Diffuse diseases of the lung parenchyma
Diffuse parenchymal lung disorders (DPLD, also referred to as interstitial lung diseases) are a heterogeneous group of disorders accounting for about 15% of respiratory clinical practice. There is diffuse lung injury and inflammation that can progress to lung fibrosis. The classification is shown in Box 15.17.
 Box 15.17
Box 15.17
Diffuse parenchymal lung diseases (DPLDs)
 Granulomatous lung disease, e.g. sarcoid
Granulomatous lung disease, e.g. sarcoid
Granulomatous lung disease with vasculitis, e.g. Wegener, Churg–Strauss, microscopic vasculitis
Pulmonary autoimmune rheumatic diseases, e.g. rheumatoid arthritis, systemic lupus erythematosus
Idiopathic interstitial pneumonias, see Box 15.18
Drugs, see Table 15.22
Granulomatous lung disease
A granuloma is a mass or nodule composed of chronically inflamed tissue formed by the response of the mononuclear phagocyte system (macrophage/histiocyte) to an insoluble or slowly soluble antigen or irritant. If the foreign substance is inert (e.g. an inhaled dust), the phagocytes turn over slowly; if the substance is toxic or reproducing, the cells turn over faster, producing a granuloma. A granuloma is characterized by epithelioid multinucleate giant cells, as seen in tuberculosis. Granulomas are also seen in other infections, including fungal and helminthic, in sarcoidosis, and in hypersensitivity pneumonitis, and can also be due to foreign bodies (e.g. talc). Granulomatous lung disease with pulmonary vasculitis is discussed on page 847.
Sarcoidosis
Sarcoidosis is a multisystem granulomatous disorder, commonly affecting young adults and usually presenting with bilateral hilar lymphadenopathy, pulmonary infiltration and skin or eye lesions. Beryllium poisoning can produce a clinical and histological picture identical to sarcoidosis, though contact with this element is now strictly controlled.
Epidemiology and aetiology
Sarcoidosis is a common disease of unknown aetiology that is often detected by routine chest X-ray. There is great geographical variation. The prevalence in the UK is approximately 19/100 000. Sarcoidosis is common in the USA but is uncommon in Japan. The course of the disease is much more severe in American blacks than in whites. The peak incidence is in the 3rd and 4th decades, with a female preponderance. There is no relation with any histocompatibility antigen, but 1st degree relatives (particularly in Caucasians) have an increased risk of developing sarcoidosis. Other proposed aetiological factors are an atypical mycobacterium or fungus, the Epstein–Barr virus, and occupational, genetic, social or other environmental factors (sarcoidosis is commoner in rural than in urban populations). None of these has been substantiated.
Immunopathology
Typical sarcoid granulomas consist of focal accumulations of epithelioid cells, macrophages and lymphocytes, mainly T cells.
There is depressed cell-mediated reactivity to tuberculin and other antigens such as Candida albicans.
There is overall lymphopenia: circulating T lymphocytes are low but B cells are slightly increased.
Bronchoalveolar lavage shows a great increase in the number of cells; lymphocytes are greatly increased (particularly CD4+ T-helper cells). The number of alveolar macrophages is increased but they represent a reduced percentage of the total number of bronchoalveolar lavage cells.
Transbronchial biopsies show infiltration of the alveolar walls and interstitial spaces with leucocytes, mainly T cells, prior to granuloma formation.
It seems likely that the decrease in circulating T lymphocytes and changes in delayed hypersensitivity responses are the result of sequestration of lymphocytes within the lung. There is no evidence to suggest that patients with sarcoidosis suffer from an overall defect in immunity, since the frequency of fungal, viral and bacterial infections is not increased and there is no evidence of any increased risk of developing malignant neoplasms.
Clinical features
Sarcoidosis can affect many different organs of the body. The most common presentation is with respiratory symptoms or abnormalities found on chest X-ray (50%). Less common presentations include fatigue or weight loss (5%), peripheral lymphadenopathy (5%) and fever (4%). Neurological presentations are rare but well recognized and can mimic a variety of conditions. Chest X-ray may be normal in up to 20% of non-respiratory cases, though pulmonary lesions may be detected later.
There are four stages of pulmonary involvement based on radiological stage of the disease, which is helpful in prognosis:
Stage I: bilateral hilar lymphadenopathy (BHL) alone
Stage II: BHL with pulmonary infiltrates
Bilateral hilar lymphadenopathy
This is a characteristic feature of sarcoidosis which is usually symptomless and only detected on chest X-ray. Occasionally, the bilateral hilar lymphadenopathy is associated with a dull ache in the chest, malaise and a mild fever.
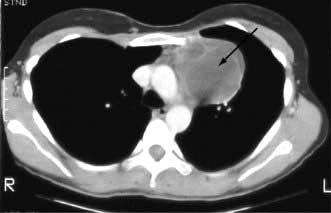
Although the lung fields may appear normal on plain chest X-ray, the lung parenchyma is nearly always involved as shown by CT scanning (Fig. 15.38), transbronchial biopsies and bronchoalveolar lavage.
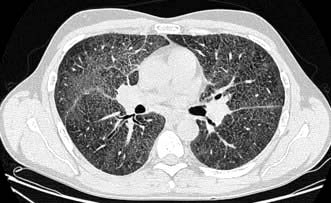
Figure 15.38 HRCT scan in sarcoidosis. Note bilateral hilar lymphadenopathy and reticular shadowing.
The differential diagnosis of bilateral hilar lymphadenopathy includes:
Pulmonary infiltration
Parenchymal sarcoidosis may be asymptomatic. The combination of pulmonary infiltration and normal lung function tests is highly suggestive of sarcoidosis. Other cases are progressive leading to increasing effort dyspnoea and eventually cor pulmonale and death. The chest X-ray shows mottling in the mid-zones evolving over time to generalized fine nodular shadows. Eventually, widespread linear shadows develop, reflecting the underlying fibrosis. A honeycomb appearance can occasionally occur. In progressive disease, lung function tests show a typical restrictive defect with reduced gas transfer (see below).
The principal differential diagnoses are tuberculosis, pneumoconiosis, idiopathic pulmonary fibrosis and alveolar cell carcinoma.
Extrapulmonary manifestations
Skin and ocular sarcoidosis are the most common extrapulmonary presentations.
Skin lesions occur in 10% of cases. Sarcoidosis is the most common cause of erythema nodosum (see p. 1216). The association of bilateral symmetrical hilar lymphadenopathy with erythema nodosum occurs only in sarcoidosis. A chilblain-like lesion known as lupus pernio is also seen, as are skin nodules (see p. 1219).
Eye lesions. Anterior uveitis is common and presents with misting of vision, pain and a red eye, but posterior uveitis may present simply as progressive loss of vision. Although ocular sarcoidosis accounts for about 5% of uveitis presenting to ophthalmologists, asymptomatic uveitis may be found in up to 25% of patients with sarcoidosis. Conjunctivitis and retinal lesions have also been reported. Uveoparotid fever is a syndrome of bilateral uveitis and parotid gland enlargement together with occasional development of facial nerve palsy and is sometimes seen with sarcoidosis.
Keratoconjunctivitis sicca and lacrimal gland enlargement also occurs.
