Cirrhosis
Cirrhosis results from the necrosis of liver cells followed by fibrosis and nodule formation. The liver architecture is diffusely abnormal and this interferes with liver blood flow and function. This derangement produces the clinical features of portal hypertension and impaired liver cell function.
Aetiology
The causes of cirrhosis are shown in Table 7.10. Alcohol is now the most common cause in the West, but viral infection is the most common cause worldwide. With the identification of HCV, and recognition of non-alcoholic fatty liver disease (NAFLD), idiopathic (cryptogenic) cirrhosis is diagnosed infrequently. Young patients with cirrhosis must be investigated to exclude treatable causes (e.g. Wilson’s disease).
Table 7.10 Causes of cirrhosis
| Common | Others |
|---|---|
Alcohol |
Biliary cirrhosis: |
Hepatitis B ± D |
Primary |
Hepatitis C |
Secondary |
Non-alcoholic fatty liver disease |
Autoimmune hepatitis |
|
Hereditary haemochromatosis |
|
Hepatic venous congestion |
|
Budd–Chiari syndrome |
|
Wilson’s disease |
|
Drugs (e.g. methotrexate) |
|
α1-Antitrypsin deficiency |
|
Cystic fibrosis |
|
Galactosaemia |
|
Glycogen storage disease |
|
Veno-occlusive disease |
|
Idiopathic (cryptogenic) |
|
? Other viruses |
Pathogenesis
Chronic injury to the liver results in inflammation, necrosis and, eventually, fibrosis (Fig. 7.22). Fibrosis is initiated by activation of the stellate cells (see p. 304). Kupffer cells, damaged hepatocytes and activated platelets are probably involved. Stellate cells are activated by many cytokines and their receptors, reactive oxygen intermediates and other paracrine and autocrine signals.
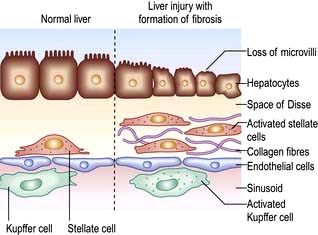
Figure 7.22 Pathogenesis of fibrosis. The normal liver is shown on the left. Activation of the stellate cell is followed by proliferation of fibroblasts and the deposition of collagen.
In the early stage of activation the stellate cells become swollen and lose retinoids with upregulation of receptors for proliferative and fibrogenic cytokines, such as platelet-derived growth factor (PDGF), and transforming growth factor β1 (TGF-β1). TGF-β1 is the most potent fibrogenic mediator identified so far. Inflammatory cells contribute to fibrosis via cytokine secretion.
In the space of Disse, the normal matrix is replaced by collagens, predominantly types 1 and 3, and fibronectin. Subendothelial fibrosis leads to loss of the endothelial fenestrations (openings) and this impairs liver function. Collagenases (matrix metalloproteinases, MMPs) are able to degrade this collagen but are inhibited by tissue inhibitors of metalloproteinases (TIMPs), which are increased in human liver fibrosis. There is accumulating evidence that liver fibrosis is reversible, particularly when inflammation is reduced on a long-term basis, e.g. by suppressing or eliminating viruses.
Pathology
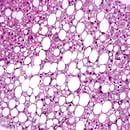
The characteristic features of cirrhosis are regenerating nodules separated by fibrous septa and loss of the normal lobular architecture within the nodules (Fig. 7.23a). Two types of cirrhosis have been described which give clues to the underlying cause:
 Micronodular cirrhosis. Regenerating nodules are usually <3 mm in size and the liver is involved uniformly. This type is often caused by ongoing alcohol damage or biliary tract disease.
Micronodular cirrhosis. Regenerating nodules are usually <3 mm in size and the liver is involved uniformly. This type is often caused by ongoing alcohol damage or biliary tract disease.
Macronodular cirrhosis. The nodules are of variable size and normal acini may be seen within the larger nodules. This type is often seen following chronic viral hepatitis.
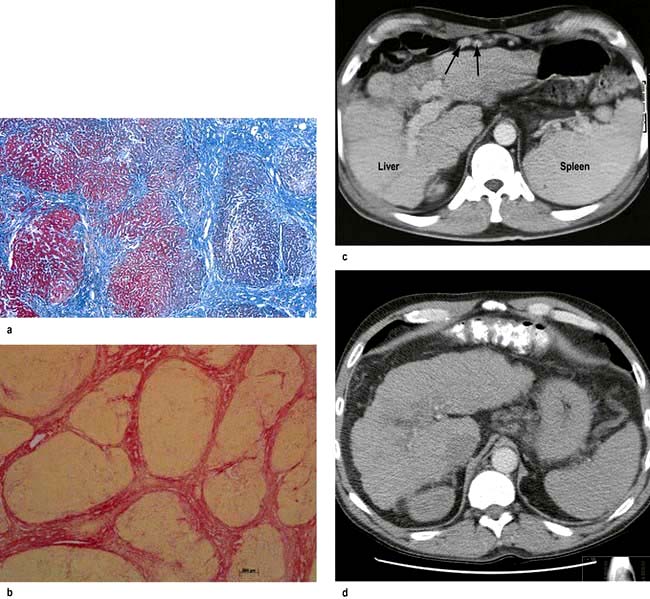
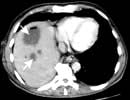
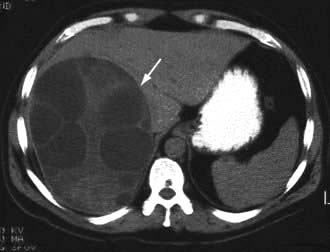
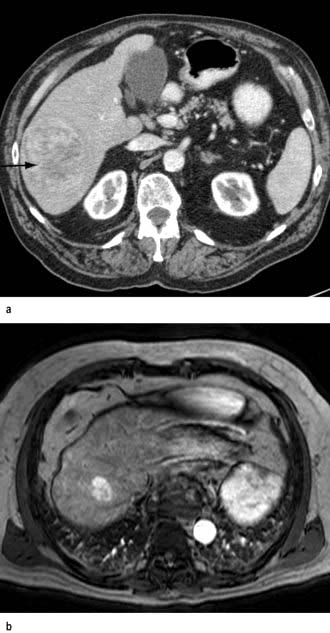
Figure 7.23 Pathology of cirrhosis. (a) Histological appearance showing nodules of liver tissue of varying size surrounded by fibrosis. (b) PicroSirius red stain of collagen used for morphometric evaluation of fibrosis. (c) CT scan showing an irregular lobulated liver. There is splenomegaly and enlargement of collateral vessels beneath the anterior abdominal wall (arrows) as a result of portal hypertension. (d) CT image showing cirrhosis, with a patent portal vein and no space-occupying lesion.
