The Child with Neuromuscular or Muscular Dysfunction
On completion of this chapter the reader will be able to:
 Discuss the nursing role in helping parents care for the child who has cerebral palsy.
Discuss the nursing role in helping parents care for the child who has cerebral palsy.
Formulate a nursing care plan for the preoperative and postoperative care of a child with myelomeningocele.
Outline a care plan for a child with Guillain-Barré syndrome.
Discuss the home care management of the child with a neuromuscular disease such as spinal muscular atrophy.
Discuss the prevention and treatment of tetanus.
RELATED TOPICS and ADDITIONAL RESOURCES
 IN TEXT
IN TEXTThe Child with Cerebral Dysfunction, Ch. 28
Genetic Evaluation and Counseling, Ch. 9
The Immobilized Child, Ch. 31
Impact of the Child’s Chronic Illness or Disability, Ch. 18
Neurologic Assessment, Ch. 6
Nursing Care of the High-Risk Newborn and Family, Ch. 9
Recommendations for Routine Immunizations: Tetanus, Ch. 10
CONGENITAL NEUROMUSCULAR OR MUSCULAR DISORDERS
A new definition proposed in 2006 describes cerebral palsy (CP) as a “group of permanent disorders of the development of movement and posture, causing activity limitation, that are attributed to nonprogressive disturbances that occurred in the developing fetal or infant brain” (Rosenbaum, Paneth, Leviton, and others, 2007). In addition to motor disorders, the condition often involves disturbances of sensation, perception, communication, cognition, and behavior; secondary musculoskeletal problems; and epilepsy (Rosenbaum, Paneth, Leviton, and others, 2007). The etiology, clinical features, and course are variable and are characterized by abnormal muscle tone and coordination as the primary disturbances. CP is the most common permanent physical disability of childhood, and the incidence is reported to be between 1.5 and 3 in every 1000 live births in the United States (Ashwal, Russman, Blasco, and others, 2004; Dabney, Lipton, and Miller, 1997; Winter, Autry, Boyle, and others, 2002). Since the 1960s the prevalence of CP has risen approximately 20%, which most likely reflects the improved survival of extremely low–birth-weight and very low—birth-weight infants.
Although the prevalent traditional hypothesis has been that CP results from perinatal problems, especially birth asphyxia, it is now believed that CP results more often from existing prenatal brain abnormalities; however, the exact cause of these abnormalities remains elusive. It has been estimated that as many as 80% of CP cases are caused by unknown prenatal factors (Krigger, 2006). Intrauterine exposure to maternal chorioamnionitis is associated with an increased risk of CP in infants of normal birth weight and preterm infants (Gibson, MacLennan, Goldwater, and others, 2003; Volpe, 2001); however, not all term infants exposed to chorioamnionitis develop CP (Grether, Nelson, Walsh, and others, 2003; Wu, Escobar, Grether, and others, 2003). The prevalence of CP in infants born before 36 weeks of gestation and weighing less than 2000 g (4.4 pounds) has been reported to be 12%; the strongest independent risk factor for development of CP was periventricular leukomalacia (Han, Bang, Lim, and others, 2002). Damage occurring as a result of shaken baby syndrome may also result in CP in survivors. Additional factors that may contribute to the development of CP postnatally include bacterial meningitis, viral encephalitis, motor vehicle accidents (MVAs), and child abuse (Krigger, 2006). A significant percentage (15% to 60%) of children with CP will also have epilepsy, thus compromising issues of self-care and progression to normalization.
Pathophysiology
It is difficult to establish a precise location of neurologic lesions based on etiology or clinical signs because no characteristic pathologic pattern exists. Some patients have gross malformations of the brain; others may have evidence of vascular occlusion, atrophy, loss of neurons, and degeneration. A few exceptions occur and are related to anatomic areas such as spastic diplegia (associated with preterm birth), caused by hypoxic infarction or hemorrhage in the area adjacent to the lateral ventricles. Ataxic CP may occur in relation to cerebral hypoplasia and, in some cases, severe hypoglycemia (Volpe, 2001). The American College of Obstetricians and Gynecologists, in conjunction with the American Academy of Pediatrics (2003), published a report that defines neonatal encephalopathy. The report affirms that approximately 70% of cases of neonatal encephalopathy occur as a result of events before the onset of labor; it establishes criteria to define events sufficiently capable of causing intrapartum asphyxia and CP. Evidence indicates that events that cause the majority of CP cases occur not as a result of intrapartum asphyxia, but as a result of other causes that have been discussed previously (American Academy of Pediatrics and American College of Obstetricians and Gynecologists, 2003).
CP has been classified in several ways. A functional classification is based on the nature and distribution of neuromuscular dysfunction (Box 32-1). Additional classifications describe the area of the brain involved, the degree of motor involvement, accompanying impairments, anatomic distribution, and cause of CP (Rosenbaum, Paneth, Leviton, and others, 2007).
BOX 32-1 Clinical Classification of Cerebral Palsy
Characterized by persistent primitive reflexes, positive Babinski, ankle clonus, exaggerated stretch reflexes, eventual development of contractures
70% to 80% of all cases of cerebral palsy
Diplegia—All extremities affected; lower more than upper (30% to 40% of spastic cerebral palsy [CP])
Quadriplegia—All four extremities involved: legs and trunk, mouth, pharynx, and tongue (10% to 15% of spastic CP)
Triplegia—Three limbs involved
Monoplegia—Only one limb involved
Hemiplegia—Motor dysfunction on one side of the body; upper extremity more affected than lower (20% to 30% of spastic CP)
DYSKINETIC (NONSPASTIC, EXTRAPYRAMIDAL)
Athetoid—Chorea (involuntary, irregular, jerking movements); characterized by slow, wormlike, writhing movements that usually involve the extremities, trunk, neck, facial muscles, and tongue
Dystonic—Slow, twisting movements of the trunk or extremities; abnormal posture
Involvement of the pharyngeal, laryngeal, and oral muscles causing drooling and dysarthria (imperfect speech articulation)
ATAXIC (NONSPASTIC, EXTRAPYRAMIDAL)
Wide-based gait
Rapid, repetitive movements performed poorly
Disintegration of movements of the upper extremities when the child reaches for objects
Combination of spastic CP and dyskinetic CP
May be labeled mixed when no specific motor pattern is dominant; however, this term is losing favor to more precise descriptions of motor function and affected area of brain involved (see Rosenbaum, Paneth, Leviton, and others, 2007)
Data from Nehring W: Cerebral palsy. In Allen PJ, Vessey JA, editors: Primary care of the child with a chronic condition, St Louis, 2004, Mosby; Jones MW, Morgan E, Shelton JE, and others: Cerebral palsy: introduction and diagnosis, part 1, J Pediatr Health Care 21(3):146-152, 2007; and National Institute of Neurologic Disorders and Stroke: Cerebral palsy: hope through research, 2006, retrieved July 9, 2007, from http://www.ninds.nih.gov/disorders/cerebral_palsy/detail_cerebral_palsy.htm.
Diagnostic Evaluation
Infants at risk according to known etiologic factors associated with CP warrant careful assessment during early infancy to identify the signs of muscular dysfunction as early as possible. The neurologic examination and history are the primary modalities for diagnosis. Neuroimaging of the child with suspected brain abnormality and CP is now recommended for diagnostic assessment, with magnetic resonance imaging (MRI) preferred to computed tomography (CT) scan. Metabolic and genetic testing is recommended if no structural abnormality is identified by neuroimaging; laboratory tests are no longer recommended in the diagnostic process for CP (Ashwal, Russman, Blasco, and others, 2004).
Early recognition is made more difficult by the lack of reliable neonatal neurologic signs. However, infants with known etiologic risk factors should be monitored and evaluated closely in the first 2 years of life. The alert observer may be suspicious when a child demonstrates some of the manifestations outlined in Boxes 32-2 and 32-3. Because cortical control of movement does not occur until later in infancy, motor impairment associated with voluntary control is usually not apparent until after 2 to 4 months of age at the earliest. More often the diagnosis cannot be confirmed until the age of 2 years because motor tone abnormalities may be indicative of another neuromuscular illness.
The persistence of primitive reflexes may be of value in establishing the diagnosis: (1) either the asymmetric tonic neck reflex or persistent Moro reflex (beyond 4 months of age), and (2) the crossed extensor reflex. The tonic neck reflex normally disappears between 4 and 6 months of age. An “obligatory” response is considered abnormal. The crossed extensor reflex, which normally disappears by 4 months, is elicited by applying a noxious stimulus to the sole of one foot with the knee extended. Normally the contralateral foot responds with extensor, abduction, and then adduction movements. The possibility of CP is suggested if these reflexes are found after the age at which they should have disappeared.
Therapeutic Management
The goals of therapy for children with CP are early recognition and promotion of optimal development to enable affected children to attain normalization and their potential within the limits of their existing health problems. The disorder is permanent, and therapy is primarily preventive and symptomatic.
1. To establish locomotion, communication, and self-help skills
2. To gain optimal appearance and integration of motor functions
3. To correct associated defects as effectively as possible
4. To provide educational opportunities adapted to the child’s needs and capabilities
5. To promote socialization experiences with other affected and unaffected children
Each child is evaluated and managed on an individual basis. The scope of the child’s needs requires multidisciplinary planning and care coordination among professionals and the child’s family. The outcome for the child and family with CP is normalization and promotion of self-care activities that empower the child and family to achieve maximum potential.
Ankle-foot orthoses (AFOs, braces) are worn by many of these children and are used to help prevent or reduce deformity, increase the energy efficiency of gait, and control alignment. Other mobilization devices include wheeled scooter boards that allow children to propel themselves while on the abdomen, wheeled go-carts that provide sitting balance and serve as early “wheelchair” experience for young children, bicycle walkers, and special devices that leave the upper extremities free (Figs. 32-1 and 32-2). Strollers can be equipped with custom seats for dependent mobilization.
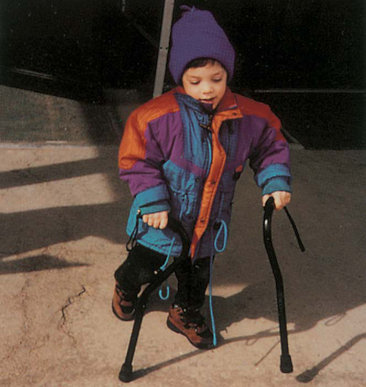
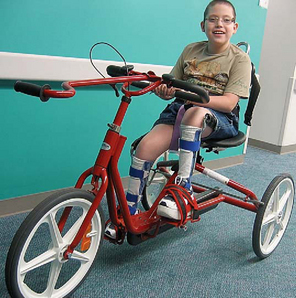
FIG. 32-2 Bike walker used to provide mobility and to enhance leg muscle strength. (Courtesy Texas Children’s Hospital, Houston.)
Orthopedic surgery may be required to correct contracture or spastic deformities, to provide stability for an uncontrollable joint, and to provide balanced muscle power. This includes tendon-lengthening procedures (especially heel-cord lengthening), release of spastic wrist flexor muscles, and correction of hip and adductor muscle spasticity or contracture to improve locomotion. Selective dorsal rhizotomy has provided marked improvement in some children with CP. The procedure involves selectively cutting dorsal column sensory rootlets that have an abnormal response to electrical stimulation. Achieving the benefits from the surgery requires intensive physical therapy and family commitment. Because the procedure results in flaccid muscles, the child must be retaught to sit, stand, and walk.
Surgical intervention is usually reserved for the child who does not respond to the more conservative measures, but it is also indicated for the child whose spasticity causes progressive deformities. Surgery is primarily used to improve function rather than for cosmetic purposes and is followed by physical therapy.
Intense pain may occur with muscle spasms in patients with CP. Pharmacologic agents given orally (dantrolene sodium, baclofen [Lioresal], and diazepam [Valium]) have had little effectiveness in improving muscle coordination in children with CP; however, they are effective in decreasing overall spasticity. The most common side effects of these agents include hepatotoxicity (dantrolene), drowsiness, fatigue, and muscle weakness; less commonly, diaphoresis and constipation may be seen with baclofen. Diazepam is used frequently but should be restricted to older children and adolescents.
Botulinum toxin A (Botox) is also used to reduce spasticity in targeted muscles. Botulinum toxin A is injected into a selected muscle (commonly the quadriceps, gastrocnemius, or medial hamstrings) after a topical anesthetic is applied. The drug acts to inhibit the release of acetylcholine into a specific muscle group, thereby preventing muscle movement. When it is administered early in the course of the illness, affected muscle contractures may be prevented, particularly in lower extremities, thus avoiding surgical procedures with possible adverse effects. The goal is to allow stretching of the muscle as it relaxes and permit ambulation with an AFO. The major reported adverse effect of botulinum toxin A injection is pain at the injection site (Roscigno, 2002). Prime candidates for botulinum toxin A injections are children with spasticity confined to the lower extremities; the drug weakens spasticity so the muscles can be stretched and the child may ambulate with or without orthoses. The onset of action occurs within 24 to 72 hours, with a peak effect observed at 2 weeks and a duration of action of 3 to 6 months (Green, Greenberg, and Hurwitz, 2003).
Children with CP may also experience pain as a result of surgical procedures intended to reduce contracture deformities, position and gastroesophageal reflux, and physical therapy (McKearnan, Kieckhefer, Engel, and others, 2004). Therefore pain management is an important aspect of care of the child with CP.
Neurosurgical and pharmacologic approach to managing the spasticity associated with CP involves the implantation of a pump to infuse baclofen directly into the intrathecal space surrounding the spinal cord to provide relief of spasticity. Intrathecal baclofen therapy is best suited for children with severe spasticity that interferes with activities of daily living (ADLs) and ambulation. Patients are screened before pump placement by the infusion of a “test dose” of intrathecal baclofen delivered via a lumbar puncture. Close monitoring for side effects (hypotonia, somnolence, seizures, nausea, vomiting, headache, and catheter- or pump-related problems [Albright, Gilmartin, Swift, and others, 2003]) and relief of spasticity occurs for several hours after the infusion. If a positive effect is noted, the patient is considered a candidate for pump placement. The implantation procedure is done in the operating room by a neurosurgeon. The pump, which is approximately the size of a hockey puck, is placed in the subcutaneous space of the midabdomen. An intrathecal catheter is tunneled from the lumbar area to the abdomen and connected to the pump. The pump is filled with baclofen and programmed to provide a set dose using a telemetry wand and a computer. Benefits of intrathecal baclofen include fewer systemic side effects than oral baclofen, dosage titration for maximizing effects, and reversibility of therapy with removal of the pump if so desired (Jacobs, 2001). The patient may remain hospitalized for 3 to 7 days to adjust the dosage and ensure proper healing. Outpatient visits to refill the pump and make dosage adjustments occur about every 4 to 6 weeks, depending on the patient’s response to the treatment. This procedure is most suited for a multidisciplinary setting where rehabilitation specialists are readily available and consistently involved in the patient’s ongoing care. Abrupt withdrawal of intrathecal baclofen, especially at high doses, may result in adverse effects such as rebound spasticity, pruritus, hyperthermia, rhabdomyolysis, disseminated intravascular coagulation, multiorgan failure, and death; in some cases intrathecal baclofen withdrawal may mimic sepsis.
