Guidelines for Opioid Drug Selection
SAFE and effective use of opioid analgesics requires the development of an individualized treatment plan. This begins with a comprehensive pain assessment, which includes clarifying the goals of treatment and discussing options with the patient and family. The need for periodic reevaluation of the goals is common and should be expected as disease progresses or, in the case of acute pain, as pain resolves (Portenoy, 1996).
Goals are stated as simply as possible and shared among the patient, family, and caregivers. Goals ordinarily include the patient’s desired pain rating and the activities that accompany this pain level. For example, a postoperative patient may identify a comfort-function goal of 3/10 to enable regular use of the incentive spirometer and may state that the mild sedation accompanying this level of analgesia is acceptable. A patient with persistent pain may identify a comfort-function goal of 2/10 as necessary for engaging in employment and may state that sedation is not compatible with this goal (see Section II).
Many factors are considered when determining the appropriate opioid analgesic for the patient with pain (Box 13-1). These include the unique characteristics of the various opioids and patient characteristics, such as pain intensity, patient age, coexisting disease, current drug regimen and potential drug interactions, prior treatment outcomes, and patient preference.
Because pain has multiple underlying mechanisms and is a multifaceted phenomenon, the use of a multimodal approach to managing all types of pain should be the rule, rather than the exception (Argoff, Albrecht, Irving, et al., 2009; Kehlet, Jensen, Woolf, 2006; Kehlet, Wilmore, 2008) (see Section I and Chapter 12). A sound treatment plan relies on the selection of appropriate analgesics from the opioid, nonopioid, and adjuvant analgesic groups.
Characteristics of Selected Mu Agonist Opioids
As discussed previously, the mu agonist opioid analgesics are capable of managing all pain intensities and are effective for many different painful conditions. They are the most common analgesics used to manage moderate to severe nociceptive pain, but they have also been shown to be effective for some neuropathic pain (Dworkin, Barbano, Tyring, et al., 2009; Dworkin, O’Connor, Backonja, et al., 2007; Eisenberg, McNicol, Carr, 2006; Gimbel, Richards, Portenoy, 2003; Maier, Hildebrandt, Klinger, et al., 2002; Watson, Moulin, Watt-Watson, et al., 2003). Mu agonists are also recommended for the management of breakthrough pain. See Table 13-1 for a summary of information on selected mu opioid analgesics. For more detail about the characteristics of the various opioid analgesics as they relate to route of administration, see Chapter 14. The equianalgesic chart in Table 16-1 on pp. 444-446 contains dosing and pharmacokinetic information on the various opioid analgesics.
Table 13-1
Characteristics of Selected Mu Opioid Agonist Drugs1
| Mu Opioid Agonist Drug | Routes Administered | Comments |
| Morphine | PO (short-acting and modified-release), SL, R, IV, IM, SC, E, I, IA | Standard for comparison. Multiple routes of administration. Several modified-release formulations available, but they are not therapeutically equivalent. Begin with lower doses in older adults. Active metabolite M6G can accumulate with repeated dosing in renal failure. 20% to 30% oral bioavailability. |
| Codeine | PO, IM, SC | Limited usefulness. Usually compounded with nonopioid (e.g., Tylenol No. 3). Used orally for mild to moderate pain, but analgesia is inferior to that of ibuprofen. IM has unpredictable absorption and high adverse effect profile; IV route not recommended, SC rarely used, and IM administration of any opioid is discouraged. |
| Fentanyl | OT, B, IV, IM, TD, E, I, IN | Fast-acting; short half-life (except TD). At steady state, slow elimination from tissues can lead to a prolonged half-life (up to 12 h). On the basis of clinical experience, fentanyl 1 mcg/h transdermally is roughly equivalent to morphine 2 mg/24 h orally2; fentanyl, 100 mcg/h parenterally and transdermally is roughly equivalent to 4 mg/h morphine parenterally.2 Opioid-naïve patients should be started on no more than 25 mcg/h transdermally. Transdermal fentanyl is not appropriate for acute pain management. OTFC and buccal fentanyl are approved for management of breakthrough pain in opioid tolerant individuals. |
| Hydrocodone | PO | Used for mild to moderate pain; available in nonopioid combination only (e.g., Vicodin, Lortab) (see Table 13-3). |
| Hydromorphone (Dilaudid) | PO, R, IV, IM, SC, E, I | Useful alternative to morphine. Metabolite may accumulate with long-term, high dose administration. Available in high-potency parenteral formulation (10 mg/mL) useful for SC infusion; 3 mg R roughly equivalent to 650 mg aspirin; oral modified-release formulation available. |
| Levorphanol (Levo-Dromoran) | PO, IV, IM, SC | Long half-life can lead to accumulation within 2 to 3 days of repetitive dosing. |
| Meperidine (Demerol) | PO, IV, IM, SC, E, I | No longer recommended for the management of any type of pain because of potential toxicity from accumulation of metabolite, normeperidine. Half-life of normeperidine is approximately 15 to 20 h; NR in older adults or patients with impaired renal function; continuous IV infusion NR. The most appropriate candidates for meperidine use are patients with acute pain who are otherwise healthy with no risk factors and are allergic to or intolerant of other opioids, such as morphine, fentanyl, and hydromorphone, or have demonstrated a more favorable outcome with meperidine than other opioid drugs. |
| Methadone (Dolophine) | PO, SL, R, IV, SC, IM, E, I | Long half-life can lead to delayed toxicity from accumulation. See text for information on methadone. |
| Oxycodone (OxyIR, OxyContin) | PO (short-acting and modified-release), IV, IM, R | Used for mild to moderate pain when combined with a nonopioid (e.g., Percocet, Tylox) (see Table 13-9). As single entity, can be used like oral morphine for severe pain. Rectal and parenteral formulation not available in the United States. Oral formulation can be administered rectally. |
| Oxymorphone (Opana, Opana ER [oral], Numorphan [parenteral, rectal]) | PO (short-acting and modified-release) IV, IM, SC, R | Used for moderate to severe pain. Available in 5 mg rectal suppositories. |
| Propoxyphene (Darvocet, Darvon) | PO | Used in combination with acetaminophen (Darvocet) and aspirin (Darvon Compound). Long half-life. Accumulation of toxic metabolite norpropoxyphene with repetitive dosing. Inappropriate for use in older adults (see Table 13-4). |
B, Buccal; E, epidural analgesia; h, hour; IM, intramuscular; I, intrathecal analgesia; IA, intraarticular; IN, intranasal; IV, intravenous; mcg, microgram; mg, milligram, mL, milliliter; M6G, morphine-6-glucuronide; NR, not recommended; OTFC, oral transmucosal fentanyl citrate; PO, oral; q, every; R, rectal; SC, subcutaneous; SL, sublingual; TD, transdermal; UK, unknown.
1See Table 16-1 on pp. 444-446, for dosing and pharmacokinetic information.
2These are the ratios used in clinical practice.
From Pasero, C., & McCaffery, M. Pain assessment and pharmacologic management, pp. 326-327, St. Louis, Mosby. Data from American Society of Health System Pharmacists. Available at http://www.ashp.org/import/news/HealthSystemPharmacyNews/newsarticle.aspx?id=3037. Accessed December 9, 2009. Barkin, R. L., Barkin, S. J., & Barkin, D. S. (2006). Propoxyphene (dextropropoxyphene), A critical review of a weak opioid analgesic that should remain in antiquity. Am J Ther, 13(6), 534-542; Burnham, R., McNeil, S., Hegedus, C., et al. (2006). Fibrous myopathy as a complication of repeated intramuscular injection for chronic headache. Pain Res Manage, 11(4), 249-252; Chamberlin, K. W., Cottle, M., Neville, R., et al. (2007). Oral oxymorphone for pain management. Ann Pharmacother, 41(7), 1144-1152; Coda, B. A. (2006). Opioids. In P. G. Barash, B. F. Cullen, & R. K. Stoelting (Eds.), Clinical anesthesia, ed 5, Philadelphia, Lippincott, Williams & Wilkins; Dale, O., Hjortkjær, R., & Kharasch, E. D. (2002). Nasal administration of opioids for pain management in adults. Acta Anaesthesiol Scand, 46(7), 759-770; Davis, M. P., Varga, J., Dickerson, D., et al. (2003). Normal-release and controlled-release oxycodone: pharmacokinetics, pharmacodynamics, and controversy. Support Care Cancer, 11(2), 84-92; De Pinto, M., Dunbar, P. J., & Edwards, W.T. (2006). Pain management. Anesthesiology Clin N Am, 24(1), 19-37; Du Pen, S., Du Pen, A., & Hillyer, J. (2006). Intrathecal hydromorphone for intractable nonmalignant pain: a retrospective study. Pain Med, 7(1), 10-15; Fick, D. M., Cooper, J. W., Wade, W. E., et al. (2003). Updating the Beers criteria for potentially inappropriate medication use in older adults: Results of a US consensus panel of experts. Arch Intern Med, 163(22), 2716-2724; Fong, H. K., Sands, L. P., & Leung, J. M. (2006). The role of postoperative analgesia in delirium and cognitive decline in elderly patients: A systematic review. Anesth Analg, 102(4), 1255-1266; Fukuda, K. (2005). Intravenous opioid anesthetics. (2005). In R. D. Miller (Ed.), Miller’s anesthesia, ed 6, St. Louis, Churchill Livingstone; Furlan, A. D., Sandoval, J. A., Mailis-Gagnon, A., et al. (2006). Opioids for chronic noncancer Pain a meta-analysis of effectiveness and side effects. Can Med Assoc J, 174(11), 1589-1594; Gupta, S., & Sathyan, G. (2007). Providing constant analgesia with OROS hydromorphone. J Pain Symptom Manage, 33(2S), S19-S24; Gutstein, H., & Akil, H. (2006). Opioid analgesics. In L. L. Brunton (Ed.), Goodman & Gilman’s the pharmacological basis of therapeutics, ed 11, New York, McGraw-Hill; Hagen, N. A., & Babul, N. (1997). Comparative clinical efficacy and safety of a novel controlled-release oxycodone formulation and controlled-release hydromorphone in the treatment of cancer pain. Cancer, 79, 1428-1437; Hale, M. E., Ahdieh, H., Ma, T., et al. (2007). Efficacy and safety of OPANA ER (oxymorphone extended release) for relief of moderate to severe chronic low back pain in opioid-experienced patients: A 12-week, randomized, double-blind, placebo-controlled study. J Pain, 8(2), 175-184; Hanks, G., Cherny, N. I., & Fallon, M. (2004). Opioid analgesics. In D. Doyle, G. Hanks, N. I. Cherny (Eds.), Oxford textbook of palliative medicine, ed 3, New York, Oxford University Press; Kalso, E. (2005). Oxycodone. J Pain Symptom Manage, 29(Suppl 5), S47-S56; Kumar, M. G., & Lin, S. (2007). Hydromorphone in the management of cancer-related pain: An update on routes of administration and dosage forms. J Pharm Sci, 10(4), 504-518; Latta, K. S., Ginsberg, B., & Barkin, R. L. (2002). Meperidine: A critical review. Am J Ther, 9(1), 53-68; Lugo, R. A., & Kern, S. E. (2004). The pharmacokinetics of oxycodone. J Pain Palliat Care Pharmacother, 18(4), 17-30; McIlwain, H., & Ahdieh, H. (2005). Safety, tolerability, and effectiveness of oxymorphone extended release for moderate to severe osteoarthritis pain. A one year study. Am J Therap, 12(2), 105-112; Miller, M. G., McCarthy, N., O’Boyle, C. A., et al. (1999). Continuous subcutaneous infusion of morphine vs. hydromorphone: A controlled trial. J Pain Symptom Manage, 18(1), 9-16; Mitchell, A., van Zanten, S. V., Inglis, K., et al. (2008). A randomized controlled trial comparing acetaminophen plus ibuprofen versus acetaminophen plus codeine plus caffeine after outpatient general surgery. J Am Coll Surg, 206(3), 472-479; Murray, A., & Hagen, N. A. Hydromorphone. (2005). J Pain Symptom Manage, 29(Suppl 5), S57-66; Prommer, E. (2006). Oxymorphone: A review. Support Care Cancer, 14(2), 109-115; Prommer, E. (2007). Levorphanol: The forgotten opioid. Support Care Cancer, 15, 259-264; Prommer, E. E. (2007). Levorphanol revisited. J Palliat Med, 10(6), 1228-1230; Quigley, C. (2002). Hydromorphone for acute and chronic pain. Cochrane Database of Systematic Reviews, issue 1. Art. No.: CD003447. DOI: 10.1002/14651858.CD003447; Quigley, C., & Wiffen, P. (2003). A systematic review of hydromorphone in acute and chronic pain. J Pain Symptom Manage, 5(2), 169-178; Riley, J., Eisenberg, E., Müller-Schwefe, G., et al. (2008). Oxycodone: A review of its use in the management of pain. Curr Med Res Opin, 24(1), 175-192; Sarhill, N., Walsh, D., & Nelson, K. A. (2001). Hydromorphone: Pharmacology and clinical applications in cancer patients. Support Care Cancer, 9(2), 84-96; Susce, M. T., Murray-Carmichael, E., & de Leon, J. (2006). Response to hydrocodone, codeine and oxycodone in a CYP2D6 poor metabolizer. Prog Neuropsychopharmacol Biol Psychiatry, 30(7), 1356-1358; United States Food and Drug Administration. Available at http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/AnestheticAndLifeSupportDrugsAdvisoryCommittee/UCM120095.pdf. Accessed December 10, 2009. Wright, A. W., Mather, L. E., & Smith, M. T. (2001). Hydromorphone-3-glucuronide: A more potent neuro-excitant than its structural analogue, morphine-3-glucuronide. Life Sci, 69(4), 409-420. Pasero C, McCaffery M. May be duplicated for use in clinical practice.
The previously used classification of opioid analgesics as “weak” or “strong” is outdated. Instead, opioid analgesics are conventionally labeled as being appropriate for the treatment of mild, moderate, or severe pain. In reality, however, all the various full mu agonists are capable of producing comparable analgesia if the dose is adjusted appropriately.
The terms short acting, immediate release, and normal release have been used interchangeably to describe oral opioids that have an onset of action of approximately 30 minutes and a relatively short duration of 3 to 4 hours. The term short acting will be used in this book to describe oral opioids with these characteristics. It should be noted that the term immediate release is misleading because none of the oral opioid analgesics have an immediate onset of analgesia. The term rapid onset may accurately be applied to drugs such as oral transmucosal fentanyl (OTFC) or buccal fentanyl because of their significantly faster onset of action compared with other short-acting opioids. The terms modified release, extended release, sustained release, and controlled release are used to describe opioids that are formulated to release over a prolonged period of time. Most often, the terms are applied to oral formulations or to transdermal formulations. The term modified release will be used in this book to describe these drugs.
Some of the generic and brand name opioid formulations have sound-alike/look-alike name similarities (e.g., hydromorphone and morphine, hydromorphone and hydrocodone, OxyContin and MSContin; Roxanol and Roxicodone), which have been blamed as a cause of medication errors (Institute for Safe Medication Practices, 2009). Some ways to avoid confusion when prescribing opioids are to use tall-man lettering (e.g., HYDROmophone, oxyCONTIN, oxyCODONE), never express doses of liquid opioids in mL alone (include mg amount), and write out opioid name modifiers (e.g., “extended release” rather than “ER” or “immediate release” rather than “IR”) (Institute for Safe Medication Practices, 2009).
The characteristics of opioid analgesics vary widely. There have been few randomized controlled trials comparing the different opioids head to head (Fine, Portenoy, 2007; Hanks, De Conno, Cherny, et al., 2001). There are also well-known, wide variations in patient response to all opioids. Therefore, no opioid can be said to be clinically superior to all others in providing analgesia across settings, indications, and populations. Understanding the unique characteristics of each helps to determine the optimal opioid analgesic for the individual patient. This chapter of the book presents a general overview of selected mu agonists that are commonly used for pain management beginning with morphine and followed by the other opioid analgesics presented in alphabetical order.
Morphine
Morphine is the prototype mu agonist opioid (Fine, Portenoy, 2007) and is the standard against which all other opioid drugs are compared (Hanks, De Conno, Cherny, et al., 2001; Inturrisi, 2002; Knotkova, Fine, Portenoy, 2009). It is the most widely used opioid throughout the world (Andersen, Christrup, Sjøgren, 2003), particularly for cancer pain (Flemming, 2010). Its role is supported by extensive research, clinical experience with its use, the availability of formulations for multiple routes of administration, and early development of modified-release formulations. In 1984, the World Health Organization designated morphine as the preferred drug for cancer pain management (WHO, 1996), and still today it is referred to as the “gold standard” for opioid analgesics (Quigley, Wiffen, 2003). This characterization can be viewed as educational; as noted previously, it is not based on comparative effectiveness or safety data. Although some guidelines continue to advocate for morphine as the preferred first-choice “strong” opioid (Hanks, De Conno, Cherny, et al., 2001; Donnelly, Davis, Walsh, et al., 2002), others recognize the lack of evidence to support a preferred status and are less definitive, instead preferring to recommend mu agonist opioids as a class (American Pain Society [APS], 2003; National Comprehensive Cancer Network, 2008). A Cochrane Collaboration Review of 54 studies of oral morphine for cancer pain concluded that well-controlled research with large numbers of patients is lacking, but that existing studies generally confirm that morphine is effective, with the most common adverse effects being constipation, nausea, and vomiting (Wiffen, McQuay, 2007).
In addition to the large role that it plays in the worldwide treatment of cancer pain (Donnelly, Davis, Walsh, et al., 2002; Hanks, Cherny, Fallon, 2004), morphine has a long history as a primary drug for acute postoperative pain management (McCartney, Niazi, 2006) and has been used to treat a wide range of other painful conditions including severe angina pectoris (Mouallem, Schwartz, Farfel, 2000), AIDS-related pain (Kaplan, Slywka, Slagle, et al., 2000), and prehospital admission trauma and medical conditions (Ricard-Hibon, Belpomme, Chollet, et al., 2008). As part of a standard anesthetic regimen, IV morphine, but not IV fentanyl, suppressed several components of the inflammatory response to cardiopulmonary bypass in patients undergoing coronary artery bypass graft surgery (Murphy, Szokol, Marymont, et al., 2007).
Although opioids are not first-line analgesics for neuropathic pain, morphine has been shown to effectively treat this type of pain, particularly in combination with first-line adjuvant analgesics for neuropathic pain, such as the gabapentinoids (i.e., gabapentin and pregabalin) and the analgesic antidepressants (Dworkin, O’Connor, Backonja, et al., 2007; Gilron, Bailey, Tu, et al., 2005; Maier, Hildebrandt, Klinger, et al., 2002). One randomized, placebo-controlled, double-blind, cross-over study (N = 76) found that opioids (morphine or methadone) and tricyclic antidepressants (despiramine or nortriptyline) were effective in treating postherpetic neuralgia with a nonsignificant trend toward greater reduction in pain with opioids (Raja, Haythornthwaite, Pappagallo, et al., 2002). Even with a higher incidence of adverse effects such as nausea and constipation, patients preferred opioid treatment. Cognitive decline during opioid administration was not observed.
Morphine has been administered by several routes of administration: oral, intranasal, intrapulmonary, rectal, IV, SC, IM, intraspinal (epidural and intrathecal), intraarticular, vaginal, sublingual/buccal, and topical (Christensen, Cohen, Mermelstein, et al., 2008; Donnelly, Davis, Walsh, et al., 2002; Hanks, Cherny, Fallon, 2004; Lavelle, Lavelle, Lavelle, 2007; Stoker, Reber, Waltzman, et al., 2008). Poor lipid solubility precludes transdermal absorption and also complicates reliable delivery through mucous membranes, such as the sublingual/buccal route (Donnelly, Davis, Walsh, et al., 2002; Reisfield, Wilson, 2007). The evidence for vaginal administration of morphine is limited to case reports (Ostrop, Lamb, Reid, 1998). Topical application of morphine is reported for painful wounds, in which case it is presumed to have a primary local action; there is a dearth of systematic research on this use, and it should not be considered an approach for systemic analgesic therapy (Paice, Von Roenn, Hudgins, et al., 2008; Zeppetella, Porzio, Aielli, 2007). Nebulized morphine has been used for dyspnea; case reports suggest a favorable local action exists, but the data overall are mixed, and there are no research reports describing this route as a means to provide systemic analgesia (see Chapters 14 and 20).
Morphine’s effectiveness as an analgesic, therefore, is established for specific systemic routes of administration—oral and parenteral—and intraspinal routes. Of the parenteral routes, IM administration is not recommended for morphine or for any other drug because of the painful injection and unreliable absorption (APS, 2003) (see Chapter 14). Short-term and long-term parenteral use can be easily accomplished with the IV or SC routes. The oral formulations of morphine are available in liquids, tablets, and capsules and in both short-acting and modified-release preparations. (See Chapter 14 for a detailed discussion of oral morphine formulations.)
Morphine is metabolized primarily in the liver. It has two main metabolites, morphine-3-glucuronide (M3G) and morphine-6-glucuronide (M6G) (Gutstein, Akil, 2006). M3G is the primary metabolite of morphine, but it is not active at the opioid receptor and does not produce analgesia (South, Smith, 2001; Andersen, Christrup, Sjogren, 2003); M6G is active at the opioid receptor and produces analgesia (Dahan, van Dorp, Smith, et al., 2008; Smith, Binning, Dahan, 2009; Vaughn, Connor, 2003). Both metabolites have been implicated in morphine toxicity in animals and in patients with advanced disease (Morita, Tei, Tsunoda, et al., 2002), but the studies in humans have not established a clear association (Andersen, Christrup, Sjøgren, 2003). The mechanism by which toxicity, which is evidenced most often by delirium or myoclonus, occurs has not been fully described, and it has been noted that these symptoms are also seen with other opioids (Harris, 2008; Okon, George, 2008) (see Chapter 19 for treatment). Renal insufficiency presumably increases the risk of morphine toxicity because both the parent compound and its major metabolites are renally excreted (Dean, 2004) and there is a direct correlation between creatinine clearance and morphine, M6G, and M3G serum levels.
Administration of M3G directly into the CNS has been shown to produce neuroexcitability and anti-analgesic effects in animals (Sharke, Geisslinger, Lotsch, 2005). This metabolite has been implicated as the cause of the neuroexcitability noted in some patients who receive large doses of morphine on a long-term basis (Inturrisi, 2002). Some have suggested that opioid-induced hyperalgesia (see Chapter 11) may be due in part to M3G activity (Hemstapat, Monteith, Smith, et al., 2003; South, Smith, 2001); however, further research in humans is needed to draw firm conclusions (Andersen, Christrup, Sjogren, 2003; Sharke, Geisslinger, Lotsch, 2005).
