The liver
Structure of the liver and biliary system
The liver
The liver is the body’s largest internal organ (1.2–1.5 kg) and is situated in the right hypochondrium. A functional division into the larger right lobe (containing caudate and quadrate lobes) and the left lobe is made by the middle hepatic vein. The liver is further subdivided into eight segments (Fig. 7.1) by divisions of the right, middle and left hepatic veins. Each segment has its own portal pedicle, permitting individual segment resection at surgery.
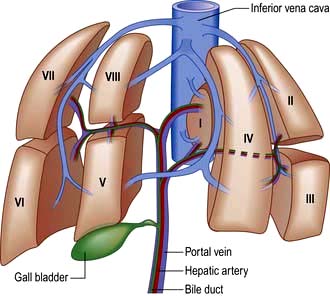
Figure 7.1 Segmental anatomy of the liver, showing the eight hepatic segments. I, caudate lobe; II–IV the left hemiliver; V–VIII the right hemiliver.
The hepatic blood supply constitutes 25% of the resting cardiac output and is delivered via two main vessels, entering via the liver hilum (porta hepatis):
 The hepatic artery, a branch of the coeliac axis, supplies 25% of the hepatic blood flow. The hepatic artery autoregulates flow ensuring a constant total blood flow.
The hepatic artery, a branch of the coeliac axis, supplies 25% of the hepatic blood flow. The hepatic artery autoregulates flow ensuring a constant total blood flow.
The portal vein drains most of the gastrointestinal tract and the spleen. It supplies 75% of hepatic blood flow. The normal portal pressure is 5–8 mmHg; flow increases after meals.
The blood from these vessels is distributed to the segments and flows into the sinusoids via the portal tracts.
Blood leaves the sinusoids, entering branches of the hepatic vein which join into three main branches before entering the inferior vena cava.
The caudate lobe is an autonomous segment as it receives an independent blood supply from the portal vein and hepatic artery, and its hepatic vein drains directly into the inferior vena cava.
Lymph, formed mainly in the perisinusoidal space, is collected in lymphatics which are present in the portal tracts. These small lymphatics enter larger vessels which eventually drain into the hepatic ducts.
The acinus is the functional hepatic unit. This consists of parenchyma supplied by the smallest portal tracts containing portal vein radicles, hepatic arterioles and bile ductules (Fig. 7.2). The hepatocytes near this triad (zone 1) are well supplied with oxygenated blood and are more resistant to damage than the cells nearer the terminal hepatic (central) veins (zone 3).
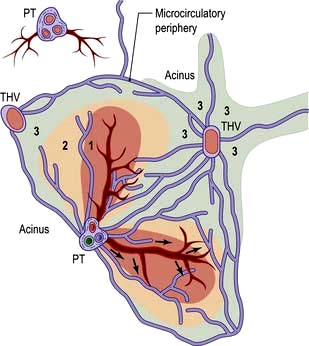
Figure 7.2 Diagram of an acinus. Zones 1, 2 and 3 represent areas supplied by blood, with zone 1 being best oxygenated. Zone 3 is supplied by blood remote from afferent vessels and is in the microcirculatory periphery of the acinus. The perivascular area (star-shaped green area around THV) is formed by the most peripheral parts of zone 3 of several adjacent acini and is the least well oxygenated. THV, terminal hepatic venule; PT, portal triad.
The sinusoids lack a basement membrane and are loosely surrounded by specialist fenestrated endothelial cells and Kupffer cells (phagocytic cells). Sinusoids are separated by plates of liver cells (hepatocytes). The subendothelial space between the sinusoids and hepatocytes is the space of Disse, which contains a matrix of basement membrane constituents and stellate cells (see Fig. 7.23).
Stellate cells store retinoids in their resting state and contain the intermediate filament, desmin. When activated (to myofibroblasts) they are contractile and probably regulate sinusoidal blood flow. Endothelin and nitric oxide play a major role in modulating stellate cell contractility. Activated stellate cells produce signal proteins for synthesis or inhibition of degradation of extracellular matrix components, including collagen, as well as cytokines and chemotactic signals (see p. 328).
The biliary system
Bile canaliculi form a network between the hepatocytes. These join to form thin bile ductules near the portal tract, which in turn enter the bile ducts in the portal tracts. These then combine to form the right and left hepatic ducts that leave each liver lobe. The hepatic ducts join at the porta hepatis to form the common hepatic duct. The cystic duct connects the gall bladder to the lower end of the common hepatic duct. The gall bladder lies under the right lobe of the liver and stores and concentrates hepatic bile; its capacity is approximately 50 mL. The common bile duct is formed at the junction of the cystic and common hepatic duct and is 8 mm in diameter or less, passing through the head of the pancreas, narrowing at its lower end to pass into the duodenum. The common bile duct and pancreatic duct open into the second part of the duodenum most often through a common channel at the ampulla of Vater, which contains the muscular sphincter of Oddi. This contracts rhythmically and prevents all of the bile from entering the duodenum, by maintaining a higher pressure than the gall bladder in the fasting state.
Functions of the liver
Protein metabolism (see also p. 197)
Synthesis and storage
The liver is the principal site of synthesis of all circulating proteins apart from γ-globulins (produced in the reticuloendothelial system). The liver receives amino acids from the intestine and muscles and, by controlling the rate of gluconeogenesis and transamination, regulates plasma levels. Plasma contains 60–80 g/L of protein, mainly albumin, globulin and fibrinogen.
Albumin has a half-life of 16–24 days, and 10–12 g are synthesized daily. Its main functions are to maintain the intravascular oncotic (colloid osmotic) pressure, and to transport water-insoluble substances such as bilirubin, hormones, fatty acids and drugs. Reduced synthesis of albumin over prolonged periods produces hypoalbuminaemia and is seen in chronic liver disease and malnutrition. Hypoalbuminaemia is also found in hypercatabolic states (e.g. trauma with sepsis) and in diseases associated with an excessive loss (e.g. nephrotic syndrome, protein-losing enteropathy).
Transport or carrier proteins such as transferrin and caeruloplasmin, acute-phase and other proteins (e.g. α1-antitrypsin and α-fetoprotein) are also produced in the liver.
The liver also synthesizes all factors involved in coagulation (apart from one-third of factor VIII) – that is, fibrinogen, prothrombin, factors V, VII, IX, X and XIII, proteins C and S and antithrombin (see Ch. 8) as well as components of the complement system. The liver stores large amounts of vitamins, particularly A, D and B12, and lesser amounts of others (vitamin K and folate), and also minerals – iron in ferritin and haemosiderin and copper.
Degradation (nitrogen excretion)
Amino acids are degraded by transamination and oxidative deamination to produce ammonia, which is then converted to urea and excreted by the kidneys. This is a major pathway for the elimination of nitrogenous waste. Failure of this process occurs in severe liver disease.
Carbohydrate metabolism
Glucose homeostasis and the maintenance of the blood sugar is a major function of the liver. It stores approximately 80 g of glycogen. In the immediate fasting state, blood glucose is maintained either by glucose release from breaking down glycogen (glycogenolysis) or by synthesizing new glucose (gluconeogenesis). Sources for gluconeogenesis are lactate, pyruvate, amino acids from muscles (mainly alanine and glutamine) and glycerol from lipolysis of fat stores. In prolonged starvation, ketone bodies and fatty acids are used as alternative sources of fuel as the body tissues adapt to a lower glucose requirement (see Ch. 5).
Lipid metabolism
Fats are insoluble in water and are transported in plasma as protein-lipid complexes (lipoproteins). These are discussed in detail on page 1005.
The liver has a major role in metabolizing of lipoproteins. It synthesizes very-low-density lipoproteins (VLDLs) and high-density lipoproteins (HDLs). HDLs are the substrate for lecithin-cholesterol acyltransferase (LCAT), which catalyses the conversion of free cholesterol to cholesterol ester (see below). Hepatic lipase removes triglyceride from intermediate-density lipoproteins (IDLs) to produce low-density lipoproteins (LDLs) which are degraded by the liver after uptake by specific cell-surface receptors (see Fig. 20.19).
Triglycerides are mainly of dietary origin but are also formed in the liver from circulating free fatty acids (FFAs) and glycerol and incorporated into VLDLs. Oxidation or de novo synthesis of FFA occurs in the liver, depending on the availability of dietary fat.
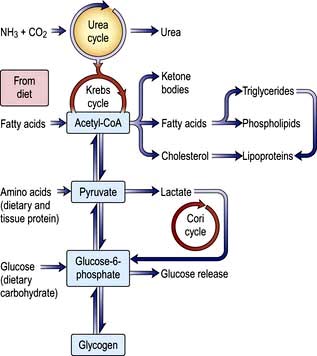
Cholesterol may be of dietary origin but most is synthesized from acetyl-CoA mainly in the liver, intestine, adrenal cortex and skin. It occurs either as free cholesterol or is esterified with fatty acids; this reaction is catalysed by LCAT. This enzyme is reduced in severe liver disease, increasing the ratio of free cholesterol to ester, which alters membrane structures. One result of this is the red cell abnormalities (e.g. target cells) seen in chronic liver disease. Phospholipids (e.g. lecithin) are synthesized in the liver. The complex interrelationships between protein, carbohydrate and fat metabolism are shown in Figure 7.3.
Formation of bile
Bile secretion and bile acid metabolism
Bile consists of water, electrolytes, bile acids, cholesterol, phospholipids and conjugated bilirubin. Two processes are involved in bile secretion across the canalicular membrane of the hepatocyte – bile salt-dependent and bile salt-independent processes – each contributing about 230 mL/day. Another 150 mL daily is produced by epithelial cells of the bile ductules.
Bile formation requires uptake of bile acids and other organic and inorganic ions across the basolateral (sinusoidal) membranes by multiple transport proteins (sodium taurocholate co-transporting polypeptide (NTCP) and sodium independent organic anion transporting polypeptide 2 (OATP2), Fig. 7.4). This process is driven by Na+/K+-ATPase in the basolateral membranes. Intracellular transport across hepatocytes is partly through microtubules and partly by cytosol transport proteins.
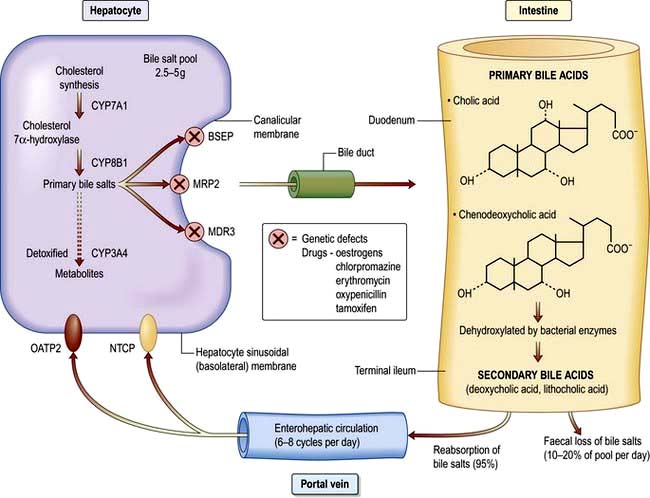
Figure 7.4 Cholesterol synthesis and its conversion to primary and secondary bile acids. All bile acids are normally conjugated with glycine or taurine. BSEP, bile salt export pump; MRP2, multidrug-resistant protein 2; MDR3, multidrug-resistant 3; OATP2, Na+ independent organic anion-transporting polypeptide 2; NTCP, Na+ taurocholate co-transporting polypeptide.
Bile acids are also synthesized in hepatocytes from cholesterol, the rate-limiting step being those catalysed mainly by cholesterol-7α-hydroxylase and the P450 enzymes (CYP7A1 and CYP8B1).
The bile acid receptor, farnesoid X, blocks bile acid formation from cholesterol and also regulates the transport proteins (NTCP, OATP2) that increase bile acid uptake by the liver. It is target for a new class of therapeutic drugs, farnesoid X receptor (FXR) agonists.
The canalicular membrane contains multispecific organic anion transporters, mainly ATPase dependent (ATP binding cassette), the multidrug-resistance protein 2 (MRP2), multidrug resistance protein (MDR3) and the bile salt excretory pump (BSEP), which carry a broad range of compounds including bilirubin diglucuronide, glucuronidated and sulphated bile acids and other organic anions against a concentration gradient into the biliary canaliculus. Na+ and water follow the passage of bile salts by diffusion across the tight junction between hepatocytes (a bile salt-dependent process). In the bile salt-independent process, water flow is due to other osmotically active solutes such as glutathione and bicarbonate.
Secretion of a bicarbonate-rich solution is stimulated mainly by secretin and is inhibited by somatostatin. This involves several membrane proteins, including the Cl−/HCO3− exchanger and the cystic fibrosis transmembrane conductance regulator which controls Cl− secretion, and water channels (aquaporins) in cholangiocyte membranes.
