Chapter 3 The immune system and disease
Anatomy and principles of the immune system
Immunity can be defined as protection from infection, whether it be due to bacteria, viruses, fungi or multicellular parasites. Like other organs involved in human physiology, the immune system is composed of cells and molecules organized into specialized tissues (Fig. 3.1).
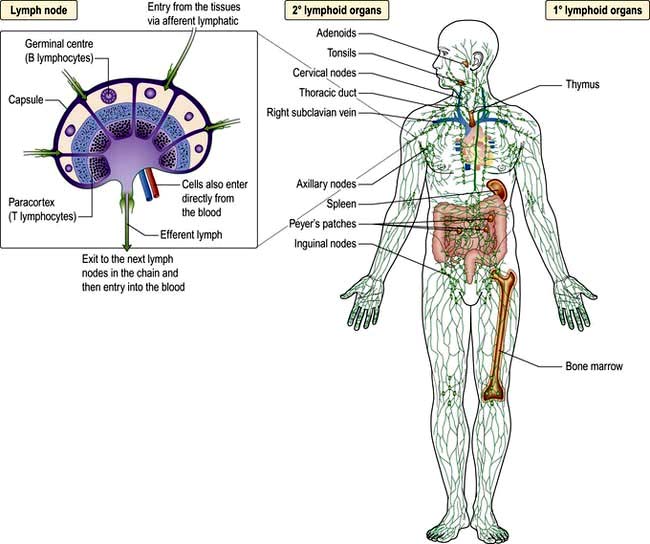
Figure 3.1 Primary and secondary lymphoid tissue. Lymphocytes are generated as precursors in the bone marrow and differentiate into T (thymus) or B (bone marrow) lymphocytes in the primary lymphoid tissue. Once differentiated, 98% of lymphocytes reside in the secondary lymphoid tissue where the adaptive immune response takes place.
The primary lymphoid organs are where the cells originate. Cells and molecules of the immune system circulate in the blood; immune responses do not take place there but are at the site of infection (typically the mucosa or skin). They are then propagated and refined in the secondary lymphoid organs (e.g. lymph nodes). After resolution of the infection, immunological memory specific for the pathogen resides in cells (lymphocytes) in the spleen and lymph nodes, as well as being widely secreted in a molecular form (antibodies).
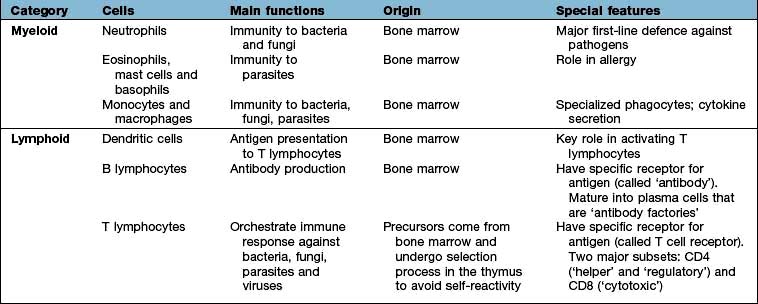
Cells involved in immune responses: origin and function
All immune cells have a common source in the pluripotent stem cells generated in the bone marrow (Fig. 3.1). They have diverse functions (Table 3.1). T lymphocytes undergo ‘education’ in the thymus to avoid self-recognition, and populate the peripheral lymphoid tissue, where B lymphocytes also reside. Both sets of lymphocytes undergo activation in the peripheral tissue, to become mature effector cells. B lymphocytes may further differentiate into antibody-secreting plasma cells. Lymphoid tissue is frequently found at mucosal surfaces in non-encapsulated patches, termed mucosa-associated lymphoid tissue (MALT).
The immune system
Cells and molecules involved in immune responses are classified into innate and adaptive systems:
 The innate immune system is inborn and operates throughout life (pp. 51–55)
The innate immune system is inborn and operates throughout life (pp. 51–55)
The adaptive immune system changes in response to the pathogens it encounters (pp. 58–62).
There are also non-immunological barriers that are involved in host protection, and very often it is the lowering of these that allows a pathogen to take a foothold (Table 3.2).
Table 3.2 Non-immunological host defence mechanisms
Normal barriers |
Events that may compromise barrier function |
Physical barriers |
|
Skin and mucous membranes |
Trauma, burns, i.v. cannulae |
Cough reflex |
Suppression, e.g. by opiates, neurological disease |
Mucosal function |
Ciliary paralysis (e.g. smoking) |
Increased mucus production (e.g. asthma) |
|
Abnormally viscid secretions (e.g. cystic fibrosis) |
|
Decreased secretions (e.g. sicca syndrome) |
|
Urine flow |
Stasis (e.g. prostatic hypertrophy) |
Chemical barriers |
Low gastric pH (gastric acid secretion inhibitors) |
Resistance to pathogens provided by commensal skin and gut organisms |
Changes in flora (e.g. broad-spectrum antibiotics) |
The immune system is immensely powerful, in terms of its ability to inflame, damage and kill, and it has a capacity to recognize a myriad of molecular patterns in the microbial world. However, immune responses are not always beneficial. They can give rise to a range of autoimmune and inflammatory diseases, known as immunopathologies. In addition, the immune system may fail, giving rise to immune deficiency states. These conditions are grouped under the umbrella of clinical immunology.
A major feature of the immune system is the complexity of the surface-bound, intracellular and soluble structures that mediate its functions. In particular, it is necessary to be aware of the CD (clusters of differentiation) classification (Box 3.1) and the functions of cytokines and chemokines.
 Box 3.1
Box 3.1
The CD classification
This is the ultimate way of defining a cell
Immune cells are distinguished by the surface receptors and proteins that they express in order to mediate their particular range of immunological functions, e.g. cell–cell signalling, cell activation
The surfaces are covered with such proteins and indicate the cell lineage or differentiation pathway. The discovery of monoclonal antibodies (proteins tailor-made to bind to a specific target) made this feasible
Surface molecules defining the origin and function of selected groups of cells are known as clusters of differentiation (CD). Over 300 CD numbers exist
A clinical example is the number of peripheral blood lymphocytes expressing CD4 (’the CD4 count’), which is used to monitor HIV infection (see p. 178)
An updated listing is available at: http://www.hcdm.org/
Cytokines
These are small polypeptides released by a cell in order to change the function of the same or another cell. These chemical messengers are found in many organ systems, but especially the immune system. Cytokines have become markers in the investigation of disease pathogenesis; therapeutic agents in their own right; and the targets of therapeutic agents (see p. 72). The key features of a cytokine are:
pleiotropy: different effects on different cells
autocrine function: modulates the cell secreting it
paracrine function: modulates adjacent cells
endocrine effects: modulates cells and organs at remote sites
synergistic activity: acting in concert with other cytokines to achieve effects greater than the summation of their individual actions.
The main immune cytokines are the interferons (IFNs) and the interleukins (IL). The IFNs are limited to a few major types (α, β and γ), whereas there are 35 interleukins.
SIGNIFICANT WEBSITE
For cytokine listings and functions of cytokines and chemokines, and related drugs in development: http://www.copewithcytokines.de/
Chemokines
The defining feature of chemokines is their function as chemotactic molecules, i.e. they attract cells along a gradient of low to high chemical concentration, particularly from the blood into the tissues and tissues into lymphatics. They also have the ability to activate immune cells. All chemokines have a similar structure relating to the configuration of cysteine residues, which gives rise to four families:
CXC: two cysteines (C) separated by any other amino acid residue (X)
Receptors on the surface are denoted by ‘R’, and are all distinctive G-coupled protein receptors with seven transmembrane spanning domains.
Anti-inflammatory drugs that block chemokine functions, or drugs that promote cell activation and migration, e.g. into tumours, are being developed.
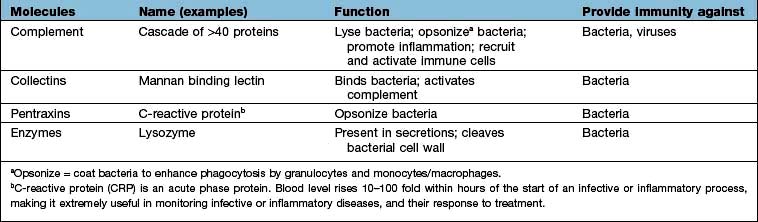
Cells and molecules of the innate immune system
Innate immunity provides immediate, first-line, host defence. The key features of this system are shown in Table 3.3. It is present at birth and remains operative at comparable intensity into old age. Innate immunity is mediated by a variety of cells and molecules (Table 3.4). Activation of innate immune responses is mediated through interaction between the:
pathogen side comprising a relatively limited array of molecules (pathogen-associated molecular patterns, PAMPs)
host side – a limited portfolio of receptors (pattern recognition receptors, PRRs).
Table 3.3 Features of the innate and adaptive immune responses
| Innate | Adaptive |
|---|---|
No memory: quality and intensity of response invariant |
Memory: response adapts with each exposure |
Recognizes limited number of non-varying, generic molecular patterns on, or made by, pathogens |
Recognizes vast array of specific antigensa on, or made by, pathogens |
Pattern recognition mediated by a limited array of receptors |
Antigen recognition mediated by a vast array of antigen-specific receptors |
Response immediate on first encounter |
Response on first encounter takes 1–2 weeks; on second encounter 3–7 days |
a Antigen is a molecular structure (protein, peptide, lipid, carbohydrate) that generates an immune response.
Activation of certain cells in the innate immune system leads to activation of the adaptive immune response (see p. 58).
The dendritic cell is especially involved in this process, and forms a bridge between innate and adaptive systems.
Complement
Complement proteins are produced in the liver. Each complement circulates in an inactive form until triggered to become enzymatically active, when it then activates several molecules of the next stage in a series. This complement cascade is initiated via three distinct pathways: alternative, classical and mannan-binding lectin (Fig. 3.2). These pathways are composed of three distinct enzyme cascades that culminate in the cleavage of C3 and C5. Cleavage of C3 has a number of biological consequences; breakdown of C5 achieves the same and, in addition, provides the triggering stimulus to the final common (‘membrane attack’) pathway, which provides most of the biological activity (Fig. 3.2).
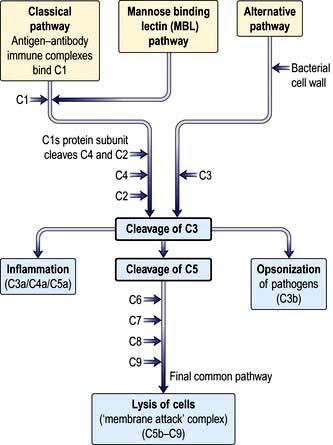
The main functions of complement activation are to:
promote inflammation (e.g. through the actions of the anaphylatoxins C3a, C4a and C5a)
recruit cells (e.g. through chemoattractants)
kill targeted cells, such as bacteria
solubilize and remove from the circulation antigen-antibody (‘immune’) complexes.
During an immune response, removal of immune complexes protects unaffected tissues from the deposition of these large, insoluble composites which could result in unwanted inflammation. Failure of this protective mechanism can result in immunopathology, e.g. in the joints, kidney and eye.
Neutrophils
Neutrophil: phagocytoses bacteria and kills them by releasing antimicrobial compounds (e.g. defensins).
Neutrophils (see p. 413) phagocytose and kill microorganisms. They are derived from the bone marrow, which can produce between 1011 (healthy state) and 1012 (during infection) new cells per day. In health, neutrophils are rarely seen in the tissues.
Neutrophil phagocytosis is activated by interaction with bacteria, either directly or after bacteria have been coated (opsonized) to make them more ingestible (Fig. 3.3). The contents of neutrophil granules are released both intracellularly (predominantly azurophilic granules) and extracellularly (specific granules) following fusion with the plasma membrane. Approximately 100 different molecules in neutrophil granules (Table 3.5) kill and digest microorganisms, for example:
Myeloperoxidase and cytochrome b558 are key components of major oxygen-dependent bactericidal systems.
Cathepsins, proteinase-3 and elastase are deadly to Gram-positive and Gram-negative organisms, as well as some Candida species.
Defensins are naturally occurring cysteine-rich antibacterial and antifungal polypeptides (29–35 amino acids).
Collagenase and elastase break down fibrous structures in the extracellular matrix, facilitating progress of the neutrophil through the tissues.
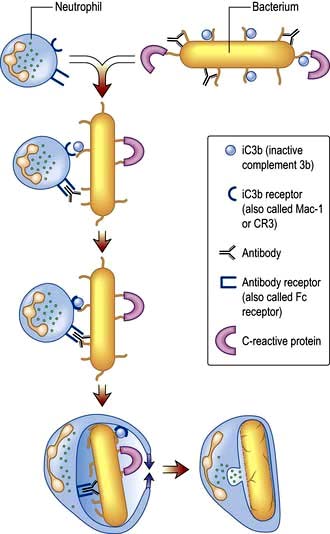
Figure 3.3 Opsonization. Bacteria are coated with a variety of soluble factors from the innate immune system (opsonins), which enhance phagocytosis. This leads to engulfment of the bacteria into phagosomes and fusion with granules to release antibacterial agents. iC3b, inactive complement 3b.