M6G is thought to produce at least some (e.g., 10%) of the analgesic effect of a dose of morphine, but potency and effectiveness studies in humans have produced mixed results (Andersen, Christup, Sjogren, 2003; Smith, South, 2001; Wittwer, Kern, 2006). M6G is absorbed and eliminated from the CNS more slowly than morphine, which may account for the observed increase in potency with long-term morphine administration (Donnelly, Davis, Walsh, et al., 2002; Smith, South, 2001). It may be that the adverse effect profile of M6G is better than that of morphine (Donnelly, Davis, Walsh, et al., 2002). Specific adverse effects that have been investigated include respiratory depression, sedation, nausea and vomiting, hyperalgesia, and myoclonus. A randomized study of 170 patients with moderate to severe postoperative pain demonstrated that M6G produced long-lasting, dose-related analgesia with minimal cardiorespiratory or opioid-like adverse effects (Smith, Binning, Dahan, 2009). In another study (N = 100), M6G was compared with morphine and was found to produce less sedation and respiratory depression, and to have a slower initial onset of effect, with no significant difference in mean pain intensity between groups at 24 hours, but higher pain intensities at 30 minutes and 1 hour after M6G administration (Hanna, Elliott, Fung, 2005). The various factors that influence blood levels of morphine, M6G, and M3G are listed in Table 13-2.
Table 13-2
Factors That Influence Blood Levels of Morphine, M6G, and M3G
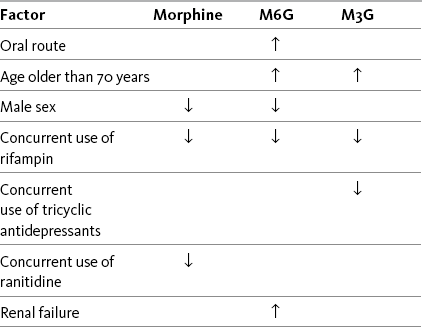
↑, Increased blood level; ↓, decreased blood level; M3G, morphine-3-glucuronide; M6G, morphine-6-glucuronide.
Morphine has two main metabolites, morphine-3-glucuronide (M3G) and morphine-6-glucuronide (M6G). M3G is the primary metabolite of morphine, but it is not active at the opioid receptor, M6G is active at the opioid receptor. With long-term oral morphine dosing, blood levels of M6G typically exceed those of morphine; the concentration ratios of M3G to morphine are inconsistent and variable. Unanticipated opioid toxicity and adverse effects are attributed to accumulation and high blood concentrations of M6G (see text regarding M3G).
From Pasero, C., & McCaffery, M. Pain assessment and pharmacologic management, p. 329, St. Louis, Mosby. Data from Buxton, I. L. O. (2006). Pharmacokinetics and pharmacodynamics. The dynamics of drug absorption, distribution, action, and elimination. In L. L. Brunton, J. S. Lazo, & K. L. Parker (Eds.), Goodman & Gilman’s the pharmacological basis of therapeutics, ed 11, New York, McGraw-Hill; Gutstein, H., & Akil, H. (2006). Opioid analgesics. In L. L. Brunton (Ed.), Goodman & Gilman’s the pharmacological basis of therapeutics, ed 11, New York, McGraw-Hill; Portenoy, R. K., & Kanner, R. M. (Eds.). (1996). Pain management: Theory and practice, Philadelphia, FA Davis; Sharke, C., Geisslinger, G., & Lotsch, J. (2005). Is morphine-3-glucuronide of therapeutic relevance? Pain, 116(3), 177-180. Pasero C, McCaffery M. May be duplicated for use in clinical practice.
Morphine is hydrophilic (soluble in aqueous solution), which contributes to its slow onset and long duration of action compared with the more lipophilic (soluble in fatty tissue) opioid drugs, such as fentanyl and sufentanil. This is not relevant after steady state is reached during continuous dosing but may be important when intermittent boluses are used systemically or intraspinally (see Chapters 15 through 17). The longer time that it takes morphine to reach its analgesic site of action must be considered when determining how quickly to administer doses during titration (IV); adequate time must be allowed to assess response to one dose before administration of another (Lotsch, Dudziak, Freynhagen, et al., 2006). Morphine has a short half-life of 2 to 4 hours; the half-life of M6G is somewhat longer (Andersen, Christup, Sjøgren, 2003). It is estimated that approximately 20% to 30% of the given dose of oral morphine is available for therapeutic effect because of first-pass effect (De Pinto, Dunbar, Edwards, 2006; Gutstein, Akil, 2006) (see Chapter 11). This is why the recommended dose of morphine by the oral route is higher than that by the parenteral route (APS, 2003) (see Table 16-1 on pp. 444-446).
IV morphine has been observed to have an increased analgesic efficacy and longer duration of action in older patients than in younger patients for reasons that are thought to be multifactorial; slow, steady titration is recommended (Villesen, Banning, Petersen, et al., 2007). A general practice is to reduce the starting dose of morphine (and other opioids) in older patients because of physiologic changes associated with aging, such as diminished first pass effect, enhanced bioavailability, and 20% to 40% decrease in clearance (Aubrun, Marmion, 2007).
Codeine
Codeine is the prototypical “weak” opioid used primarily for short-term acute pain. It is usually prescribed as an oral combination product that also contains aspirin or acetaminophen. Codeine combination products may also include caffeine or a muscle relaxant.
Combination preparations that include codeine are not appropriate for moderate to severe or escalating pain because of the dosing limitations inherent in the nonopioid constituent. The ceiling on the maximum safe daily doses of acetaminophen (4000 mg) and aspirin (4000 mg) limits dose increases for inadequate pain control (see Section III). In addition, aspirin is contraindicated for patients with a number of underlying conditions, such as those with a bleeding disorder or history of asthma. See Patient Education Form VI-2 (pp. 547-548) on codeine with acetaminophen at the end of Section IV.
Although a single-entity codeine formulation could theoretically undergo dose escalation sufficient to manage severe persistent pain, this is not pursued in practice. At the customary doses used, codeine provides analgesia for mild to moderate pain. A common oral dose of 60 mg produces analgesia equal to 600 mg of aspirin (less than two 325 mg tablets) (Gutstein, Akil, 2006). A systematic literature search concluded that acetaminophen/codeine (e.g., 300 mg/30 mg) was less efficacious and associated with more adverse effects than NSAIDs (e.g., ibuprofen, naproxen) for postpartum and postlaparotomy analgesia (Nauta, Landsmeer, Koren, 2009). One double-blind study randomized patients to receive codeine (30 mg) plus acetaminophen (300 mg) and caffeine (15 mg) per dose or ibuprofen (400 mg) plus acetaminophen (325 mg) per dose four times daily for 7 days or until pain free following outpatient hernia repair or laparascopic cholecystectomy (Mitchell, van Zanten, Inglis, et al., 2008). Those who received ibuprofen plus acetaminophen had lower pain ratings and fewer adverse effects throughout the treatment period and were more satisfied and less likely to discontinue treatment due to adverse effects or ineffectiveness compared with those who received codeine. A Cochrane Collaboration Review concluded that single doses of dihydrocodeine, a synthetic opioid with structure and pharmacokinetics very similar to codeine, is not sufficient for postoperative pain relief and that 400 mg of ibuprofen was superior to 30 or 60 mg of dihydrocodeine (Edwards, McQuay, Moore, 2004).
There also is evidence that higher doses of codeine would be relatively less effective than other opioids. Doses above 65 mg have been described as providing diminishing incremental analgesia but continued increase in adverse effects (Miaskowski, Cleary, Burney, et al., 2005).
The IM route has been used to administer codeine, but absorption is unreliable and is associated with a five-fold variation in peak blood level; the peak occurs approximately 30 to 60 minutes after IM administration. Nine-fold differences in minimum effective analgesic concentration have been found by this route, and late respiratory depression can occur. These properties make the IM route unfavorable for use in postoperative pain management (Cousins, Umedaly, 1996). It has also long been regarded as inappropriate for IV administration, with low doses of IV morphine recommended instead (Semple, Macintyre, Hooper, 1993) (see Table 13-1).The dose ratio for total analgesic effect between IM and oral codeine is 0.6:1, and a comparison between parenteral codeine and oxycodone found an equianalgesic dose ratio of 10:1; however, its relative potency varies with the extent to which it is converted to its active metabolite (Knotkova, Fine, Portenoy, 2009).
Codeine is a prodrug and is approximately 60% bioavailable orally (as compared, for example, with morphine, which has oral bioavailability of 20% to 30%). This is because codeine, like levorphanol, oxycodone, and methadone, undergoes less first-pass metabolism than morphine (Gutstein, Akil, 2006) (see Chapter 11). Once absorbed, 10% of codeine is metabolized in the liver to morphine, its active form (Somogyi, Barratt, Coller, 2007), which probably provides the bulk of its analgesic effect. However, as with other opioids, extremely wide variations exist between individuals in terms of absorption and analgesic requirements of codeine.
The metabolism of codeine to morphine depends on the presence of the enzyme cytochrome P450 2D6 (Fine, Portenoy, 2007) (see Chapter 11 for a detailed discussion of this enzyme system). There is population variation in phenotype of cytochrome P450 2D6, distinguishing patients intermediate, extensive (or rapid), ultra-rapid metabolizers, or poor metabolizers. Extensive metabolizers are the norm and are able to perform catalyzed biotransformation of codeine (and other drugs). Approximately 10% of Caucasians and varying frequencies in other ethnic groups are poor metabolizers. These individuals have a very limited ability to convert codeine to morphine, and as a consequence, are relatively less responsive to codeine’s analgesic effect (Palmer, Giesecke, Body, et al., 2005). In contrast, ultra-rapid metabolizers, who biotransform codeine more rapidly or more completely and most of the population, may have an exaggerated (i.e., toxic) response to codeine (Voronov, Przybylo, Jagannathan, 2007; United States Food and Drug Administration [U.S. FDA], 2007a; Palmer, Giesecke, Body, et al., 2005).
The impact of the biotransformation via P450 2D6 can vary according to circumstances. Although biotransformation at this isoenzyme occurs at the same rate in neonates and adults, neonates can develop toxicity from codeine because the clearance of morphine is relatively reduced and it can accumulate in the blood. In 2007, the U.S. FDA issued a warning that nursing mothers who are ultra-rapid metabolizers of codeine can transfer sufficient morphine to their breast-feeding infants to cause life-threatening or fatal adverse effects (U.S. FDA, 2007a) (see Chapter 20 for more on opioid use during breast- feeding). In adults, the efficiency of the enzyme can be affected by certain drugs, leading to changes in the production of morphine from codeine. Drugs such as paroxetine (Paxil) and fluoxetine (Prozac), for example, may inhibit cytochrome P450 2D6, and therefore, could potentially interfere with the metabolism of codeine. Case reports suggest that hydrocodone may be effective in CPY2D6 poor metabolizers for whom codeine was ineffective (Susce, Murray-Carmichael, de Leon, 2006).
Codeine Post-Craniotomy
The incidence of moderate-to-severe postoperative pain after craniotomy is common, and the surgical procedure is associated with the development of persistent postsurgical pain (Gottschalk, 2009). Pain management is complicated by concerns about opioid-related adverse effects, such as sedation, miosis, nausea, and vomiting, in this patient population. Codeine has been used for the treatment of postcraniotomy and other types of neurosurgical pain for decades, but its unpredictable absorption, variability in demethylation, and high incidence of nausea and sedation at effective doses make it a particularly poor choice in this population (Roberts, 2004). A review of randomized controlled trials revealed two studies that showed more consistent pain control with morphine compared with codeine and no differences in respiratory depression, sedation, pupillary size, and cardiovascular (CV)effects in patients following craniotomy (Nemergut, Durieux, Missaghi, et al., 2007). A study comparing IV morphine PCA, IV tramadol PCA, and IM codeine in postcraniotomy patients found that morphine produced significantly better analgesia with less vomiting than the other two drugs (Sudheer, Logan, Terblanche, et al., 2007). Roberts (2004) points out that patients are admitted to the intensive care setting where close monitoring is standard following craniotomy. This and thoughtful titration to minimize adverse effects help to ensure the safety of morphine in this population (see also discussion of remifentanil later in this chapter and Chapter 26 for discussion of gabapentin as a component of a multimodal analgesic regimen for craniotomy pain).
Fentanyl
Fentanyl is the prototype in a subset of mu agonists which includes sufentanil, alfentanil, and remifentanil (see separate discussions for the latter three). All of these drugs are characterized by high potency and high lipophilicity (fat solubility). When administered parenterally to the opioid-naïve patient, the effects are characterized by rapid onset and short duration of action. The injectable formulations of these drugs are administered via the IV, epidural, and intrathecal routes and typically are used for acute pain in the perioperative and procedural settings, often in conjunction with anesthetic or sedating agents; they are the most commonly used opioids in anesthesia (Coda, 2006). They also may be administered transmucosally, e.g., by the buccal, sublingual, or intranasal routes (see Chapter 14).
As a class, fentanyl and comparable drugs are versatile, although none are commercially available in an oral or rectal formulation. There are pharmacologic and cost differences that are considered in drug selection for each therapeutic application. With rapid IV administration of high doses, these drugs can produce chest wall rigidity and subsequent difficult ventilation (Lalley, 2005; Fukuda, 2005); this is a concern when fentanyl is used for intraoperative anesthesia (see Chapter 16 for more on speed of injection). Likely related is cough, which has been reported as a complication of both fentanyl and sufentanil (Agarwal, Gautam, Nath, et al., 2007); this effect has been successfully suppressed with IV lidocaine 0.5 mg/kg (Pandey, Raza, Ranjan, et al., 2005).
Fentanyl differs in many ways from morphine. Its lipophilicity means that there is wide and rapid distribution after IV administration, as well as ready passage through the blood-brain barrier. When given as a single IV bolus, fentanyl’s onset (within 1 to 5 minutes) is faster and its duration (sometimes less than 1 hour) is shorter than morphine as the drug moves from blood to lungs, muscle, and fat (Taylor, 2005). It also is approximately 100 times more potent than morphine, so that a single IV bolus of 100 mcg produces roughly the same analgesia as morphine 10 mg; however, caution is recommended when converting to and from fentanyl as studies demonstrate considerable variability in conversion ratios (see Knotkova, Fine, Portenoy, 2009 for a discussion of this research).
The lipophilicity and potency of fentanyl makes it an excellent candidate for transdermal and oral transmucosal formulation. The fentanyl transdermal patch is commonly used in long-term pain treatment (see Chapter 14). Oral transmucosal fentanyl formulations are used in the treatment of breakthrough pain: oral transmucosal fentanyl and buccal fentanyl; a buccal patch was recently approved in the United States, and a sublingual formulation and an intranasal formulation are available in some other countries (see Chapter 14 for oral transmucosal formulations).
After repetitive dosing or continuous infusion of fentanyl, a steady state is approached. Although some clinicians believe that fentanyl has a very short half-life, this is a misconception. When fentanyl or some other very lipophilic drug is administered to the patient who is not receiving regular dosing, the blood levels decline quickly as the drug redistributes into fatty tissue. This redistribution, or “alpha” phase, is associated with a short half-life and a brief duration of clinical effects. With fentanyl, it is typically only minutes long. In contrast, regular dosing of fentanyl or any other lipophilic drug eventually leads to a steady state in which there is equilibrium between the blood and fatty tissues. A bolus injection in this setting still has a redistribution phase, but most of the elimination time, the “beta” phase, results from metabolism and redistribution of drug from fat back into blood. As a result, the half-life, and the duration of effect after the bolus, is much longer. The so-called terminal elimination half-life is the half-life that is obtained after the redistribution has taken place.
Given these kinetics, the half-life of fentanyl varies in the literature depending on whether the study that yielded the value measured the decline in concentration in a steady-state situation or not. Although it has been reported that fentanyl has a terminal half-life of approximately 3 to 4 hours, it is much longer—four to five times longer—after steady state has been approached (Dershwitz, Landow, Joshi-Ryzewicz, 2003; Liu, Gropper, 2003). After steady state is achieved using transdermal fentanyl, half-life also is affected by continued absorption from the skin depot under the patch; the half-life is therefore even longer, typically over 24 hours (see Table 13-1).
Fentanyl’s lipophilicity and storage in fatty tissue has significant implications for obese patients in the perioperative setting. If perioperative dosing is based on body weight alone, obese patients are likely to receive too high a dose. Dosing based on a calculated “pharmacokinetic mass” (i.e., for patients weighing 140 to 200 kg, dosing weights of 100 to 108 kg are projected) has been shown in two clinical studies to provide safe and effective intraoperative and postoperative analgesia at lower doses than would be predicted by actual weight (Shibutani, Inchiosa, Sawada, et al., 2004, 2005). This is a result of a nonlinear relationship between total body weight and fentanyl clearance.
Fentanyl is metabolized in the liver, has no active metabolites, and produces minimal hemodynamic effects (Fukuda, 2005). These characteristics have made fentanyl a favorite in the critically ill, including older critically ill adults, and especially patients who are hemodynamically unstable or have renal failure (Graf, Puntillo, 2003; Jacobi, Fraser, Coursin, et al., 2002). It is recommended for patients with end-stage renal disease who need opioid analgesia (Dean, 2004; Johnson, 2007; Murtagh, Chai, Donohoe, et al., 2007). Though more research is needed, fentanyl appears safe in patients with hepatic dysfunction as well (Johnson, 2007).
When used for pain, fentanyl typically is given either parenterally (by the IV route usually) or intraspinally (see Chapter 15). Its rapid onset and short duration in the non–steady state situation make fentanyl the most commonly used opioid in combination with benzodiazepines for procedural analgesia and sedation. Along with morphine and hydromorphone, fentanyl has become a first-line choice for postoperative pain management via IV PCA in many institutions (Pasero, 2005) (see Chapter 17 for PCA dosing). There have been no randomized controlled trials comparing these drugs. A retrospective analysis of medical records compared adverse effects associated with morphine (N = 93), hydromorphone (N = 89), and fentanyl (N = 72) via postoperative IV PCA and found lower mean rates of nausea, pruritus, urinary retention, and sedation with fentanyl; there were no differences among the opioids in incidence of respiratory depression, headache, agitation, confusion, and hallucinations (Hutchison, Chon, Tucker, et al., 2006). However, well-controlled research is needed to draw conclusions regarding differences.
Parenteral fentanyl can be administered by SC infusion (Anderson, Shreve, 2004). A small study comparing continuous SC infusions of morphine followed by fentanyl (N = 13) and fentanyl followed by morphine (N = 10) in hospice cancer patients revealed that fentanyl is as efficacious as morphine with no differences in adverse effects except less constipation in patients receiving fentanyl (Hunt, Fazekas, Thorne, et al., 1999). The researchers recommended a conversion ratio of morphine 10 mg to fentanyl 150 mcg as appropriate but emphasized that further research is needed in this area.
Although fentanyl’s properties of high lipophilicity, rapid onset, and short duration in the non–steady state situation make it an attractive option for pain management in a variety of settings, populations, and conditions, these properties also necessitate careful patient selection, appropriate monitoring, and adherence to the safety warnings that accompany fentanyl products, especially the patches and oral transmucosal products. A number of fatalities have occurred due to improper prescribing and use. See Chapter 14 for specific information on indications, patient selection, administration, monitoring, and precautions for the various fentanyl products.
Hydrocodone
Hydrocodone is available in several proprietary products (e.g., Lortab, Vicodin, Lorcet, Hydrocet, and Norco) and generic preparations, and in several different fixed-dose combinations with acetaminophen, aspirin, and ibuprofen. Most are available in tablet form, while others are available in capsule or liquid form (Table 13-3). Combination drugs containing hydrocodone and a nonopioid drug can provide more effective relief than either drug alone. Ibuprofen combined with a variety of hydrocodone doses was shown to increase the effectiveness of hydrocodone seven-fold in animal research (Kolesnikov, Wilson, Pasternak 2003). See Patient Education Form IV-6 on hydrocodone with acetaminophen on pp. 556-557 at the end of Section IV.
Table 13-3
Commercially Available Combinations of Hydrocodone and Nonopioids
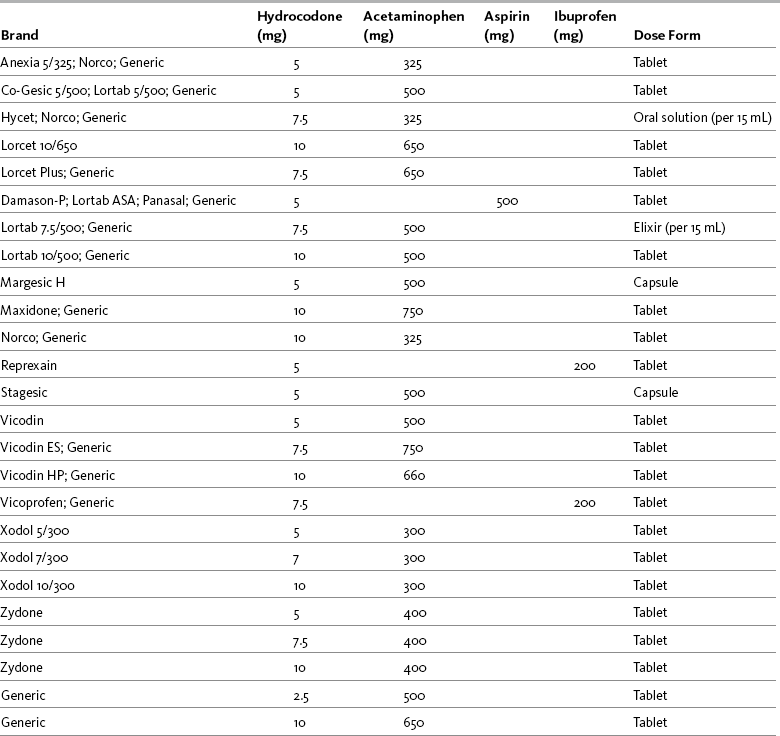
From Pasero, C., & McCaffery, M. Pain assessment and pharmacologic management, p. 333, St. Louis, Mosby. Pasero C, McCaffery M. May be duplicated for use in clinical practice.
Hydrocodone is not available in single-entity form. The dose is limited by the ceiling on safety and efficacy inherent in the nonopioid constituent. The dose limitation, in turn, typically means that the drug is useful for the management of mild to moderate pain in the opioid-naïve patient. Use in persistent pain (except for breakthrough dosing) should be carefully evaluated. Long-acting analgesics without a fixed dose co-analgesic are preferred for moderate to severe persistent cancer or noncancer pain. A modified-release (12-hour) oral formulation of hydrocodone 15 mg plus acetaminophen 500 mg has been studied in clinical trials but has not been approved in the United States (Coddings, Levinsky, Hale, et al., 2008; Golf, Robson, Pollak, et al., 2008).
Hydrocodone with acetaminophen is not only the most commonly prescribed analgesic in the United States, it is by far the most commonly prescribed medication in all classes (Lamb, 2008). In 2008, hydrocodone plus acetaminophen was first in the list of the 200 most commonly prescribed drugs (Drug Topics, 2008). It is critical to avoid prescribing or administering amounts that would exceed the daily maximum dose for acetaminophen (4000 mg), aspirin (4000 mg), or ibuprofen (3200 mg) and teach the patient the dangers of exceeding these amounts (see Section III).
Hydrocodone has an onset of action of approximately 20 minutes, reaches peak effectiveness by 60 minutes, and has a half-life of 3.8 hours (Gutstein, Akil, 2006). It is metabolized by the cytochrome P450 2D6 (CYP2D6) enzyme. Case reports suggest that hydrocodone may be effective in CPY 2D6 poor metabolizers for whom codeine was ineffective (Susce, Murray-Carmichael, de Leon, 2006). (See Chapter 11 for more on the cytochrome P450 enzyme system and drug-drug interactions.)