The bile acids are excreted into bile and then pass via the common bile duct into the duodenum. The two primary bile acids – cholic acid and chenodeoxycholic acid (Fig. 7.4) – are conjugated with glycine or taurine, which increases their solubility. Intestinal bacteria convert these acids into secondary bile acids, deoxycholic and lithocholic acid. Figure 7.5 shows the enterohepatic circulation of bile acids.
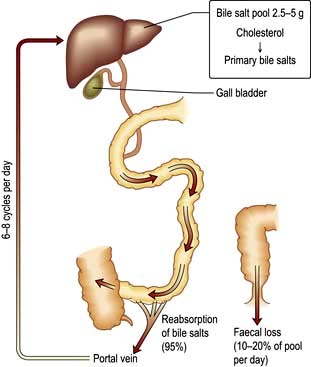
Figure 7.5 Recirculation of bile acids. The bile salt pool is relatively small and the entire pool recycles six to eight times via the enterohepatic circulation. Synthesis of new bile acids compensates for faecal loss.
The average total bile flow is 600 mL/day. When fasting half flows into the duodenum and half is diverted into the gall bladder. The gall bladder mucosa absorbs 80–90% of the water and electrolytes, but is impermeable to bile acids and cholesterol. Following a meal, the I cells of the duodenal mucosa secrete cholecystokinin, which, stimulates contraction of the gall bladder and relaxation of the sphincter of Oddi, allowing bile to enter the duodenum. An adequate bile flow is dependent on bile salts being returned to the liver by the enterohepatic circulation.
Bile acids act as detergents; their main function is lipid solubilization. Bile acid molecules have both a hydrophilic and a hydrophobic end. In aqueous solutions they form micelles, with their hydrophobic (lipid-soluble) ends in the centre. Micelles are expanded by cholesterol and phospholipids (mainly lecithin), forming mixed micelles.
Bilirubin metabolism
Bilirubin is produced mainly from the breakdown of mature red cells by Kupffer cells in the liver and reticuloendothelial system; 15% of bilirubin is formed from catabolism of other haem-containing proteins, such as myoglobin, cytochromes and catalases.
Normally, 250–300 mg (425–510 mmol) of bilirubin are produced daily. The iron and globin are removed from haem and are reused. Biliverdin is formed from haem and then reduced to form bilirubin. The bilirubin produced is unconjugated and water-insoluble, due to internal hydrogen bonding, and is transported to the liver attached to albumin. Bilirubin dissociates from albumin and is taken up by hepatic cell membranes and transported to the endoplasmic reticulum by cytoplasmic proteins, where it is conjugated with glucuronic acid and excreted into bile. The microsomal enzyme, uridine diphosphoglucuronosyl transferase, catalyses the formation of bilirubin monoglucuronide and then diglucuronide. This conjugated bilirubin is water-soluble and is actively secreted into biliary canaliculi and excreted into the intestine within bile (Fig. 8.5). It is not absorbed from the small intestine because of its large molecular size. In the terminal ileum, bacterial enzymes hydrolyse the molecule, releasing free bilirubin which is then reduced to urobilinogen, some of which is excreted in the stools as stercobilinogen. The remainder is absorbed by the terminal ileum, passes to the liver via the enterohepatic circulation, and is re-excreted into bile. Urobilinogen bound to albumin enters the circulation and is excreted in urine via the kidneys. When hepatic excretion of conjugated bilirubin is impaired, a small amount is strongly bound to serum albumin and is not excreted by the kidneys; it accounts for the persistent hyperbilirubinaemia for a time after cholestasis has resolved.
Hormone and drug inactivation
The liver catabolizes hormones such as insulin, glucagon, oestrogens, growth hormone, glucocorticoids and parathyroid hormone. It is also the prime target organ for many hormones (e.g. insulin). It is the major site for the metabolism of drugs (see p. 348) and alcohol (see p. 225). Fat-soluble drugs are converted to water-soluble substances that facilitate their excretion in the bile or urine. Cholecalciferol is converted to 25-hydroxycholecalciferol.
Immunological function
The hepatic reticuloendothelial system contains many immunologically active cells. The liver acts as a ‘sieve’ for bacterial and other antigens carried to it by the portal vein from the gastrointestinal tract. These antigens are phagocytosed and degraded by the Kupffer cells, which have specific membrane receptors for ligands and are activated by several factors, such as infection. They are part of the innate immune system and secrete interleukins, tumour necrosis factor (TNF), collagenase and lysosomal hydrolases. Antigens are degraded without the production of antibody, as there is very little lymphoid tissue and thus, they are prevented from reaching antibody-producing sites and thereby prevent generalized adverse immunological reactions. The reticuloendothelial system also plays a role in tissue repair, T and B lymphocyte interaction, and cytotoxic activity in disease processes. Following stimulation by, for example, endotoxin, the Kupffer cells release IL-6, IL-8 and TNF-α. These cytokines stimulate sinusoidal, stellate, and natural killer, cells to release pro-inflammatory cytokines. The stimulated hepatocytes themselves express adhesion molecules and release IL-8, which is a potent neutrophil chemoattractant. Homing of mucosal lymphocytes (enterohepatic circulation) has been proposed. These exogenous leucocytes again release more cytokines – all damaging the function of the hepatocyte, including bile formation which leads to cholestasis. Cytokines also stimulate hepatic apoptosis.
Investigations
Investigative tests can be divided into:
Urine tests – for bilirubin and urobilinogen
Blood tests ordered for ‘liver function’ are usually processed by an automated multichannel analyser to produce serum levels of bilirubin, aminotransferases, alkaline phosphatase, γ-glutamyl transpeptidase (γ-GT) and total proteins. These routine tests are markers of liver damage, but not actual tests of ‘function’ per se. Subsequent investigations are often based on these tests.
Blood tests
Useful blood tests for certain liver diseases are shown in Table 7.1.
Table 7.1 Useful blood and urine tests for certain liver diseases
| Test | Disease |
|---|---|
Anti-mitochondrial antibody |
Primary biliary cirrhosis |
Anti-nuclear, smooth muscle (actin), liver/kidney microsomal antibody |
Autoimmune hepatitis |
Raised serum immunoglobulins: |
|
IgG |
Autoimmune hepatitis |
IgM |
Primary biliary cirrhosis |
Viral markers |
Hepatitis A, B, C, D, E and others |
α-Fetoprotein |
Hepatocellular carcinoma |
Serum iron, transferrin saturation, serum ferritin |
Hereditary haemochromatosis |
Serum and urinary copper, serum caeruloplasmin |
Wilson’s disease |
α1-Antitrypsin |
Cirrhosis (± emphysema) |
Anti-nuclear cytoplasmic antibodies |
Primary sclerosing cholangitis |
Markers of liver fibrosis |
Non-alcoholic fatty liver disease |
|
Hepatitis C |
Genetic analyses |
e.g. HFE gene (hereditary haemochromatosis) |
Liver function tests
This is a marker of synthetic function and is useful to gauge the severity of chronic liver disease: a falling serum albumin is a bad prognostic sign. In acute liver disease initial albumin levels may be normal.
This is also a marker of synthetic function. Because of its short half-life, it is a sensitive indicator of both acute and chronic liver disease. Vitamin K deficiency should be excluded as the cause of a prolonged PT by giving an intravenous bolus (10 mg) of vitamin K. Vitamin K deficiency commonly occurs in biliary obstruction, as the low intestinal concentration of bile salts results in poor absorption of vitamin K.
Prothrombin times vary in different laboratories depending upon the thromboplastin used in the assay. The International normalized ratio (INR) was developed to standardize anticoagulation with coumarin derivatives, but is very variable in liver disease, and causes large differences when included in prognostic scores for cirrhosis across different centres.
Liver biochemistry
Serum bilirubin is normally almost all unconjugated. In liver disease, increased serum bilirubin is usually accompanied by other abnormalities in liver biochemistry. Differentiation between conjugated or unconjugated bilirubin is only necessary in congenital disorders of bilirubin metabolism (see below) or to exclude haemolysis.
These enzymes (often referred to as transaminases) are contained in hepatocytes and leak into the blood with liver cell damage. Two enzymes are measured:
Aspartate aminotransferase (AST) is primarily a mitochondrial enzyme (80%; 20% in cytoplasm) and is also present in heart, muscle, kidney and brain. High levels are seen in hepatic necrosis, myocardial infarction, muscle injury and congestive cardiac failure.
Alanine aminotransferase (ALT) is a cytosol enzyme, more specific to the liver so that a rise only occurs with liver disease.
This is present in hepatic canalicular and sinusoidal membranes, but also in bone, intestine and placenta. If necessary, its origin can be determined by electrophoretic separation of isoenzymes or bone-specific monoclonal antibodies. In clinical practice, if the γ-GT is also abnormal the ALP is presumed to come from the liver.
Serum ALP is raised in both intrahepatic and extrahepatic cholestatic disease of any cause, due to increased synthesis. In cholestatic jaundice, levels may be four to six times the normal limit. Raised levels also occur with hepatic infiltrations (e.g. metastases), and in cirrhosis, frequently in the absence of jaundice. The highest serum levels due to liver disease (>1000 IU/L) are seen with hepatic metastases and primary biliary cirrhosis.
This is a microsomal enzyme present in liver, but also in many tissues. Its activity can be induced by drugs such as phenytoin and by alcohol. If the ALP is normal, a raised serum γ-GT can be a useful guide to alcohol intake (see p. 1182). However, mild elevations of γ-GT are common, even with a small alcohol consumption and is also raised with fatty liver disease. It does not necessarily indicate liver disease if the other liver biochemical tests are normal. In cholestasis the γ-GT rises in parallel with the ALP as it has a similar pathway of excretion. This is also true of 5-nucleotidase, another microsomal enzyme that can be measured in blood.
Additional blood investigations
Haematological
A full blood count may show anaemia. The red cells are often macrocytic and can have abnormal shapes – target cells and spur cells – owing to membrane abnormalities. Vitamin B12 levels are normal or high, while folate levels are often low owing to poor dietary intake. Other changes are caused by the following:
Bleeding produces a hypochromic, microcytic picture.
Alcohol causes macrocytosis, sometimes with leucopenia and thrombocytopenia.
Hypersplenism results in pancytopenia.
Cholestasis can often produce abnormal-shaped cells, and also deficiency of vitamin K.
Haemolysis may accompany acute liver failure and jaundice.
Aplastic anaemia occurs in up to 2% of patients with acute viral hepatitis.
A raised serum ferritin with transferrin saturation (>60%) is seen in hereditary haemochromatosis.
Biochemical
a1-Antitrypsin. A deficiency of this enzyme can produce cirrhosis.
α-Fetoprotein. This is normally produced by the fetal liver. Its reappearance in increasing and high concentrations in adults indicates hepatocellular carcinoma. Increased concentrations in pregnancy in blood and amniotic fluid suggest fetal neural tube defects. Blood levels are also slightly raised with regenerative liver tissue in patients with hepatitis, chronic liver disease and also in teratomas.
Raised urinary copper, and low serum cooper and caeruloplasmin in Wilson’s disease (see p. 341).
Immunological tests
Increased γ-globulins are thought to result from reduced phagocytosis by sinusoidal and Kupffer cells of the gut absorbed antigens. These antigens then stimulate antibody production in the spleen, lymph nodes and portal tract lymphoid and plasma cell infiltrates. In primary biliary cirrhosis, the predominant raised serum immunoglobulin is IgM, while in autoimmune hepatitis it is IgG. IgG4 is helpful in autoimmune pancreatitis.
Anti-mitochondrial antibody (AMA) in serum is found in over 95% of patients with primary biliary cirrhosis (PBC) (p. 338). Several different AMA subtypes are described, depending on their antigen specificity, and are also found in autoimmune hepatitis and other autoimmune diseases. AMA is demonstrated by an immunofluorescent technique and is neither organ- nor species-specific. M2 subtype is specific for PBC.
Nucleic, smooth muscle (actin), liver/kidney microsomal antibodies can be found in serum, often in high titre, in patients with autoimmune hepatitis. These serum antibodies can also be found in other autoimmune conditions and other liver diseases.
Anti-nuclear cytoplasmic antibodies (ANCA) can be found in serum of patients with primary sclerosing cholangitis.
Fibrosis plays a key role in the outcome of many chronic liver diseases. Blood tests are being used to detect and quantify fibrosis and thereby decrease the need of liver biopsy. To date, these have been developed mainly for chronic hepatitis C, and less so for non-alcoholic fatty liver disease.
Commercial tests are available which measure α2-macroglobulin, α2-haptoglobulin, γ-globulin, apoprotein A1, γ-GT and total bilirubin. Some are based on age, blood ferritin, glucose, AST, ALT, platelet count and bodyweight. These results are formulated to determine a fibrosis index. The indices are sensitive and specific (>90%) for the absence of fibrosis, and have 80% sensitivity and specificity for severe fibrosis, but cannot reliably assess the severity of fibrosis.
Markers of matrix deposition include procollagen I and III peptide and type IV collagen. Markers of matrix degradation, e.g. matrix metalloproteinase (MMP) 2 and 9, and tissue inhibitors of metalloproteinases (TIMPS), e.g. TIMP1 and 2, all are being used as markers of fibrosis.