Antiepileptic drugs (AEDs) such as carbamazepine (Tegretol) and divalproex (valproate sodium and valproic acid; Depakote) are prescribed routinely for children who have seizures. Gabapentin (Neurontin) has been used in adults with spinal cord injury (SCI) to decrease spasticity with success; no studies are available on the effectiveness of the drug in children. The α2-adrenergic agonists clonidine (Catapres) and tizanidine (Zanaflex) have been used to decrease spasticity in adults with SCI and multiple sclerosis, but use in children with CP has yet to be reported (Krach, 2001). All medications should be monitored for maintenance of therapeutic levels and avoidance of subtherapeutic or toxic levels.
Dental hygiene is especially important. Regular visits to the dentist and prophylaxis, including brushing, fluoride, and flossing, should be instituted as soon as the teeth erupt. Dental care is especially important for children being given phenytoin, since they often develop gum hyperplasia. Additional problems common among children with CP include constipation caused by neurologic deficits and lack of exercise; poor bladder control and urinary retention; chronic respiratory tract infections and aspiration pneumonia, which occur as a result of gastroesophageal reflux, abnormal muscle tone, immobility, and altered positioning; and skin problems as a result of altered positioning, poor nutrition, and immobility.
A wide variety of technical aids is available to improve the functioning of children with CP. These include electromechanical toys that employ the concept of biofeedback and operate from a head unit. The toy is manipulated only when the head and trunk are in correct alignment. Eye-hand coordination can also be enhanced by computerized toys and games. Microcomputers combined with voice synthesizers aid children with speech difficulties to “speak.” These and other devices print messages onto screen monitors and paper.
Many other electronic devices allow independent functioning. Sensors can be activated and deactivated by using a head-stick or tongue, or other voluntary muscle movement over which the child has control. Voice-activated computer technology may also allow increased mobility and ambulation with specially designed devices such as wheelchairs. The application of this technology makes it possible for persons with CP to function eventually in their own residences and can be extended into the workplace.
Physical therapy is one of the most frequently used conservative treatment modalities. It requires the specialized skills of a qualified therapist with an extensive repertoire of exercise methods who can design a program to stimulate each child to achieve his or her functional goals.
An active therapy program involves the family; the physical therapist; and often other members of the health team, including the nurse. The most common approach uses traditional types of therapeutic exercises that consist of stretching, passive, active, and resistive movements applied to specific muscle groups or joints to maintain or increase range of motion, strength, or endurance.
Prognosis.: In general, the more severe the functional disability, the worse the prognosis. Children with a severe physical disability, mental retardation, tube feedings, and severe seizures are known to have a shortened life expectancy. According to available data, approximately 30% to 50% of individuals with CP are mentally retarded, and an even higher percentage have mild cognitive and learning deficits (Green, Greenberg, and Hurwitz, 2003); however, many children with severe spastic quadriplegic CP have normal intelligence. Growth is affected in children with spastic quadriplegia, and many children remain below the 5th percentile for age and gender. As children with CP transition to adulthood, about 30% remain in the home and are cared for by a parent or caregiver; 50% of individuals with spastic quadriplegia live in independent settings and function at appropriate social levels considering their disability (Green, Greenberg, and Hurwitz, 2003). Vocational rehabilitation and higher education are possible for adults with CP, and one study found that 53% of all persons with CP were able to work outside the home in regular jobs; one third of the severely disabled adults with CP worked outside the home (Murphy, Molnar, and Lankasky, 2000).
Nursing Care Management
The nursing process in the care of the child with CP is outlined in the Nursing Process box.
Because children with CP are being identified and treated at an earlier age, parents are participating earlier in treatment programs for their disabled child. They are taught the proper handling and home care of young children with CP and need a carefully planned program so that their change of role from parent to caregiver can be melded into the already established relationship. Close work with other multidisciplinary team members is essential. Nurses reinforce the therapeutic plan and assist the family in devising and modifying equipment and activities to continue the therapy program in the home (see Nursing Care Plan).
Because children with CP expend so much energy in their efforts to accomplish ADLs, more frequent rest periods should be arranged to avoid the fatigue that may tax their limited capabilities. The diet should be tailored to the child’s activity and metabolic needs. Gastrostomy feedings may be necessary to supplement regular feedings and ensure adequate weight gain, particularly in the child who is at risk for growth failure and chronic malnutrition. In children with severe CP and subsequent oral feeding difficulties, a feeding gastrostomy should be considered (Rogers, 2004). Gastrostomy feeding as a supplement to oral feeding is often recommended, especially when illness and decreased fluid or medication intake affect the child’s well-being (Rogers, 2004). Oral feedings may be continued to maintain oral motor skills. Weight gain is perceived as an important measure of adequate oral feeding efficiency.
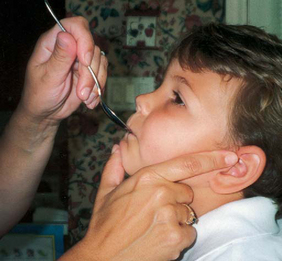
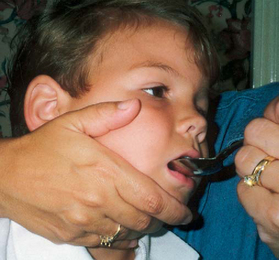
Parents may need assistance and advice with medication administration through a gastrostomy tube to prevent clotting. A skin-level gastrostomy is particularly suited for the child with CP. Because jaw control is often compromised, more normal control can be achieved if the feeder provides stability of the oral mechanism from the side or front of the face. When directed from the front, the middle finger of the nonfeeding hand is placed posterior to the body portion of the chin, the thumb is placed below the bottom lip, and the index finger is placed parallel to the child’s mandible (Fig. 32-3). Manual jaw control from the side assists with head control, correction of neck and trunk hyperextension, and jaw stabilization. The middle finger of the nonfeeding hand is placed posterior to the bony portion of the chin, the index finger is placed on the chin below the lower lip, and the thumb is placed obliquely across the cheek to provide lateral jaw stability (Fig. 32-4).
Safety precautions are implemented, such as having children wear protective helmets if they are subject to falls or capable of injuring their heads on hard objects. Because the child with CP is at risk for altered proprioception and subsequent falls, the home and play environment should be adapted to the child’s needs to prevent bodily harm. Appropriate immunizations should be administered to prevent childhood illnesses and protect against respiratory tract infections such as influenza. Depending on the level of involvement, dental problems may be more common in children with CP, which creates a need for meticulous attention to all aspects of dental care.
nursingprocess: The Child with Cerebral Palsy
Nursing assessment includes risk identification of infants with etiologic factors that are associated with cerebral palsy (CP). Early recognition of CP is important so early interventions may be implemented (see Box 32-3). Ongoing assessment of infants for abnormal muscle tone, inability to achieve developmental milestones, and persistence of neonatal reflexes alert the nurse to investigate further.
BOX 32-3 Early Signs of Cerebral Palsy
Failure to meet any developmental milestones such as rolling over, raising head, sitting up, crawling
Persistent primitive reflexes such as Moro, atonic neck
Poor head control (head lag) and clenched fists after 3 months of age
Stiff or rigid arms or legs; scissoring legs
Pushing away or arching back; stiff posture
Floppy or limp body posture, especially while sleeping
Inability to sit up without support by 8 months
Using only one side of the body, or only the arms to crawl
Persistent gagging or choking when fed
After 6 months of age, tongue pushing soft food out of the mouth
Data from Pathways Awareness Foundation: Parents … if you see any of these warning signs … don’t delay, Chicago, 1991, The Foundation; Nehring W: Cerebral palsy. In Allen PJ, Vessey JA, editors: Primary care of the child with a chronic condition, St Louis, 2004, Mosby; and Jones MW, Morgan E, Shelton JE, and others: Cerebral palsy: introduction and diagnosis, part 1, J Pediatr Health Care 21(3):146-152, 2007.
DIAGNOSIS (PROBLEM IDENTIFICATION)
After a thorough assessment a number of nursing diagnoses are evident (see Nursing Care Plan, p. 1151). Other nursing diagnoses include:
The effectiveness of nursing interventions for the family and child with CP is determined by continual reassessment and evaluation of care based on the following observational guidelines:
Child’s movements and use of mobilization devices
Child’s speech and ability to use communication devices
Child’s activities, especially those related to self-care
Family perception regarding child’s activities and school attendance
Child’s interactions with others and choice of activities; child’s feelings and concerns
The involvement of physical therapy, speech therapy, and occupational therapy is particularly important in establishment and maintenance of muscle function, development of adequate speech and phonation, and identification of modifications necessary for the child’s environment so that ADLs can be performed to the child’s satisfaction.

As in all aspects of care, educational requirements are determined by the child’s needs and potential. Children with mild to moderate involvement are generally able to participate, for varying amounts of time, in regular classes. Resource rooms are available in most schools to provide more individualized attention. Integration of children with CP into regular classrooms should be the initial goal. For those who are unable to benefit from formal education, a vocational training program may be appropriate. At adolescence, prevocational and vocational counseling and guidance are arranged. At any phase or in any setting, education is geared toward the child’s assets.
Recreational outlets and after-school activities should be considered for the child who is unable to participate in the regular athletic programs and other peer activities. Some children can compete in athletic and artistic endeavors, and many games and pastimes are suited to their capabilities. Competitive sports are also becoming increasingly available to children with disabilities and offer an added dimension to physical activities. Information on training programs and competition on local, state, regional, and national levels can be obtained from the National Disability Sports Alliance.*
Recreational activities serve to stimulate children’s interest and curiosity, help them adjust to their disability, improve their functional abilities, and build self-esteem. Any accomplishment that helps children approach a “normal” way of life enhances their self-concept.
Support Family.: Probably the nursing interventions most valuable to the family are support and help in coping with the emotional aspects of the disorder, many of which are discussed in relation to the child with a disability (see Chapter 18). Initially the parents need supportive counseling directed toward understanding the implications of the diagnosis and all of the feelings that it engenders. Later they need clarification regarding what they can expect from the child and from health professionals. Educating families in the principles of family-centered care and parent-professional collaboration is essential. The family may require assistance in modifying the home environment for care of the child (see also Chapter 20). Transportation to the practitioner’s office and other health care agencies often requires special considerations.
Care management for the child and family with CP is an important nursing role. In many cases the family assumes complete care of the child and becomes adept at meeting his or her individual needs. The home health nurse or care manager has an important role in the support and encouragement for families who assume the primary care of a child with CP. Having a child with CP implies numerous problems of daily management and changes in family life, and the nurse can stress principles of normalization.
The nurse needs to support the parents in their frustration, problem solving, concerns, approaches to helping the child, and lack of gratification, as well as the positive approaches they use. All these aspects must be explored and discussed. Parents and other family members require much support and counseling. Siblings of a child with a disability are affected and may respond to the child’s presence with overt or less evident behavioral problems. The family needs a relationship with nurses who can provide continued contact, support, and encouragement through the long process of habilitation.
 FAMILY FOCUS
FAMILY FOCUSThe Reality of Acceptance of Cerebral Palsy
Acceptance is rarely achieved in the length of time implied in the literature.
In the first place, what is acceptance? To me, it is the end of comparing my son with every other child I see. I focus on his gains, not society’s expectations.
It is also being able to laugh periodically at his “clumsiness.” It is “gallows humor” as he achieves adulthood; jokes about CP can be funny now.
The bitterness is gone; I am now happy for people who have children without CP.
I no longer feel sorry for my son, but rather for the people who cannot see him for the great person he is; the CP does not come first.
He is now a young man of 25 years and I am learning to accept his independence.
Parents may also find help and comfort from parent groups, with whom they can share problems and concerns and from whom they can derive comfort and practical information. Parent support groups are most helpful through sharing experiences and accomplishments. For example, parents can learn from others what it is like to have a child with CP, which is generally not possible from professionals (see Family Focus box). The national organization United Cerebral Palsy* has branches in most communities. The association provides a variety of services for children and families. A number of excellent books also are available to guide parents and nurses who work with the child with CP.
Support Hospitalized Child.: CP is not a disorder that requires hospitalization; therefore, when children with CP are hospitalized, they are usually admitted for another reason or for corrective surgery. Nursing care of the child with CP is the same as with any other child with a disability. Children with CP should be approached the same as any child in the hospital. Speech impairment is common in children with CP. To facilitate the care and management of these children, the therapy program should be continued, insofar as their condition allows, during the time they are hospitalized. This should be incorporated into the nursing care plan with every effort expended to make certain that the ground that has been so laboriously gained is not lost. Encouraging the parent to room-in and actively participate in the child’s care facilitates a continuation of the home therapy program and helps the child adjust to an unfamiliar environment. However, it is equally important to remember that hospitalization may be the first time a parent can defer care to a nurse and not be the primary caregiver. This respite may be crucial to the parent’s well-being.
SPINA BIFIDA (MYELOMENINGOCELE)
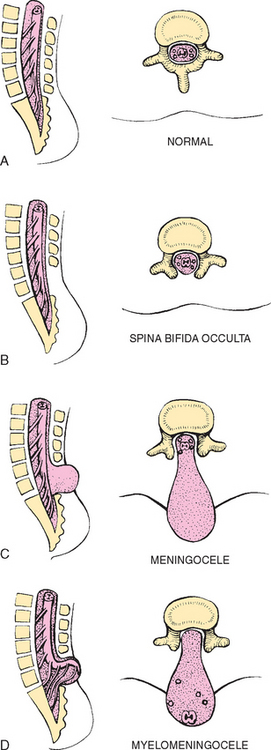
Abnormalities that derive from the embryonic neural tube (neural tube defects [NTDs]) constitute the largest group of congenital anomalies that are consistent with multifactorial inheritance. Normally the spinal cord and cauda equina are encased in a protective sheath of bone and meninges (Fig. 32-5, A). Failure of neural tube closure produces defects of varying degrees (Box 32-4). They may involve the entire length of the neural tube or may be restricted to a small area.
In the United States, rates of NTDs have declined from 1.3 per 1000 births in 1970, to 0.3 per 1000 births after the introduction of mandatory food fortification with folic acid in 1998 (Honein, 2001). Further data indicate that the incidence of NTDs showed a 23% decline from 1996 to 2001 (Matthews, Honein, and Erickson, 2002). Increased use of prenatal diagnostic techniques and termination of pregnancies have also affected the overall incidence of NTDs (see also Prevention, p. 1157).
Myelodysplasia refers broadly to any malformation of the spinal canal and cord. Midline defects involving failure of the osseous (bony) spine to close are called spina bifida (SB), the most common defect of the central nervous system. SB is categorized into two types: spina bifida occulta and spina bifida cystica.
Spina bifida occulta refers to a defect that is not visible externally. It occurs most frequently in the lumbosacral area (L5 and S1) (see Fig. 32-5, B). SB occulta may not be apparent unless there are associated cutaneous manifestations or neuromuscular disturbances.
