Physiology and Pharmacology of Opioid Analgesics
THIS chapter expands on the underlying mechanisms of the opioid analgesics presented in Section I of this book. Several pharmacokinetic and pharmacodynamic concepts are explained. Terms such as opioid naïve and opioid tolerant are distinguished, and addiction, pseudoaddiction, physical dependence, tolerance, and cross tolerance are defined. The research and recommended diagnostic and treatment approaches for opioid-induced hyperalgesia are presented.
Groups of Opioids
The opioid agonist drugs can be divided into two major groups. The largest group is the morphine-like agonists. The terms morphine-like drugs, mu agonists, pure agonists, and full agonists are used interchangeably. Throughout this book, the term mu agonist will be used when referring to opioid drugs in this group. The other group of opioids is the agonist-antagonist group and is further divided into the mixed agonist-antagonists and the partial agonists. Opioid agonist drugs can be distinguished from opioid compounds more generally. Opioid compounds include both drugs and endogenous chemicals (generically called the endorphins); they also refer to both agonists, which produce effects at opioid receptors, and antagonists, which block or reverse effects produced by the agonists.
Underlying Mechanisms of Opioid Analgesia and Adverse Effects
To understand the appropriate use of opioids in the treatment of pain, it is valuable to review the mechanisms that are described by the term nociception. Nociception refers to the normal functioning of physiologic systems that lead to the perception of noxious stimuli as painful. In short, it means “normal” pain transmission. Nociception is described in Section I of this book. Review of Section I is advised so that the following discussion of the underlying mechanisms of the analgesia and adverse effects of opioids is clear.
Endogenous Opioid System
As explained in the discussion of nociception in Section I, the modulation (inhibition) of pain involves the release of dozens of neurochemicals by peripheral and central systems. Endogenous opioids (internal naturally-occurring), for example, are found throughout the periphery and central nervous system (CNS), including in the cardiovascular (CV) and gastrointestinal (GI) systems, pituitary gland, and in immune cells (Machelska, 2007; Mousa, 2003; Murphy, 2006; Rittner, Brack, 2007). It is thought that opioids given therapeutically activate endogenous pain-modulating systems and produce analgesia and other effects by binding to opioid receptor sites and mimicking the action of endogenous opioid compounds. Endogenous opioids are composed of three distinct families of peptides (naturally occurring compounds of two or more amino acids), all pharmacologically related to morphine: enkephalins, dynorphins, and β-endorphins (Gutstein, Akil, 2006; Inturrisi, 2002). Other endogenous peptides that have been more recently discovered—the endomorphins and orphanin-FQ—also appear to be important in pain processing, but their roles are yet poorly understood; the endomorphins bind selectively to the mu receptor, and orphanin-FQ is a ligand for another receptor, which is known as opioid receptor–like 1 (ORL1).
Although discovered in the 1970s (Gutstein, Akil, 2006), not everything is known about the physiologic role of endogenous opioids (Fine, Portenoy, 2007). They may serve as neurotransmitters, neuromodulators, and neurohormones (Inturrisi, 2002). Research is ongoing to identify more endogenous opioid compounds and add to the understanding of this role and the potential for new analgesics (Noble, Roques, 2007; Wisner, Dufour, Messaoudi, et al., 2006). Among other important activities, they are involved in hemostasis and the stress response.
Opioid Receptors
Drugs exert their effects on the body by interacting with specialized macromolecular components in cells called drug receptors. Drug receptors usually are cellular proteins, but can be enzymes, carbohydrate residues, and lipids. The binding of drug molecules to their specific receptor molecules often is described as similar to a key fitting a lock (Figure 11-1). Binding affinity refers to the strength of attachment of a drug to the receptor site, and drugs bind with varying strength. The electromagnetic forces produced by the bond between a drug and receptor distort the configuration of the receptor molecule, changing its biochemical properties and functions. The body’s responses to the drug are a result of these changes (Bateman, Eddleston, 2007).
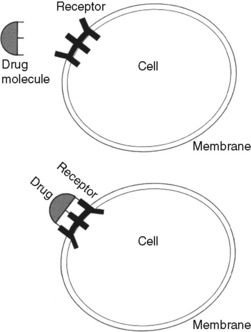
Figure 11-1 Drug and receptor interaction. The binding of drug molecules to their specific receptor molecules, often described as similar to a key fitting a lock, is shown. The electromagnetic forces produced by the bond between a drug and receptor distort the configuration of the receptor molecule, changing its biochemical properties and functions. The body’s responses to the drug are a result of these changes. From Spencer, R. T. Pharmacodynamics and pharmacokinetics. In Clinical pharmacology and nursing management, ed 4, Philadelphia, 1993, Lippincott Williams & Wilkins.) As appears in Pasero, C., & McCaffery, M. Pain assessment and pharmacologic management, p. 284, St. Louis, Mosby. May be duplicated for use in clinical practice.
Researchers think that receptors evolved for the purpose of interacting with endogenous compounds. The endogenous opioid system is an excellent example of this interaction. Opioid receptors are particularly abundant in the periacqueductal gray (PAG) and dorsal horn of the spinal cord. They are also located in the brainstem, thalamus, and cortex. Their presence in the midbrain PAG, nucleus raphe magnus, and the rostral ventral medulla help to inhibit pain via the descending modulatory system (Inturrisi, 2002). (See Section I and Figure I-2, D on pp. 4-5.)
Nociceptors—the primary afferent neurons that carry information about noxious stimuli from the periphery—terminate in the dorsal horn of the spinal cord. These cells release neurotransmitters, such as adenosine triphosphate, glutamate, and substance P, to further pain transmission. This is one of the sites at which endogenous and exogenous opioids play an important role in pain control by binding with opioid receptors, reducing the influx of calcium at the cellular level, and, among other functions, blocking the release of presynaptic neurotransmitters, principally substance P (Inturrisi, 2002). They also increase potassium influx, resulting in a decrease in synaptic transmission.
Opioids also reduce pain transmission by activating inhibitory pathways that originate segmentally (in the spinal cord) and supraspinally (Inturrisi, 2002). For example, the gamma aminobutyric acid (GABA) pathway is one of the major inhibitory neurotransmitter systems (see Section I), and opioids can activate the GABA system, leading to inhibition of pain transmission (Bridges, Thompson, Rice, 2001).
For many years, opioid analgesics were thought to produce analgesia only through the CNS. However, more recently, opioid receptors have been found also on peripheral terminals of sensory nerves and cells of the immune system (Fine, Portenoy, 2007; Inturrisi, 2002; Machelska, 2007; Mousa, 2003; Rittner, Brack, 2007). Their location in peripheral tissues has led to research suggesting that opioids also can produce analgesia following local administration by binding to the peripheral opioid receptors (Inturrisi, 2002; Zajaczkowska, Wlodzimierz, Wordliczek, et al., 2004). This may account for the antiinflammatory actions of opioid drugs on peripheral tissues (Nunez, Lee, Zhang, et al., 2007).
Classes of Opioid Receptor Sites
Three major classes or types of opioid receptor sites are involved in analgesia: mu, delta, and kappa. The pharmacologic differences in the various opioids are the result of their interaction with these three opioid receptor types (Inturrisi, 2002). A fourth receptor, structurally similar to the opioid receptor and designated as ORL-1 (opioid receptor–like 1), has been identified (Fine, Portenoy, 2007). The ligand for ORL-1, orphanin FQ or OFQ, induces spinal analgesia and appears to be involved in the modulation of pain but is not associated with respiratory depression. Research is needed to develop drugs that take advantage of this receptor site (Murphy, 2006). Subtypes of each of the main opioid receptor types have also been identified and may account for the wide variability in patient response to the various opioid analgesics (Fine, Portenoy, 2007; Pasternak, 2005).
When an opioid binds to the mu, delta, or kappa opioid receptor sites as an agonist, it produces analgesia as well as unwanted effects, such as nausea, constipation, and respiratory depression. Antagonists are drugs that also bind to opioid receptors but produce no analgesia. If an antagonist is present, it competes with opioid molecules for binding sites on the receptors. When a drug binds to any of the opioid receptor sites as an antagonist, analgesia and other effects are blocked. For example, naloxone, an opioid antagonist, can bind to the mu site and reverse analgesia and other opioid adverse effects, such as respiratory depression and sedation (Gutstein, Akil, 2006). See Table 11-1 for a summary of actions at opioid receptor type.
Table 11-1
Summary of Actions at Opioid Receptor Type
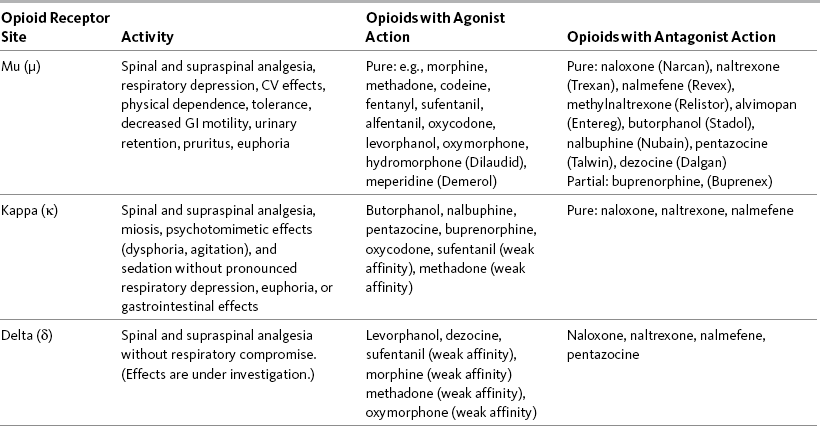
CV, Cardiovascular; GI, gastrointestinal.
