Disorders in the male
Hypogonadism
Clinical features
Male hypogonadism may be a presenting complaint or an incidental finding, such as during investigation for subfertility. The testes may be small and soft and there may be gynaecomastia. Except with subfertility, the symptoms are usually of androgen deficiency, primarily poor libido, erectile dysfunction and loss of secondary sexual hair (Table 19.20) rather than deficiency of semen production. Sperm makes up only a very small proportion of seminal fluid volume; most is prostatic fluid.
Table 19.20 Effects of androgens and consequences of androgen deficiency in the male
| Physiological effect | Consequences of deficiency |
|---|---|
General |
|
Maintenance of libido |
Loss of libido |
Deepening of voice |
High-pitched voice (if prepubertal) |
Frontotemporal balding |
No temporal recession |
Facial, axillary and limb hair |
Decreased hair |
Maintenance of erectile and ejaculatory function |
Loss of erections/ejaculation |
Pubic hair |
|
Maintenance of male pattern |
Thinning and loss of pubic hair |
Testes and scrotum |
|
Maintenance of testicular size/consistency (needs gonadotrophins as well) |
Small soft testes |
Rugosity of scrotum |
Poorly developed penis/scrotum |
Stimulation of spermatogenesis |
Subfertility |
Musculoskeletal |
|
Epiphyseal fusion |
Eunuchoidism (if prepubertal) |
Maintenance of muscle bulk and power |
Decreased muscle bulk |
Maintenance of bone mass |
Osteoporosis |
Causes of male hypogonadism are shown in Table 19.21.
Table 19.21 Causes of male hypogonadism
Reduced gonadotrophins (hypothalamic-pituitary disease) |
Hyperprolactinaemia |
Primary gonadal disease (congenital) |
Primary gonadal disease (acquired) |
Androgen receptor deficiency/abnormality |
Investigations
Testicular disease may be immediately apparent but basal levels of testosterone, LH and FSH should be measured. These will allow the distinction between primary gonadal (testicular) failure and hypothalamic-pituitary disease to be made. Depending on the causes, semen analysis, chromosomal analysis (e.g. to exclude Klinefelter’s syndrome) and bone age estimation are required.
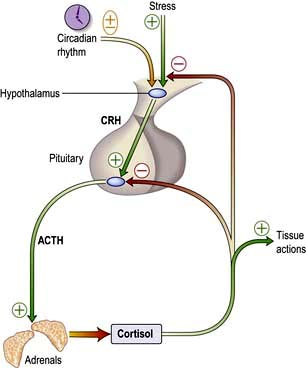
In clear-cut gonadotrophin deficiency, pituitary MRI scan, prolactin levels and other pituitary function tests are needed. However, equivocal lowering of serum testosterone (7–10 nmol/L) without elevation of gonadotrophins is a relatively common biochemical finding, and is a frequent cause of referral in men with poor libido or erectile dysfunction. Such tests are compatible with mild gonadotrophin deficiency, but may also be seen in acute illness of any cause and often simply represent the lower end of the normal range or the normal circadian rhythm of testosterone when bloods are checked in afternoon or evening surgeries. ‘Anabolic’ steroid (i.e. androgen) abuse causes similar biochemical findings, and is likely if the patient appears well virilized. People with obesity and diabetes mellitus commonly have low circulating SHBG levels associated with insulin resistance and therefore low total testosterone levels.
A therapeutic trial of testosterone replacement is often justified and forms part of the investigation in some patients; full pituitary evaluation may be required in such cases to exclude other pituitary disease.
Treatment
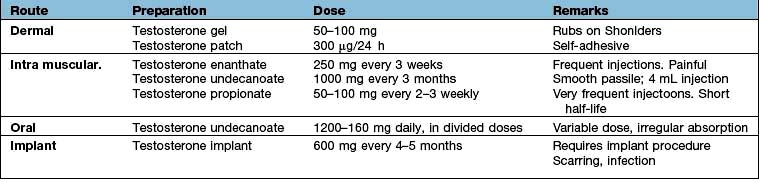
The cause of hypogonadism can rarely be reversed and testosterone replacement therapy should be commenced to control current symptoms and prevent osteoporosis in the long term. Replacement is usually given by transdermal gel or by intramuscular injection (Table 19.22). In gonadotrophin deficiency LH and FSH (purified or synthetic) or pulsatile GnRH may be used when fertility is required
Special instances of hypogonadism
Cryptorchidism
Cryptorchidism (or undescended testes) is usually treated by surgical exploration and orchidopexy in early childhood. After that age, the germinal epithelium is increasingly at risk, and lack of descent by puberty is associated with subfertility. A short trial of HCG occasionally induces descent: an HCG test with a testosterone response 72 h later excludes anorchia. Intra-abdominal testes have an increased risk of developing malignancy; if presentation is after puberty, orchidectomy is advised. Patients may present in adulthood with primary testicular failure (due to the testicular damage before or during surgery) or gonadotrophin deficiency (presumably the cause of maldescent initially).
Klinefelter’s syndrome
Klinefelter’s syndrome is a common congenital abnormality, affecting 1 in 1000 males. It is a chromosomal disorder (47XXY and variants, e.g. 46XY/47XXY mosaicism), i.e. a male with an extra X chromosome. There is both a loss of Leydig cells and seminiferous tubular dysgenesis. Patients usually present in adolescence with poor sexual development, small or undescended testes, gynaecomastia or infertility. In 47XXY there is long leg length with tall stature – the androgen deficiency leads to lack of epiphyseal closure in puberty. Patients occasionally have behavioural problems and learning difficulties. There is also a predisposition to diabetes mellitus, breast cancer, emphysema and bronchiectasis; these are all unrelated to the testosterone deficiency.