Spina bifida cystica refers to a visible defect with an external saclike protrusion. The two major forms of SB cystica are meningocele, which encases meninges and spinal fluid but no neural elements (see Fig. 32-5, C), and myelomeningocele (or meningomyelocele), which contains meninges, spinal fluid, and nerves (see Fig. 32-5, D). Meningocele is not associated with neurologic deficit, which occurs in varying, often serious, degrees in myelomeningocele. Clinically the term spina bifida is used to refer to myelomeningocele.
Pathophysiology
Most authorities believe that the primary defect in NTDs is a failure of neural tube closure during the embryo’s early development (the first 3 to 5 weeks). However, evidence also implicates a multifactorial etiology, including drugs, radiation, maternal malnutrition, chemicals, and possibly a genetic mutation in folate pathways in some cases, which may result in abnormal development. There is also evidence of a genetic component in the development of SB; myelomeningocele may occur in association with syndromes such as trisomy 18, PHAVER syndrome, and Meckel-Gruber syndrome (Shaer, Chescheir, and Schulkin, 2007). Additional factors predisposing children to an increased risk of NTDs include prepregnancy maternal obesity, maternal diabetes mellitus, previous NTD pregnancy, low maternal vitamin B12 status, maternal hyperthermia, and the use of AEDs in pregnancy.
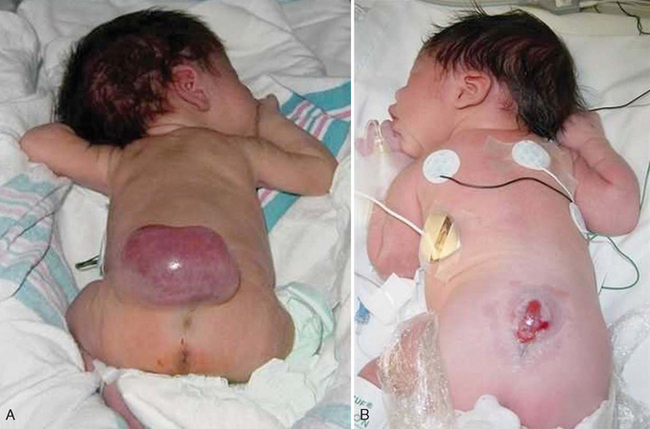
The degree of neurologic dysfunction depends on where the sac protrudes through the vertebrae, the anatomic level of the defect, and the amount of nerve tissue involved. Most myelomeningoceles involve the lumbar or lumbosacral area (Fig. 32-6). Hydrocephalus is a frequently associated anomaly in 80% to 90% of the children.
Diagnostic Evaluation
The diagnosis of SB is made on the basis of clinical manifestations (Box 32-5) and examination of the meningeal sac. Diagnostic measures used to evaluate the brain and spinal cord include MRI, ultrasound, CT, and myelography. Laboratory examinations are used primarily to determine causative organisms for common complications associated with myelomeningocele: meningitis and urinary tract infections.
BOX 32-5 Clinical Manifestations of Spina Bifida
Sensory disturbances usually parallel to motor dysfunction
Prenatal Detection.: It is possible to determine the presence of some major open NTDs prenatally. Ultrasonographic scanning of the uterus and elevated maternal concentrations of α-fetoprotein (AFP, or MS-AFP), a fetal-specific γ1-globulin, in amniotic fluid may indicate anencephaly or myelomeningocele. The optimum time for performing these diagnostic tests is between 16 and 18 weeks of gestation, before AFP concentrations normally diminish and in sufficient time to permit a therapeutic abortion. It is recommended that such diagnostic procedures and genetic counseling be considered for all mothers who have borne an affected child, and testing is offered to all pregnant women (Kirkham, Harris, and Grzybowski, 2005). In addition, elective prelabor cesarean birth may result in less motor dysfunction. Chorionic villus sampling is also a method for prenatal diagnosis of NTDs; however, it carries certain risks (skeletal limb depletion) and is not recommended before 10 weeks of gestation.
Therapeutic Management
Early surgical closure of the myelomeningocele sac through fetal surgery has been evaluated in relation to prevention of injury to the exposed spinal cord tissue and improvement of neurologic and urologic outcomes in the affected child. Initial fetal surgical success and survival rates appear to be positive; however, reports vary in relation to the success of fetal surgery in the actual reduction of urologic problems, improvement of lower leg function, and prevention of hydrocephalus in the postnatal period. The overall mortality rate from fetal surgery is 4%, and complications include oligohydramnios, preterm delivery, and a smaller birth weight (Kaufman, 2004). The Management of Myelomeningocele Study, a clinical trial supported by the National Institutes of Health, is currently investigating the outcome of fetal surgery.
Management of the child who has a myelomeningocele requires a multidisciplinary approach involving the specialties of neurology, neurosurgery, pediatrics, urology, orthopedics, rehabilitation, physical therapy, and social services, along with intensive nursing care in a variety of specialty areas. The collaborative efforts of these specialists are focused on (1) the myelomeningocele and the problems associated with the defect—hydrocephalus, paralysis, orthopedic deformities, and genitourinary abnormalities; (2) possible acquired problems that may or may not be associated, such as meningitis, hypoxia, and hemorrhage; and (3) other conditions, such as cardiac or gastrointestinal malformations.
Infancy.: Initial care of the newborn involves preventing infection; performing a neurologic assessment, including observing for associated anomalies; and dealing with the impact of the anomaly on the family. Although meningoceles are repaired early, especially if there is danger of rupture of the sac, the philosophy regarding skin closure of myelomeningocele varies. Most authorities believe that early closure, within the first 24 to 72 hours, offers the most favorable outcome. Early closure, preferably in the first 12 to 18 hours, not only prevents local infection and trauma to the exposed tissues, but also avoids stretching of other nerve roots (which may occur as the meningeal sac expands during the first hours after birth), thus preventing further motor impairment. Broad-spectrum antibiotics are initiated, and neurotoxic substances such as povidone-iodine are avoided at the malformation.
Associated problems are assessed and managed by appropriate surgical and supportive measures. Shunt procedures provide relief from imminent or progressive hydrocephalus (see Chapter 28). Meningitis, urinary tract infection, and ventriculitis are treated with vigorous antibiotic therapy and supportive measures. Surgical intervention for Chiari malformation (a downward herniation of the brain into the brainstem) or for tethered cord (scar tissue binding the spinal cord) is indicated only when the child is symptomatic.
Improved surgical techniques do not alter the major physical disability, spinal defect, or chronic urinary tract infections that affect the quality of life for these children. Superimposed on the physical problems are the effects that the disorder has on family life and finances, including the need for long-term specialized school and health care services.
Orthopedic Considerations.: According to most orthopedists, musculoskeletal problems that will affect later locomotion should be evaluated early, and treatment, where indicated, should be instituted without delay. Neurologic assessment will determine the neurosegmental level of the lesion and enable recognition of spasticity and progressive paralysis, potential for deformity, and functional expectations. Orthopedic management includes prevention of joint contractures, correction of the existing deformity, prevention or minimization of the effects of motor and sensory deficits, prevention of skin breakdown, and acquisition of the best possible function of affected lower extremities. Common orthopedic problems requiring attention in SB include deformities of the knees, hips, feet, and spine; fractures and insensate skin further complicate orthopedic care. Other problems that may occur later include kyphosis and scoliosis (Brown, 2001). Because children with this condition often have decreased sensitivity in lower extremities, preventive skin care is important. A high percentage (60%) of children seen in a wound clinic for skin breakdown had myelomeningocele (Samaniego, 2003). The status of the neurologic deficit remains the most important factor in determining the child’s ultimate functional abilities.
With technologic advances, a variety of lightweight orthoses, including braces, special “walking” devices, and custom-built wheelchairs, are available to provide mobility to children with spinal cord lesions (see also Chapter 19). Early in infancy, intervention with passive range-of-motion exercises, positioning, and stretching exercises may help decrease the incidence of muscle contractures (Brown, 2001). Corrective surgical procedures, when indicated, are best initiated at an early age so that the child will not lag significantly behind age-mates in developmental progress. Where there is little hope for lower extremity functioning, surgery is seldom recommended unless it will improve sitting position in a wheelchair and function for ADLs and mobility.
Management of Genitourinary Function.: Myelomeningocele is one of the most common causes of neuropathic (neurogenic) bladder dysfunction among children. In infants the goal of treatment is to preserve renal function. In older children the goal is to preserve renal function and achieve optimal urinary continence. Urinary incontinence is a chronic, often debilitating problem for the child. In addition, the neuropathic bladder may produce urinary system distress, characterized by symptomatic urinary tract infections, ureterohydronephrosis, and vesicoureteral reflux or renal insufficiency. The characteristics of bladder dysfunction in children vary according to the level of the neurologic lesion and the influence of bony growth and development on the spine. Therefore ongoing urologic monitoring is essential. Evidence is growing that early intervention, based on evaluation during the neonatal period and before complications occur, serves to improve bladder function, reduces the risk of subsequent urinary system distress, and decreases the need for reconstructive surgery of the lower urinary tract (Snodgrass and Adams, 2004; Tarcan, Onol, Ilker, and others, 2006).
Treatment of renal problems includes (1) regular urologic care with prompt and vigorous treatment of infections; (2) a method of regular emptying of the bladder, such as clean intermittent catheterization (CIC) taught to and performed by parents and self-catheterization taught to children; (3) medications to improve bladder storage and continence, such as oxybutynin chloride (Ditropan) and tolterodine (Detrol); and (4) surgical procedures such as vesicostomy (bladder surgically brought out to the abdominal wall, allowing continuous urinary drainage) and augmentation enterocystoplasty (using a segment of bowel or stomach to increase bladder capacity, thereby reducing high bladder pressures).
However, despite the combined efforts of CIC, medication, and surgical intervention, some children with myelodysplasia may continue to experience debilitating urinary incontinence. Many of these children are able to attain social continence with a continent urinary diversion commonly referred to as a Mitrofanoff procedure. In this procedure, a catheterizable channel is surgically created from appendix, ureter, or tapered bowel. The proximal end of the channel is connected to the bladder with the distal end brought out as a small stoma on the abdominal wall, usually near the umbilicus. The bladder neck may be sutured to prevent urinary leakage from the urethra. CIC through the easily accessible abdominal route fosters greater independence in children, especially in those unable to transfer from wheelchair to toilet to perform CIC.
Bowel Control.: Some degree of fecal continence can be achieved in most children with myelomeningocele with diet modification, regular toilet habits, and prevention of constipation and impaction. It is frequently a lengthy process. Dietary fiber supplements (recommended 10 g/day), laxatives, suppositories, or enemas aid in producing regular evacuation. Older children and adolescents seeking more independence may attain bowel continence and higher quality of life after undergoing an antegrade continence enema (ACE) procedure (Doolin, 2006). In a procedure similar to the Mitrofanoff, the appendix or ileum is used to create a catheterizable channel with attachment of the proximal end to the colon. The distal end of the channel exits through a small abdominal stoma. Every 1 or 2 days, a catheter is passed through the stoma, allowing enema solution to be instilled directly into the colon. After administration of the enema solution, the child sits on the toilet for 30 to 60 minutes as stool is flushed out through the rectum. Frequency of enemas and volume of solution used to completely evacuate the bowel vary among individuals.
Prognosis.: The early prognosis for the child with myelomeningocele depends on the neurologic deficit present at birth, including motor ability, bladder innervation, and associated neurologic anomalies. Early surgical repair of the spinal defect, antibiotic therapy to reduce the incidence of meningitis and ventriculitis, prevention of urinary system dysfunction, and early detection and correction of hydrocephalus have significantly increased the survival rate and quality of life in such children. Many children with SB achieve partial independent living and gainful employment. Reports of survival rates vary, and many include adults who were born before medical advances and surgical techniques seen in the past 25 years. Researchers have noted that as adolescents with SB transition to young adulthood, they have increased difficulty obtaining centralized health care for the different health problems associated with SB (Lazzaretti and Pearson, 2004). This chronic condition has an array of associated complications, including hydrocephalus and shunt malfunctions, scoliosis, bowel and bladder management issues, latex allergy, and epilepsy. However, based on current medical knowledge and ethical considerations, aggressive, early management is favored for the child with myelomeningocele.
Prevention.: The widespread use of folic acid among women of childbearing age is expected to significantly decrease the incidence of SB. It has been estimated that a daily intake of 0.4 mg of folic acid in women of childbearing age will prevent 50% to 70% of all cases of NTDs (Centers for Disease Control and Prevention, 2004). Preliminary data show a 24% decrease in cases of SB between 1996 and 2001 (Matthews, Honein, and Erickson, 2002). Although folic acid intake increased in 1999 to 2000 (compared with intake from 1988 to 1994), recent data indicate that serum folate concentrations among women of childbearing age decreased 16% from 2003 to 2004 in all ethnic groups studied. Lowest serum folate levels were seen in non-Hispanic whites in 2003 to 2004; however, overall serum folate levels remained below recommended levels in non-Hispanic blacks during all three periods studied (Centers for Disease Control and Prevention, 2007). These results indicate that nurses and other health care workers have an important task in disseminating information that may decrease the incidence of birth defects in children by promoting maternal consumption of folic acid.*
For women who have had a previous pregnancy affected by NTDs, folic acid intake is increased to 4 mg/day under supervision of a practitioner beginning 1 month before a planned pregnancy and continuing during the first trimester. Supplementation of 4 mg of folate should not be given in multivitamin preparations because of the risk of overdose of other vitamins.
To ensure adequate daily intake of the recommended amount of folic acid, women must take a folic acid supplement, eat a fortified breakfast cereal containing 100% of the recommended dietary allowance (RDA) of folic acid (e.g., Kellogg’s Product 19, General Mills Total, Multigrain Cheerios Plus), or increase their consumption of other fortified foods (cereal, bread, rice, grits, pasta) and foods naturally rich in folate (green leafy vegetables and citrus fruits). The only population in which folic acid has not proved to be effective in decreasing the incidence of NTDs is epileptic women taking antiepileptic medications during pregnancy.
Nursing Care Management
At birth an examination is performed to assess the intactness of the membranous cyst. During transport to the nursery, every effort is made to prevent trauma to this protective covering. In addition to the routine assessment of the newborn (see Chapter 8), the infant is assessed for the level of neurologic involvement. Movement of extremities or skin response, especially an anal reflex, that might provide clues to the degree of motor or sensory impairment is noted. It is important to observe the infant’s behavior in conjunction with the stimulus, since limb movements can be induced in response to spinal cord reflex activity that has no connection with the higher centers. Observation of urinary output, especially if a diaper remains dry, may indicate urinary retention. Abdominal assessment revealing bladder distention, even with a wet diaper, may indicate urinary overflow in a retentive bladder. The head circumference is measured daily (see Chapter 6), and the fontanels are examined for signs of tension or bulging.