AST to platelet ratio index (APRI) is less accurate than the tests mentioned above.
Genetic analysis
These tests are performed routinely for haemochromatosis (HFE gene) and for α1-antitrypsin deficiency. Markers are also available for the most frequent abnormal genes in Wilson’s disease (see p. 341).
Urine tests
Dipstick tests are available for bilirubin and urobilinogen. Bilirubinuria is due to the presence of conjugated (soluble) bilirubin, is found in patients with jaundice due to hepatobiliary disease, but is absent if unconjugated bilirubin is the major cause of jaundice. The presence of urobilinogen in urine in practice is of little value but suggests haemolysis or hepatic dysfunction.
Imaging techniques
Ultrasound examination
This is a non-invasive, safe and relatively cheap technique. It involves the analysis of the reflected ultrasound beam detected by a probe moved across the abdomen. The normal liver appears as a relatively homogeneous structure. The gall bladder, common bile duct, pancreas, portal vein and other structures in the abdomen can be visualized. Abdominal ultrasound is useful in:
a jaundiced patient (p. 312)
the detection of gallstones (Fig. 7.6)
focal liver disease – lesions >1 cm
general parenchymal liver disease
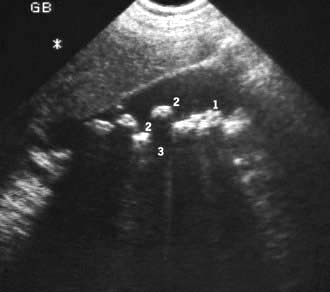
Figure 7.6 Gall bladder ultrasound with multiple echogenic gallstones causing well-defined acoustic shadowing. (1) gall bladder; (2) gallstones; (3) echogenic shadow.
Other abdominal masses can be delineated and biopsies obtained under ultrasonic guidance.
Colour Doppler ultrasound will demonstrate vascularity within a lesion and the direction of portal and hepatic vein blood flow (Fig. 7.7).
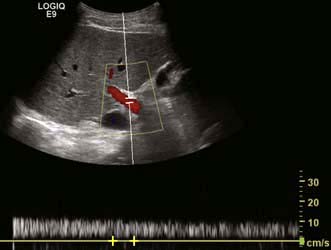
Figure 7.7 Doppler signal of the portal vein is shown as a trace and the visual counterpart in red showing patency and forward flow into the liver.
Ultrasound contrast agents, mostly based on production of microbubbles within flowing blood, enhance the detection of vascularity, allowing the detection of abnormal circulation within liver nodules, giving a more specific diagnosis of hepatocellular carcinoma.
Hepatic stiffness (transient elastography). Using an ultrasound transducer, a vibration of low frequency and amplitude is passed through the liver, the velocity of which correlates with hepatic stiffness. Stiffness (measured in kPa) increases with worsening liver fibrosis (sensitivity and specificity 80–95% compared to liver biopsy). It is not accurate enough to diagnose cirrhosis, and less accurate for less severe fibrosis. It cannot be used in the presence of ascites and morbid obesity, and it is affected by inflammatory tissue and congestion. Acoustic radiation force impulse is incorporated into standard B mode ultrasonography and has similar physical principles to transient elastography.
A small high-frequency ultrasound probe is incorporated into the tip of an endoscope and placed by direct vision into the lumen of the gut. The close proximity of the probe to the pancreas and biliary tree permits high-resolution ultrasound imaging. It allows accurate staging of small, potentially operable, pancreatic tumours and offers a less invasive method for bile duct imaging. It has a high accuracy in detection of small neuroendocrine tumours of the pancreas. EUS-guided fine-needle aspiration of tumours provides cytological/histological tissue for confirmation of malignancy. EUS is also used to place transmural tubes to drain pancreatic and peripancreatic fluid collections.
Computed tomography (CT) examination
CT during or immediately after i.v. contrast shows both arterial and portal venous phases of enhancement, enabling more precise characterization of a lesion and its vascular supply (Fig. 7.8). Retrospective analysis of data allows multiple overlapping slices to be obtained with no increase in the radiation dose, providing excellent visualization of the size, shape and density of the liver, pancreas, spleen, lymph nodes and lesions in the porta hepatis. Multi-planar and three-dimensional reconstruction in the arterial phase can create a CT angiogram, often making formal invasive angiography unnecessary. CT also provides guidance for biopsy. It has advantages over US in detecting calcification and is useful in obese subjects, although US is usually the imaging modality used first to investigate liver disease.
Magnetic resonance imaging (MRI)
MRI produces cross-sectional images in any plane within the body and does not involve radiation. MRI is the most sensitive investigation of focal liver disease. Diffuse liver disease alters the T1 and T2 characteristics. Other fat-suppression modes such as STIR allow good differentiation between haemangiomas and other lesions. Contrast agents such as intravenous gadolinium, which allow further characterization of lesions, are suitable for those with iodine allergy, and provide angiography and venography of the splanchnic circulation. This has superseded direct arteriography.
Magnetic resonance cholangiopancreatography (MRCP)
This technique involves the manipulation of data acquired by MRI. A heavily T2-weighted sequence enhances visualization of the ‘water-filled’ bile ducts and pancreatic ducts to produce high-quality images of ductal anatomy. This non-invasive technique is replacing diagnostic (but not therapeutic) ERCP (see below), and is usually the next test if a biliary abnormality is present on US examination.
Radionuclide imaging – scintiscanning
In a 99mTc-IODIDA scan, technetium-labelled iododiethyl IDA is taken up by the hepatocytes and excreted rapidly into the biliary system. Its main uses are in the diagnosis of:
Endoscopy
Upper GI endoscopy is used for diagnosis and treatment of varices, detection of portal hypertensive gastropathy, and for associated lesions such as peptic ulcers. Colonoscopy may show portal hypertensive colopathy. Capsule endoscopy can identify small intestinal varices.
Endoscopic retrograde cholangiopancreatography (ERCP)
This technique outlines the biliary and pancreatic ducts. It involves the passage of an endoscope into the second part of the duodenum and cannulation of the ampulla. Contrast is injected into both systems and the patient is screened radiologically. Contrast medium with a low iodine content of 1.5 mg/mL is used for the common bile duct so that gallstones are not obscured; a higher iodine content of 2.8 mg/mL is used for the pancreatic duct. Diagnostic ERCP has been replaced by MRCP in nearly all clinical settings. Therapeutic ERCP involves the following:
Common bile duct stones can be removed after performing a diathermy cut of the sphincter to facilitate their withdrawal. Sphincterotomy has a morbidity rate of 3–5%: acute pancreatitis is the commonest, severe haemorrhage is rare. There is an overall mortality of 0.4%.
The biliary system can be drained by passing a tube (stent) through an obstruction, or placement of a nasobiliary drain.
Brachytherapy can be administered after placement at ERCP for therapy of cholangiocarcinoma.
A raised serum amylase is often seen following ERCP and pancreatitis is the most common complication. Cholangitis with or without septicaemia is also seen, and broad-spectrum antibiotics (e.g. 500 mg ciprofloxacin × 2) should be given prophylactically to all patients with suspected biliary obstruction, or a history of cholangitis.
Percutaneous transhepatic cholangiography (PTC)
Under a local anaesthetic, a fine flexible needle is passed into the liver. Contrast is injected slowly until a biliary radicle is identified and then further contrast is injected to outline the whole of the biliary tree. In patients with dilated ducts, the success rate is near 100%. PTC is performed if ERCP fails or is likely to be technically difficult.
In difficult cases ERCP and PTC are sometimes combined, PTC showing the biliary anatomy above the obstruction, with ERCP showing the more distal anatomy. If an obstruction in the bile ducts is seen, a bypass stent can usually be inserted with or without temporary external biliary drainage. Contraindications of PTC are as for liver biopsy (see below). The main complications are bleeding and cholangitis with septicaemia, and prophylactic antibiotics should be given as for ERCP.
Angiography
This is performed by selective catheterization of the coeliac axis and hepatic artery. It outlines the hepatic vasculature and the abnormal vasculature of hepatic tumours, but spiral CT and magnetic resonance scanning have replaced diagnostic angiography. The portal vein can be demonstrated with increased definition using subtraction techniques replacing splenoportography (by direct splenic puncture).
In digital vascular imaging (DVI), contrast given intravenously or intra-arterially can be detected in the portal system using computerized subtraction analysis.
Hepatic venous cannulation allows abnormal hepatic veins to be diagnosed in patients with Budd–Chiari syndrome and is also used to measure portal pressure indirectly. There is a 1:1 relationship of occluded (by balloon) hepatic venous pressure, with portal pressure in patients with alcoholic or viral-related cirrhosis. The height of portal pressure has prognostic value for survival in cirrhosis, and a difference of the occluded minus the free hepatic venous pressure (hepatic venous pressure gradient HVPG) of 20% or more from baseline values or <12 mmHg, has been associated with protection from rebleeding, and prevention of other complications of cirrhosis.
Retrograde CO2 portography is used when there is doubt about portal vein patency and can be combined with transjugular biopsy and hepatic venous pressure measurement.
Liver biopsy (Practical Box 7.1)
Histological examination of the liver is valuable in the differential diagnosis of diffuse or localized parenchymal disease. Liver biopsy can be performed on a day-case basis. The indications and contraindications are shown in Table 7.2. The mortality rate is less than 0.02% when performed by experienced operators.
 Practical Box 7.1
Practical Box 7.1
Needle biopsy of the liver
This should be performed only by experienced doctors and with sterile precautions. Patient consent must be obtained following explanation of the procedure.
The patient’s coagulation status (prothrombin time, platelets) is checked.
The patient’s blood group is checked and serum saved for crossmatching.
The patient lies on their back at the edge of the couch.
Ultrasound examination can be used to confirm liver margins and position of the gall bladder. Alternatively, the liver margins are delineated using percussion.
Local anaesthetic is injected at the point of maximum dullness in the mid-axillary line through the intercostal space during expiration. Anaesthetic (1% lidocaine, approximately 5 mL) should be injected down to the liver capsule.
A tiny cut is made in the skin with a scalpel blade.
A special needle (Menghini, Trucut or Surecut) is used to obtain the liver biopsy while the patient holds his breath in expiration.
The biopsy is laid on filter paper and placed in 10% formalin. If a culture of the biopsy is required it should be placed in a sterile pot.
The patient should be observed, with pulse and blood pressure measurements taken regularly for at least 6 h.
Table 7.2 Indications and contraindications for liver biopsy
|
|
Liver biopsy guided by ultrasound or CT is performed when specific lesions need to be biopsied. Laparoscopy with guided liver biopsy is performed through a small incision in the abdominal wall under local anaesthesia (general anaesthesia is preferred in some centres). A transjugular approach is used when liver histology is essential for management but coagulation abnormalities or ascites prevent the percutaneous approach.
Most complications of liver biopsy occur within 24 h (usually in the first 2 h). They are often minor and include abdominal or shoulder pain which settles with analgesics. Minor intraperitoneal bleeding can occur, but this settles spontaneously. Rare complications include major intraperitoneal bleeding, haemothorax and pleurisy, biliary peritonitis, haemobilia and transient septicaemia. Haemobilia produces biliary colic, jaundice and melaena within 3 days of the biopsy.
Symptoms of liver disease
Acute liver disease
This may be asymptomatic and anicteric. Symptomatic disease, which is often viral, produces generalized symptoms of malaise, anorexia and fever. Jaundice may appear as the illness progresses.
Chronic liver disease
Patients may be asymptomatic or complain of nonspecific symptoms, particularly fatigue. Specific symptoms include:
Right hypochondrial pain due to liver distension
Abdominal distension due to ascites
Ankle swelling due to fluid retention
Haematemesis and melaena from gastrointestinal haemorrhage
Pruritus due to cholestasis – this is often an early symptom of primary biliary cirrhosis
Breast swelling (gynaecomastia), loss of libido and amenorrhoea due to endocrine dysfunction
Confusion and drowsiness due to neuropsychiatric complications (portosystemic encephalopathy).
Signs of liver disease
Acute liver disease
There may be few signs apart from jaundice and an enlarged liver. Jaundice is a yellow coloration of the skin and mucous membranes and is best seen in the conjunctivae and sclerae. In the cholestatic phase of the illness, pale stools and dark urine are present. Spider naevi and palmar erythema usually indicate chronic disease but they can occur in severe acute disease.
Chronic liver disease
The physical signs are shown in Figure 7.9. However, the physical examination is sometimes normal in patients with advanced chronic liver disease.
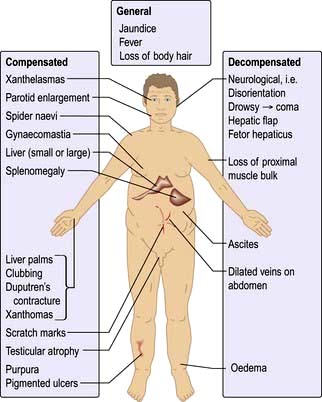
The chest and upper body may show spider naevi. These are telangiectases that consist of a central arteriole with radiating small vessels. They are found in the distribution of the superior vena cava (i.e. above the nipple line) – commonly more than five is taken as diagnostic. They are also found in pregnancy. In haemochromatosis the skin may have a slate-grey appearance.