Table 3.5 Contents and function of key neutrophil granules
| Function | Primary or azurophilic granules | Secondary or specific granules |
|---|---|---|
Antibacterial |
Lysozyme |
Respiratory burst components (e.g. cytochrome b558) producing reactive oxygen metabolites, such as hydrogen peroxide, hydroxyl radicals and singlet oxygen |
Cathepsins |
Lysozyme |
|
Bactericidal/permeability increasing protein (BPI) |
Lactoferrin |
|
Cell movement |
|
Collagenase |
|
CD11b/CD18 (adhesion molecule) |
|
|
N-formyl-methionyl-leucylphenylalanine receptor (FMLP-R) |
Granule release is initiated by the products of bacterial cell walls, complement proteins (e.g. inactive complement 3b, iC3b), leukotrienes (LTB4) and chemokines (e.g. CXCL8, also known as IL-8) and cytokines such as tumour necrosis factor α (TNF-α).
Eosinophils
In contrast to neutrophils, several hundred times more eosinophils are present in the tissues than in the blood, particularly at epithelial surfaces where they survive for several weeks. The main role of eosinophils is protection against multicellular parasites such as worms (helminths). This is achieved by the release of pro-inflammatory mediators, which are toxic, cationic proteins. In populations and societies in which such parasites are rare, eosinophils contribute mainly to allergic disease, particularly asthma (see p. 827). Eosinophils have two types of granules:
Specific granules (95%) contain the cationic proteins, of which there are four main types: major basic protein (MBP), eosinophil cationic protein (ECP), eosinophil neurotoxin, which are all potently and exquisitely toxic to helminths (ECP also has some bactericidal properties), and eosinophil peroxidase, which has similar activity to neutrophil myeloperoxidase.
Primary granules (5%) synthesize and release leukotrienes C4 and D4 and platelet-activating factor (PAF) which alter airway smooth muscle and vasculature (see p. 828).
Eosinophils are activated and recruited by a variety of mediators via specific surface receptors, including complement factors and leukotriene (LT) B4. In addition, the CC chemokines eotaxin-1 (CCL11) and eotaxin-2 (CCL24) are highly selective in eosinophil recruitment. Receptors are also present for the cytokines IL-3 and IL-5, which promote the development and differentiation of eosinophils.
Mast cells and basophils
Mast cells and basophils share features in common, especially in containing:
high-affinity receptors for immunoglobulin E (IgE; an antibody type that is involved in allergic disease, see p. 68, 69).
Mast cells are found in tissues (especially skin and mucosae) and basophils in the blood. Both mast cells and basophils release pro-inflammatory mediators which are either pre-formed or synthesized de novo (Table 3.6).
Table 3.6 Mast cell and basophil mediators
| Mediators | Effects |
|---|---|
Pre-formed: |
|
Histamine |
Vasodilatation |
Vascular permeability ↑ |
|
Smooth muscle contraction in airways |
|
Proteases |
Digestion of basement membrane causes ↑vascular permeability and aids migration |
Proteoglycans (e.g. heparan) |
Anticoagulant activity |
Synthesized de novo: |
|
Platelet-activating factor (PAF) |
Vasodilator |
LTB4, LTC4, LTD4 |
Neutrophil and eosinophil activators and chemoattractants |
Vascular permeability ↑ |
|
Bronchoconstrictors |
|
Prostaglandins (mainly PGD2) |
Vascular permeability ↑ |
|
Bronchoconstrictors |
|
Vasodilators |
Histamine is a low-molecular-weight amine (111 Da) with a blood half-life of less than 5 minutes; it constitutes 10% of the mast cell’s weight. When injected into the skin, histamine induces the typical ‘weal and flare’ or ‘triple’ response: reddening (erythema) due to increased blood flow, swelling (weal) due to increased vascular permeability, and distal vascular changes (flare) due to effects on local axons.
The complement-derived anaphylatoxins C3a, C4a and C5a activate basophils and mast cells, as does IgE. The mast cell also has a role in the early response to bacteria through release of TNF-α, in cell recruitment to inflammatory sites such as arthritic joints, in promotion of tumour growth by enhancing neovascularization and in allograft tolerance.
Monocytes and macrophages
Monocyte (blood)/macrophage (tissue): ingests and kills bacteria; releases pro-inflammatory molecules; presents antigen to T lymphocytes; necessary in immunity to intracellular pathogens, e.g. mycobacteria.
Cells of the monocyte/macrophage lineage are highly sophisticated phagocytes. Monocytes are the blood form of a cell that spends a few days in the circulation before entering into the tissues to differentiate into macrophages, and possibly some types of dendritic cells.
Blood monocytes
Blood monocytes can be divided into two subsets: those expressing CD14 (a receptor for lipopolysaccharide, a bacterial cell wall component) and those expressing CD14 and CD16 (a receptor for IgG antibodies). In vitro, both subsets have the potential to differentiate into myeloid dendritic cells (after culture with IL-4 and granulocyte-monocyte colony simulating factor, GM-CSF) and into macrophages, which may exist in specialized forms (e.g. alveolar and gut macrophages and osteoclasts).
Tissue macrophages
A key role of tissue macrophages is the maintenance of tissue homeostasis, through clearance of cellular debris, especially following infection or inflammation. They are responsive to a range of pro-inflammatory stimuli, using their pattern recognition receptors (PRR) to recognize pathogen-associated molecular patterns (PAMPs). Once activated, they engulf and kill microorganisms, especially bacteria and fungi. In doing so they release a range of pro-inflammatory cytokines and have the capacity to present fragments of the microorganisms to T lymphocytes (see below) in a process called antigen presentation. Recent evidence suggests that evolutionarily conserved molecular patterns in mitochondria (organelles that originally derived from bacteria) can also activate monocytes. These damage-associated molecular patterns (DAMPs) could play a major role in the systemic inflammatory response that follows extensive tissue damage (e.g. following ischaemic injury).
It has been observed that some PAMPs induce the cytoplasmic assembly of large oligomeric structures of PRRs termed inflammasomes. There are numerous examples: members of the Nod-like receptor (NLR) family can be activated by stimuli such as viruses, bacterial toxins, and interestingly, crystallized endogenous molecules, including urate. Inflammasomes have potent effects in activating caspases, leading to processing and secretion of pro-inflammatory cytokines such as IL-1β and IL-18.
Macrophages have pro-inflammatory and microbicidal capabilities similar to those of neutrophils. Under activation conditions, antigen presentation (see p. 57, 58) is enhanced and a range of cytokines secreted, notably TNF-α, IL-1 and IFN-γ. These are necessary for the removal of certain pathogens that live within mononuclear phagocytes (e.g. mycobacteria). Macrophages and related cells may also undergo a process termed autophagy (p. 32, Ch. 2). This self-cannibalization is a critical property of many cell types under starvation conditions, but is used by the immune system to destroy intracellular pathogens such as Mycobacterium tuberculosis, which otherwise persist within cells and block normal antibacterial processes. Autophagy is also a means of enhancing antigen presentation pathways.
Tissue macrophages involved in chronic inflammatory foci may undergo terminal differentiation into multinucleated giant cells, typically found at the site of the granulomata characteristic of tuberculosis and sarcoidosis (see p. 845).
Dendritic cells
The major function of dendritic cells (DCs) is activation of naive T lymphocytes to initiate adaptive immune responses; they are the only cells capable of this. The definition of a dendritic cell is one that has:
dendritic morphology (Fig. 3.4)
machinery for sensing pathogens
the ability to process and present antigens to CD4 and CD8 T lymphocytes, coupled with the ability to activate these T lymphocytes from a naive state
the ability to dictate the T lymphocyte’s future function and differentiation.
Figure 3.4 Mature dendritic cell: ingests pathogens; migrates to lymph nodes and presents pathogen-derived antigen to T lymphocytes to enable adaptive immunity.
This is a powerful cell type that functions as a critical bridge between the innate and adaptive immune systems.
Types of dendritic cell
The major types are the myeloid DC (mDC), the plasmacytoid DC (pDC) and a variety of specialized DCs found in tissues that resemble mDCs (e.g. the Langerhans cell in the skin, see Fig. 24.1). DCs have several distinctive cell surface molecules, some of which have pathogen-sensing activity (e.g. the antigen uptake receptor DEC205 on mDCs) whilst others are involved in interaction with T lymphocytes (Table 3.7). Immature mDCs and pDCs are present in the blood, but at very low levels (<0.5% of lymphocyte/monocyte cells).
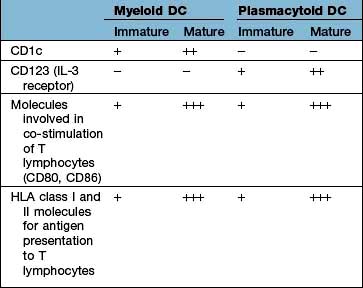
Pathogen sensing is a key component of the function of immature DCs, as well as monocytes/macrophages, and is achieved through expression of a limited array of specialized PRR molecules capable of binding to structures common to pathogens, aided by long cell dendrites and pinocytosis (constant ingestion of soluble material).
Mannan-binding lectin, which initiates complement activity inducing opsonization (p. 51, 52).
Signalling receptors such as the PRR known as TLR4 (for toll-like receptor 4), which binds lipopolysaccharide, a molecular pattern found in the cell walls of many Gram-negative bacteria (Table 3.8), whilst others bind double-stranded and single-stranded RNA from viruses. Innate immunity critically depends on toll-like receptor signalling. These receptors act through a critical adaptor molecule, myeloid differentiation factor 88 (MyD88), to regulate the activity of nuclear factor kappa B (NF-κB) pathways.
Endocytic pattern recognition receptors, which act by enhancing antigen presentation on macrophages, by recognizing microorganisms with mannose-rich carbohydrates on their surface or by binding to bacterial cell walls and scavenging bacteria from the circulation. All lead to phagocytosis.
TREM-1 (triggering receptor expressed on myeloid cells) is a cell surface receptor which, when bound to its ligand, triggers secretion of pro-inflammatory cytokines. It is upregulated by bacterial lipopolysaccharides but not in non-infective disorders.
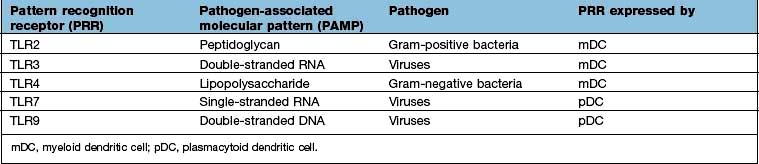
The key principle at play here is that the immune system has devised a means of identifying most types of invading microorganisms by using a limited number of PRRs recognizing common molecular patterns, or PAMPs. This recognition event has been termed a ‘danger signal’: it alerts the immune system to the presence of a pathogen. Sensing danger is a key role of the DC and a key first step towards activation of the adaptive immune system.
DCs and T cell activation
In a sequence of events that spans 1–2 days, immature DCs are activated by PAMPs or DAMPs in the tissues binding to a PRR on DCs. The immature pDC is a small rounded cell that develops dendrites upon activation and secretes enormous quantities of IFN-α, a potent antiviral and pro-inflammatory cytokine. On activation, the DC migrates to the local lymph node with the engulfed pathogen. During migration the DC matures, changing its shape, gene and molecular profile and function within a matter of hours to take on a mature form, with altered functions (Table 3.9, Fig. 3.5), in particular upregulating machinery required to activate T lymphocytes. Once in the lymph node, the mature DC interacts with naive T lymphocytes (antigen presentation), resulting in two key outcomes:
1. Activation of T cells with the ability to recognize peptide fragments (termed epitopes) of the pathogen
2. Polarization of the T cell towards a functional phenotype (see below) that is tailored to the particular pathogen.
Table 3.9 Myeloid dendritic cell (DC) maturation
| Immature mDC | Mature mDC |
|---|---|
Highly pinocytotic |
Ceases pinocytosis |
Low level expression of molecules required for T lymphocyte activation |
Upregulates CD80, CD86 and HLA molecules |
Low level expression of machinery required to process and present microbial antigens |
Begins to process microbial antigens (break down into small peptides) in readiness to present them to T lymphocytes (using HLA molecules) |
Generally localized and sedentary |
Begins active migration to local lymph node |
Minimal secretion of cytokines |
Active secretion of cytokines in readiness to stimulate T lymphocytes; in particular IL-12 |
mDC, myeloid dendritic cell.