The effects (activity) a drug produces depends on the type(s) of opioid receptor(s) to which the drug binds and whether the drug acts as an agonist or an antagonist at that opioid receptor type. When a drug binds to any of these receptor sites as an agonist, it produces analgesia and other effects. When a drug binds to any of the opioid receptor sites as an antagonist, analgesia and other effects are blocked. Table 11-1 summarizes the activity of drugs when they bind to any of three opioid receptor types that are involved in analgesia.
From Pasero, C., & McCaffery, M. Pain assessment and pharmacologic management, p. 285, St. Louis, Mosby. Data from Fine, P., & Portenoy, R. K. (2007). A clinical guide to opioid analgesia. New York, Vendome Group, LLC; Gutstein, H. B., & Akil, H. (2006). Opioid analgesics. In L. L. Brunton, J. S. Lazo, & K. L. Parker (Eds.), Goodman & Gilman’s the pharmacological basis of therapeutics, ed 11, New York, McGraw-Hill; Hanks, G., Cherny, N. I., & Fallon, M. Opioid analgesic therapy. In D. Doyle, G. Hanks, N. I. Cherny, et al. (Eds.), Oxford textbook of palliative medicine, ed 3, New York, Oxford Press. Pasero C, McCaffery M. May be duplicated for use in clinical practice.
Opioid drugs that produce analgesia all have agonist effects at one or more of the opioid receptor site types and are classified based on the receptors to which they bind and act (Pasternak, 2005). Most of the clinically useful opioid analgesics bind primarily to mu opioid receptor sites (Gutstein, Akil, 2006). These are the mu agonist opioid analgesics, which are considered the mainstay of analgesia for acute pain and cancer pain. Examples of mu agonist opioid analgesics are morphine, hydromorphone (Dilaudid), fentanyl, oxycodone, hydrocodone, codeine, methadone (Dolophine), and meperidine (Demerol). Mu agonists can be administered by numerous routes, and onset of analgesia is within minutes by some routes. They can be combined with almost any of the nonopioid and adjuvant analgesics.
The mixed agonist-antagonist opioid analgesics are designated as mixed because they bind to more than one opioid receptor site. They bind as agonists, producing analgesia at the kappa opioid receptor sites, and as weak antagonists at the mu opioid receptor sites (Gutstein, Akil, 2006). Mixed agonist-antagonists opioid analgesics include butorphanol (Stadol), nalbuphine (Nubain), pentazocine (Talwin), and dezocine (Dalgan). Their clinical usefulness is limited because of undesirable adverse effects (Gutstein, Akil, 2006). Some of the mixed agonist-antagonists produce more dysphoria and psychotomimetic effects than the pure mu agonists, and all appear to have a ceiling effect to the respiratory depression effects.
Although buprenorphine (Buprenex) has some kappa agonist activity, it is known primarily as a partial mu agonist drug. It is referred to as partial because it binds as an agonist at the mu opioid receptors but has limited intrinsic efficacy (Gutstein, Akil, 2006). In the clinical setting, this means that analgesia plateaus as the dose is increased. Buprenorphine also has very high affinity for the mu receptor. It is not readily reversed by opioid antagonists, such as naloxone, which suggests the drug dissociates very slowly from the mu opioid receptor sites (Gutstein, Akil, 2006). The drug is used most often as a maintenance drug for the treatment of addictive disease (Suboxone, Subutex) (see Chapter 13).
Opioid Receptors and Adverse Effects
The type of opioid receptor site and its location determine the effects an opioid drug produces. As mentioned, in addition to producing analgesia, opioid drugs produce a number of other effects, including constipation, nausea and vomiting, sedation, respiratory depression, and urinary retention (Gutstein, Akil, 2006) (see Table 11-1).
The main GI effect of opioid drugs is inhibition of GI peristalsis and diminished GI, biliary, and pancreatic secretions, which can lead to constipation and predispose to ileus and other adverse effects as a result of opioid binding to receptors located in the GI tract and CNS (Gutstein, Akil, 2006; Kraft, 2007; Thomas, 2008). Opioid bowel syndrome is described as a “constellation” of undesirable outcomes, including but not limited to constipation (McNicol, Boyce, Schumann, et al., 2008). Nausea and vomiting are the result of opioid binding to receptors located in the fourth ventricle of the brain and direct stimulation of the chemoreceptor trigger zone in the area postrema of the medulla (Freye, 2008). Urinary retention may occur when opioid binding leads to inhibition of the release of acetylcholine (Freye, 2008). Respiratory depression may follow binding in the pontine and ventral medulla of the brainstem (Freye, 2008), and sedation occurs from binding to receptors in the brain (Gutstein, Akil, 2006) (see Chapter 19 for discussion of opioid adverse effects).
Pharmacologic Concepts
After systemic (oral or parenteral) administration, an opioid drug is absorbed into the vascular system. For the drug to produce a pharmacologic effect, it must leave the plasma, diffuse into tissue, reach opioid receptors, and activate them (Gutstein, Akil, 2006). Topical administration may rely on both peripheral and systemic absorption (LeBon, Zeppetella, Higginson, et al., 2009; Ribeiro, Joel, Zeppetella, 2004; Sawynok, 2003). When administered by intraspinal routes of administration, opioids are carried via the cerebrospinal fluid (CSF) to opioid receptor sites in the spinal cord and brain (Freye, 2008). Appropriate use of opioid analgesics requires an understanding of these processes and some important pharmacologic concepts. The following is a discussion of pharmacokinetics (the movement of the drug through the body) and pharmacodynamics (what effects are produced). Tolerance, cross-tolerance, opioid-induced hyperalgesia, physical dependence, addiction, pseudoaddiction, and equianalgesia also are discussed.
Pharmacokinetics
Pharmacokinetics is the science of what the body does to a drug after its administration. Pharmacokinetic processes include absorption, distribution, metabolism, and elimination and are discussed in the following sections along with other related key concepts.
Absorption, Bioavailability, First Pass Effect, and Solubility
Absorption is the rate and extent to which a drug leaves its site of administration and moves to plasma or other tissues. A more clinically important concept than absorption is bioavailability, which is the extent to which a dose of a drug reaches its site of action (Buxton, 2006), or how much drug is available for therapeutic effect. Opioid drugs are 100% bioavailable when administered intravenously because they are introduced directly into the systemic circulation. After oral administration, opioids are absorbed from the GI tract and transported by the portal vein to the liver, the primary site of drug metabolism, before they reach systemic circulation. This process reduces the bioavailability of an opioid when administered orally (Buxton, 2006). Oral bioavailability depends on how much of the drug is absorbed in the GI tract and inactivated as it passes through the liver. This is called first pass effect. First pass effect is why the dose of an opioid drug by the oral route must be much larger than by the parenteral route to produce equal analgesia (Buxton, 2006). For example, the bioavailability of morphine when given orally usually is between 20% and 30% because of first pass losses (De Pinto, Dunbar, Edwards, 2006; Gutstein, Akil, 2006; Stevens, Ghazi, 2000) (Figure 11-2).
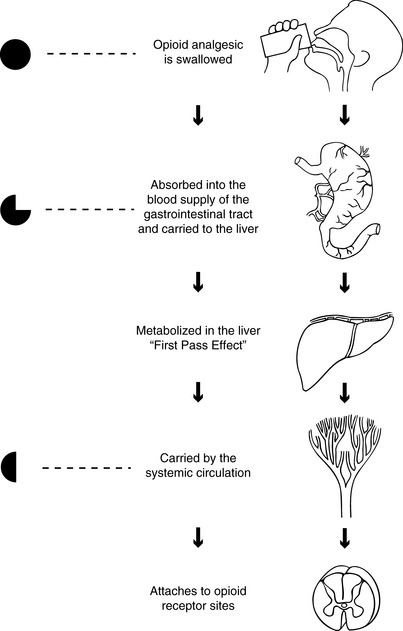
Figure 11-2 Drug absorption and metabolism by the oral route. The solid sphere represents an opioid analgesic pill. The decreasing size of the pill represents loss through absorption to the gastrointestinal tract and metabolism in the liver. The half sphere represents the dose of opioid available for analgesia at the opioid receptor sites (bioavailability) after it has been diminished by absorption and metabolism. From Pasero, C., & McCaffery, M. Pain assessment and pharmacologic management, p. 287, St. Louis, Mosby. Pasero C, McCaffery M. May be duplicated for use in clinical practice.
Many factors influence a drug’s absorption and bioavailability besides route of administration. The site of absorption, including its surface area and vascularity, is important. Drugs are absorbed rapidly from large surface areas, such as the intestinal mucosa, and when there is increased blood flow at the site (Buxton, 2006). A high concentration of drug in a small volume leads to faster absorption compared with a low concentration in a large volume. The presence of a pathologic condition also affects bioavailability. For example, bioavailability is increased in hepatic dysfunction because the liver cannot metabolize and excrete the drug efficiently (Johnson, 2007).
Characteristics of the drug itself also help to determine its bioavailability (Buxton, 2006). A drug’s bioavailability will be decreased if it is a drug for which the liver has a great capacity to metabolize and excrete, such as hydromorphone. When given intravenously, hydromorphone is 100% bioavailable and the recommended starting adult dose for severe pain is 1.5 mg given over a 4-hour period; when given orally, which subjects the drug to a significant first-pass effect, the equianalgesic dose is approximately five times greater at 7.5 mg (see later in this chapter for more on equianalgesia).
A drug’s solubility also influences its bioavailability. The more lipid soluble (also referred to as lipophilic, meaning readily dissolved into fatty tissues) the drug, the more readily it moves through membranes, which may increase bioavailability. Lipid solubility also is related to other pharmacokinetic parameters, which may be more important in practice (Buxton, 2006). For example, when highly lipid soluble opioids, such as fentanyl and sufentanil, are administered to opioid-naïve patients, the analgesic and other effects have a rapid onset and a short duration; in contrast, comparable doses of drugs that are less lipid soluble, like morphine and hydromorphone, lead to effects that have a slower onset of action and longer duration; meperidine is intermediate between these drugs. Lipophilicity and all the other factors discussed in this section can affect the efficacy and toxicity of a drug and therefore must be considered when establishing an opioid analgesic regimen.