The adverse effects of hydrocodone are comparable to that of other opioids. It has been shown to have similar efficacy (when titrated to effect) and a lower incidence of adverse effects compared with tramadol and codeine (Rodriguez, Bravo, Castro, et al., 2007; Rodriguez, Castillo, Del Pilar Castillo, et al., 2007). Several cases of an unusual adverse effect (hearing loss) have been reported in patients taking hydrocodone and in hydrocodone abusers (Ho, Vrabec, Burton, 2007). No causative mechanism, if it exists, has been identified for this association.
To ensure safety, it should be specifically noted that the name hydrocodone is similar in appearance and sound to oxycodone, oxymorphone, and hydromorphone. An increased level of alertness is required to prevent medication errors with these opioids.
Hydromorphone
Hydromorphone is often considered an alternative to morphine, especially for acute pain (Chang, Bijur, Meyer, et al., 2006; Rapp, Egan, Ross, et al., 1996). Although there are few head-to-head studies, morphine and hydromorphone appear to provide equivalent analgesic effects and very similar adverse effect profiles (Ripamonti, Bandieri, 2009; Quigley, Wiffen, 2003); there is some evidence that hydromorphone may be associated with less nausea and pruritus (Chang, Bijur, Meyer, et al., 2006).
IV hydromorphone is a first- or second-choice opioid (after morphine) for postoperative pain management via PCA (Quigley, 2002). When given IV as a bolus, its onset of action is 5 minutes, its peak effect occurs in 8 to 20 minutes, and its duration is approximately 4 hours (Sarhill, Walsh, Nelson, 2001).
Oral short-acting hydromorphone is available in 2, 4, and 8 mg tablets and in a 1 mg/mL oral solution. It is approximately 60% bioavailable with an onset of action of 30 minutes via the oral route (Kumar, Lin, 2007) and a duration of approximately 3 to 4 hours; maximum plasma concentrations are reached within 1 hour of dosing (Gupta, Sathyan, 2007). Modified-release formulations of oral hydromorphone are available in Canada and Europe and most recently on the U.S. market (Gupta, Sathyan, 2007) (see Chapter 14 for modified-release hydromorphone). See Patient Education Form IV-3 on short-acting hydromorphone at the end of Section IV.
Hydromorphone also has been administered via a variety of other routes. Its greater potency compared with morphine, as well as its availability in concentrated parenteral form (10 mg/mL), has made it attractive for SC administration, especially when high doses are needed. It was found to be comparable to morphine by SC continuous infusion for persistent cancer pain in terminally ill patients (Miller, McCarthy, O’Boyle, et al., 1999). Absorption via the IM route is erratic and not recommended (Golembiewski, 2003). The epidural and intrathecal routes have been utilized for acute and persistent cancer and noncancer pain (DuPen, DuPen, Hillyer, 2006). Hydromorphone given rectally (3 mg suppository) is as effective as by the oral route; it is not, however, absorbed well by the oral mucosa (Kumar, Lin, 2007; Sarhill, Walsh, Nelson, 2001) (see Table 13-1).
Hydromorphone is metabolized in the liver and eliminated via the kidneys (Sarhill, Walsh, Nelson, 2001). It has a neuroexcitatory metabolite, hydromorphone-3-glucuronide (H3G), and it may be speculated that neurotoxicity at high doses is related to this molecule (Thwaites, McCann, Broderick, 2004; Wright, Mather, Smith, 2001; Smith, 2000). Although neurotoxic symptoms can occur in advanced disease and decreased renal clearance, hydromorphone appears to be a safer choice than morphine under these conditions (Dean, 2004). Hydromorphone is often recommended as the first alternative for opioid rotation when these symptoms occur during morphine administration (Hanks, Reid, 2005). Some clinicians will use hydromorphone in older adults as a first-line opioid instead of morphine because of the theoretically improved tolerance in the presence of decreased renal function; however, this practice has not been studied and has not been recommended by published guidelines. A prospective, randomized study (N = 50) comparing morphine and hydromorphone via postoperative IV PCA found no difference in efficacy, adverse effects, or patient satisfaction (Hong, Flood, Diaz, 2008). A retrospective analysis of medical records found the mean rates of nausea, pruritus, urinary retention, and sedation with postoperative IV PCA hydromorphone to be similar to that of IV PCA morphine and more common than with IV PCA fentanyl; there were no differences among the three opioids in incidence of respiratory depression, headache, agitation, confusion, and hallucinations (Hutchison, Chon, Tucker, et al., 2006). Well-controlled research is needed to draw conclusions regarding differences among the various opioids.
The equianalgesic dose conversion between morphine and hydromorphone is unclear and likely varies with the length of time a patient has been on one drug or the other (Berdine, Nesbit, 2006; Knotkova, Fine, Portenoy, 2009). Published equianalgesic tables typically show oral hydromorphone to be 5 times more potent than oral morphine. This is the most common ratio used when preparing equianalgesic solutions for PCA administration (e.g., 0.2 mg hydromorphone per 1 mL solution is considered approximately equal to 1 mg morphine per 1 mL solution) (Golembiewski, 2003). However, these data are generally derived from acute pain treatment in opioid-naïve patients or healthy volunteers (APS, 2003). An early study of morphine-hydromorphone equivalence showed that after a week of PCA treatment, the ratio was 3:1 (morphine 10 mg to hydromorphone 3.3 mg) (Dunbar, Chapman, Buckley, et al., 1996). Subsequent research found that when switching from long-term dosing of either oral or parenteral morphine to hydromorphone, the ratio was approximately 5.5:1 (morphine 10 mg to hydromorphone 2 mg) (Lawlor, Turner, Hanson, et al., 1997). However, when switching from hydromorphone to morphine, the ratio was 3.7:1 (morphine 10 mg to hydromorphone 2.7 mg). Still others suggest that an equianalgesic dose conversion of parenteral morphine to hydromorphone for long-term dosing is probably 4:1 (morphine 10 mg to hydromorphone 2.5 mg) (Hanks, Cherny, Fallon, 2004). A systematic review of hydromorphone for various types of pain concluded that there is insufficient evidence to recommend specific ratios of hydromorphone (Quigley, Wiffen, 2003) (see also Knotkova, Fine, Portenoy, 2009). (See Table 16-1).
When using equianalgesic dosing to switch a patient from another opioid to hydromorphone, the general condition of the patient and the severity of pain must be considered when choosing a starting dose (Fine, Portenoy, the Ad Hoc Expert Panel on Evidence Review and Guidelines for Opioid Rotation, 2009). In general, it is safest to use a conservative conversion ratio then titrate to effect. No matter what ratio or method for conversion is used, it is vital to take into account that hydromorphone is significantly more potent than morphine, there is great interindividual variability (Murray, Hagen, 2005), and individualization and monitoring are essential elements of prescribing and administering these agents. Deaths have occurred because of confusion between the two agents and a failure to take their inherent differences into account (Institute for Safe Medication Practices, 2004a).
It also should be noted that the name hydromorphone is similar in appearance and sound to oxycodone, oxymorphone, and hydrocodone. An increased level of alertness is required to prevent medication errors with these opioids.
Levorphanol
Like methadone, levorphanol (Levo-Dromoran) is considered a second-line drug for cancer pain (Hanks, Cherny, Fallon, 2004). It has not been widely used clinically since the modified-release formulations of morphine and oxycodone became available in the 1990s (McNulty, 2007). Most clinicians are unfamiliar with its pharmacology, which is somewhat different than the more commonly used opioids (McNulty, 2007; Prommer, 2007a; Prommer, 2007b). It is an agonist at both the mu and kappa opioid receptor sites and, like methadone, it also is an N-methyl-d-aspartate (NMDA) antagonist. In addition, it is an inhibitor of serotonin and norepinephrine reuptake (see Section I).
Levorphanol is available for oral (2 mg/tablet) and parenteral administration (2 mg/mL) and has a parenteral-to-oral ratio of 1:2 (Prommer, 2007a). Its metabolism is similar to morphine’s. It undergoes glucuronidation in the liver and is not affected by the CYP 450 system. For this reason, it has fewer potential drug-drug interactions than methadone but is subject to the effects of inducers and inhibitors of glucuronidation (Prommer, 2007a).
Levorphanol’s duration of analgesia is reported to range from a low of 3 hours to a high of 15 hours (Fine, Portenoy, 2007; Hanks, Cherny, Fallon, 2004; Prommer, 2007a) with both IV and oral dosing. Importantly, levorphanol has a longer half-life (15 hours) than morphine (2 to 4 hours), and it is likely that at least some patients can attain sustained analgesia with a relatively long dosing interval. Like methadone, the discrepancy between analgesic duration and half-life can predispose levorphanol to accumulation. Although outliers with half-lives that can extend to as much as 30 hours (Prommer, 2007a) are likely to pose substantial risk, accumulation overall appears to be less of a problem than it can be with methadone (Hanks, Cherny, Fallon, 2004). Excretion is by the kidneys.
A study using levorphanol at two dose levels established that neuropathic pain is responsive to opioid treatment (Rowbotham, Twilling, Davies, et al., 2003). In fact, all types of neuropathic pain responded to levorphanol except central poststroke pain in the study. In the higher-dose arm (mean was approximately 9 mg/day) apparent CNS toxicity occurred (irritability, mood changes, confusion, weakness). The reasons and risk factors for these changes were unknown. Close monitoring as well as dose and interval changes are indicated for older adults and those with impaired renal function.
Levorphanol is available in the United States orally only in 2 mg tablets, which can make titration difficult. It is no longer available in the United Kingdom or Canada (Hanks, Cherny, Fallon, 2004). (See Table 16-1 on pp. 444-446.)
Meperidine
Meperidine (Demerol) was once the most widely used opioid analgesic. In recent years, it has been either removed from or severely restricted on hospital formularies, the result of concerted efforts to improve patient safety during opioid use (Gordon, Jones, Goshman, et al., 2000; Raymo, Camejo, Fudin, 2007) (see the paragraphs that follow). The Beers Criteria of inappropriate medication use in older individuals, originally developed in 1991 (Beers, Ouslander, Rollingher, et al., 1991), described meperidine as having many disadvantages and continues to advise against the use of the drug in older adults (Beers, 1997; Fick, Cooper, Wade, et al., 2003) (Table 13-4). A refinement of the 1996 Medical Expenditure Panel Survey designated the drug as one to “always avoid” in older adults (Zhan, Sangl, Bierman, et al., 2001). Meperidine has some positive attributes, but it continues to be overused (Kornitzer, Manace, Fischberg, 2006) and misused (Hubbard, Wolfe, 2003) because of lack of knowledge about its pharmacology. Numerous misconceptions about meperidine persist (Table 13-5).
Table 13-4
Beers Criteria for Inappropriate Medication Use in Older Adults: Selected Analgesics
| Drug | Concerns | Severity Rating |
| Propoxyphene (Darvon, Darvocet, Darvon Compound) | Offers no advantages over other opioids; toxic metabolite; high adverse effect profile. | Low |
| Meperidine (Demerol) | Offers few if any advantages over other opioids; toxic metabolite that can cause CNS disturbances. | High |
| Pentazocine (Talwin) | Low analgesic efficacy; high incidence of CNS adverse effects (e.g., hallucinations, delirium). | High |
| Short-acting benzodiazepines (e.g., Ativan, Restoril, Serax, Xanax) | Increased sensitivity in older adults; dose-related adverse CNS effects. | High |
| Long-acting benzodiazepines (e.g., Librium, Valium) | Increased sensitivity in older adults; long half-life; dose-related adverse CNS effects; associated with falls and fractures. Short-acting is preferred if a benzodiazepine is needed. | High |
| Flurazepam (Dalmane) | Very long half-life in older adults (often days); prolonged. | High |
| Anticholinergics and antihistamines (e.g., Benadryl, Atarax, Vistaril) | Most antihistamines have potent anticholinergic effects. Noncholinergic antihistamines are preferred. Hydroxyzine (Vistaril, Atarax) and diphenhydramine (Benadryl) can cause confusion and sedation. If antihistamine is necessary, use low dose. | High |
| Amitriptyline (Elavil) | High incidence of anticholinergic and sedative adverse effects; rarely appropriate in older adults. | High |
| Daily fluoxetine (Prozac) | Long half-life; can produce CNS stimulation, sleep disturbances, and agitation. | High |
| Clonidine (Catapres) | Can cause orthostatic hypotension and CNS adverse effects. | Low |
| Orphenadrine (Norflex) | Can cause significant sedation and anticholinergic effects. | High |
| Muscle relaxants (e.g., Soma, Flexeril, Skelaxin) | Most have anticholinergic effects and are poorly tolerated by older adults. Can cause sedation and muscle weakness, which may contribute to falls. | High |
| Long-term use of full-dose, longer half-life, nonselective NSAIDs (e.g., Naprosyn, Aleve, Feldene) | Potential GI, renal, CV adverse effects. | High |
| Ketorolac (Toradol) | High incidence of GI adverse effects in older adults. | High |
| Indomethacin (Indocin) | High incidence of CNS adverse effects. | High |
CNS, Central nervous system; CV, cardiovascular; GI, gastrointestinal,
From Pasero, C., & McCaffery, M. Pain assessment and pharmacologic management, p. 336, St. Louis, Mosby. Data from Beers, M. H. (1997). Explicit criteria for determining potentially inappropriate medication use by the elderly. An update. Arch Intern Med, 157(14), 1531-1536; Beers, M. H., Ouslander, J. G., Rollingher, I, et al. (1991). Explicit criteria for determining inappropriate medication use in nursing home residents. UCLA Division of Geriatric Medicine. Arch Intern Med, 151(9), 1825-1832; Fick, D. M., Cooper, J. W., Wade, W. E., et al. (2003). Updating the Beers criteria for potentially inappropriate medication use in older adults: Results of a US consensus panel of experts. Arch Intern Med, 163(22), 2716-2724; Zhan, C., Sangl, J., Bierman, A. S., et al. (2001). Potentially inappropriate medication use in the community-dwelling elderly: Findings from the 1996 Medical Expenditure Panel Survey. JAMA, 286(22), 2823-2829. Pasero C, McCaffery M. May be duplicated for use in clinical practice.
Table 13-5
| Misconception | Correction |
| Meperidine causes less respiratory depression than morphine. | At equianalgesic doses, opioid analgesics produce equal respiratory depression. |
| Meperidine is less likely than morphine to cause addiction. | The abuse liability for meperidine is at least as high as that for morphine. In other words, people addicted to opioids find morphine and meperidine equally attractive. Several early reports suggested meperidine may be the more addictive of the two. |
| Meperidine causes less constriction of the sphincter of Oddi and the biliary tract than does morphine. | Both meperidine and morphine cause constriction of the sphincter of Oddi and the biliary tract. Laboratory studies show that morphine may cause more constriction in animals, but this has never been shown to be clinically relevant in humans. In humans, morphine and meperidine caused a rise in bile duct pressure of 52.7% and 61.3%, respectively. |
| Meperidine is less constipating than morphine. | Meperidine may be less constipating but only when used on a long-term basis, and long-term use is not recommended. |
| Long-term clinical experience with meperidine proves it is safe and effective. | Meperidine prescribing has declined, but the drug continues to be used despite ample evidence that it has no advantages over other opioids and has toxicities that make it undesirable for almost any use. Historically, therapeutic doses (e.g., 100 mg IM for adults) were seldom used, and studies show that during decades of use, many patients were undertreated for pain. Furthermore, problems may have gone unnoticed because the existence of the metabolite normeperidine was not known and patients were not assessed for signs of neurotoxicity. Meperidine cannot be used safely if pain is treated aggressively. |
| Meperidine is the only drug effective for treatment of perioperative and postdelivery shivering. | Although low-dose meperidine is widely used to treat perioperative and post-delivery shivering, other drugs are also effective. These include clonidine, ondansetron, and tramadol. |
IM, Intramuscular; mg, milligram.
Meperidine (Demerol) continues to be used despite sufficient evidence that it is not appropriate as a first-line opioid analgesic for the management of any type of pain.
From Pasero, C., & McCaffery, M. Pain assessment and pharmacologic management, p. 337, St. Louis, Mosby. Data from Austin, K. L., Stapleton, J. V., & Mather, L. E. (1980). Relationship between blood meperidine concentrations and analgesic response: A preliminary report. Anesthesiology, 53(6), 460-466; Beaulé, P. E., Smith, M. I., & Nguyen, V. N. (2004). Meperidine-induced seizure after revision hip arthroplasty. J Arthroplasty, 19(4), 516-519; Burnham, R., McNeil, S., Hegedus, C., et al. (2006). Fibrous myopathy as a complication of repeated intramuscular injection for chronic headache. Pain Res Manage, 11(4), 249-252; Coelho, J. C., Senninger, N., Runkel, N., et al. (1986). Effect of analgesic drugs on electromyographic activity of the gastrointestinal tract and sphincter of Oddi and on biliary pressure. Ann Surg, 204(1), 53-58; Fong, H. K., Sands, L. P., & Leung, J. M. (2006). The role of postoperative analgesia in delirium and cognitive decline in elderly patients: A systematic review. Anesth Analg 102(4), 1255-1266; Hubbard, G. P., & Wolfe, K. R. (2003). Meperidine misuse in a patient with sphincter of Oddi dysfunction. Ann Pharmacother, 37(4), 534-537; Kornitzer, B. S., Manace, L. C., Fischberg, D. J., et al. (2006). Prevalence of meperidine use in older surgical patients. Arch Surg, 141(8), 76-81; Latta, K. S., Ginsberg, B., & Barkin, R. L. (2002). Meperidine: A critical review. Am J Ther, 9(1), 53-68; Lee, F., & Cundiff, D. (1998). Meperidine vs morphine in pancreatitis and cholecystitis. Arch Intern Med, 158(21), 2399; Mohta, M., Kumari, N., Tyagi, A., et al. (2009). Tramadol for prevention of postanaesthestic shivering: A randomised double-blind comparison with pethidine. Anaesthesia, 64(2), 141-146; Kranke, P., Eberhart, L. H., Roewer, N., et al. (2004). Single-dose parenteral pharmacological interventions for prevention of postoperative shivering: A quantitative systematic review of randomized controlled trials. Anesth Analg, 99(3), 718-727; Radnay, P. A., Brodman, E., Mankikar, D., et al. (1980). The effect of equianalgesic doses of fentanyl, morphine, meperidine, and pentazocine on common bile duct pressure. Anaesthetist 29, 26-29; Schwarzkopf, K. R. G., Hoff, H., Hartmann, M., et al. (2001). A comparison between meperidine, clonidine and urapidil in the treatment of postanesthetic shivering. Anesth Analg, 92(1), 257-260. Pasero C, McCaffery M. May be duplicated for use in clinical practice.
Meperidine has a rapid onset and short duration of action, which would seem to make it an attractive option for limited use, such as IV analgesia for short procedures. Indeed, this is now its most commonly approved use in some institutions, although other opioids, such as fentanyl, hydromorphone, or morphine are usually favored for procedural pain. In low doses (12.5 to 25 mg IV), meperidine also has a role in alleviating the shivering associated with general anesthesia and drugs, such as amphoteracin and some biologic agents; other agents, including clonidine, ondansetron, and tramadol, among others, are also effective for treatment of shivering (Kranke, Eberhart, Roewer, et al., 2004; Mohta, Kumari, Tyagi, et al., 2009; Schwarzkopf, Hoff, Hartmann, et al., 2001).
Meperidine is one fourth as potent orally as parenterally (APS, 2003). By the SC and IM routes, it has an onset of action of 10 minutes, a peak effect of 30 minutes, and a duration of up to 4 hours. When given orally, the analgesic effects of meperidine are felt within 30 minutes. Its peak effect is within 1 to 2 hours, and its duration of action is approximately 3 hours (Latta, Ginsberg, Barkin, 2002). Regardless of route, it has a half-life of 2 to 3 hours. Although absorbed by all routes of administration, the rate of absorption is erratic after IM injection, with a wide range of peak plasma concentrations (Gutstein, Akil, 2006; Latta, Ginsberg, Barkin, 2002). In addition, repeated IM injection is associated with fibrous myopathy (Burnham, McNeil, Hegedus, et al., 2006; Latta, Ginsberg, Barkin, 2002) (see Tables 13-1 and 13-5; also see Table 16-1 on pp. 444-446).
A major drawback to the use of meperidine is its active metabolite, normeperidine (Latta, Ginsberg, Barkin, 2002). Normeperidine is a CNS stimulant and can cause delirium, irritability, tremors, myoclonus, muscle twitches, shaky feelings, and generalized seizures (Simopoulos, Smith, Peeters-Asdourian, et al., 2002). Because normeperidine is eliminated by the kidneys, meperidine should not be used in patients with decreased renal function (APS, 2003; Latta, Ginsberg, Barkin, 2002; Simopoulos, Smith, Peeters-Asdourian, et al., 2002). It is a particularly poor choice in older patients and individuals with sickle cell disease because most have some degree of renal insufficiency. Because normeperidine has a half-life that is usually more than four times longer than meperidine itself, repeated dosing leads to an initial accumulation of normeperidine. This accumulation increases the risk of normeperidine toxicity during the early period (first several days) of repetitive dosing with meperidine. The effects of normeperidine have been observed even in young, otherwise healthy patients given sufficiently high doses of meperidine postoperatively (Simopoulos, Smith, Peeters-Asdourian, et al., 2002). The risk of toxicity overall, particularly risk that increases in the setting of progressive renal insufficiency, has led to the recommendation that meperidine should not be prescribed for patients requiring long-term opioid treatment, such as those with persistent cancer or noncancer pain (Hanks, Cherny, Fallon, 2004; Miaskowski, Cleary, Burney, et al., 2005).
Research shows that meperidine is more likely than other opioid drugs to cause delirium in postoperative patients of all ages (Fong, Sands, Leung, 2006). In a case-control study (N = 91 with 1 to 2 controls) meperidine more than doubled the risk of delirium when given either epidurally or IV (Marcantonio, Juarez, Goldman, et al., 1994). It has also been suggested to have a relatively severe negative impact on mood (Latta, Ginsberg, Barkin, 2002), which is sometimes the first sign of neurotoxicity. A prospective, randomized controlled study found that IV meperidine produced more nausea and vomiting than IV morphine in 200 women following gynecologic surgery (Ezri, Lurie, Stein, et al., 2002).
The most appropriate candidates for meperidine use are patients with acute pain who are otherwise healthy and do not tolerate other opioids, such as morphine and hydromorphone (Simopoulos, Smith, Peeters-Asdourian, et al., 2002), or those with normal renal function who already have demonstrated a favorable outcome with meperidine. If meperidine is used in these patients, frequent high doses should be avoided, the course of treatment should be restricted to no more than 2 days if possible, and the total daily dose should be limited to 600 mg (APS, 2003). A similar guideline, based on a retrospective chart review, is proposed when meperidine is given via PCA to patients unable to take morphine or hydromorphone: 10 mg/kg/day for no more than 3 days in patients with normal renal function (Simopoulos, Smith, Peeters-Asdourian, et al., 2002).
Patients who are taking meperidine should be evaluated frequently, probably every 8 to 12 hours, for signs of CNS irritability, specifically restlessness, shakiness, tremors, twitching, and jerking. Tremors are assessed by asking patients to stretch out their arms in front and noting a postural tremor in the hands. Patients should also be questioned about being awakened at night by twitching or jerking. If symptoms are present and have occurred after the initiation of meperidine doses, they may be due to normeperidine toxicity. The patient should be switched to another opioid analgesic, such as morphine or hydromorphone. Further accumulation of normeperidine may result in seizures (Beaule, Smith, Nguyen, 2004).