The hands may show palmar erythema, which is a nonspecific change indicative of a hyperdynamic circulation; it is also seen in pregnancy, thyrotoxicosis or rheumatoid arthritis. Clubbing occasionally occurs, and a Dupuytren’s contracture is often seen in alcoholic cirrhosis.
Xanthomas (cholesterol deposits) are seen in the palmar creases or above the eyes in primary biliary cirrhosis.
Initial hepatomegaly will be followed by a small liver in well-established cirrhosis. Splenomegaly is seen with portal hypertension.
Gynaecomastia (occasionally unilateral) and testicular atrophy may be found in males. The cause of gynaecomastia is complex, but it is probably related to altered oestrogen metabolism or to treatment with spironolactone.
In decompensated cirrhosis, additional signs that can be seen are shown in Figure 7.9.
Jaundice
Jaundice (icterus) is detectable clinically when the serum bilirubin is >50 µmol/L (3 mg/dL). It is useful to divide jaundice into:
Haemolytic jaundice – increased bilirubin load for the liver cells
Congenital hyperbilirubinaemias – defects in conjugation
Cholestatic jaundice, including hepatocellular (parenchymal) liver disease and large duct obstruction.
Haemolytic jaundice
The increased breakdown of red cells (see p. 375) leads to an increase in production of bilirubin. The resulting jaundice is usually mild (serum bilirubin of 68–102 µmol/L or 4–6 mg/dL) as normal liver function can easily handle the increased bilirubin derived from excess haemolysis. Unconjugated bilirubin is not water-soluble and therefore does not pass into urine; hence the term ‘acholuric jaundice’. Urinary urobilinogen is increased.
The causes of haemolytic jaundice are those of haemolytic anaemia (p. 375). The clinical features depend on the cause; anaemia, jaundice, splenomegaly, gallstones and leg ulcers may be seen.
Investigations show features of haemolysis (p. 375). The level of unconjugated bilirubin is raised but the serum ALP, transferases and albumin are normal. Serum haptoglobulins are low. The differential diagnosis is from other forms of jaundice.
Congenital hyperbilirubinaemias (non-haemolytic)
Unconjugated
This is the most common familial hyperbilirubinaemia and affects 2–7% of the population. It is asymptomatic and is usually detected as an incidental finding of a slightly raised bilirubin (17–102 µmol/L or 1–6 mg/dL) on a routine check. All the other liver biochemistry is normal and there are no signs of liver disease. There is a family history of jaundice in 5–15% of patients. Hepatic glucuronidation is approximately 30% of normal, resulting in an increased proportion of bilirubin monoglucuronide in bile. Most patients have reduced levels of UDP-glucuronosyl transferase (UGT-1) activity, the enzyme that conjugates bilirubin with glucuronic acid. Mutations occur in the gene (HUG-Br1) encoding this enzyme, with an expanded nucleotide repeat consisting of two extra bases in the upstream 5′ promoter element. This abnormality appears to be necessary for the syndrome, but is not in itself sufficient for the clinical manifestation (phenotypic expression).
Establishing this diagnosis is necessary to inform the patient that this is not a serious disease and to prevent unnecessary investigations. The raised unconjugated bilirubin is diagnostic and rises on fasting and during a mild illness. The reticulocyte count is normal, excluding haemolysis, and no treatment is necessary.
This is very rare. Only patients with type II (autosomal dominant) with a decrease rather than absence (type I – autosomal recessive) of UDP-glucuronosyl transferase can survive into adult life. Mutation of the HUG-Br1 gene for UDP-glucuronosyl transferase has been demonstrated in the coding region. Liver histology is normal. Transplantation is the only effective treatment.
Conjugated
Dubin–Johnson (autosomal recessive) and Rotor syndromes are due to defects in hepatic bilirubin handling. The prognosis is good in both. In the Dubin–Johnson syndrome there are mutations in both MRP2 (p. 305) transporter genes; the liver is black owing to melanin deposition.
Benign recurrent intrahepatic cholestasis
This is rare and presents in early adulthood. Recurrent attacks of acute cholestasis occur without progression to chronic liver disease. Jaundice, severe pruritus, steatorrhoea and weight loss develop. Serum γ-GT is normal. The gene has been mapped to the FIC1 locus, but the precise relation to cholestasis is unclear. It may be associated with intrahepatic cholestasis of pregnancy (p. 346).
Progressive familial intrahepatic cholestasis (PFIC) syndromes
This is a heterogeneous group of conditions defined by defective secretion of bile acids (see Figs 7.4 and 7.29). They are autosomal recessive. In type 1 (PFIC1), with cholestasis in the first weeks of life, the γ-GT is normal. The gene is on the familial intrahepatic cholestasis-1 gene (FIC1) locus, and has been mapped to a region encoding P type ATPases (ATP8BI) on chromosome 18q21. Type 2 (PFIC2) has been mapped to the bile salt export pump gene (BSEP, also called ABCB1). The protein is located in the canalicular domain of the hepatocyte plasma membrane. The phenotypic expression frequently is a nonspecific giant cell hepatitis progressing to cholestasis – in both types, the γ-GT is normal. Type 3 is due to a multidrug resistance protein 3-P-glycoprotein PGY3 (MDR-3) gene mutation (also called ABCB4 gene) leading to deficient canalicular phosphatidylcholine transport and thus toxic bile acids causing liver damage, which can lead to cirrhosis. Liver transplantation is the only cure for these syndromes.
Cholestatic jaundice (acquired)
This can be divided into extrahepatic and intrahepatic cholestasis. The causes are shown in Figure 7.10.
Extrahepatic cholestasis is due to large duct obstruction of bile flow at any point in the biliary tract distal to the bile canaliculi.
Intrahepatic cholestasis occurs owing to failure of bile secretion. A number of cellular mechanisms in cholestasis have been described in animal models, including inhibition of the Na+/K+-ATPase in the basolateral membranes, decreased fluidity of the sinusoidal plasma membrane, disruption of the microfilaments responsible for canalicular tone, and damage to the tight junctions. In addition, inflammatory change in ductular cells interferes with bile flow.
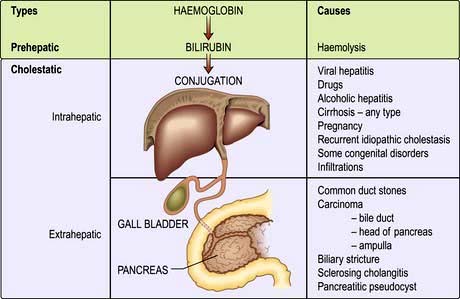
Clinically in both types, there is jaundice with pale stools and dark urine, and the serum bilirubin is conjugated. However, intrahepatic and extrahepatic cholestatic jaundice must be differentiated as their clinical management is entirely different.
Differential diagnosis of jaundice
The history often gives a clue to the diagnosis. Certain causes of jaundice are more likely in particular categories of people. For example, a young person is more likely to have hepatitis, so questions should be asked about drug and alcohol use, and sexual behaviour. An elderly person with gross weight loss is more likely to have a carcinoma. All patients may complain of malaise. Abdominal pain occurs in patients with biliary obstruction by gallstones, and sometimes with an enlarged liver there is pain resulting from distension of the capsule.
Questions should be appropriate to the particular situation, and the following aspects of the history should be covered.
Country of origin. The incidence of hepatitis B virus (HBV) infection is increased in many parts of the world (p. 318).
Duration of illness. A history of jaundice with prolonged weight loss in an older patient suggests malignancy. A short history, particularly with a prodromal illness of malaise, suggests a hepatitis.
Recent outbreak of jaundice. An outbreak in the community suggests hepatitis A virus (HAV).
Recent consumption of shellfish. This suggests HAV infection.
Intravenous drug use, or recent injections or tattoos. These all increase the chance of HBV and hepatitis C virus (HCV) infection.
Men having sex with men. This increases the chance of HBV infection.
Female sex workers. This increases the chance of HBV infection.
Blood transfusion or infusion of pooled blood products. Increased risk of HBV and HCV. In developed countries all donors are screened for HBV and HCV.
Alcohol consumption. A history of drinking habits should be taken, although many patients often understate their consumption.
Drugs taken (particularly in the previous 2–3 months). Many drugs cause jaundice (see p. 348).
Travel. Certain areas have a high risk of HAV infection as well as hepatitis E (HEV) infection (this has a high mortality in pregnancy), but HAV is common in the UK.
A recent anaesthetic. Halothane (named patient basis only in UK) and occasionally isoflurane, and sevoflurane may cause jaundice, particularly in those already sensitive to halogenated anaesthetics. The risk with desflurane appears remote.
Family history. Patients with, for example, Gilbert’s disease may have family members who get recurrent jaundice.
Recent surgery on the biliary tract or for carcinoma.
Environment. People engaged in recreational activities in rural areas, as well as farm and sewage workers, are at risk for leptospirosis, hepatitis E and exposure to chemicals.
Fevers or rigors. These are suggestive of cholangitis or possibly a liver abscess.
Clinical features
The signs of acute and chronic liver disease should be looked for (p. 312). Certain additional signs may be helpful:
Hepatomegaly. A smooth tender liver is seen in hepatitis and with extrahepatic obstruction, but a knobbly irregular liver suggests metastases. Causes of hepatomegaly are shown in Table 7.3.
Splenomegaly. This indicates portal hypertension in patients when signs of chronic liver disease are present. The spleen can also be ‘tipped’ occasionally in viral hepatitis.
Ascites. This is found in cirrhosis but can also be due to carcinoma (particularly ovarian) and many other causes (see Table 7.14).
Table 7.3 Causes of hepatomegaly
A palpable gall bladder occurs with a carcinoma of the pancreas obstructing the bile duct. Generalized lymphadenopathy suggests a lymphoma.
Investigations
Jaundice is not itself a diagnosis and the cause should always be sought. The two most useful tests are the viral markers for HAV, HBV and HCV (in high-risk groups), with an ultrasound examination. Liver biochemistry confirms the jaundice and may help in the diagnosis.
An ultrasound examination should always be performed to exclude an extrahepatic obstruction, and to diagnose any features compatible with chronic liver disease except when hepatitis A is strongly suspected in a young patient. Ultrasound will demonstrate:
the size of the bile ducts, which are dilated in extrahepatic obstruction (Fig. 7.11)
the cause of the obstruction in virtually all patients with tumours and in 75% of patients with gallstones.
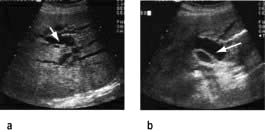
Figure 7.11 Liver ultrasound showing (a) dilated intrahepatic bile ducts (arrow), (b) common bile duct (arrow). The normal bile duct measures 6 mm at the porta hepatis.
The pathological diagnosis of any mass lesion can be made by fine-needle aspiration cytology (sensitivity approximately 60%) or by needle biopsy using a spring-loaded device (sensitivity approximately 90%).
A flow diagram for the general investigation of the jaundiced patient is shown in Figure 7.12.
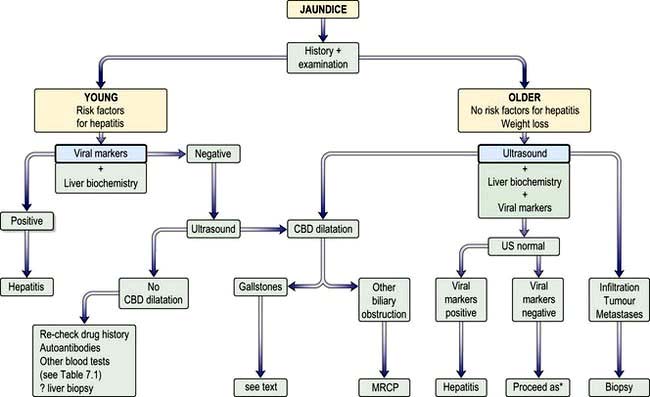
Figure 7.12 Approach to patient with jaundice. CBD, common bile duct; US, ultrasound; MRCP, magnetic resonance cholangiopancreatography. *Proceed as in bottom left box (Re-check drug history …)
In hepatitis, serum AST and ALT tend to be high early in the disease with only a small rise in serum ALP. Conversely, in extrahepatic obstruction the ALP is high with a smaller rise in aminotransferases. However, these findings cannot be relied upon alone to make a diagnosis in an individual case. The prothrombin time (PT) is often prolonged in longstanding liver disease, and the serum albumin is also low.
In haemolytic jaundice the bilirubin is raised and the other liver biochemistry is normal. A raised white cell count may indicate infection (e.g. cholangitis). A leucopenia often occurs in viral hepatitis, while abnormal mononuclear cells suggest infectious mononucleosis and a Monospot test should be performed.