Protein Binding
Many drugs are bound to plasma proteins, primarily albumin (Buxton, 2006). Plasma protein binding can be viewed as a transport mechanism that delivers drugs to their sites of metabolism. Plasma protein binding limits a drug’s concentration in tissues and at its site of action because only unbound drug is in equilibrium across membranes and can move freely. Bound drug is devoid of pharmacologic activity; drug responses, whether efficacious or toxic, are a function of unbound concentrations (Buxton, 2006).
Metabolism, Metabolites, and Prodrugs
When a drug passes through the liver, and often through other tissues, it is subjected to multiple biochemical processes and reactions (metabolism) that change part of the drug into different compounds. Enzymes mediate most of these processes and reactions. The resulting products are called metabolites. Many opioid analgesics have metabolites, including morphine (morphine-6-glucuronide [M6G] and morphine-3-glucuronide [M3G]), meperidine (normeperidine), and propoxyphene (norpropoxyphene). Metabolites are referred to as being active (having pharmacologic action) or inactive (having no pharmacologic action) (Buxton, 2006).
Metabolites often have properties and characteristics different from their parent drug. Sometimes their pharmacologic actions are indistinguishable from the parent drug, but their biologic activity may be increased, decreased, or eliminated. For example, the major metabolite of morphine, M6G, is analgesic like its parent but is significantly more potent (Buxton, 2006) (see Chapter 13).
Prodrugs are pharmacologically inactive compounds (sometimes called inactive precursors) that are converted quickly to active metabolites following administration (Buxton, 2006). Prodrugs are sometimes used to maximize the amount of the active drug that reaches the site of action. Codeine is an example of a prodrug. To produce analgesia, it must be catalyzed by the cytochrome (CY) P450 enzyme CYP2D6 to morphine.
Cytochrome P450 Enzyme System: Two major hepatic enzyme systems are responsible for metabolism of opioids: CYP450 and, to a lesser extent, the UDP-glucuronosyltransferases (UGTs) (Holmquist, 2009). The UGTs are involved in the formation of glucuronides and the metabolism of hydromorphone, morphine, and oxymorphone. The CYP450 enzyme system is important to the metabolism of codeine, fentanyl, methadone, oxycodone, and oxymorphone (Holmquist, 2009). CYP450 enzymes also are located in other tissues of the body and are the primary enzymes involved in drug metabolism and the production of cholesterol, steroids, prostacylins, and other essential substances (Holmquist, 2009).
Drugs interact with the CYP450 enzyme system by acting as a substrate (metabolized by one or more of the CYP450 enzymes); an inhibitor (slowing the activity of CYP450 enzyme metabolism); or an inducer (boosting the activity of CYP450 metabolism) (Holmquist, 2009). There are more than 50 different CYP450 enzymes, but not all are involved in drug metabolism. The CYP2D6 and CYP3A4 are the most important to opioid metabolism (Fine, Portenoy, 2007). Patients may lack normal levels of these enzymes as a result of genetics, hepatic disease, or competition with other medications that are metabolized by the same enzymes (Fine, Portenoy, 2007).
There are four major CYP2D6 phenotypes, which render patients poor, intermediate, extensive, or ultra-rapid metabolizers (Holmquist, 2009). There are interethnic variations in phenotypes (Palmer, Giesecke, Body, et al., 2005); it is estimated that 7% to 10% of Caucasians and 1% of Asians are poor metabolizers (Holmquist, 2009; Paice, 2008). Patients who are poor metabolizers have altered pharmacokinetics of 2D6-metabolized drugs, which may cause clinically important effects in some. For example, whereas hydromorphone, morphine, and oxymorphone are not metabolized by the CYP450 enzymes to a great extent (UGT is their primary metabolic pathway) and genetic polymorphisms of the CYP450 enzymes have little effect, the prodrug codeine relies on 2D6 to become the active compound morphine, and slow metabolizers may not respond well to codeine as a result (Fine, Portenoy, 2007; Palmer, Giesecke, Body, et al., 2005). Ultra-rapid metabolizers at 2D6 may have an exaggerated (i.e., toxic) response to codeine (Voronov, Przybylo, Jagannathan, et al., 2007; United States Food and Drug Administration [U.S. FDA], 2007a; Palmer, Giesecke, Body, et al., 2005).
Some drugs compete with opioids for metabolism by specific enzymes, which may result in drug-drug interactions. Table 11-2 lists potential drug interactions for the CYP2D6 and CYP3A4 enzymes. Box 11-1 provides websites where information can be found regarding drugs that are metabolized by the cytochrome P450 enzyme system and their potential drug-drug interactions.
Table 11-2
Potential Drug Interactions for Major Cytochrome P-450 Enzymes CYP3A4 and CYP2D6
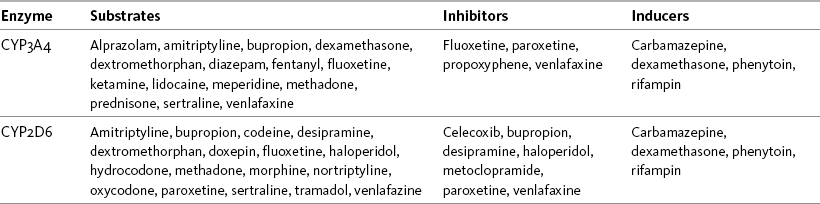
From Pasero, C., & McCaffery, M. Pain assessment and pharmacologic management, p. 289, St. Louis, Mosby. Data from Clinical Pharmacology Online. Gold Standard, Inc. Available at http://clinicalpharmacology.com; Cytochrome P450 Interactions ©GlobalRPh Inc. Available at http://www.globalrph.com/cytochrome.htm; Cytochrome P450 and Drug Interactions (Drugs that Induce or Inhibit Various Cytochrome P-450 Systems). Available at http://www.edhayes.com/CYP450-3.html. Pasero C, McCaffery M. May be duplicated for use in clinical practice.
Half-Life, Clearance, Steady State, and Accumulation
Drugs are eliminated from the body either unchanged or as metabolites. The kidney is the primary organ for elimination of drugs and metabolites. Drugs are also excreted in the feces, breast milk, sweat, saliva, tears, hair, and skin. The pulmonary route of excretion is important mainly for anesthetic gases and vapors (Buxton, 2006).
Terminal half-life provides an estimate of how fast a drug leaves the body. By definition, half-life is the time it takes for the amount of drug in the body to be reduced by 50% (Buxton, 2006). Half-life varies significantly from one drug to another. For example, the half-life of morphine is 2 to 4 hours, whereas the half-life of methadone ranges from 4.2 hours to 130 hours in some individuals (Lynch, 2005). Terminal half-life is different from, but sometimes confused with, distribution half-life, which reflects the time necessary for a drug to move from the blood and plasma to other tissues.
Clearance is also a measure of the body’s ability to eliminate a drug from the body. The clearance of a drug depends on the organs of elimination coming in contact with the blood or plasma containing the drug (Buxton, 2006). Because the kidney is the major organ of elimination, renal insufficiency can alter drug clearance. Creatinine clearance analysis is a measure of kidney function and a tool to determine the body’s ability to handle drugs that are primarily eliminated by the kidney. A high creatinine level may be an indication of reduced kidney function and, therefore, a reduced ability to eliminate a drug. Normally, men tend to have higher creatinine levels than women. Aging is associated with decreased body mass and total body water and an increased proportion of body fat which can alter the rates of clearance and elimination (American Geriatrics Society [AGS], 2009; Cook, Rooke, 2003). Box 11-2 provides serum creatinine and creatinine clearance values for all age groups and the formula for calculating creatinine clearance.
Box 11-2 Creatinine Values and Formula for Calculating Creatinine Clearance
Creatinine clearance can be easily estimated using the following formula*:

*For women, multiply this amount by 0.85.
From Pasero, C., & McCaffery, M. Pain assessment and pharmacologic management, p. 290, St. Louis, Mosby. Pasero C, McCaffery M. May be duplicated for use in clinical practice.
A drug’s half-life changes according to the body’s ability to clear the drug; therefore, both half-life and clearance are important to consider when developing a regimen for long-term opioid analgesic administration. Both are influenced by age, gender, disease, and body composition. For example, because clearance decreases with age, half-life can be expected to increase (AGS, 2009).
A concept closely related to half-life is steady state. A steady state is approached when the rate of excretion of a drug equals the rate at which the drug enters the system. Long-term opioid analgesic treatment is intended to maintain a steady state of opioid within the therapeutic range.
Half-life determines how long it will take for a given opioid drug to reach steady state. For example, the full effects of a change in dose of an opioid drug will not be seen until the time equal to four to five times the half-life of the opioid drug has passed (Bateman, Eddleston, 2007; Buxton, 2006).
Accumulation (or cumulation) of drug occurs when doses are administered but steady state has not yet been approached; less drug is excreted than is absorbed. When this happens, serum drug levels rise. The phenomenon of accumulation occurs whenever dosing is initiated or the dose is increased during the course of therapy; it also can occur if drug metabolism or excretion changes and clearance declines as a result. Because four to five half-lives are required to approach steady state, opioids that have a relatively long half-life can require a prolonged period of time to stabilize in the blood. During this period, the drug concentration is slowly rising and patients must be carefully monitored. If the dose is increased in an effort to produce analgesia, but the blood level continues to rise for a while after the dose is stable (until four to five half-lives pass), it is possible that unanticipated toxic effects can occur. This scenario can occur with long half-life opioids, like methadone. Problems also can occur if a drug has an active metabolite with a half-life longer than the parent compound. For example, meperidine’s metabolite, normeperidine, has a long half-life (15 to 20 hours) compared with the half-life of its parent, meperidine (2 to 3 hours). If meperidine is administered for a few days, accumulation of the normeperidine may lead to adverse effects, which include irritability, tremors, and seizures (see Chapter 13).