A mixed picture with small and large nodules is sometimes seen.
Symptoms and signs are described on page 312.
Investigations
These are performed to assess the severity and type of liver disease.
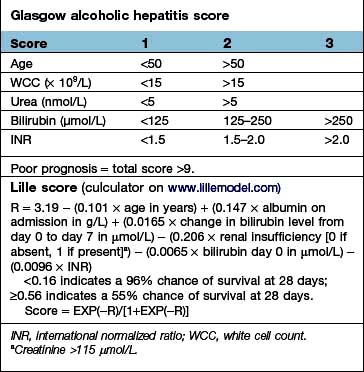
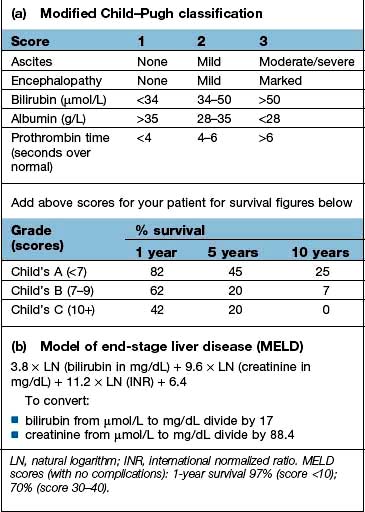
Liver function. Serum albumin and prothrombin time are the best indicators of liver function: the outlook is poor with an albumin level below 28 g/L. The prothrombin time is prolonged commensurate with the severity of the liver disease (Box 7.3).
Liver biochemistry. This can be normal, depending on the severity of cirrhosis. In most cases there is at least a slight elevation in the serum ALP and serum aminotransferases. In decompensated cirrhosis all biochemistry is deranged.
Serum electrolytes. A low sodium indicates severe liver disease due to a defect in free water clearance or to excess diuretic therapy.
Serum creatinine. An elevated concentration >130 µmol/L is a marker of worse prognosis.
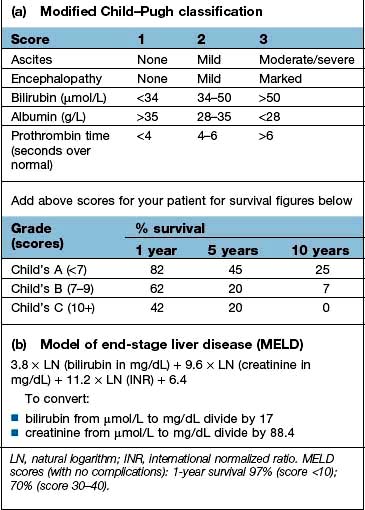
 Box 7.3
Box 7.3
Scoring systems in cirrhosis
In addition, serum α-fetoprotein if >200 ng/mL is strongly suggestive of the presence of a hepatocellular carcinoma.
Serum copper and serum α1-antitrypsin should always be measured in young cirrhotics. Total iron-binding capacity (TIBC) and ferritin should be measured to exclude hereditary haemochromatosis; genetic markers are also available (p. 339).
Ultrasound examination. This can demonstrate changes in size and shape of the liver. Fatty change and fibrosis produce a diffuse increased echogenicity. In established cirrhosis there may be marginal nodularity of the liver surface and distortion of the arterial vascular architecture. The patency of the portal and hepatic veins can be evaluated. It is useful in detecting hepatocellular carcinoma. Elastography is being used in diagnosis and follow-up to avoid liver biopsy (p. 309).
CT scan (see p. 310). Figure 7.23c,d shows hepatosplenomegaly, and dilated collaterals are seen in chronic liver disease. Arterial phase-contrast-enhanced scans are useful in the detection of hepatocellular carcinoma.
Endoscopy is performed for the detection and treatment of varices, and portal hypertensive gastropathy. Colonoscopy is occasionally performed for colopathy.
MRI scan. This is useful in the diagnosis of both malignant and benign tumours such as haemangiomas. MR angiography can demonstrate the vascular anatomy and MR cholangiography the biliary tree.
This is usually necessary to confirm the type and severity of liver disease. The core of liver often fragments and sampling errors may occur in macronodular cirrhosis. Special stains are required for iron and copper, and various immunocytochemical stains can identify viruses, bile ducts, angiogenic structures and oncogenic markers. Chemical measurement of iron and copper is necessary to confirm diagnosis of iron overload or Wilson’s disease. Adequate samples in terms of length and number of complete portal tracts are necessary for diagnosis and for staging/grading of chronic viral hepatitis. Digital image analysis of picroSirius red staining can be used to quantitate collagen in biopsy specimens (Fig. 7.23b).
Management
Management is that of the complications seen in decompensated cirrhosis. Patients should have 6-monthly ultrasound to detect the early development of a hepatocellular carcinoma (see p. 347), as all therapeutic strategies work best with small single tumours.
Treatment of the underlying cause may arrest or occasionally reverse the cirrhotic changes (see below). Patients with compensated cirrhosis should lead a normal life. The only dietary restriction is to reduce salt intake. Aspirin and NSAIDs should be avoided. Alcohol should be avoided, although if the cirrhosis is not due to alcohol and not due to viral hepatitis, small amounts not taken on a regular basis are probably not harmful.
Course and prognosis
This is extremely variable, depending on many factors, including the aetiology and the presence of complications. Poor prognostic indicators are given in Table 7.11. Development of any complication usually worsens the prognosis. In general, the 5-year survival rate is approximately 50%, but this also varies depending on the aetiology and the stage at which the diagnosis is made.