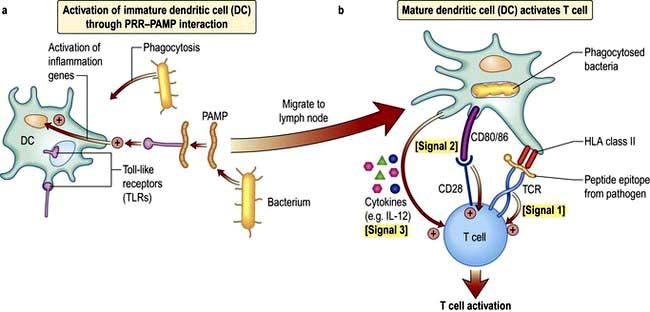
Figure 3.5 (a) Immature dendritic cells (DCs) in the tissues are activated by pathogens through PAMP–PRR interaction. (b) Multiple rapid changes in gene expression lead to migration to the lymph node as the DC takes on the mature phenotype. During migration there is synthesis of the machinery required for activation of T lymphocytes, shown here in response to signals 1–3.
The mature DC provides three major signals to naive T cells Fig. 3.5):
Hla molecules and antigen presentation
On the short arm of chromosome 6 is a collection of genes termed the major histocompatibility complex (MHC; known as the human leucocyte antigens, or HLA in man), which plays a critical role in immune function. MHC genes code for proteins expressed on the surface of a variety of cell types that are involved in antigen recognition by T lymphocytes. The T lymphocyte receptor for antigen recognizes its ligand as a short antigenic peptide embedded within a physical groove at the extremity of the HLA molecule (Fig. 3.6).
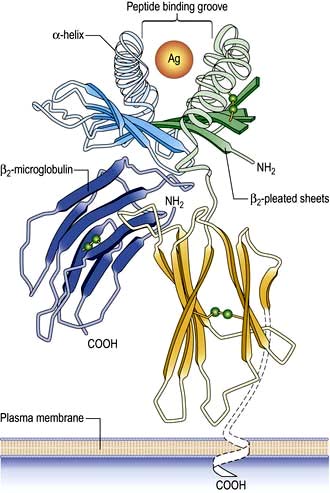
Figure 3.6 Structure of HLA class I molecule showing the peptide binding groove that is used to present antigen (Ag) to T lymphocytes.
The HLA genes are particularly interesting for clinicians and biologists. First, differences in HLA molecules between individuals are responsible for tissue and organ graft rejection (hence the name ‘histo’(tissue)-compatibility). Second, possession of certain HLA genes is linked to susceptibility to particular diseases (Table 3.10).
Table 3.10 HLA associations with immune-mediated and infectious diseases
| Disease process | Disease | HLA type |
|---|---|---|
Autoimmunity |
Type 1 diabetes |
Class II: |
|
DQA1*0301/DQB1*0302 (susceptibility) |
|
DQA1*501/DQB1*201 (susceptibility) |
||
DQA1*0102/DQB1*0602 (protection) |
||
|
Class I: |
|
|
HLA-A24; HLA-B*18; HLA-B*39 |
|
Multiple sclerosis |
DRB1*1501 (susceptibility) |
|
Rheumatoid arthritis |
DRB1*0404 (susceptibility) |
|
Autoimmune hepatitis |
DRB1*03 and DRB1*04 (susceptibility) |
|
Goodpasture’s syndrome (anti-glomerular basement membrane disease) |
HLA-DRB1*1501 (susceptibility) |
|
Pemphigus vulgaris |
DRB1*0402; DQB1*0503 (susceptibility) |
|
Inflammatory |
Coeliac disease |
DQA1*0501/DQB1*0201 (susceptibility) |
Ankylosing spondylitis |
HLA-B27 (susceptibility) |
|
Psoriasis |
HLA-Cw*0602 (susceptibility) |
|
Infectious |
Human immunodeficiency virus infection |
HA-B27; HLA-B*51; HLA-B*57 (associated with slow progression of disease) |
HLA-B*35 (associated with rapid progression) |
The human major histocompatibility complex
The human MHC comprises three major classes (I, II and III) of genes involved in the immune response (Fig. 3.7).
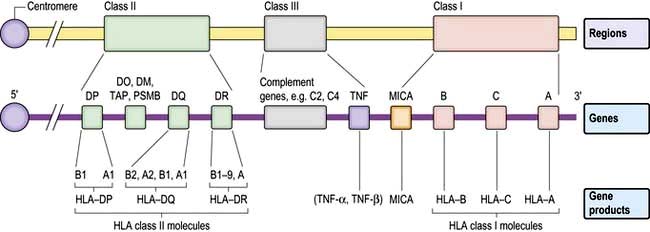
Figure 3.7 The HLA system in man. On chromosome 6 are three major HLA regions (classes I–III) including genes that encode the HLA class I and II molecules, complement genes, the cytokine TNF and other genes involved in antigen presentation (HLA-DO, -DM; transporter associated with processing, TAP; proteasome subunit beta, PSMB; and the non-classical HLA class Ic molecule, MICA).
HLA classes
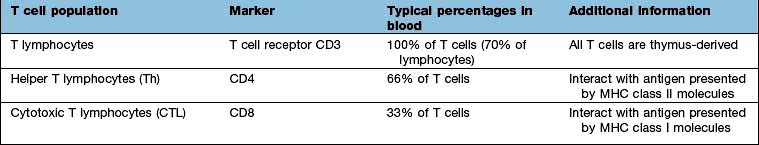
Classical class I HLA genes (also termed Ia), are designated HLA-A, HLA-B and HLA-C. Each encodes a class I α chain, which combines with a β chain to form the class I HLA molecule (Fig. 3.6). While there are several types of α chains, there is only one type of β chain, β2 microglobulin. The HLA class I molecule has the role of presenting short (8–10 amino acids) antigenic peptides to the T cell receptor on the subset of T lymphocytes that bear the co-receptor CD8. As an example of HLA polymorphism, there are nearly 200 allelic forms at the A gene locus. Class I HLA molecules are expressed on all nucleated cells.
Non-classical HLA class I genes are less polymorphic, have a more restricted expression on specialized cell types, and present a restricted type of peptide or none at all. These are the HLA-E, F and G (Ib genes) and MHC class I-related (MIC, or class Ic) genes, A and B. The products of these genes are predominantly found on epithelial cells, signal cellular stress and interact with lymphoid cells, especially natural killer cells (see p. 61, 62).
The class II genes have three major subregions, DP, DQ and DR. In these subregions are genes encoding A and B genes that combine to form dimeric αβ molecules that present short (12–15 amino acid) peptides to T lymphocytes that bear the CD4 co-receptor. Class II HLA genes (apart from DRA) are highly polymorphic. Other genes in this region encode proteins with key roles in antigen presentation (e.g. TAP, HLA-DM, HLA-DO, proteasome subunits; see below). Class II HLA genes are expressed on a restricted cohort of cells that go by the general term of antigen presenting cells (APCs; DCs, monocyte/macrophages, B lymphocytes).
HLA class III genes encode proteins that can regulate/modify immune responses, e.g. tumour necrosis factor (TNF), heat shock protein (HSP) and complement protein (C2, C4).
HLA genotypes and the range of their protein products
HLA genotype is denoted first by the letters that designate the locus (e.g. HLA-A, HLA-DR, HLA-DQ). For class I alleles, this is followed by an asterisk and then a 2- to 4-digit number defining the allelic variant at that locus, often called the HLA type (e.g. HLA-A*02 is the 02 variant of the HLA-A gene). The class II nomenclature is the same, except that both A and B genes are named (although HLA-DR molecules only require the name of the B gene, because the A gene is the same in all of us).
Some general principles apply to the HLA genes and their protein products:
The presence of multiple genes on each chromosome, and the fact that both maternal and paternal genes are co-dominantly expressed, allows considerable breadth in the number of HLA molecules that an individual expresses.
The existence of polymorphisms at each locus provides great breadth in the number of HLA molecules expressed at a population level. The polymorphic forms of HLA molecules differ predominantly in the peptide-binding groove (Fig. 3.6).
Overall, then, each human can bind a range of peptide epitopes from pathogens to enhance individual protection, and similarly the population has an even greater range of protection, to ensure population survival.
Antigen presentation
HLA molecules bind short peptide fragments which are processed (‘chopped up’) from larger proteins (antigens) derived from pathogens. The peptide–HLA complex is presented on antigen presenting cells (APCs) for recognition by T cell receptors (TCRs) on T lymphocytes. There are three major routes to antigen processing and presentation:
The endogenous route (Fig. 3.8) is a property of all nucleated cells: the internal milieu is sampled to generate peptide–HLA class I complexes for display (‘presentation’) on the cell surface. In a healthy cell, the peptides are derived from self-proteins in the cytoplasm (Fig. 3.8) and are ignored by the immune system. In a virus-infected cell, viral proteins are processed and presented. The resulting viral peptide–HLA class I complex is presented to CD8 T lymphocytes that have cytotoxic (killer) function. In an immune response against a virus infection, CD8 T lymphocytes recognizing viral peptide–HLA complexes on the surface of an infected cell will kill it as a means to limit and eradicate infection.
The exogenous route (Fig. 3.9) is a property of APCs: the external milieu is sampled. Antigens are internalized, either in the process of phagocytosis of a pathogen, through pinocytosis, or through specialized surface receptors (e.g. for antigen/antibody/complement complexes). The antigen is broken down by a combination of low pH and proteolytic enzymes for ‘loading’ into HLA class II molecules. At the APC surface the pathogen peptide–HLA class II complex is presented to and able to interact with CD4 T lymphocytes. Presentation by DCs can initiate an adaptive immune response by activating a naive, pathogen-specific CD4 T lymphocyte. Presentation by monocyte/macrophages and B lymphocytes can maintain and enhance this response by activating effector and memory pathogen-specific CD4 T lymphocytes.
Cross-presentation refers to the ability of some APCs (mainly DCs) to internalize exogenous antigens and process them through the endogenous route (Fig. 3.8). This is an essential component in the activation of CD8 cytotoxic T cell responses against a virus.
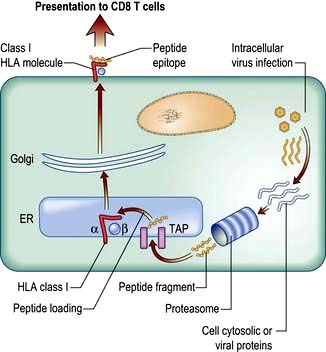
Figure 3.8 The endogenous route of antigen presentation to CD8 T lymphocytes. Cytosolic proteins derived from self (resting cells) or viruses (virus-infected cells) are broken down by the proteasome (a sort of ‘protein recycler’) into fragments 2–25 amino acids long. Some of these are taken into the endoplasmic reticulum (ER) by a transporter (TAP), trimmed and loaded into empty HLA class I molecules. These are exported to the surface membrane for presentation to CD8 T lymphocytes.
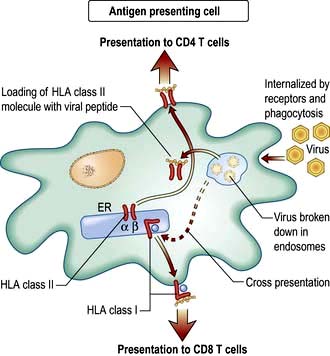
Figure 3.9 The exogenous route of antigen presentation and cross-presentation. External material (e.g. virus particles) is taken into an antigen presenting cell and broken down into specialized compartments by a combination of low pH and proteolysis. Peptides are then loaded into HLA class II molecules for presentation to CD4 T lymphocytes. Material may also be transferred across the cell so that it is loaded onto HLA class I molecules for presentation to CD8 T lymphocytes. This process of cross-presentation is restricted to specialized APCs such as DCs.
Cells and molecules of the adaptive immune system
The information gained by DCs that interact with a pathogen is passed on, in the form of signals 1–3 (Fig. 3.5b). These activate T lymphocytes in the adaptive immune system, which recognize the same pathogen. T lymphocytes may be involved in pathogen removal directly (e.g. by killing) or indirectly (e.g. by recruiting B lymphocytes to make specific antibody).
Antigen receptors on T and B lymphocytes
One of the key features of the adaptive immune system is specificity for antigen. For example, if you are immunized against the measles virus, you do not have immunity to hepatitis B, and vice-versa. Specificity is conferred by two types of receptor: the TCR on T lymphocytes and an equivalent on B lymphocytes, the BCR. BCRs are also termed surface immunoglobulin (sIg) and differ from TCRs in also being secreted in large quantities by end-stage B lymphocytes (plasma cells) as soluble immunoglobulins, also known as antibodies.