Metabolic manifestations. It is rare for sarcoidosis to present with problems of calcium metabolism, though hypercalcaemia is found in 10% of established cases. Hypercalcaemia and hypercalciuria can lead to the development of renal calculi and nephrocalcinosis. The cause of the hypercalcaemia is an increase in circulating 1,25-dihydroxyvitamin D3, with 1 α-hydroxylation occurring in sarcoid macrophages in the lung in addition to that taking place in the kidney.
Central nervous system. CNS involvement is rare (2%) but can lead to severe neurological disease (see p. 1130).
Bone and joint involvement. Arthralgia without erythema nodosum is seen in 5% of cases. Bone cysts are found, particularly in the digits, with associated swelling. In the absence of swelling, routine X-rays of the hands are unnecessary.
Hepatosplenomegaly. Sarcoidosis is a cause of hepatosplenomegaly, though it is rarely of any clinical consequence. Liver biopsy is occasionally performed when the diagnosis is in doubt and will show granulomas.
Cardiac involvement is rare (3%) but can be serious. Ventricular dysrhythmias, conduction defects and cardiomyopathy with congestive cardiac failure are seen.
Investigations
Imaging. Chest X-ray (see above). High-resolution CT is useful for assessment of diffuse lung parenchymal involvement.
Full blood count. There may be a mild normochromic, normocytic anaemia with raised ESR.
Serum biochemistry. Serum calcium is often raised and there is hypergammaglobulinaemia.
Transbronchial biopsy is the most useful investigation, with positive results in 90% of cases of pulmonary sarcoidosis with or without X-ray evidence of lung parenchymal involvement. Pulmonary non-caseating granulomas are found in approximately 50% of patients with extrapulmonary sarcoidosis in whom the chest X-ray is normal.
Serum angiotensin-converting enzyme (ACE) level is raised by two standard deviations above the normal mean value in over 75% of patients with untreated sarcoidosis. Raised (but lower) levels are also seen in patients with lymphoma, pulmonary tuberculosis, asbestosis and silicosis, limiting the diagnostic value of the test. However, the test is useful in assessing disease activity and response to treatment. Reduction of serum ACE during corticosteroid treatment does not, however, imply complete resolution of the disease.
Lung function tests show a restrictive lung defect in patients with pulmonary infiltration or fibrosis. There is a decrease in TLC, FEV1 and FVC, and gas transfer. Lung function is usually normal in patients who present with extrapulmonary disease or who only have hilar adenopathy on chest X-ray.
Treatment
Both the need to treat and the value of corticosteroid therapy are contested in many aspects of this disease. Hilar lymphadenopathy alone does not require treatment. Persisting infiltration visible on the chest X-ray with normal lung function tests should be monitored carefully. Patients with abnormal lung function tests are unlikely to improve without corticosteroid treatment. If the disease is not improving spontaneously 6 months after diagnosis, treatment should be started with prednisolone 30 mg for 6 weeks, reducing to alternate-day treatment with prednisolone 15 mg for 6–12 months. Although there have been no controlled trials of corticosteroids, they are indicated when there is continuing deterioration of lung function. Eye involvement or persistent hypercalcaemia are mandatory indications for systemic steroids.
If the erythema nodosum of sarcoidosis is severe or persistent it will respond rapidly to a 2-week course of prednisolone 5–15 mg daily, as will patients with uveoparotid fever. Myocardial sarcoidosis and neurological manifestations are also treated with prednisolone.
Prognosis
Sarcoidosis is a much more severe disease in certain racial groups, particularly American blacks, where death rates of up to 10% have been recorded. It is probable that the disease is fatal in fewer than 5% of cases in the UK, either as a result of respiratory failure and cor pulmonale or, more rarely, from myocardial sarcoidosis and renal damage. The initial chest X-ray provides a guide to prognosis. The disease remits within 2 years in over two-thirds of patients with hilar lymphadenopathy alone (stage I), in approximately one-half with hilar lymphadenopathy plus chest X-ray evidence of pulmonary infiltration (stage II), but in only one-third of patients with X-ray evidence of infiltration without any demonstrable lymphadenopathy (stage III). Lung volumes and gas transfer are the most useful way to monitor progression.
Granulomatous lung disease with vasculitis
The classification of pulmonary vasculitis and granulomatous disorders is unsatisfactory. In broad terms there are two main groups: the respiratory manifestations of systemic diseases, and disorders associated with the presence of anti-neutrophil cytoplasmic antibodies (ANCAs).
Anti-neutrophil cytoplasmic antibodies (ANCAs) (see also p. 587) are found in the acute phase of vasculitides, particularly Wegener’s granulomatosis, Churg–Strauss syndrome and microscopic polyangiitis (polyarteritis) associated with neutrophil infiltration of the vessel wall.
Two major ANCA reactivities are recognized: proteinase-3 (PR3) ANCA and myeloperoxidase (MPO) ANCA.
Some 10–15% cases of progressive glomerulonephritis with anti-glomerular basement membrane (GBM) antibodies Goodpasture’s syndrome (see below) are also MPO-ANCA-positive and these are the most likely to suffer pulmonary haemorrhage.
Wegener’s granulomatosis (granulomatosis with polyangiitis)
This granulomatous disease (see p. 544) of unknown aetiology predominantly affects small arteries. It is characterized by lesions involving the upper respiratory tract, lungs and kidneys. It often starts with severe rhinorrhoea, with subsequent nasal mucosal ulceration followed by cough, haemoptysis and pleuritic pain. Occasionally, the skin and nervous system are involved. Single or multiple nodular masses or pneumonic infiltrates with cavitation are seen on chest X-ray. These appear to migrate, with large lesions clearing in one area and new lesions appearing elsewhere. The typical histological changes are usually best seen on renal biopsy, which shows necrotizing microvascular glomerulonephritis. This disease responds well to treatment with cyclophosphamide 150–200 mg daily. Rituximab is also being used. A variant of Wegener’s granulomatosis called ‘midline granuloma’ affects the nose and paranasal sinuses and is particularly mutilating; it has a poor prognosis.
Churg–Strauss syndrome
This condition classically occurs in males in their 4th decade, who present with rhinitis and asthma, eosinophilia and systemic vasculitis. The aetiology is uncertain, with some believing that it is an unusual progression of allergic disease while others regard it as a primary vasculitis which presents like asthma because of the involvement of eosinophils.
There is an eosinophilic infiltration with a characteristic high blood eosinophil count, vasculitis of small arteries and veins, and extravascular granulomas. Typically, it involves the lungs, peripheral nerves and skin, but renal involvement is uncommon. Transient patchy pneumonia-like shadows may occur, but sometimes these can be massive and bilateral. Skin lesions include tender subcutaneous nodules as well as petechial or purpuric lesions. ANCA is usually positive. The disease responds well to corticosteroids. Occasionally, Churg–Strauss syndrome is revealed when oral steroids are withdrawn in patients being treated for asthma. There is no evidence that anti-asthma drugs precipitate the condition.
Pulmonary autoimmune rheumatic disease
Rheumatoid disease (see also p. 521)
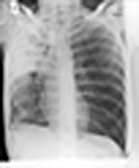
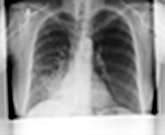
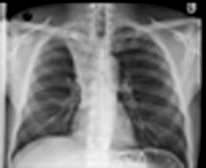
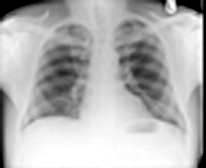
The lungs can be affected by rheumatoid disease and also by some anti-rheumatic drugs used in its treatment (Fig. 15.39).