Care of the Myelomeningocele Sac.: The infant is usually placed in an incubator or warmer so that temperature can be maintained without clothing or covers that might irritate the spinal lesion. When an overhead warmer is used, the dressings over the defect require more frequent moistening because of the dehydrating effect of the radiant heat.
Before surgical closure the myelomeningocele is prevented from drying by the application of a sterile, moist, nonadherent dressing over the defect. The moistening solution is usually sterile normal saline. Dressings are changed frequently (every 2 to 4 hours), and the sac is closely inspected for leaks, abrasions, irritation, or any signs of infection. The sac must be carefully cleansed if it becomes soiled or contaminated. Sometimes the sac ruptures during delivery or transport, and any opening in the sac greatly increases the risk of infection to the central nervous system.
One of the most important and challenging aspects in the early care of the infant with myelomeningocele is positioning. Before surgery the infant is kept in the prone position to minimize tension on the sac and the risk of trauma. The prone position allows for optimal positioning of the legs, especially in cases of associated hip dysplasia. The infant is placed prone with the hips slightly flexed and supported to reduce tension on the defect. The legs are maintained in abduction with a pad between the knees to counteract hip subluxation, and a small roll is placed under the ankles to maintain a neutral foot position. A variety of aids, including diaper rolls, pads, small foam pads, or specially designed frames and appliances, can be used to maintain the desired position.
Prevent Complications.: The prone position affects other aspects of the infant’s care. For example, in this position the infant is more difficult to keep clean, pressure areas are a constant threat, and feeding becomes a problem. The infant’s head is turned to one side for feeding. Fortunately, most defects are repaired early, and the infant can be held for feeding soon after surgery. Special care must be taken to avoid pressure on the operative site.
Diapering the infant may be contraindicated until the defect has been repaired and healing is well advanced or epithelialization has taken place. The padding beneath the diaper area is changed as needed to keep the skin dry and free of irritation. When urinary retention is detected, CIC is employed. Because the bowel sphincter is frequently affected, there is continual passage of stool, often misinterpreted as diarrhea, which is a constant irritant to the skin and a source of infection to the spinal lesion.
Areas of sensory and motor impairment are subject to skin breakdown and therefore require meticulous care. Placing the infant on a special mattress or mattress overlay reduces pressure on the knees and ankles. Periodic cleansing, application of lotion, and gentle massage aid circulation.
Gentle range-of-motion exercises are carried out to prevent contractures, and stretching of contractures is performed when indicated. However, these exercises may be restricted to the foot, ankle, and knee joint. When the hip joints are unstable, stretching against tight hip flexors or adductor muscles, which act much like bowstrings, may aggravate a tendency toward subluxation. Consultation with a physical therapist is an important aspect of the short- and long-term management of infants with myelomeningocele.
Infants with unrepaired myelomeningocele may be held in the arms and cuddled as unaffected infants are, so their need for tactile stimulation is met by caressing, stroking, and other comfort measures. Individualized developmental care with age-appropriate stimulation is provided (see Developmental Outcome, Chapter 9).
Provide Postoperative Care.: Postoperative care of the infant with myelomeningocele involves the same basic care as that of any postsurgical infant: monitoring vital signs, monitoring intake and output, providing nourishment, observing for signs of infection, and managing pain as needed. Care of the operative site is carried out under the direction of the surgeon and includes close observation for signs of leakage of cerebrospinal fluid (CSF). General care is continued as preoperatively.
The prone position is maintained after surgical closure, although many neurosurgeons allow a side-lying or partial side-lying position unless it aggravates a coexisting hip dysplasia or permits undesirable hip flexion. This offers an opportunity for position changes, which reduces the risk of pressure sores and facilitates feeding. If permitted, the infant can be held upright against the body, with care taken to avoid pressure on the operative site. After the effects of anesthesia have subsided and the infant is alert, feedings may be resumed unless there are other anomalies or associated complications.
Support Family and Educate About Home Care.: As soon as the parents are able to cope with the infant’s condition, they are encouraged to become involved in care. They need to learn how to continue at home the care that has been initiated in the hospital—positioning, feeding, skin care, and range-of-motion exercises when appropriate. They are taught CIC technique when it is prescribed. Parents also need to know the signs of complications (urinary, neurologic, orthopedic) and how to obtain assistance when needed.
The mother who wishes to breastfeed the infant is encouraged to do so, since this will be beneficial. Shortly after delivery the mother is started on a program of pumping to initiate and maintain milk supply until the infant is stable enough to begin breastfeeding (Hurtekant and Spatz, 2007). This process may require considerable support from nurses, physicians, and family members because of separation from the infant for surgical care and recovery.
The long-range planning with and support of the parents and newborn begin in the hospital and continue throughout childhood and even into young adulthood. The life expectancy of children with SB extends well into adulthood; therefore planning should involve long-term goals and plans for optimum function as an adult. Discussion about aspects of adulthood such as receiving educational or vocational training and education, living independently, having a mate, having sexual relationships, and bearing and rearing children is important and should not be overlooked. The unique service needs of adolescents with SB as they attempt to gain independence from family and establish a life of their own have not been adequately addressed in the literature (Buran, McDaniel, and Brei, 2002). Nurses assume an important role as a central member of the health team. As a care manager and coordinator, the nurse reviews information with the family, takes responsibility for family teaching, and acts as a liaison between inpatient and outpatient services. The child will need numerous hospitalizations over the years, and each one will be a source of stress to which the younger child is especially vulnerable (see Chapter 18 for a discussion of care of the child with a disability).
Habilitation involves not only solving problems of self-help and locomotion, but also solving the most distressing problem of urinary or bowel incontinence, which threatens the child’s social acceptability. Assistance in preparing the child and the school regarding the child’s special needs helps provide a better initial adjustment to this broader social experience. The Spina Bifida Association of America* is organized to provide services and support for families of children with spinal lesions.
Latex Allergy
Latex allergy was identified as a serious health hazard when a report linked intraoperative anaphylaxis with latex in children with SB. The high prevalence of latex allergy (up to 80%) in children with SB has been attributed to the repeated exposure to latex products during surgery and numerous bladder catheterizations and possible disease-associated factors. Some evidence suggests that children with SB are at increased risk for latex allergy as a result of the disease itself rather than repeated latex exposures (Eiwegger, Dehlink, Schwindt, and others, 2006). Allergic reactions range from urticaria, wheezing, watery eyes, and rashes to anaphylactic shock. More severe reactions tend to occur when latex comes in contact with mucous membranes, wet skin, the bloodstream, or an airway. There also can be cross-reactions to a number of foods (e.g., banana, avocado, kiwi, chestnut). Latex allergy has been diagnosed in infants; symptoms manifested include wheezing, facial swelling, facial rash, and anaphylaxis (Kimata, 2004). In addition to patients with SB, high-risk populations include patients with urogenital anomalies or multiple surgeries and health care workers (see Box 32-6 for medical conditions associated with SB).
BOX 32-6 Medical Conditions Associated with Risk of Latex Allergy
Does your child have any symptoms, such as sneezing, coughing, rashes, or wheezing, when handling rubber products (e.g., balloons, tennis or Koosh balls, adhesive bandage strips) or when in contact with rubber hospital products (e.g., gloves, catheters)?
Has your child ever had an allergic reaction during surgery?
Does your child have a history of rashes; asthma; or allergic reactions to medication or foods, especially milk, kiwi, bananas, or chestnuts?
How would you identify or recognize an allergic reaction in your child?
What would you do if an allergic reaction occurred?
Has anyone ever discussed latex or rubber allergy or sensitivity with you?
Has your child had any allergy testing?
When did your child last come in contact with any type of rubber product? Were you present?
Modified from Romanczuk A: Latex use with infants and children: it can cause problems, MCN 18(4):208-212, 1993.
The most important goals are prevention of latex allergy and identification of children with a known hypersensitivity (see Nursing Care Guidelines box). High-risk and latex-allergic individuals must be managed in a latex-free environment. Care must be taken so that they do not come in direct or secondary contact with products or equipment containing latex at any time during medical treatment. Allergy testing has been used to identify latex allergy with varying success. Skin prick testing and provocation testing carry the risk of allergic reaction or anaphylaxis. The radioallergosorbent test (RAST) has been used to measure the serum level of latex-specific immunoglobulin E. The RAST has been shown to be 90% to 95% sensitive (Kellett, 1997). Pretreatment with an antihistamine and steroids (dexamethasone) before and after surgery to reduce the possibility of a serious reaction remains controversial, since it may interfere with healing.
Because children who have SB are prone to develop an allergy to latex, reducing exposure, from birth on, may decrease the chance of allergy development. Many health care facilities are establishing latex-safe environments when patients and health care workers are at risk. In the health care arena it is important to use products with the lowest potential risk of sensitizing patients and staff members.
Lists have been published of products, such as vinyl gloves, that may be substituted for latex (see footnote to Box 32-7). The U.S. Food and Drug Administration has proposed user labeling for latex-containing devices that come into contact, directly or indirectly, with live human tissue.*
The identification of those sensitive to latex is best accomplished through careful screening of all patients (see Nursing Care Guidelines box for questions related to latex allergy). Children with latex allergy should carry or wear some form of medical identification; those who have had serious reactions should also carry an injectable epinephrine pen and a pair of latex-free gloves for emergencies. Education programs regarding latex hypersensitivity are aimed at those who care for high-risk groups, such as children with SB, and may include relatives, school nurses, teachers, childcare workers, and baby-sitters. In addition to educating caregivers about the child’s exposure to medical products that contain latex, nurses need to inform them of common nonmedical latex objects (Box 32-7). Parents should also be given literature explaining signs and symptoms of latex hypersensitivity and appropriate emergency treatment (see Anaphylax, Chapter 25).
SPINAL MUSCULAR ATROPHY, TYPE 1 (WERDNIG-HOFFMANN DISEASE)
Spinal muscular atrophy (SMA) type 1 (Werdnig-Hoffmann disease) is a disorder characterized by progressive weakness and wasting of skeletal muscles caused by degeneration of anterior horn cells. It is inherited as an autosomal recessive trait and is the most common paralytic form of the floppy infant syndrome (congenital hypotonia). The sites of the pathologic condition are the anterior horn cells of the spinal cord and the motor nuclei of the brainstem, but the primary effect is atrophy of skeletal muscles. The age of onset is variable, but the earlier the onset, the more disseminated and severe the motor weakness. The disorder may be manifested early—often at birth—and almost always before 2 years of age; death may occur as a result of respiratory failure by age 2 years (Iannaccone and Burghes, 2002). The manifestations (Box 32-8) and prognosis are categorized according to the age of onset, severity of weakness, and clinical course; some children may fluctuate between exhibiting symptoms of types 1 and 2, or types 2 and 3, in regard to clinical function (Sarnat, 2007). A severe rare fetal form of SMA, classified as type 0, is reported to be quite lethal in the perinatal period (Sarnat, 2007).
BOX 32-8 Clinical Manifestations of Spinal Muscular Atrophy (SMA)
TYPE 1 (WERDNIG-HOFFMANN DISEASE)
Clinical manifestations within first few weeks or months of life
Inactivity the most prominent feature
Infant lying in a frog-leg position with legs externally rotated, abducted, and flexed at knees
Limited movements of shoulder and arm muscles
Active movement usually limited to fingers and toes
Diaphragmatic breathing with sternal retractions (diaphragmatic paralysis may occur)
Abnormal tongue movements (at rest)
Tiring quickly during feedings (if breastfed, may lose weight before noticeable)
Normal sensation and intellect
Onset before age 18 months
Early—Weakness confined to arms and legs
Legs usually involved to greater extent than arms
Movements absent during complete relaxation or sleep
Some infants able to sit if placed in position, but few can ambulate
Life span from 7 months to 7 years, although many have normal life expectancy
TYPE 3 (KUGELBERG-WELANDER SYNDROME; MILD SMA)
Onset of symptoms after 18 months of age
Normal head control and ability to sit unassisted by 6 to 8 months of age
note: These classifications are general, but some research suggests there may be variations in life span and other characteristics (Iannaccone and Burghes, 2002; Russman, Iannaccone, Buncher, and others, 1992; Russman, 1996).
Diagnostic Evaluation and Therapeutic Management
The diagnosis is based on the molecular genetic marker for the SMN (survival motor neuron) gene, which is located on chromosome 5q13. Prenatal diagnosis may be made by genetic analysis of circulating fetal cells in maternal blood (Beroud, Karliova, Bonnefont, and others, 2003) or circulating fetal cells in amniotic fluid. The risk of subsequent affected offspring in carriers of the mutant gene or in families with known cases of SMA may also be evaluated genetically. Further diagnostic studies include muscle electromyography (EMG), which demonstrates a denervation pattern, and muscle biopsy; however, the genetic analysis has become the gold standard for diagnosis of the condition.
There is no cure for the disease, and treatment is symptomatic and preventive, primarily preventing joint contractures and treating orthopedic problems, the most serious of which is scoliosis. Hip subluxation and dislocation may also occur. Many children benefit from powered chairs, lifts, special pressure-adjustable mattresses, and accessible environmental controls. Muscle and joint contractures require careful attention and care to prevent further complications. Nutritional growth failure may occur in infants and toddlers as a result of poor feeding; supplemental gastrostomy feedings may be required to maintain adequate nutritional status and maintain weight gain. The use of lower extremity orthoses may assist with ambulation, but eventually the child may be confined to a wheelchair as muscle atrophy progresses. Sleep-disordered breathing is common in children with SMA and often requires nocturnal mechanical ventilation. Noninvasive ventilation methods such as bilevel positive airway pressure (BiPAP) have decreased the morbidity and increased the survival rate of children with SMA types 1 and 2. Upper respiratory tract infections may occur and are treated with antibiotic therapy.
Prognosis.: Prognosis varies according to age of onset or group as described in Box 32-8. Individuals with SMA type 1 may succumb to respiratory infections or failure between 1 and 24 months of age (Iannaccone and Burghes, 2002); however, some may live into their third or fourth decade of life. A significant number of infants with SMA require a tracheotomy, and associated medical conditions in survivors include gastroesophageal reflux, scoliosis, early onset puberty, hip dysplasia, and recurrent oral candidiasis (Bach, 2007).
Nursing Care Management
The infant or small child with progressive muscle weakness requires nursing care similar to that of the immobilized patient (see Chapter 31). However, the underlying goal of treatment should be to assist the child and family in dealing with the illness while progressing toward a life of normalization within the child’s capabilities. Special attention should be directed to preventing muscle and joint contractures, promoting independence in performance of ADLs, and becoming incorporated into the mainstream of school when possible. In addition, parents need support and resources to be able to provide for the child and remain an intact family. Because children with neuromuscular disease have abnormal breathing patterns that often contribute to early death, it is important to assess adequate oxygenation, especially during the sleep phase when shallow breathing occurs and hypoxemia may develop. Home pulse oximetry may be used to assess the child during sleep and provide noninvasive ventilation as necessary (Young, Lowe, Fitzgerald, and others, 2007; Bush, Fraser, Jardine, and others, 2005) (see Duchenne [Pseudohypertrophic] Muscular Dystrophy, below, for respiratory management). Supportive care also includes management of orthoses and other orthopedic equipment as required. Because children with SMA are intellectually normal, verbal, tactile, and auditory stimulation are important aspects of developmental care. Supporting them so that they can see the activities around them and transporting them in appropriate conveyances (e.g., wagon, power wheelchair) for a change of environment provide stimulation and a broader scope of contacts.