Table 7.11 Poor prognostic indicators in cirrhosis
|
|
There are a number of prognostic classifications based on modifications of Child’s grading (A, B and C; see Box 7.3) and the model for end-stage disease (MELD), based on serum bilirubin, creatinine and INR, which is widely used as a predictor of mortality in patients awaiting liver transplantation.
Liver transplantation
This is an established treatment for a number of liver diseases. Shortage of donors is a major problem in all developed countries and in some, such as Japan, living related donors form the majority of transplant operations. Indications include the following:
Acute liver disease. Patients with fulminant hepatic failure of any cause, including acute viral hepatitis (p. 318).
Chronic liver disease. The indications for transplantation are usually for complications of cirrhosis, no longer responsive to therapy. Timing of the transplant depends on donor availability. All patients with end-stage (Child’s grade C) cirrhosis should be referred to a transplant centre and also those with debilitating symptoms. In addition specific extrahepatic complications of cirrhosis, even with preserved liver function, such as hepatopulmonary syndrome (shunting in the lung leading to hypoxia) and porto-pulmonary hypertension, can be reversed by liver transplantation.
Primary biliary cirrhosis. Patients with this disease should be transplanted when their serum bilirubin is persistently >100 µmol/L or symptoms such as itching are intolerable.
Chronic hepatitis B if HBV DNA negative or levels falling under therapy. Following transplantation, recurrence of hepatitis is prevented by hepatitis B immunoglobulin and nucleoside analogues in combination to prevent escape mutants (see this chapter).
Chronic hepatitis C is the most common indication. Universal HCV reinfection occurs with chronic hepatitis of varying severity and cirrhosis occurs in 10–20% at 5 years. Antiviral agents may delay this progression if sustained viral response occurs.
Autoimmune hepatitis. In patients who have failed to respond to medical treatment or have major side-effects of corticosteroid therapy. It can reoccur.
Alcoholic liver disease. Well-motivated patients who have stopped drinking without improvement of liver disease are offered a transplant, with concomitant and frequent counselling before and after transplant.
Primary metabolic disorders. Examples are Wilson’s disease, hereditary haemochromatosis and α1-antitrypsin deficiency.
Other conditions, such as primary sclerosing cholangitis (PSC), polycystic liver disease, NASH and metabolic diseases in which the defect is in the liver, e.g. primary oxaluria.
Contraindications
Absolute contraindications include active sepsis outside the hepatobiliary tree, malignancy outside the liver, liver metastases (except neuroendocrine), and if the patient is not psychologically committed.
Relative contraindications are mainly anatomical considerations that would make surgery more difficult, such as extensive splanchnic venous thrombosis. With exceptions, patients aged 70 years or over are not usually transplanted. In hepatocellular carcinoma the recurrence rate is high unless there are fewer than three small (<3 cm) lesions or a solitary nodule of <5 cm.
Preparation for surgery
Pretransplant work-up includes confirmation of the diagnosis, ultrasound and cross-sectional imaging, radiological demonstration of the hepatic arterial and biliary tree as well as assessment of cardiorespiratory and renal status. Because of the ethical and financial implications of this operation, regular psychosocial support is vital, and psychiatric counselling may be necessary in some cases.
The donor should be ABO compatible (but no HLA matching is necessary) and have no evidence of active sepsis, malignancy, HIV, HBV or HCV infection. Younger donors (<50 years) result in better graft function. The donor liver is cooled and stored on ice; its preservation time can be up to 20 h. The recipient operation takes approximately 8 hours and rarely requires a large blood transfusion, and sometimes none at all. Cadaveric donor livers may consist of whole graft, split grafts (for two recipients) or reduced grafts or from non-heart-beating donors. Live donors may be healthy individuals or patients with, for example, familial amyloid polyneuropathy, whose livers can then be transplanted into others (domino transplant). Right lobe donors have a mortality between 1 in 200 and 1 in 400.
The operative mortality is low. Most postoperative deaths occur in the first 3 months. Sepsis and haemorrhage can be serious complications. Opportunistic infections are still a problem owing to immunosuppression. Various immunosuppressive agents have been used, but microemulsified ciclosporin, tacrolimus in combination with either azathioprine or mycophenolate mofetil, steroids and sirolimus are the most common. A pretransplant serum creatinine above 160 µmol/L (2 mg/dL) is the best predictor of post-transplant death.
Rejection
Acute or cellular rejection is usually seen 5–10 days post-transplant; it can be asymptomatic or there may be a fever. Histologically, there is a pleomorphic portal infiltrate with prominent eosinophils, bile duct damage and endothelialitis of the blood vessels. This type of rejection responds to immunosuppressive therapy.
Chronic ductopenic rejection is seen from 6 weeks to 9 months post-transplant, with disappearing bile ducts (vanishing bile duct syndrome, VBDS) and an arteriopathy with narrowing and occlusion of the arteries. Early ductopenic rejection may rarely be reversed by immunosuppression, but often requires retransplantation.
Prognosis
Elective liver transplantation in low-risk patients has a 90% 1-year survival. Five-year survivals are now as high as 70–85%. Patients require lifelong immunosuppression, although the doses can be reduced over time without significant problems. Transplantation for HCV cirrhosis, PSC and HCC, are the major diseases in which long term survival is compromised by disease recurrence.
Complications and effects of cirrhosis
These are shown in Table 7.12.
Table 7.12 Complications and effects of cirrhosis
Portal hypertension
The portal vein is formed by the union of the superior mesenteric and splenic veins. The pressure within it is normally 5–8 mmHg with only a small gradient across the liver to the hepatic vein in which blood is returned to the heart via the inferior vena cava. Portal hypertension can be classified according to the site of obstruction:
Prehepatic – due to blockage of the portal vein before the liver
Intrahepatic – due to distortion of the liver architecture, which can be presinusoidal (e.g. in schistosomiasis) or post-sinusoidal (e.g. in cirrhosis)
Post-hepatic – due to venous blockage outside the liver (rare).
As portal pressure rises above 10–12 mmHg, the compliant venous system dilates and collaterals occur within the systemic venous system. The main sites of the collaterals are at the gastro-oesophageal junction, rectum, left renal vein, diaphragm, retroperitoneum and the anterior abdominal wall via the umbilical vein.