Pleural adhesions, thickening and effusions are the most common lesions. The effusion is often unilateral and tends to be chronic. It has a low glucose content but this is not specific.
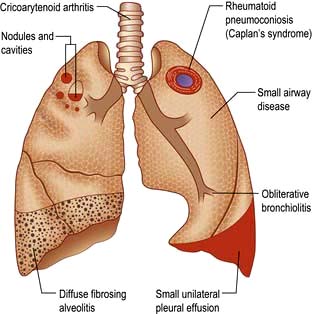
Figure 15.39 Respiratory manifestations of rheumatoid disease. Many drugs affect the lungs, see text, page 1231.
Several forms of parenchymal disease can occur in patients with rheumatoid arthritis. These include fibrosing alveolitis, rheumatoid nodules, cryptogenic organizing pneumonia, lymphoid interstitial pneumonia and bronchiectasis; other pulmonary problems include pulmonary hypertension and fibrosis (Fig. 15.39). Some patients will have modified presentations because they are already on disease-modifying drugs such as prednisolone or methotrexate for their arthritis.
Fibrosing alveolitis occurring in rheumatoid arthritis can be considered as a variant of the cryptogenic form of the disease (see p. 848). The clinical features and gross appearance are the same but the disease is often more chronic.
Rheumatoid nodules appear on the chest X-ray as single or multiple nodules ranging in size from a few millimetres to a few centimetres. The nodules frequently cavitate. They usually produce no symptoms but can give rise to a pneumothorax or pleural effusion.
Obliterative disease of the small bronchioles is rare. It is characterized by progressive breathlessness and irreversible airflow limitation. Corticosteroids may prevent progression.
Cricoarytenoid joint involvement by rheumatoid arthritis gives rise to dyspnoea, stridor, and hoarseness. Occasionally severe obstruction necessitates tracheostomy.
Caplan’s syndrome is due to a combination of dust inhalation and the disturbed immunity of rheumatoid arthritis. It occurs particularly in coal-worker’s pneumoconiosis but it can occur in individuals exposed to other dusts, such as silica and asbestos. Typically the lesions appear as rounded nodules 0.5–5.0 cm in diameter, though sometimes they become incorporated into large areas of fibrosis that are indistinguishable radiologically from progressive massive fibrosis. There may not be much evidence of simple pneumoconiosis prior to the development of the nodule. These lesions may precede the development of the arthritis. Rheumatoid factor is always present in the serum.
Drugs used in the treatment of rheumatoid arthritis can cause pulmonary problems: e.g. pneumonitis with methotrexate, gold, NSAIDs; fibrosis, with methotrexate; bronchospasm with NSAIDs, aspirin; infections due to corticosteroids, methotrexate; reactivation of tuberculosis with anti-TNF therapy.
FURTHER READING
King TE, Pards A, Selman M. Idiopathic pulmonary fibrosis. Lancet 2011; 378:1949–1961.
Systemic lupus erythematosus (see also p. 536)
The most common respiratory manifestation of this disease is pleurisy, occurring in up to two-thirds of cases, with or without an effusion. Effusions are usually small and bilateral. Basal pneumonitis is often present, perhaps as a result of poor movement of the diaphragm, or restriction of chest movements because of pleural pain. Pneumonia also occurs because of either infection or the disease process itself. In contrast to rheumatoid arthritis, diffuse pulmonary fibrosis is rare.
Systemic sclerosis (see pp.538 and 1218)
Autopsy studies have indicated that there is almost always some diffuse fibrosis of alveolar walls and obliteration of capillaries and the alveolar space. Severe changes result in nodular then streaky shadowing on the chest X-ray, followed by cystic changes, ending up with a honeycomb lung. Lung function tests reveal a restrictive defect and poor gas transfer. Dilation of the oesophagus increases the risk of aspiration pneumonia (see p. 243). Breathlessness may be worsened by restriction of chest wall movement owing to thickening and contraction of the skin and trunk.
Idiopathic interstitial pneumonia (IIP)
A current international classification is shown in Box 15.18. IIP is characterized by diffuse inflammation and fibrosis in the lung parenchyma.
Box 15.18
Classification of idiopathic interstitial pneumonias
| Clinical diagnosis | Pathological pattern |
|---|---|
Idiopathic pulmonary fibrosis (IPF) |
Usual interstitial pneumonia (UIP) |
Desquamative interstitial pneumonia (DIP) |
Desquamative interstitial pneumonia (DIP) |
Respiratory bronchiolitis interstitial lung disease (RBILD) |
Respiratory bronchiolitis interstitial lung disease (RBILD) |
Acute interstitial pneumonia (AIP) |
Diffuse alveolar damage (DAD) |
Nonspecific interstitial pneumonia (NSIP) |
Nonspecific interstitial pneumonia (NSIP) |
Cryptogenic organizing pneumonia (COP) |
Organizing pneumonia (OP) |
Lymphoid interstitial pneumonia (LIP) |
Lymphoid interstitial pneumonia (LIP) |
ATS/ERS. American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. American Journal of Respiratory and Critical Care Medicine 2002; 165:277.
Idiopathic pulmonary fibrosis (IPF)
This is also known as usual interstitial pneumonia (UIP) and was previously known as cryptogenic fibrosing alveolitis (CFA).
It is relatively rare with a prevalence of about 20/100 000 population, mean onset is in the late 60s and it is more common in males. The cause is unknown but possible contributory factors include cigarette smoking, chronic aspiration, antidepressants, wood and metal dusts and infections, e.g. Epstein–Barr virus.
Pathology
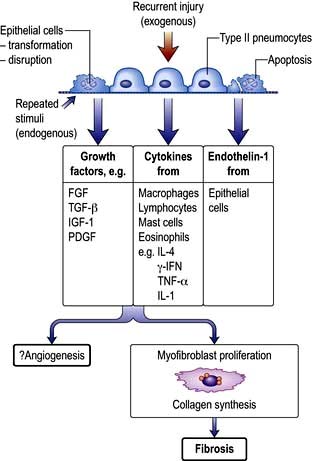
The key features are patchy fibrosis of the interstitium (often with intervening normal lung), subpleural and paraseptal changes, minimal or absent inflammation, acute fibroblastic proliferation and collagen deposition (fibroblastic foci) and honeycombing.
Pathogenesis
Several hypotheses have been proposed but it seems likely that injury results from repeated exogenous and endogenous unknown stimuli. It is now understood that inflammation plays little or no part. Multiple micro-injuries to the alveolar cells cause them to secrete growth factors that recruit fibroblasts in a fibrotic environment. These fibroblasts differentiate into myofibroblasts under the influence of TGF-β, synthesize collagen and aggregate to form ‘fibrotic foci’ (Fig. 15.40). In familial pulmonary fibrosis, several mutations have been identified, suggesting a genetic disposition. Genes over-expressed in IPF include matrix metalloproteinases (MMP1, 7), cyclin A2 (CLNA2), α-defensins, surfactant protein A1 and the gene encoding mucinSB (MUCSB). There is an absence of type 1 pneumocytes with a lack of differentiation of type 2 into type 1 pneumocytes resulting in a dysfunctional alveolar epithelium.