Hepatitis
Acute parenchymal liver damage can be caused by many agents (Fig. 7.13).
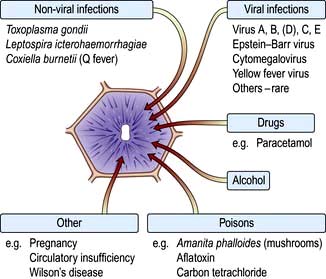
Chronic hepatitis is defined as any hepatitis lasting for 6 months or longer and is classified according to the aetiology (Table 7.4). Chronic viral hepatitis is the principal cause of chronic liver disease, cirrhosis and hepatocellular carcinoma worldwide.
Table 7.4 Causes of chronic hepatitis
Acute hepatitis
Pathology
Although some histological features are suggestive of the aetiological factor, most changes are similar whatever the cause. Hepatocytes show degenerative changes (swelling, cytoplasmic granularity, vacuolation), undergo necrosis (becoming shrunken, containing eosinophilic Councilman bodies) and are rapidly removed. The distribution of these changes may vary with aetiology, but necrosis is usually maximal in zone 3. The extent of the damage is very variable between individuals even when affected by the same agent: at one end of the spectrum, single and small groups of hepatocytes die (spotty or focal necrosis), while at the other end there is multiacinar necrosis involving a substantial part of the liver (massive hepatic necrosis) resulting in fulminant hepatic failure. Between these extremes there is limited confluent necrosis with collapse of the reticulin framework resulting in linking (bridging) between the central veins, the central veins and portal tracts, and between the portal tracts. The extent of the inflammatory infiltrate is also variable, but portal tracts and lobules are infiltrated mainly by lymphocytes. Other variable features include cholestasis in zone 3 and fatty change, the latter being prominent in hepatitis that is due to alcohol or certain drugs.
Chronic hepatitis
Pathology
Chronic inflammatory cell infiltrates comprising lymphocytes, plasma cells and sometimes lymphoid follicles are usually present in the portal tracts. The amount of inflammation varies from mild to severe. In addition, there may be:
loss of definition of the portal/periportal limiting plate – interface hepatitis (damage is due to apoptosis rather than necrosis)
lobular change, focal lytic necrosis, apoptosis and focal inflammation
fibrosis which may be mild, bridging (across portal tracts) or severe cirrhosis.
The overall severity of the hepatitis is judged by the degree of the hepatitis and inflammation (grading) and the severity of the fibrosis or cirrhosis (staging). In chronic viral hepatitis there are various scoring systems. For example, the Knodell Scoring System (histological activity index) uses the sum of four factors (periportal or bridging necrosis, intralobular degeneration and focal necrosis, portal inflammation and fibrosis). The Ishak score stages fibrosis from 0 (none) to 6 (cirrhosis). The METAVIR system has four stages. Scoring systems are used for drug trials and for assessing progression of disease, but are descriptions of architectural changes and not quantitative measures of fibrosis.
Viral hepatitis
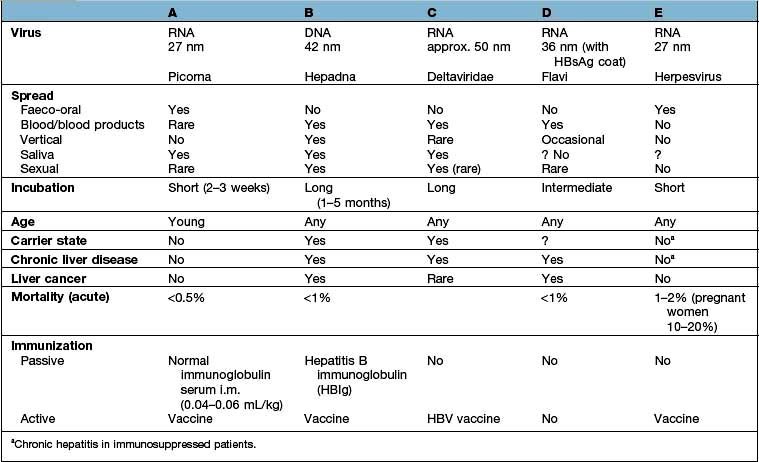
The differing features of the common forms of viral hepatitis are summarized in Table 7.5.
Hepatitis A
Epidemiology
Hepatitis A is the most common viral hepatitis occurring worldwide, often in epidemics. The disease is commonly seen in the autumn and affects children and young adults. Spread of infection is mainly by the faeco-oral route and arises from the ingestion of contaminated food or water (e.g. shellfish). Overcrowding and poor sanitation facilitate spread. There is no carrier state. In the UK it is a notifiable disease.
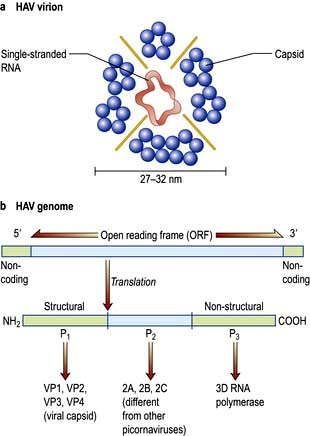
Hepatitis A virus (HAV)
HAV is a picornavirus, having the structure shown in Figure 7.14. It has a single serotype as only one epitope is immuno-dominant. It replicates in the liver, is excreted in bile and then excreted in the faeces for about 2 weeks before the onset of clinical illness and for up to 7 days after. The disease is maximally infectious just before the onset of jaundice. HAV particles can be demonstrated in the faeces by electron microscopy.
Clinical features
The viraemia causes the patient to feel unwell with nonspecific symptoms that include nausea, anorexia and a distaste for cigarettes. Many recover at this stage and remain anicteric.
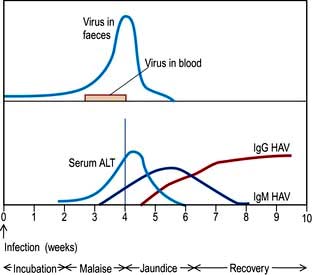
After 1 or 2 weeks, some patients become jaundiced and symptoms often improve. Persistence of nausea, vomiting or any mental confusion warrants assessment in hospital. As the jaundice deepens, the urine becomes dark and the stools pale owing to intrahepatic cholestasis. The liver is moderately enlarged and the spleen is palpable in about 10% of patients. Occasionally, tender lymphadenopathy is seen, with a transient rash in some cases. Thereafter the jaundice lessens and in the majority of cases the illness is over within 3–6 weeks. Extrahepatic complications are rare but include arthritis, vasculitis, myocarditis and acute kidney injury. A biphasic illness occasionally occurs, with the return of jaundice. Rarely the disease may be very severe with fulminant hepatitis, liver coma and death. The typical sequence of events after HAV exposure is shown in Figure 7.15.
Investigations
Prodromal stage: the serum bilirubin is usually normal. However, there is bilirubinuria and increased urinary urobilinogen. A raised serum AST or ALT, which can sometimes be very high, precedes the jaundice.
Icteric stage: the serum bilirubin reflects the level of jaundice. Serum AST reaches a maximum 1–2 days after the appearance of jaundice, and may rise above 500 IU/L. Serum ALP is usually less than 300 IU/L.
After the jaundice has subsided, the aminotransferases may remain elevated for some weeks and occasionally for up to 6 months.
There is leucopenia with a relative lymphocytosis. Very rarely there is a Coombs’-positive haemolytic anaemia or an associated aplastic anaemia. The prothrombin time (PT) is prolonged in severe cases. The erythrocyte sedimentation rate (ESR) is raised.
Course and prognosis
The prognosis is excellent, with most patients making a complete recovery. The mortality in young adults is 0.1% but it increases with age. Death is due to fulminant hepatic necrosis. During convalescence, 5–15% of patients may have relapse of the hepatitis but this settles spontaneously. Occasionally, a more severe jaundice with cholestasis will run a prolonged course of 7–20 weeks and is called ‘cholestatic viral hepatitis’.
There is no reason to stop alcohol consumption other than for the few weeks when the patient is ill. Patients may complain of debility for several months following resolution of the symptoms and biochemical parameters. This is known as the post-hepatitis syndrome; it is a functional illness. Treatment is by reassurance. HAV hepatitis never progresses to chronic liver disease.
Treatment
There is no specific treatment, and rest and dietary measures are unhelpful. Corticosteroids have no benefit. Admission to hospital is not usually necessary.
Prevention and prophylaxis
Control of hepatitis depends on good hygiene. The virus is resistant to chlorination but is killed by boiling water for 10 min.
A formaldehyde-inactivated HAV vaccine is given to people travelling frequently to endemic areas, patients with chronic liver disease, people with haemophilia, and workers in frequent contact with hepatitis cases (e.g. in residential institutions for patients with learning difficulties). Community outbreaks can be interrupted by vaccination. A single dose produces antibodies that persist for at least 1 year, with immunity lasting beyond 10 years. This obviates the need for a booster injection in healthy individuals. Universal vaccination has been suggested.
Hepatitis B
Epidemiology
The hepatitis B virus (HBV) is present worldwide with an estimated 360 million carriers. The UK and the USA have a low carrier rate (0.5–2%), but it rises to 10–20% in parts of Africa, the Middle and the Far East.
Vertical transmission from mother to child in utero, during parturition or soon after birth, is the usual means of transmission worldwide. This is related to the HBV replicative state of the mother (90% HbeAg+, 30% HbeAg−ve) and is uncommon in Africa where horizontal transmission (sib to sib) is common. HBV is not transmitted by breast-feeding.
Horizontal transmission occurs particularly in children through minor abrasions or close contact with other children, and HBV can survive on household articles, e.g. toys, toothbrushes, for prolonged periods.
HBV spread also occurs by the intravenous route (e.g. by transfusion of infected blood or blood products, or by contaminated needles used by drug users, tattooists or acupuncturists), or by close personal contact, such as during sexual intercourse, particularly in men having sex with men (25% of cases in the USA). The virus can be found in semen and saliva.
FURTHER READING
Amon JJ. Hepatitis in drug users: time for attention, time for action. Lancet 2011; 378:543–544.
Dienstag JL. Hepatitis B viral infection. N Engl J Med 2008; 359:1486–1500.
Nelson PK, Mathers BM, Cowie B et al. Global epidemiology of hepatitis B and hepatitis C in people who inject drugs: results of systematic reviews. Lancet 2011; 378:578–583.
Hepatitis B virus (HBV)
The complete infective virion or Dane particle is a 42 nm particle comprising an inner core or nucleocapsid (27 nm) surrounded by an outer envelope of surface protein (HBsAg). This surface coat is produced in excess by the infected hepatocytes and can exist separately from the whole virion in serum and body fluid as 22 nm particles or tubules.
HBsAg contains a major ‘a’ antigenic determinant as well as several subtypes: ‘d’, ‘y’, ‘w’ and ‘r’. Combinations of these subdeterminants (e.g. adr, adw, ayw and ayr) are used to classify HBV genotypes A-H, of which the main types are type A (35%), B (22%), C (31%) and D (10%). There is a strong correlation between genotypes and geographical areas. Genotype A in north-west Europe, North America and Central Africa; B in South-east Asia (including China, Taiwan and Japan); genotype C in South-east Asia; D in southern Europe, India and the Middle East; E in West Africa; F in South and Central America, in American Indians and in Polynesia; G in France and USA; and H in Central and South America. These genotypes have a bearing on, for example, the time to HBeAg seroconversion (B < C), response to interferon treatment (A > B; C > D) and the development of chronic liver disease (A < D).
The core or nucleocapsid is formed of core protein (HBcAg) containing incompletely double-stranded circular DNA and DNA polymerase/reverse transcriptase. One strand is almost a complete circle and contains overlapping genes that encode both structural proteins (pre-S, surface (S), core (C)) and replicative proteins (polymerase and X). The other strand is variable in length. DR1 and DR2 are direct repeats necessary for HBV synthesis during viral replication (Fig. 7.16).
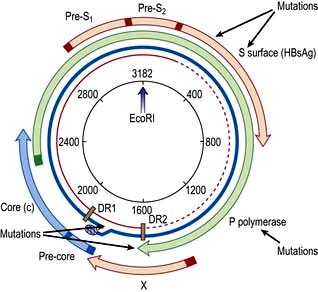
Figure 7.16 Hepatitis B virus (HBV) genome. The viral DNA is partially double-stranded (red incomplete circle and blue circle). The long strand (blue) encodes seven proteins from four overlapping reading frames (S, surface (Pre-S1, Pre-S2, S); c, core (Pre-C, C) P, polymerase (P) and X gene (X)). EcoRI restriction-enzyme-binding site is included as a reference point. DR, direct repeats.
HBeAg is a protein formed via specific self-cleavage of the pre-core/core gene product which is secreted separately by the cell.
Hepatitis B mutants
Mutations occur in the various reading frames of the HBV genome (Fig. 7.16). These mutants can emerge in patients with chronic HBV infection (escape mutants) or can be acquired by infection.