Clinical examination shows a wide spectrum of features with small pea-sized but firm testes, usually gynaecomastia and other signs of androgen deficiency. Some patients have a normal puberty and may present later with infertility. Confirmation is by chromosomal analysis. Treatment is androgen replacement therapy unless testosterone levels are normal. No treatment is possible for the abnormal seminiferous tubules and infertility.
Kallmann’s syndrome
This is isolated GnRH deficiency. It is associated with decreased or absent sense of smell (anosmia), and sometimes with other bony (cleft palate), renal and cerebral abnormalities (e.g. colour blindness). It is often familial and is usually X-linked, resulting from a mutation in the KAL1 gene which encodes anosmin-1 (producing loss of smell); one sex-linked form is due to an abnormality of a cell adhesion molecule. Management is that of secondary hypogonadism (see p. 976). Fertility is possible.
Normosmic idiopathic hypogonadotropic hypogonadism
This refers to isolated GnRH deficiency in the absence of anosmia. Known mutations account for less than 15% of normosmic idiopathic hypogonadotropic hypogonadism (nIHH). Mutations include the KISS1 gene which codes for kisspeptin, the protein which acts on GPR54 receptor, and the FGFR1 gene.
Oligospermia or azoospermia
These may be secondary to gonadotrophin deficiency and can be corrected by gonadotrophin therapy. More often they result from primary testicular diseases, in which case they are rarely treatable.
Azoospermia with normal testicular size and low FSH levels suggests a vas deferens block, which is sometimes reversible by surgical intervention.
Lack of libido and erectile dysfunction
Lack of libido is a loss of sexual desire; erectile dysfunction (ED) is inability to achieve or maintain erection; they may occur together or separately, and each can precipitate the other. Both are common symptoms in hypogonadism, but most people with either symptom have normal hormones and many have no definable organic cause. ED may be psychological, neurogenic, vascular, endocrine or related to drugs, and often includes contributions from several causes. Vascular disease is a common aetiology, especially in smokers, and is often associated with vascular problems elsewhere. Autonomic neuropathy, most commonly from diabetes mellitus, is a common contributory cause (see p. 1025) and many drugs produce ED. The endocrine causes are those of hypogonadism (see above) and can be excluded by normal testosterone, gonadotrophin and prolactin levels. The presence of nocturnal emissions and frequent satisfactory morning erections make endocrine disease unlikely.
Psychogenic erectile dysfunction is frequently a diagnosis of exclusion, though complex tests of penile vasculature and function are available in some centres.
Treatment. Offending drugs should be stopped. Phosphodiesterase type-5 inhibitors (sildenafil, tadalafil, vardenafil) which increase penile blood flow (see p. 1027) are first choice for therapy. Other treatments include apomorphine, intracavernosal injections of alprostadil, papaverine or phentolamine, vacuum expanders and penile implants.
If no organic disease is found, or if there is clear evidence of psychological problems, the couple should receive psychosexual counselling.
Gynaecomastia
Gynaecomastia is development of breast tissue in the male. Causes are shown in Table 19.23. It is due to an imbalance between free oestrogen and free androgen effects on breast tissue.
Table 19.23 Causes of gynaecomastia
Physiological |
Drugs |
Oestrogenic: |
Pubertal gynaecomastia occurs in perhaps 50% of normal boys, often asymmetrically. It usually resolves spontaneously within 6–18 months, but after this duration may require surgical removal, as fibrous tissue will have been laid down. The cause is thought to be relative oestrogen excess, and the oestrogen antagonist tamoxifen is occasionally helpful.
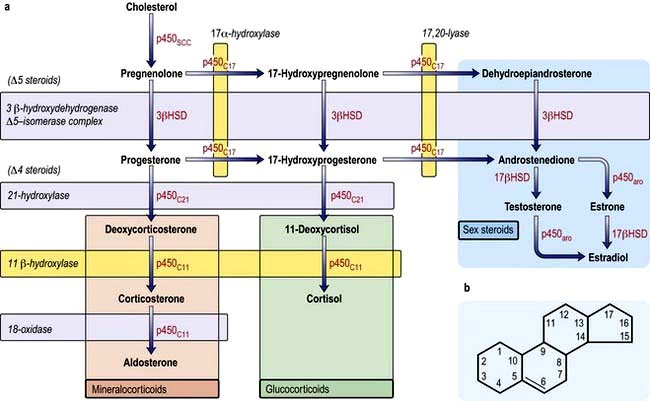
Gynaecomastia in the older male requires a full assessment to exclude potentially serious underlying disease, such as bronchial carcinoma and testicular tumours (e.g. Leydig cell tumour). However, aromatase activity (p. 972) increases with age and may be the cause of gynaecomastia in this group. Aromatase is an enzyme of the cytochrome P450 family and converts androgens to produce oestrogens. Drug effects are common (especially digoxin and spironolactone), and once these and significant liver disease are excluded most cases have no definable cause. Surgery is occasionally necessary.
 Box 19.8
Box 19.8

 Emergency Box 19.1
Emergency Box 19.1