Pharmacodynamics
Pharmacodynamics refers to the effects of drugs on the body after their administration. Pharmacodynamic phenomena include potency, efficacy, tolerance, cross tolerance, and physical dependence and are discussed in the following sections along with other conditions, such as opioid-induced hyperalgesia and addiction.
Opioid Responsiveness: Potency and Efficacy
Terms that are sometimes confused when discussing opioid analgesics and their patients’ responses to opioid analgesic treatment include efficacy, potency, responsiveness, and resistance. The meaning of efficacy varies, depending on the context in which it is used. Intrinsic efficacy is a pharmacology term defined as the proportion of opioid receptors that must be occupied by a drug to produce a given effect. The receptor occupancy required for an agonist to produce a response is inversely proportional to its intrinsic efficacy.
In the clinical setting, efficacy refers to the extent to which a drug or another treatment “works” and can produce the effect in question—analgesia in this context. To determine whether this is the case, the treatment must be compared with another, typically a placebo, but sometimes an active comparator. Maximal efficacy refers to the maximum effect that can be produced by a drug, and comparative efficacy refers to the relative effects of two or more treatments compared at comparable treatment intensities.
The term efficacy often is contrasted and confused with the term potency, which refers to the dose of a drug required to produce a specified effect (Fine, Portenoy, 2007). Relative potency is the ratio of doses required to produce the same analgesic effect (Freye, 2008). For example, parenteral hydromorphone is more potent than parenteral morphine because the dose of hydromorphone required to achieve the same analgesia as morphine is about one sixth that of the morphine dose. On the basis of single-dose studies, the relative potency of parenteral hydromorphone and parenteral morphine is 1.5:10 (see later in this chapter for a discussion on equianalgesia).
A common misconception is that the more potent a drug is, the more therapeutically superior it is. In reality, all opioid analgesics are theoretically capable of producing the same degree of analgesia if doses are appropriately adjusted. Increased potency alone does not provide any advantage because the more potent drugs also exhibit a parallel increase in their ability to produce undesirable effects (Bateman, Eddleston, 2007).
The clinical relevance of efficacy and potency and why potency is less important than efficacy in the clinical setting is better understood by comparing the differences between butorphanol, a mixed agonist-antagonist opioid analgesic, and morphine, a mu agonist opioid analgesic. Although butorphanol is five times more potent than morphine (2 mg of butorphanol produces analgesia comparable to 10 mg of morphine), it has a maximal efficacy far lower than that of morphine. That is, butorphanol’s analgesic effect plateaus as the dose is increased, and further dose increases for unrelieved pain will produce no increase in analgesia (analgesic ceiling). On the other hand, morphine, like other mu agonist opioid analgesics, has no analgesic ceiling, and the only limiting factor in increasing its dose is the incidence and severity of adverse effects (Fine, Portenoy, 2007).
The term efficacy often is used when clinicians discuss the patient’s response to opioid analgesic treatment. A better term to describe clinical response is opioid responsiveness. Opioid responsiveness refers to the probability that adequate analgesia (an overall favorable balance between satisfactory pain relief and adverse effects that are considered tolerable and manageable) can be achieved during dose titration. This is influenced by patient characteristics and the particular type of pain or pain syndrome being treated.
It is a common misconception that certain patient characteristics, such as advancing age (AGS, 2009), or types of pain, such as neuropathic (Finnerup, Otto, McQuay, et al., 2005; Rowbotham, Twilling, Davies, et al., 2003), render a group of patients “opioid-resistant.” No evidence exists that this phenomenon of opioid-resistance occurs, and clinicians should not withhold a trial of an opioid on the basis of a priori certainty that an opioid drug will be inefficacious. It is more accurate to proceed on the assumption that all individuals and types of pain are potentially opioid responsive, but they vary in the degree to which they respond, i.e., in the probability that a favorable balance between analgesia and adverse effects will occur if an effort is made to individualize the dose. In short, it is best to view opioid responsiveness on a continuum rather than as an all-or-none phenomenon.
Opioid analgesics should be considered for the treatment of all types of pain. Presumed responsiveness should be one consideration among many in making a decision to proceed with a treatment trial (see the paragraphs that follow). For example, nociceptive somatic pains, such as cancer pain, usually are highly responsive to opioid analgesics, and this observation combined with a worldwide consensus that pain associated with active cancer should be treated with an opioid supports the view that opioid therapy is first-line in this context (Miaskowski, Cleary, Burney, et al., 2005). In contrast, although opioids clearly work in neuropathic pain, guidelines consider them as second-line approaches in most settings (Dworkin, O’Connor, Backonja, et al., 2007). This has many reasons, but one relates to the view that neuropathic pain may be relatively less responsive to opioid therapy than other types of pain and that the opioid doses needed to control neuropathic pain may be more likely to cause intolerable adverse effects (Rowbotham, Twilling, Davies, et al., 2007).
Opioid responsiveness is affected by both the inherent response of the pain to the therapy and also to the predisposition to opioid-related adverse effects. Any factor that increases the risk of dose-limiting toxicity, such as advanced age or major organ dysfunction, will reduce responsiveness.
Significant variation exists in patients’ responsiveness to opioid analgesics (interindividual differences). With the same opioid, one patient may achieve excellent analgesia with few adverse effects, whereas another patient experiences intolerable adverse effects with minimal or no analgesia. Patients can vary also in their individual responsiveness to different opioid analgesics (intraindividual differences). For example, a patient may experience unacceptable nausea and poor analgesia with one opioid analgesic and no nausea but unacceptable somnolence and lesser, equal, or better analgesia with another.
Tolerance
In a consensus statement developed by the American Academy of Pain Medicine (AAPM), the American Pain Society (APS), and the American Society of Addiction Medicine (ASAM), tolerance is defined as “a state of adaptation in which exposure to a drug induces changes that result in a diminution of one or more of the drug’s effects over time” (ASAM, 2001). In other words, tolerance is a state of adaptation characterized by decreasing effects of the drug at a constant dose or, conversely, the need for a higher dose of drug to maintain an effect (Fine, Portenoy, 2007). The development of acute tolerance, such as may occur when remifentanil is used in surgical patients, has recently been recognized, but the implications of this phenomenon remain uncertain (Fukuda, 2005). In humans, it is likely that tolerance to a variety of opioid effects, including analgesia, can begin to develop after the first dose of opioid, but this is seldom clinically significant (Webster, Dove, 2007). The focus in the clinical setting is on tolerance that develops following continued exposure to opioids, i.e. continued exposure to the drug is the primary cause.
Clinicians often incorrectly conclude that patients with chronic disease who experience diminished analgesia after a period of stable dosing have developed tolerance. A diagnosis of tolerance cannot be made until other causes of decreased pain relief, such as new pathology, disease progression, or lack of adherence to the medication treatment plan, can be ruled out (Fine, Portenoy, 2007). A differential diagnosis is required.
The underlying mechanism of tolerance is not well understood. Several theories involving changes in opioid receptors have been proposed (DuPen, Shen, Ersek, 2007). For example, one theory proposes that opioid receptors become desensitized. However, no agreement has been reached, and the issue remains complex (Angst, Chu, Tingle, et al., 2009; Simonnet, 2009).
Tolerance should not be confused with addiction or physical dependence (described in the following paragraphs), and it is not a predictor of abuse (Fine, Portenoy, 2007). Further, opioid-induced hyperalgesia (discussed in the following paragraphs) and tolerance are two distinct, albeit related, phenomena (Chu, Angst, Clark, 2008).
Although many clinicians suggest that opioid tolerance should be expected with long-term opioid treatment (Miaskowski, Cleary, Burney, et al., 2005), tolerance to the analgesic effects of an opioid may or may not be evident during long-term therapy. Patients who do not have progressive painful disease often achieve a stable dose after titration and require no increase in opioid doses, unless other reasons occur, as mentioned earlier in the chapter (Portenoy, Foley, 1986; Portenoy, Maldonado, Fitzmartin, et al., 1989). There is no specific timeframe for the development of tolerance (Wu, 2005), and, although there may be patient characteristics associated with an increased likelihood of analgesic tolerance (e.g., tolerance may develop more quickly in younger individuals than in older individuals [Buntin-Mushock, Phillip, Moriyana, et al., 2005]), there is great variation among individuals (Webster, Dove, 2007), and clinicians cannot predict those patients who will or will not develop a pattern of opioid response consistent with this phenomenon.
Tolerance develops to both the desirable and undesirable effects of opioids. Tolerance to the various effects of opioids differs, and the rate at which tolerance develops varies (Webster, Dove, 2007). Tolerance to the nonanalgesic effects of an opioid drug (i.e., adverse effects) occurs and can be beneficial in achieving a balance between analgesia and adverse effects. Tolerance usually develops quickly to respiratory depression (ASAM, 2001), and tolerance to respiratory depression may precede or be more profound than tolerance to sedation; as a result, observations of sedation level are a good way to monitor for impending opioid-induced respiratory depression (see Chapter 19).
Although tolerance to respiratory depression is never complete, it allows patients to escalate doses to a level required for analgesia. Studies have shown that patients with cancer pain may require and safely receive daily doses ranging from 30 to 7000 mg of morphine (or equivalent) (Foley, 1995). In a study of 435 home-care hospice patients, 7 required doses equal to or exceeding 600 mg of oral morphine a day (Bercovitch, Adunsky, 2004). In a study of patients on very high doses of IV methadone, one patient received 1920 mg per day (Vadalouca, Moka, Argyra, et al., 2008). Obviously, these high doses are not common, but they demonstrate the degree of tolerance to respiratory depression that some patients develop. Although there appears to be no limit to the degree of tolerance to respiratory depression that may develop, there is always a lethal dose for the individual.