The collaterals at the gastro-oesophageal junction (varices) are superficial in position and tend to rupture. Portosystemic anastomoses at other sites seldom give rise to symptoms. Rectal varices are found frequently (30%) if carefully looked for and can be differentiated from haemorrhoids, which are lower in the anal canal. The microvasculature of the gut becomes congested giving rise to portal hypertensive gastropathy and colopathy, in which there is punctate erythema and sometimes erosions, which can bleed.
Pathophysiology
Portal vascular resistance is increased in chronic liver disease. During liver injury, stellate cells are activated and transform into myofibroblasts. In these cells there is de novo expression of the specific smooth muscle protein α-actin. Under the influence of mediators, such as endothelin, nitric oxide or prostaglandins, the contraction of these activated cells contributes to abnormal blood flow patterns and increased resistance to blood flow. In addition the balance of fibrogenic and fibrolytic factors is shifted towards fibrogenesis. This increased resistance leads to portal hypertension and opening of portosystemic anastomoses in both precirrhotic and cirrhotic livers. Neoangiogenesis also occurs. Patients with cirrhosis have a hyperdynamic circulation. This is thought to be due to the release of mediators, such as nitric oxide and glucagon, which leads to peripheral and splanchnic vasodilatation. This effect is followed by plasma volume expansion due to sodium retention (see the discussion on ascites, p. 335), and this has a significant effect in maintaining portal hypertension.
Causes (Table 7.13)
The most common cause is cirrhosis. Other causes include the following.
Table 7.13 Causes of portal hypertension
|
|
Prehepatic causes
Extrahepatic blockage is due to portal vein thrombosis. The cause is often unidentified, but some cases are due to portal vein occlusion secondary to congenital portal venous abnormalities or neonatal sepsis of the umbilical vein. Many are due to inherited defects causing prothrombotic conditions, e.g. factor V Leiden.
Patients usually present with bleeding, often at a young age. They have normal liver function and, because of this, their prognosis following bleeding is excellent.
The portal vein blockage can be identified by ultrasound with Doppler imaging; CT and MR angiography are also used.
Treatment for variceal bleeding is usually repeated endoscopic therapy or non-selective beta-blockade. Splenectomy is only performed if there is isolated splenic vein thrombosis. Anticoagulation prevents further thrombosis and intestinal infarction, and does not increase the risk of bleeding and prevents intestinal infarction.
Intrahepatic causes
Although cirrhosis is the most common intrahepatic cause of portal hypertension, there are other causes:
Non-cirrhotic portal hypertension. Patients present with portal hypertension and variceal bleeding but without cirrhosis. Histologically, the liver shows mild portal tract fibrosis. The aetiology is unknown, but arsenic, vinyl chloride, antiretroviral therapy and other toxic agents have been implicated. A similar disease is found frequently in India. The liver lesion does not progress and the prognosis is therefore good.
Schistosomiasis with extensive pipe-stem fibrosis is the commonest cause, but is confined to endemic areas such as Egypt and Brazil. However, often there may be concomitant liver disease such as HCV infection.
Other causes include congenital hepatic fibrosis, nodular regenerative hyperplasia and partial nodular transformation (the last two conditions are rare). They all share the common features of hyperplastic liver cell growth in the form of nodules. A wedge liver biopsy is usually required to establish the diagnosis. In none of these conditions are hormones implicated in aetiology or progression.
Post-hepatic causes
Prolonged severe heart failure with tricuspid incompetence and constrictive pericarditis can both lead to portal hypertension. The Budd–Chiari syndrome is described on page 343.
Clinical features
Patients with portal hypertension are often asymptomatic and the only clinical evidence of portal hypertension is splenomegaly. Clinical features of chronic liver disease are usually present (see p. 312). Presenting features include:
Variceal haemorrhage
Approximately 90% of patients with cirrhosis will develop gastro-oesophageal varices, over 10 years, but only one-third of these will bleed from them. Bleeding is likely to occur with large varices, red signs on varices (diagnosed at endoscopy) and in severe liver disease.
Management
Management can be divided into:
Prophylactic measures to prevent the first haemorrhage. Despite all the therapeutic techniques available, the prognosis depends on the severity of the underlying liver disease, with an overall mortality from variceal haemorrhage of 15% to 25%, reaching 50% in Child’s grade C.
Initial management of acute variceal bleeding (Fig. 7.24)
See also the discussion of the general management of gastrointestinal haemorrhage on page 327.
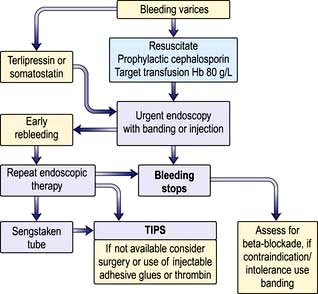
Figure 7.24 Management of gastrointestinal haemorrhage due to oesophageal varices. TIPS, transjugular intrahepatic portosystemic shunt.
Assess the general condition of the patient – pulse and blood pressure.
Insert an intravenous line and obtain blood for grouping and crossmatching, haemoglobin, PT/INR, urea, electrolytes, creatinine, liver biochemistry and blood cultures.
Restore blood volume with plasma expanders or, if possible, blood transfusion. These measures are discussed in more detail in the treatment of shock (p. 886). Prompt correction, but not over-correction, of hypovolaemia is necessary in patients with cirrhosis as their baroreceptor reflexes are diminished. Target haemoglobin only needs to be 80 g/L, as this lessens the likelihood of early rebleeding.
Monitor for alcohol withdrawal. Give thiamine i.v.
Start prophylactic antibiotics – third-generation cephalosporins, e.g. cefotaxime. These treat and prevent infection and early rebleeding and reduce mortality.
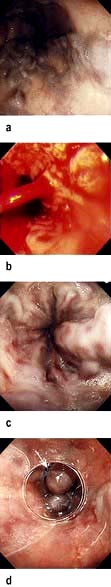
Endoscopic pictures of oesophageal varices. (a) Gross varices. (b) Blood spurting from a varix. (c) Following a recent bleed. (d) Varices with a band in place (arrow).