Clinical features
The main features are progressive breathlessness, a non-productive cough and cyanosis, which eventually lead to respiratory failure, pulmonary hypertension and cor pulmonale. Fine bilateral end-inspiratory crackles are heard on auscultation and gross finger clubbing occurs in two-thirds of cases. An acute form known as the Hamman–Rich syndrome occasionally occurs. Various autoimmune diseases are associated with IPF (see also Box 15.17). IPF has also been reported in association with coeliac disease, ulcerative colitis and renal tubular acidosis.
Investigations
Chest X-ray initially shows a ground glass appearance, followed by irregular reticulonodular shadowing (often maximal in the lower zones) and finally a honeycomb lung.
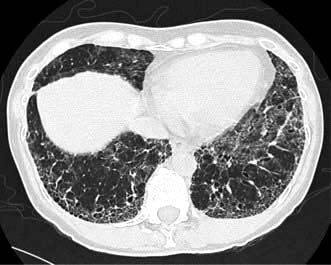
High-resolution CT scan (HRCT) shows characteristic bilateral changes mainly involving the lower lobes (Fig. 15.41). There may be subpleural reticular abnormalities with minimal or no ground glass changes, honeycombing, i.e. thick-walled cysts 0.5–2 cm in diameter in terminal and respiratory bronchioles, and traction bronchiectasis.
Respiratory function tests show a restrictive ventilatory defect – lung volumes are reduced, the FEV1 to FVC ratio is normal to high (with both values being reduced), and carbon monoxide gas transfer is reduced. Peak flow rates may be normal.
Blood gases show arterial hypoxaemia, caused by a combination of alveolar-capillary block and ventilation–perfusion mismatch with normal or low PaCO2 owing to hyperventilation.
Blood tests. Anti-nuclear antibodies and rheumatoid factors are present in one-third of patients. The ESR and immunoglobulins are mildly elevated.
Bronchoalveolar lavage shows increased numbers of cells, particularly neutrophils and macrophages.
Histological confirmation is necessary in some patients. Transbronchial lung biopsy is rarely diagnostic, but can exclude other conditions which present similarly, e.g. sarcoidosis or lymphangitis carcinomatosa. Video-assisted thoracoscopic lung biopsy is used to obtain a larger specimen, which will allow a clear histological diagnosis to be made.
Differential diagnosis
The diagnosis of IPF is usually made in a patient presenting with the above signs and characteristic HRCT changes. The differential diagnosis of the chest X-ray appearance includes hypersensitivity pneumonitis, bronchiectasis, chronic left heart failure, sarcoidosis, industrial lung disease and lymphangitis carcinomatosa.
Prognosis and treatment
The median survival time for patients with IPF is approximately 5 years, although mortality is very high in the more acute forms. Treatment with prednisolone (30 mg daily) is usually prescribed for disabling disease although it produces little benefit. Azathioprine or cyclophosphamide is added if there is no response. A number of other treatments have been tried. Pirfenidone, an anti-fibrotic anti-inflammatory drug, has reduced lung funtion deterioration in two randomized controlled trials. A tyrosine kinase inhibitor, BIBF1120, has shown the same benefit in a phase II study. Supportive treatment includes domiciliary oxygen therapy. In younger patients with severe disease, lung transplantation is offered.
Desquamative interstitial pneumonia (DIP)
This occurs mainly in middle-aged male smokers and is less severe but similar to usual interstitial pneumonia (UIP). Pathologically there are more mononuclear cells than in UIP, due to smoking (pigmented macrophages). The prognosis is good with corticosteroid therapy.
Respiratory bronchiolitis interstitial lung disease (RBILD)
Clinically, RBILD is like DIP. Pathologically, pigmented macrophages are seen in the lumen of respiratory bronchioles. It has a better prognosis than UIP.
Acute interstitial pneumonia (AIP)
There is a very acute onset of pneumonia, often preceded by an upper respiratory tract infection, and progressive respiratory failure. Diffuse alveolar damage (DAD) is seen on lung biopsy. The prognosis is poor.
Nonspecific interstitial pneumonia (NSIP)
The onset is subacute with a fever in 30%. Men and women are equally affected and finger clubbing does not occur. Biopsy reveals a chronic interstitial pneumonia with mononuclear inflammatory cells and some fibrosis. Prognosis is variable (depending on amount of fibrosis).
Cryptogenic organizing pneumonia (COP)
This condition, previously called bronchiolitis obliterans organizing pneumonia (BOOP), is an organizing pneumonia of unknown aetiology. Typically, patients present with single or recurrent episodes of malaise associated with cough, breathlessness and fever. Pleuritic chest pain is sometimes present but finger clubbing is very rare. Chest X-rays show confluent bilateral parenchymal shadowing. Lung function tests are usually normal but may show a restrictive defect. The white blood count is normal, but the ESR may be raised. The diagnosis is usually made on history and X-ray appearances. Lung biopsy reveals characteristic buds of connective tissue (Masson’s bodies) in respiratory bronchioles and in alveolar ducts. Open biopsy has now been replaced by video-assisted thorascopic lung biopsy. COP responds rapidly to corticosteroid treatment but can recur episodically, especially in older women.
Lymphoid interstitial pneumonia
This condition is more common in children than in adults and is characterized by infiltration with lymphocytes, plasma cells and immunoblasts. It is thought to be a viral pneumonia and causes diffuse reticulonodular infiltrates on the chest X-ray. Corticosteroid therapy appears to be of benefit, as is zidovudine.
Drugs
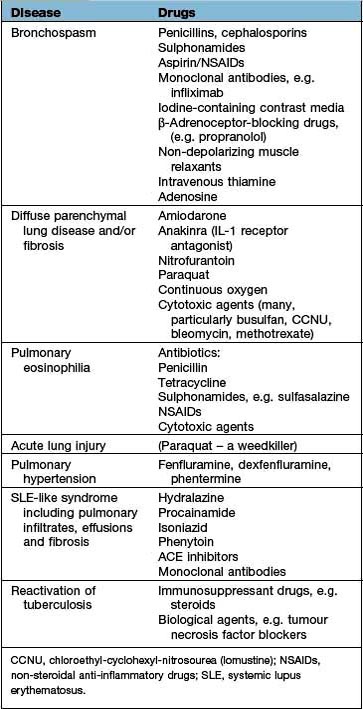
Drugs causing diffuse parenchymal lung disease are shown on page 854 and in Table 15.22.
