often affected.
When LEMS is related to cancer, treatment of the malignancy leads to resolution. The potassium channel blocker amifampridine is the first choice drug for symptom relief. It delays repolarisation of the nerve terminal after an action potential giving more time for Ca2+ to accumulate in the neuron, and the prolonged depolarisation allows greater ACh release. Pyridostigmine can help the symptoms by prolonging the action of released ACh at the motor end plate. Immunosuppression is relatively ineffective in the treatment of LEMS.
True/false questions
1. In myasthenia gravis autoantibodies develop to nicotinic N1 receptors.
2. Acetylcholinesterase inhibitors may cause bronchoconstriction.
3. Pyridostigmine produces fewer muscarinic effects than neostigmine.
4. In myasthenia gravis there is increased sensitivity to suxamethonium.
5. A cholinergic crisis should be confirmed by giving pyridostigmine.
One-best-answer (OBA) question
Which statement about myasthenia gravis is the most accurate?
A People diagnosed with myasthenia gravis invariably have a thymoma.
B Acetylcholinesterase (AChE) inhibitors must cross the blood–brain barrier to be effective in treating myasthenia gravis.
C Glucocorticoids can be of benefit in myasthenia gravis because of their anti-inflammatory actions.
D The unwanted effects of AChE inhibitors include diarrhoea, urination, miosis, bradycardia, nausea, lacrimation and salivation.
E Plasmapheresis reduces plasma levels of pseudocholinesterase, thereby reducing breakdown of acetylcholine.
Case-based questions
A 35-year-old woman with no previous illness noticed that she had ptosis and occasional diplopia. Over a period of time she became aware that she suffered from leg weakness on exertion, although her coordination was normal. Following a sustained upward gaze for a minute, ptosis and diplopia could be elicited. Myasthenia gravis was suspected.
1. False. Skeletal muscle weakness in myasthenia gravis is due to autoimmunity to nicotinic N2 receptors in the neuromuscular junction.
2. True. Increased ACh at muscarinic receptors in the parasympathetic nervous system can cause bronchoconstriction; such effects can be blocked with antimuscarinic drugs such as propantheline.
3. True. Neostigmine has greater potency and a shorter duration of action than pyridostigmine so is more likely to cause muscarinic effects; pyridostigmine is therefore preferred in the treatment of myasthenia gravis.
4. False. Due to the loss of nicotinic N2 receptor function people with myasthenia gravis are less sensitive to the depolarising neuromuscular junction blocker suxamethonium but more sensitive to competitive blockers such as vecuronium.
5. False. The cholinergic crisis results from the excessive effects of an AChE inhibitor and would be exacerbated by the long-acting pyridostigmine; assisted ventilation and withdrawal of the treatment should be performed.
OBA answer
Answer D is the most accurate.
A Incorrect. About 15% have a thymoma and 60–80% have hyperplasia of the thymus.
B Incorrect. The main anticholinesterases used in treating myasthenia gravis are quaternary amines and do not cross the blood–brain barrier; they act at the neuromuscular junction.
C Incorrect. Glucocorticoids work by suppressing production of autoantibodies to nicotinic N2 receptors.
D Correct. These parasympathomimetic effects are caused by excess ACh activity at muscarinic receptors.
E Incorrect. Plasmapheresis reduces the levels of circulating autoantibodies to nicotinic N2 receptors.
Case-based answers
A Tests include electromyography (Jolly test), single muscle fibre electromyography (SFEMG), anti-acetylcholine receptor (AChR) antibody titres and injection of a short-acting inhibitor of AChE (the Tensilon test, named after the proprietary name of edrophonium).
B Autoantibody blocks nicotinic N2 receptors; receptors are destroyed by complement activation and receptors are crosslinked, which causes them to be destroyed more rapidly. The decrease in functional receptors impairs motor endplate potentials and reduces the likelihood of the muscle contracting.
C It is short-acting, giving an improvement in muscle strength within 30–60 s, subsiding in 4–5 min.
D An AChE inhibitor, most commonly pyridostigmine, is used for symptomatic treatment; an antimuscarinic drug may be needed to reduce unwanted effects. Immunosuppression may be required with a corticosteroid, or with immunosuppressants such as ciclosporin or azathioprine. Plasmapheresis, intravenous immunoglobulins or thymectomy may also be considered.
Conti-Fine, BM, Milani, M, Kaminski, HJ. Myasthenia gravis: past, present, and future. J Clin Invest. 2006;116:2843–2854.
Hart, IK, Sathasivam, S, Sharshar, T. Immunosuppressive agents for myasthenia gravis. Cochrane Database Syst Rev. (4):2007.
Newsom-Davis, J. Therapy in myasthenia gravis and Lambert–Eaton myasthenic syndrome. Semin Neurol. 2003;23:191–197.
Schwendimann, RN, Burton, E, Minagar, A. Management of myasthenia gravis. Am J Ther. 2005;12:262–268.