Because the half-life of the normeperidine is much longer than that of the meperidine, and the depressant effects of the latter drug may be partially suppressing the effects of the former, symptoms of toxicity may paradoxically worsen as the dose of meperidine is initially decreased. Naloxone should be avoided because it does not reverse the action of normeperidine and may even exacerbate the CNS hyperexcitability by decreasing the level of the depressant meperidine (Gordon, Jones, Goshman, et al., 2000). If meperidine has been used repeatedly and there are signs of toxicity, careful monitoring is necessary if the dose is suddenly lowered or if, in rare circumstances, naloxone is needed; if increased agitation or tremulousness appears, consideration should be given to administration of a sedative-hypnotic or an anticonvulsant.
Meperidine is frequently dosed inadequately, which may be one of the reasons it has been considered a safe drug despite ample evidence to the contrary (Latta, Ginsberg, Barkin, 2002). The habitual under-dosing is somewhat surprising since meperidine has relatively low potency and a very short duration of action (Latta, Ginsberg, Barkin, 2002). In one study, a 75 mg parenteral dose was found to be effective for only 30 minutes (Fairlie, Marshall, Walker, et al., 1999). In another study, a 50 mg parenteral dose was found to be no more effective than placebo (Austin, Stapleton, Mather, 1980a). The initial optimal dose of meperidine recommended for adults with moderate to severe pain is 75 to 100 mg, with some adults requiring 150 mg. The effective interval between doses ranges from 2 to 4 hours, with 3 hours as the average. These doses would quickly exceed the 600 mg/24 h maximum recommended by the APS and produce numerous adverse effects.
As mentioned, meperidine can be administered by the oral route, but just as with the parenteral route, it is rarely dosed appropriately. Oral meperidine is less than one fourth as potent as parenteral meperidine. This means that if a patient is receiving 75 mg of meperidine by the IV or IM route over a 3- to 4-hour period, 300 mg orally would be required to produce equianalgesia (see Table 16-1 on pp. 444-446). Even if the patient’s pain had decreased by 50% at the time the switch was made from parenteral to oral, 150 mg would be required orally. Obviously, doses required for analgesia by the oral route produce a significant risk of accumulation of normeperidine (Latta, Ginsberg, Barkin, 2002), making oral meperidine inappropriate for any type of pain management.
Meperidine is contraindicated in children, older adults, patients with impaired renal function, and those who have taken MAOIs in the past 14 days (APS, 2003; Gutstein, Akil, 2006). It is also contraindicated in patients with untreated hypothyroidism, Addison’s disease, benign prostatic hypertrophy, or urethral stricture. It should be used with extreme caution in patients with preexisting convulsive disorders and in patients with atrial flutter or other supraventricular tachycardias (Antonopolous, Bollinger, Goshman, 1996).
Meperidine continues to be commonly used for procedural analgesia, particularly for GI procedures, based on the misconception that it produces less biliary spasm than other opioids (Lee, Cundiff, 1998) (see Table 13-5). It is important to note that all opioids are capable of causing constriction of the sphincter of Oddi and the biliary tract, and there is no clinical advantage to using meperidine from this perspective (see Chapter 19 for a detailed discussion).
In summary, meperidine has no advantages over any other opioid (Latta, Ginsberg, Barkin, 2002) and it has no place in the treatment of persistent pain or in delivery systems such as PCA. Oral meperidine undergoes extensive first-pass metabolism, leaving the patient with poor analgesia but even more rapid normeperidine accumulation than the IV preparation (Latta, Ginsberg, Barkin, 2002). In healthy adults, meperidine should not be used for more than 48 hours or at doses exceeding 600 mg/24 h (APS, 2003). Given the multiple alternative opioids available, there is little rationale to continue using meperidine as an analgesic except in extraordinary cases.
Meperidine Use in Sickle Cell Patients
Although meperidine has been used for many years to treat pain in patients with sickle cell disease, it is a particularly poor choice in this population because the toxic metabolite is renally excreted and most individuals with sickle cell disease have renal insufficiency. Current guidelines recommend morphine as the opioid of choice for treatment of sickle cell pain (Platt, Eckman, Beasley, et al., 2002; Rees, Olujohungbe, Parker, et al., 2003). The Georgia Comprehensive Sickle Cell Center, well known for its research and clinical treatment of sickle cell disease, provides a protocol for rapid management of pain that includes IV morphine 3 to 5 mg every 10 minutes until pain is controlled plus IV ketorolac, followed by IV PCA morphine (with basal rate as needed) or ATC oral morphine dosing (Platt, Eckman, Beasley, et al., 2002). A common and recommended practice is to store a specified number of PCA pumps, drug reservoirs, and infusion tubings (number is dependent on the size of the institution) in the emergency department so that the therapy can be initiated without delay in sickle cell patients as well as other patients who are admitted for treatment of severe pain crises.
The British Committee for Standards in Haematology General Haematology Task Force by the Sickle Cell Working Party guideline calls for rapid assessment and treatment of painful episodes, a designated nursing staff who are trained and experienced with the management of patients with sickle cell disease, and multidisciplinary consultation as needed (Rees, Olujohungbe, Parker, et al., 2003). The guideline recommends treatment of acute pain based on pain intensity (see Chapter 12 for the WHO Ladder) and a multimodal approach that includes IV morphine titration (0.1 mg/kg every 20 minutes until pain is controlled) plus acetaminophen and NSAIDs, such as IV ketorolac. Ongoing analgesia is also based on pain intensity and may include nonopioids for mild to moderate pain and long-acting opioids with short-acting opioid breakthrough doses for more severe pain. Anticonvulsants are considered if neuropathic pain is present.
Patients who have received meperidine in the past may be resistant to changing from meperidine to safer analgesics, such as morphine. Understandably, patients will request what has been effective in relieving their pain in the past. It is important for clinicians to have patience and realize that change can be frightening especially when it occurs during a painful episode. Providing pain relief by the method preferred by the patient and then discussing changes in the treatment plan after pain is controlled is apt to be met with more patient acceptance (see Section II).
Methadone
Methadone (Dolophine; Methadose) is a unique opioid analgesic that may have advantages over other opioids in carefully selected and closely monitored patients (Table 13-6). In all countries except Germany, methadone is sold as a racemic mixture containing two mirror-image molecules—the l-isomer and the d-isomer. The l-isomer of methadone is a mu receptor agonist that has properties similar to other mu agonists but also has a long and variable half-life. The d-isomer of methadone is not an opioid compound, but instead, is an antagonist at the N-methyl-d-aspartate (NMDA) receptor. Based on extensive preclinical science, it is believed that NMDA receptor antagonism has the potential to produce analgesic effects independent of the opioid effect, at least in some neuropathic pain states; it also has been shown in animal models to reduce opioid tolerance. The combined effects of the l-isomer and d-isomer lead to effects from methadone that may be different than other opioids and potentially may make it a useful choice as a second- or third-line opioid for syndromes that have been poorly responsive to other opioids, including neuropathic pain syndromes (Dworkin, O’Connor, Backonja, et al., 2007). In other words, these effects may make methadone a favorable drug to consider when planning opioid rotation (switching) to an alternative opioid in the setting of poor opioid responsiveness due to inadequate analgesia (Mannino, Coyne, Swainey, et al., 2006) or when unacceptable adverse effects occur (Manfredi, Houde, 2003) (see Chapter 18).
Table 13-6
Methadone Advantages and Disadvantages
*Available dose forms/strengths may vary in different countries.
From Pasero, C., & McCaffery, M. (2011). Pain assessment and pharmacologic management, p. 340, St. Louis, Mosby. Data from Daeninck, P. J., & Bruera, E. (1999). Reduction in constipation and laxative requirements following opioid rotation to methadone: A report of four cases. J Pain Symptom Manage, 18(4), 303-309; Davis, M. P., & Walsh, D. (2001). Methadone for relief of cancer pain: A review of pharmacokinetics, pharmacodynamics, drug interactions and protocols of administration. Support Care Cancer, 9(2), 73-83; Krantz, M. J., Martin, J., Stimmel, B., et al. (2009). QTc interval screening in methadone treatment. Ann Intern Med, 150(6), 387-395; Lynch, M. E. (2005). A review of the use of methadone for the treatment of chronic noncancer pain. Pain Res Manag, 10(3), 133-144; Mannino, R., Coyne, P., Swainey, C., et al. (2006). Methadone for cancer-related neuropathic pain: A review of the literature. J Opioid Manage, 2(5), 269-276; Weschules, D. J., & Bain, K. T. (2008). A systematic review of opioid conversion ratios used with methadone for the treatment of pain. Pain Med, 9(5), 595-612. Pasero C, McCaffery M, Quinn TE. May be duplicated for use in clinical practice.
Given the observation that up to 80% of cancer patients will require rotation to another opioid during the course of pain treatment and that 44% may need more than one switch (Foley, Houde, 1998), methadone could potentially assume an important role in long-term opioid therapy. In a prospective controlled trial, 25% of patients were considered nonresponders to morphine, but after one, two, or three switches to alternative opioids, 96% of patients were able to achieve adequate analgesia with tolerable adverse effects (Riley, Ross, Rutter, et al., 2006). A 10-year review of opioid switching showed a dozen studies in which the range of success in switching from another opioid to methadone was 75% to 100% (Mercadante, Bruera, 2006).
Methadone is most often used by the oral route for persistent cancer and noncancer-related pain. Although usually considered a second- or third-line treatment (Mannino, Coyne, Swainey, et al., 2006; Fredheim, Moksnes, Borchgrevink, et al., 2008), some experts suggest that methadone can be used as a first-line opioid for cancer pain (Bruera, Palmer, Bosnjak, et al., 2004; Mercadante, Porzio, Ferrera, et al., 2008) and some types of neuropathic pain (Dworkin, O’Connor, Backonja, et al., 2007; Mannino, Coyne, Swainey, et al., 2006; Raja, Haythornthwaite, Pappagallo, et al., 2002).
For many people, the more familiar use of methadone is as maintenance therapy to reduce craving and prevent withdrawal in patients with addiction. Despite significantly increased use of methadone as an analgesic, some patients are reluctant to take it because of the perceived stigma of taking a medication that is used by opioid addicts (Davis, Walsh, 2001; Fredheim, Moksnes, Borchgrevink, et al., 2008). It is important to elicit patient and family concerns and provide clarification. The indication (“For Pain”) should be written on the prescription to avoid misunderstanding at the pharmacy.
It is extremely important that acute pain episodes, such as following trauma or surgery, be treated adequately in individuals who are taking methadone, including those who are receiving it for treatment of addictive disease. Individuals with addiction have been observed to have exaggerated pain responses (Savage, Schofferman, 1995), which may necessitate frequent dose adjustments if pain is treated with an opioid (Peng, Tumber, Gourlay, 2005; Savage, Schofferman, 1995). (See further discussion of pain in individuals with addictive disease in Chapter 20.)
After an oral dose, methadone has an onset of analgesia comparable to morphine (30 to 60 minutes). Plasma concentrations peak at approximately 2 hours after oral administration (Davis, Walsh, 2001). Methadone is extensively metabolized in the liver via the cytochrome P450 enzyme system, which has important implications for drug-drug interactions (see the paragraphs that follow and Box 11-1). Its oral bioavailability is 80% to 85%, which is significantly higher than that of morphine. This is reflected in a lower parenteral/oral potency ratio of 1:2 compared with 1:6 for morphine for initial dosing in an opioid-naïve patient. The elimination half-life of methadone is long and highly variable (5 to 130 hours; mean 20 to 35) (Lugo, Satterfield, Kern, 2005).
Methadone does not have neurotoxic metabolites, unlike morphine and hydromorphone. It may be less likely to cause severe constipation than other opioids, but this has not been confirmed (Daeninck, Bruera, 1999; Leppert, 2009). Methadone is synthetic and structurally unrelated to other opioids, and for this reason, it may be an alternative for the rare patient with a true allergy to another opioid.
Although methadone usually is administered orally (tablets and liquid are available), it can be given intravenously, intramuscularly, subcutaneously, sublingually, rectally, and topically. A guideline for the use of parenteral methadone in pain and palliative care has been published (Shaiova, Berger, Blinderman, et al., 2008). Most published reports on continuous SC administration indicate that local irritation is a significant management problem (Makin, 2000; Mathew, Storey, 1999; Centeno, Vara, 2005); flushing of the access site with normal saline has been reported to minimize irritation so that sites can be maintained for prolonged periods without the need for dose limitation or medications added to prevent irritation (Hum, Fainsinger, Bielech, 2007) (see Chapter 14 for the SC route). Although a commercial product is not available, rectal administration of methadone is very effective. The rectal to oral dose ratio is 1:1, with rapid absorption (Davis, Walsh, 2001). Methadone has been investigated for breakthrough pain via both oral and sublingual routes. In a pilot study, all patients reported that oral methadone was at least as effective as their usual breakthrough opioid, and remarkably, some reported onset of analgesia in as short as 10 minutes. The authors acknowledged that an explanation for such rapid relief is uncertain (Fisher, Stiles, Hagen, 2004). A pilot study of liquid sublingual methadone for breakthrough pain demonstrated a median time to meaningful pain reduction of 5 minutes (Hagen, Fisher, Stiles, 2007).
As described previously, the racemic mixture of methadone, which is available in the United States and most other countries, has multiple mechanisms of action. The opioid constituent, which is primarily an agonist at the mu opioid receptor, also has agonist effects at the delta and kappa receptors, which may contribute to analgesia (Davis, Walsh, 2001). In morphine-insensitive mice, methadone provided analgesia via the mu receptor, indicating that methadone and morphine occupy the mu receptor differently (Chang, Emmel, Rossi, et al., 1998). It also suggests a partial explanation for the observed incomplete cross-tolerance between methadone and morphine (and other opioids) (Pasternak, 2001). Clinically this means that when switching from another opioid to methadone, the effective methadone dose will be lower than predicted by equianalgesic calculations (Davis, Walsh, 2001).
Methadone’s analgesia also may be related to its antagonism of the NMDA receptor (Manfredi, Houde, 2003; Trafton, Ramani, 2009). This mechanism, which may involve both a direct analgesic effect and a partial reversal of opioid tolerance, is likely to be another factor that produces analgesia at lower than expected doses. The proposed mechanisms for reduced development of tolerance by methadone compared with other opioids is suggested by animal studies but have not yet been conclusively demonstrated in humans (Trafton, Ramani, 2009; Leppert, 2009).
Finally, the analgesic effects of methadone could be related to its inhibition of the reuptake of serotonin and norepinephrine at central synapses. Serotonin/ norepinephrine reuptake inhibitors have been found to be effective in alleviating neuropathic pain, similar to NMDA receptor antagonism (Manfredi, Houde, 2003) (see Section I and Chapter 22). Although there is no evidence that methadone is more effective for neuropathic pain than other opioid drugs, it has been speculated that these unique mechanisms on other receptors may yield such an effect. More research is needed to determine whether there are drug-selective differences in opioid efficacy for neuropathic pain.
Another important methadone characteristic is its lipophilicity. Methadone readily crosses the blood-brain barrier, is widely distributed throughout the body, and is highly protein-bound (Lynch, 2005). Lipophilicity makes methadone a potential candidate for sublingual (Hagen, Fisher, Stiles, 2007) and topical (Gallagher, Arndt, Hunt, 2005) administration.
The duration of analgesia after an oral dose of methadone usually is just 3 to 6 hours, particularly in those who have not been receiving regular doses (Davis, Walsh, 2001; Lugo, Satterfield, Kern, 2005; Bruera, Palmer, Bosnjak, et al., 2004); it may extend to 8 to 12 hours (or longer) with continued dosing. The half-life, as noted previously, typically is about 24 hours and has an upper limit more than 6 times this duration. These characteristics impact clinical management, especially during the initial titration phase. After dosing begins, or is increased, accumulation may occur for many days, or even weeks, before a steady state is approached. Clearance can increase with long-term dosing (Davis, Walsh, 2001). Even after accumulation, some patients may continue to need dosing four times a day (Peng, Tumber, Stafford, et al., 2008). These characteristics are manageable, but require close attention, as described in the following paragraphs and in Box 13-2. Some authors suggest that a few “priming” (Mercadante, Casuccio, Fulfaro, et al., 2001) or “loading” (Ayonrinde, Bridge, 2000; Blackburn, Somerville, Squire, 2002) doses on the first day or two of treatment will decrease the amount of time it takes to distribute the drug and reach steady state.
Elimination of methadone is primarily via feces. Unlike morphine and hydromorphone, there are no methadone toxic metabolites to accumulate in renal failure, and dose adjustment in the presence of renal insufficiency is generally not necessary (Davis, Walsh, 2001). However, as with other opioids, initial doses for older adults should be lower and dosing intervals longer than for healthy younger adults (Lugo, Satterfield, Kern, 2005); the half-life in older adults is likely to be relatively longer than in younger patients (Leppert W, 2009). Some methadone metabolism occurs in the intestine rather than the liver. Therefore, methadone doses may not need to be adjusted in cirrhosis and stable chronic liver disease (Lugo, Satterfield, Kern, 2005).
Drug-Drug Interactions
One of the major safety and clinical management limitations of methadone is the large number of medications with which it interacts. However, these drug interactions are still poorly understood in that many of the reports are theoretical and not clinically supported with case reports or clinical trials (Weschules, Bain, Richeimer, 2008; Gourevitch, Friedland, 2000). It is important to remember as well that these pharmacokinetic interactions may be compounded by pharmacodynamic interactions, or effects that are overlapping but have nothing to do with changes in blood levels. For example, methadone and benzodiazepines both cause sedation and should be used together with caution (Webster, Choi, Desai et al., 2008).
The monoamine oxidase inhibitors (MAOIs) should be avoided in patients receiving methadone because of the risk of inducing serotonin syndrome (Weschules, Bain, Richeimer, 2008; Leavitt, 2005, 2006) (see Chapter 22). Methadone is partially metabolized in the liver via multiple cytochrome P450 (CYP450) pathways, primarily CYP3A4, but also CYP1A2 and CYP2D6. Most methadone drug-drug interactions are related to CYP3A4 (Davis, Walsh, 2001). Methadone has the potential to induce or inhibit the metabolism of other drugs that use the same pathways. In turn, methadone metabolism can be induced or inhibited by drugs that use these enzyme pathways (see Chapter 11). Inducing metabolism has the effect of lowering serum concentration and decreasing the intended effect. Inhibiting metabolism has the effect of increasing serum concentration and increasing the intended effect or causing toxicity (Armstrong, Cozza, Sandson, 2003; Weschules, Bain, Richeimer, 2008). In the case of methadone, decreasing serum concentration may cause an increase in pain or even cause withdrawal syndrome. Increasing methadone serum concentration can cause adverse effects such as sedation and respiratory depression. The potential for either toxicity or undermedication and withdrawal from the previous opioid may be substantial in some instances (Lugo, Satterfield, Kern, 2005). A known food interaction (grapefruit juice inhibits gut-wall CYP3A4) and herb interaction (St. John’s wort induces CYP3A4 metabolism) further underscores the complexity of methadone and reinforces the need for thorough history taking and patient teaching (Armstrong, Wynn, Sandson, 2009; Zhou, Chan, Pan, et al., 2004). A drug interaction should be suspected whenever a new drug or remedy is introduced and there is a sudden increase in pain or increase in sedation when the methadone dose is stable.
Sharing a metabolic pathway is not an absolute contraindication for two drugs being taken concurrently, but close observation is necessary and dose adjustment of one medication may be required. Alternative medications with the same intended effect and which are metabolized differently should also be considered (Weschules, Bain, Richeimer, 2008) (Table 13-7). A particularly complex population is patients with HIV/AIDS. Research has shown that interactions are common between methadone and antiretroviral therapies and certain other medications commonly used in this population (Gourevitch, Friedland, 2000; Bruce, Altice, Gourevitch et al., 2006).
Table 13-7
Methadone: Drug-Drug Interactions1
| Medication Class | Selected Medications with Known or Theoretical Interaction with Methadone | Potential Alternatives (Low or No Pharmacokinetic Interaction Potential) |
| Anticonvulsants | Phenytoin, phenobarbital, carbamazepine | Valproic acid, gabapentin, lamotrigine, levetiracetam |
| Antidepressants | Fluvoxamine, fluoxetine, paroxetine, desipramine, amitriptyline, imipramine | Sertraline, citalopram, escitalopram, venlafaxine, mirtazapine |
| Neuroleptics | Risperidone | Olanzapine |
| Anxiolytics | Alprazolam, diazepam | Buspirone, lorazepam, oxazepam, triazolam |
| H2 receptor blockers | Cimetidine | Famotidine, ranitidine |
| Antituberculosis antibiotics | Rifamycins (e.g., rifampin) | Rifabutin |
| Antifungals | Ketoconazole, fluconozole, itraconozole | Terbinafine, caspofungin |
| Macrolide antibiotics | Erythromycin, clarithromycin | Azithromycin |
| Quinolone antibiotics | Ciprofloxacin | Levofloxacin |
| Antiretrovirals | Most | See Bruce et al. and Gourevitch et al. below for extensive discussion of management |
1This table contains selected drugs and their potential for interaction with methadone. It is not a comprehensive list and is meant to be a quick reference for clinicians treating multiple diagnoses.
From Pasero, C., & McCaffery, M. (2011). Pain assessment and pharmacologic management, p. 343, St. Louis, Mosby. Data from Bruce, R. D., Altice, F. L., Gourevitch, M. N., et al. (2006). Pharmacokinetic drug interactions between opioid agonist therapy and antiretroviral medications: implications and management for clinical practice. J Acquir Immune Defic Syndr, 41(5), 563-572; Gourevitch, M. N., & Friedland, G. H. (2000). Interactions between methadone and medications used to treat HIV infection: A review. Mt Sinai J Med, 67(5-6), 429-436; Leavitt, S. B. (2005). Methadone-drug interactions, ed 3. Addiction Treatment Forum. Available at http://www.atforum.com/SiteRoot/pages/addiction_resources/Drug_Interactions.pdf. Accessed December 20, 2009; Leppert, W. (2009). The role of methadone in cancer pain treatment—A review. Int J Clin Pract, 63(7), 1095-1109; Weschules, D. J., Bain, K.T., & Richeimer, S. (2008). Actual and potential drug interactions associated with methadone. Pain Med, 9(3), 315-344. Quinn TE, Pasero C, McCaffery M. May be duplicated for use in clinical practice.
Renal excretion of methadone is affected by urine pH. Methadone renal clearance is increased three-fold in a low pH environment (Davis, Walsh, 2001). Medications that lower urine pH such as furosemide, and drugs that increase urine pH such as sodium bicarbonate and acetazolamide (Diamox) must therefore be used with caution.
An additional potential concern with methadone and drug-drug interaction that is only partially related to its metabolic pathway is an association with cardiac arrhythmia, specifically, prolongation of the QTc interval seen on electrocardiogram (ECG). A critically prolonged QTc interval—one greater than 500 ms—can lead to the potentially fatal cardiac arrhythmic disorder torsades de pointes (Krantz, Martin, Stimmel, et al., 2009). Many drugs, including methadone, can prolong the interval and potentially increase this risk (University of Arizona, 2009).