In contrast to the tolerance to respiratory depression, somnolence and mental clouding, and nausea, all of which usually occur within days or weeks (Foley, 1995; Portenoy, 1996), tolerance to constipation more typically occurs far more slowly, or not at all. It is generally accepted that patients, especially those who are otherwise predisposed to constipation, e.g., older adults, should be routinely treated with a preventative bowel regimen if long-term opioid analgesic therapy is undertaken (Coyle, Cherny, Portenoy, 1995) (see Chapter 19).
Tolerance to analgesia may be evident after a few days of treatment. The first indication of tolerance is most commonly a decrease in the duration of analgesia for a given opioid dose (Miaskowski, Cleary, Burney, et al., 2005) followed by a decrease in analgesic effect. This can be treated easily, usually by increasing the opioid dose or decreasing the interval between doses.
After the opioid is titrated to an acceptable dose, the dose stabilizes if the pain is stable. Further dose escalation in patients with stable pain syndromes is unusual. Thus, stable pain leads to stable doses (Foley, 1993; Levy, 1989; Miaskowski, Cleary, Burney, et al., 2005). If analgesia does decline, it is handled easily, usually by increasing the dose. “There is no arbitrary ceiling beyond which a dose of opioids is unsafe.” (p. 23, Webster, Dove, 2007). Further, as stated by ASAM (2001), “Tolerance to the analgesic effects of opioids is variable in occurrence but is never absolute; thus no upper limit to dosage of pure opioid agonists can be established.” For these reasons, clinicians should not withhold opioid analgesia from patients with long life expectancies or delay initiating opioid analgesic treatment for fear of producing tolerance or reaching a dose beyond which no further analgesia can be obtained (see Chapter 20).
It is important to reinforce information about tolerance and lack of a ceiling effect to patients and families. They are frequently reluctant to begin opioid analgesic treatment because they are concerned that the effectiveness of opioid analgesics will diminish over time and that the patient will be subjected to severe pain in later stages of disease if the opioid is started in the early stages (Portenoy, 1996). The patient and family should be reassured that as pain increases, the opioid dose probably can be increased and other analgesics, such as nonopioids and adjuvants, can be added.
Pain itself may diminish the adverse effects of opioids. Evidence exists that patients thought to be tolerant to the adverse effects of an opioid drug can experience a return of adverse effects if the pain is lessened or eliminated. Consider a patient who is tolerant to opioid-induced sedation and nausea associated with high doses of morphine taken for pain related to a malignant lesion. The patient may once again experience all of the adverse effects of morphine, including respiratory depression, if an intervention such as cordotomy suddenly eliminates the pain. Under these circumstances, patients may develop signs and symptoms of overdose if the opioid dose is not reduced promptly after the intervention (Portenoy, 1996).
The treatment of acute pain in those receiving long-term opioid therapy for persistent (chronic) pain may be a challenge. It is impossible to predict the degree of tolerance to analgesia and to other effects such as respiratory depression. In opioid-tolerant patients with acute pain, such as postoperative pain, high pain ratings are common (Patanwala, Jarzyna, Miller, et al., 2008). The clinician needs to treat pain based on these pain ratings but also on the observation of safety factors such as sedation levels and respiratory status. Ideally a treatment plan is created preoperatively that assures continuation of the patient’s baseline opioid requirements and also considers the use of additional drugs, such as nonopioids and local anesthetics (Patanwala, Jarzyna, Miller, et al., 2008; Wu, 2005). This is discussed in more detail later in the section (see Chapters 16 and 20).
Incomplete Cross-Tolerance
Drugs that act at the same receptor can produce different levels of tolerance, in part because similar drugs (such as the mu agonists) have different intrinsic efficacies. Additionally, each opioid drug interacts with a different group of receptor subtypes (de Leon-Casasola, 2008; Pasternak, 2001, 2005). For these reasons, a switch from one opioid to another similar drug (e.g., from one mu agonist to another) is associated with a variable degree of tolerance to different effects. For example, an animal can be made tolerant to morphine’s analgesic effect, but partial analgesia will reappear on switching to another mu agonist such as hydromorphone.
Clinicians have wondered why analgesics that act at the same mu opioid peptide receptor can result in such a range of responses in individuals, such as differences in adverse effects, and why switching from one mu agonist to another results in incomplete cross-tolerance. One reason is related to the presence of multiple mu opioid peptide receptor subtypes that may be genetically different (de Leon-Casasola, 2008; Pasternak, 2001, 2005). Thus, incomplete-cross-tolerance among mu opioids may be explained in part by their different selectivities for the receptor subtypes.
In opioid rotation, or switching from one mu agonist to another, the principle of incomplete cross-tolerance is said to be the keystone to success. Patients with persistent pain who have been on high-dose opioid therapy are likely to be significantly sensitive to a new opioid (Knotkova, Fine, Portenoy, 2009; Vadalouca, Moka, Argyra, et al., 2008). Therefore, when switching to another opioid, it is vital for clinicians to assume that cross-tolerance exists but will be incomplete (Foley, 1995). This means that the starting dose of the new opioid must be reduced by at least 25% to 50% of the calculated equianalgesic dose to prevent overdosing; otherwise the full calculated equianalgesic dose of the new opioid could lead to effects such as sedation that would be greater than expected (Fine, Portenoy, Ad Hoc Expert Panel on Evidence Review and Guidelines for Opioid Rotation, 2009; Knotkova, Fine, Portenoy, 2009; Indelicato, Portenoy, 2002; Vadalouca, Moka, Argyra, et al., 2008). A methadone dose may need to be reduced by as much as 90% (Fine, Portenoy, Ad Hoc Expert Panel on Evidence Review and Guidelines for Opioid Rotation, 2009) (see Chapter 13 for conversion to methadone). Then the dose is gradually increased as needed to the point of pain relief and tolerable, manageable adverse effects (Coyle, Cherny, Portenoy, 1995; Levy, 1993; Indelicato, Portenoy, 2002).
An example of a clinical implication of incomplete cross-tolerance is that a patient receiving morphine may have developed tolerance to nausea, but when switched to the new opioid drug may experience severe nausea. Thus surveillance of all of the opioid’s effects is warranted when switching to an alternative opioid (see Chapter 18).
Opioid-Tolerant versus Opioid-Naive
The terms opioid-tolerant and opioid-naïve are used to distinguish between patients who have, or have not, respectively, been taking opioid drugs regularly. Whereas an opioid-naïve individual has not recently taken enough opioid on a regular basis to become opioid-tolerant (i.e., tolerant to the effects of an opioid), an opioid-tolerant individual has taken opioids long enough at doses high enough to develop tolerance to many of the effects of the opioid, including analgesia and sedation. Unfortunately, as mentioned earlier in the chapter, there is no set time for the development of tolerance (Wu, 2005), and there is great variation among individuals, with some not developing tolerance (Webster, Dove, 2007). Therefore, it is difficult to determine if and when an individual on regular doses of opioids has become tolerant. Consequently, there is no widely accepted definition for classifying a patient as opioid-tolerant (Patanwala, Jarzyna, Miller, et al., 2008).
Clinicians appear to agree that if a patient has been on long-term opioid therapy, opioid tolerance should be expected (Miaskowski, Cleary, Burney, et al., 2005). By convention, many clinicians will consider a patient who has used opioids regularly for approximately seven days or more to be opioid-tolerant. For example, in a study of patients undergoing total knee arthroplasty, patients were considered opioid-tolerant if they had taken 30 mg or more of oral morphine a day, or its equivalent, for one week prior to surgery (Patanwala, Jarzyna, Miller, et al., 2008). Patients who took 10 mg or less of oral morphine a day, or its equivalent, for the week preceding surgery were considered opioid-naïve. Grades of opioid tolerance are seldom discussed, but a few clinicians have suggested that patients requiring the equivalent of 1 mg or more of intravenous (IV) morphine or 3 mg or more of oral morphine per hour for longer than 1 month may be considered to have a high grade of opioid tolerance (Mitra, Sinatra, 2004).
Failing to appreciate the differences between opioid-tolerant and opioid-naïve individuals can lead to overdosing opioid-naïve patients and underdosing opioid-tolerant patients (Pantanwala, Jarzyna, Miller, et al., 2008). This is especially evident with the use of IV patient-controlled analgesia (PCA). A continuous infusion postoperatively is recommended for opioid-tolerant patients (Rozen, DeGaetano, 2006). In the use of opioids to manage persistent cancer pain, patients often receive the major portion of their opioid requirement as a continuous infusion without experiencing adverse effects such as sedation (APS, 2003). Many clinicians have discovered that the doses of opioid analgesics tolerated well by postoperative opioid-tolerant patients can produce significant adverse effects in opioid-naïve postoperative patients (Parker, Holtmann, White, 1991). For example, compared with opioid-tolerant patients, some studies have shown that opioid-naïve postoperative patients may be more likely to experience opioid-induced sedation with a continuous opioid infusion (APS, 2003). However, others have found that the addition of a low continuous infusion to IV PCA can be done safely in many opioid-naïve patients (Pasero, McCaffery, 2004) (see Chapter 17).
Clinicians unfamiliar with caring for opioid-tolerant patients are likely to be fearful of the high doses often required, and as a result, the patient may be underdosed. However, the opioid-tolerant patient has developed tolerance to most of the opioid adverse effects. Tolerance to the adverse effects of opioids develops more rapidly than to analgesia, meaning that opioids may be safely titrated to high doses to provide adequate analgesia (Mehta, Langford, 2006). Most importantly, the occurrence of respiratory depression is rare in opioid-tolerant individuals whose doses are carefully titrated. The fear of producing respiratory depression in these individuals usually is overstated. This fear should not interfere with adequate opioid dosing. In addition, compared with opioid-naïve individuals, opioid-tolerant individuals are generally able to tolerate faster escalation of larger doses of opioid drugs without experiencing life-threatening adverse effects (Foley, 1995).