Children who are able to sit require proper support and attention to alignment to prevent deformities and other complications. Children who survive beyond infancy will need attention to educational needs and opportunities for social interaction with other children. The parents of a child who is chronically ill require much support and encouragement* (see Chapter 18). Parents who have not sought genetic counseling should be encouraged to do so to evaluate further risk potential.
SPINAL MUSCULAR ATROPHY, TYPE 3 (KUGELBERG-WELANDER DISEASE)
SMA type 3 (Kugelberg-Welander disease) is a result of anterior horn cell and motor nerve degeneration. The disease is characterized by a pattern of muscular weakness similar to that of infantile SMA (see Box 32-8). Several modes of inheritance have been reported for the disease: autosomal recessive, autosomal dominant, and X-linked recessive.
The onset occurs from younger than 1 year of age into adulthood, with symptoms resembling group 3 infantile SMA. Proximal muscle weakness (especially of the lower limbs) and muscular atrophy are the predominant features. The disease runs a slowly progressive course. Some children lose the ability to walk 8 to 9 years after the onset of symptoms, but many can still walk after 30 years or more. Many affected persons have a normal life expectancy (Iannaccone, 1998).
Therapeutic Management and Nursing Care Management
The management is primarily symptomatic and supportive and is related to maintaining mobility as long as possible, preventing complications such as skin breakdown, optimizing and maintaining respiratory function, and providing support to the child and family.
MUSCULAR DYSTROPHIES
Muscular dystrophies (MDs) constitute the largest and most important single group of muscle diseases of childhood. The MDs have a genetic origin in which there is gradual degeneration of muscle fibers, and they are characterized by progressive weakness and wasting of symmetric groups of skeletal muscles, with increasing disability and deformity. In all forms of MD there is insidious loss of strength, but each type differs in regard to muscle groups affected (Fig. 32-7), age of onset, rate of progression, and inheritance pattern. The most common form, Duchenne muscular dystrophy (DMD), is considered separately in the next section.
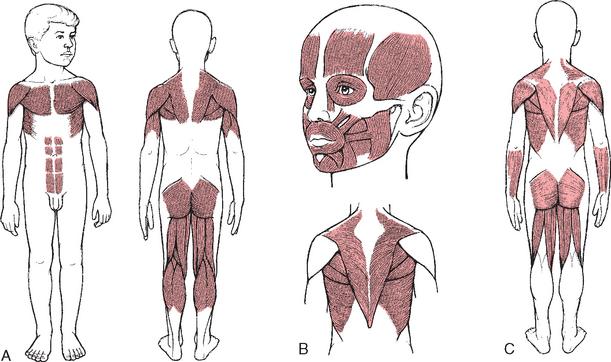
FIG. 32-7 Initial muscle groups involved in muscular dystrophies. A, Pseudohypertrophic. B, Fascioscapulohumeral. C, Limb girdle.
Facioscapulohumeral (Landouzy-Dejerine) muscular dystrophy is inherited as an autosomal dominant disorder with onset in early adolescence. It is characterized by difficulty in raising the arms over the head, lack of facial mobility, and a forward slope of the shoulders. The progression is slow, and the life span is usually unaffected.
Limb-girdle muscular dystrophy is an autosomal recessive disease of later childhood, adolescence, or early adulthood with variable but usually slow progression. It is characterized by weakness of proximal muscles of the pelvic and shoulder girdles.
Treatment of the MDs consists mainly of supportive measures, including physical therapy, orthopedic procedures to minimize deformity, ventilation support, and assistance for the affected child in meeting the demands of daily living.
DUCHENNE (PSEUDOHYPERTROPHIC) MUSCULAR DYSTROPHY
DMD is the most severe and the most common MD of childhood. It is inherited as an X-linked recessive trait, and the single-gene defect is located on the short arm of the X chromosome. DMD has a reportedly high mutation rate, with a positive family history in 65% of all cases (Thompson and Berenson, 2001); therefore genetic counseling is an important aspect of the care of the family.
As in all X-linked disorders, males are affected almost exclusively. At the genetic level, both DMD and Becker MD (a milder variant) result from mutations of the gene that encodes dystrophin, a protein product in skeletal muscle. Dystrophin is absent from the muscle of children with DMD and is reduced or abnormal in children with Becker MD. Children with Becker MD have a later onset of symptoms, which are usually not as severe as those seen in DMD. The incidence is approximately 1 in 3600 male births for the Duchenne form and approximately 1 in 30,000 live births for the Becker type (Sarnat, 2007; Thompson and Berenson, 2001). Box 32-9 describes the characteristics of DMD.
Most children with DMD reach the appropriate developmental milestones early in life, although they may have mild, subtle delays. Evidence of muscle weakness usually appears during the third to seventh year, although there may have been a history of delay in motor development, particularly walking. Difficulties in running, riding a bicycle, and climbing stairs are usually the first symptoms noted. Later, abnormal gait on a level surface becomes apparent. In the early years, rapid developmental gains may mask the progression of the disease. Questioning the parents may reveal that the child has difficulty in rising from a sitting or supine position. Typically, affected boys have a waddling gait and lordosis, fall frequently, and develop a characteristic manner of rising from a squatting or sitting position on the floor (Gower sign; Fig 32-8). Parents may also notice the child has enlarged calves (Box 32-10).
BOX 32-10 Clinical Manifestations of Duchenne Muscular Dystrophy
Relentless progression of muscle weakness; possible death from respiratory or cardiac failure
Gower sign (child turns onto side or abdomen, flexes knees to assume a kneeling position, then with knees extended gradually pushes torso to an upright position by “walking” the hands up the legs)
Enlarged (hypertrophied) muscles (especially calves, thighs, and upper arms); feel unusually firm or woody on palpation
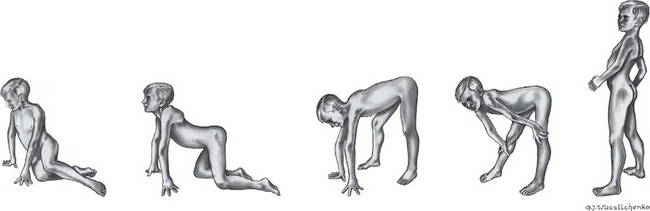
FIG. 32-8 Child with Duchenne muscular dystrophy attains standing posture by kneeling, then gradually pushing his torso upright (with knees straight) by “walking” his hands up his legs (Gower sign). Note marked lordosis in upright position.
Pseudohypertrophy is a term applied to muscular enlargement caused by fatty infiltration. Profound muscular atrophy occurs in later stages, and as the disease progresses, contractures and deformities involving large and small joints are common complications. Ambulation usually becomes impossible by 12 years of age. Facial, oropharyngeal, and respiratory muscles are often spared until the terminal stages of the disease. Ultimately the disease process involves the diaphragm and auxiliary muscles of respiration, and cardiovascular involvement (cardiomyopathy, dysrhythmias, and heart failure) is common. Mild mental delay is common in roughly 30% of all individuals with MD, and many will have permanent learning disabilities; however, children with DMD should be transitioned into early learning programs and eventually into regular classrooms as much as possible. The eventual cause of death is usually respiratory tract infection or cardiac failure; however, much progress has been made in providing ventilatory methods to prolong and maintain quality of life.
Diagnostic Evaluation
The diagnosis of DMD is primarily established by blood polymerase chain reaction (PCR) for the dystrophin gene mutation (Sarnat, 2007). Prenatal diagnosis is also possible as early as 12 weeks of gestation. Serum enzyme measurement, muscle biopsy, and EMG may also be used in establishing the diagnosis. Serum creatine kinase levels are extremely high in the first 2 years of life, before the onset of clinical weakness. If the child demonstrates the usual characteristics, has a positive family history for DMD, and the PCR is positive, the muscle biopsy may be deferred.
Therapeutic Management
No effective treatment exists for childhood MD. The use of the corticosteroids prednisone and deflazacort has been evaluated as a treatment for DMD. Several clinical trials demonstrated increased muscle strength and improved performance and pulmonary function, with significant decrease in the progression of weakness, when prednisone was administered for 6 months. The American Academy of Neurology has published a practice parameter for the administration of corticosteroids in the treatment of DMD (Moxley, Ashwal, Pandya, and others, 2005). Major side effects in these studies included weight gain and a cushingoid facial appearance. Deflazacort caused less weight gain than prednisone and was effective in decreasing muscle wasting in some children (Angelini, 2007). Deflazacort is not available in the United States but is in Canada.
Maintaining optimal function in all muscles for as long as possible is the primary goal; secondary is the prevention of contractures. Children with DMD who remain as active as possible are able to avoid wheelchair confinement for a longer time. Maintenance of function often involves stretching exercises, strength and muscle training, breathing exercises to increase and maintain vital lung capacity, range-of-motion exercises, surgery to release contracture deformities, bracing, and performance of ADLs.
Parents should always be involved in making decisions about the child’s care, and teaching regarding home safety and prevention of falls is important as well. Parents should also be encouraged to have the child keep follow-up appointments for medical care and physical and occupational therapy. Because respiratory tract infections are most troublesome in these children, influenza and pneumococcal vaccines are encouraged, and contact with persons with respiratory tract infections should be avoided.
Eventually respiratory and cardiac problems become the central focus of the debilitating illness. Children with neuromuscular disease have abnormal breathing patterns, particularly during rapid-eye-movement sleep, and hypoxia may occur as a result of inadequate oxygenation (Birnkrant, 2002). The child and parents should be involved in a discussion of long-term ventilation options. Cardiac and respiratory assessment during wake-sleep cycles is imperative. Respiratory care for children with neuromuscular conditions such as SMA and DMD may involve the use of noninvasive ventilation with BiPAP on a temporary or full-time basis, mechanically assisted coughing (MAC), or tracheotomy and relief of airway obstruction with coughing and suctioning devices; the tracheotomy, however, is associated with more complications (Simonds, 2006; Young, Lowe, Fitzgerald, and others, 2007). Home pulse oximetry may be used to monitor oxygenation during sleep or to aid in decision making regarding the use of MAC to clear the airways.
Several devices are available for children with neuromuscular disease to assist in clearing the airway when the cough reflex is ineffective or diminished. The mechanical cough in-exsufflator (MIE) has been evaluated and found to be safe and effective in the daily management of respiratory function (Miske, Hickey, Kolb, and others, 2004). The MIE delivers positive inspiratory pressures at a set rate, followed by negative pressure exsufflation coordinated with the patient’s own breathing rhythm; the exsufflation is designed to mimic a cough reflex so mucus can be effectively cleared. Airway suctioning after exsufflation is accomplished as necessary to clear the airways. In children the MIE may be connected directly to a tracheostomy or used with a mouthpiece or face mask.
Survival in individuals with DMD may be prolonged several years with the use of noninvasive ventilation and MAC as alternatives to tracheotomy and airway suctioning (Simonds, 2006). The American Thoracic Society (2004) has published extensive guidelines for respiratory monitoring and care of children and adults with DMD.
The American Academy of Pediatrics (2005) recommends an extensive cardiac evaluation of the child diagnosed with either DMD or Becker MD. Patients with neuromuscular conditions may not be seen with the typical signs and symptoms of cardiac dysfunction. Therefore symptoms such as weight loss, nausea and vomiting, cough, increased fatigue on performance of ADLs, and orthopnea should be carefully evaluated to detect early signs of cardiomyopathy.
Genetic counseling is also recommended for parents, female siblings, and maternal aunts and their female offspring. Long-term care, end-of-life directives, and palliative care options are issues that must be discussed with the child and family affected by MD. Professional counseling may be required to allow frank discussion of these issues in some cases, and referrals should be made as appropriate.
Research evaluating a number of treatments for DMD is in progress. These include clinical trials with glutamine and creatine monohydrate to preserve muscle strength; utrophin, a protein that is similar to dystrophin and in large quantities may counteract the effects of the deficiency of dystrophin (Chakkalakal, Thompson, Parks, and others, 2005); and the enzyme CT GalNAc transferase, which blocks muscle wasting in mice (Metules, 2002).
Nursing Care Management
The major emphasis of nursing care is to help the child and family cope with a chronic, progressive, incapacitating disease; to help design a program that will afford maximal independence and reduce the predictable and preventable disabilities associated with the disorder; and to help the child and family deal constructively with the limitations the disease imposes on their daily lives. Because of advances in technology, children with MD may live into early adulthood; therefore the goals of care should also involve decisions regarding quality of life, achievement of independence, and transition to adulthood.
Working closely with other team members, nurses assist the family in developing the child’s self-help skills to give the child the satisfaction of being as independent as possible for as long as possible. This requires continual evaluation of the child’s capabilities, which are often difficult to assess. Fortunately, most children with MD instinctively recognize the need to become as independent as possible and strive to do so.
Practical difficulties faced by families are physical limitations of housing and mobility. Some families live in houses or apartments that are unsuited to wheelchairs. Transportation may also be a barrier for families of children with MD. Assisting with these challenges requires team problem solving. Diet, nutritional needs, and nutrition modification are discussed according to the needs of the individual child and family.
The parents’ social activities are also restricted, and the family’s activities must be continually modified to accommodate the needs of the affected child. When the child becomes increasingly incapacitated, the family may consider home-based care, an assisted living facility, or respite care. Unless the child is severely incapacitated, he should also be involved in the decisions regarding such care. Nurses can assist with decision making by exploring all available options and resources and support the child and family in the decision. Older boys with MD may also need psychiatric or psychologic counseling to deal with issues such as depression, anger, and quality of life. Parents also need to be encouraged to become involved in support groups because there is evidence that adequate social support from family, community, and other parents is crucial to appropriate coping in families with children with chronic illness.
Regardless of how successful the program or how well the family adapts to the disorder, superimposed on the physical and emotional problems associated with a child with a long-term disability is the constant knowledge of the ultimate outcome of the disease. All the manifestations seen in the child with a chronic fatal illness are encountered in these families (see Chapter 18).
Nurses are especially valuable health professionals as they come to know the family and the family’s problems. Nurses can be alert to the family’s problems and needs and make necessary referrals when supplementary services are indicated. The Muscular Dystrophy Association—USA* has branches in most communities to assist families in which there is a member with MD.
ACQUIRED NEUROMUSCULAR DISORDERS
GUILLAIN-BARRÉ SYNDROME (INFECTIOUS POLYNEURITIS)
Guillain-Barré syndrome (GBS), also known as infectious polyneuritis, is an uncommon acute demyelinating polyneuropathy with a progressive, usually ascending flaccid paralysis. Children are less often affected than adults; among children, those between ages 4 and 10 years have higher susceptibility. The male/female ratio is reported to be 1.5:1. Two peak time periods have been identified with an increased incidence of GBS: late adolescence and young adulthood. The incidence is also increased in pregnant and postpartum women (Newswanger and Warren, 2004).