The U.S. FDA issued an FDA Alert in 2006 to warn health professionals about the potential for cardiac arrhythmias with methadone use (United States Food and Drug Administration, 2006). The risk of QTc prolongation is presumably dose-dependent and related to other factors, including co-morbid heart disease, the use of other drugs with effects on cardiac conduction, and genetic factors. The degree of risk must be balanced with severity of pain and goals of care (Moryl, Coyle, Foley, 2008; Sekine, Obens, Coyle, et al., 2007).
There is no consensus on routine screening with ECG prior to starting methadone (Schmittner, Krantz, 2006; Cruciani, 2008). It is prudent to obtain an ECG if the patient has a history of significant heart disease or if the patient is older. Routine serial ECG is recommended by one recent guideline (Krantz, Martin, Stimmel, et al., 2009), but is considered controversial (Gourevitch, 2009). Periodic ECG monitoring of patients on doses of methadone greater than 100 mg/day also has been recommended (Krantz, Martin, Stimmel, et al., 2009).
Rotating to Methadone
Safely switching a patient from another opioid to methadone is a clinical challenge (Fine, Portenoy, Ad Hoc Expert Panel on Evidence Review and Guidelines for Opioid Rotation, 2009) (Box 13-3; also see Patient Examples). The most common reason for making the switch is adverse effects from the previous opioid. Another reason is ineffectiveness of another opioid despite appropriate titration. Because of its long half-life, the titration of methadone takes at least several days, during which strict adherence to the dose and schedule are required and close monitoring of the patient is essential. A short-acting opioid may be used for breakthrough pain (Lugo, Satterfield, Kern, 2005). Careful patient selection and clear instructions are required.
Determining the starting morphine to methadone dose ratio and using a systematic method of patient selection and monitoring are related but distinct elements of the conversion process. Factors to consider in planning the switch:
• Onset of action is comparable to other oral opioids, about 30 to 60 minutes.
• Duration of analgesia with acute dosing (e.g., the beginning of the titration period) is 4 to 6 hours.
• Duration of analgesia with long-term dosing may be 8 to 12 hours or longer.
• Peak effect is at about 2.5 hours.
• Steady state will not be reached for several days, and in occasional patients with longer half-lives, as long as several weeks.
• Drug accumulation occurs during initial titration when the concentration in the blood continues to increase above the effective analgesic level and into the toxic range as a result of the relatively prolonged period required to approach steady state. Toxic accumulation is more likely if a relatively high dose or short dosing interval is used in an effort to quickly identify an analgesic dose; accumulation-related adverse effects can include excessive sedation and respiratory depression.
• Clinical experience indicates that the greatest incidence of serious adverse effects occurs on days 3 to 5. There is great interindividual pharmacokinetic variability.
• There is no fixed equianalgesic ratio between methadone and other opioids, contrary to the experience with most other opioids.
It is now widely agreed among pain specialists that published equianalgesic dose tables that include methadone do not reflect the pharmacology of methadone and the clinical reality of patient management (Knotkova, Fine, Portenoy, 2009; Pereira, Lawlor, Vigano, et al., 2001). Several attempts have been made in the past decade to determine the equianalgesic dose ratio of other opioids, principally morphine, to methadone (Mercadante, Bruera, 2006; Benítez-Rosario, Salinas-Martín, Aguirre-Jaime, et al., 2009). It is clear that the new dose of methadone is dependent on the prior dose of morphine, but not in a fixed ratio. As the dose of morphine increases, the equianalgesic dose of methadone decreases (see the discussion of tolerance in Chapter 11 for possible explanations of this phenomenon). This has been described as a “dynamic inverse potency relationship between methadone and other opioids” (Weschules, Bain, 2008). For example, with a daily morphine dose of 80 mg, the morphine to methadone ratio was found to be 4:1 (Ripamonti, Groff, Brunelli, et al., 1998). As the daily morphine dose rises, the morphine to methadone ratio increases to as much as 14:1 or higher (Pereira, Lawlor, Vigano, et al., 2001; Soares, 2005; Ayonrinde, Bridge, 2000). Clinical research-derived tables that match morphine dose ranges to morphine-methadone ratios are in close, but not complete, agreement (see examples in Table 13-8). Use of these tables is intended to find a safe starting dose, but does not predict the final effective methadone dose, which varies widely across patients (Benítez-Rosario, Salinas-Martín, Aguirre-Jaime, et al., 2009).
Table 13-8
Comparison of Selected Methods of Converting Morphine to Methadone

From Pasero, C., & McCaffery, M. (2011). Pain assessment and pharmacologic management, pp. 346-347, St. Louis, Mosby. Data from Ayonrinde, O. T., & Bridge, D. T. (2000). The rediscovery of methadone for cancer pain management. Med J Aust, 173(10), 536-540; Blackburn, D., Somerville, E., & Squire, J. (2002). Methadone: An alternative conversion regime. Eur J Palliat Care, 9(3), 92-96; Bruera, E., & Sweeney, C. (2002). Methadone use in cancer patients with pain: A review. J Palliat Med, 5(1), 127-138; Indelicato, R. A., & Portenoy, R. K. (2002). Opioid rotation in the management of refractory cancer pain. J Clin Oncol, 20(1), 348-352; Mercadante, S., Casuccio, A., Fulfaro, F., et al. (2001). Switching from morphine to methadone to improve analgesia and tolerability in cancer patients: A prospective study. J Clin Oncol, 19(11), 2898-2904; Morley, J. S., & Makin, M. K. (1997). Comments on Ripamonti et al. Pain, 73(1), 114; Ripamonti, C., Groff, L., Brunelli, C., et al. (1998). Switching from morphine to oral methadone in treating cancer pain: What is the equianalgesic dose ratio? J Clin Oncol, 16(10), 3216-3221. Quinn TE, Pasero C, McCaffery M. May be duplicated for use in clinical practice.
Attempts have been made to simplify the process of determining the morphine to methadone dose ratio that is necessary to calculate the starting dose of methadone for a particular patient. Nomograms (Toombs, 2008; New Hampshire Hospice and Palliative Care Organization at http://www.nhhpco.org/opioid.htm) and formulas (Plonk, 2005) have been derived using values from published research. These methods yield more precise values than matching ratios to a table of dose ranges, but it is not known whether such precision has clinical relevance. In their extensive review, Weschules and Bain (2008) conclude that the actual dose ratio used to determine the starting dose of methadone is probably less important than careful patient selection and close systematic monitoring of the patient during the titration period.
An additional safety consideration is found in the setting of rapid dose escalation of the previous opioid. If the previous opioid dose is relatively stable, any of the morphine-methadone ratio tables described above could be used. However, if the previous opioid has recently been escalated, it may be prudent to use the dose prior to escalation as the starting point for calculating the starting methadone dose in order to avoid methadone adverse effects (Zimmermann, Seccareccia, Booth, et al., 2005).
The dose ratio between methadone and other opioids is not bidirectional. In other words, the ratio used to switch a patient to methadone cannot be used to switch a patient from methadone to another opioid (Bhimji, 2005; Prommer, 2006a; Walker, Palla, Pei, et al., 2008). Clearly, this area of practice requires clinical expertise, individualization of care, and close monitoring.
There are widely differing approaches to systematically switch a patient to methadone from another opioid (see Table 13-8). There is no evidence of superiority of one conversion method over another (Weschules, Bain, 2008). In general, these regimens can be divided into two groups: a gradual substitution of methadone for the other opioid over 3 days (Bruera, Sweeney, 2002; Ripamonti, Groff, Brunelli, et al., 1998) and completely stopping the previous opioid while initiating titration of methadone (“stop and go” method) (Morley, Makin, 1997; Ayonrinde, Bridge, 2000; Mercadante, Casuccio, Calderone, 1999; Mercadante, Casuccio, Fulfaro, et al., 2001). The latter approach is particularly appropriate if the reason for switching is opioid toxicity and the previous opioid must be stopped quickly.
A consensus guideline (Fine, Portenoy, the Ad Hoc Expert Panel on Evidence Review and Guidelines for Opioid Rotation, 2009) concludes that a two-step approach guided by a careful clinical assessment is a safe strategy when switching from any pure mu agonist opioid to methadone. The first step involves calculation of the equianalgesic dose using a generally accepted equianalgesic table, followed by reduction in this calculated dose by 75% to 90% (with high dose therapy and some other factors suggesting the use of the upper bound of the calculated reduction). After this first step, a possible second step is undertaken, which involves another dose adjustment of 15% to 30%, either decrease or increase, depending on whether the assessment reveals any of a set of clinical characteristics (e.g., presence of severe pain would suggest adding to the calculation, whereas presence of medical comorbidities would suggest subtracting further). Studies are needed to provide an empirical basis to this and other conversion strategies. Pending these studies, however, the most important consideration must be safety, which requires significant reduction in the calculated equianalgesic dose, followed by dose titration.
Some experts have concluded that the risk associated with a switch to methadone is sufficient to institute additional requirements for safety. These include limiting any single dose to 30 mg during the initial titration, even if the calculated dose is higher (Morley, Makin, 1997) and switching gradually over 3 days (Lawlor, Turner, Hanson, et al., 1998).
Generally, an opioid other than methadone, specifically one with a short half-life, is desirable for the treatment of severe, escalating pain, when rapid titration is needed. The risk of accumulation of drug levels into the toxic range is greater with an opioid drug with a long half-life, such as methadone. Deaths have been reported among patients in the community experiencing too-rapid dose increases without adequate follow up (Institute for Safe Medication Practices, 2008).
A variety of additional dosing schemes have been suggested in an effort to provide analgesic doses while avoiding toxic accumulation (Mercadante, Bruera, 2006). These include using a higher “loading” or “priming” dose (so that steady state can be reached sooner) (Mercadante, Casuccio, Calderone, 1999; Ayonrinde, Bridge, 2000; Blackburn, Somerville, Squire, 2002); the use of a PRN methadone regimen to start, which is then converted to fixed dosing after titration; and the use of an initial fixed dosing interval of 3 or 6 hours, which is lengthened to 8 to 12 hours after analgesia is obtained (Ayonrinde, Bridge, 2000; Morley, Makin, 1997; Lugo, Satterfield, Kern, 2005). Additionally, a second, short-acting drug, such as morphine, oxycodone, or hydromorphone, also can be provided for rescue doses (Lugo, Satterfield, Kern, 2005). The rescue dose may require titration in terms of size or frequency during the initial period of methadone titration.
The factors outlined above strongly support the use of a systematic procedure and the assistance of a clinician experienced in the use of methadone when switching from another opioid (Leppert, 2009). In addition, the patient and family must be willing and able to follow the regimen, and a process for regular monitoring must be incorporated in the procedure.
Methadone is a complex analgesic. To be used optimally and safely, a clinician well versed in its characteristics should be involved in care planning and implementation. Patient selection and close monitoring during the titration phase are critical elements in pain management with this drug. The reader is referred to an excellent website for information on methadone safety, Pain Treatment Topics, http://pain-topics.org/opioid_rx/methadone.php. See Patient Education Form IV-8 on methadone on pp. 560-561 at the end of Section IV.
Following are two patient examples to illustrate how patients may be switched from other opioids to methadone using morphine-methadone dose ratios and other pointers from Box 13-3 and Table 13-8.
Oxycodone
Oxycodone is used to treat acute cancer and noncancer-related persistent pain. It is commercially available in single entity oral formulations and a variety of fixed combinations with acetaminophen, aspirin, and ibuprofen (Table 13-9). When taken in fixed combination with a nonopioid, it is critical to patient safety and treatment efficacy to establish exactly what dose of the nonopioid the patient is taking. Care should be taken to avoid prescribing or administering amounts that would exceed the daily maximum dose for acetaminophen (4000 mg), aspirin (4000 mg), or ibuprofen (3200 mg). Patients should also be told not to take additional nonopioid that could result in excess dosing (see Section III).
Table 13-9
Commercially Available Fixed Combination Doses of Oxycodone and Nonopioids
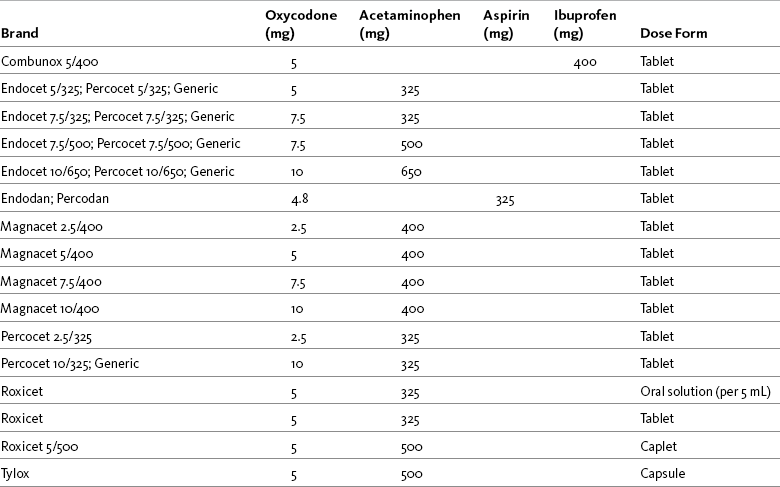
From Pasero, C., & McCaffery, M. (2011). Pain assessment and pharmacologic management, p. 351, St. Louis, Mosby. Pasero C, McCaffery M. May be duplicated for use in clinical practice.
As a single agent, oxycodone is available as a tablet, capsule, and elixir, and also in a modified-release (every 12-hour) formulation (OxyContin) (see Chapter 14 for a detailed discussion of specific oral oxycodone formulations). A once-daily oxycodone was in development at the time of publication. Oxycodone is not available in the United States in suppository form for rectal administration; however, oral preparations are commonly administered rectally. It is given via the IV route outside of the United States and has been shown to be effective for postoperative pain by this route (Lenz, Sandvik, Qvigstad, et al., 2009).
Oxycodone is metabolized in the liver primarily by the CYP450 2D6 enzyme and excreted by the kidneys. The starting dose should be reduced and the dosing interval may need to be increased for patients with hepatic or renal insufficiency (Riley, Eisenberg, Müller-Schwefe, et al., 2008). One of the metabolic products of oxycodone is oxymorphone, but it does not account for a significant proportion of oxycodone’s analgesic effect (Kalso, 2007; Lalovic, Kharasch, Hoffer, et al., 2006). Although it is known that there is significant genetic polymorphism at P450 2D6—leading to intermediate, extensive (or rapid), ultra-rapid metabolizers, or poor metabolizers (Palmer, Giesecke, Body, et al., 2005)—CYP450 polymorphism probably is not responsible when pain in an individual seems not to be oxycodone-responsive. Unchanged oxycodone and multiple metabolites produced by other pathways appear to produce oxycodone’s pharmacodynamic effects, although the role of the several metabolites has not been well described (Lalovic, Kharasch, Hoffer, et al., 2006). Nonetheless, interactions at this enzyme potentially could be responsible for adverse effects and interactions with other drugs affected by the same metabolic pathway (Foster, Mobley, Wang, 2007). For example, rifampin was shown to greatly reduce the plasma concentrations of both IV and oral oxycodone (Nieminen, Hagelberg, Saari, et al., 2009). Drug-drug interaction lists (see Table 11-2 and Box 11-1 on p. 289) should be consulted when oxycodone is initiated or other medications, especially selective serotonin reuptake inhibitor (SSRI) antidepressants, are started in a patient taking oxycodone (Davis, Varga, Dickerson, et al., 2003) (see Chapter 11 for more on the cytochrome P450 enzyme system and drug-drug interactions).
Compared with morphine, oxycodone is more potent (Boström, Hammarlund-Udenaes, Simonsson, 2008; Davis, Varga, Dickerson, et al., 2003; Kalso 2007), has greater oral bioavailability (~60%) (Kalso 2007), comparable onset of action (30 to 60 minutes) and half-life (3 to 4 hours), and a similar adverse effect profile (see Table 16-1 on pp. 444-446). Older equianalgesic tables used a 1:1 ratio for morphine to oxycodone, but more typically a 1.5:1 ratio is now used (APS, 2003). However, similar to hydromorphone, the ratio between oxycodone and morphine may be influenced by the direction of the change in opioid. Some research suggests that when converting from morphine to oxycodone, a ratio of 2:1 might be used, but when converting from oxycodone to morphine, a ratio of 1:1 might be better (Knotkova, Fine, Portenoy, 2009).
Absorption and peak blood levels by the rectal route appear to be comparable to that of the oral route, but maximum blood concentration is prolonged. Bioavailability by the rectal route is estimated to be 45% to 60% (Lugo, Kern, 2004). Although further research is needed, morphine may cause more nausea and oxycodone more constipation (Riley, Eisenberg, Müller-Schwefe, et al., 2008; Davis, Varga, Dickerson, et al., 2003). Women may have a greater analgesic response to oxycodone than men (Davis, Varga, Dickerson, et al., 2003). A study comparing the adverse effects of short-acting oxycodone found no gender- or age-related differences between the adverse effects experienced by healthy older and middle-aged adults without pain, which led the researchers to reinforce to clinicians that they should not avoid prescribing oral opioids to older adults based on the belief that older adults are at higher risk of adverse effects than younger adults (Cherrier, Amory, Ersek, et al., 2009).
In contrast to morphine, oxycodone binds to the kappa receptor as well as the mu receptor (Lauretti, Oliveira, Pereira, 2003). This kappa affinity is relatively weak, compared to its mu affinity, but the dual action and interaction between receptor types theoretically may provide clinical advantages in some pain states. For example, it may be speculated that the kappa activity could render oxycodone relatively more effective than morphine for visceral pain and some types of neuropathic pain (Riley, Eisenberg, Muller-Schwefe, et al., 2008; Nielsen, Ross, Lotfipour, et al., 2007). Further research will be needed to determine whether these differences exist and are clinically meaningful.
There is also evidence of synergy between morphine and oxycodone (Ross, Wallis, Smith, 2000). A combined morphine-oxycodone tablet is currently in clinical trials. A small study of patients with cancer pain (N = 22) compared modified-release formulations of oxycodone and morphine (Lauretti, Oliveira, Pereira, 2003). Those who took oxycodone consumed 38% less short-acting morphine for breakthrough pain to achieve the primary outcome of achieving and maintaining a VAS of 4 or less and reported less nausea and vomiting compared with those who took morphine only. The researchers concluded that a combination of opioids with different receptor action sites (i.e., morphine and oxycodone) may produce better analgesia than the use of an opioid that has one action site.
There is a lack of head-to-head studies comparing oxycodone with other analgesics (Ripamonti, Bandieri, 2009). A large (N = 456) randomized, placebo-controlled, single-dose study of women with moderate to severe pain after abdominal or pelvic surgery found that the combination of oxycodone (5 mg) plus ibuprofen (400 mg) provided significantly better pain relief than either agent taken alone (Singla, Pong, Newman, et al., 2005). A meta-analysis identified just four randomized controlled trials that compared oral oxycodone with either oral morphine (N = 3) or oral hydromorphone (N = 1) and concluded that there were no significant differences in efficacy and tolerability between the opioids (Reid, Martin, Sterne, et al., 2006). A more recent Cochrane Collaboration Review concluded that single oxycodone doses higher than 5 mg are effective for postoperative pain and two to three times stronger than codeine (Gaskell, Derry, Moore, et al., 2009). Efficacy was increased when oxycodone was combined with acetaminophen. Oxycodone 10 mg plus acetaminophen (650 mg) provided good analgesia to half of those treated, comparable to NSAIDs but with a longer duration of action.
It should be noted that the name oxycodone is similar in appearance and sound to oxymorphone, hydrocodone, and hydromorphone. An increased level of alertness is required to prevent medication errors with these opioids.
Oxymorphone
Oxymorphone is a semi-synthetic opioid indicated for moderate to severe cancer and noncancer pain (Smith, 2009). It is an agonist at both the mu and delta opioid receptors (Prommer, 2006b). Although the clinical implications of these interactions with different types of opioid receptors are unclear, it is possible that delta receptor agonism may potentiate mu receptor analgesic effects (Chamberlin, Cottle, Neville, et al., 2007; Fishbain, 2009; Smith, 2009).
Oxymorphone is available in parenteral and rectal forms (Numorphan) (Smith, 2009). In 2006, it was released in short-acting (Opana) and modified-release (Opana ER) oral formulations (see Chapter 14 for discussion of these formulations of oral oxymorphone).
Parenterally, oxymorphone is 10 times more potent than IV morphine (Fukuda, 2005). When given IV, it has a quick onset (5 to 10 minutes). Its peak time is 15 minutes, and its duration is 3 to 6 hours. It sometimes is used as a preoperative medication and a supplement for balanced anesthesia but is less popular for this purpose than the faster-acting opioids such as fentanyl and sufentanil (White, Freire, 2005). Oymorphone has been shown to provide effective pain relief via IV PCA (Farragher, Laffey, 2006), and it is well absorbed subcutaneously and intramuscularly, although IM administration of any opioid is discouraged (see Chapter 14).
The safety, efficacy, and adverse effect profiles of oral oxymorphone are similar to that of other mu agonist opioids (Chamberlin, Cottle, Neville, et al., 2007). When converting from morphine or oxycodone, the suggested oral conversion ratios are 3:1 and 2:1, respectively (Smith, 2009). Oxymorphone is more lipophilic than morphine, which may account for its slightly faster onset of action (30 to 45 minutes for oral short-acting) (Smith, 2009). A mean time to peak effect of 30 minutes has been associated with all doses of short-acting oxymorphone (Smith, 2009). Its half-life (7 to 11 hours) is longer than morphine’s (2 to 4 hours) (Chamberlin, Cottle, Neville, 2007) and significantly longer than parenteral oxymorphone’s (2 hours) (Smith, 2009). The oral bioavailability of oxymorphone is 10% (Prommer, 2006b); therefore, parenteral oxymorphone is 10 times more potent than oral oxymorphone.
Food, particularly food with a high fat content, can increase the plasma concentration of oral oxymorphone by as much as 50%, so the drug should be taken on an empty stomach (1 hour before or 2 hours after a meal). Alcohol ingestion at the time of dosing can accelerate the drug delivery from the modified-release formulation and lead to increased serum levels (Smith, 2009). These are important considerations when selecting an opioid; oral oxymorphone would not be a good choice in those who are unable to follow these restrictions concerning the timing of food consumption or alcohol intake. Given an upper bound of half-life measurements that approaches 11 hours, time to steady state with both short-acting and modified-release oxymorphone could be as long as almost 3 days (Smith, 2009) (see Chapter 14 for pharmacokinetic information on modified-release oxymorphone).
Besides morphine and hydromorphone, oxymorphone is the only other opioid analgesic commercially available in the United States as a rectal suppository (Numorphan). By the rectal route, its onset of action is 15 to 30 minutes, with a peak time of 120 minutes. In one study, rectal oxymorphone was found to be one-tenth as potent as IM oxymorphone but one twentieth as potent in terms of peak effect (Beaver, Frise, 1977).