Table 15.22 Some drug-induced respiratory reactions
| Disease | Drugs |
|---|---|
Bronchospasm |
Penicillins, cephalosporins |
Sulphonamides |
|
Aspirin/NSAIDs |
|
Monoclonal antibodies, e.g. infliximab |
|
Iodine-containing contrast media |
|
β-Adrenoceptor-blocking drugs, (e.g. propranolol) |
|
Non-depolarizing muscle relaxants |
|
Intravenous thiamine |
|
Adenosine |
|
Diffuse parenchymal lung disease and/or fibrosis |
Amiodarone |
Anakinra (IL-1 receptor antagonist) |
|
Nitrofurantoin |
|
Paraquat |
|
Continuous oxygen |
|
Cytotoxic agents (many, particularly busulfan, CCNU, bleomycin, methotrexate) |
|
Pulmonary eosinophilia |
Antibiotics: |
Penicillin |
|
Tetracycline |
|
Sulphonamides, e.g. sulfasalazine |
|
NSAIDs |
|
Cytotoxic agents |
|
Acute lung injury |
(Paraquat – a weedkiller) |
Pulmonary hypertension |
Fenfluramine, dexfenfluramine, phentermine |
SLE-like syndrome including pulmonary infiltrates, effusions and fibrosis |
Hydralazine |
Procainamide |
|
Isoniazid |
|
Phenytoin |
|
ACE inhibitors |
|
Monoclonal antibodies |
|
Reactivation of tuberculosis |
Immunosuppressant drugs, e.g. steroids |
Biological agents, e.g. tumour necrosis factor blockers |
CCNU, chloroethyl-cyclohexyl-nitrosourea (lomustine); NSAIDs, non-steroidal anti-inflammatory drugs; SLE, systemic lupus erythematosus.
Other types of diffuse lung disease
Langerhans’ cell histiocytosis (LCH)
This rare disease (prevalence 1/50 000) is characterized histologically by proliferation of Langerhans’ cells, identified by the presence of Birbeck granules on electron microscopy or the CD1a antigen on the surface of the cells. There is a wide variation in clinical presentation, from unifocal bone lesions in older children (which may regress spontaneously), to more disseminated disease in younger children (with a high mortality). Pulmonary LCH occurs almost exclusively in smokers. Chest X-rays (and HRCT) show multiple small cysts (honeycomb lung), fibrosis or widespread nodular shadows. Treatment involves stopping smoking, with regression of disease, corticosteroids and immunosuppressive therapy. For advanced progressive disease, lung transplantation is the only option. Five-year and 10-year survivals are 75% and 65%, respectively.
Goodpasture’s syndrome
This disease (see also p. 585) often starts with symptoms of an upper respiratory tract infection followed by cough and intermittent haemoptysis, tiredness and eventually anaemia, although massive bleeding may occur. The chest X-ray shows transient blotchy shadows that are due to intrapulmonary haemorrhage. These features usually precede the development of an acute glomerulonephritis by several weeks or months. The course of the disease is variable: some patients spontaneously improve while others proceed to renal failure.
The disease usually occurs in individuals over 16 years of age. It is due to a type II cytotoxic reaction driven by antibodies directed against the basement membrane of both kidney and lung. It has been proposed that there is a shared antigen. ANCA may be positive. An association with influenza A2 virus has been reported.
Treatment is with corticosteroids, but in some cases dramatic improvement has been seen with plasmapheresis to remove the autoantibodies.
Diffuse alveolar haemorrhage
This is clinically similar to Goodpasture’s syndrome, but anti-basement-membrane antibodies are absent and the kidneys are less frequently involved. Most cases occur in children under 7 years of age. The child develops a chronic cough and anaemia. The chest X-ray shows diffuse shadows that are due to intrapulmonary bleeding, and eventually miliary nodulation. Characteristically, haemosiderin-containing macrophages are found in the sputum. There is an association with sensitivity to cow’s milk, and an appropriate diet is usually tried.
The general prognosis is poor but treatment with corticosteroids or azathioprine is usually given.
Lymphangioleiomyomatosis
This is a rare disorder of young women with hamartomatous smooth muscle infiltration of the lungs. Gene mutations in the hamartin–tuberin complex are present; the gene products regulate the activity of rapamycin complex 1. Patients present with dyspnoea, chylous pleural effusions, haemoptysis and pneumothorax. Treatment with hormones/oophorectomy has shown little benefit. Sirolimus (rapamycin) treatment has shown benefit.
Pulmonary alveolar proteinosis
This is a rare disease in which there is accumulation of lipoproteinaceous material within the alveoli. It can be congenital, but most cases are acquired and appear to have an autoimmune basis, with antibodies directed against the cytokine GM-CSF. The disease mostly affects men and presents with progressive exertional dyspnoea and cough. Inspiratory crackles are present in only about 50%. Diagnosis is made by bronchial lavage, which reveals a milky appearance and many large, foamy macrophages but few other inflammatory cells.
FURTHER READING
Chen M, Kallenberg CG. ANCA-associated vasculitides–advances in pathogenesis and treatment. Nat Rev Rheumatol 2010; 6:653–664.
Danoff SK, Terry PB, Horton MR. A clinician’s guide to the diagnosis and treatment of interstitial lung disease. South Med J 2007; 100:579–587.
King TE. Clinical advances in the diagnosis and therapy of interstitial lung disease. Am J Respir Crit Care Med 2005; 172:268–279.
Tazi A. Adult pulmonary Langerhans’ cell histiocytosis. Eur Respir J 2006; 27:1272–1285.
Pulmonary infiltration with eosinophilia
The common types and characteristics of these diseases are shown in Table 15.23. They range from very mild, simple, pulmonary eosinophilias to the often fatal hypereosinophilic syndrome.
Simple and prolonged pulmonary eosinophilia
Simple pulmonary eosinophilia is a relatively mild illness with a slight fever and cough and usually lasts for less than 2 weeks. Occasionally, the disease becomes more prolonged, with a high fever lasting for over a month. There is usually an eosinophilia in the blood and this condition is then called prolonged pulmonary eosinophilia. In both conditions the chest X-ray shows either localized or diffuse opacities. The simple form is probably due to a transient allergic reaction in the alveoli. Many allergens have been implicated, including Ascaris lumbricoides, Ankylostoma, Trichuris, Trichinella, Taenia and Strongyloides. Drugs such as aspirin, penicillin, nitrofurantoin and sulphonamides have been implicated. Often, no allergen is identified. The disease is self-limiting and no treatment is required, apart from treating the cause. In the more chronic form all unnecessary treatment should be withdrawn and, where appropriate, worms are treated. Corticosteroid therapy is indicated, with resolution of the disease over the ensuing weeks.
Asthmatic bronchopulmonary eosinophilia
This is characterized by the presence of asthma, transient fleeting shadows on the chest X-ray, and blood or sputum eosinophilia. By far the most common cause worldwide is allergy to A. fumigatus (see below), although Candida albicans and other mycoses may be the allergen in a small number of patients. In many, no allergen can be identified. Whether these cases are intrinsic or driven by an unidentified extrinsic factor is uncertain.
Diseases caused by Aspergillus fumigatus
The various types of lung disease caused by A. fumigatus are illustrated in Figure 15.42.
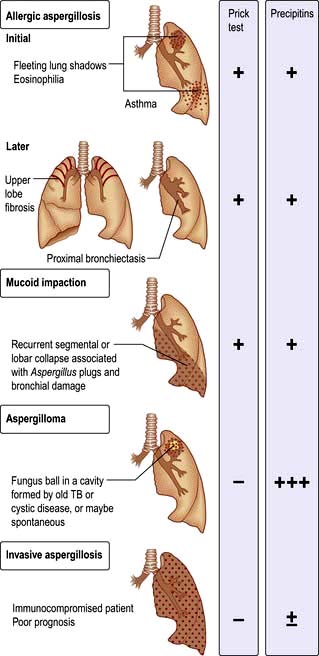
Figure 15.42 Diseases caused by Aspergillus fumigatus. Allergic aspergillosis (initial and later) and three other forms.