Congenital GBS is rare yet may be seen in the neonatal period and consists of hypotonia, weakness, and decreased or absent reflexes; maternal neuromuscular disease may or may not be present. Diagnosis is established by the same criteria as in older children, but the symptoms gradually subside over the first few months of life and disappear by 12 months (Sarnat, 2007).
Pathophysiology
GBS is an immune-mediated disease often associated with a number of viral or bacterial infections or the administration of certain vaccines. It has been associated with infectious mononucleosis, measles, mumps, Campylobacter jejuni (gastroenteritis), cytomegalovirus, Borrelia burgdorferi (Lyme disease), Epstein-Barr virus, Helicobacter pylori, and Mycoplasma and Pneumocystis infections. Onset of GBS symptoms usually occurs within 10 days of the primary infection. Pathologic changes in spinal and cranial nerves consist of inflammation and edema with rapid, segmented demyelination and compression of nerve roots within the dural sheath. Nerve conduction is impaired, producing ascending partial or complete paralysis of muscles innervated by the involved nerves. GBS has three phases:
1. Acute—Phase starts when symptoms begin and continues until new symptoms stop appearing or deterioration ceases; it may last as long as 4 weeks.
2. Plateau—Symptoms remain constant without further deterioration; it may last from days to weeks.
3. Recovery—Patient begins to improve and progress to optimal recovery; it usually lasts a few weeks to months depending on the deficits incurred by the illness.
Diagnostic Evaluation
Diagnosis of GBS is based on clinical manifestations (Box 32-11), CSF analysis, and EMG findings. CSF analysis reveals an increased protein concentration, and EMG shows evidence of acute muscle denervation; other laboratory studies are usually noncontributory. The symmetric nature of the paralysis helps differentiate this disorder from spinal paralytic poliomyelitis, which usually affects sporadic muscles.
BOX 32-11 Clinical Manifestations of Guillain-Barré Syndrome
Muscle tenderness
Paresthesia and cramps (sometimes)
Proximal symmetric muscle weakness
Ascending paralysis from lower extremities
Frequently involves muscles of trunk and upper extremities and those supplied by cranial nerves (especially facial)
Flaccid paralysis with loss of reflexes
May involve facial, extraocular, labial, lingual, pharyngeal, and laryngeal muscles
Therapeutic Management
Treatment of GBS is primarily supportive. In the acute phase patients are hospitalized because respiratory and pharyngeal involvement may require assisted ventilation, sometimes with a temporary tracheotomy. Treatment modalities include aggressive ventilatory support, intravenous administration of immunoglobulin (IVIG), and steroids; plasmapheresis and immunosuppressive drugs may also be used. Plasmapheresis has been shown to decrease the length of recovery in patients with severe GBS yet is expensive, and side effects include hypotension, fever, bleeding disorders, chills, urticaria, and bradycardia. Further evidence reports equal benefits to treatment of GBS with IVIG administration or plasmapheresis; both sped up recovery time in studies reviewed (Hughes and Cornblath, 2005). Corticosteroids alone did not decrease the symptoms or shorten the duration of the disease. CSF filtration has been used in patients with GBS with some success; CSF is removed, filtered, and replaced into the patient. This process presumably removes toxins and bacteria; however, further studies are needed to evaluate this treatment modality (Atkinson, Carr, Maybee, and others, 2006).
Medications that may be administered during the acute phase include a low-molecular-weight heparin to prevent deep vein thrombosis, a mild laxative or stool softener to prevent constipation, pain medication such as acetaminophen, and a histamine-antagonist to prevent stress ulcer formation. Chronic neuropathic pain following GBS may be treated with gabapentin, which is reported to be more effective than carbamazepine (Sarnat, 2007).
Course and Prognosis.: Better outcomes are associated with younger age, no requirement for mechanical respiratory assistance, slower progression of disease, normal peripheral nerve function on EMG, and treatment with either IVIG or plasmapheresis. Recovery usually begins within 2 to 3 weeks, and most patients regain full muscle strength. The recovery of muscle strength progresses in the reverse order of onset of paralysis, with lower extremity strength being the last to recover. Poor prognosis with subsequent residual effects in children is reportedly associated with cranial nerve involvement, extensive disability at time of presentation, and intubation.
Most deaths associated with GBS are caused by respiratory failure; therefore early diagnosis and access to respiratory support are especially important. The rate of recovery is usually related to the degree of involvement and may extend from a few weeks to months. The greater the degree of paralysis, the longer the recovery phase.
Nursing Care Management
Nursing care is essentially supportive and is the same as that required for the child with immobilization and respiratory depression. The emphasis of care is on close observation to assess the extent of paralysis and on prevention of complications, including autonomic dysfunction (hypertension, orthostatic hypotension, syndrome of inappropriate antidiuretic hormone secretion, life-threatening dysrhythmias), respiratory dysfunction, fear and anxiety, and pain management.
During the acute phase of GBS the child’s condition should be carefully observed for possible difficulty in swallowing and respiratory involvement. The child’s respiratory function is closely monitored, and oxygen source, appropriate-sized resuscitation bag and mask, endotracheal intubation and suctioning equipment, tracheotomy tray, and vasoconstrictor drugs are kept available. Vital signs, including neurologic signs and level of consciousness, are monitored frequently. For the child who develops respiratory impairment, the care is the same as that for any child with respiratory distress requiring mechanical ventilation.
Throughout the recovery phase, special emphasis is placed on prevention of complications, including proper postural alignment, frequent change of position, assessment of skin at pressure points, and passive range-of-motion exercises. Respiratory care, should intubation be required, requires close monitoring of oxygenation status, usually by pulse oximetry and arterial blood gases; maintenance of an open airway with suctioning; and postural changes to prevent pneumonia. Children with oral and pharyngeal involvement may be fed via a nasogastric tube to ensure adequate feeding. Immobilization, which occurs with GBS, decreases gastrointestinal function; therefore attention to problems such as decreased gastric emptying and constipation requires nursing assessment and appropriate collaborative interventions.
Prevention of deep vein thrombosis is accomplished with antiembolism devices, administration of a low-molecular-weight heparin, and early mobilization and ambulation. Temporary urinary catheterization may be required; urinary retention is not uncommon, and appropriate assessment of urinary output is vital. Sensory impairment and paralysis in the lower extremities makes the child susceptible to skin breakdown; therefore attention should be given to meticulous skin care. Although the child may have a generalized paralysis, cognitive function remains intact; therefore it is important for nursing care to involve communication with the child regarding procedures and treatments that may be frightening, especially if mechanical ventilation is required. Parents are encouraged to talk to the child and make eye and physical contact as much as possible to reassure the child during the illness. Psychosocial care of the child with GBS focuses on dealing with the child’s anxiety and fear related to the disease itself and the unknown prognosis. In addition, the child’s parents are allowed to express feelings and are encouraged to be involved in the child’s daily care.
Pain management is crucial in the care of children with GBS. Although neuromuscular impairment may make pain perception more difficult to accurately evaluate, objective pain scales should be used.
Physical therapy is limited to passive range-of-motion exercises during the evolving phase of the disease. Later, as the disease stabilizes and recovery begins, an active physical therapy program is implemented to prevent contracture deformities and facilitate muscle recovery. This may include active exercise, gait training, and bracing.
Throughout the course of the illness, support of the child and parents is paramount. The usual rapidity of the paralysis and the long period of recovery greatly tax the emotional reserves of all family members. The parents and child benefit from repeated reassurance that recovery is occurring and from realistic information regarding the possibility of permanent disability. In the event of a residual disability, the family needs assistance in accepting and adjusting to the loss of function (see Chapter 18). The GBS/CIDP Foundation International* is a nonprofit organization devoted to support, education, and research. It provides support to families from recovered persons, publishes informational literature and a newsletter, and maintains a list of practitioners experienced with the disease.
TETANUS
Tetanus, or lockjaw, is an acute, preventable, but often fatal disease caused by an exotoxin produced by the anaerobic spore-forming, gram-positive bacillus Clostridium tetani. It is characterized by painful muscular rigidity primarily involving the masseter and neck muscles. There are four requirements for the development of tetanus: (1) presence of tetanus spores or vegetative forms of the bacillus, (2) injury to the tissues, (3) wound conditions that encourage multiplication of the organism, and (4) a susceptible host.
Tetanus spores are found in soil, dust, and the intestinal tracts of humans and animals, especially herbivorous animals. The organisms are more prevalent in rural areas but are readily carried to urban areas by the wind. The organisms are not invasive but enter the body by way of wounds, particularly a puncture wound, burn, or crushed area. They may enter through a minor, unnoticed break in the skin, such as a thorn or needle prick, bee sting, or scratch. In the newborn, infection may occur through the umbilical cord, usually in situations in which infants are delivered in severely contaminated surroundings or the mother is not adequately immunized. The disease has the greatest incidence in months when persons are more involved in outdoor activities.
Pathophysiology
When prevention efforts are not effective and conditions are favorable, the organisms proliferate and form potent exotoxins, one of which is tetanospasmin. Tetanospasmin affects the central nervous system to produce the clinical manifestations of the disease. The ideal conditions for the organisms’ growth are devitalized tissues without access to air, such as wounds that have not been washed or kept clean and those that have crusted over, trapping pus beneath. The exotoxin appears to reach the central nervous system by way of either the neuron axons or the vascular system. The toxin becomes fixed on nerve cells of the anterior horn of the spinal cord and the brainstem. The toxin acts at the myoneural junction to produce muscular stiffness and lower the threshold for reflex excitability.
The incubation period for tetanus varies from 2 days to months and averages 8 days; most cases occur within 14 days. In neonates it is usually 5 to 14 days. Shorter incubation periods have been associated with more heavily contaminated wounds, more severe disease, and a worse prognosis (American Academy of Pediatrics, 2006).
The manner of onset varies, but the initial symptoms are usually a progressive stiffness and tenderness of the muscles in the neck and jaw. Eventually all voluntary muscles are affected (Box 32-12). As the child recovers from the disease, the paroxysms become less frequent and gradually subside. Survival beyond 4 days usually indicates recovery, but complete recovery may require weeks.
BOX 32-12 Clinical Manifestations of Tetanus
Progressive stiffness and tenderness of muscles in neck and jaw
Characteristic difficulty in opening the mouth (trismus)
Risus sardonicus (sardonic smile) caused by facial muscle spasm
Opisthotonic positioning
Boardlike rigidity of abdominal and limb muscles
Extreme sensitivity to external stimuli (slight noise, gentle touch, or bright light):
Therapeutic Management
Preventive measures are based on the child’s immune status and the nature of the injury. Specific prophylactic therapy after trauma is administration of tetanus toxoid (tetanus antitoxin is not available in the United States) (see Immunizations, Chapter 10, for age-specific recommendations).
The unprotected or inadequately immunized child who sustains a “tetanus-prone” wound (including wounds contaminated with dirt, feces, soil, and saliva; puncture wounds; avulsions; and wounds resulting from missiles, crushing, burns, and frostbite) should receive tetanus immunoglobulin (TIG). Concurrent administration of both TIG and tetanus toxoid at separate sites is recommended both to provide protection and to initiate the active immune process. Once the individual has received primary tetanus immunization, antitoxin is believed to provide protection for at least 10 years and for a longer period after booster immunization (American Academy of Pediatrics, 2006). Completion of active immunization is carried out according to the usual pattern. Antibiotic treatment with oral or intravenous metronidazole (Flagyl) (alternatively parenteral penicillin G) is important in the management of tetanus as an adjunct against vegetative forms of clostridia (American Academy of Pediatrics, 2006).
Aggressive supportive care is necessary to treat tetanus in the acute phase. The acutely ill child is best treated in an intensive care facility where close and constant observation and equipment for monitoring and respiratory support are readily available. A quiet environment is preferred to reduce external stimuli.
General supportive care is indicated, including maintaining adequate airway and fluid and electrolyte balance, managing pain, and ensuring adequate caloric intake. Indwelling oral or nasogastric feedings may be required to maintain adequate fluid and caloric intake; continued laryngospasm may necessitate total parenteral nutrition or gastrostomy feeding. Severe or recurrent laryngospasm or excessive secretions may require advanced airway management such as endotracheal intubation.
TIG therapy to neutralize toxins is the most specific therapy for tetanus. Antibiotics are administered to control the proliferation of the vegetative forms of the organism at the site of infection. Local care of the wound by surgical débridement and cleansing helps reduce the numbers of proliferating organisms at the site of injury. The cleansing should be repeated several times during the first 48 hours, and deep, infected lacerations are usually exposed and débrided.
Diazepam is the drug of choice for seizure control and muscle relaxation (Arnon, 2007b), but lorazepam (Ativan) may be used in some cases. Other AEDs may be administered as well. Intrathecal baclofen, magnesium sulfate, dantrolene sodium, and midazolam may also be used in the management of tetanus. Patients with severe tetanus and those who do not respond to other muscle relaxants may require the administration of a neuromuscular blocking agent, such as rocuronium or vecuronium; intrathecal baclofen may be used as a muscle relaxant but only in the intensive care unit because it often induces apnea. Because of their paralytic effect on respiratory muscles, use of these drugs requires mechanical ventilation with endotracheal intubation or tracheotomy and constant cardiopulmonary monitoring. Endotracheal tube insertion or tracheostomy is often indicated and should be performed before severe respiratory distress develops. Despite the absence of pain manifestation with these drugs, it is important to administer adequate analgesia. The administration of corticosteroids has met with success in some cases.
Nursing Care Management
The care of the child with tetanus requires supportive management with particular attention to airway and breathing. Respiratory status is carefully evaluated for any signs of distress, and appropriate emergency equipment is kept available at all times. The location, extent, and severity of muscle spasms are important nursing observations. Muscle relaxants, opioids, and sedatives that may be prescribed can also cause respiratory depression; therefore the child must be assessed for excessive central nervous system depression. Attention to hydration and nutrition involve monitoring an intravenous infusion, monitoring nasogastric or gastrostomy feedings, and suctioning oropharyngeal secretions when indicated.
In caring for the child with tetanus during the acute phase, the nurse makes every effort to control or eliminate stimulation from sound, light, and touch. Although a darkened room is ideal, sufficient light is essential so that the child can be carefully observed; light appears to be less irritating than vibratory or auditory stimuli. The infant or child is handled as little as possible, and extra effort is expended to avoid any sudden or loud noise to prevent seizures.
If a potent muscle relaxant such as vecuronium is used, the total paralysis makes oral communication impossible. The drug is not a sedative, however, and anxiety should be considered in children who are intubated. Therefore all the child’s needs must be anticipated and procedures carefully explained beforehand.
Because their mental status is clear, children are aware of what is happening to them and are often extremely anxious. They should not be left alone, and all efforts should be made to reduce anxiety, which can contribute to muscle spasms. Parents are encouraged to stay with the child to offer security and support. They also need support, information and reassurance from the nurse.