HBsAg mutants are produced by alterations in the ‘a’ determinants of the HbsAg proteins with usually a substitution of glycine for arginine at position 145. This results in changes in the antibody binding domain and the usual tests for HBsAg may be affected.
A mutation in the pre-core region when a guanosine (G) to adenosine (A) change creates a stop codon (TAG) prevents the production of HBeAg, but the synthesis of HBcAg is unaffected. To detect infectivity, HBV DNA must always be measured as no eAg will be present.
DNA polymerase mutants occur, particularly with directly-acting antiviral drugs.
Pathogenesis
Pre-S1 and pre-S2 regions are involved in attachment to an unknown receptor on the hepatocyte. After penetration into the cell, the virus loses its coat and the virus core is transported to the nucleus without processing. The transcription of HBV into mRNA takes place by the HBV DNA being converted into a closed circular form (Yc DNA), which acts as a template for RNA transcription.
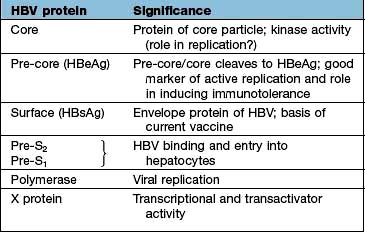
Translation into HBV proteins (Table 7.6) as well as replication of the genome takes place in the endoplasmic reticulum; they are then packaged together and exported from the cell. There is an excess production of non-infective HbsAg particles which are extruded into the circulation.
The HBV is not directly cytopathic and liver damage is produced by the host cellular immune response.
HBV-specific cytotoxic CD8 T cells recognize the viral antigen via HLA class I molecules on the infected hepatocytes. However, suppressor or regulatory T cells inhibit these cytotoxic cells, leading to viral persistence and chronic HBV infection. Th1 responses (interleukin-2, γ-interferon) are thought to be associated with viral clearance and Th2 (interleukins 4, 5, 6, 10, 13) responses with the development of chronic infection and disease severity. Viral persistence in patients with a very poor cell-mediated response leads to asymptomatic inactive chronic HBV infective state. However, a better response, results in continuing hepatocellular damage with the development of chronic hepatitis.
Chronic HBV infection progresses through a replicative and an integrated phase. In the replicative phase there is active viral replication with hepatic inflammation and the patient is highly infectious with HBeAg and HBV DNA positivity. At some stage the viral genome becomes integrated into the host DNA and the viral genes are then transcribed along with those of the host. At this stage, the level of HBV DNA in the serum is low and the patient is HBeAg negative and HBe antibody positive. The aminotransferases are now normal or only slightly elevated and liver histology shows little inflammation, often with cirrhosis. Hepatocellular carcinoma (HCC) develops in patients with this late-stage disease, but the mechanism is still unclear. The REVEAL Study (Risk Evaluation of Viral load Elevation and Associated Liver disease) showed that the risk of HCC was related to levels of HBV DNA rather than a raised aminotransferase (ALT). Integration of the viral DNA with the host-cell chromosomal DNA does appear to have a major role in carcinogenesis. There is evidence to implicate inactivation of p53-induced apoptosis by protein X (Table 7.6), allowing accumulation of abnormal cells and, eventually, carcinogenesis.
Clinical features of acute hepatitis
The sequence of events following acute HBV infection is shown in Figure 7.17. However, in many, the infection is subclinical. When HBV infection is acquired perinatally, an acute hepatitis usually does not occur as there is a high level of immunological tolerance and the virus persists in over 90%. If there is an acute clinical episode the virus is cleared in approximately 99% of patients as there is a good immune reaction. The clinical picture is the same as that found in HAV infection, although the illness may be more severe. In addition, a serum sickness-like immunological syndrome may be seen. This consists of rashes (e.g. urticaria or a maculopapular rash) and polyarthritis affecting small joints occurring in up to 25% of cases in the prodromal period. Fever is usual. Extrahepatic immune complex-mediated conditions such as an arteritis or glomerulonephritis are occasionally seen.
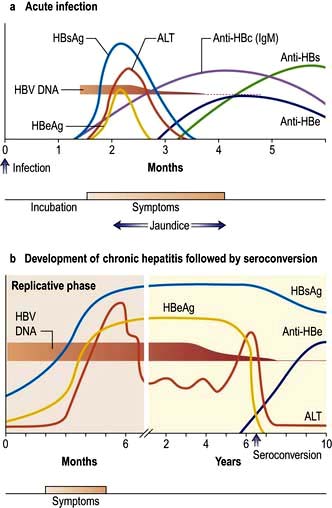
Figure 7.17 Time course of the events and serological changes seen following infection with hepatitis B virus. (a) Acute infection. Antigens: HBsAg appears in the blood from about 6 weeks to 3 months after an acute infection and then disappears. HBeAg rises early and usually declines rapidly. Antibodies: Anti-HBs appears late and indicates immunity. Anti-HBc is the first antibody to appear and high titres of IgM anti-HBc suggest an acute and continuing viral replication. It persists for many months. IgM anti-HBc may be the only serological indicator of recent HBV infection in a period when HBsAg has disappeared and anti-HBs is not detectable in the serum. Anti-HBe appears after the anti-HBc and its appearance relates to a decreased infectivity, i.e. a low risk. (b) Development of chronic hepatitis followed by seroconversion. HBsAg persists and indicates a chronic infection (or carrier state). HBeAg persists and correlates with increased severity and infectivity and the development of chronic liver disease. When anti-HBe develops (seroconversion) the Ag disappears and there is a rise in ALT. HBV DNA suggests continual viral replication. For mutants, see text.
Investigations
These are generally the same as for hepatitis A.
Specific tests. The markers for HBV are shown in Table 7.7. HBsAg is looked for initially; if it is found, a full viral profile is then performed. In acute infection, as HBsAg may be cleared rapidly, anti-HBc IgM is diagnostic. HBV DNA is the most sensitive index of viral replication. HBV DNA has been shown to persist (using polymerase chain reaction (PCR) techniques) even when the e antibody has developed.
Table 7.7 Significance of viral markers in hepatitis B
Antigens |
|
HBsAg |
Acute or chronic infection |
HBeAg |
Acute hepatitis B |
Persistence implies: |
|
Continued infectious state development of chronicity |
|
Increased severity of disease |
|
HBV DNA |
Implies viral replication |
Found in serum and liver |
|
Levels indicate response to treatment |
|
Antibodies |
|
Anti-HBs |
Immunity to HBV; previous exposure; vaccination |
Anti-HBe |
Seroconversion |
Anti-HBc |
|
IgM |
Acute hepatitis B (high titre) |
Chronic hepatitis B (low titre) |
|
IgG |
Past exposure to hepatitis B (HBsAg-negative) |
Course
The majority of patients recover completely, fulminant hepatitis occurring in up to 1%. Some patients go on to develop chronic hepatitis (p. 321), cirrhosis (p. 328) and hepatocellular carcinoma (p. 347) or have inactive chronic HBV infection. The outcome depends upon several factors, including the virulence of the virus and the immuno-competence and age of the patient. Some genetic factors, e.g. the presence of MHC class II genotype, may alter host defence to HBV.
Treatment for acute hepatitis
This is mainly symptomatic. However, patients should have their HBV markers monitored. Several experts suggest that entecavir or tenofovir should be given for the persistent presence of HbeAg beyond 12 weeks, and in some patients who are very ill.
Prevention and prophylaxis
Prevention depends on avoiding risk factors (see above). These include not sharing needles and having safe sex. Vertical transmission is discussed below. Infectivity is highest in those with the e antigen and/or HBV DNA in their blood. These patients should be counselled about their infection. In developing countries, blood and blood products are still a hazard. Standard safety precautions in laboratories and hospitals must be enforced strictly to avoid accidental needle punctures and contact with infected body fluids.
Passive and active immunization
Vaccination is obligatory in most developed countries (but not the UK) as well as countries with high endemicity. Groups at high risk are: all healthcare personnel; members of emergency and rescue teams; morticians and embalmers; children in high-risk areas; people with haemophilia; patients in some psychiatric units; patients with chronic kidney disease/on dialysis units; long-term travellers; men who have sex with men (MSM), bisexual men and sex workers; intravenous drug users.
Combined prophylaxis (i.e. vaccination and immunoglobulin) should be given to: staff with accidental needle-stick injury; all newborn babies of HBsAg-positive mothers; regular sexual partners of HBsAg-positive patients, who have been found to be HBV-negative.
For adults a dose of 500 IU of specific hepatitis B immunoglobulin (HBIG) (200 IU to newborns) is given and the vaccine (i.m.) is given at another site.
This is with a recombinant yeast vaccine produced by insertion of a plasmid containing the gene of HBsAg into a yeast.
Dosage regimen. Three injections (at 0, 1 and 6 months) are given into the deltoid muscle; this gives short-term protection in over 90% of patients. People who are over 50 years of age or clinically ill and/or immunocompromised (including those with HIV infection or AIDS) have a poor antibody response; more frequent and larger doses are required. Antibody levels should be measured at 7–9 months after the initial dose in all at-risk groups. Antibody levels fall steadily after vaccination and booster doses may be required after approximately 3–5 years. It is not cost-effective to check antibody levels prior to active immunization. There are few side-effects from the vaccine.
Chronic HBV infection
Following an acute HBV infection, which may be subclinical, approximately 1–10% of patients will not clear the virus and will develop a chronic HBV infection. This occurs more readily with neonatal (90%) or childhood (20–50% below the age of 5 years) infection than when HBV is acquired in adult life (<10%).
Immune tolerant chronic HBV infection
Patients are asymptomatic and HBsAg and HBeAg positive with very high levels of serum HBV DNA, but normal liver function tests. Most have been infected perinatally so that this is common in Asians. They do not require treatment, but often in the third to fifth decade in life there is a change in host immunity with lymphocytes recognizing infected hepatocytes so that an acute hepatitis develops and chronic hepatitis can follow.
Inactive carrier with chronic HBV infection
There is a vast geographical variation in the incidence of inactive carriers of chronic hepatitis B. In the UK, patients are usually discovered incidentally by blood tests, such as when they are screened for donating blood for transfusion or when attending genital medicine or antenatal clinics. Patients have HBsAg in their serum and are HBeAg negative, HBe antibody positive, with levels of HBV DNA, which are below 400 iu/L in their serum. They have normal aminotransferase levels. Most remain HBsAg positive, but do not develop progressive liver disease, although rarely some patients have episodes of reactive hepatitis such as during chemotherapy for cancer or with bone marrow transplantation: antiviral prophylaxis is indicated. There is an annual spontaneous clearance rate of HBsAg of 1–2%.
Clinical features and investigations of active chronic hepatitis B
Chronic hepatitis is more frequent in men and it is often not preceded by an acute attack. The condition may be asymptomatic or may present as a mild, slowly progressive hepatitis; 50% present with established chronic liver disease. Clinical relapses or ‘flares’ occur, sometimes associated with seroconversion (see below) of HBeAg to anti-HBe or vice versa.
Investigations show a moderate rise in aminotransferases and a slightly raised ALP. The serum bilirubin is often normal. HBsAg and HBV DNA are found in the serum, usually with HBe antigen, unless a mutant virus is involved (see p. 319).
Histologically, there is a full spectrum of changes from near normal with only a few lymphocytes and interface hepatitis to a full-blown cirrhosis. HBsAg may be seen as a ‘ground-glass’ appearance in the cytoplasm on haematoxylin and eosin staining, and this can be confirmed on orcein staining or more specifically with immunohistochemical staining. HBcAg can also be demonstrated in hepatocytes by appropriate immunohistochemical staining.
Treatment for chronic hepatitis B
Indications for therapy are similar for HBeAg positive or negative patients with chronic hepatitis. Three criteria are used: serum HBV DNA levels, serum ALT levels and histological grade and stage:
Patients with moderate to severe active necroinflammation and/or fibrosis in the liver biopsy with HBV DNA above 2000 iu/mL (approximately 10 000 copies/mL) and/or ALT above the upper limit of normal. Age and co-morbidities also affect the decision to treat and with which agents.
If cirrhosis is present, treatment should be given independent of ALT or HBV DNA levels. Patients with decompensated cirrhosis can also be treated with oral antiviral agents, but liver transplantation may be required.
Immunotolerant patients, usually young with persistently normal ALT and high HBV DNA levels, without evidence of liver disease, and without a family history of cirrhosis or hepatocellular cancer do not need therapy, but must receive follow-up.
The aim of treatment is the seroconversion of HBeAg when present to anti-HBe, and the reduction of HBV DNA to 400 iu/L or less measured by sensitive PCR techniques. In addition normalization of serum ALT, histological improvement in inflammation and fibrosis, and loss of HBsAg reflect a good response.
If HBeAg disappears, remission is usually sustained for many years. Patients usually remain HBsAg positive, but there is a small, but incremental loss of HBsAg/annum.
Interferon, entecavir and tenofovir are the most commonly used drugs (p. 93). Response to therapy is judged by a reduction in the HBV DNA level, and if HBeAg is present, by sero-conversion to anti-HBe.