To maintain protection against the multiplicity of pathogens in our environment, each of us generates great diversity amongst TCRs and antibodies. This is achieved through a mechanism shared by both TCR and antibody, in which distinct families of genes are encoded in the germline, each family (called constant, variable, diversity and joining) contributing a sequence to part of the receptor. Recombination between randomly selected members of each family ensures diversity in the end product. Recombination frequently involves base deletions and additions, adding to the diversity. In the case of antibodies, the result is a potential capacity of >1014 different antibody molecules; for TCRs it may be as high as 1018.
Immunoglobulins
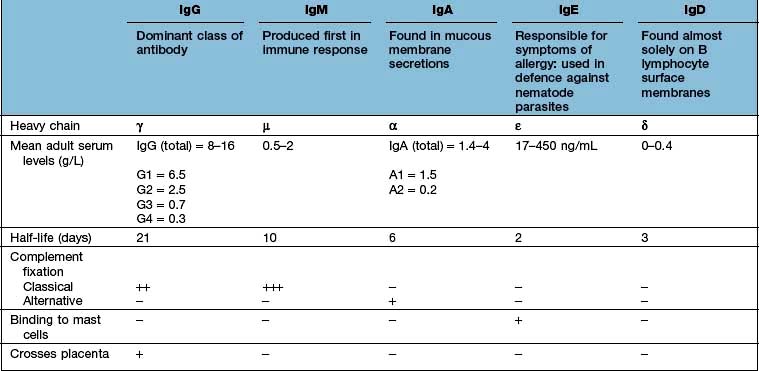
In structural terms, antibodies have four chains; two identical heavy and two identical light chains (Fig. 3.10). Each chain contains both highly variable and essentially constant regions. The variable parts of the heavy and light chains pair to form the potentially diverse part of the antibody molecule that binds antigen. The constant region of the heavy chain dictates the function of the antibody, and belongs to one of the classes M, G1–4, A1–2, D and E, giving rise to antibodies called IgM, IgG1–4, IgA1–2, IgD and IgE. The characteristics of these different isotypes are shown in Table 3.11.
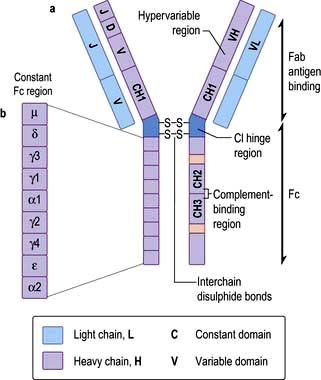
Figure 3.10 Immunoglobulin structure. (a) Basic subunit consisting of two heavy and two light chains. (b) Genes on the Fab and Fc regions of an immunoglobulin. The chain is made up of a V (variable) gene, which is translocated to the J (joining) chain. The VJ segment is then spliced to the C (constant) gene. Heavy chains have an additional D (diversity) segment which forms the VDJ segment that bears the antigen-binding site determinants.
Antibody production
Essential points about antibody production are:
IgM is the first isotype to be made in a primary immune response and thus measurement of pathogen specific IgM is a useful diagnostic test for recent infection.
IgG dominates in the second exposure to antigen.
IgG and IgM are the most efficient complement activators when bound to antigen in an immune complex.
IgG antibodies cross the placenta, and can carry both immunity and disease to the unborn fetus.
IgA antibodies are present in secretions (tears, saliva, GI tract) to give protection to the mucosae.
As it matures, and under the instruction of T lymphocytes, a B lymphocyte may change the class (class switching), but never the specificity, of the antibody it makes.
As B lymphocytes mature and are stimulated to undergo further division, minor changes in antibody gene sequence can arise (somatic mutation) potentially allowing antibodies with higher affinity to arise and be selected for the effector response (affinity maturation).
Antibody function
Essential points about antibody function are:
In host defence, antibodies target, neutralize and remove from the circulation and tissues infectious organisms and toxins, often through recruitment of innate host effector mechanisms such as complement, phagocytes and mast cells (by binding to specific surface receptors on these cells).
In clinical medicine, specific anti-pathogen antibody levels are used in diagnosing/monitoring infectious disease, and may also be administered as serum pools to passively provide host protection.
Antibodies can be raised in animals to generate monoclonal antibodies, which are commonly used in diagnostic immunology tests and increasingly used as therapeutics (e.g. to target cancer cells), often after ‘humanization’ (see below).
TCR genes and receptor diversity
The genomic organization of TCR genes and principles of generation of receptor diversity are similar to those of immunoglobulin genes. The TCR exists as a heterodimer, with a similar overall structure as the antibody molecule. There are two TCR types:
α and β chains (αβ TCR; expressed on all CD4 T lymphocytes and ~90% of CD8 T lymphocytes play a role in adaptive immune responses)
γ and δ chains (γδ TCR; fewer in number, mainly on intraepithelial lymphocytes; involved in epithelial defence).
The chains of each type of TCR are divided into variable and constant domains, each domain being encoded by separate gene pools. Like the B lymphocyte producing a single clone of immunoglobulin molecules, the T lymphocyte expresses only one form of TCR once the genes have been rearranged. Unlike antibodies, TCRs do not undergo somatic hypermutation and are not secreted.
SIGNIFICANT WEBSITE
Source of antibody resources for educators and researchers: http://www.antibodyresource.com/
T lymphocyte development and activation
T lymphocytes are generated from precursors in the bone marrow, which migrate to the thymus (Fig. 3.11). Only 1% of the cells that enter the thymus will leave it as naive T lymphocytes to populate the lymph nodes. This process (termed thymic selection) leads to a cohort of cells (Table 3.12) with:
functionally rearranged genes allowing surface expression of a receptor for antigen (the T cell receptor, TCR) alongside the CD3 accessory molecule involved in transducing the antigen-specific signal
selection of a co-receptor, either CD4 or CD8, to stabilize the interaction between TCR and peptide-HLA:
a reduced or absent tendency of the selected TCR to recognize self antigens (thus avoiding autoimmunity).
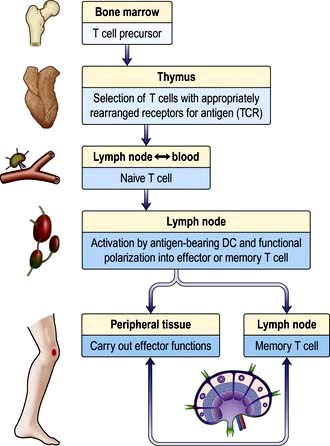
Figure 3.11 Development of T lymphocytes in the thymus and lifecycle in the secondary lymphoid tissue. DC, dendritic cell; TCR, T cell receptor.
Thus, during thymic education most TCRs are rejected for further use (negative selection), either because they are unable to bind self HLA molecules, or because they bind with too strong an affinity, which would run the risk of self-reactivity and autoimmune disease. The chosen TCRs (positive selection) have low/intermediate affinity for self HLA molecules. During post-thymic activation of T lymphocytes in the lymph node, TCR interaction with HLA has to be bolstered by additional signals (co-stimulation) provided by DCs. This ensures that T lymphocytes are only activated when the checkpoint of DC maturation has been passed, which will only happen in the presence of pathogens.
Most naive T lymphocytes are resident in the lymph nodes or spleen, whilst 2% are present in the blood, representing a recirculating pool. Naive T lymphocytes are activated for the first time in the lymph node by antigens presented to their TCRs as short peptides bound to MHC molecules on the surface of DCs (Fig. 3.5). Provision of signals 1–3 (see p. 55) sets off an intracellular cascade of signalling molecule activation, leading to induction of gene transcription in T lymphocytes.
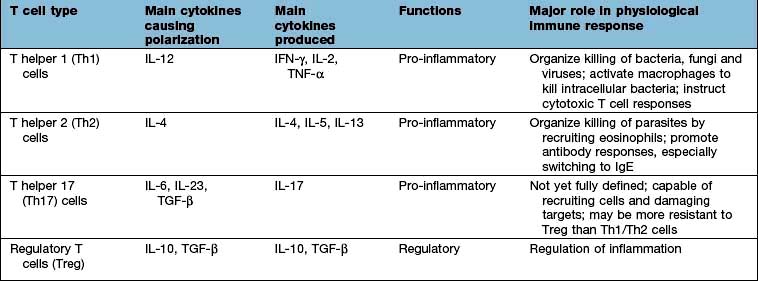
Nuclear factor kappa B (NF-κB) is a pivotal transcription factor in chronic inflammatory diseases and malignancy. It is found in the cytoplasm bound to an inhibitor IκB which prevents it from entering the nucleus (see Fig. 2.9). It is released from IκB on stimulation of the cell and passes into the nucleus, where it binds to promoter regions of target genes involved in inflammation. It is stimulated by, for example, cytokines, protein C activators and viruses. The outcome is T lymphocyte activation, cell division and functional polarization, which is the acquired ability to promote a selected type of adaptive immune response. These processes take several days to achieve. The best described polarities of T cell responses (Table 3.13) are:
CD4+ pro-inflammatory T lymphocytes; T helper 1 (Th1), Th2 and Th17
Through cell division, a proportion of the T lymphocytes that are activated in response to a pathogen undertake these effector or regulatory functions, whilst a proportion is assigned to a memory pool. Once established, effector and memory T lymphocytes have lesser requirements for subsequent activation, which can be mediated by monocytes, macrophages and B lymphocytes.
FURTHER READING
Bousso P. T cell activation by dendritic cells in the lymph node: lessons from the movies. Nat Rev Immunol 2008; 8:675–684.
Breart B, Bousso P. Cellular orchestration of T cell priming in lymph nodes. Curr Opin Immunol 2006; 18:483–490.
Corthay A. A three-cell model for activation of native T helper cells. Scand J Immunol 2006; 64:93–96.
CD4 T lymphocyte functions
As the pivotal cell in immune responses, the CD4 T lymphocyte influences most aspects of immunity, either through the release of cytokines, or via direct cell–cell interaction, a process often termed ‘licensing’. Licensing critically involves the pairing of CD40 on the APC and CD40 ligand (CD154) on the CD4 T lymphocyte; defects in this process result in antibody deficiency (see below).
Major functions of CD4 T lymphocytes are:
Licensing of DCs during antigen presentation to activate CD8 T lymphocytes and generate cytotoxic cells
Licensing of B lymphocytes to initiate and mature antibody responses, leading to class switching, affinity maturation of antibodies and generation of plasma cells or memory cells
Secretion of cytokines responsible for growth and differentiation of a range of cell types, especially other T lymphocytes, macrophages and eosinophils
T helper 17 cells
The Th17 subset of T lymphocytes derives its name from the secretion of IL-17. The role of Th17 cells in the immune response is not yet entirely clear. They are capable of secreting numerous pro-inflammatory cytokines (e.g. IL-6, IL-8) and growth factors (GM-CSF). Studies in mice suggest that these cells are critically involved in the protection from pathogens and inflammation associated with the murine versions of diseases such as multiple sclerosis, rheumatoid arthritis and ankylosing spondylitis; it is unknown whether this is also true in man but they do appear to have some role in Crohn’s disease. In psoriasis, the full blown disease has been shown to be due to an interaction of Th17 and IL-22.
Regulatory T lymphocytes
The generation of B and T lymphocytes provides a potentially vast array of rearranged antigen receptors. Although there are selection processes to remove lymphocytes with ‘dangerous’ avidity for ‘self’ these are not foolproof and the potential for autoreactivity remains. The fact that there is no self-destruction in the vast majority of people implies a state of immunological self tolerance; the controlled inability to respond to self. Several mechanisms operate to maintain this state, including CD4 T lymphocytes that respond to antigenic stimulation by suppressing ongoing immune responses. These regulatory T lymphocytes (Tregs) have different origins, phenotypes and modes of action:
CD4+ CD25hi Tregs; express high levels of CD25, the receptor for IL-2; low levels of the IL-7 receptor (CD127); regulate other T lymphocytes by cell-cell contact and also secrete the immune suppressive cytokines IL-10 and TGF-β; can be generated in the thymus or post-thymically in the periphery; have high expression of the transcription factor Foxp3.
Tr1 Tregs; generated naturally or induced (e.g. by repeated antigen injection); regulate through production of IL-10.
Evidence that Tregs are clinically relevant is given by the example of immune deficiency states in which they are defective. For example, genetic defects in the Foxp3 gene give rise to IPEX (Immune dysregulation, Polyendocrinopathy, Enteropathy, X-linked syndrome). Patients with this rare syndrome have defective Tregs and develop a range of conditions soon after birth, including organ-specific autoimmune disease such as type 1 diabetes. Much research effort is directed at harnessing this natural regulatory potential, for example to control organ graft rejection and autoimmune disease.