The spores of A. fumigatus (diameter 5 mm) are readily inhaled and are present in the atmosphere throughout the year, though they are at their highest concentration in the late autumn. They can be grown from the sputum in up to 15% of patients with chronic lung disease in whom they do not produce disease. They are a cause of extrinsic asthma in atopic individuals.
Allergic bronchopulmonary aspergillosis (asthmatic pulmonary eosinophilia)
This rare disease is caused by a hypersensitivity reaction when the bronchi are colonized by Aspergillus. It can complicate asthma and cystic fibrosis. Proximal bronchiectasis occurs.
Clinical features. There are episodes of eosinophilic pneumonia throughout the year, particularly in late autumn and winter. The episodes present with a wheeze, cough, fever and malaise. They are associated with expectoration of firm sputum plugs containing the fungal mycelium, which results in the clearing of the pulmonary infiltrates on the chest X-ray. Occasionally large mucus plugs obliterate the bronchial lumen, causing collapse of the lung.
Left untreated, repeated episodes of eosinophilic pneumonia can result in progressive pulmonary fibrosis that usually affects the upper zones and can give rise to a chest X-ray appearance similar to that produced by tuberculosis.
Investigations. The peripheral blood eosinophil count is usually raised, and total levels of IgE are usually extremely high, at >1000 ng/mL (both that specific to Aspergillus and nonspecific). Skin-prick testing to protein allergens from A. fumigatus gives rise to positive immediate skin tests. Sputum may show eosinophils and mycelia. Precipitating antibodies in the serum are usually, but not always, found in the serum.
Lung function tests show a decrease in lung volumes and gas transfer in more chronic cases, but there is evidence of reversible airflow limitation in all cases.
Treatment is with prednisolone 30 mg daily, which causes rapid clearing of the pulmonary infiltrates. Frequent episodes of the disease can be prevented by long-term treatment with prednisolone, but doses of 10–15 mg daily are usually required. Antifungal agents (itraconazole, voriconazole) should be used in patients on high doses of steroids; there is evidence that treatment with itraconazole improves pulmonary function. The asthma component responds to inhaled corticosteroids, although these do not influence the occurrence of pulmonary infiltrates. Omalizumab, a humanized monoclonal antibody against IgE, is being trialled.
Aspergilloma and invasive aspergillosis
Aspergilloma is the growth of A. fumigatus within previously damaged lung tissue where it forms a ball of mycelium within lung cavities. Typically the chest X-ray shows a round lesion with an air ‘halo’ above it. Continuing antigenic stimulation gives rise to large quantities of precipitating antibody in the serum. The aspergilloma itself causes little trouble, though occasionally massive haemoptysis may occur, requiring resection of the area of damaged lung containing the aspergilloma. The antifungal agent voriconazole is the drug of choice. Amphotericin is used if patients are intolerant of voriconazole. Invasive aspergillosis is a well-recognized complication of immunosuppression and requires aggressive antifungal therapy; immunosuppression should be reduced if possible.
Tropical pulmonary eosinophilia
This term is reserved for an allergic reaction to microfilaria from Wuchereria bancrofti. The condition is seen in the Indian subcontinent and presents with cough and wheeze together with fever, lassitude and weight loss. The typical appearance of the chest X-ray is of bilateral hazy mottling that is often uniformly distributed in both lung fields. Individual shadows may be as large as 5 mm or may become more confluent, giving the appearance of pneumonia.
The disease is characterized by a very high eosinophil count in peripheral blood. The filarial complement fixation test is positive in almost every case, although the microfilaria are seldom found. The treatment of choice is diethylcarbamazine (see p. 154 for details).
The hypereosinophilic syndrome
This disease is characterized by eosinophilic infiltration in various organs, sometimes associated with an eosinophilic arteritis. The heart muscle is particularly involved, but pulmonary involvement in the form of a pleural effusion or interstitial lung disease occurs in about 40% of cases. Typical features are fever, weight loss, recurrent abdominal pain, persistent non-productive cough and congestive cardiac failure. Corticosteroid treatment may be of value in some cases.
Hypersensitivity pneumonitis
In this disease there is a widespread diffuse inflammatory reaction in both the small airways of the lung and the alveoli. It is due to the inhalation of a number of different antigens, the most common being microbial spores contaminating vegetable matter (e.g. straw, hay, mushroom compost). Some examples are illustrated in Table 15.24. By far the most common of these diseases worldwide is farmer’s lung, which affects up to 1 in 10 of the farming community in disadvantaged, wet communities around the world. Cigarette smokers have a lower risk of developing the disease due to decreased antibody reaction to the antigen.
Table 15.24 Hypersensitivity pneumonitis – some causes
| Disease | Situation | Antigens |
|---|---|---|
Farmer’s lung |
Forking mouldy hay or any other mouldy vegetable material |
Thermophilic actinomycetes, e.g. Micropolyspora faeni |
Fungi, e.g. Aspergillus umbrasus |
||
Bird fancier’s lung |
Handling pigeons, cleaning lofts or budgerigar cages |
Proteins present in the ‘bloom’ on the feathers and in excreta |
Maltworker’s lung |
Turning germinating barley |
Aspergillus clavatus |
Humidifier fever |
Contaminated humidifying systems in air conditioners or humidifiers in factories (especially in printing works) |
Possibly a variety of bacteria or amoeba (e.g. Naegleria gruberi) |
Thermoactinomyces |
||
Mushroom workers |
Turning mushroom compost |
Thermophilic actinomycetes |
Cheese washer’s lung |
Mouldy cheese |
Penicillin casei |
Aspergillus clavatus |
||
Winemaker’s lung |
Mould on grapes |
Botrytis |
Pathogenesis
Histologically there is an initial infiltration of the small airways and alveolar walls with neutrophils followed by T lymphocytes and macrophages, leading to the development of small non-caseating granulomas. These comprise multinucleated giant cells, occasionally containing the inhaled antigenic material. The allergic response to the inhaled antigens involves both cellular immunity and the deposition of immune complexes causing foci of inflammation through the activation of complement via the classical pathway. Some of the inhalant materials may also lead to inflammation by directly activating the alternative complement pathway. These mechanisms attract and activate alveolar and interstitial macrophages, so that continued antigenic exposure results in the progressive development of pulmonary fibrosis.
Clinical features
Typically fever, malaise, cough and shortness of breath come on several hours after exposure to the causative antigen. Thus, a farmer forking hay in the morning may notice symptoms during the late afternoon and evening with resolution by the following morning. On examination, the patient may have a fever, tachypnoea, and coarse end-inspiratory crackles and wheezes throughout the chest. Cyanosis caused by ventilation–perfusion mismatch may be severe even at rest. Continued exposure leads to a chronic illness characterized by severe weight loss, effort dyspnoea and cough as well as the features of idiopathic pulmonary fibrosis (see p. 848).
Investigations
Chest X-ray shows fluffy nodular shadowing with the subsequent development of streaky shadows, particularly in the upper zones. In very advanced cases, honeycomb lung occurs.
High-resolution CT shows reticular and nodular changes with ground glass opacity, which can be further categorized with multislice CT.
Lung function tests show a restrictive ventilatory defect with a decrease in carbon monoxide gas transfer.
Polymorphonuclear leucocyte count is raised in acute cases. Eosinophilia is not a feature.