(a, c and d Courtesy of Dr Peter Fairclough.)
Endoscopy should be performed to confirm the diagnosis of varices. It also excludes bleeding from other sites (e.g. gastric ulceration) or portal hypertensive (or congestive) gastropathy. The latter term is used for chronic gastric congestion, punctate erythema and gastric erosions. It is a source of bleeding but varices may or may not be present. Propranolol (see below) is the best treatment for this gastropathy.
Variceal banding or injection sclerotherapy
Varices can be banded by mounting a band on the tip of the endoscope, sucking the varix just into the end of the scope and dislodging the band over the varix using a trip-wire mechanism. Alternatively, varices can be injected with a sclerosing agent that may arrest bleeding by producing vessel thrombosis. A needle is passed down the biopsy channel of the endoscope and a sclerosing agent is injected into the varices.
Acute variceal banding and sclerotherapy are the treatments of choice; they arrest bleeding in more than 80% of cases and reduce early rebleeding. Between 15% and 20% of bleeding comes from gastric varices and results of sclerotherapy and banding are poor. Injection of tissue glue is preferable.
Other measures available
The main use of this is for emergency control of bleeding while waiting for endoscopy and in combination with endoscopic techniques. The aim of vasoconstrictor agents is to restrict portal inflow by splanchnic arterial constriction.
Terlipressin. This is the only vasoconstrictor shown to reduce mortality. The dose is 2 mg 6-hourly, reducing to 1 mg 4-hourly after 48 h if a prolonged dosage regimen is used. It should not be given to patients with ischaemic heart disease. The patient will complain of abdominal colic, will defecate and have facial pallor owing to the generalized vasoconstriction.
Somatostatin. This drug has few side-effects. An infusion of 250–500 µg/hour appears to reduce bleeding, but has no effect on mortality. It should be used if there are contraindications to terlipressin.
Balloon tamponade is used mainly to control bleeding if endoscopic therapy or vasoconstrictor therapy has failed or is contraindicated or if there is exsanguinating haemorrhage. The usual balloon tube is a four-lumen Sengstaken–Blakemore, which should be left in place for no more than 12 hours and removed in the endoscopy room prior to the endoscopic procedure. The tube is passed into the stomach and the gastric balloon is inflated with air and pulled back. It should be positioned in close apposition to the gastro-oesophageal junction to prevent the cephalad variceal blood flow to the bleeding point. The oesophageal balloon should be inflated only if bleeding is not controlled by the gastric balloon alone.
This technique is successful in up to 90% of patients and is very useful in the first few hours of bleeding. However, it can have serious complications such as aspiration pneumonia, oesophageal rupture and mucosal ulceration, which lead to a 5% mortality. The procedure is very unpleasant for the patient.
A self-expanding covered metal stent with a wire loop to enable removal, introduced orally or endoscopically, can be placed over the varices, is effective and has the advantage that swallowing is not impaired. It is removed 7 days after insertion.
Additional management of acute episode
Measures to prevent encephalopathy. Portosystemic encephalopathy (PSE) can be precipitated by a large bleed (since blood contains protein). The management is described on page 337.
Nursing. Patients require high-dependency/intensive care nursing. They should be nil by mouth until bleeding has stopped.
Reduction in acid secretion. Ranitidine is preferable to proton pump inhibitors as it lessens the risk of C. difficile infection. Sucralfate 1 g four times daily can reduce oesophageal ulceration following endoscopic therapy.
Management of an acute rebleed
Rebleeding occurs in about 15–20% within 5 days after a single session of therapeutic endoscopy. The source of rebleeding should be established by endoscopy. It is sometimes due to a sclerotherapy-induced ulcer or slippage of a ligation band. Management starts with repeat endoscopic therapy – once only to control rebleeding (further sessions of sclerotherapy or banding are not advisable).
Transjugular intrahepatic portocaval shunt (TIPS)
TIPS is used when bleeding cannot be stopped or early rebleeding occurs after endoscopic therapy within 5 days. In this technique, a guidewire is passed from the jugular vein into the liver and into the portal vein. After a balloon expansion of the tract between hepatic and portal vein, an expandable covered metal shunt is placed over the wire to form a channel between the systemic and portal venous systems. It reduces the hepatic sinusoidal and portal vein pressure by creating a total shunt. There is an increased risk of portal systemic encephalopathy. Recurrent portal hypertension due to stent stenosis or thrombosis is far less frequent with ‘covered’ compared to ‘bare’ stents. Collaterals arising from the splenic or portal veins can be selectively embolized.
This is used when other measures fail or if TIPS is not available and, particularly, if the rebleeding is from gastric fundal varices. Oesophageal transection and ligation of the feeding vessels to the bleeding varices is the most common surgical technique. Acute portosystemic shunt surgery (see below) is infrequently performed.
Prevention of recurrent variceal bleeding
The risk of recurrence of bleeding without prophylaxis is 60–80% over a 2-year period with an approximate mortality of 20% per episode.
Non-selective beta-blockade. Oral propranolol in a dose sufficient to reduce resting pulse rate by 25% has been shown to decrease portal pressure. Portal inflow is reduced by two mechanisms: by a decrease in cardiac output (β1), and by the blockade of β2 vasodilator receptors on the splanchnic arteries, leaving an unopposed vasoconstrictor effect. This decreases the frequency of rebleeding, and is as effective as sclerotherapy and ligation as it also prevents bleeding from portal hypertensive gastropathy. It is the treatment of first choice, combined with endoscopic ligation (see below), but a substantial number of patients either have contraindications or are intolerant of beta-blockers. Significant reduction of hepatic venous pressure gradient (HVPG, measured by hepatic vein catheterization) is associated with very low rates or absence of rebleeding, particularly if <12 mmHg. Assessment of HVPG target reduction has prognostic specificity but poor sensitivity and thus poor clinical applicability, and as combined ligation and beta-blockers are the established treatment, monitoring of HVPG is redundant.
Endoscopic treatment. The use of repeated courses of banding at 2-weekly intervals leads to obliteration of varices. This markedly reduces rebleeding, most instances occurring before the varices have been fully obliterated. Between 30% and 40% of varices return per year, so follow-up endoscopy with ablation should be performed. Banding is superior to sclerotherapy, and should be used combined with beta-blockers.