Unfortunately, there are no evidence-based guidelines for predicting postoperative opioid requirements on the basis of the opioid dose consumed before surgery. A small prospective observational study (N = 29) classified patients as opioid tolerant or opioid naïve based on their preoperative daily opioid requirements; those taking 10 mg or less of oral morphine equivalent were designated opioid naïve, and those taking at least 30 mg of oral morphine equivalent were designated opioid tolerant (Pantanwala, Jarzyna, Miller, et al., 2008). Significantly higher pain scores and postoperative opioid consumption were noted in the opioid tolerant patients compared with the opioid naïve patients; median IV morphine equivalents were 56 mg, 108 mg, and 152.3 mg for the opioid tolerant patients and 8.2 mg, 20.5 mg, and 25 mg in opioid naïve in the PACU, during the first 24 hours after discharge from the PACU, and 24 to 48 hours after discharge from the PACU, respectively. One suggestion is to expect opioid requirements postoperatively in the opioid-tolerant patient to be two to four times the dose required in an opioid-naïve individual (Carroll, Angst, Clark, 2004). Clearly, more research is needed on opioid dose requirements in opioid tolerant patients in the postoperative setting and those who require high postoperative opioid doses in addition to their usual preoperative dose must be watched closely for sedation and respiratory depression (see Chapter 19).
Doses of opioid required by the opioid-tolerant patient with cancer pain also are sometimes extremely high. In one study, the maximum doses required for acceptable analgesia were 80 mg/h of morphine, 50 mg/h of meperidine (use of this drug is not recommended; see Chapter 13), and 60 mg/h of hydromorphone. Adverse effects and safety profiles were acceptable (Kerr, Sone, DeAngelis, et al., 1988). Some cancer patients have required as much as 40,000 mg of IV morphine per 24 hours to achieve adequate analgesia (Weinstein, 1994). These findings reinforce that in many patients no ceiling on analgesia exists when mu agonist opioid analgesics are used, and “high” doses are safe and appropriate in many opioid-tolerant individuals. It is prudent to note, however, that new information about the phenomenon of opioid-induced hyperalgesia obligates the clinician to be particularly careful when doses need to be escalated to relatively high levels.
Opioid-Induced Hyperalgesia (OIH)
Hyperalgesia means increased sensitivity to pain. Opioid-induced hyperalgesia (OIH) is a paradoxical situation in which increasing doses of opioid result in increasing sensitivity to pain (Compton, 2008). OIH has only recently been identified as a clinical reality (Mitra, 2008). The incidence of clinically significant OIH has not been determined, but it appears to be a rare but serious consequence of opioid administration (Angst, Clark, 2006; Chu, Angst, Clark, 2008). Compton (2008) has hypothesized that the incidence of OIH may be much greater, since the large number of patients with persistent noncancer pain who fail to get relief from opioids or discontinue opioid therapy may actually have undiagnosed OIH. As yet, it is not possible to predict who will develop OIH as a result of opioid exposure.
Some experts characterize OIH and analgesic tolerance as “opposite sides of the coin.” In tolerance, increasing doses of opioid are needed to provide the same level of pain relief because opioid exposure induces neurophysiologic changes that reverse analgesia; in OIH, opioid exposure induces neurophysiologic changes that produce pain or increase sensitivity to noxious input (Angst, Clark, 2006). In other words, tolerance may be inferred clinically when opioid treatment leads to decreased sensitivity to opioid analgesia over time (in the absence of another process that would explain this), whereas OIH may be inferred clinically when opioid treatment leads to increased pain or sensitivity to pain over time (DuPen, Shen, Ersek, 2007).
Individuals with OIH may not have increased sensitivity to all types of pain. One study showed that individuals with OIH had increased sensitivity to cold but not heat (Chu, Clark, Angst, 2006). Another study of methadone-maintained patients found more pronounced hyperalgesia to cold than electrical pain (Doverty, White, Somogyi, et al., 2001).
The mechanisms underlying OIH remain largely unknown. In general, OIH is thought to be the result of changes in the central and peripheral nervous systems that upregulate pain-processing mechanisms, which results in increased transmission of nociceptive signals (Compton, 2008). The mechanism presumably involves neuroexcitatory and possibly pronociceptive (pain facilitation) effects of specific molecules in the brain and spinal cord (Compton, 2008; Mitra, 2008). The best studied OIH mechanism involves the upregulation of excitatory N-methyl-d-aspartate (NMDA) receptors in dorsal horn neurons (Compton, 2008). Another mechanism involves neuroplastic changes that result in part from the activation of descending pain facilitation mechanisms arising in the brain (Mitra, 2008).
A small prospective study of opioid-naïve patients with moderate to severe persistent low back pain suggested that OIH could occur within 4 weeks after exposure to moderate doses (median dose 75 mg/day) of oral morphine (Chu, Clark, Angst, 2006). A few studies have examined the occurrence of OIH after acute perioperative opioid exposure, but the results have been mixed (Chu, Angst, Clark, 2008).
Although the majority of case reports of OIH concern systemic or intrathecal administration of morphine, raising the possibility that morphine metabolites may be involved in OIH (Chu, Angst, Clark, 2008), hyperalgesia has also been demonstrated in patients with opioid addiction receiving methadone maintenance therapy (Chu, Angst, Clark, 2008), and it has been observed anecdotally during treatment with other opioids. Development of OIH may vary between different opioid medications, but which opioids are most likely to contribute to OIH is as yet unknown (Compton, 2008). Some evidence suggests that not all opioids produce OIH, and efforts are being made to identify these (Yaksh, Harty, 1988).
OIH is suspected when increasing doses (usually high, rapidly escalating doses) of opioid fail to relieve pain and actually make pain worse at the original site of pain or at other sites (Chu, Angst, Clark, 2008; Mitra, 2008). OIH may involve unexplained pain, diffuse pain (even the whole body), and diffuse allodynia (Compton, 2008; Chu, Angst, Clark, 2008; Mitra, 2008). Allodynia may be so severe that gentle touch or movement causes generalized tenderness of the skin and soft tissue (Angst, Clark, 2006). OIH may be accompanied by generalized neuroexcitation such as agitation, multifocal myoclonus, seizures, and even delirium (Mitra, 2008).
Similar to the suspicion of tolerance, the possibility that OIH is occurring demands a careful re-evaluation to determine whether other reasons for failure of the opioid to relieve pain exist. These include increase in pain pathology, opioid or other types of withdrawal, opioid addiction or pseudoaddiction, and other phenomena (Compton, 2008). Table 11-3 gives a brief description of these conditions compared with OIH and suggests some distinguishing characteristics. More detail is presented in the paragraphs that follow. Each of these conditions merits a different response or treatment. In all of these conditions, except OIH, pain will improve with increasing opioid doses.
Guidelines
Table 11-3
Differential Assessment of Opioid-Induced Hyperalgesia vs. Other Conditions
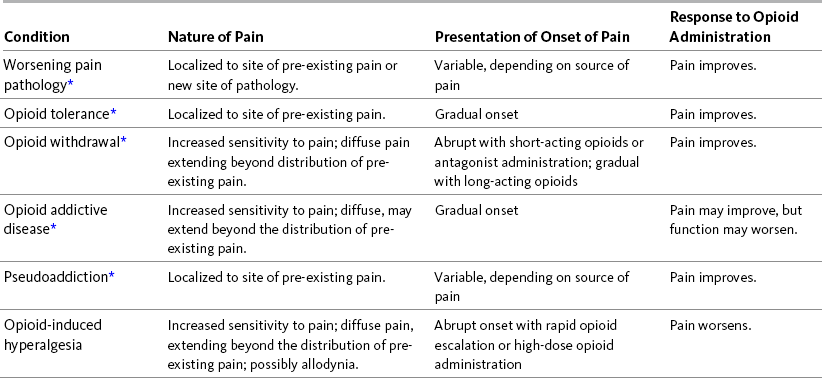
*If careful evaluation reveals that none of the above conditions is causing increased pain sensitivity and the following characteristics are largely descriptive of the current presentation, then it is reasonable to suspect the existence of OIH.
As appears in Pasero, C., & McCaffery, M. Pain assessment and pharmacologic management, p. 296, St. Louis, Mosby. Modified from Compton, P. (2008). The OIH paradox: Can opioids make pain worse? Pain Treatment Topics. Available at: http://pain-topics.org/pdf/Compton-OIH-Paradox.pdf. Accessed February 16, 2009. Pasero C, McCaffery M. May be duplicated for use in clinical practice.
When an opioid analgesic appears to lose its effectiveness, one obvious reason is that the pain pathology has worsened. This situation requires a careful evaluation of possible causes of an increase in existing pain or new sites of pain. In a patient with persistent cancer-related pain, this may involve increased tumor growth or a new tumor site. This simple example illustrates how important it is to the treatment of this patient to determine if OIH exists or if pain pathology underlies the appearance of decreasing effectiveness of current opioid treatment. In this patient example, appropriate responses may include adding or adjusting adjuvant analgesics, radiologic treatment, surgical excision, or increase in opioid dose.
Although the phenomena may be intricately related mechanistically, OIH should be distinguished from opioid tolerance. Initially it may seem that a patient with OIH has developed tolerance to analgesia, and that the best course of action is to increase the opioid dose. Careful evaluation is needed to exclude the possibility that the increases in opioid dose are actually worsening pain. If adverse effects of the original opioid are absent, manageable, or tolerable, a practical approach to making the differential diagnosis is to continue to increase the dose of the same opioid until pain is finally controlled with higher or more frequent doses of opioid, signifying tolerance that can be overcome by dose escalation, or until it is obvious that increased doses result in increased pain, indicating OIH. Opioid tolerance may be treated by increasing the dose or frequency of opioid, by opioid rotation, or by combining opioids (Compton, 2008; Mitra, 2008). Opioid rotation or combining opioids is also used in the treatment of OIH but must be accompanied by a decrease in the dose of the original opioid (Mitra, 2008).