CD8 T lymphocyte functions
Cytotoxic CD8 T lymphocytes (CTL) are involved in defence against viruses. CTLs kill virus-infected cells following recognition of viral peptide-HLA class I complexes. CTLs must be activated first in the lymph node by a DC cross-presenting the same viral peptide and licensed by a CD4 T lymphocyte recognizing viral peptide-HLA class II complexes. The same defence mechanism may also apply in tumour surveillance. This is a checkpoint that ensures that CTL responses, which have great destructive power, are only activated against a target for which there is also a CD4 T cell response. CTLs kill via three mechanisms:
cytotoxic granule proteins (cytolysins such as perforin, granzyme B)
toxic cytokines (e.g. IFN-γ, TNF-α)
death-inducing surface molecules (e.g. Fas ligand binds Fas on target cells mediating apoptosis via caspase activation) (see p. 33).
Natural killer (NK) cells
These are bone marrow-derived, present in the blood and lymph nodes and represent 5–10% of lymphoid cells. The name is derived from two features. Unlike B and T lymphocytes, NK cells are able to mediate their effector function spontaneously (i.e. killing of target cells through release of perforin, a pore-forming protein) in the absence of previous known sensitization to that target. Also, unlike B and T lymphocytes, NK cells achieve this with a very limited repertoire of germline-encoded receptors that do not undergo somatic recombination. Despite their close resemblance to T and B lymphocytes in morphology, the lack of requirement for sensitization and the absence of gene rearrangement to derive receptors for target cells mean that NK cells are also categorized as a part of the innate immune system. For identification purposes, the main surface molecules associated with NK cells are CD16 (see below) and CD56 (note, NK cells are CD3- and TCR-negative).
The role of NK cells is to kill ‘abnormal’ host cells, typically cells that are virus-infected, or tumour cells. Killing is achieved in similar ways to CTLs. NK cells also secrete copious amounts of IFN-γ and TNF-α, through which they can mediate cytotoxic effects and activate other components of the innate and adaptive immune system. To become activated, NK cells integrate the signal from a potential target cell through a series of receptor-ligand pairings (Table 3.14). These pairings provide activating and inhibitory signals, and it is the overall balance of these that determines the outcome for the NK cell. The balance can be abnormal on a virus-infected or tumour cell, which might have altered expression of HLA molecules, for example, that mark them out for NK cytotoxicity.
Table 3.14 Examples of natural killer (NK) cell receptors
| Receptors on NK cells | Ligand |
|---|---|
Inhibitory: |
|
KIR2DL1 |
HLA-C molecules |
KIR3DL1 |
HLA-B molecules |
KIRDL2 |
HLA-A molecules |
NKG2A |
HLA-E molecules |
Activating: |
|
CD16 (low-affinity receptor for IgG) |
IgG |
NKG2D |
MHC class I chain related gene A (MICA) |
KIR2DS1 |
HLA-C |
KIR, killer cell immunoglobulin-like receptor.
In addition, through CD16, which is the low-affinity receptor for IgG (FcgRIIIA), NK cells can kill IgG-coated target cells in a process termed antibody-dependent cellular cytotoxicity (ADCC).
One of the targets of NK cell activating receptors, MICA (non-classical HLA class Ic molecule), is induced under conditions of cellular ‘stress’, such as might arise during infection or neoplastic transformation. Many viruses have developed immunoevasion strategies that avoid presentation of viral proteins to CTLs by interfering with the MHC class I presentation pathway, or circumvent target cell recognition by reducing MICA expression. NK cells detect the reduced levels of MHC class I molecules, which renders infected cells susceptible to killing by NK cells. Likewise, tumours that escape immune surveillance by CTLs through the outgrowth of daughter cells that have low MHC expression also become NK cell targets. That NK cells are vital to antiviral immunity, is indicated by the associations between KIR and susceptibility to chronic infection with viruses, notably the human immunodeficiency virus (see p. 173).
Cell migration
Immune cells are mobile. They can migrate into the lymph node to participate in an evolving immune response (e.g. a pathogen-loaded DC from the skin, or a recirculating naive T lymphocyte), or can migrate from the lymph node, via the blood, to the site of a specific infection in the tissues. Such migration takes place along blood and lymphatic vessels, and is a highly regulated process.
Taking the example of a DC migrating into the lymph node from the tissues via the lymphatics (Fig. 3.12), this is highly dependent upon expression of the chemokine receptor CCR7 by the migrating cell and of its ligand (secondary lymphoid tissue chemokine, SLC or CCL21) by the target tissue. Likewise, circulating naive lymphocytes are CCR7+ and migrate with ease into the lymph nodes from the blood or via tissue recirculation. In addition, naive lymphocytes express L-selectin which binds a glycoprotein cell adhesion molecule (GlyCAM-1) found on the high endothelial venules of lymph nodes. This system can be upregulated in an inflamed lymph node, leading to an influx of naive lymphocytes and the typical symptom of a swollen node.
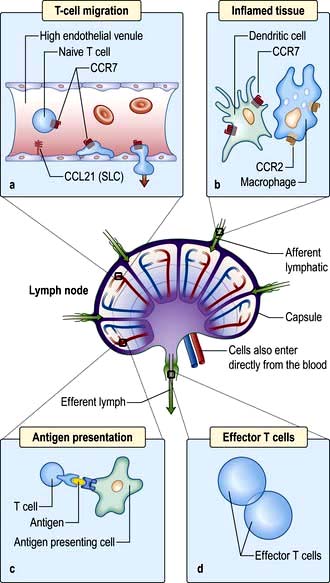
Figure 3.12 Migration of an immune cell through a lymph node. (a) T lymphocytes migrate through endothelial venules, which express the chemokine CCL21 (secondary lymphoid tissue chemokine or SLC). (b) Dendritic cells (expressing CCR7), macrophages (expressing CCR2) enter lymph nodes via afferent lymphatics and localize in the paracortex. (c) Antigen presentation (see also Figs 3.8 and 3.9) results in activation of T lymphocytes. (d) Effector T lymphocytes leave lymph node via efferent lymphatic.
Migration into inflamed tissue requires:
(a) that an affected organ or tissue signals that there is a focus of injury/infection and
(b) that responding immune cells bind and adhere specifically to that tissue.
This process is highly organized and has a similar basis for all immune cells, involving three basic steps: rolling, adhesion and trans-migration. Each of these is dependent on specialized adhesion molecules, as shown in Figure 3.13.
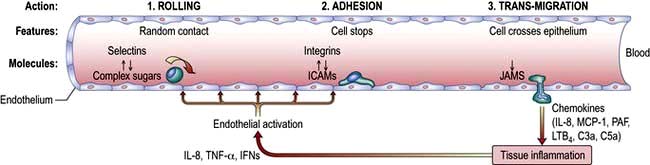
Figure 3.13 Neutrophil migration. Neutrophils arrive at the site of the inflammation, attracted by various chemoattractants. (1) They roll along the blood vessel wall through interaction between L-selectin, on their surface, binding to a carbohydrate structure (e.g. sialyl-LewisX) on the endothelium. E-selectin on the endothelium mediates a similar effect. P-, E- and L-selectin (Platelet, Endothelium and Leucocyte, respectively) are named after the predominant cell type that expresses them and bind to carbohydrate moieties such as sialyl-LewisX (CD15s) on immune cells, inducing them to slow and roll along the vessel lumen. (2) Firm adhesion is then mediated via interaction between integrins on the neutrophil and intercellular and vascular cell adhesion molecules (ICAM-I and VCAM-1) on the endothelium. The best known integrin is the heterodimer of CD11a/CD18 (leucocyte function associated antigen-1 – LFA-1) which binds ICAM-1. (3) Trans-migration (diapedesis) then occurs: this is a complex process that involves platelet-endothelial cell adhesion molecule-1 (PECAM-1) and junctional adhesion molecules (JAMs). Following transmigration (diapedesis), mediators such as interleukin-8, macrophage-chemotactive factor and TNF-α attract and activate the neutrophil to the infected tissue where it phagocytoses and destroys the C3b-coated bacteria. PAF, platelet-activating factor; LTB4, leukotriene B4.
The controlled regulation of these molecules (e.g. LFA-1 expression) is upregulated on T lymphocytes after activation in the lymph node. ICAM-1 expression on tissue endothelium is sensitive to numerous pro-inflammatory molecules and allows immune cells to be guided from the blood into the tissues. Once there, cells move along a gradient of increasing concentration of mediators such as chemokines in the process of chemotaxis.
The immune system in concert
Acute inflammation: events and symptoms
This is the early and rapid host response to tissue injury. Taking a bacterial infection as the classic example:
Local expansion of pathogen numbers leads to direct activation of complement in the tissues, with ensuing degranulation of mast cells.
Inflammatory mediators (from mast cells and complement) change the blood flow and attract and activate granulocytes.
Concomitantly, there are the local symptoms of heat, pain, swelling and redness, and perhaps more systemic symptoms such as fever due to the effect of circulating cytokines (IL-1, IL-6, TNF-α) on the hypothalamus. Indeed, gene mutations that lead to excessive actions of IL-1 (e.g. mutation of the IL1RN gene, which encodes a natural IL-1 antagonist) give rise to rare diseases with just these symptoms, as well as bone erosion and skin rashes, that are treatable with IL-1 blockade using soluble IL-1 receptor antagonist (anakinra) or monoclonal anti-IL-1β antibody.
Systemically active mediators (especially IL-6) also initiate the production of C-reactive protein (CRP) in the liver.
Bacterial lysis follows through the actions of complement and neutrophils, leading to formation of fluid in the tissue space containing dead and dying bacteria and host granulocytes (‘pus’).
At the site of pathogen entry there is often relative tissue hypoxia. The low oxygen tension has the effect of amplifying the responses of innate immune cells and suppressing the response of adaptive immune cell. This is probably an effective means of preventing excessive immune activation, which can result in collateral damage (Fig. 3.14).
The inflammation may become organized and walled off through local fibrin deposition to protect the host.
Antigens from the pathogen travel via the lymphatics (which may become visible as red tracks in the superficial tissues – lymphangitis) in soluble form or are carried by dendritic cells to establish an adaptive immune response which, at the first host-pathogen encounter, takes approximately 7–14 days.
The adaptive immune response leads to activation of pathogen-specific T lymphocytes, B lymphocytes and production of pathogen-specific antibody, initially of the IgM class and of low-moderate affinity and subsequently of the IgG class (or IgA if the infection is mucosal) and of high affinity.
Resolution of the infection is aided by the scavenging activity of tissue macrophages.
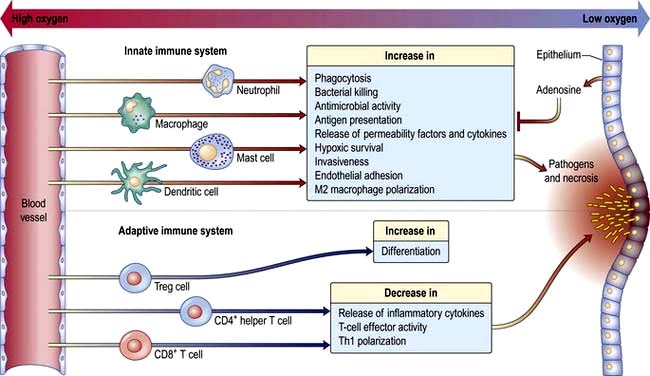
Figure 3.14 Hypoxia and inflammation. At the site of pathogen invasion and necrosis, the oxygen tension is low. Hypoxia acts upon the inflammatory process by amplifying innate immune cell responses and dampening down the adaptive response. Overall, this benefits the host by avoiding an excessive uncontrolled immune response.
Chronic inflammation: events and symptoms
Inflammation arising in response to immunological insults that cannot be resolved in days/weeks gives rise to chronic inflammation. Examples include infectious agents (chronic viral infections such as hepatitis B and C or intracellular bacteria such as mycobacteria) and environmental toxins such as asbestos and silicon. At the intracellular level, key processes of inflammasome generation and autophagy serve to enhance the chronic inflammatory process. Chronic inflammation is also a hallmark of some forms of allergic disease, autoimmune disease and organ graft rejection.
The common feature of these pathological processes is that the inciting stimulus is not easy to remove. For example, some viruses and mycobacteria remain hidden intracellularly; antigens that incite allergy (allergens) may be constantly present in the host’s environment; self or donated organs are a resident source of antigens. In many ways, the pathology that results is thus inadvertent; the immune system is caught between the repercussions of not dealing with the infection/insult, and the tissue damage that is caused by chronic activation of lymphoid and mononuclear cells.
Chronic inflammation may lead to permanent organ damage or impaired vascular function and can be fatal. If the inciting stimulus is removed, inflammation resolves. However, inflammation returns rapidly (24–48 hours) on re-exposure. This rapid recall response is the basis for patch testing to identify the cause of contact dermatitis, another form of chronic inflammation, and also for the Mantoux test of tuberculosis immunity.