BOTULISM
Botulism is an acute flaccid paralysis caused by the preformed toxin produced by the anaerobic bacillus Clostridium botulinum. In classic, or foodborne, botulism the most common source of the toxin is a contaminated food source. In addition to foodborne botulism, other forms include wound botulism; infant botulism; and man-made botulism, usually a result of bioterrorism (Arnon, 2007a). In foodborne illness, central nervous system symptoms appear abruptly within a few hours or gradually over several days after ingestion of contaminated food and may not be preceded by acute digestive disturbance (Box 32-13).
Treatment consists of intravenous administration of botulism antitoxin and general supportive measures, primarily respiratory and nutritional. Toxins vary in protein-binding capacity. Some have a relatively short half-life and do not bind to tissues firmly; therefore therapy is continued until paralysis abates. Other toxins appear to bind irreversibly to nerve endings and are therefore not amenable to neutralization.
Infant Botulism
Infant botulism, unlike the disease in older persons, is caused by ingestion of spores or vegetative cells of C. botulinum and the subsequent release of the toxin from organisms colonizing the gastrointestinal tract. C. botulinum types A and B are the most common causative strains of infant botulism. This form of botulism has become more prevalent than any other form. Many cases of infant botulism occur in breastfed infants who are being introduced to nonhuman milk substances (American Academy of Pediatrics, 2006). There appears to be no common food or drug source of the organisms; however, the C. botulinum organisms have been found in honey. Botulism may occur in infants as young as 1 week of age up to 12 months of age with peak incidence between 2 and 4 months of age.
The severity of the disease varies widely, from mild constipation to progressive sequential loss of neurologic function and respiratory failure (see Box 32-13). The affected infant is usually well before the onset of symptoms. Constipation is a common presenting symptom, and almost all infants exhibit generalized weakness and a decrease in spontaneous movements. Deep tendon reflexes are usually diminished or absent. Cranial nerve deficits are common, as evidenced by loss of head control, difficulty in feeding, weak cry, and reduced gag reflex. SMA type 1 and metabolic disorders are often mistaken for infant botulism in the initial diagnostic phase because of the similarities in clinical manifestations of hypotonia, lethargy, and poor feeding (Francisco and Arnon, 2007). Presenting clinical signs also often mimic those of sepsis in young infants. Botulism toxin exerts its effect by inhibiting the release of acetylcholine at the myoneural junction, thereby impairing motor activity of muscles innervated by affected nerves.
Diagnosis is made on the basis of the clinical history, physical examination, and laboratory detection of the organism in the patient’s stool and, less commonly, blood. However, isolation of the organism may take several days; therefore suspicion of botulism by clinical presentation should require emergent treatment (Arnon, 2007a). EMG may be helpful in establishing the diagnosis; however, results may be normal early in the course of the illness.
Treatment consists of immediate administration of botulism immune globulin intravenously (BIG-IV [BabyBIG]) (Francisco and Arnon, 2007), without delaying for laboratory diagnosis. Early administration of BIG-IV neutralizes the toxin and stops the progression of the disease. The human-derived botulism antitoxin (BIG-IV) has been evaluated and is now available nationwide for use only in infant botulism. In one study, infants treated with BIG-IV experienced a mean length of hospitalization decrease from 5.7 to 2.6 weeks and also had decreased time spent in intensive care, decreased mean duration of mechanical ventilation, and decreased mean duration of tube or intravenous feeding. In addition, the infants did not experience any adverse events related to BIG-IV; most infants received BIG-IV treatment within 3 to 18 days of hospitalization for botulism (Arnon, Schechter, Maslanka, and others, 2006). Approximately 50% of affected infants will require intubation and mechanical ventilation; therefore respiratory support is crucial, as is nutritional support since the infant is unable to feed. Trivalent equine botulinum antitoxin and bivalent antitoxin, used in adults and older children, is not administered to infants. Antibiotic therapy is not part of the management because the botulinum toxin is an intracellular molecule and antibiotics would not be effective; aminoglycosides in particular should not be administered because they may potentiate the blocking effects of the neurotoxin (Arnon, 2007a).
The prognosis is generally good if the patient is adequately treated, although recovery may be slow, requiring a few weeks after severe illness; the average length of stay for infant botulism is 44 days, and the fatality rate is reported to be less than 2%. Untreated patients may require a longer hospitalization.
Nursing Care Management
Nursing responsibilities include observing for and reporting signs of neuromuscular weakness or impairment and providing intensive nursing care when the infant is hospitalized (see Nursing Care of the High-Risk Newborn and Family, Chapter 9). Parental support and reassurance are important. Most infants recover when the disorder is recognized and therapy is implemented. Parents should be aware that, during recovery, patients tire easily when muscular action is sustained. This has important implications for timing the resumption of feedings because of the risk of aspiration. Parents should also be advised that normal bowel action may not return for several weeks; therefore a stool softener can be beneficial.
SPINAL CORD INJURIES
SCIs with major neurologic involvement traditionally have not been a common cause of physical disability in children. However, a sufficient number of children with these injuries are admitted to major medical centers, and because of the increased survival rate as a result of improved management, nurses are often involved in the care and rehabilitation of children with SCI.
Mechanisms of Injury
The most common cause of serious spinal cord damage in children is trauma involving MVCs (including automobile-bicycle, all-terrain vehicles, and snowmobiles), sports injuries (especially from diving, trampoline activities, gymnastics, and football), birth trauma, and nonaccidental trauma. The increased use of recreational activities involving motorized vehicles such as jet water skis, all-terrain vehicles, and motorcycles has also increased the incidence of SCIs in children. Congenital defects of the spine such as myelomeningocele also may in some cases produce the effects of SCI.
Transverse myelitis (inflammation of the spinal cord) has also been reported to develop from inadvertent intraarterial administration of long-acting penicillin injected into the buttocks. Damage can be extensive enough to result in paraplegia or even lower limb amputation.
In MVCs most SCIs in children are a result of indirect trauma caused by sudden hyperflexion or hyperextension of the neck, often combined with a rotational force. Trauma to the spinal cord without evidence of vertebral fracture or dislocation (SCIWORA) is particularly likely to occur in an MVC when proper safety restraints are not used. An unrestrained child becomes a projectile during sudden deceleration and is subject to injury from contact with a variety of objects inside and outside the vehicle. Individuals who use only a lap seat belt restraint are at greater risk of SCI than those who use a combination lap and shoulder restraint. High cervical spine injuries have been reported in children younger than 2 years of age who are improperly restrained in forward-facing car seats. Infants who are improperly restrained in an infant car seat may experience cervical trauma in a car crash. Small children may also be severely injured by deploying front seat air bags.
Falling from heights occurs less often in children than in adults, but vertebral compression from blows to the head or buttocks can occur in water sports (diving and surfing), falls from horses, or other athletic activities. Birth injuries may occur in breech deliveries from traction force on the spinal cord during delivery of the head and shoulders. When shaken, infants commonly sustain cervical cord damage, as well as subdural hematoma and retinal hemorrhage; mental retardation and death may occur subsequent to the traumatic event. Infants have weak neck muscles, and during vigorous shaking their large and heavy heads rapidly wobble back and forth. A significant number of adolescents receive SCIs secondary to gunshot wounds, stabbings, or other violent inflicted injury.
The injury sustained can affect any of the spinal nerves, and the higher the injury, the more extensive the damage. The child can be left with complete or partial paralysis of the lower extremities (paraplegia) or with damage at a higher level and without functional use of any of the four extremities (quadriplegia). A high cervical cord injury that affects the phrenic nerve paralyzes the diaphragm and leaves the child dependent on mechanical ventilation.
A mild but equally frightening form of cord trauma is spinal cord compression, a temporary neural dysfunction without visible damage to the cord. Complete quadriplegia can result but initially may not be differentiated from serious cord injury.
Therapeutic Management
The management of the child with SCI has changed dramatically in the past two decades as a result of improved technology, surgical procedures, and research into the complexity of the spinal cord and its neurologic components. Initial care begins at the scene of the accident; therefore education and training of first responder personnel in spinal immobilization, stabilization, and transfer techniques to prevent or reduce the severity of injury are critically important. Because of the complexity of these injuries, it is usually recommended that those with injuries be transported to a spinal injury center for care by specially trained health care personnel as soon as possible after the injury for appropriate diagnostic evaluation and intervention.
A number of special pediatric immobilization devices are now available that are physiologically appropriate for children’s unique characteristics. A large head (proportionately), weak neck musculature, and weak tracheal cartilage predispose small children to airway compromise if placed supine; hence a spinal board with a built-in head drop may serve to protect the child’s spine and neck and provide better airway management than the traditional flat board (DeBoer and Seaver, 2004).
Treatment with functional electrical stimulation (an implantable electrical stimulator with leads attached to the paralyzed muscles or nerves) has allowed children with certain SCIs greater mobility and functional use of paralyzed muscles to sit, stand, and walk with the aid of crutches, a walker, or other orthoses. Administration of pharmacologic agents such as clonidine hydrochloride may improve ambulation in patients with partial SCI, and exercise therapy through interactive locomotor training has been beneficial in helping some individuals with SCI regain ambulatory function.
SCI management guidelines and standards of care have recently been published for adult and pediatric patients with spinal injuries by the American Association of Neurological Surgeons and the Congress of Neurological Surgeons. For information regarding these guidelines and a more in-depth review of SCI care, the reader is referred to the Barker and Saulino (2002) reference for a synopsis of the guidelines, or see “Guidelines for the Management of Acute Cervical Spine and Spinal Cord Injuries” by Hadley, Walters, Grabb, and others (2002).
Nursing Care Management
The nursing care of the paraplegic or quadriplegic child is complex and challenging. A multidisciplinary SCI team is equipped to manage the acute phase of the injury, and some members, including the nurse, may follow the patient to eventual recovery. Nursing management is concerned with ensuring adequate initial stabilization of the entire spinal column with a rigid cervical collar with supportive blocks on a rigid backboard. During the acute phase of the injury it is imperative that airway patency be maintained and respiratory function monitored. It is important to evaluate the extent of the neurologic damage early to establish a baseline for neurologic functioning; continual assessment of function should occur to prevent further deterioration of neurologic status as a result of spinal cord edema. The American Spinal Injury Association (ASIA) Impairment Scale may be used to assess neurologic function on a routine basis during the patient’s recovery (Barker and Saulino, 2002). Once the patient is admitted, further evaluation of the patient’s abilities to perform ADLs can be made with the Functional Independence Measure (FIM) scale; this scale measures the patient’s abilities during recovery and need for assistance (Barker and Saulino, 2002).
The nursing care of the child with an SCI is, in most respects, the same as that of any immobilized child (see The Immobilized Child, Chapter 31). Additional aspects of care that should be addressed on an individual basis include hypercalcemia in adolescent males, deep vein thrombosis, latex sensitization, and sleep-disordered breathing (Vogel, Hickey, Klaas, and others, 2004).
Respiratory care often focuses on maintaining an adequate airway and effective ventilation. The child with a high-level cervical injury (C3 and above) requires continuous ventilatory assistance. In most instances a tracheostomy is the method of choice for greater ease in clearing secretions and for less trauma to tissues during long-term ventilatory dependence. In some children breathing pacemaker devices (phrenic nerve stimulators) are implanted to stimulate the phrenic nerve and produce diaphragmatic contractions and lung expansion without assisted ventilation. In the child who does not require mechanical ventilation, special attention to clearance of secretions is vital because of decreased pulmonary function. In addition to chest physiotherapy, the child may require a cough-assist device to clear secretions effectively (see Duchenne Muscular Dystrophy: Therapeutic Management).
Temperature is often poorly regulated in children with SCI; therefore body temperature must be monitored closely for fluctuations. Response to environmental temperature changes may be slow or absent, and the ability to dissipate heat through the process of shivering may be compromised.
Children with SCI have unique needs in relation to skin care. Because of decreased sensation and impaired mobility, they depend on others to assess and assist in the management of intact skin. Skin care practices are the same as those for any child who is immobilized. A skin score scale such as the Braden Q Scale should be used to objectively evaluate risks for skin breakdown and skin conditions (Curley, Razmus, Roberts, and others, 2003). An alternating-pressure mattress or other pressure relief or reduction device is kept underneath the child, and the skin is thoroughly inspected at least once a day (or more often if there is increased risk) for signs of pressure and breakdown, especially over bony prominences.
Bowel and bladder function is often affected in the child with SCI. CIC may be required to regularly empty the neurogenic bladder and prevent urinary tract infections. A regular bowel management program is tailored to the child’s needs.
All adaptive devices help children increase their mobility, function, and endurance. The child with some lower extremity function progresses to parallel bars and then to a walker; the child with quadriplegia learns to use a wheelchair—among the most valuable aids available to the child with an SCI (Fig. 32-9). The wheelchair should be selected carefully in relation to where it will be used, the architectural barriers, and the child’s functional capacity. For children with severe upper extremity paralysis, a variety of motorized wheelchairs are used; however, the more complex they are, the greater their cost, weight, and tendency to break down. Wheelchair tolerance is gained over time and is accompanied by measures to prevent orthostatic hypotension and pressure ulcers.
FIG. 32-9 Wheelchair allows adolescent mobility and independence. (Courtesy Texas Children’s Hospital, Houston.)
A variety of orthoses and other appliances can be adapted for use by many children. The primary purpose of lower extremity bracing in the child with an SCI is for ambulation, although correction of deformities may be attempted.
During the recovery and rehabilitation phase, patients with SCI must be carefully monitored for complications of immobility such as deep vein thrombosis and pulmonary embolus.
The child and family with SCI are prepared for the eventual discharge from the acute care facility to a rehabilitation center. The major aims of physical rehabilitation are to prepare the child and family to achieve normalization and resume life at home and in the community. Additional goals of rehabilitation in children with SCI are to promote independence in mobility and self-care skills, academic achievement, independent living, and employment.
The nurse is a crucial member of the health care team in relation to helping the family cope with the magnitude of the injury and disability, understand the extent of the disability, verbalize expected outcomes, and move toward eventual rehabilitation and normalization within the child’s capabilities. The goals of rehabilitation include preparing the child and family to live at home and function as independently as possible.
Clinical manifestations of CP include delayed gross motor development, altered motor performance, alterations of muscle tone and subsequent muscle contractures, abnormal posture, and associated disabilities such as seizures and sensory impairment.
Therapy for CP takes into account the nature of the physical disability, defects associated with the disorder, and interpersonal and social influences encountered by the affected child.
Care of the infant and child with myelomeningocele is directed toward protecting the meningeal sac, preventing infection and skin breakdown, observing for signs of urologic and bowel complications, promoting early parent-infant interaction, and planning appropriate interventions to optimize the child’s development.
SMA type 1 (Werdnig-Hoffmann disease) is characterized by progressive weakness and wasting of skeletal muscles caused by degeneration of anterior horn cells of the spinal cord.
MDs are the most prevalent cause of neuromuscular dysfunction of childhood.
Children with neuromuscular disease require careful monitoring of respiratory function and prompt interventions to prevent hypoxia and hypercarbia.