Pegylated α-2a interferon (180 µg once a week subcutaneously) gives response rates of 25–45% (depending on genotype – A and B respond best) after 48 weeks of treatment. Patients with higher serum aminotransferase values (3× the upper limit of normal), who are younger, with viral loads <107 IU/mL respond best. Patients with concomitant HIV respond poorly and those with cirrhosis should not receive interferon.
Side-effects of treatment are many, with an acute flu-like illness occurring 6–8 h after the first injection. This usually disappears after subsequent injections, but malaise, headaches and myalgia are common; depression, reversible hair loss and bone marrow depression and infection may also occur. The platelet count should be monitored. These drug reactions occur in up to 30% of patients, and the dose may have to be lowered.
Overall, the response rate (i.e. disappearance of HbeAg) is 25–40%. The success rate depends on factors shown in Table 7.8.
Table 7.8 Factors predictive of a sustained response to treatment in patients with chronic hepatitis B
Duration of disease |
Short |
Liver biochemistry |
High serum aminotransferases |
Histology |
Active liver disease (mild to moderate) |
Viral levels |
Low HBV DNA levels |
Other |
Absence of immunosuppression |
Female gender |
|
Adult acquired |
|
Delta virus negative |
|
Rapidity of response to oral therapy |
In older patients HbeAg usually disappears (sometimes due to changes in the viral genome). Many such patients have inactive disease but some reactivate without regaining HbeAg (HbeAg negative disease). They respond poorly to interferon but can be treated with nucleoside and nucleotide analogues. Patients who have developed viral mutations should be treated with a two-drug combination.
Oral therapy
Entecavir is very effective and reduces HBV DNA quickly, and there is little viral resistance. Serum HBV DNA becomes negative in 67% (HBeAg-positive patients) and 90% (HBeAg-negative patients) by 48 weeks. It should not be used in patients with lamivudine-induced mutants, but can be used if adefovir mutations have occurred.
Tenofovir is also very effective, has a similar potency to entecavir, and as yet no resistance mutants have been described. It is used for HIV patients with HBV infection. It can also be used in patients with lamivudine mutations, but not on its own for adefovir mutations.
Lamivudine, 100 mg/day given orally, is well tolerated. However, by 4 years, 80% develop viral resistance due to YMDD mutant (tyrosine (Y), methionine (M), aspartate (D)), which itself causes hepatitis. Lamivudine monotherapy is no longer recommended.
The duration of all treatment is probably lifelong and is still being assessed.
Prognosis
The clinical course of hepatitis B is very variable and treatments have improved survival, and stopped progression of fibrosis and enabled regression of fibrosis to occur. Progression from the acute to the chronic phase depends on the age at which infection is acquired. Established cirrhosis is associated with a poor prognosis. Hepatocellular carcinoma is a frequent association and is one of the most common carcinomas in HBV-endemic areas such as the Far East. Surveillance for HCC must continue even when HBV DNA is negative in patients who have HBsAg and are not treated, and also in these rendered negative by therapy. The incidence of HCC is being reduced by routine HBV vaccination of all children.
Hepatitis D
This is caused by the hepatitis D virus (HDV or delta virus) which is an incomplete RNA particle enclosed in a shell of HbsAg and belongs to the Deltaviridae family. It is unable to replicate on its own but is activated by the presence of HBV. It is particularly seen in intravenous drug users but can affect all risk groups for HBV infection. Hepatitis D viral infection can occur either as a:
Co-infection of HDV and HBV which is clinically indistinguishable from an acute icteric HBV infection, but a biphasic rise of serum aminotransferases may be seen. Diagnosis is confirmed by finding serum IgM anti-HDV in the presence of IgM anti-HBc. IgM anti-delta appears at 1 week and disappears by 5–6 weeks (occasionally 12 weeks) when serum IgG antidelta is seen. The HDV RNA is an early marker of infection. The infection may be transient but the clinical course is variable.
Superinfection which results in an acute flare-up of previously quiescent chronic HBV infection. A rise in serum AST or ALT may be the only indication of infection. Diagnosis is by finding HDV RNA or serum IgM anti-HDV at the same time as IgG anti-HBc. Active HBV DNA synthesis is reduced by delta superinfection and patients are usually negative for HBeAg with low HBV DNA.
Fulminant hepatitis can follow both types of infection but is more common after co-infection. HDV RNA in the serum and liver can be measured and is found in acute and chronic HDV infection.
Chronic D hepatitis
This is a relatively infrequent chronic hepatitis, but spontaneous resolution is rare. Between 60% and 70% of patients will develop cirrhosis, and more rapidly than with HBV infection alone. In 15% the disease is rapidly progressive with development of cirrhosis in only a few years. The diagnosis is made by finding anti-delta antibody in a patient with chronic liver disease who is HBsAg positive. It can be confirmed by finding HDV in the liver or HDV RNA in the serum by reverse transcription PCR. Treatment for patients with active liver disease (raised ALT levels and/or inflammation on biopsy) is with pegylated α-2a interferon and adefovir for 12 months, with a response rate of about 30%.
Hepatitis C
Epidemiology
The prevalence rate of infection in healthy blood donors is about 0.02% in Northern Europe, 1–3% in Southern Europe (possibly linked to intramuscular injections of vaccines or other medicines), 6% in Africa, and in Egypt the rates are as high as 19% owing to parenteral antimony treatment for schistosomiasis. The virus is transmitted by blood and blood products and was common in people with haemophilia treated before screening of blood products was introduced. The incidence in intravenous drug users is high (50–60%). The low rate of hepatitis C (HCV) infection in high-risk groups – such as men who have sex with men, sex workers and attendees at STI clinics – suggests a limited role for sexual transmission. Vertical transmission from a healthy mother to child can occur, but is very rare. Other routes of community-acquired infection (e.g. close contact) are extremely rare. In 20% of cases the exact mode of transmission is unknown. An estimated 240 million people are infected with this virus worldwide.
Hepatitis C virus (HCV)
HCV is a single-stranded RNA virus of the Flaviviridae family. The RNA genome is approximately 10 kb in length, encoding a polyprotein product consisting of structural (capsid and envelope) and non-structural viral proteins (Fig. 7.18). Comparisons of subgenomic regions, such as E1, NS4 or NS5, have allowed variants to be classified into six genotypes. Variability is distributed throughout the genome with the non-structural gene of different genotypes showing 30–50% nucleotide sequence disparity. Genotypes 1a and 1b account for 70% of cases in the USA and 50% in Europe. There is a rapid change in envelope proteins, making it difficult to develop a vaccine. Antigens from the nucleocapsid regions have been used to develop enzyme-linked immunosorbent assays (ELISA). The current assay, ELISA-3, incorporates antigens NS3, NS4 and NS5 regions.
Clinical features
Most acute infections are asymptomatic, with about 10% of patients having a mild flu-like illness with jaundice and a rise in serum aminotransferases. Most patients will not be diagnosed until they present, years later, with evidence of abnormal transaminase values at health checks or with chronic liver disease. Extrahepatic manifestations are seen, including arthritis, cryoglobulinaemia with or without glomerulonephritis and porphyria cutanea tarda. There is a higher incidence of diabetes, and associations with lichen planus, sicca syndrome and non-Hodgkin’s lymphoma.
Diagnosis
This is frequently by exclusion in a high-risk individual with negative markers for HAV, HBV and other viruses. A drug cause for hepatitis should be excluded if possible. HCV RNA can be detected from 1 to 8 weeks after infection. Anti-HCV tests are usually positive 8 weeks from infection.
Treatment
Interferon has been used in acute cases to prevent chronic disease. Needle-stick injuries must be followed and treated early if there is evidence of HCV viraemia, usually re-tested for, at 4 weeks.
Course
Some 85–90% of asymptomatic patients develop chronic liver disease. A higher percentage of symptomatic patients ‘clear’ the virus with only 48–75% going on to chronic liver disease (p. 312). Cirrhosis develops in about 20–30% within 10–30 years and of these patients between 7% and 15% will develop hepatocellular carcinoma. The course is adversely affected by co-infection with HBV and/or HIV, and by alcohol consumption, which should be discouraged.
Chronic hepatitis C infection
Pathogenesis
As with hepatitis B infection, cytokines in the Th2 phenotypes are profibrotic and lead to the development of chronic infection. A dominant CD4 Th2 response with a weak CD8 γ-interferon response may lead to rapid fibrosis. Th1 cytokines are antifibrotic and thus a dominant CD4 Th1 and CD8 cytolytic response may cause less fibrosis. Viral load and viral genotype do not affect rate of fibrosis; persistence of HCV infection has been shown to be associated with HLA-DRB1*0701 and DRB4*0101. Other factors also have an effect on the development of fibrosis, particularly male gender, high alcohol intake, a fatty liver and diabetes.
Clinical features
Patients with chronic hepatitis C infection are usually asymptomatic, the disease being discovered only following a routine biochemical test when mild elevations in the aminotransferases (usually ALT) are noticed (50%). The elevation in ALT may be minimal and fluctuating (Fig. 7.19) and some patients have a persistently normal ALT (25%), the disease being detected by checking HCV antibodies (e.g. in blood donors).
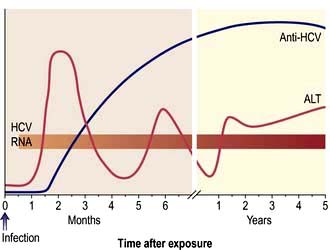
Figure 7.19 Time course of the events and serological changes seen following infection with hepatitis C virus.
Severe chronic hepatitis (25%) and even cirrhosis can be present with only minimal elevation in aminotransferases, but progression is very uncommon in those with a persistently normal ALT. Fatigue is the commonest symptom with sometimes nausea, anorexia and weight loss, which do not correlate with disease activity.
Diagnosis
This is made by finding HCV antibody in the serum using third-generation ELISA-3 tests. HCV RNA should be assayed using quantitative HCV-RNA PCR. The viraemia is usually variable; less than 600 000 iu/mL signifies a greater likelihood of response to antiviral therapy.
The HCV genotype should be characterized in patients who are to be given treatment (see below).
Liver biopsy is indicated if treatment is being considered, especially for genotypes 1 and 4. Non-invasive methods for the diagnosis of fibrosis such as serum markers and elastography (p. 309) can replace the need for biopsy in many cases and are useful in follow-up. The changes on liver biopsy are highly variable. Sometimes only minimal inflammation is detected, but in most cases the features of chronic hepatitis are present, as described (p. 316). Lymphoid follicles are often present in the portal tracts, and fatty change is frequently seen. Histological scoring systems such as METAVIR and Ishak evaluate the inflammation and fibrosis and are used to guide therapy.
Treatment
Treatment (Fig. 7.20) is appropriate for patients with chronic hepatitis on liver histology and/or who have HCV RNA in their serum whether or not serum aminotransferases are raised. The presence of cirrhosis is not a contraindication, but therapeutic responses are less likely. Patients with decompensated cirrhosis should be considered for transplantation. The aim of treatment is to eliminate the HCV RNA from the serum in order to:
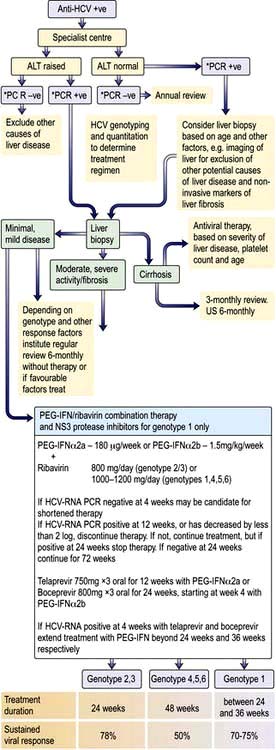
Figure 7.20 Approach to a patient with hepatitis C. ALT, alanine aminotransferase; PCR, polymerase chain reaction. *RNA PCR.
(Adapted from: Dhumeaux D, Marcellin P, Lerebours E. Treatment of hepatitis C. The 2002 French consensus. Gut 2003; 52, with permission from the BMJ Publishing Group.)
A clinical cure is determined by a sustained virological response (SVR), which is defined by a negative HCV-RNA by PCR, 6 months after the end of therapy.
Current treatment is combination therapy with pegylated interferon (Peg), which is interferon with a polyethyleneglycol tail (α-2a 180 µg/week or α-2b 1.5 µg/kg/week), and ribavirin (R) (1000–1200 mg/day for genotype 1 and 4, 800 mg/day for genotype 2 or 3). For genotype 1 only, Peg/R is combined with either of two NS3 protease inhibitors, telaprevir and boceprevir. The combination increases SVR rates compared with Peg/R, to 70–75% or more, in treatment naive patients, or previous relapsers to Peg/R, 55–60% in previous partial responders, and to 30% in previous null responders. Other drugs, NS5B protease inhibitors, cyclophilin inhibitors, new types of interferon and ribavirin analogues are all being developed which will change future treatment algorithms. Treatment duration with Peg/R alone is 12 months for genotypes 4 and 6, and 6 months for genotypes 2 or 3, and as short as 6 months for some genotype 1 patients receiving triple therapy.