The main immunological event is the presence of a proinflammatory focus comprising T and B lymphocytes and APCs, especially macrophages. If antigen persists, inflammation becomes chronic and the macrophages in the lesion fuse to form giant cells and epithelioid cells. Both Th1 and Th2 reactivity is recognized, but specific syndromes may be polarized towards one or the other (e.g. chronic mycobacterial or viral infection engenders Th1 responses, chronic allergic inflammation Th2).
When the inflammation is sufficiently chronic it may take on the appearance of organized lymphoid tissue resembling a lymph node germinal centre (e.g. in the joints in rheumatoid arthritis, p. 517). There is massive cytokine production by T lymphocytes and APCs, which contributes to local tissue damage. Granulomata, which ‘wall off’ the inciting stimulus, may also arise and result in fibrosis and calcification. Symptoms typically relate to the site of the inflammation and the type of pathology, but there may also be systemic effects such as fever and weight loss.
Crohn’s disease Chronic inflammation is a hallmark of several immune-mediated and autoimmune diseases, but it is often unclear what kick-starts or maintains the inflammatory process. Large scale studies that identify the genetic basis for these disorders (genome-wide association studies, GWAS) are beginning to provide some clues. A good example is Crohn’s disease (CD). Several of the polymorphisms associated with CD reside in genes known to be involved in inflammasome induction, such as NOD2. Cytokine regulation has also emerged as a critical disease pathway in GWAS studies on CD: most notably the IL-23R (IL-23 receptor gene), which influences Th17 cell differentiation. Clues like these point to patients with CD having impaired ability to control or terminate inflammasome activity, as well as a predisposition to make polarized pro-inflammatory cytokine responses. This information can now be exploited to devise novel therapeutic approaches.
Laboratory investigations of the immune system
In the clinical immunology laboratory, proteins and cells can be measured to ascertain the status of the immune system. The results may indicate an undiagnosed inflammatory or infectious disease (e.g. through high C-reactive protein level); a state of immune deficiency (e.g. low concentration of IgG); or a state of immune pathology (e.g. the presence of autoantibodies or allergen-specific IgE).
Examples of the commoner tests and their interpretation are shown in Table 3.15.
Table 3.15 Examining the immune system in the clinical immunology laboratory
| Measurement | Interpretation | |
|---|---|---|
Proteins |
C-reactive protein |
Raised levels indicate infection or inflammation |
Immunoglobulins |
Low levels indicate antibody deficiency, usually a result of underlying disease or primary immunodeficiency. High levels, e.g. ↑ IgM seen in acute viral infection (hepatitis A) |
|
Complement C3 and C4 |
Low levels indicate consumption of complement in immune complex disease |
|
IgE |
Raised levels in allergy; allergen-specific IgE useful to pinpoint the inciting stimulus (e.g. pollen, grass) |
|
Cells |
Neutrophils |
High levels in bacterial infection; low levels in secondary immune deficiency |
Eosinophils |
High levels in allergic or parasitic disease |
|
CD4 T lymphocytes |
Low levels in HIV infection |
|
Function |
Neutrophil respiratory burst |
Absent in the immune deficiency chronic granulomatous disease |
T lymphocyte proliferation |
Abnormally low in primary T cell immune deficiency disease |
|
Autoantibodies (see also Table 11.3 and Box 11.16) |
Rheumatoid factor, anti-cyclic citrullinated peptide antibodies (ACPA) |
Rheumatoid arthritis |
Double-stranded DNA autoantibodies |
Systemic lupus erythematosus |
|
Acetylcholine receptor antibodies |
Myasthenia gravis |
|
Anti-neutrophil cytoplasmic antibodies (ANCA) |
Vasculitis |
|
Mitochondrial |
Primary biliary cirrhosis |
Clinical immunodeficiency
Secondary (acquired) versus primary immunodeficiency
Most forms of immunodeficiency are secondary to infection (mainly HIV) or therapy (e.g. corticosteroids, anti-TNF-α monoclonal antibody therapy, cytotoxic anticancer drugs, bone marrow ablation pre-transplant). Examples of other secondary immunodeficiencies are:
Acquired neutropenias, which are common (e.g. due to myelosuppression by disease or drugs or the increased rate of destruction in hypersplenism or autoimmune neutropenia) and carry a high risk of infection once the neutrophil count falls below 0.5 × 109/L.
Acquired reductions in levels of immunoglobulins (hypogammaglobulinaemia), which are seen in patients with myeloma and chronic lymphocytic leukaemia or lymphoma.
Impairment of defence against capsulated bacteria, especially pneumococcus, following splenectomy; such patients should receive pneumococcal, meningococcal and Hib vaccinations as a matter of course (see p. 406).
Primary immunodeficiency is rare and arises at birth as the congenital effect of a developmental defect or as a result of genetic abnormalities (Table 3.16). Gene defects may not become manifest until later in infancy or childhood, and some forms of immunodeficiency typically present in adolescence or adulthood.
Table 3.16 Classification of immunodeficiencies and the main diseases in each category: apart from AIDS, all diseases shown here are primary immunodeficiencies
| Immune component | Examples of diseases |
|---|---|
T lymphocyte deficiency |
DiGeorge’s syndrome |
Acquired immunodeficiency syndrome/HIV infection |
|
T cell activation defects (e.g. CD3γ chain mutation) |
|
X-linked hyper-IgM syndrome (XHIM; CD40L deficiency) |
|
B lymphocyte deficiency |
X-linked agammaglobulinaemia (XLA) |
Common variable immunodeficiency (CVID) |
|
Selective IgA deficiency (IgAD) |
|
Combined T and B cell defects |
Severe combined immunodeficiency (SCID) (e.g. due to defects in common γ chain receptor for IL-2, -4, -7, -9, -15) |
T cell–APC interactions |
IFN-γ receptor deficiency |
IL-12 and IL-12 receptor deficiency |
|
Neutrophil defects |
Chronic granulomatous disease (CGD) |
Leucocyte adhesion deficiency (LAD) |
|
Deficiency of complement components |
Classical pathway |
Alternative pathway |
|
Common pathway |
|
Regulatory proteins |
|
Mannan binding lectin |
Clinical features of immunodeficiency
The infections associated with immunodeficiency have several typical features:
They are often chronic, severe or recurrent
They resolve only partially with antibiotic therapy or return soon after cessation of therapy
The organisms involved are often unusual (‘opportunistic’ or ‘atypical’).
The pattern of infection, in terms of the type of organism involved, is indicative (Table 3.17):
‘Opportunistic’ organisms are of low virulence but become invasive in immunodeficient states, e.g. atypical mycobacteria, Pneumocystis jiroveci, Staphylococcus epidermis.
Phagocyte defects cause, e.g. deep skin infections, abscesses, osteomyelitis.
Defective antibody producers experience infections with pyogenic (‘pus-forming’) bacteria.
T lymphocyte deficiency causes infection with fungi, protozoa and intracellular microorganisms.
Congenital deficiencies of antibody production are not revealed for several months after birth, due to the 28-day half-life of maternal IgG.
Table 3.17 Immune defects and associated infections
The family history may reveal unexplained sibling death; only females of the family affected; or consanguinity, each of which make a primary genetic syndrome more likely. Graft-versus-host disease (GVHD) may arise as a complication of primary or secondary T lymphocyte immunodeficiency. For GVHD to arise, there must be impaired T lymphocyte function in the recipient and the transfer of immunocompetent T lymphocytes from an HLA non-identical donor (see below). GVHD usually arises from therapeutic interventions such as transfusions or transplantation.
Primary immunodeficiency
T lymphocyte deficiency
DiGeorge’s syndrome
This is due to T lymphocyte deficiency. The third and fourth pharyngeal arches, which normally give rise to the parathyroid glands, aortic arch and the thymus, fail to develop. DiGeorge arises in 1–5 per 100 000 of the population and presents at birth with dysmorphic facies, hypoparathyroidism and cardiac defects, followed by infections (fungi, protozoa) in later months. The lack of a location for T lymphocyte development, i.e. the thymus, means that affected children have reduced/absent T lymphocyte number and proliferation responses. Apart from calcium supplementation, correction of cardiac abnormalities and prophylactic antibiotics, cure has been reported with thymic transplantation using fetal tissue or stem cell transplant (SCT) from HLA identical siblings.
Other T lymphocyte deficiencies
Other T lymphocyte deficiencies caused by single gene defects have been characterized and typically present from the age of 3 months with candidal infections of the mouth and skin, protracted diarrhoea, fever and failure to thrive. Examples include deficiency of CD3 itself; defects in signal transduction pathways; and deficiency of IL-2. These disorders are similar in presentation and management to the combined (T and B lymphocyte) immune deficiencies.
T and B lymphocyte deficiency
Severe combined immunodeficiencies (SCID)
These are a heterogeneous group of rare (1–2 live births per 100 000), genetically determined disorders resulting from impaired T, NK and B lymphocyte immunity. The most common form of SCID is an X-linked defect in the IL-2 receptor γ chain, interfering with the function of not just IL-2 but also IL-4, IL-7, IL-9 and IL-15 which share this component in their receptors. Clinical features are similar to pure T lymphocyte deficiency and in the blood T and NK cells are lacking whilst B lymphocyte numbers may be normal. The most successful treatment for all forms of SCID is SCT, preferably from HLA-identical donors such as siblings.
Hyper IgM syndrome
This is a milder mixed deficiency, which may not be diagnosed until later in life. It results from an X-linked defect in the CD40 ligand (CD40L (ligand); CD154) gene. Signalling between CD40 on B lymphocytes and CD40L on T lymphocytes is necessary for the generation of class switched B lymphocytes bearing high-affinity immunoglobulin, as well as for the maturation of the T cell response. In CD40L deficiency, B lymphocytes are trapped at the immature level producing only IgM. Levels of other types of immunoglobulin IgG and IgA are absent/low and there are mild defects in T cell function.
Opportunistic infections occur, for example Pneumocystis, Cryptosporidium, herpes viral infections, Candida and Cryptococcus.
Wiskott–Aldrich syndrome
This is an X-linked defect (at Xp11–23) in a gene involved in signal transduction and cytoskeletal function with associated eczema and thrombocytopenia; a mainly cell-mediated defect with falling immunoglobulins is seen, and autoimmune manifestations and lymphoreticular malignancy may develop.
Ataxia telangiectasia
These patients have defective DNA repair mechanisms and have cell-mediated defects with low IgA and IgG2; lymphoid malignancy is again common.
Epstein–Barr Virus (EBV) associated immunodeficiency (Duncan’s syndrome)
Apparently normal, but genetically predisposed (usually X-linked) individuals develop overwhelming EBV infection, polyclonal EBV-driven lymphoproliferation, combined immunodeficiency, aplastic anaemia and lymphoid malignancy. EBV appears to act as a trigger for the expression of a hitherto silent immunodeficiency.
B lymphocyte deficiency
X-linked agammaglobulinaemia (XLA)
This presents soon after maternal IgG protection falls, with recurrent infections of the upper and lower respiratory tract with pyogenic organisms, enteritis and malabsorption. Morbidity and mortality are high, mainly due to chronic lung disease and CNS infections with enteroviruses. IgG levels are usually much less than 2.0 g/L (a 1–2-year-old child will usually have an IgG level of 3.5–14 g/L) and all five classes of immunoglobulins are affected, with total levels of less than 2.5 g/L (typically >4 g/L at age 1–2 years). B lymphocyte development is arrested at the pre-B stage and B lymphocytes are absent in the blood. The cause is a loss of function mutation on chromosome Xq22 affecting the gene for a tyrosine kinase involved in cell activation and maturation.
Treatment is by i.v. immunoglobulins (IVIg) to a level that controls infections. Trough IgG concentrations should remain within normal limits (i.e. >5–6 g/L). Prognosis has improved in recent years as more patients survive into adulthood, but chronic lung disease and lymphomas are life-threatening complications.
Selective IgA deficiency
Selective IgA deficiency (IgAD; serum IgA <0.05 g/L), with normal levels of IgG and IgM, is found in approximately 1/600 northern Europeans, making it the most common primary immunodeficiency, although the underlying defect is unknown. Patients typically present at any age with recurrent infections caused by pyogenic organisms and affecting mucosal sites.
Treatment mainly comprises antibiotics. Some IgAD patients produce anti-IgA antibodies of the IgG and IgE class. Infusion of exogenous IgA (e.g. during a blood transfusion) could therefore result in anaphylaxis. IgAD patients should therefore be screened for anti-IgA antibodies, and transfused if necessary with washed red cells, blood from an IgAD donor or with stored aliquots of their own blood.