Oxymorphone is extensively metabolized in the liver and produces clinically inert metabolites (Smith, 2009). A major metabolite, noroxymorphone, is a potent mu opioid receptor agonist when administered intrathecally but lacks systemic efficacy most likely because of its inability to penetrate the blood-brain barrier (Lemberg, Siiskonen, Kontinen, et al., 2008). Though further research is needed, dose adjustments are likely necessary in patients with renal and hepatic disease (Smith, 2009); Guay (2007) recommends avoiding oxymorphone entirely in patients with moderate-to-severe hepatic impairment. The drug was shown in one study to be removed by hemodialysis (Smith, 2009). There appears to be a low risk for interaction with concurrent medications that are metabolized by the CYP450 enzyme system, which may be a significant benefit in patients who are poor metabolizers or those who take multiple medications that rely on this enzyme system for metabolism, such as some antidepressants, beta blockers, antipsychotics, chemotherapeutic agents, and some other opioids (Adams, Pieniaszek, Gammaitoni, et al., 2005; Chamberlin, Cottle, Neville, 2007; McIlwain, Ahdieh, 2005) (see Chapter 11 for more on cytochrome P450 enzymes and drug-drug interactions).
It should be noted that the name oxymorphone is similar in appearance and sound to oxycodone, hydrocodone, and hydromorphone. An increased level of alertness is required to prevent medication errors with these opioids.
Propoxyphene
Propoxyphene (Darvon) is prescribed for mild to moderate pain but has fallen out of favor over the years (Barkin, Barkin, Barkin, 2006) ( discussed later in this section). It is often used in combination with acetaminophen (Darvocet) or aspirin (Darvon Compound). Propoxyphene is one half to one third as potent as codeine. Its recommended dose of 100 mg is equal in analgesic effect to 60 mg of codeine, which is known to be equal to 600 mg of aspirin (less than two 325 mg tablets). A systematic review of 26 randomized trials showed that adding propoxyphene to acetaminophen improved analgesia by an average of only 7.3% compared with acetaminophen alone (Li Wan Po, Zhang, 1997). Despite having antagonist effects at the NMDA receptor, it is no better for neuropathic pain than any comparable drug (Scott, 2005; Patt, 1998). In a meta-analysis of opioids for all types of noncancer pain, the so-called “weak” opioids, including propoxyphene, were found to be better than placebo but less effective than acetaminophen, NSAIDs, and strong opioids (Furlan, Sandoval, Mailis-Gagnon, et al., 2006).
At equianalgesic doses, propoxyphene has the same incidence of adverse effects as codeine and a longer half-life (6 to 12 hours) (see Table 13-1; also see Table 16-1 on pp. 444-446). It also is metabolized in the liver by the CYP2D6 enzyme system, and therefore, has the potential for interaction with many other drugs. Serious interactions are also possible with several other drugs that may have additive adverse effects (Barkin, Barkin, Barkin, 2006). (See Chapter 11 for more on the CYP2D6 enzyme system and drug-drug interactions.)
Propoxyphene has an active metabolite, norpropoxyphene, which has a half-life of 30 to 36 hours and can accumulate with repeated dosing, particularly in patients with renal insufficiency (Strassels, McNicol, Suleman 2008). Norpropoxyphene can produce pulmonary edema and cardiotoxicity, including arrhythmias (Strassels, McNicol, Suleman 2008). Apnea, cardiac arrest, and death have been reported. Seizures and other CNS toxicities have occurred (Barkin, Barkin, Barkin, 2006). These effects are not reversed by naloxone (Barkin, Barkin, Barkin, 2006), and even dialysis may not help (Bailie, Johnson, 2002).
Because of the toxicity related to norpropoxyphene, propoxyphene is no longer favored for the routine treatment of pain. It is an especially poor choice for use in older individuals because most have some degree of renal insufficiency. It has caused adverse events in residents in nursing homes (Perri, Menon, Deshpande, et al., 2005) and community-dwelling older adults (Gallagher, Barry, Ryan, et al., 2008; Kamal-Bahl, Stuart, Beers, 2006) and has been associated with hip fractures, possibly because of the CNS adverse effects of dizziness and sedation (Kamal-Bahl, Stuart, Beers, 2006). Since 1991, it has been listed in the Beers Criteria as an inappropriate medication for older adults (Beers, 1997; Beers, Ouslander, Rollingher, et al., 1991; Fick, Cooper, Wade, et al., 2003) (see Table 13-4). Others have similarly designated the drug as “rarely appropriate” in older adults (Zhan, Sangl, Bierman, et al., 2001). In Europe, propoxyphene was associated with accidental deaths and suicide and was withdrawn in the United Kingdom (Committee on the Safety of Medicines of the UK, 2006). A review of prescribing data in England and Wales reported that changes in prescribing after the withdrawal of co-proxamol (propoxyphene plus acetaminophen) resulted in a reduction in mortality involving the drug and little evidence of substitution of suicide method related to increased prescribing of other analgesics (Hawton, Bergen, Simkin, et al., 2009).
Unfortunately, despite its inappropriateness and published guidelines recommending against its use in older adults (APS, 2003; American Geriatrics Society [AGS], 2002; Fick, Cooper, Wade, et al., 2003; Zhan, Sangl, Bierman, et al., 2001), propoxyphene continues to be commonly prescribed for this population group (Singh, Sleeper, Seifert, 2007; Kamal-Bahl, Stuart, Beers, 2006; Liu, Christensen, 2002). Most recently, the U.S. FDA decided to require manufacturers to strengthen their warning to emphasize the risk of overdose with propoxyphene but allowed continued marketing of the drug despite agreement among a majority of FDA advisers that propoxyphene-containing products offer little benefit and should be removed from the market (Traynor, 2009).
In summary, despite a history of widespread use and occasional anecdotal reports in its favor (Scott 2005; Patt, 1998), the overwhelming evidence strongly suggests that this drug should not be a first-line analgesic and should be avoided in older adults, those with renal insufficiency, and those likely to need multiple medications for co-morbidities. Oxycodone (Mercadante, Arcuri, 2007) and tramadol (Mullins, Wild, 2003) have been suggested as safer alternatives for older adults. See Patient Education Form IV-5 on propoxyphene at the end of Section IV.
Other Mu Opioid Analgesics
Some mu opioid analgesics are seldom used for pain but may offer options for selected patients. They include alfentanil, remifentanil, and sufentanil.
Alfentanil
Alfentanil is rarely used for pain management today (Gutstein, Akil, 2006). It is the least potent of the fentanils: one-fortieth as potent as fentanyl (Mildh, Scheinin, Kirvelä, 2001). It is lipophilic and has a faster onset but a shorter duration of action (30 minutes) than fentanyl (Duncan, 2002; Murphy, 2006; Upton, 2007). It is used primarily in the intraoperative and procedural settings via the IV route (Fukuda, 2005). Patients who received a mixture of alfentanil and morphine in the PACU achieved equal but faster comfort with no increase in opioid-induced adverse effects, compared with those who received morphine alone (Alkhazrajy, Macintyre, Upton, et al., 2007).
Given its wide distribution in the body, alfentanil is like fentanyl in having a short duration of effect when administered in a non–steady state situation when the decline in blood level after the dose is largely determined by redistribution into fat, rather than by metabolism. In this setting, given rapid clearance from its site of analgesic action in the brain (Upton, 2007) and its resultant short duration of action, it is considered a poor choice for postoperative analgesia. SC alfentanil infusions in palliative care settings have been reported (Urch, Carr, Minton, 2004), and there are anecdotal reports of its use for breakthrough pain in palliative care via intranasal and buccal administration, although there are no commercial preparations available for this use (Dale, Hjortkjær, Kharasch, 2002; Duncan, 2002). The drug is metabolized in the liver, has no clinically significant metabolites (Murphy, 2006), and is one of the few drugs (along with fentanyl and methadone) to have been recommended for patients with end-stage renal disease who need opioid analgesia (Murtagh, Chai, Donohoe, et al., 2007).
Remifentanil
Remifentanil is in the same mu agonist subclass as fentanyl, sufentanil, and alfentanil but has unique pharmacokinetic properties. It is available for IV use only. Current formulations of the drug contain glycine and are not approved for intrathecal or epidural use (Fukuda, 2005).
Remifentanil is unique in that its structure includes an ester linkage, which is broken down in the blood. As a result, it is rapidly cleared from the blood after a dose. It is not metabolized in the liver and is minimally affected by renal and hepatic function; accumulation does not occur (Beers, Camporesi, 2004; Pitsiu, Wilmer, Bodenham, et al., 2004). Its principle metabolite, remifentanil acid, has low potency and is excreted by the kidneys. Although remifentanil acid clearance is markedly reduced in patients with renal failure, increasing the half-life of the metabolite from 2 hours to 26 hours, this has not been shown to produce adverse effects (Hoke, Shlugman, Dershwitz, et al., 1997). For example, clearance of remifentanil acid was reduced in critically ill patients (N = 40) with moderate to severe renal impairment to about 25% that of patients with no to mild renal impairment; however, prolonged opioid effects were not noted (Pitsiu, Wilmer, Bodenham, et al., 2004). With these pharmacokinetics, the elimination half-life of remifentanil is only 3.2 minutes (25% longer in older adults), and this short half-life exists even after a long-duration (more than 8 hours) infusion (Beers, Camporesi, 2004).
Remifentanil is slightly more potent than fentanyl (Servin, Billard, 2008), quickly crosses the blood-brain barrier (Beers, Camporesi, 2004), and has an ultra rapid onset of action (1.5 minutes in young adults, increasing with age). The pharmacokinetic profile appears to be unaltered by obesity (Beers, Camporesi 2004). A study comparing remifentanil pharmacokinetics in 12 obese and 12 lean patients undergoing surgery demonstrated no significant differences between the 2 groups (Egan, Huizinga, Gupta, et al., 1998). The researchers recommended dosing regimens based on ideal body weight, or lean body mass, rather than total body weight.
Remifentanil’s characteristics allow easy titration for intraoperative sedation and analgesia; it has been given via patient-controlled sedation (Fong, Kwan, 2005) and target-controlled infusions (titrated according to respiratory rate) (Beers, Camporesi 2004). The drug is capable of providing extremely rapid and intense analgesia with minimal effect on cognition, making it suitable for procedures during which patient response to instruction is required, such as fiberoptic intubation (Beers, Camporesi 2004; Johnson, Swenson, Egan, et al., 2002). Several studies have found remifentanil to be safe and effective for analgesia and sedation during extracorporeal shock-wave lithotripsy (ESWL) (Beloeil, Corsia, Coriat, et al., 2002; Cortinez, Munoz, De al Fuente, et al., 2005; Medina, Galvin, Dirckx, et al., 2005) and a variety of other painful procedures. A comparison of infusion doses led one group of researchers to recommend a regimen of 0.05 mcg/kg/minute with 10 mcg PCA doses for ESWL (Medina, Galvin, Dirckx, et al., 2005). It has been used successfully for sedation and analgesia in critically ill patients after organ transplantation as well (Evans, Park, 1997).
Similar to fentanyl and sufentanil, remifentanil allows rapid extubation in critically ill medical and surgical patients (Cheng, Newman, Duke, et al., 2001; Dahaba, Grabner, Rehak, et al., 2004; Engoren, Luther, Fenn-Buderer, 2001). In these populations, it has been shown to be superior to morphine in terms of ease of titration, hours of optimal sedation, amount of supplemental sedation required, and time to extubation with comparable adverse effects (Muellejans, Lopez, Cross, et al., 2004). Remifentanil has been shown to allow more rapid awakening and shorter time to neurologic testing after cessation of infusion compared with other short-acting opioids (Soltesz, Biedler, Silomon, et al., 2001; Wilhelm, Schlaich, Harrer, et al., 2001). This has led researchers to suggest it as an option for intermittent, rapid neurologic assessment in those with intracranial disease (Soletz, Biedler, Silomon, et al., 2001). By continuous infusion, the drug was not found to be effective in blocking cough response to endotracheal suctioning, resulting in increased intracranial pressure in mechanically-ventilated head-injured patients (Leone, Albanese, Viviand, et al., 2004).
The short duration of action of remifentanil after a dose, or after discontinuation of an infusion, is a major disadvantage when the need for analgesia is anticipated following painful medical or surgical procedures. Patients consistently report severe pain shortly after cessation of remifentanil administration (Litman, 2000; Soltesz, Biedler, Silomon, et al., 2001). In such cases, a proactive, preventive approach that includes administration of local or regional anesthesia, nonopioids, or the use of a longer-acting opioid is essential (Beers, Camporesi, 2004).
Like other mu opioids, remifentanil produces dose-related analgesia and adverse effects; nausea and pruritus are common. Of more concern is the high incidence of adverse respiratory events (Joo, Perks, Kataoka, et al., 2001; Litman, 2000). Incidence of respiratory depression has been reported to be as high as 29% in the postoperative setting (Bowdle, Camporesi, Maysick, et al., 1996), and apnea with oxygen desaturation is common (Bowdle, Camporesi, Maysick, et al., 1996; Joo, Perks, Kataoka, et al., 2001; Litman, 2000). Like the other fentanils, muscle rigidity, in particular chest wall rigidity requiring resuscitation, can occur with rapid bolus administration (Beers, Camporesi, 2004). A randomized, double-blind study of 64 healthy volunteers (16 over 60 years old) led researchers to conclude that bolus doses of remifentanil (up to 200 mcg) can be highly effective for clinical situations requiring intense analgesia; however, older adults experienced more respiratory depression than younger subjects, and 4 older subjects experienced significant but short-lived and easily managed respiratory depression at 75 mcg (Egan, Kern, Muir, et al., 2004). The researchers reinforced the need for careful monitoring of respiratory function when this drug is used. In addition, remifentanil bolus dosing was associated with severe hemodynamic instability and 1 case of myocardial infarction (MI) in a group of patients scheduled for coronary artery bypass graft (CABG) surgery, leading the researchers to terminate the study after 8 patients and recommend remifentanil administration only by slow infusion in this population (Elliott, O’Hare, Bill, et al., 2000). (See the following on PCA use after CABG.)
For severe postoperative pain treatment, a continuous infusion of 0.1 mcg/kg/min of remifentanil (N = 15) resulted in the need for less rescue medication than 0.05 mcg/kg/min (N = 15) (Calderon, Pernia, De Antonio, et al., 2001). The 2 infusion rates yielded 1 case of nausea and vomiting each and no cases of respiratory depression. Despite low dosing recommendations, several have advised against the use of remifentanil for postoperative pain management outside of a monitored setting, such as the intensive care unit (ICU), citing the absence of around-the-clock, readily available anesthesia providers and a potential for life-threatening dose errors or equipment malfunction as barriers to safe use (Beers, Camporesi, 2004).
A randomized controlled trial compared IV PCA remifentanil (0.05 mcg/kg/min basal rate, 0.25 mcg/kg PCA bolus, lockout 5 minutes) with IV PCA morphine (0.3 mg/h, 1 mg PCA bolus, lockout 5 minutes) in 60 patients after CABG and found better analgesia with cough and movement in those receiving remifentanil and concluded that remifentanil is a safe alternative to morphine in this patient population (Baltali, Turkoz, Bozdogan, et al., 2009). Although remifentanil has been found to produce effective pain relief via PCA during labor and delivery (Blair, Hill, Fee 2001; Douma, Verwey, Kam-Endtz, et al., 2010; Evron, Glezerman, Sadan, et al., 2005), wide variation in analgesic requirements and adverse effects, such as maternal oxygen desaturation and nausea, and the need for very close monitoring (Saunders, Glass 2002; Volmanen, Akural, Raudaskoski, et al., 2002) would seem to limit its use in this setting. However, an observational study of 205 women in active labor who received remifentanil via a titrated continuous IV infusion (0.025 mcg/kg/minute increased in a stepwise approach to a maximum dose of 0.15 mcg/kg/minute) reported reductions in mean visual analog scores from 9.4 to 5.1 after 5 minutes and to 3.6 after 30 minutes of infusion (D’Onofrio, Novelli, Mecacci, et al., 2009). This was accomplished with minimal maternal opioid-related adverse effects (e.g., 3 patients reported nausea), no hemodynamic instability, and no fetal or neonatal adverse effects. The researchers recommended this stepwise titration regimen and suggested that fixed-dose regimens of remifentanil in this setting may have led to the underdosing and inadequate analgesia or overdosing and maternal desaturation reported in other studies.
Remifentanil has provided some observations intended to elucidate the controversial clinical effect of opioid-induced hyperalgesia. For many years, animal and healthy human volunteer studies have documented secondary hyperalgesia after acute and long-term opioid exposure (Angst, Koppert, Pahl, et al., 2003; Joly, Richebe, Guignard, et al., 2005) (see Chapter 11 for a detailed discussion of opioid-induced hyperalgesia). Postoperative secondary hyperalgesia has also been reported in the clinical setting, as evidenced by increased pain and morphine consumption in patients who received high intraoperative doses of remifentanil compared with those who did not (Guignard, Bossard, Coste, et al., 2000; Joly, Richebe, Guignard, et al., 2005). Other studies have not confirmed this (Lahtinen, Kokki, Hynynen 2008). The underlying mechanisms of opioid-induced hyperalgesia may involve a pain facilitator system mediated by the NMDA receptor (Angst, Koppert, Pahl, et al., 2003; Joly, Richebe, Guignard, et al., 2005) (see Section I). Indeed, co-administration of the NMDA receptor antagonist ketamine with remifentanil has been shown to abolish hyperalgesia (Angst, Koppert, Pahl, et al., 2003; Joly, Richebe, Guignard, et al., 2005).
Sufentanil
Sufentanil is a very potent, lipophilic mu agonist opioid analgesic usually used in perioperative or procedural settings as an IV, epidural, or intrathecal infusion. It is the most lipid-soluble opioid (twice that of fentanyl) and, being approximately 1000 times more potent than morphine (Gutstein, Akil, 2006), is the most potent of the fentanyl analogs (White, Hardy, Boyd, et al., 2008) (see Knotkova, Fine, Portenoy, 2009 for research on potency ratios). It has no active metabolites and, like other highly lipophilic drugs, is fast acting (IV peak analgesic effect is ~5 minutes) and has a short duration of action (approximately 1 hour), because it moves rapidly from plasma to opioid receptor action sites (Fukuda, 2005; Gutstein, Akil, 2006; Murphy, 2006). These qualities have attracted investigators and clinicians to consider it for alternative routes that require high potency and low volume, such as intranasal administration for acute postoperative pain (Mathieu, Cnudde, Engelman, et al., 2006; Dale, Hjortkjaer, Kharasch, 2002) and SC infusion in palliative care settings (Urch, Carr, Minton, 2004; White, Hardy, Boyd, et al., 2008). These characteristics also make the drug suitable for breakthrough pain. Indeed, sufentanil has been given via the oral transmucosal route (Gardner-Nix, 2001a) and the intranasal route (Jackson, Ashby, Keech, 2002) to provide rapid and effective analgesia for breakthrough cancer pain. Intraarticular administration of a single bolus of sufentanil at the end of arthroscopic knee surgery produced effective pain relief for up to 24 hours; the addition of methylprednisolone (e.g., Solu-Medrol, Depo-Medrol) further prolonged analgesia (Kizilkaya, Yildirim, Dogan, et al., 2004). A transdermal sufentanil patch for long-term pain treatment was in development at the time of publication (Freye, 2008).
Sufentanil is sometimes combined with a local anesthetic when administered epidurally (Bauer, Hentz, Ducrocq, et al., 2007). In the postoperative setting, sufentanil is reported to be more potent via the intrathecal route than the IV route of administration. A randomized, double-blind study of 40 patients older than 75 years undergoing total hip replacement showed that those who received intrathecal sufentanil had significantly faster pain relief and required less rescue medication than those who received IV sufentanil (Fournier, Weber, Gamulin, 2005). Whereas oxygen desaturation was common with IV sufentanil, pruritus was common with intrathecal sufentanil. In another study (N = 20) comparing epidural and IV sufentanil, approximately 50% more sufentanil was required epidurally than intravenously to produce comparable pain relief (Menigaux, Guignard, Fletcher, et al., 2001). The researchers suggested that the bioavailability of epidural sufentanil is reduced and much of the lipophilic drug may be absorbed into the epidural fat (see Chapter 15 for more on intraspinal sufentanil). The effects of sufentanil’s lipophilicity could also be seen in a small study comparing 8 obese and 8 control patients in which IV sufentanil’s half-life was 208 minutes and 135 minutes, respectively (Schwartz, Matteo, Ornstein, et al., 1991).
As with fentanyl, there is a wide volume of distribution, but clearance is much faster. When given by continuous IV infusion as part of a sedation/analgesia protocol, distribution and clearance were extended compared with bolus or short infusion in the perioperative setting (Ethuin, Boudaoud, Leblanc, et al., 2003). Sufentanil has been shown to be sufficiently hypnotic to be used as a long-term (12 days) sedative agent in critically ill mechanically-ventilated patients without renal or hepatic disease (Ethuin, Boudaoud, Leblanc, et al., 2003). In critically ill trauma patients, analgesia was easier to maintain following discontinuation of sufentanil/propofol (N = 10) infusion compared with remifentanil/propofol (N = 10) infusion, although awakening was more rapid with the latter (Soltesz, Biedler, Silomon, et al., 2001). Sufentanil does not interfere with hemodynamic stability (Fukuda, 2005); however, adverse effects, such as respiratory depression and nausea and vomiting, were more common compared with remifentanil when sufentanil was used for procedural pain management (Beloeil, Corsia, Coriat, et al., 2002).
Characteristics of Selected Agonist-Antagonist Opioids
Historically, agonist-antagonist opioids have been discouraged as first-line drugs for any type of pain (APS, 2003) and contraindicated in patients receiving a mu agonist opioid, because of their ability to antagonize at the mu opioid receptor and reverse analgesia and precipitate withdrawal (Miaskowski, Cleary, Burney, et al., 2005). This perspective continues to be held with regard to the mixed agonist-antagonist opioids, such as pentazocine (Butorphanol), but has been evolving with the advent of new formulations of the partial mu agonist buprenorphine. Some clinicians use agonist-antagonist opioid analgesics believing that they cause no respiratory depression or less than the mu agonist opioids (see the misconceptions table in the Introduction to Section IV). Rather, they have a ceiling on the respiratory depression; they can produce respiratory depression comparable to morphine at a relatively low dose (e.g., 10 mg of parenteral morphine), and patients who would be at risk from morphine at this dose presumably also would be at risk from an agonist-antagonist opioid. The agonist-antagonists also have a ceiling on their analgesic effects (see discussion in the following buprenorphine section) (APS, 2003), which is why they are especially inappropriate for severe, escalating pain.
All agonist-antagonist opioids are available parenterally. Pentazocine is the only agonist-antagonist available in oral formulation, and this is in combination with naloxone (Talwin NX). Buprenorphine is formulated for sublingual administration as a single-entity drug (Subutex) and in combination with naloxone (Suboxone); these drugs are indicated for the office-based treatment of opioid addiction. Buprenorphine also is available in some countries as a transdermal patch. Butorphanol (Stadol) is available by nasal spray.
The division of the agonist-antagonists into partial agonists (buprenorphine) and mixed agonist-antagonists (butorphanol, nalbuphine [Nubain], and pentazocine) is clinically important. This is apparent in reviewing the pharmacology of the individual drugs.