Although a reduction in bleeding episodes occurs, the effect on survival is controversial and probably small. Complications include oesophageal ulceration, mediastinitis and rarely strictures.
Transjugular portosystemic stent shunts. These reduce rebleeding rates compared to endoscopic techniques, but do not improve survival and increase encephalopathy. They are used if endoscopic or medical therapy fails.
Surgical portosystemic shunting is associated with an extremely low risk of rebleeding, and is used if TIPS is not available. Hepatic encephalopathy is a significant complication. Operative mortality is low in patients with Child’s grade A (0–5%) but rises with worsening liver disease. The ‘shunts’ performed are usually an end-to-side portocaval anastomosis or a selective distal splenorenal shunt (Warren shunt), which transiently maintains hepatic blood flow via the superior mesenteric vein.
Devascularization procedures including oesophageal transection do not produce encephalopathy, and can be used when there is splanchnic venous thrombosis.
Liver transplantation (p. 331) is the best option when there is poor liver function.
Prophylactic measures
Patients with cirrhosis and varices that have not bled should be prescribed non-selective beta-blockers (e.g. propranolol or carvedilol). This reduces the chances of upper GI bleeding, may increase survival and is cost-effective. If there are contraindications or intolerance, variceal banding is an option. Beta-blockers do not prevent development of varices.
Ascites
Ascites is fluid within the peritoneal cavity and is a common complication of cirrhosis. The pathogenesis of ascites in liver disease is secondary to renal sodium and water retention. Several factors are involved.
Sodium and water retention results from peripheral arterial vasodilatation and consequent reduction in the effective blood volume. Nitric oxide and other substances (e.g. atrial natriuretic peptide and prostaglandins) act as vasodilators. The reduction in effective blood volume activates various neurohumoral pressor systems such as the sympathetic nervous system and the renin–angiotensin system, thus promoting salt and water retention (see Fig. 13.5).
Portal hypertension exerts a local hydrostatic pressure and leads to increased hepatic and splanchnic production of lymph and transudation of fluid into the peritoneal cavity.
Low serum albumin (a consequence of poor synthetic liver function) may further contribute by a reduction in plasma oncotic pressure.
In patients with ascites, urine sodium excretion rarely exceeds 5 mmol in 24 hours. Loss of sodium from extrarenal sites accounts for approximately 30 mmol in 24 hours. The normal daily dietary sodium intake may vary between 120 and 200 mmol, resulting in a positive sodium balance of approximately 90–170 mmol in 24 h (equivalent to 600–1300 mL of fluid retained).
Clinical features
The abdominal swelling associated with ascites develops over many weeks or as rapidly as a few days. Precipitating factors include a high sodium diet or the development of a hepatocellular carcinoma or splanchnic vein thrombosis. Mild generalized abdominal pain and discomfort are common but, if more severe, should raise the suspicion of spontaneous bacterial peritonitis (see below). Respiratory distress accompanies tense ascites, and also causes difficulty in eating.
The presence of fluid is confirmed by demonstrating shifting dullness. Many patients also have peripheral oedema. A pleural effusion (usually on the right side) may infrequently be found and arises from the passage of ascitic fluid through congenital diaphragmatic defects.
Investigations
A diagnostic aspiration of 10–20 mL of fluid should be obtained and the following performed:
Cell count. A neutrophil count above 250 cells/mm3 is indicative of an underlying (usually spontaneous) bacterial peritonitis.
Gram stain and culture – for bacteria and acid-fast bacilli.
Protein. A high serum-ascites albumin gradient of >11 g/L suggests portal hypertension, and a low gradient <11 g/L is associated with abnormalities of the peritoneum, e.g. inflammation, infections, neoplasia (Box 7.4).
Box 7.4
The serum-ascites albumin gradient
Modified from: Chung RT, Iafrate AJ, Amrein PC et al. Case records of the Massachusetts General Hospital. New England Journal of Medicine 2006; 354: 2166–2175.
The differential diagnosis of ascites is listed in Table 7.14.
Table 7.14 Causes of ascites divided according to the type of ascitic fluid
|
|
Management
The aim is to both reduce sodium intake and increase renal excretion of sodium, producing a net reabsorption of fluid from the ascites into the circulating volume. The maximum rate at which ascites can be mobilized is 500–700 mL in 24 h (see below). The management is as follows:
Check serum electrolytes, creatinine and eGFR at the start and every other day; weigh patient and measure urinary output daily.
Bed rest alone will lead to a diuresis in a small proportion of people by improving renal perfusion, but in practice is not helpful.
By dietary sodium restriction it is possible to reduce sodium intake to 40 mmol in 24 h and still maintain an adequate protein and calorie intake with a palatable diet.
Drugs: many contain significant amounts of sodium (up to 50 mmol daily). Examples include antacids, antibiotics (particularly the penicillins and cephalosporins) and effervescent tablets. Sodium-retaining drugs (non-steroidals, corticosteroids) should be avoided.
Fluid restriction is probably not necessary unless the serum sodium is under 128 mmol/L (see below).
The diuretic of first choice is the aldosterone antagonist spironolactone, starting at 100 mg daily. Chronic administration produces gynaecomastia. Eplerenone 25 mg once daily does not cause gynaecomastia.
The aim of diuretic therapy should be to produce a net loss of fluid approaching 700 mL in 24 hours (0.7 kg weight loss or 1.0 kg if peripheral oedema is present). Although 60% of patients respond with this regimen, diuresis is often poor and the spironolactone can be increased gradually to 400 mg daily providing there is no hyperkalaemia. A loop diuretic, such as furosemide 20–40 mg or bumetanide 0.5 mg or 1 mg daily, is added if response is poor. These loop diuretics have several potential disadvantages, including hyponatraemia, hypokalaemia and volume depletion.
Ascitic fluid is mobilized more slowly than interstitial fluid, and diuretics should be given with great care in those without peripheral oedema.