Precipitating antibodies are present in the serum. One-quarter of pigeon fanciers have precipitating IgG antibodies against pigeon protein and droppings in their serum, but only a small proportion have lung disease. Precipitating antibodies are evidence of exposure, not disease.
Bronchoalveolar lavage shows increased T lymphocytes and granulocytes.
Differential diagnosis
Although hypersensitivity pneumonitis due to inhalation of the spores of Micropolyspora faeni is common among farmers, it is probably more common for these individuals to suffer from asthma related to inhalation of antigens from a variety of mites that infest stored grain and other vegetable material. These include Lepidoglyphus domesticus, L. destructor and Acarus siro. Symptoms of asthma resulting from inhalation of these allergens are often mistaken for farmer’s lung. Lung function tests will effectively discriminate between the disorders. Pigeon fancier’s lung is quite common, but alveolitis from budgerigars, parrots and parakeets is very rare.
Management
Prevention is the aim. This can be achieved by changes in work practice, with the use of silage for animal fodder and the drier storage of hay and grain. Pigeon fancier’s lung is more difficult to control since affected individuals remain strongly attached to their hobby. Prednisolone, initially in large doses of 30–60 mg daily, may achieve regression during the early stages of the disease. Established fibrosis will not resolve and in some patients the disease may progress inexorably to respiratory failure in spite of intensive therapy. Farmer’s lung is a recognized occupational disease in the UK and sufferers are entitled to compensation, depending upon their degree of disability.
Humidifier fever
Humidifier fever (Table 15.24), one cause of building-related illnesses (p. 936), may present with the typical features of hypersensitivity pneumonitis without any radiographic changes. This disease has occurred in outbreaks in factories in the UK, particularly in printing works. In North America it is more commonly found in office blocks with contaminated air-conditioning systems. Humidifier fever may be effectively prevented by sterilization of the re-circulating water used in large humidifying plants.
Drug and radiation-induced respiratory reactions
Drugs may produce a wide variety of disorders of the respiratory tract (Table 15.22). The mechanisms are varied and include direct toxicity (e.g. bleomycin), immune complex formation with arteritis, hypersensitivity (involving both T cell and IgE mechanisms) and autoimmunity. Tuberculosis reactivation is seen with immunosuppressive drugs.
Pulmonary infiltrates with fibrosis may result from the use of a number of cytotoxic drugs used in the treatment of cancer. The most common cause of these reactions is bleomycin. The pulmonary damage is dose-related, occurring when the total dosage is >450 mg, but will regress in some cases if the drug is stopped. The most sensitive test is a decrease in carbon monoxide gas transfer, and therefore gas transfer should be measured repeatedly during treatment with the drug. The use of corticosteroids may help resolution. Drugs affecting the respiratory system are shown in Table 15.22, together with the types of reaction they produce. Anaphylaxis with bronchospasm can occur with many drugs. The list is not exhaustive; e.g. over 20 different drugs are known to produce a systemic lupus erythematosus-like syndrome, sometimes complicated by pulmonary infiltrates and fibrosis. Paraquat ingestion causes severe pulmonary oedema and death, and pulmonary fibrosis develops in many of the few who survive.
Irradiation of the lung during radiotherapy can cause a radiation pneumonitis. Patients complain of breathlessness and a dry cough. Radiation pneumonitis results in a restrictive lung defect. Corticosteroids should be given in the acute stage.
Occupational lung disease
Exposure to dusts, gases, vapours and fumes at work can cause several different types of lung disease:
Acute bronchitis and even pulmonary oedema from irritants such as sulphur dioxide, chlorine, ammonia or the oxides of nitrogen
Pulmonary fibrosis due to mineral dust
Occupational asthma (see Table 15.13) – this is now the commonest industrial lung disease in the developed world
Hypersensitivity pneumonitis (see Table 15.24)
Bronchial carcinoma due to industrial agents (e.g. asbestos, polycyclic hydrocarbons, radon in mines).
The degree of fibrosis that follows inhalation of mineral dust varies. While iron (siderosis), barium (baritosis) and tin (stannosis) lead to dramatic dense nodular shadowing on the chest X-ray, their effect on lung function and symptoms is minimal. In contrast, exposure to silica or asbestos leads to extensive fibrosis and disability. Coal dust has an intermediate fibrogenic effect and used to account for 90% of all compensated industrial lung diseases in the UK. The term ‘pneumoconiosis’ means the accumulation of dust in the lungs and the reaction of the tissue to its presence. The term is not wide enough to encompass all occupational lung disease and is now generally used only in relation to coal dust and its effects on the lung.
Coal-worker’s pneumoconiosis
The disease is caused by dust particles approximately 2–5 µm in diameter that are retained in the small airways and alveoli of the lung. The incidence of the disease is related to total dust exposure, which is highest at the coal face, particularly if ventilation and dust suppression are poor. Improved ventilation and working conditions have reduced the risk of this disease.
Two very different syndromes result from the inhalation of coal.
Simple pneumoconiosis
This simply reflects the deposition of coal dust in the lung. It produces fine micronodular shadowing on the chest X-ray and is by far the most common type of pneumoconiosis. It is graded on the chest X-ray appearance according to standard categories set by the International Labour Office (see below). Considerable dispute remains about the effects of simple pneumoconiosis on respiratory function and symptoms. In many cases, the symptoms are due to COPD related to cigarette smoking, but this is not always the case. Changes to UK workers’ compensation legislation means that coal miners who develop COPD are compensated for their disability regardless of their chest X-ray appearance.
Categories of simple pneumoconiosis are as follows:
1. Small round opacities definitely present but few in number
2. Small round opacities numerous but normal lung markings still visible
3. Small round opacities very numerous and normal lung markings partly or totally obscured.
Simple pneumoconiosis can progress to the development of progressive massive fibrosis (PMF) (see below). PMF virtually never occurs on a background of category 1 simple pneumoconiosis but occurs in about 7% of those with category 2 and in 30% of those with category 3. Miners with category 1 pneumoconiosis are unlikely to receive compensation unless they also have evidence of COPD. Those with more extensive radiographic changes are compensated solely on the basis of their X-ray appearances.
Progressive massive fibrosis
In PMF, patients develop round fibrotic masses several centimetres in diameter, almost invariably in the upper lobes and sometimes having necrotic central cavities. The pathogenesis of PMF is still not understood, though it seems clear that some fibrogenic promoting factor is present in individuals developing the disease, leading to the formation of immune complexes, analogous to the development of large fibrotic nodules in coal miners with rheumatoid arthritis (Caplan’s syndrome). Rheumatoid factor and anti-nuclear antibodies are both often present in the serum of patients with PMF, and also in those suffering from asbestosis or silicosis. Pathologically there is apical destruction and disruption of the lung, resulting in emphysema and airway damage. Lung function tests show a mixed restrictive and obstructive ventilatory defect with loss of lung volume, irreversible airflow limitation and reduced gas transfer.
The patient with PMF suffers considerable effort dyspnoea, usually with a cough. The sputum may be black. The disease can progress (or even develop) after exposure to coal dust has ceased and may lead to respiratory failure.