Factors affecting response are an HCV RNA viral load of >600 000 IU/L, abnormal body mass index (BMI), older age, male gender, insulin resistance, non-genotype 2 or 3, and CC homozygosity for the IL-28 polymorphism in genotype 1 patients (confers a 70% chance of SVR with Peg/R, compared with TT homozygosity for which SVR is about 20%), which is related to an interferon response gene.
This polymorphism is more common in ethnic Asians than in Caucasians, and is less common in African American and Afro-Caribbeans, and has less influence in genotype-2 or -3 patients or triple therapy with protease inhibitors.
Side-effects of interferon are described on page 321. Ribavirin is usually well tolerated but side-effects include a dose-related haemolysis, pruritus and nasal congestion. Telaprevir causes a rash and anaemia, and boceprevir causes dysgeusia and anaemia. Pregnancy must be avoided with antiviral therapy.
Monitoring results. A rapid virological response is a negative HCV-RNA by PCR at 4 weeks (RVR), which is a very good surrogate of SVR. In about 80% of patients tolerating full dosage of Peg/R, an early virological response (EVR) occurs which is defined as becoming HCV RNA negative or having at least a 2 log reduction in the first 12 weeks. If this is not achieved, the patient is a null responder, and so unlikely to respond that Peg/R should be stopped. If the HCV RNA is positive at 24 weeks, having been below the threshold at 12 weeks, Peg/R should be stopped; if negative, treatment should continue for 72 weeks (genotype 4). For genotype 2 and 3, an absent RVR and/or a positive HCV RNA (but more than a 2 log drop) at 12 weeks leads to 48 weeks of therapy. If HCV RNA is undetectable at 4 weeks, treatment for patients with genotype 2 can be stopped at 12–16 weeks and for genotype 1 (if <600 000 iu/mL at baseline) at 6 months.
A sustained response is clearance of HCV RNA at 6 months after the end of therapy. It is a good surrogate marker for the resolution of the hepatitis. This is achieved in 70–75% of genotype 1 patients with triple therapy, and with Peg/R alone in 50% of patients with genotype 4, and 80% in genotype 2 or 3. In sustained responders relapse is unlikely and histological progression is halted. Best results are obtained in young patients with low HCV RNA levels and genotype 2 or 3.
Oral daclatasvir, a highly selective HCV NS5A replication complex inhibitor, with oral asunaprevir, a highly active HCV NS3 protease inhibitor, have shown good sustained responses in patients with HCV genotype 1 who have been resistant to other therapy. An oral two drug therapy with SVRs approaching are likely in the future.
Hepatitis E
Hepatitis E virus (HEV) is an RNA virus (herpesvirus) (Fig. 7.21) causing a hepatitis clinically very similar to hepatitis A. It is enterally transmitted, usually by contaminated water, with 30% of dogs, pigs and rodents carrying the virus. Epidemics have been seen in many developing countries and sporadically in developed countries, in patients who have had contacts with farm animals or travel abroad. It has a mortality from fulminant hepatic failure of 1–2%, which rises to 20% in pregnant women. There is no carrier state and it does not progress to chronic liver disease except in some immunosuppressed patients. An ELISA for IgG and IgM anti-HEV is available for diagnosis. HEV RNA can be detected in the serum or stools by PCR. Prevention and control depend on good sanitation and hygiene; a vaccine has been developed and used successfully in China.
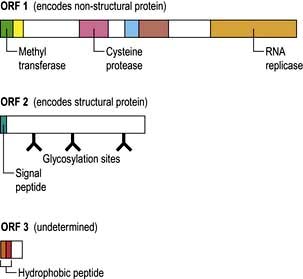
Acute hepatitis due to other infectious agents
Abnormal liver biochemistry is frequently found in a number of acute infections. The abnormalities are usually mild and have no clinical significance.
Infectious mononucleosis (see also p. 99). This is due to the Epstein–Barr (EB) virus. Mild jaundice associated with minor abnormalities of liver biochemistry is extremely common, but ‘clinical’ hepatitis is rare. Hepatic histological changes occur within 5 days of onset; the sinusoids and portal tracts are infiltrated with large mononuclear cells but the liver architecture is preserved. A Paul–Bunnell or Monospot test is usually positive, and atypical lymphocytes are present in the peripheral blood. Treatment is symptomatic.
Cytomegalovirus (CMV) (see also p. 99). This can cause acute hepatitis, usually a glandular fever type syndrome in healthy individuals, but is more severe in those with an impaired immune response. Only the latter need treatment with valganciclovir or ganciclovir.
CMV DNA is positive in blood; CMV IgM is also positive, but there are false-positive reactions.
The liver biopsy shows intranuclear inclusions and giant cells.
Herpes simplex (see also p. 97). Very occasionally the herpes simplex virus causes a generalized acute infection, particularly in the immunosuppressed patient, and occasionally in pregnancy. Aminotransferases are usually massively elevated. Liver biopsy shows extensive necrosis. Aciclovir is used for treatment.
Toxoplasmosis (see also p. 149). The clinical picture is similar to infectious mononucleosis, with abnormal liver biochemistry, but the Paul–Bunnell test is negative.
Yellow fever (see also p. 329). This viral infection is carried by the mosquito Aedes aegypti and can cause acute hepatic necrosis. There is no specific treatment.
Fulminant hepatic failure (FHF)
This is defined as severe hepatic failure in which encephalopathy develops in under 2 weeks in a patient with a previously normal liver (occasionally in some patients with previous liver damage; e.g. D virus superinfection in a previous carrier of HBsAg, Budd–Chiari syndrome or Wilson’s disease). Cases that evolve at a slower pace (2–12 weeks) are called subacute or subfulminant hepatic failure. FHF is a rare but often life-threatening syndrome that is due to acute hepatitis from many causes (Table 7.9). The causes vary throughout the world; most cases are due to viral hepatitis, but paracetamol overdose is common in the UK (50% of cases). HCV does not usually cause FHF although exceptional cases have been reported from Japan and India.
Table 7.9 Causes of fulminant hepatic failure
|
|
Histologically, there is multiacinar necrosis involving a substantial part of the liver. Severe fatty change is seen in pregnancy (p. 346), Reye’s syndrome (p. 348) or following tetracycline administration intravenously.
Clinical features
Examination shows a jaundiced patient with a small liver and signs of hepatic encephalopathy. The mental state varies from slight drowsiness, confusion and disorientation (grades I and II) to unresponsive coma (grade IV) with convulsions. Fetor hepaticus is common, but ascites and splenomegaly are rare. Fever, vomiting, hypotension and hypoglycaemia occur. Neurological examination shows spasticity and hyperreflexia; plantar responses remain flexor until late. Cerebral oedema develops in 80% of patients with FHF but is far less common with subacute failure and its consequences of intracranial hypertension and brain herniation account for about 25% of the causes of death. Other complications include bacterial and fungal infections, gastrointestinal bleeding, respiratory arrest, kidney failure (hepatorenal syndrome and acute tubular necrosis) and pancreatitis.
Investigations
There is hyperbilirubinaemia, high serum aminotransferases and low levels of coagulation factors, including prothrombin and factor V. Aminotransferases are not useful indicators of the course of the disease as they tend to fall along with the albumin with progressive liver damage. An EEG is sometimes helpful in grading the encephalopathy. Ultrasound will define liver size and may indicate underlying liver pathology.
Treatment
There is no specific treatment, but patients should be managed in a specialized unit. Transfer criteria to such units are shown in Box 7.1. Supportive therapy as for hepatic encephalopathy is necessary (see p. 337). When signs of raised intracranial pressure (which is sometimes measured directly) are present, 20% mannitol (1 g/kg bodyweight) should be infused intravenously; this dose may need to be repeated. Dexamethasone is of no value. Hypoglycaemia, hypokalaemia, hypomagnesaemia, hypophosphataemia and hypocalcaemia should be anticipated and corrected with 10% dextrose infusion (checked by 2-hourly dipstick testing), potassium, calcium, phosphate and magnesium supplements. Hyponatraemia should be corrected with hypertonic saline. Coagulopathy is managed with intravenous vitamin K, platelets, blood or fresh frozen plasma. Haemorrhage may be a problem and patients are given a proton pump inhibitor (PPI) to prevent gastrointestinal bleeding. Prophylaxis against bacterial and fungal infection is routine, as infection is a frequent cause of death and may preclude liver transplantation. Suspected infection should be treated immediately with suitable antibiotics. Renal and respiratory failure should be treated as necessary. Liver transplantation has been a major advance for patients with FHF. It is difficult to judge the timing or the necessity for transplantation, but there are guidelines based on validated prognostic indices of survival (see below).
 Box 7.1
Box 7.1Course and prognosis
In mild cases (grades I and II encephalopathy with drowsiness and confusion), two-thirds of the patients will survive. The outcome of severe cases (grades III and IV encephalopathy with stupor or deep coma) is related to the aetiology. In special units, 70% of patients with paracetamol overdosage and grade IV coma survive, as do 30–40% patients with HAV or HBV hepatitis. Poor prognostic variables indicating a need to transplant the liver are shown in Box 7.2.
Autoimmune hepatitis
This condition occurs most frequently in women. In type I (see below) there is an association with other autoimmune diseases (e.g. pernicious anaemia, thyroiditis, coeliac disease and Coombs’-positive haemolytic anaemia) and 60% of cases are associated with HLA-DR3, DR52a loci, HLA-DRB1*0301 and HLA-DRB2*0401. In Asians, the condition is associated with HLA-DR4.
Pathogenesis
The cause is unknown. It is proposed, in a genetically predisposed person, that an environmental agent (perhaps a virus) causes a sequence of T cell mediated events against liver antigens, producing a progressive necroinflammatory process which results in fibrosis and cirrhosis. In vitro observations have shown that there is a defect of suppressor (regulatory) T cells which may be primary or secondary. However, no clear mechanism causing the inflammation has been found.
FURTHER READING
Czaja AJ. Genetic factors affecting the occurrence, clinical phenotype, and outcome of autoimmune hepatitis. Clin Gastroenterol Hepatol 2008; 6(4):379–388.
Manns MP, Woynarowski M, Kreisel W et al. Budesonide induces remission more effectively than prednisolone in a controlled trial of patients with autoimmune hepatitis. Gastroenterology 2010; 139:1198–1206.
Clinical features
There are two peaks in presentation. In the peri- and post-menopausal group, patients may be asymptomatic or present with fatigue, the disease being discovered by abnormalities in liver biochemistry or because of signs of chronic liver disease on routine examination. In the teens and early 20s, the disease (often type II) presents as an acute hepatitis with jaundice and very high aminotransferases, which do not improve with time. This age group often has clinical features of cirrhosis with hepatosplenomegaly, cutaneous striae, acne, hirsutes, bruises and, sometimes, ascites. An ill patient can also have features of an autoimmune disease with a fever, migratory polyarthritis, glomerulonephritis, pleurisy, pulmonary infiltration or lung fibrosis.
There are rare overlap syndromes with primary biliary cirrhosis and primary sclerosing cholangitis, existing concomitantly or developing consecutively.
Investigations
The serum aminotransferases are high, with lesser elevations in the ALP and bilirubin. The serum γ-globulins are high, frequently twice normal, particularly the IgG. The biochemical pattern is similar in both types.
A mild normochromic normocytic anaemia with thrombocytopenia and leucopenia is present, even before portal hypertension and splenomegaly. The prothrombin time is often high.
Two types of autoimmune hepatitis have been recognized:
Type I with antibodies (titres >1 : 80)
Type II with antibodies: anti-liver/kidney microsomal (anti-LKM1). The main target is cytochrome P4502D6 (CYP2D6) on liver cell plasma membranes.
Approximately 13% of patients lack the above autoantibodies.
This shows the changes of chronic hepatitis described previously. The amount of interface hepatitis is variable, but tends to be high in untreated patients. Lymphoid follicles are less often seen than in hepatitis C, and plasma cell infiltration is frequent. Approximately one-third of patients have cirrhosis at presentation.
Treatment
Budesonide 3 mg Ö 2 or 3 daily has fewer side-effects than prednisolone and is now the preferred treatment. Alternatively, prednisolone 30 mg is given daily for at least 2 weeks, followed by a slow reduction and then a maintenance dose of 10–15 mg daily. Azathioprine should be added, 1–2 mg/kg daily, as a steroid-sparing agent and in some patients as sole long-term maintenance therapy. Levels of thiopurine methyltransferase should be obtained. Mycophenolate, ciclosporin and tacrolimus have been used in resistant cases.
Course and prognosis
Steroid and azathioprine therapy induce remission in over 80% of cases. This response forms part of the diagnostic criteria for autoimmune hepatitis. Treatment is lifelong in most cases. Those with initial cirrhosis are more likely to relapse following treatment withdrawal and require indefinite therapy. Liver transplantation is performed if treatment fails, although the disease may recur. Hepatocellular carcinoma occurs less frequently than with viral-induced cirrhosis.