Diuretics should be temporarily discontinued if a rise in serum creatinine level occurs, representing overdiuresis and hypovolaemia, or if there is hyperkalaemia or the development of precoma. Hyponatraemia occurring during therapy almost always represents haemodilution secondary to a failure to clear free water (usually a marker of reduced renal perfusion) and should be treated by stopping the diuretics if the sodium level falls below approximately 128 mmol/L as well as introducing water restriction. Vaptans (p. 645), a class of drugs that increase free water clearance by inhibition of vasopressin receptors, are being evaluated in cirrhosis.
This is used to relieve symptomatic tense ascites or when diuretic therapy is insufficient to control accumulation of fluid. The main complication is hypovolaemia and renal dysfunction (post-paracentesis circulatory dysfunction) as the ascites reaccumulates at the expense of the circulating volume; this is more likely with >5 L removal and worse liver function. In patients with normal renal function and without hyponatraemia, this is overcome by infusing albumin (8 g/L of ascitic fluid removed). In practice, up to 20 L can be removed over 4–6 hours, with albumin infusion.
A transjugular intrahepatic portosystemic shunt (TIPS) is used for resistant ascites providing there is no spontaneous portosystemic encephalopathy and minimal disturbance of renal function. Frequency of paracentesis and diuretic use is usually reduced and nutrition improves. Survival may improve. The use of a peritoneo-venous shunt has been abandoned in most centres due to a high rate of blockage.
Spontaneous bacterial peritonitis (SBP)
This represents a serious complication of ascites with cirrhosis and occurs in approximately 8%. The infecting organisms gain access to the peritoneum by haematogenous spread; most are Escherichia coli, Klebsiella or enterococci. The condition should be suspected in any patient with ascites who clinically deteriorates. Features such as pain and pyrexia are frequently absent. Diagnostic aspiration should always be performed (see above). A raised neutrophil count in ascites is alone sufficient evidence to start treatment immediately. A third-generation cephalosporin, such as cefotaxime or ceftazidime, is used and is modified on the basis of culture results. Mortality is 10–15%. Recurrence is common (70% within a year) and an oral quinolone, e.g. norfloxacin 400 mg daily, is given for prevention, prolonging the survival. Primary prophylaxis of SBP in patients with ascites protein <10 g/L or severe liver disease also prevents hepatorenal syndrome and improves survival.
Portosystemic encephalopathy (PSE)
This is a chronic neuropsychiatric syndrome secondary to cirrhosis. Acute encephalopathy can occur in acute hepatic failure (see p. 326). PSE can occur in portal hypertensive patients due to spontaneous ‘shunting’, or in those with surgical or TIPS shunts. Encephalopathy is potentially reversible.
Pathogenesis
The mechanism is unknown but several factors are involved. In cirrhosis, the portal blood bypasses the liver via the collaterals and the ‘toxic’ metabolites pass directly to the brain to produce the encephalopathy.
Many ‘toxic’ substances may be causative factors, principally ammonia, but also free fatty acids, mercaptans and accumulation of false neurotransmitters (octopamine) or activation of the γ-aminobutyric acid (GABA) inhibitory neurotransmitter system. Increased blood levels of aromatic amino acids (tyrosine and phenylalanine) and reduced branched-chain amino acids (valine, leucine and isoleucine) also occur. Ammonia has a major role; ammonia-induced alteration of brain neurotransmitter balance – especially at the astrocyte-neurone interface – is the leading pathophysiological mechanism. Ammonia is produced by intestinal bacteria breaking down protein. The factors precipitating PSE are shown in Table 7.15.
Table 7.15 Factors precipitating portosystemic encephalopathy
TIPS, transjugular intrahepatic portocaval shunt.
Clinical features
An acute onset often has a precipitating factor (Table 7.15). The patient becomes increasingly drowsy and comatose.
Chronically, there is a disorder of personality, mood and intellect, with a reversal of normal sleep rhythm. These changes may fluctuate, and a history from a relative must be obtained. The patient is irritable, confused, disorientated and has slow slurred speech. General features include nausea, vomiting and weakness. Coma occurs as the encephalopathy becomes more marked, but there is always hyperreflexia and increased tone. Convulsions are so very rare that other causes must be looked for.
Fetor hepaticus (a sweet smell to the breath)
A coarse flapping tremor seen when the hands are outstretched and the wrists hyperextended (asterixis)
Constructional apraxia, with the patient being unable to write or draw, e.g. a five-pointed star
Decreased mental function, which can be assessed by using the serial-sevens test. A trail-making (or connection) test (the ability to join numbers and letters (in chronological order) with a pen within a certain time – a standard psychological test for brain dysfunction) is prolonged.
Diagnosis is clinical. Routine liver biochemistry merely confirms the presence of liver disease, not the presence of encephalopathy.
Electroencephalography (EEG) shows a decrease in the frequency of the normal α-waves (8–13 Hz) to α-waves of 1.5–3 Hz. These changes occur before coma supervenes.
Visual evoked responses (see p. 1090) also detect subclinical encephalopathy.
Arterial blood ammonia can be useful for the differential diagnosis of coma and to follow a patient with PSE, but is not readily available.
Management
Identify and remove the possible precipitating cause, such as drugs with cerebral depressant properties, constipation or electrolyte imbalance due to overdiuresis.
Give purgation and enemas to empty the bowels of nitrogenous substances. Lactulose (10–30 mL three times daily) is an osmotic purgative that reduces the colonic pH and limits ammonia absorption. Lactilol (β-galactoside sorbitol 30 g daily) is metabolized by colonic bacteria and is comparable in efficacy to lactulose.
Maintain nutrition with adequate calories, given if necessary via a fine-bore nasogastric tube, and do not restrict protein for more than 48 h.
Give antibiotics. Rifaximin is mainly unabsorbed and well tolerated long term, and prevents further episodes of PSE. Metronidazole (200 mg four times daily) is also effective in the acute situation. Neomycin should be avoided. Stop or reduce diuretic therapy.
Give intravenous fluids as necessary (beware of too much sodium).
Increase protein in the diet to the limit of tolerance as the encephalopathy improves.
FURTHER READING
Rodriguez-Roisin R, Krowka MJ. Hepatopulmonary syndrome. N Engl J Med 2008; 358:2378–2387.
Schuppan D, Afdhal NH. Liver cirrhosis. Lancet 2008; 371:838–851.