Buprenorphine
Buprenorphine is the only partial mu agonist with high affinity for the mu opioid receptor. As such, it binds tightly to the mu opioid receptor but does not “turn on” that receptor as completely as a full mu agonist opioid, such as morphine (Heit, Gourlay, 2008; Johnson, Strain, Amass, 2003; Johnson, Fudala, Payne, 2005). It has a complex pharmacology and probably also has antagonist effects at other opioid receptors, such as the kappa receptor. In opioid-naïve individuals, low-dose buprenorphine acts like a potent mu agonist opioid. As the dose is increased, the drug has a ceiling effect (Johnson, Strain, Amass, 2003). As mentioned, this is believed to limit its usefulness for the treatment of severe, escalating pain. If it is administered to a patient who already has physical dependence to another mu agonist drug, it has the potential to produce acute withdrawal.
Buprenorphine can be given parenterally (Buprenex) and, being highly lipophilic, is absorbed well by the oral mucosa (Subutex, Suboxone) (Center for Drug Evaluation and Research, 2002) and can be administered transdermally. The transdermal buprenorphine patch currently is available in several European and other countries and is used for the treatment of persistent pain. Research and clinical experience is lacking on transdermal buprenorphine, but a large multicenter (110 centers) study of cancer patients (N = 1801) demonstrated that the formulation was well tolerated and produced analgesia similar to mu opioid agonists (Apolone, Corli, Negri, et al., 2009). A clinical trial that compared pain relief and adverse effects in 82 adults ages 65 years and older with equal numbers of adults between the ages of 51 and 64 and 50 years or younger found that transdermal buprenorphine was equally effective, tolerable, and safe in all age groups for moderate to severe noncancer pain (Likar, Vadlau, Breschan, et al., 2008). Buprenorphine is not offered in an oral formulation because of low oral bioavailability from significant first-pass metabolism (Johnson, Fudala, Payne, 2005) (see Chapter 11).
Sublingual absorption of a liquid buprenorphine formulation, which is not available in the United States, occurs within 5 minutes. In contrast, sublingual tablets, approved for treatment of opioid addiction (discussed in the next section), require at least 20 minutes, with peak plasma concentrations measured over a wide range of 60 to 360 minutes. Bioavailability is 51% to 55% with the sublingual formulations, but with wide interpatient variability (Johnson, Fudala, Payne, 2005).
IV buprenorphine has a slow onset of action with a peak effect that can be as long as 3 hours and a terminal half-life of 5 to 6 hours (Johnson, Fudala, Payne, 2005). The duration of buprenorphine varies significantly depending on route of administration. Acceptable postoperative pain was attained via every-3-hour dosing with sublingual buprenorphine, while patients receiving IM buprenorphine reported analgesia lasting for approximately 12 hours (Johnson, Fudala, Payne, 2005).
Buprenorphine’s principle metabolite norbuprenorphine has not been studied extensively in humans but has been found to be a potent partial agonist in animals (Huang, Kehner, Cowan, et al., 2001), though less potent and with a lower affinity for the mu opioid receptor than buprenorphine (Fukuda, 2005). Norbuprenorphine’s analgesic effect is one-fiftieth that of buprenorphine via IV administration, and it has a slower elimination than buprenorphine (Johnson, Strain, Amass, 2003).
Buprenorphine has been used for postoperative pain, administered by IM and IV injection and by IV PCA (Wu, 2005). It also has been given epidurally (Johnson, Fudala, Payne, 2005) and intrathecally (Vadivelu, Hines, 2007). It has some local anesthetic effects and has been added to local anesthetic blocks to prolong the effect (Candido, Franco, Khan, et al., 2001; Candido, Winnie, Ghaleb, et al., 2002). Neuropathic pain and cancer-related pain have been successfully treated with buprenorphine by all routes of administration (Johnson, Fudala, Payne, 2005; Davis, 2005; Hans, 2007). Concern about the ability to reverse this high affinity drug with typical doses of naloxone has led to the recommendation that it not be used in laboring patients (APS, 2003).
Sedation, nausea, vomiting, dizziness, sweating, and headache are the most common adverse effects of parenteral buprenorphine. It may be less constipating and less likely to alter the levels of sex hormones than other opioids (Davis, 2005). Like other opioids, it may produce dysphoric and psychomimetic effects (Johnson, Fudala, Payne, 2005). In contrast to butorphanol and pentazocine, buprenorphine does not produce adverse cardiac effects (Fukuda, 2005). It also has a lower incidence of prolonged QT interval than methadone (Wedam, Bigelow, Johnson, et al., 2007). Safety in renal failure and in older adults has not been studied. Neither buprenorphine nor its active metabolites are excreted by the kidneys, so some speculate that dose reductions may not be necessary in those with renal insufficiency (Davis, 2005).
Parenteral buprenorphine is about 30 times more potent than morphine, but at usual doses is less likely to cause respiratory depression (Johnson, Fudala, Payne, 2005). A ceiling on respiratory depression after 0.15 to 1.2 mg IV in adults is reported (Fukuda, 2005). Dahan and colleagues (2005) found a respiratory ceiling effect at IV doses equal to or greater than 2.9 mcg kg−1; the administration of IV doses as high as 7 mg without respiratory depression have also been described (Johnson, Fudala, Payne, 2005). Peak respiratory depression has been found to occur between 150 to 180 minutes after IV infusion (Dahan, Yassen, Romberg, et al., 2006). Respiratory depression plateaus have also been noted between 0.4 and 0.8 mg sublingually (Dahan, Yassen, Bijl, et al., 2005). High doses of buprenorphine may stimulate respiration via its antagonist characteristic (Fukuda, 2005).
Some studies provide empirical support for the conclusion that the ceiling dose for respiratory depressant effects is relevant in the context of acute pain management (Dahan, Yassan, Bijl, et al., 2005; Dahan, Yassen, Romberg, et al., 2006). Researchers administered 0.2 mg and 0.4 mg IV doses to 20 healthy male volunteers (age 22 to 35 years, weight 62 to 92 kg) for the purpose of examining buprenorphine’s respiratory and analgesic effects during experimental pain induced by electrical current via surface electrodes (Dahan, Yassen, Romberg, et al., 2006). Doubling the buprenorphine dose (from 0.2 mg to 0.4 mg) increased its peak analgesic effect by a factor of 3.5, but did not change the respiratory effects. These findings suggest that the doses that are effective for pain are in a range that yields a plateau on respiratory depression. Further research in patients in the clinical setting is warranted.
Though clinically significant respiratory depression is rare with buprenorphine and associated most often with the use of other sedating drugs, such as benzodiazepines (Dahan, Yassen, Bijl, et al., 2005), the unusual times that it happens may pose a significant challenge in management because buprenorphine is not readily reversed by naloxone. In fact, very high doses (10 to 35 mg) of naloxone may be required to reverse respiratory depression due to buprenorphine’s strong affinity for the mu opioid receptor (Johnson, Fudala, Payne, 2005).
Buprenorphine for Treatment of Opioid Addiction
There are two sublingual buprenorphine formulations (Subutex and Suboxone). They are the only agents approved for treatment of opioid addictive disease by the Drug Addiction Treatment Act of 2000 (DATA) (methadone maintenance programs are approved and licensed under a different law). Suboxone contains buprenorphine and the opioid antagonist naloxone, a combination intended as an abuse deterrent. Suboxone is available in two tablet strengths: 2 mg buprenorphine plus 0.5 mg naloxone and 8 mg buprenorphine plus 2 mg naloxone. Subutex contains buprenorphine alone in 2 and 8 mg strength tablets. Sublingual buprenorphine tablets are placed under the tongue until dissolved, for about 2 to 10 minutes (Center for Drug Evaluation and Research, 2002) (see Chapter 14). Because buprenorphine is approved as a treatment for addiction and clinicians who prescribe it for this purpose must indicate on the prescription that they have been certified to do so, it is best that the words “For Pain” be written on the prescription if the drug is to be used as an analgesic (Heit, Gourlay, 2008). This is frequent practice when using methadone for pain as well.
It is extremely important that acute pain episodes, such as following trauma or surgery, be treated adequately in individuals who are taking buprenorphine for addictive disease. There is very little experience in the use of morphine or some other pure mu agonist drug to augment the effects of buprenorphine. Theoretically, relatively high doses of a pure mu opioid would be needed because buprenorphine binds so tightly to the opioid receptor that fewer receptors will be available for activation. In addition, there is concern that patients with the disease of addiction may be predisposed to experience relatively more pain after injury as a result of pain facilitation through mechanisms linked to withdrawal (Savage, Schofferman, 1995). Whatever the reason, patients who are receiving buprenorphine, or methadone, and require opioid administration for the management of acute severe pain, may need a relatively aggressive treatment protocol, with quick dose escalation based on observed response to the therapy. (See Chapter 20 for more discussion of treatment of pain in individuals with addictive disease.)
Butorphanol
Butorphanol is a mixed agonist-antagonist opioid with an affinity for mu, delta, and kappa receptors. It is an agonist at the kappa receptor and either an antagonist or partial agonist at the mu opioid receptor (Fukuda, 2005). As discussed, the antagonistic characteristic can lead to withdrawal symptoms if butorphanol is administered to an individual who is physically dependent on a mu opioid.
Butorphanol is available for IV (Stadol) and intranasal (Stadol NS) administration. Though there is no clear advantage of butorphanol over the first-line mu agonist opioids, it has been used in perioperative (Wu, 2005) and labor settings (Birnbach, Browne, 2005) and as a treatment for migraine (Davis, Rudy, Archer, et al., 2005; Rapoport, Bigal, Tepper, et al., 2004). There are experimental data suggesting that women may achieve better analgesia than men with butorphanol (Miller, Ernst, 2004).
The onset, peak, and duration of analgesic action of IV butorphanol are similar to that of IV morphine, and it has a plasma half-life of approximately 3 hours (Gutstein, Akil, 2006). It has been shown to be effective for IV PCA following abdominal hysterectomy (Thakore, D’Mello, Saksena, et al., 2009). Dose adjustment is not needed in renal or hepatic insufficiency or in the older patient, but extending the dosing interval may be advisable (Asenjo, Brecht, 2005; Vachharajani, Shyu, Garnett, et al., 1996).
Intranasal butorphanol is absorbed rapidly, detectable in serum in 5 minutes or less, but onset of analgesia may be as much as 15 minutes (Asenjo, Brecht, 2005). It is approximately 60% to 70% bioavailable (Cashman, 2008). Adverse effects are typical of opioids, with the addition of an unpleasant taste (Davis, Rudy, Archer, et al., 2005). In opioid-naïve patients who are taking occasional mu agonist opioids, such as codeine or oxycodone, the addition of butorphanol nasal spray may provide additive analgesia. However, in opioid-tolerant patients, such as those receiving ATC morphine, the addition of butorphanol nasal spray should be avoided because it may reverse analgesia and precipitate withdrawal.
Adverse effects of butorphanol include headache, vertigo, lethargy, and lightheadedness. Psychomimetic effects are possible and include a feeling of floating, confusion, hallucinations, unusual dreams, and depersonalization (Dawn, Yosipovitch, 2006). Parenteral butorphanol is associated with a high degree of sedation (Coda, 2006). Therapeutic doses of butorphanol can produce levels of respiratory depression equal to mu opioid agonist drugs until a ceiling for respiratory depression is reached, typically at doses of 30 to 60 mcg/kg (Fukuda, 2005). Butorphanol by any route is not recommended for use in the treatment of MI or patients with hypertension because it produces multiple adverse effects on cardiac function, including increased pulmonary arterial pressure and cardiac workload (Fukuda, 2005; Gutstein, Akil, 2006).
Butorphanol and other agonist-antagonist opioids are sometimes used at low doses for treatment of opioid-induced adverse effects, particularly pruritus. However, studies on the effectiveness of butorphanol on opioid-induced pruritus have been mixed. Two small uncontrolled trials suggest that intranasal butorphanol may be effective against pruritus related to opioid agonists and to skin conditions (Dawn, Yosipovitch, 2006; Dunteman, Karanikolas, Filos, 1996). Others have reported that IV butorphanol reduced analgesia but not itching (Sakai, Fukano, Sumikawa, 2001). The risk of adding adverse effects, and inadvertently reversing analgesia and inducing withdrawal, must be weighed against the benefit of a small reduction in the severity of a minor adverse effect that quite often can be treated effectively with opioid dose reduction or other methods (see Chapter 19 for more on management of pruritus).
Dezocine
Approved in 1989, dezocine (Dalgan) has not been widely used for the treatment of pain, and there is very little clinical research on the drug. It was discontinued in the United States in 1999 (APS, 2003). Although it is classified as an agonist-antagonist opioid, some researchers question the existence of antagonist properties. Experimental research using human embryonic kidney cells concluded that it is an antagonist at the kappa opioid receptor, rather than an agonist like butorphanol (Gharagozlou, Hashemi, DeLorey, et al., 2006), and earlier studies in animals showed that large doses of naloxone failed to antagonize the drug (Picker, 1997). It has been administered with mu opioid agonists and other agonist-antagonists without precipitating withdrawal symptoms (Barkin Lubenow, Bruehl, et al., 1996; Wilson, Cohen, Kezer, et al., 1995). These are confusing data, and most experts continue to classify dezocine as a mixed agonist-antagonist, with properties similar to other drugs of this type.
Dezocine undergoes extensive first-pass hepatic metabolism and is without active metabolites. An early review of dezocine described the drug as having extensive distribution, high clearance, and short half-life (3 hours) over a range of IV doses and being well absorbed by all parenteral routes (Locniskar, Greenblatt, Zinny, 1986). Peak effect was 0.6 hours, and bioavailability was 97% following IM injection. Doses should be reduced if used in patients with hepatic or renal dysfunction. No evidence exists of changes in pressure within the common bile duct or ampulla of Vater in patients after biliary surgery. Mental status changes and delirium have been reported with use of dezocine in older adults (Barkin, Lubenow, Bruehl, et al., 1996).
Nalbuphine
Nalbuphine (Nubain) is a mixed agonist-antagonist opioid and like butorphanol is a kappa receptor agonist and a mu receptor antagonist (Fukuda, 2005). Similar to the other drugs in this subclass, nalbuphine can reverse analgesia or precipitate withdrawal if administered to an individual who is physically dependent on a mu opioid, and it has a ceiling dose for both analgesia and respiratory depressant effects (Gunion, Marchionne, Anderson, 2004). It is available for administration only by the parenteral route (IM, SC, IV).
Although there is no advantage over the first-line mu agonist opioids, nalbuphine is occasionally used for postoperative pain (Coda, 2006; Gunion, Marchionne, Anderson, 2004; Wu, 2005). It also has been administered for labor pain (Birnbach, Browne, 2005), but a warning was issued by the manufacturer in 2005 noting a risk of serious fetal and neonatal adverse effects (including fetal bradycardia, respiratory depression at birth, apnea, cyanosis, and hypotonia) associated with its use during labor and delivery. As a result, the drug should not be used during labor and delivery unless clearly indicated and only if the benefits outweigh the risks (http://www.fda.gov/medwatch/safety/2005/aug_PI/Nubain_PI.pdf). (See Chapter 20 for opioid use during pregnancy and breast-feeding.)
Nalbuphine has an IV onset of 2 to 3 minutes, a duration of 4 to 6 hours (Birnbach, Browne, 2005), and a half-life of 2 to 3 hours (Guinon, Marchionne, Anderson, 2004). It has no clinically significant metabolites (Guinon, Marchionne, Anderson, 2004). In contrast to butorphanol and pentazocine, nalbuphine does not produce adverse cardiac effects, so it is acceptable for use when MI is suspected (Fukuda, 2005; Gutstein, Akil, 2006).
Nalbuphine may cause less nausea and vomiting than other opioids (Birnbach, Browne, 2005); however, compared with fentanyl for outpatient anesthesia, nalbuphine produced more unpleasant dreams during surgery and more postoperative nausea, anxiety, and sedation (White, Freire, 2005). At equianalgesic doses, nalbuphine and morphine produce a similar degree of respiratory depression. The ceiling for respiratory depression is reached at doses of more than 30 mg of nalbuphine; however, no increase in analgesia beyond 30 mg occurs (Gutstein, Akil, 2006). Like other agonist-antagonists, this property means that there is no safeguard against respiratory depression if this adverse effect occurs at a dose below the ceiling dose.
Single low dose (2.5 to 5 mg) and continuous infusions of parenteral nalbuphine sometimes are used to prevent or treat opioid-induced adverse effects, primarily pruritus, associated with mu agonist opioid epidural analgesia; however, reversal of analgesia can occur (Coda, 2006). Although nalbuphine has been reported to reverse morphine-induced respiratory depression while maintaining analgesia (Coda, 2006), others report that nalbuphine does not reverse and may actually increase respiratory depression after morphine (Fukuda, 2005). Even more report severe pain, hypertension, and tachycardia requiring pharmacologic intervention following administration of nalbuphine for treatment of opioid-induced adverse effects (Fukuda, 2005). In an attempt to reduce adverse effects, a nalbuphine and naloxone mixture was administered by slow IV push upon awakening from gynecologic surgery in a study of 12 patients (Gordon, Levine, Dubois, et al., 2007); all but two of the patients required rescue analgesia within 50 minutes of receiving the mixture. The risk of adding adverse effects and inadvertently reversing analgesia must be weighed against the benefit of a small reduction in the severity of a minor adverse effect (see Chapter 19 for more on management of pruritus and other adverse effects).
Pentazocine
Pentazocine is the only agonist-antagonist opioid to have an oral formulation. Although originally intended as an opioid with low abuse potential, abuse occurred after the drug was marketed, and this was addressed by reformulating it into a combination product containing pentazocine (50 mg) and naloxone (0.5 mg) (Talwin Nx). This markedly reduced crushing and IV injection of the oral formulation (Gutstein, Akil, 2006). A generic pentazocine combined with acetaminophen is also available.
Pentazocine has one-fourth the potency of morphine and a duration of action of 3 hours. As with other agonist-antagonist opioids, it has a ceiling effect on analgesia and respiratory depression; this probably occurs in the 30 to 70 mg range (Fukuda, 2005). When given to patients with respiratory depression from morphine, respiratory depression is not reversed, but patients who are dependent on other opioids may experience withdrawal symptoms (Gutstein, Akil, 2006). Pentazocine is associated with a relatively high incidence of psychotomimetic effects, and doses above 60 mg are associated with dysphoria (Gutstein, Akil, 2006), especially in older adults (Fukuda, 2005). The dysphoric effects are reversible with naloxone. Pentazocine also can produce negative CV effects, and it should not be used for analgesia in patients with MI (Fukuda, 2005). It has been used most often in labor and delivery and perioperative settings in the past, but it is very rarely used today.
Dual Mechanism Analgesics
Analgesics with a dual mechanism of action are relatively new to pain management. They bind weakly to the mu opioid receptor site and, similar to antidepressants, inhibit serotonin and/or norepinephrine. The two commercially available at the time of publication, tramadol and tapentadol, are discussed here.
Tramadol
Tramadol is a synthetic atypical opioid analgesic, indicated for moderate to some moderately severe pain. As mentioned, it has a dual mechanism of action: binding weakly to the mu opioid receptor site and inhibiting serotonin and norepinephrine reuptake, which in turn activates the descending inhibitory spinal pathway (Leppert, Luczak, 2005; Scott, Perry, 2000) (see Section I).
Tramadol has 68% oral bioavailability, is subject to first-pass effect, and is metabolized via the CYP2D6 pathway. Both tramadol and its active metabolite, M1, are excreted by the kidneys. M1 has 200 times stronger affinity for the mu receptor, is 2 to 4 times more potent, and has a longer half-life than tramadol (Sinatra, 2009). The onset of action of the short-acting formulation is as long as 60 minutes, which is slower than morphine (Sarbu, Radulescu, Robertson, et al., 2007), and its duration is longer (6 hours).
Tramadol is equipotent with codeine and five times less potent than morphine (Leppert, Luczak, 2005). A study comparing tramadol/acetaminophen to hydrocodone/acetaminophen and placebo in adults with partial ankle ligament tear showed 1 to 2 capsules of 37.5 mg tramadol/325 mg acetaminophen provided equivalent analgesia and comparable adverse effects to 7.5 mg hydrocodone/650 mg acetaminophen (Hewitt, Todd, Xiang, et al., 2007).
Tramadol is one of the most widely used analgesics throughout the world (Raffa, Stone, 2008). Despite reports of abuse, it is not a controlled substance under the Controlled Substances Act in the United States (Drug Enforcement Administration, 2008). In the United States, tramadol is available in short-acting (Ultram) and modified-release tablets (Ultram ER, Ryzolt) and in a combination tablet with acetaminophen (Ultracet). Acetaminophen combined with tramadol appears to enhance effectiveness beyond the degree expected, demonstrating a “supra-additive effect” (Filitz, Ihmsen, Gunther, et al., 2007; McClellan, Scott, 2003). There are conflicting findings regarding synergism with other opioids (Marinangeli, Ciccozzi, Aloisio, et al., 2007; Marcou, Marque, Mazoit, et al., 2005; Webb, Leong, 2005). In Europe and Australia, in addition to the oral formulations, tramadol is used intravenously and epidurally for perioperative pain, for postoperative pain via IV PCA, and to relieve postoperative shivering (Kaya, Sariyildiz, Karakus, et al., 2003). It is also available in Europe for rectal administration (Mercadante, Arcuri, Fusco, et al., 2005).
Short-acting tramadol is available in 50 mg tablets. Modified-release tramadol (100, 200, and 300 mg) is effective for 24 hours. The maximum recommended dose of tramadol is 400 mg/day (Dworkin, O’Connor, Backonja, et al., 2007). A maximum dose limit of 300 mg/day for older adults over age 75 (AGS, 2002; Dworkin, O’Connor, Backonja, et al., 2007) and in the acute pain setting (Sinatra, 2009) is recommended.
Tramadol has been found to be effective in a variety of pain states, including postoperative pain (McCartney, Niazi, 2006; Scott, Perry, 2000), minor musculoskeletal trauma (Hewitt, Todd, Xiang, et al., 2007), migraine/headache (Alemdar, Pekdemir, Selekler 2007), cancer-related pain (Prommer, 2005; Leppert, Luczak, 2005), and pain from rheumatoid arthritis (Lee, Lee, Park, et al., 2006) and osteoarthritis (OA) (Vorsanger, Xiang, Jordan, et al., 2007). It is regarded as a first- or second-line analgesic in guidelines for neuropathic pain (Argoff, Backonja, Belgrade, et al., 2006; Dworkin, O’Connor, Backonja, et al., 2007; Finnerup, Otto, McQuay, et al., 2005; Moulin, Clark, Gilron, et al., 2007), although it is acknowledged that it may not be effective against severe neuropathic pain (Dworkin, O’Connor, Backonja, et al., 2007). It is not considered appropriate for severe pain associated with major surgical procedures either (Sinatra, 2009). Tramadol has been used for treatment of mild to moderate acute pain associated with sickle cell disease (Varadarajan, Weisman, 2009) and is listed as an option for persistent pain in older adults (AGS, 2002, 2009). The drug is widely prescribed to older adults with moderate to severe pain. In the only study to stratify patients by age (younger than 65, 65, younger than 75, 75 or older), the drug was well tolerated and effective for moderate to severe cancer and noncancer pain, and there were no significant differences in tolerability and effectiveness across age groups (Likar, Wittels, Molnar, et al., 2006). A Cochrane Collaboration Review and a later systematic review and meta-analysis by the same researchers concluded that the analgesic and functional outcome benefits are small and adverse events are reversible and not life-threatening but are often a cause of treatment cessation for OA (Cepeda, Camargo, Zea, et al., 2006, 2007).