Silicosis
This disease is uncommon though it may still be encountered in stonemasons, sand-blasters, pottery and ceramic workers and foundry workers involved in fettling (removing sand from metal castings made in sand-filled moulds). Silicosis is caused by the inhalation of silica (silicon dioxide). This dust is highly fibrogenic. For example, a coal miner can remain healthy with 30 g of coal dust in his lungs but 3 g of silica is sufficient to kill. Silica seems particularly toxic to alveolar macrophages and readily initiates fibrogenesis (see Fig. 15.40). The chest X-ray appearances and clinical features of silicosis are similar to those of PMF, but distinctive thin streaks of calcification may be seen around the hilar lymph nodes (‘eggshell’ calcification).
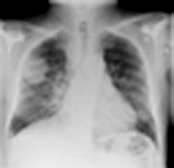
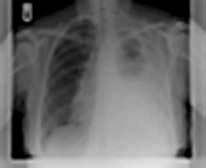
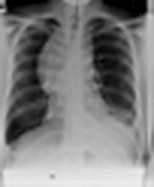
Asbestos
Asbestos is a mixture of silicates of iron, magnesium, nickel, cadmium and aluminium, and has the unique property of occurring naturally as a fibre. It is remarkably resistant to heat, acid and alkali, and has been widely used for roofing, insulation and fireproofing. Asbestos has been mined in southern Africa, Canada, Australia and Eastern Europe. Several different types of asbestos are recognized: about 90% of asbestos is chrysotile, 6% crocidolite and 4% amosite. Chrysotile or white asbestos is the softest asbestos fibre. Each fibre is often as long as 2 cm but only a few microns thick. It is less fibrogenic than crocidolite.
Crocidolite (blue asbestos) is particularly resistant to chemical destruction and exists in straight fibres up to 50 mm in length and 1–2 µm in width. Crocidolite is the type of asbestos most likely to produce asbestosis and mesothelioma. This may be due to the fact that it is readily trapped in the lung. Its long, thin shape means that it can be inhaled, but subsequent rotation against the long axis of the smaller airways, particularly in turbulent airflow during expiration, causes the fibres to impact. Crocidolite is also particularly resistant to macrophage and neutrophil enzymatic destruction.
Exposure to asbestos occurred particularly in shipbuilding yards and in power stations, but it was used so widely that low levels of exposure were very common. Up to 50% of city dwellers have asbestos bodies (asbestos fibres covered in protein secretions) in their lungs at post mortem. Regulations in the UK prohibit the use of crocidolite and severely restrict the use of chrysotile. Careful dust control measures are enforced, which should eventually abolish the problem. Workers continue to be exposed to blue asbestos in the course of demolition or in the replacement of insulation, and it should be remembered that there is a considerable time lag between exposure and development of disease, particularly mesothelioma (20–40 years).
The risk of primary lung cancer (usually adenocarcinoma) is increased in people exposed to asbestos, even in non-smokers. This risk is about 5–7-fold greater in those who have parenchymal asbestosis and about 1.5-fold in those with pleural plaques without parenchymal fibrosis. A synergistic relationship exists between asbestosis and cigarette smoking with the risk of bronchial carcinoma multiplied about five-fold above the risk attributable to smoking alone.
Diseases caused by asbestos are summarized in Table 15.25. Bilateral diffuse pleural thickening, asbestosis, mesothelioma and asbestos-related carcinoma of the bronchus are all eligible for industrial injuries benefit in the UK.
Asbestosis
Asbestosis is defined as fibrosis of the lungs caused by asbestos dust, which may or may not be associated with fibrosis of the parietal or visceral layers of the pleura. It is a progressive disease characterized by breathlessness and accompanied by finger clubbing and bilateral basal end-inspiratory crackles. Minor degrees of fibrosis that are not seen on chest X-ray are often revealed on high-resolution CT scan. No treatment is known to alter the progress of the disease, though corticosteroids are often prescribed.
Mesothelioma
The number of cases of mesothelioma has increased progressively since the mid-1980s and has now reached 2100 deaths/year in the UK, which has the highest per capita death rate from this condition. Rates of mesothelioma in the UK are expected to peak around 2020 at about 2300/year. The most common presentation of mesothelioma is a pleural effusion, typically with persistent chest wall pain, which should raise the index of suspicion even if the initial pleural fluid or biopsy samples are non-diagnostic. Video-assisted thoracoscopic lung biopsy is often needed to obtain sufficient tissue for diagnosis. Some early promise is emerging from clinical trials of chemotherapy, sometimes combined with surgery, but the outlook for most patients remains very limited.
Byssinosis
This disease occurs worldwide but is declining rapidly in areas where the number of people employed in cotton mills is falling. Typically symptoms start on the first day back at work after a break (Monday sickness), with improvement as the week progresses. Tightness in the chest, cough and breathlessness occur within the first hour in dusty areas of the mill, particularly in the blowing and carding rooms where raw cotton is cleaned and the fibres are straightened.
The exact nature of the disease and its aetiology remain disputed. Pure cotton does not cause the disease, and cotton dust has some effect on airflow limitation in all those exposed. Individuals with asthma are particularly badly affected by exposure to cotton dust. The most likely aetiology is endotoxins from bacteria present in the raw cotton causing constriction of the airways of the lung. There are no changes on the chest X-ray and there is considerable dispute as to whether the progressive airflow limitation seen in some patients with the disease is due to cotton dust or to other factors such as cigarette smoking or co-existent asthma.
Berylliosis
Beryllium-copper alloy has a high tensile strength and is resistant to metal fatigue, high temperature and corrosion. It is used in the aerospace industry, in atomic reactors and in many electrical devices.
When beryllium is inhaled, it can cause a systemic illness with a clinical picture similar to sarcoidosis. Clinically there is progressive dyspnoea with pulmonary fibrosis. However, strict control of levels in the working atmosphere has made this disease a rarity.
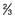 upper limit of normal for serum (105–333 IU/L)
upper limit of normal for serum (105–333 IU/L)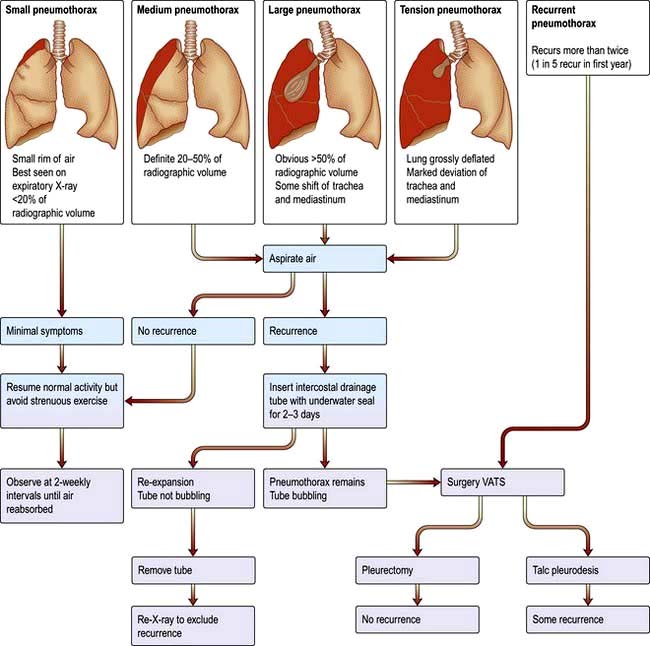
 Practical Box 15.4
Practical Box 15.4