Withdrawal symptoms that occur when the opioid dose is decreased or when an opioid antagonist is given result in increased pain in patients with physical dependence. This pain is similar to that of OIH in that increased sensitivity to pain may be diffuse and may extend beyond the pre-existing pain. This increase in pain needs to be differentiated from OIH. Pain associated with opioid withdrawal may be abrupt if the opioid is short acting or an antagonist is given, or it may be gradual if the opioid is long acting. Obviously, when the opioid dose is increased, pain improves, and this observation distinguishes it from OIH (Compton, 2008).
Opioid addictive disease must also be distinguished from OIH. As with pain associated with opioid withdrawal and OIH, increased sensitivity to pain in individuals with opioid addictive disease may be diffuse and extend beyond the distribution of pre-existing pain. This pain may have a gradual onset (Compton, 2008). Patients may request an increased dose, more frequent doses, or a specific opioid. This behavior is usually attributed to the addictive disease, and clinicians are reluctant to provide what the patient requests. However, a trial of increased opioid doses may result in pain relief. Unfortunately in some patients, function may worsen, and, despite their admissions of satisfactory pain relief, they may demonstrate behaviors typical of addiction such as inability to control opioid use, requests for opioids from multiple prescribers, or repeated unauthorized escalations in dose despite discussions with the patients about these behaviors.
OIH must also be differentiated from pseudoaddiction, defined as a mistaken diagnosis of addictive disease in patients with undertreated pain who manifest behaviors very similar to those typical of addictive disease, described in the previous paragraph (Weissman, Haddox, 1989). Pseudoaddiction can be distinguished from true addictive disease in that the behaviors resolve when pain is effectively treated.
If the above conditions have been ruled out and OIH is suspected, several strategies may be considered to manage it. These strategies overlap those used to address poor opioid responsiveness and include opioid rotation and efforts to reduce the opioid dose by concurrent administration of a nonopioid analgesic (Mitra, 2008), an adjuvant analgesic, or a nonpharmacologic treatment. OIH is not reversed by administration of an opioid antagonist (Angst, Clark, 2006).
Opioid rotation is one possible strategy for treating OIH. The original opioid dose is reduced, and another opioid is introduced. Incomplete cross-tolerance between opioids often allows comparable analgesia with another opioid at a lower equianalgesic opioid dose, in effect lowering the amount of opioid required for satisfactory analgesia (Compton, 2008).
In case reports of patients receiving large doses of morphine for intractable pain who had developed opioid-induced allodynia, reduction or elimination of the allodynia was achieved by reducing the morphine dose or substituting another opioid (fentanyl, sufentanil, methadone) (Angst, Clark, 2006). However, in this overview of case reports, it was not clear whether opioid-induced allodynia was always associated with OIH. The authors discussed the two as potentially separate entities.
Methadone may be particularly useful for opioid rotation because its NMDA antagonist activity reduces neuronal excitability (Compton, 2008). Many clinicians choose methadone for opioid rotation, and several case reports have shown that this is an effective way to reduce OIH (Chu, Angst, Clark, 2008). However, methadone is not a panacea and has actually worsened the patient’s condition in some situations. Note that switching from one opioid to methadone is a complex process (Fine, Portenoy, Ad Hoc Expert Panel on Evidence Review and Guidelines for Opioid Rotation, 2009; Knotkova, Fine, Portenoy, 2009), which is discussed in Chapter 13. Several conversions involving ratios depending upon the dose of the first opioid have been developed, but research has not established which is the most reliable.
Studies of the use of the NMDA antagonist dextromethorphan in OIH have shown conflicting results (Compton, 2008). A trial of this drug may be helpful for some patients, but it is not a first-line choice. In a study using ketamine, another NMDA antagonist, OIH was completely reversed, but the potential for unpleasant adverse effects with ketamine may make it a less attractive alternative (Mitra, 2008) (see Chapter 23). Although there is some evidence that perioperative use of ketamine modulates OIH or opioid tolerance and that it reduces postoperative wound hyperalgesia, some researchers think that the clinical significance of ketamine’s benefits requires larger prospective studies (Chu, Angst, Clark, 2008).
Research has begun to identify potential approaches to preventing OIH. These include proper timing of COX-2 selective NSAIDs in postoperative patients, NMDA antagonists, opioid dose-sparing medications such as certain adjuvant analgesics, and avoidance of rapid opioid dose escalation when possible. Studies have shown that COX-2 selective NSAIDs may partially reduce OIH and that proper timing of the administration is critical in producing this effect (Mitra, 2008); in postoperative pain, 40 mg of parenteral parecoxib (a COX-2 selective drug not available in the United States) 30 minutes prior to the opioid, may be helpful in preventing OIH (Troster, Sittl, Singler, et al., 2006) (see Section III). Continued use of COX-2 selective NSAIDs, as in persistent pain, is not only opioid dose-sparing but also may help prevent OIH because these medications reduce the spinal release of excitatory neurotransmitters (Mitra, 2008). Additional opioid dose-sparing approaches include gabapentin for both acute pain and neuropathic pain. Other useful adjuvants for neuropathic pain may involve other anticonvulsants and antidepressants (see Section V).
Rapid escalations in opioid plasma levels have been associated with the development of OIH. For that reason, the use of long-acting opioids seems preferable to short-acting opioids for persistent pain. The more gradual onset and offset of long-acting opioids helps avoid rapid escalations in opioid plasma levels and may minimize the development of OIH (Compton, 2008). On the other hand, one study comparing around-the-clock (ATC) opioid dosing with PRN (“as needed”) dosing in patients with persistent pain found that scheduled opioid dosing resulted in 12.4 times more opioid than the PRN dosing group (Miaskowski, Mack, Dodd, et al., 2002). No differences in pain relief were found between the two groups. Since higher doses are associated with OIH, PRN versus scheduled dosing needs to be evaluated as a strategy to prevent OIH (Chu, Angst, Clark, 2008).
It has been proposed that use of very low doses of an opioid antagonist in combination with opioid agonists may counteract the development of OIH. This approach is in the investigational stages (Compton, 2008; Sloan, Harmann, 2006). Some evidence suggests that clonidine preoperatively reduces development of opioid tolerance, but no evidence has substantiated its usefulness in OIH (Mitra, 2008).
In summary, the knowledge that long-term opioid therapy (especially if doses escalate rapidly) may actually worsen pain for selected patients adds to the list of reasons to be cautious about the use of opioids and particularly vigilant if a patient appears to require relatively high doses. Prescribers need to be aware of ways to prevent or minimize OIH and implement these when possible. They should also be alert to symptoms of OIH and know how to make a differential diagnosis. Fortunately, if correctly diagnosed, OIH can be treated and satisfactory pain relief restored in most patients.
Physical Dependence
Physical dependence is defined in a consensus statement published by the ASAM (2001) as “a state of adaptation that is manifested by a drug class specific withdrawal syndrome that can be produced by abrupt cessation, rapid dose reduction, decreasing blood level of the drug, and/or administration of an antagonist.” (p. 2). The focus here is on opioids, but other drugs that patients may become physically dependent on include all of the sedative-hypnotics (including alcohol and benzodiazepines), beta blockers, corticosteroids, and antidepressants.
Physical dependence is one of the most frequently misunderstood terms and is often confused with addiction or “dependence” (Webster, Dove, 2007). The term dependence should be avoided since it is used by specialists in addiction medicine as a synonym for addiction and creates confusion between physical dependence and the type of psychological dependence that is associated with addictive disease (Fine, Portenoy, 2007). Because of this confusion, patients should not be referred to as dependent.
One source of confusion over the term dependence is related to definitions in the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revised (DSM-IV-TR) (American Psychiatric Association [APA], 2000). The manual lists withdrawal symptoms and tolerance as “defining features” of addiction, thus failing to distinguish addictive disease from the normal physiologic responses of physical dependence and tolerance. Tolerance and physical dependence are likely to be present in an individual addicted to opioids, but neither tolerance nor physical dependence is required for addiction to be present, and either phenomenon may occur independently of addictive disease. In short, physical dependence and addiction are distinct; the term physical dependence should only be applied to the overt or potential capacity to have withdrawal, the term addiction should never be used when the term physical dependence is appropriate, and the term dependence should not be used at all.
To reduce the fear that may prevent patients and families from seeking appropriate pain management, it is important that caregivers understand and reinforce to patients and families the differences between addiction and physical dependence. Patients on long-term opioid therapy are appropriately told not to stop taking their opioid medication. This offers an opportunity to discuss the reason for not stopping their medication and assure the patient and family that it does not indicate possible addiction. The patient needs to understand that the potential for withdrawal symptoms is not a problem when opioid doses are tapered as opposed to suddenly stopped (APS, 2003).
Physical dependence is an expected result of opioid use. However, how long it takes to develop physical dependence and the dose of drug required is unknown (Fine, Portenoy, 2007). Certainly in long-term therapy with opioids, physical dependence should be expected (Miaskowski, Cleary, Burney, et al., 2005). It may also occur after opioids have been administered for only a few days (Fine, Portenoy, 2007).
The presence of physical dependence is not evident until some event such as abrupt cessation of the opioid occurs that causes the withdrawal, or abstinence, syndrome. Thus, physical dependence is not problematic as long as measures are taken to avoid abrupt cessation or administration of an opioid antagonist (Fine, Portenoy, 2007).
Symptoms of acute withdrawal include anxiety, irritability, salivation, rhinorrhea, insomnia, chills alternating with hot flashes, joint and muscle pain, tearing of the eyes, sweating, nausea, vomiting, diarrhea, and abdominal cramps (APS, 2003; Miaskowski, Cleary, Burney, et al., 2005). Acute withdrawal has been likened to having the “flu” and may be mild or severe and may manifest with any combination of the precipitated phenomena (e.g., abrupt cessation of the opioid or administration of an antagonist) or with all of them. Although as a rule, higher opioid doses and longer duration of opioid treatment are associated with a more severe syndrome, there also is substantial individual variation, with some patients experiencing only mild withdrawal symptoms, even if the opioid dose is relatively high, and some experiencing such a degree of physical dependence that they have withdrawal between doses of opioid and find it difficult to stop a low-dose PRN regimen.