The most common adverse effects of tramadol are nausea and vomiting, dizziness, drowsiness, and dry mouth. The risk of respiratory depression is lower than with pure mu agonist opioids (Scott, Perry, 2000). Because tramadol is largely dependent on the CYP2D6 metabolic pathway for analgesic activation, drug interactions must be taken into consideration. The selective serotonin reuptake inhibitors (SSRIs) (e.g., fluoxetine, paroxetine) may inhibit tramadol metabolism (see Chapter 11 for more on the CYP2D6 enzyme system and drug-drug interactions). During prolonged therapy at high doses, combined tramadol and SSRIs has led to life-threatening serotonin syndrome, evidenced by shivering, nausea, low-grade fever, sweating, mental confusion, and delirium. It should not be administered with MAOIs, as this has been associated with the development of psychosis (Sinatra, 2009). As with other opioids, additive toxicity is seen with concurrent use of CNS depressants (Hersh, Pinto, Moore, 2007). Starting with a low dose (12.5 mg to 25 mg) and titrating to effect may limit the development of adverse effects.
Early reports of a significant risk of tramadol-induced seizures have been questioned (Raffa, Stone, 2008; Marquardt, Alsop, Albertson, 2005; Gasse, Derby, Vasilakis-Scaramozza, et al., 2000). There may not be a greater risk than that associated with other opioids (Gasse, Derby, Vasilakis-Scaramozza, et al., 2000), and attempts to induce seizures with tramadol in mice were unsuccessful (Raffa, Stone, 2008). However, tramadol should be taken within the recommended dose range and used with caution in patients with a history of seizure and when concurrently taking SSRIs and other drugs that can lower the seizure threshold (AGS, 2009). There have been anecdotal reports of the need to gradually taper doses to prevent withdrawal if the drug is to be discontinued.
Tapentadol
Tapentadol (Nucynta) is another centrally-acting analgesic with a dual mechanism of action, binding as an agonist to the mu opioid receptor site and also blocking the reuptake of norepinephrine. Tapentadol is a Schedule II drug indicated for moderate to severe pain and available in short-acting formulation in 50, 75, and 100 mg tablets. Dosing is recommended every 4 to 6 hours, and doses higher than 600 mg are not recommended because they have not been studied. A modified-release formulation of tapentadol was in development at time of publication. One phase III study showed that when compared with placebo and modified-release oxycodone (20 to 50 mg), significantly more patients taking modified-release tapentadol (100 to 250 mg) experienced 30% or greater and 50% or greater pain relief and a lower incidence of adverse effects and discontinuations due to adverse effects (Etropolski Rauschkolb-Loffler, Shapiro, et al., 2009).
The pharmacokinetics of a single 100 mg dose of tapentadol were studied in 4 healthy males (Terlinden, Ossig, Fliegert, et al., 2007). Absorption of the drug was described as rapid (time to peak plasma concentration was 1.25 to 1.5 hours), and elimination was almost exclusively by the renal system; it was excreted in the urine in the form of unconjugated metabolites (no clinically relevant metabolites). Other research revealed an oral bioavailability of 32% and a time to steady state of 25 to 30 hours with 4-hour dosing (Tzschentke, De Vry, Terlinden, et al., 2006). Although the manufacturer warns of possible interactions with drugs that are metabolized by the cytochrome P450 enzyme system (Pricara, 2008), research that evaluated tapentadol for protein binding and induction and inhibition of cytochrome P450 enzymes concluded that no clinically relevant drug-drug interactions are likely to occur through these mechanisms (Kneip, Terlinden, Beier, et al., 2008). Tapentadol has not been evaluated for use in patients with renal or hepatic impairment; the manufacturer advises not to use the drug in patients with these conditions.
Because this is a relatively new drug, clinical experience is lacking and there were only a few clinical trials at the time of publication. A single-dose (25, 50, 75, 100, or 200 mg) study of 400 patients with moderate to severe pain following dental surgery demonstrated that 75 mg or more of the drug produced effective dose-related analgesia and was well tolerated (Kleinert, Lange, Steup, et al., 2008). Tapentadol 200 mg produced higher total pain relief scores and a faster onset of action than 60 mg of oral morphine. Nausea and vomiting were also less common with all doses of tapentadol than with morphine. Another large trial randomized 901 patients to receive tapentadol 50 or 75 mg, oxycodone 10 mg, or placebo every 4 to 6 hours for 72 hours following bunionectomy (Daniels, Casson, Stegmann, et al., 2009). Acetaminophen was allowed during the study. Both doses of tapentadol and oxycodone were superior to placebo, and both doses of tapentadol produced similar pain relief as oxycodone. Nausea and vomiting were statistically significantly lower with tapentadol 50 mg and numerically lower (not statistically significant) with tapentadol 75 mg compared with oxycodone.
The drug has also been studied in patients with persistent pain. A 10-day, phase III, randomized, placebo-controlled study found tapentadol 50 and 75 mg produced similar analgesia as oxycodone 10 mg in patients awaiting joint replacement for uncontrolled arthritis pain (Hartrick, Van Hove, Stegmann, et al., 2009). As in other studies, GI adverse effects, such as nausea, vomiting, and constipation, were less frequent with both doses of tapentadol than with oxycodone. Another phase III study randomized 849 patients with lower back pain or hip or knee OA to receive tapentadol or oxycodone using a flexible dosing regimen for 90 days (Hale, Upmalis, Okamoto, et al., 2009). Pain relief was similar among the groups. CNS symptoms such as dizziness and somnolence were similar, but, again, GI adverse effects were less likely with tapentadol than with oxycodone. The researchers noted that dose tapering after treatment with tapentadol for 90 days did not appear to be necessary.
Tapentadol differs from tramadol in that it does not directly block the reuptake of serotonin; however, elevated norepinephrine levels can result in elevated levels of serotonin, which can lead to the development of serotonin syndrome evidenced by shivering, nausea, low-grade fever, sweating, mental confusion, and delirium. The same precautions for tramadol apply to tapentadol (i.e., avoid combining tapentadol with other drugs that increase serotonin level, such as tramadol and SSRIs). Tapentadol should also not be administered with MAOIs. As with other opioids, additive toxicity may occur with concurrent use of CNS depressants.
Effects of Patient Characteristics on Opioid Drug Selection
Other important factors to consider when selecting the optimum opioid analgesic are intrinsic to the patient. These factors include pain intensity, patient age, coexisting disease, current drug regimen and potential drug interactions, prior treatment outcomes, patient preference and convenience, and cost.
Pain Intensity
Evaluation of the patient’s report of pain intensity is critical to the development of an individualized opioid analgesic treatment plan. A variety of pain rating scales exist for the purpose of assessing pain intensity (see Section II). For some types of mild to moderate pain, the options to consider typically include the dual-mechanism drugs tramadol or tapentadol, or one of the pure mu agonists. There also is extensive experience with the agonist-antagonist drugs in some types of pain, such as the use of the agonist-antagonist butorphanol (Stadol nasal spray) for migraine headache pain (Rapoport, Bigal, Tepper, et al., 2004). For moderate to severe persistent pain that already has been treated with an opioid, the preferred drug is a pure mu agonist; the preferred route is oral or transdermal. Severe acute pain is treated with pure mu agonist opioids.
Guidelines usually underscore the conclusion that most patients with mild pain do not require an opioid. A nonopioid such as ibuprofen or celecoxib may be sufficient (see Section III). If an opioid is appropriate, mild pain theoretically could be treated with any of the opioids, even morphine, if the dose is kept low; however, it is far more customary to use an opioid-nonopioid combination, such as oxycodone or hydrocodone compounded with a nonopioid such as acetaminophen (see Tables 13-3 and 13-9), or one of the dual-mechanism drugs.
Some pain of moderate intensity may also be satisfactorily relieved with a nonopioid (e.g., IV ketorolac). If an opioid is required, an opioid-nonopioid combination may be selected; however, as mentioned previously, the dose of the opioid will be restricted by the dose of the nonopioid with which it is compounded. In patients with moderate pain associated with progressive illness and in whom it may be predicted that pain will worsen over time, it is reasonable to select a single-entity opioid because it will allow the opioid dose to be titrated upward without concern about exceeding the recommended total daily dose of a nonopioid.
In addition to providing valuable information needed to select the correct analgesic for a patient, evaluating the severity of pain may suggest the underlying mechanism and pain syndrome. For example, the pain associated with radiation-induced nerve injury usually is not severe. The occurrence of severe pain in a previously irradiated area suggests recurrent or new pathology in the patient with cancer (Coyle, Cherny, Portenoy, 1995; Portenoy, 1996).
In the case of postoperative pain management, research and clinical experience determine the expected or usual pain intensity associated with surgical procedures and are used as guides for opioid selection and determining appropriate starting doses for pain relief. For example, pain associated with a thoracotomy is expected to be severe. Preplanning for severe postthoracotomy pain may include the placement of an epidural catheter for intraoperative and postoperative pain management using a mu agonist opioid analgesic and a local anesthetic. (See Chapter 16 for determining an appropriate starting dose.)
Patient Age
For the younger adult with no major organ failure, any of the available mu agonist opioids can be selected. For the older adult and those with major organ failure, changes in metabolism and elimination of drugs must be considered. Opioids with a short half-life are recommended, such as morphine, hydromorphone, and oxycodone. These drugs will achieve stable plasma concentrations within 24 hours (4 to 5 half-lives) and are, therefore, simpler to titrate and monitor (Coyle, Cherny, Portenoy, 1995). Mu agonists with a long half-life, such as methadone and levorphanol, are more challenging to dose and monitor in older patients and may be avoided on this basis. Drugs with active metabolites, such as meperidine and propoxyphene, are also avoided in older adults (see Table 13-4) and in all patients with renal dysfunction.
Coexisting Disease
Major organ failure, particularly cardiac, hepatic, and renal failure, influences the distribution, clearance, and excretion of opioids (Wellington, Chia, 2009) (see Chapter 11). Opioids, in turn, have adverse effects that may interact with dysfunctional organ systems. For example, opioids decrease the cough reflex, dry secretions, and release histamine (leading rarely to bronchial constriction) (Gutstein, Akil, 2006)—effects that increase the need for caution in patients with compromised respiratory function, including those with obstructive sleep apnea syndrome (American Society of Anesthesiologists, 2006) (also see Chapter 19).
When an opioid is prescribed to a patient with major organ failure, it is appropriate to start with less than the usual recommended dose and titrate the dose gradually. Extended dosing intervals are also advisable in some patients, particularly when multiple daily doses are required (Johnson, 2007).
All opioid drugs are metabolized to some extent by the liver. In patients with liver disease, clearance is decreased and bioavailability and half-life of opioids are increased (Wellington, Chia, 2009). This can lead to adverse effects from higher than expected plasma concentrations of these drugs. Higher plasma concentrations can also occur following liver surgery (Rudin, Lundberg, Hammarlund-Udenaes, et al., 2007). Administration of reduced doses (e.g., 50% less) or increased interval (e.g., twice the usual time period) and close monitoring of sedation and respiratory status is advised in the setting of liver dysfunction (Johnson, 2007). Propoxyphene, codeine, and meperidine should be avoided in patients with hepatic disease (Johnson, 2007). Johnson (2007) recommends also avoiding methadone in patients with hepatic disease, but others suggest that methadone may be used and doses may not need to be adjusted in cirrhosis and stable chronic liver disease because some methadone metabolism occurs in the intestine rather than the liver (Lugo, Satterfield, Kern, 2005).
Fentanyl is considered relatively safer in patients with hepatic dysfunction, and dose adjustment is not usually needed (Johnson, 2007). Remifentanil has also been suggested as an option for those with hepatic impairment (Wellington, Chia, 2009), but further research and clinical experience with this drug in patients with this condition is warranted.
Patients with renal disease may accumulate the active metabolites of drugs. For example, patients with renal insufficiency can develop relatively high concentrations of normeperidine during meperidine treatment, norpropoxyphene during propoxyphene treatment, and the glucuronidated metabolites during morphine treatment. Metabolite accumulation happens with all routes of administration, but the concentrations of the metabolites are greatest during oral administration. Metabolite accumulation is most problematic with meperidine (Marcantonio, Juarez, Goldman, et al., 1994). The accumulation of M6G and M3G can produce clinical effects (Dean, 2004), and a trial of an alternative opioid (e.g., hydromorphone or fentanyl) often is recommended if morphine toxicity occurs in a patient with renal disease (Portenoy, 1996). Morphine should be avoided in patients with end-stage renal disease (Dean, 2004). See Table 13-10 for recommendations regarding the use of selected mu opioids in patients with renal failure or undergoing dialysis.
When indicated by the type and severity of pain, the use of alternative routes may be a solution for some patients with major organ failure. Opioids can be given in lower doses by the epidural and intrathecal routes compared with the doses needed when given by the oral and parenteral routes. Because of this, patients with major organ failure sometimes are able to tolerate opioids by an intraspinal route that they were unable to tolerate by the oral or parenteral routes. Lipophilic drugs, such as fentanyl and sufentanil, can accumulate, particularly when administered on a long-term basis or by continuous infusion (Kurella, Bennett, Chertow, 2003); morphine and hydromorphone may be better choices by the epidural route in patients with hepatic dysfunction and CNS depression. Continuous peripheral nerve block may be an alternative for postoperative patients in whom opioids must be avoided entirely (see Chapter 26).
Current Drug Regimen and Potential Drug Interactions
Many patients with pain, especially those with co-existing disease and continuous cancer or persistent noncancer pain, take several medications. Co-administration of two or more drugs often can result in a change in the metabolism, clearance, or both of the drugs (Pasero, Portenoy, McCaffery, 1999). It is extremely important when developing an opioid treatment plan to know all the medications the patient is taking and how they will interact with the opioid, and to monitor on an ongoing basis for drug interactions.
As discussed in Chapter 11, many opioid drugs are metabolized by the liver’s cytochrome P450 (CYP) system, and the most important enzymes responsible for opioid metabolism are CYP2D6 and CYP3A4. Concomitant administration of a drug that induces one of these enzymes can lead to decreased levels of the opioid, and treatment with a drug that inhibits one of these enzymes can lead to increased levels (Fine, Portenoy, 2007) (see Tables 11-2 and 11-3, pp. 289 and 296).
The tricyclic antidepressants, including clomipramine (Anafranil) and amitriptyline (Elavil), may increase the bioavailability and half-life of morphine. This would cause a rise in plasma morphine levels (Coyle, Cherny, Portenoy, 1995). Phenytoin and the antitubercular drug, rifampin, increase the metabolism of methadone, and phenobarbital and phenytoin increase the metabolism of meperidine. This can result in decreased plasma levels of these opioids. Treatment with rifampin can result in complete loss of the analgesic effects of morphine (Fromm, Eckhardt, Li, et al., 1997). In the presence of MAOIs, meperidine can precipitate excitation, hyperpyrexia, convulsions, and death (Coyle, Cherny, Portenoy, 1995).
When drugs with anticholinergic effects, such as some antihistamines, phenothiazines, tricyclic antidepressants, and antiparkinsonian drugs, are administered with opioids, adverse effects, including dry mouth and constipation, may increase. It is important to emphasize the need for a bowel management regimen in patients taking any of these drugs with opioids (Coyle, Cherny, Portenoy, 1995) (see Chapter 19).
Seizures have been identified as a risk associated with the use of tramadol. Although the severity of this risk is uncertain, it is presumably enhanced when patients are taking tricyclic antidepressants, MAOIs, neuroleptics, and other drugs that reduce the seizure threshold. Serotonin syndrome can also occur when drugs that increase the serotonin level, such as tramadol, tapentadol, and SSRIs, are taken concomitantly.
Additive effects when opioid drugs are combined with other drugs also must be considered when implementing a pain treatment plan. Close monitoring of sedation is indicated when opioids are combined with antidepressants, phenothiazines, benzodiazepines, or neuroleptics. Because of their sedative and respiratory depressive effects, the amount of opioid that can be safely administered may be limited (APS, 2003; Anwari, Iqbal, 2003; Coyle, Cherny, Portenoy, 1995; Webster, Choi, Desai, et al., 2008).
Combining drugs can also have a positive effect on analgesia and adverse effects (APS, 2003; Inturrisi, 2002). For example, a review of the literature between 1996 and 2002 concluded that methylphenidate (Ritaline) attenuated opioid-induced somnolence, augmented opioid analgesia, treated depression, and improved cognitive function in cancer patients and recommended its use in palliative care (Rozans, Dreisbach, Lertora, et al., 2002). Co-administration of amphetamine has been reported to enhance the analgesic effects while counteracting the sedative effects of both morphine and meperidine (APS, 2003). (See Chapter 19 for treatment of opioid-induced sedation and Section V for more on the effects of adjuvant drugs.) NSAIDs combined with opioids can improve pain relief, and if this allows reduction in the opioid dose, the combination can reduce opioid-induced adverse effects (Ashburn, Caplan, Carr, et al., 2004; Kim, Kim, Nam, et al., 2008; Marret, Kurdi, Zufferey, et al., 2005; Schug, 2006; Schug, Manopas, 2007; White, 2005) (see Section III).
Prior Treatment Outcomes
In some cases, the prior experiences with opioids can be used to predict the response to future therapy. If a patient reports having previously experienced unmanageable adverse effects with an opioid, explore with the patient the occurrence, severity, and management of the adverse effects. An attempt should be made to determine whether the adverse effects were really unmanageable (e.g., nausea unresponsive to antiemetics) or simply unmanaged (e.g., no attempt made to relieve nausea). A drug that has been associated with intensely negative effects in the past usually is not preferred during later therapy. This is comparable to the decision to switch to another opioid if intolerable and unmanageable adverse effects occur during therapy, the strategy known as opioid rotation (Hanks, Cherny, Fallon, 2004) (see Chapter 18).
A true allergy to an opioid is extremely rare (Amabile, Bowman, 2006; Hanks, Cherny, Fallon, 2004; Woodall, Chiu, Weissman, 2008). Generalized erythema and bronchospasm within 4 hours of application of transdermal fentanyl were reported in an allergic patient (Dewachter, Lefebvre, Kalaboka, et al., 2009). Often, patients erroneously report being “allergic” to an opioid after experiencing adverse effects from it in the past. For example, patients commonly mistake nausea and vomiting after an opioid bolus dose as an allergy to the opioid. Patients also frequently assume that flushing, itching, or hives after initiation of opioid therapy reflects allergy. In most cases, this effect is believed to be related to a direct histamine-releasing effect of these drugs, an effect distinct from true hypersensitivity. In the absence of evidence of allergy, patients should be educated about the outcomes they experience and reassured that an adverse effect such as nausea or itching is an adverse effect and not an allergy.
Patient Preferences and Convenience
Often, patients have preferences when it comes to the choice of opioid, route of administration, and scheduling of doses. Unfortunately, a patient’s request for a specific opioid is sometimes misinterpreted as a sign of addiction (see Section II). Respecting patients’ preferences whenever feasible and making the opioid treatment regimen as convenient as possible may help the patient adhere better to the plan. Occasionally preferences are based on myths and misconceptions that can be corrected by providing factual information. During the initial interview with the patient, time can be taken to determine whether this is the case and ensure that the patient has an accurate understanding of pain management.
The oral and transdermal routes are the preferred routes of administration for continuous cancer and noncancer pain management. Occasionally, an alternative is needed. Although the rectal route is safe and effective, some patients with family caregivers are uncomfortable with the thought of a family member administering rectal medications to them. Occasionally patients who can self-administer their medications object to doing so by the rectal route. In these cases, alternative opioid delivery systems, such as continuous subcutaneous infusion, may provide a solution (see Chapter 14 for these alternative routes of administration).
Complying with the established opioid treatment regimen can become a problem for some patients, especially if the regimen requires the patient to take several pills several times a day. Older patients in particular may forget to take their pills at the prescribed intervals or object to taking so many pills. For these patients, a modified-release preparation of the opioid may be ideal. This will allow the patient to take pills once or twice daily instead of several times a day required by short-acting preparations. Scheduling doses along with other medications the patient is already accustomed to taking also is recommended. For example, individuals can take their morning opioid dose with their daily vitamin. Nonopioids and opioids may be given at the same time (staggered dosing is not necessary). The fentanyl transdermal patch, which is changed only every 3 days in most individuals (see Chapter 14), can be considered also for patients who are unreliable in taking oral medications.
Cost
The cost of an opioid is an important consideration in selecting the optimal opioid for the pain treatment plan, especially for long-term opioid therapy. The cost of medications can vary greatly. Several years ago, an analysis of equianalgesic doses of opioids revealed a 19-fold price difference among opioid prescriptions (Ferrell, Griffith, 1994). A later cost comparison of routes of administration showed the cost for one day of opioid treatment; oral or rectal MS Contin 120 mg twice daily was $18.16; IV morphine at 3.3 mg/h was $17.08; SC hydromorphone at 0.9 mg/h was $24.33; and transdermal fentanyl at 50 mcg/h was $8.16 (Stevens, Ghazi, 2000). The authors pointed out that IV and SC infusions also incur the expense of supplies and equipment (e.g., tubings and pumps).
A number of factors influence the cost of drugs, including packaging of the drug, wholesale prices, and pharmacy dispensing fees. The cost of prescription opioids varies from one pharmacy to another, but the newer opioids without generic equivalents tend to be more expensive (Chamberlin, Cottle, Neville, et al., 2007). There are numerous websites, such as PharmacyChecker.com (http://www.pharmacychecker.com/), that compare drug costs worldwide. Some pain guidelines include tables displaying the costs of analgesics at the time of publication. Although listed prices in guidelines are likely to be different from current prices, they provide an idea of relative cost.
As a rule, morphine and especially methadone are significantly less expensive than other opioids, bulk containers of drugs are less expensive than prefilled syringes and unit dose or blister packs, and large hospitals or urban chain pharmacies that can purchase drugs in large quantities are likely to charge less than small independent pharmacies (Kunz, 1994). The use of multiple prescriptions (polypharmacy) to manage pain is more costly than single prescriptions. Although combined modalities are recommended, single opioids in sufficient doses may provide adequate pain control and certainly should be tried before adding adjuvant analgesics (Ferrell, Griffith, 1994).
Today, most individuals assume all or at least part of the cost of their medications. The insured patient’s ability to pay depends on the amount of their co-payment. Great care should be taken to ensure cost savings for patients with end-stage disease so that appropriate pain management is not prohibitive and does not overburden patients and families with excessive costs. Many pharmaceutical companies have financial assistance programs that allow reduced purchase prices for patients who qualify. Prescribers can find information about this in the Physician’s Desk Reference and at most pharmaceutical company websites. If drug costs are a significant issue, prices should be checked with the pharmacy.
Conclusion
There are many opioid analgesics to choose from when developing a pain treatment plan. The first-line opioids are mu agonist opioids; in most cases, there is no advantage to using an agonist-antagonist opioid for pain management. Selection of the optimal opioid analgesic is influenced by a number of patient factors, including age, coexisting disease, potential for drug-drug interaction, and previous experience with opioids. Cost of the drug is also a major consideration.