Common variable immunodeficiency (CVID)
This is a heterogeneous disease affecting 1/50 000 and arising during late childhood and early adulthood, with IgG levels <0.5 g/L. Presenting with recurrent upper and lower respiratory tract infections is typical and may progress to chronic bronchiectatic lung disease, malabsorption and diarrhoea. There may be additional features of T lymphocyte deficiency. Genetic defects underlying the condition have revealed a mutation in the gene encoding ICOS-L (ligand for inducible co-stimulator) on activated T lymphocytes in some cases. IVIg and antibiotics are the treatments of choice.
Defects in antigen presenting cell function
A series of rare genetic defects have been uncovered in which APCs demonstrate an inability to mount protective responses to intracellular bacteria, particularly low virulence mycobacteria and salmonella. The axis affected is the interplay between CD4 T lymphocytes and APCs which drives Th1 responses, and therefore in turn activates mononuclear cells such as macrophages to kill and eradicate intracellular pathogens. Defective genes so far identified include those encoding a component chain of IL-12; a component chain of the IL-12 receptor; and IFN-γ receptor chains 1 or 2.
Neutrophil defects
Chronic granulomatous disease (CGD)
This is a rare (1/250 000) immunodeficiency due to a defect in neutrophil killing and characterized by deep-seated infections. The functional defect is an inability to generate antibacterial metabolites through the respiratory burst (Table 3.5). Typical onset is at toddler age. Neutrophil numbers are normal or increased. A simple respiratory burst test of neutrophil function is diagnostic.
Treatment of infections and prophylactic antibiotic and antifungal therapy are required. Immunotherapy with interferons may have a role in patients with intractable infections, and in some cases SCT is required.
Leucocyte adhesion deficiency (LAD)
This results from defects in integrins (see p. 23). Numerous underlying defects in the gene encoding one of the component chains, CD18, have been described in LAD-I. LAD-I has an autosomal recessive inheritance presenting almost immediately after birth, with delayed umbilical cord separation. Recurrent infections similar to those in chronic granulomatous disease appear during the first decade of life. Blood neutrophil levels are high but cells are absent from the sites of infection, which require aggressive antimicrobial and antifungal treatment, and SCT for cure.
Hyper IgE syndrome (or Job’s syndrome)
This is an autosomal dominant (occasionally sporadic) immune disorder with high serum IgE levels, dermatitis, boils, pneumonias with cyst formation and bone and dental abnormalities. Mutations in STAT3 have been found.
Shwachman’s syndrome
This can resemble cystic fibrosis clinically with exocrine pancreatic insufficiency and pyogenic infections. A mild neutropenia is associated with a defect of neutrophil migration.
Chédiak–Higashi syndrome
This is a rare, recessive disorder due to a mutation in the lysosomal trafficking regulator gene (LYST) on chromosome 1q42–45. There are defects in neutrophil function with defective phagolysosome fusion and large lysosome vesicles are seen in phagocytes. Patients have recurrent infections, neutropenia, anaemia and hepatomegaly. Similar abnormalities in melanocytes cause partial oculocutaneous albinism.
Complement deficiency
The consequences of deficiency of complement proteins can be predicted from their functions (Fig. 3.2).
Failures in innate response components (e.g. the alternative pathway) lead to impaired non-specific immunity with an increase in bacterial infections.
Genetically determined low levels of mannose binding lectin (MBL) are associated with a number of inflammatory and infectious diseases.
Failure of the classical pathway results in a tendency towards infection but also towards diseases in which immune complex deposition causes inflammation, such as systemic lupus erythematosus (SLE; e.g. C1 and C4 deficiency), vasculitis and glomerulonephritis.
An unexpected finding is that neisserial infections (e.g. meningitis due to N. meningitidis) are often encountered in patients with complement defects of the membrane attack complex.
Complement regulatory proteins
Deficiency of C1 inhibitor (C1 esterase) deficiency (see also p. 1211) is relatively rare. Since this enzyme is involved in regulation of several plasma enzyme systems (e.g. the kinin system) and is continuously consumed, a single parental chromosome defect resulting in 50% of normal production barely copes with the demand and fails under stress (hence an autosomal dominant effect). As a result, uncontrolled activation of complement and the kinins may occur, leading to oedema of the deep tissues affecting the face, trunk, viscera and the airway, hence the alternative name of hereditary angio-oedema (HAE).
Type I hypersensitivity and allergic disease
Normally host defence can cope with potentially harmful cells and molecules. Under some circumstances, a harmless molecule can initiate an immune response that can lead to tissue damage and death. Such exaggerated, inappropriate responses are termed hypersensitivity reactions or allergic disease.
In Type I (immediate) hypersensitivity, the binding of an antigen to specific IgE bound to its high-affinity receptor on a mast cell surface results in massive and rapid cell degranulation and the inflammatory response outlined on page 53. The antigens involved are typically inert molecules present in the environment (termed allergens; see p. 824).
The immediate effects of allergen exposure are often very florid (early phase response). Allergic disorders also have a second phase, occurring a few hours after exposure and lasting up to several days. These ‘late phase responses’ (LPR) are mediated by Th2 cells recognizing peptide epitopes of the allergen. Recruitment of eosinophils is often a prominent feature.
From a pathological and therapeutic viewpoint, the LPR gives rise to chronic inflammation which is difficult to control. In asthma the LPR gives rise to the prolonged wheezing that can be fatal. Immediate hypersensitivity is usually responsive to antihistamines but the LPR is not, requiring powerful immune modulators such as corticosteroids.
In immunopathological terms, in the LPR:
Neutrophils and eosinophils are prominent in the first 6–18 hours and may persist for 2–3 days.
Th2 cells accumulate around small blood vessels and persist for 1–2 days.
Mediators responsible for the cellular infiltrate include platelet-activating factor and leukotrienes (Table 3.18).
Th2 cytokines IL-4 and IL-5 and chemokines such as eotaxin act as growth and activation stimuli for eosinophils, which are capable of extensive tissue damage. Th2 cytokines are also responsible for the class switch of Ig production towards IgE, maintaining the cycle of immediate and late responses.
Table 3.18 Mediators involved in the allergic response
|
|
What makes an allergen so powerful? Several allergens are proteolytic enzymes allowing them to cross skin and mucosal barriers. They are often contained within small, aerodynamic particles (e.g. pollen grains) that gain access to nasal and bronchial mucosa.
Why do some people react and others not? The tendency to develop allergic responses (known as atopy) shows strong heritability. Between 20% and 30% of the UK population is atopic and two, one or no atopic parents pass on the atopic trait to their children with a risk of 75%, 50% and 15%, respectively. In developing nations the tendency to allergy is estimated at one-tenth of the rate in industrialized countries. Amongst the predisposing genes are those encoding the β chain of the high-affinity receptor for IgE and IL-4, both strongly associated with Th2 pathways. The presence of Th2 cells recognizing allergens is the pathological hallmark of allergy.
What environmental factors are involved? Early exposure to allergens (even in utero) may be a factor in developing atopy. Over-zealous attention to cleanliness (the hygiene hypothesis) in developed societies (use of antibiotics, reduced exposure to pathogens which might favour a Th1-like environment) may favour a reduction in Treg activity. This environmental factor is shown by the rapid increase of allergy in the Eastern part of Germany following reunification.
In clinical terms, approximately two-thirds of atopic individuals (who can be identified as those with circulating allergen-specific IgE) have clinical allergic disease (equating to 15–20% of the UK population). Allergy accounts for up to one-third of school absences because of chronic illness. Other allergic disorders include allergic rhinitis (hay fever), allergic eczema, bee and wasp venom allergy and some forms of food allergy, urticaria and angio-oedema.
Diagnosis
Diagnosis of allergic disease is usually made on the history and backed up by skin-prick testing (insertion of a tiny quantity of allergen under the skin and measurement of the size of the weal) and/or measurement of allergen-specific IgE. Mast cell tryptase serum levels peak 1–2 hours after an event, remaining high for 24 hours.
Treatment
Avoidance is the first-line of therapy.
Antihistamines are effective for many immediate hypersensitivity reactions (but have no role in the treatment of asthma). A monoclonal antibody that mops up serum IgE (omalizumab) is also used (see p. 831).
Corticosteroids have several well-identified modifying actions in the allergic process: production of prostaglandin and leukotriene mediators is suppressed, inflammatory cell recruitment and migration is inhibited and vasoconstriction leads to reduced cell and fluid leakage from the vasculature.
Cysteinyl leukotriene receptor antagonists (LTRAs) inhibit leukotrienes (LTs) by blocking the type I receptor (e.g. montelukast, used in asthma, particularly aspirin induced).
Omalizumab is a monoclonal antibody that binds IgE. It is used in severe asthma that cannot be controlled with a corticosteroid plus a long-acting β2 agonist. Treatment must be initiated in a specialist centre with experience of treating severe persistent asthma.
Desensitization (allergen immunotherapy) can be used. The principle is that allergy can be prevented by inoculation by giving the allergen in a controlled way. It is indicated for disorders in which the hypersensitivity is IgE mediated, e.g. life-threatening allergy to insect stings, drug allergy and allergic rhinitis. An induction course of subcutaneous injections of increasing doses of the allergen extract, given once every 1–2 weeks, is followed by maintenance injections monthly for 2–3 years. A systematic review of 51 published randomized placebo-controlled clinical trials, enrolling a total of nearly 3000 participants, showed a low risk of adverse events with consistent clinical benefit. From an immunological viewpoint, desensitization seems capable of modifying the allergic response at several levels (Table 3.19). Sublingual allergen immunotherapy (using grass pollen extract tablets of Phl p 5 from Timothy grass; see p. 810) is used in hay fever that has not responded to anti-allergic drugs (the first dose is given under medical supervision).
Table 3.19 Mechanism of action of desensitization for allergy
| Probable mechanism | Effect |
|---|---|
IgG blocking antibodies |
During repeated exposure to desensitizing allergen, IgG class antibodies develop (especially IgG4 subclass); these compete with the pathogenic IgE for allergen binding, and/or prevent IgE-allergen complexes binding to mast cell high-affinity IgE receptors |
Regulation |
Exposure to repeated desensitizing allergen induces Treg cells which recognize allergen but invoke regulatory immune responses, dampening down migration, infiltration and inflammation |
Immune deviation |
A shift away from Th2- to Th1-producing CD4 cells results in the generation of cytokines (e.g. IFN-γ) which are inhibitory to IgE production |
Anaphylaxis
Anaphylaxis is a serious allergic reaction that is rapid in onset and may cause death. It arises as an acute, generalized IgE-mediated immune reaction involving specific antigen, mast cells and basophils. The reaction requires priming by the allergen, followed by re-exposure. To provoke anaphylaxis, the allergen must be systemically absorbed, either after ingestion or parenteral injection. A range of allergens that provoke anaphylaxis has been identified (Table 3.20).
Table 3.20 Sources of allergens known to provoke anaphylaxis
Foods |
Nuts: peanuts (protein-arachis hypogaea Ara h 1–3), Brazil, cashew |
Shellfish: shrimp (allergen Met e 1), lobster |
|
Dairy products |
|
Egg |
|
More rarely: citrus fruits, mango, strawberry, tomato |
|
Venoms |
Wasps, bees, yellow-jackets, hornets |
Medications |
Antisera (tetanus, diphtheria), dextran, latex, some antibiotics |
Anaphylaxis is rare, and the symptom/sign constellation ranges from widespread urticaria to cardiovascular collapse, laryngeal oedema, airway obstruction and respiratory arrest leading to death:
Fatal reactions to penicillin occur once every 7.5 million injections.
Between 1 in 250 and 1 in 125 individuals have severe reactions to bee and wasp stings, and a death takes place every 6.5 million stings, with 60–80 deaths per year in North America, and 5–10 in the UK.
Central to the pathogenesis of anaphylaxis is the activation of mast cells and basophils, with systemic release of some mediators and generation of others. The initial symptoms may appear innocuous: tingling, warmth and itchiness. The ensuing effects on the vasculature give vasodilatation and oedema. The consequence of these may be no more than a generalized flush, with urticaria and angio-oedema. More serious sequelae are hypotension, bronchospasm, laryngeal oedema and cardiac arrhythmia or infarction. Death may occur within minutes.
Serum platelet-activating factor (PAF) levels correlate directly with the severity of anaphylaxis whereas PAF acetylhydrolase (the enzyme that inactivates PAF) correlated inversely and was significantly lower in peanut sensitive patients with fatal anaphylactic reactions.
Treatment
Early recognition and treatment are essential (Emergency Box 3.1).
 Emergency Box 3.1
Emergency Box 3.1
Treatment of acute anaphylaxis
Management
ABCDE (Airway Breathing Circulation Disability Exposure)
Position the patient lying flat with feet raised
Administer 0.5 mg intramuscular adrenaline (epinephrine) and repeat every 5 min if shock persists.