The onset of withdrawal symptoms after abrupt cessation of an opioid depends upon the half-life of the opioid being used. For example, codeine, hydrocodone, morphine, and hydromorphone have short half-lives, and symptoms may begin within 6 to 12 hours and peak at 24 to 72 hours. With a long half-life opioid, such as methadone or levorphanol, the onset of withdrawal symptoms may not occur for 24 hours or longer, and the symptoms are usually less severe (APS, 2003; Miaskowski, Cleary, Burney, et al., 2005).
When opioids become unnecessary because pain has been resolved, the withdrawal syndrome can be avoided by slowly tapering the dose, i.e., weaning the patient off the regimen. This process should not be referred to as detoxification because of the use of the latter term in relation to the treatment of addiction (Webster, Dove, 2007).
Several guidelines for tapering the opioid dose are available, but most offer similar plans for gradual decreases in opioid doses. The longer the patient has been taking the opioid, the longer it takes to complete the weaning process. One suggested approach to tapering the opioid in an adult is to start with one-half the previous total daily dose, and administer this for 2 days. Then decrease the dose by 25% every two days until the patient reaches a total daily dose of 30 mg a day of oral morphine or its equivalent. After 2 days at this minimum dose, the opioid may be discontinued (Miaskowski, Cleary, Burney, et al., 2005).
If anxiety, sweating, or other withdrawal symptoms are problematic, the tapering can be slowed, medications can be added to treat the symptoms (e.g., a drug for nausea or diarrhea), the patient can be “rotated” to another opioid and tapering recommenced, or clonidine (a specific treatment for opioid withdrawal) can be added, starting at a dose of 0.1 mg to 0.2 mg per day (APS, 2003).
Addiction and Pseudoaddiction
No universally accepted definition of opioid addiction exists. A consensus statement published by ASAM (2001), defines addiction as “a primary chronic, neurobiologic disease, with genetic, psychosocial, and environment factors influencing its development and manifestations. It is characterized by behaviors that include one or more of the following: impaired control over drug use, compulsive use, continued use despite harm, and craving.” (p. 2) An important message in this definition is that taking opioids for pain relief is not addiction, no matter how long an individual takes opioids or at what doses. Individuals taking opioid drugs for relief of pain are using them therapeutically. Addiction is discussed at length in Section II. A brief summary of important points is presented here.
How likely is it that addiction will occur as a result of using opioids for pain relief? This is a difficult question to frame and to answer. The specifics of the question must be clarified. Is the phenomenon being considered a diagnosis of “substance dependence disorder” as described by the APA’s Diagnostic and Statistical Manual of Mental Disorders (DSM)? This diagnosis, which requires compulsive use of a substance, is usually said to be synonymous with addiction and is distinguished from substance abuse disorder. In non-DSM language, the lack of an international consensus on the definition of addiction obviously complicates questions related to the incidence of addiction during medical treatment of pain.
During long-term therapy, the clinical concern relates not only to the occurrence of addiction per se, but also to the development of drug abuse, or even nonadherence behaviors that are too “mild” to be labeled abuse, but still raise a “red flag” for clinicians. The more clinically relevant question, therefore, is not limited to the incidence of addiction but rather to the broader occurrence of nonadherence and the extent to which this can be related to the abuse liability of these drugs.
The question is further complicated by the heterogeneity of the population. Is the issue the de novo appearance of an addictive pattern of medication use among the population of patients who have no prior history of addiction? If so, the question revolves around the incidence of iatrogenic addiction rather than relapse, and the data used to answer this must include a careful screening for prior drug abuse. Should a patient with a prior history of drug abuse, but not addiction, be in the denominator of patients treated for pain who are evaluated for the incidence of addiction?
Finally, the question is complicated by treatment issues. The incidence of addiction after short-term treatment of pain following surgery is very unlikely to be relevant to the incidence during the long-term treatment of persistent pain.
Given the uncertainties surrounding the nature of the question, the data extant to evaluate the incidence of addiction are very limited and difficult to interpret (see Section II). In an evidence-based review of all available studies on the development of drug abuse behaviors, addiction, and aberrant drug-related behavior in patients with persistent noncancer pain being treated with opioids, only four studies preselected patients for no past or present history of abuse/addiction (Fishbain, Cole, Lewis, et al., 2008). In these studies, the percentage of abuse/ addiction following opioid therapy was calculated at 0.19%. These data are reassuring, suggesting that patients with no past or present history of abuse/addiction usually remain responsible medication users over time.
A recent registry study of oxycodone (OxyContin)-treated patients who were followed for up to 3 years after participating in a clinical trial also showed a very low occurrence of problematic drug-related behavior (of 227 cases, just 6 cases of misuse and none of addiction) (Portenoy, Farrar, Backonja, et al., 2007). Because this registry included only those patients who had previously been in trials that had excluded patients with significant drug abuse, the data can only be used to evaluate the incidence of addiction among those with no (or very limited) history of abuse. Again, this number is very reassuring in terms of the rate of iatrogenic addiction among those with no history of abuse, but is not relevant to the general population of patients.
Pseudoaddiction, as the name implies, is a mistaken diagnosis of addictive disease. The term was first used and the behaviors described in a case report by Weissman and Haddox (1989). When a patient’s pain is not well controlled, the patient may begin to manifest symptoms suggestive of addictive disease. In an effort to obtain adequate pain relief, the patient may respond with demanding behavior, escalating demands for more or different medications, repeated requests for opioids before the prescribed interval between doses has elapsed, and frequent visits to the emergency department. (See Table 2-2 p. 36, for other behaviors that may be mistaken as addictive disease but actually reflect undertreated pain.)
As an example, patients who receive opioid doses that are too low or at intervals greater than the opioid’s duration of action may understandably try to manipulate the staff into giving them more analgesic. Pain relief typically eliminates these behaviors and is often accomplished by increasing opioid doses, decreasing intervals between doses, or providing an extra prescription in case it is needed.
Although pseudoaddiction can be distinguished from true addictive disease in that the behaviors resolve when pain is effectively treated, the challenge in the clinical setting typically relates to patients who may be manifesting both pseudoaddiction and drug abuse (unrelieved pain may be driving a relapse of addiction, or increased abuse among those with a history of drug abuse, and it may be exceedingly difficult to know whether an effort to relieve pain by adding medication will both lessen pain and lessen newly established abuse behaviors). Another challenge exists in the decision to call more severe behaviors pseudoaddiction, rather than addiction. Although it is probable that the patient with a history of addiction may be more likely to buy heroin if pain is unrelieved, it is obviously difficult to address this behavior as a case of pseudoaddiction. These challenges indicate that a careful assessment must be done before consideration is given to labeling a patient’s behavior as pseudoaddiction, that there must be a willingness to diagnosis both pseudoaddiction and addiction (and to treat accordingly), and that an effort should always be made to determine whether the unrelieved pain can be addressed using a treatment other than an opioid.
Equianalgesia
The term equianalgesia means approximately “equal analgesia.” Equianalgesic doses refer to the doses of various opioid analgesics that provide approximately the same pain relief. An equianalgesic chart (see Table 16-1, pp. 444-446) provides a list of analgesic doses, both oral and parenteral, that are approximately equal to each other in ability to provide pain relief. These doses are also referred to as equianalgesic dose units. Most of the doses in equianalgesic charts are based on single-dose studies, often conducted with opioid-naïve surgical patients, using morphine, 10 mg IM, for comparison (Knotkova, Fine, Portenoy, 2009). The parenteral doses listed are typical of IM doses given approximately every 3 to 4 hours. Equianalgesic dose calculation provides a basis for selecting the appropriate starting dose when changing from one opioid drug or route of administration to another. However, these calculations are just estimates and vary with repeated dosing and opioid rotation (Knotkova, Fine, Portenoy, 2009; Shaheen, Walsh, Lasheen, et al., 2009). The optimal dose for the patient is determined always by titration (Fine, Portenoy, 2007).
Conversion Charts: Conversion charts for opioid analgesics are provided by drug manufacturers to assist clinicians in converting from various other opioids to the opioid being marketed. They usually provide a conservative estimate of the dose of the new opioid that will be required. The charts often assume that the switch to the new opioid is likely to be taking place in an opioid-tolerant patient. The reasoning behind using conversion charts is that cross tolerance is not complete and patients respond differently to different opioids as discussed earlier in this chapter. Adverse effects and overdosing can be more readily avoided by using conservative estimates of the dose of the new drug. Thus conversion charts are not the same as equianalgesic charts. Rather they take into consideration equianalgesic doses and reduce that estimate.
Because conversion charts are conservative in their estimate of what the initial dose of the new drug should be, many patients require titration upward to achieve satisfactory analgesia. For example, one manufacturer of transdermal fentanyl cautions that when using the estimates from its conversion chart, 50% of the patients switched from other opioids by other routes to transdermal fentanyl will require an increase in dose after the initial dose of transdermal fentanyl (Janssen, 2008). The manufacturer of OxyContin provides a table with multiplication factors to determine the starting dose when converting from a variety of opioids to OxyContin; however, it appropriately cautions that no fixed conversion ratio will fit all patients, and the doses are a starting point and should be titrated as needed (Purdue Pharma, 2007). It is also important to note that the doses recommended in conversion tables do not yield equianalgesic doses (see Chapter 16 for more on equianalgesia).
Conclusion
This chapter lays the foundation for the content presented in the rest of the chapters in this section. An understanding of the underlying mechanisms of opioids as well as the key concepts related to pharmacokinetics and pharmacodynamics of drugs as presented here is essential to initiating and maintaining a sound pain treatment plan.