Major complications of DMD include joint contractures, disuse atrophy, obesity, cardiomyopathy, sleep-disordered breathing, and respiratory failure as the condition progresses.
Nursing care of the child with GBS consists of maintaining a patent airway; monitoring vital signs and neurologic signs; ensuring proper body alignment and positioning; and providing physical therapy, reassurance, and support to the child and family.
Tetanus occurs when tetanus spores or vegetative bacilli enter a wound and multiply in a susceptible host.
Clinical manifestations of infant botulism include constipation, decreased activity, poor feeding, and lethargy.
Therapeutic management of SCI is directed toward immobilizing the entire spinal column at the scene of the traumatic event, transporting safely by health care personnel trained to transport possible spinal trauma victims, evaluating neurologic damage, preventing further neurologic damage, and implementing an aggressive rehabilitation program designed to help achieve independence and movement.
REFERENCES
Albright, AL, Gilmartin, R, Swift, D, et al. Long-term intrathecal baclofen therapy for severe spasticity of cerebral origin. J Neurosurg. 2003;98(2):291–295.
American Academy of Pediatrics, Committee on Infectious Diseases, Pickering L, eds. Red book: 2006 report of the Committee on Infectious Diseases, ed 27, Elk Grove Village, Ill: The Academy, 2006.
American Academy of Pediatrics. Cardiovascular health supervision for individuals affected by Duchenne or Becker muscular dystrophy. Pediatrics. 2005;116(6):1569–1573.
American Academy of Pediatrics, American College of Obstetricians and Gynecologists. Neonatal encephalopathy and cerebral palsy: defining the pathogenesis and pathophysiology. Washington, DC: The College, 2003.
American Thoracic Society. Respiratory care of the patient with Duchenne muscular dystrophy. Am J Respir Crit Care Med. 2004;170(4):456–465.
Angelini, C. The role of corticosteroids in muscular dystrophy: a critical appraisal. Muscle Nerve. 2007;36(4):424–435.
Arnon, SS. Anaerobic bacterial infections: botulism (Clostridium botulinum). In Kliegman RM, Behrman RE, Jenson HB, et al, eds.: Nelson textbook of pediatrics, ed 18, Philadelphia: Saunders, 2007.
Arnon, SS. Tetanus (Clostridium tetani). In Kliegman RM, Behrman RE, Jenson HB, et al, eds.: Nelson textbook of pediatrics, ed 18, Philadelphia: Saunders, 2007.
Arnon, SS, Schechter, R, Maslanka, SE, et al. Human botulism immune globulin for the treatment of infant botulism. N Engl J Med. 2006;354(5):462–471.
Ashwal, S, Russman, BS, Blasco, PA, et al. Practice parameter: diagnostic assessment of the child with cerebral palsy: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2004;62(6):851–863.
Atkinson, SB, Carr, RL, Maybee, P, et al. The challenge of managing and treating Guillain-Barré syndrome during the acute phase. Dimensions Crit Care Nurs. 2006;25(6):256–263.
Bach, JR. Medical considerations of long-term survival of Werdnig-Hoffmann disease. Am J Phys Med Rehabil. 2007;86(5):349–355.
Barker, E, Saulino, MF. Special report: first-ever guidelines for spinal cord injuries. RN. 2002;65(10):32–37.
Beroud, C, Karliova, M, Bonnefont, JP, et al. Prenatal diagnosis of spinal muscular atrophy by genetic analysis of circulating fetal cells. Lancet. 2003;361(9362):1013–1014.
Birnkrant, DJ. The assessment and management of the respiratory complications of pediatric neuromuscular diseases. Clin Pediatr. 2002;41(5):301–308.
Brown, JP. Orthopaedic care of children with spina bifida: you’ve come a long way, baby!. Orthop Nurs. 2001;20(4):51–58.
Buran, CF, McDaniel, AM, Brei, TJ. Needs assessment in a spina bifida program: a comparison of the perceptions by adolescents with spina bifida and their parents. Clin Nurs Spec. 2002;16(5):256–262.
Bush, A, Fraser, J, Jardine, E, et al. Respiratory management of the infant with type 1 spinal muscular atrophy. Arch Dis Child. 2005;90(7):709–711.
Centers for Disease Control and Prevention. Botulism. http://www.cdc.gov/nczved/dfbmd/disease_listing/botulism_gi.html, 2008. [retrieved July 18, 2007, from].
Centers for Disease Control and Prevention. Folate status in women of childbearing age, by race/ethnicity—United States, 1999-2000, 2001-2002, and 2003-2004. MMWR. 2007;55(51):1377–1380.
Centers for Disease Control and Prevention. Spina bifida and anencephaly before and after folic acid mandate—United States, 1995-1996 and 1999-2000. MMWR. 2004;53(17):362–365.
Chakkalakal, JV, Thompson, J, Parks, RJ, et al. Molecular, cellular, and pharmacological therapies for Duchenne/Becker muscular dystrophies. FASEB J. 2005;19(8):880–891.
Curley, MAQ, Razmus, IS, Roberts, KE, et al. Predicting pressure ulcer risk in pediatric patients: the Braden Q Scale. Nurs Res. 2003;52(1):22–33.
Dabney, KW, Lipton, GE, Miller, F. Cerebral palsy. Curr Opin Pediatr. 1997;9(1):81–88.
DeBoer, SL, Seaver, M. Pediatric spinal immobilization: C-spines, car seats, and color-coded collars. J Emerg Nurs. 2004;30(5):481–484.
Doolin, E. Bowel management for patients with myelodysplasia. Surg Clin North Am. 2006;86(2):505–514.
Eiwegger, T, Dehlink, E, Schwindt, J, et al. Early exposure to latex products mediates latex sensitization in spina bifida but not in other diseases with comparable latex exposure rates. Clin Exp Allergy. 2006;36(10):1242–1246.
Francisco, AM, Arnon, SS. Clinical mimics of infant botulism. Pediatrics. 2007;119(4):826–828.
Gibson, CS, MacLennan, AH, Goldwater, PN, et al. Antenatal causes of cerebral palsy: associations between inherited thrombophilias, viral and bacterial infection, and inherited susceptibility to infection. Obstet Gynecol Surv. 2003;58(3):209–220.
Green, L, Greenberg, GM, Hurwitz, E. Primary care of children with cerebral palsy. Clin Fam Pract. 2003;5(2):1–21.
Grether, JK, Nelson, KB, Walsh, E, et al. Intrauterine exposure to infection and risk of cerebral palsy in very preterm infants. Arch Pediatr Adolesc Med. 2003;157(1):26–32.
Hadley, MN, Walters, BC, Grabb, PA, et al. Guidelines for management of acute cervical spine and spinal cord injuries. Neurosurgery. 2002;50(3 Suppl):S21–S29.
Han, TR, Bang, MS, Lim, JY, et al. Risk factors of cerebral palsy in preterm infants. Am J Phys Med Rehabil. 2002;81(4):297–303.
Henshaw, SK. Unintended pregnancy in the United States. Fam Plan Perspect. 1998;30:24–29.
Honein, MA. Impact of folic acid fortification of the US food supply and occurrence of neural tube defects. JAMA. 2001;285(23):2981–2986.
Hughes, RA, Cornblath, DR. Guillain-Barré syndrome. Lancet. 2005;366(9497):1653–1666.
Hurtekant, KM, Spatz, DL. Special considerations for breastfeeding the infant with spina bifida. J Perinat Neonatal Nurs. 2007;21(1):69–75.
Iannaccone, ST. Spinal muscular atrophy. Semin Neurol. 1998;18(1):19–26.
Iannaccone, ST, Burghes, A. Spinal muscular atrophies. Adv Neurol. 2002;88:83–98.
Jacobs, JM. Management options for the child with spastic cerebral palsy. Orthop Nurs. 2001;20(3):53–59.
Kaufman, BA. Neural tube defects. Pediatr Clin North Am. 2004;51(2):389–419.
Kellett, PB. Latex allergy: a review. J Emerg Nurs. 1997;23(1):27–36.
Kimata, H. Latex allergy in infants younger than 1 year. Clin Exp Allergy. 2004;34(12):1910–1915.
Kirkham, C, Harris, S, Grzybowski, S. Evidence-based prenatal care, part I, General prenatal care and counseling issues. Am Fam Physician. 2005;71(7):1307–1316.
Krach, LE. Pharmacotherapy of spasticity: oral medications and intrathecal baclofen. J Child Neurol. 2001;16(1):31–36.
Krigger, KW. Cerebral palsy: an overview. Am Fam Physician. 2006;73(1):91–100. [101-102,].
Lazzaretti, CC, Pearson, C. Myelodysplasia. In Allen PJ, Vessey JA, eds.: Primary care of the child with a chronic condition, ed 4, St Louis: Mosby, 2004.
Matthews, TJ, Honein, MA, Erickson, JD. Spina bifida and anencephaly prevalence—United States, 1991-2001. MMWR Recomm Rep. 2002;51(RR-13):9–11.
McKearnan, KA, Kieckhefer, GM, Engel, JM, et al. Pain in children with cerebral palsy: a review. J Neurosci Nurs. 2004;36(5):252–259.
Metules, T. Duchenne muscular dystrophy. RN. 2002;65(10):39–47.
Miske, LJ, Hickey, EM, Kolb, SM, et al. Use of the mechanical in-exsufflator in pediatric patients with neuromuscular disease and impaired cough. Chest. 2004;125(4):1406–1412.
Moxley, RT, Ashwal, S, Pandya, S, et al. Practice parameter: corticosteroid treatment of Duchenne dystrophy. Neurology. 2005;64(1):13–20.
Murphy, KP, Molnar, GE, Lankasky, K. Employment and social issues in adults with cerebral palsy. Arch Phys Med Rehabil. 2000;81:807–811.
Newswanger, DI, Warren, CR. Guillain-Barré syndrome. Am Fam Physician. 2004;69(10):2405–2410.
Rogers, B. Feeding method and health outcomes of children with cerebral palsy. J Pediatr. 2004;145(2 Suppl):S28–S32.
Roscigno, CI. Addressing spasticity-related pain in children with spastic cerebral palsy. J Neurosci Nurs. 2002;34(3):123–131.
Rosenbaum, P, Paneth, N, Leviton, A, et al. A report: the definition and classification of cerebral palsy April 2006. Dev Med Child Neurol. 2007;49(S109):1–44.
Russman, BS. Function changes in spinal muscular atrophy II and III: the DCN/SMA group. Neurology. 1996;47(4):973–976.
Russman, BS, Iannaccone, ST, Buncher, CR, et al. Spinal muscular atrophy: new thoughts on the pathogenesis and classification schema. J Child Neurol. 1992;7(4):347–353.
Samaniego, IA. A sore spot in pediatrics: risk factors for pressure ulcers. Pediatr Nurs. 2003;29(4):278–282.
Sarnat, HB. Neuromuscular disorders. In Kliegman RM, Behrman RE, Jenson HB, et al, eds.: Nelson textbook of pediatrics, ed 18, Philadelphia: Saunders, 2007.
Shaer, CM, Chescheir, N, Schulkin, J. Myelomeningocele: a review of the epidemiology, genetics, risk factors for conception, prenatal diagnosis, and prognosis for affected individuals. Obstet Gynecol Surv. 2007;62(7):471–479.
Simonds, AK. Recent advances in respiratory care for neuromuscular disease. Chest. 2006;130(6):1879–1886.
Snodgrass, WT, Adams, R. Initial urologic management of myelomeningocele. Urol Clin North Am. 2004;31(3):427–434. [viii,].
Tarcan, T, Onol, FF, Ilker, Y, et al. The timing of primary neurosurgical repair significantly affects neurogenic bladder prognosis in children with myelomeningocele. J Urol. 2006;176(3):1161–1165.
Thompson, GH, Berenson, FR. Other neuromuscular disorders. In Morrissy RT, Weinstein SL, eds.: Lovell and Winter’s pediatric orthopaedics, ed 5, Philadelphia: Lippincott Williams & Wilkins, 2001.
Vogel, LC, Hickey, KJ, Klaas, SJ, et al. Unique issues in pediatric spinal cord injury. Orthop Nurs. 2004;23(5):300–308.
Volpe, JJ. Neurology of the newborn, ed 4. Philadelphia: Saunders, 2001.
Winter, S, Autry, A, Boyle, C, et al. Trends in the prevalence of cerebral palsy in a population-based study. Pediatrics. 2002;110(6):1220–1225.
Wu, YW, Escobar, GJ, Grether, JK, et al. Chorioamnionitis and cerebral palsy in term and near-term infants. JAMA. 2003;290(20):2677–2684.
Young, HK, Lowe, A, Fitzgerald, DA, et al. Outcome of noninvasive ventilation in children with neuromuscular disease. Neurology. 2007;68(3):198–201.
*25 W. Independence Way, Kingston, RI 02881; (401) 792-7130; fax: (401) 792-7132; e-mail: info@ndsaonline.org; http://www.ndsaonline.org. (note: The National Disability Sports Alliance is the corporate name [0]of the U.S. Cerebral Palsy Athletic Association, Inc.)
*1660 L St., NW, Suite 700, Washington, DC 20036; (800) 872-5827; fax: (202) 776-0414; e-mail: info@ucp.org; http://www.ucpa.org. The website also has links to each state’s United Cerebral Palsy organization.
*Information is available from Division of Birth Defects and Pediatric Genetics, NCBDDD, CDC, 1600 Clifton Road NE, Mailstop E-86, Atlanta, GA 30333; (404) 498-3800; e-mail: flo@cdc.gov; http://www.cdc.gov/ncbddd/folicacid And from March of Dimes Resource Center, 1275 Mamaroneck Ave., White Plains, NY 10605; http://www.marchofdimes.com.
*4590 MacArthur Blvd. NW, Suite 250, Washington, DC 20007-4226; (202) 944-3285, (800) 621-3141; http://www.spinabifidaassociation.org.
*Additional information regarding latex allergy may be found at http://www.latex-allergy.org and http://latexallergylinks.tripod.com. For a list of latex products and alternative products, see Latex List on the Spina Bifida Association home page, http://www.spinabifidaassociation.org.
*Family resources for SMA include Families of Spinal Muscular Atrophy, PO Box 196, Libertyville, IL 60048; (800) 886-1762; http://www.fsma.org; and Muscular Dystrophy Association-USA, 3300 E. Sunrise Drive, Tucson, AZ 85718; (800) 572-1717; http://www.mda.org. In Canada: Families of Spinal Muscular Atrophy Canada, PO Box 97, Rivers, MB R0K 1X0; (800) 866-0016; http://www.curesma.ca.
*3300 E. Sunrise Drive, Tucson, AZ 85718; (800) 572-1717; http://www.mda.org. In Canada: Muscular Dystrophy Canada, 2345 Yonge St., Suite 900, Toronto, ON M4P 2E5; [0](866) MUSCLE-8; fax: (416) 488-7523; http://www.muscle.ca.
*The Holly Building, 104½ Forrest Ave., Narberth, PA 19072; (610) 667-0131, (866) 224-3301; http://gbs-cidp.org.