Administer intravenous antihistamine (e.g. 10–20 mg chlorphenamine) slowly
If hypotension persists, give 1–2 L of intravenous fluid.
The best treatment is prevention. Avoidance of triggering foods, particularly nuts and shellfish, may require almost obsessive self-discipline. Patient education is necessary and many are instructed in the self-administration of adrenaline (epinephrine) and carry pre-loaded syringes. Desensitization has a well-established place in the management of this disorder, particularly if exposure is unavoidable or unpredictable, as in insect stings.
Autoimmune disease
Autoimmunity is when the immune response turns against self, i.e. recognizes ‘self’ antigens. The vast array of possible TCRs and antibodies that can be generated by the host make it highly probable that at least a small proportion can recognize self (i.e. are autoreactive). Moreover, a degree of autoreactivity is physiological – the TCR is designed to interact both with the peptide epitope in the HLA molecule binding groove, and with the HLA molecule itself.
The critical event in the development of autoimmune disease is when T and B lymphocytes bearing these receptors become activated. The following are the major checkpoints that the immune system has in place to prevent this:
1. Removal of TCRs with very strong affinity for ‘self’ in the thymus
2. The presence of naturally arising regulatory T lymphocytes (Tregs)
3. The requirement for a danger signal to license dendritic cells to activate CD4 T lymphocytes.
Autoimmune diseases affect 5% of the population at some stage of their life.
Failure of Checkpoint 1, thymic education
During thymic education, TCRs with dangerously high affinity for self are deleted. It has become apparent that this relies upon the thymic expression of self antigens. Situations which compromise the expression of a self protein would be expected to favour the development of autoimmunity. Indeed, in a rare group of patients who develop multiple autoimmune disorders affecting the adrenal and parathyroid glands (autoimmune polyglandular syndrome type 1; see p. 997), there is a defect in the AIRE gene (for autoimmune regulator), which controls thymic expression of a host of self genes. When the gene malfunctions, there is reduced expression of self proteins in the thymus and autoimmune disease is a consequence.
Failure of Checkpoint 2, regulatory T lymphocytes
An example of Treg failure is the defect in the gene encoding Foxp3, a critical transcription factor in Tregs (see p. 61) that leads to IPEX (see p. 61). IPEX is very rare, but it serves to indicate how Treg defects can lead to autoimmune disease. Laboratory studies in this area are revealing subtle Treg defects in several autoimmune diseases (e.g. type 1 diabetes, multiple sclerosis, rheumatoid arthritis).
Failure of Checkpoint 3, CD4 T lymphocyte activation against an autoantigen (or its mimic)
For an autoimmune disease to develop, there must be presentation of autoantigens to a naive, potentially autoreactive CD4 T lymphocyte by activated DCs. This could happen in one of two ways:
Tissue damage due to infection leads to both the release of hidden self antigens and the provision of sufficient danger signals to activate DCs, which in turn activate autoreactive CD4 T lymphocytes as well as the pathogen-specific ones. This is often termed ‘bystander activation’.
A pathogen mimics a self antigen. In the process of making an entirely appropriate immune response against the pathogen, T or B lymphocytes are generated that also have the capacity to recognize self. This is termed molecular mimicry.
It is unlikely that for the common autoimmune diseases (Table 3.21) there is a ‘single checkpoint’ explanation. Rather, it is likely that multiple subtle defects, at various checkpoints, are at play.
Table 3.21 Some autoimmune diseases and their autoantigens
| Disease | Antigens |
|---|---|
Addison disease |
21 α-hydroxylase |
Goodpasture’s syndrome |
Type IV collagen (located in GBM) |
Graves’ thyroiditis |
Thyroid-stimulating hormone receptor |
Hashimoto’s thyroiditis |
Thyroid peroxidase, thyroglobulin |
Multiple sclerosis |
Myelin basic protein |
Myelin oligodendrocyte glycoprotein |
|
Myasthenia gravis |
Acetylcholine receptor |
Pemphigus vulgaris |
Desmoglein-3 |
Pernicious anaemia |
H+/K+-ATPase, intrinsic factor |
Polymyositis/dermatomyositis |
tRNA synthases |
Primary biliary cirrhosis |
Pyruvate dehydrogenase complex |
Rheumatoid arthritis |
Citrullinated cyclic peptide, IgM |
Scleroderma |
Topoisomerase |
Sjögren’s syndrome |
Ro/La ribonuclear proteins |
Systemic lupus erythematosus |
Sm/RNP, Ro/La (SS-A/SS-B), histone and native DNA |
Type 1 diabetes |
Proinsulin, glutamic acid decarboxylase, IA-2, ZNT8 |
Vitiligo |
Pigment cell antigens |
Wegener’s granulomatosis (Granulomatosis with polyangiitis). |
Neutrophil proteinase 3 |
GBM, glomerular basement membrane.
Mechanisms of tissue damage in autoimmune disease
Figure 3.15 illustrates potential mechanisms of immune damage in autoimmune disease.
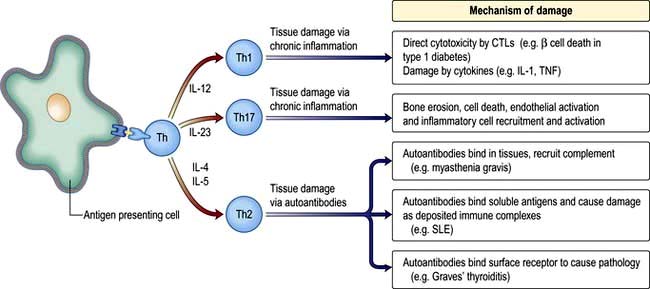
Tissue damage via chronic inflammation
In a number of autoimmune diseases (e.g. type 1 diabetes, multiple sclerosis) autoreactive T lymphocytes are likely to be the main drivers of disease and tissue damage. Th1 cells, CTLs and Th17 cells induce chronic inflammation and release cytokines that recruit other effector cells (macrophages) or are directly toxic (e.g. insulin-producing β-cells are susceptible to the combined effects of IL-1β, IFN-γ and IL-17 in type 1 diabetes).
Tissue damage via autoantibodies
Examples of damage through direct binding to a target cell or structure, with recruitment of complement and other destructive processes, include:
myasthenia gravis (autoantibodies against the acetylcholine receptor cause damage to the neuromuscular junction)
autoimmune haemolytic anaemia (autoantibody targets red blood cell autoantigens leading to lysis)
Goodpasture’s syndrome (antiglomerular basement membrane autoantibodies damage glomerular integrity).
Autoantibodies can also bind their antigen in the circulation to form immune complexes. When these are deposited in the tissues, complement and cell-mediated immune reactions are initiated. Immune complexes preferentially deposit in sites such as the kidney glomerulus, leading to chronic kidney disease. This is a feature of SLE, in which the autoantigen within the immune complexes is DNA.
Autoantibody binding may act by binding to surface receptors. In Graves’ thyroiditis, for example, the antithyroid stimulating hormone receptor antibody stimulates follicular cells to produce thyroid hormones, leading to hyperthyroidism.
Common autoimmune diseases
There are now over 80 diseases classified as autoimmune. Some of the diseases are very clear cut in their autoimmune origin, but others are not and both major and minor factors must be defined.
Major criteria
Evidence of autoreactivity (e.g. activated or memory autoreactive T lymphocytes or autoantibodies)
Clinical response to immune suppression
Passive transfer of the putative immune effector (e.g. autoreactive T lymphocyte or autoantibody) causes the disease (hardest criterion to satisfy in man but most stringent).
Minor criteria
An animal model exists that resembles the human condition, and in which there is a similar loss of immunological tolerance to self
Evidence that, in the animal model, passive transfer of the putative immune effectors reproduces the disease in a naive animal
HLA association (frequent indicator that a disease is autoimmune).
Organ rejection in clinical transplantation
The outcome of an allograft (i.e. a graft between genetically non-identical members of the same species) in the absence of adequate immunosuppressive therapy is immunological rejection. Histological analysis of rejected organs shows a range of immunological processes in action (Table 3.22). With modern tissue-matching approaches, hyperacute rejection is rare and acute rejection can usually be prevented or treated with immunosuppression. The process of chronic rejection typically takes place over several years and is the main reason for organ graft failure.
The antigens recognized in acute and chronic graft responses are donor HLA molecules (alloantigens):
In acute rejection, it is thought that the predominant response is against intact HLA molecules (direct allorecognition) and is mediated by CD4+ and CD8+ T lymphocytes.
As the rejection process becomes more chronic, peptides from donor HLA molecules are processed and presented to T lymphocytes by host HLA molecules (indirect allorecognition).
Immune-based therapies
Manipulating the immune response in a therapeutic setting has seen many successes, as evidenced by the control of organ rejection in clinical transplantation through targeted immunosuppression (Table 3.22). Monoclonal antibodies offer the opportunity to neutralize the unwanted effects of cytokines, or to direct immune responses, drugs, toxins or irradiation against a specific target, whether it be a tumour cell or an immune cell involved in a damaging autoimmune response. Natural antiviral mediators, such as the interferons, are already in the clinic as therapies for, amongst other things, chronic viral infection.
Monoclonal antibody therapy (targeted therapy)
The combined power of monoclonal antibody (MAb) and recombinant DNA technology has led to a series of ‘designer’ drugs, which have been engineered so that they are:
(b) have optimal effector function
(c) do not carry antigenic segments that may incite a neutralizing response in the host.
In general, this has meant a process of ‘humanizing’ antibodies of mouse origin and selecting an appropriate effector function. For example, a MAb designed to remove a subpopulation of lymphocytes from the patient should have good complement-fixing ability or bind well to receptors on phagocytes, whereas a MAb designed to ‘modulate’ a cell without depletion should have these functions removed. An example of the latter is an anti-CD3 MAb that has modified Fc regions and is designed to functionally downregulate, but not deplete, T lymphocytes. Many examples of MAbs in current use are described in individual chapters.
Immunosuppressive drugs
Glucocorticosteroids (cortisone, hydrocortisone, prednisone and prednisolone) are the most commonly used steroids and have a variety of effects on immune function, including:
potent effects on monocyte production of the pro-inflammatory cytokines IL-1 and TNF-α
blockade of T lymphocyte production of IL-2 and IFN-γ
reduced activation and migration of a range of innate and adaptive immune cells.
Ciclosporin, tacrolimus and rapamycin (sirolimus) have similar effects on T lymphocyte function. Ciclosporin and tacrolimus are calcineurin inhibitors and inhibit Ca2+-dependent second messenger signals in T lymphocytes following activation via TCR. By contrast, sirolimus achieves a similar effect but acts at the level of post-activation events in the nucleus.
Purine analogues such as azathioprine are also frequently used as anti-inflammatory drugs in conjunction with steroids and act by inhibiting DNA synthesis in dividing adaptive immune cells. Similar in mode of action, but more powerful, is mycophenolate mofetil (MMF).
Alkylating agents that interfere with DNA synthesis, such as cyclophosphamide, are also used for immunosuppression.
Cytokines and anticytokines
Cytokines are pleiotropic agents with powerful pro-inflammatory and immunosuppressive effects and are attractive targets for therapies that inhibit or enhance their function. TNF-α targeting agents (cytokine modulators) have been tested in rheumatoid arthritis, as has the recombinant IL-1 receptor antagonist anakinra, although with much less success. Nonetheless, the good safety profile of anakinra has prompted its use in other diseases, and a beneficial effect has been shown in type 2 diabetes, which may have an innate inflammatory component. IL-2 and its receptor (CD25) are also obvious candidates for immune therapies. Interferons are successfully used to boost pro-inflammatory immunity in chronic virus infection.
Restoring tolerance in autoimmune diseases and allergy
One of the goals for immune-based therapies for autoimmune disease is not to simply achieve immunosuppression, but also to restore immunological tolerance against the relevant autoantigens. In animal models an effective means of achieving this is to administer the autoantigen itself, or key peptide epitopes from it. This is known as antigen-specific or peptide immunotherapy and is under trial in several autoimmune diseases, where it appears to induce Tregs. The diseases that have the most advanced clinical data are in the field of allergy. For example, administration of cocktails of peptides of the cat allergen Fel d 1 has led to a reduction in detectable skin-prick responses and improved clinical scores.
Intravenous immunoglobulin
Intravenous immunoglobulin (IVIg) is a preparation of polyspecific IgG chemically purified from the plasma of large numbers (>20 000) of healthy donors. IVIg is used as a replacement therapy in patients with primary and secondary antibody deficiencies. However, when used for inflammatory conditions there is also a therapeutic benefit, although randomized placebo-controlled studies are few. In the USA, IVIg is only recommended for a small number of diseases in addition to antibody deficiency. These include: immune-mediated thrombocytopenia, Kawasaki’s syndrome, chronic inflammatory demyelinating polyneuropathy and post-transfusion purpura.