Shawcross DL, Jalan R. Dispelling myths in the treatment of hepatic encephalopathy. Lancet 2005; 365:431–433.
Sidhu SS, Goyal O, Mishra BP et al. Rifaximin improves psychometric performance and health-related quality of life in patients with minimal hepatic encephalopathy (the RIME trial). Am J Gastroenterol 2011; 106:307–316.
Wong F, Nadim MK, Kellum JA et al. Working Party proposal for a revised classification system of renal dysfunction in patients with cirrhosis. Gut 2011; 60: 702–709.
Course and prognosis
Acute encephalopathy in acute liver failure has a very poor prognosis as the disease itself has a high mortality. In cirrhosis, chronic PSE is very variable and adversely affects prognosis. Very rarely with chronic portosystemic shunting, an organic syndrome with cerebellar signs or choreoathetosis can develop, as well as a myelopathy leading to a spastic paraparesis due to demyelination. Patients should be referred to a liver transplant centre.
Renal failure (hepatorenal syndrome)
The hepatorenal syndrome occurs typically in a patient with advanced cirrhosis, portal hypertension with jaundice and ascites. The urine output is low with a low urinary sodium concentration, a maintained capacity to concentrate urine (i.e. tubular function is intact) and almost normal renal histology. The renal failure is described as ‘functional’. It is sometimes precipitated by overvigorous diuretic therapy, NSAIDs, diarrhoea or paracentesis, and infection, particularly spontaneous bacterial peritonitis.
The mechanism is similar to that producing ascites. The initiating factor is thought to be extreme peripheral vasodilatation, possibly due to nitric oxide, leading to an extreme decrease in the effective blood volume and hypotension (p. 305). This activates the homeostatic mechanisms, causing a rise in plasma renin, aldosterone, noradrenaline (norepinephrine) and vasopressin, leading to vasoconstriction of the renal vasculature. There is an increased preglomerular vascular resistance causing the blood flow to be directed away from the renal cortex. This leads to a reduced glomerular filtration rate and plasma renin remains high. Salt and water retention occur with reabsorption of sodium from the renal tubules. There is also a decrease in cardiac output inappropriate to the degree of systemic vasodilatation, which further exacerbates the haemodynamic abnormalities.
Other mediators have been incriminated in the pathogenesis of the hepatorenal syndrome, in particular the eicosanoids. This has been supported by the precipitation of the syndrome by inhibitors of prostaglandin synthase such as non-steroidal anti-inflammatory drugs (NSAIDs).
Diuretic therapy should be stopped and intravascular hypovolaemia corrected, preferably with albumin. Terlipressin or noradrenaline with intravenous albumin improves renal function in one-third of patients. Liver transplantation is the best option.
Hepatopulmonary syndrome
This is defined as a hypoxaemia occurring in patients with advanced liver disease. It is due to intrapulmonary vascular dilatation with no evidence of primary pulmonary disease. The patients have features of cirrhosis with spider naevi and clubbing as well as cyanosis. Most patients have no respiratory symptoms, but with more severe disease, patients are breathless on standing. Transthoracic ECHO shows intrapulmonary shunting, and arterial blood gases confirm the arterial oxygen desaturation. These changes are improved with liver transplantation.
Porto-pulmonary hypertension
This must be distinguished from the hepatopulmonary syndrome as in this group there is pulmonary hypertension. It occurs in 1–2% of patients with cirrhosis related to portal hypertension. It may respond to medical therapy. Severe pulmonary hypertension is a contraindication for liver transplantation.
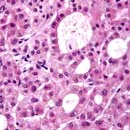
Types of cirrhosis
Primary biliary cirrhosis
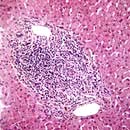
Primary biliary cirrhosis showing portal tract inflammation with lymphocytes and plasma cells (×10).
Primary biliary cirrhosis (PBC) is a chronic disorder in which there is a progressive destruction of the small bile ducts, eventually leading to cirrhosis. Of those affected, 90% are women in the age range 40–50 years. PBC is frequently being diagnosed in its milder forms. The prevalence is approximately 7.5 per 100 000, with a 1–6% increase in first-degree relatives. PBC has been called ‘chronic non-suppurative destructive cholangitis’; this term is more descriptive of the early lesion and emphasizes that true cirrhosis occurs only in the later stages of the disease.
Aetiology
The aetiology is unknown, but immunological mechanisms play a part. Serum anti-mitochondrial antibodies (AMA) are found in almost all patients with PBC, and of the mitochondrial proteins involved, the antigen M2 is specific to PBC.
Five M2-specific antigens have been further defined using immunoblot techniques, of which the E2 component of the pyruvate dehydrogenase complex (PDC) is the major M2 autoantigen (72 kDa E2 subunit (PDC, E2)). The presence of AMA in high titre is unrelated to the clinical or histological picture and its role in pathogenesis is unclear. Antibodies against nuclear antigens, e.g. anti gp210, are present in 50% of patients and correlate with progression towards liver failure.
It seems likely that an environmental factor acts on a genetically predisposed host via molecular mimicry initiating autoimmunity. E. coli and N. aromaticivorans antibodies are present in high titre. Halogenated hydrocarbons mimic the PDC autoepitopes.
Although damage to bile ducts is a feature, antibodies to bile ductules are not specific to PBC. Biliary epithelium from patients with PBC expresses aberrant class II HLAs, but it is not known whether this expression is the cause or result of the inflammatory response. Cell-mediated immunity is impaired (demonstrated both in vitro and by skin testing); cytotoxic CD4+ and CD8+ T lymphocytes directly produce biliary epithelium damage. They recognize the inner lipoyl domain and lipoic acid also recognized by AMA. There is an increased synthesis of IgM, thought to be due to a failure of the switch from IgM to IgG antibody synthesis. No specific associated class II MHC loci have been found.
Clinical features
Asymptomatic patients are discovered on routine examination or screening and may have hepatomegaly, a raised serum alkaline phosphatase or autoantibodies.
Pruritus is often the earliest symptom, preceding jaundice by a few years. Fatigue, which is often disabling, may accompany pruritus, particularly in progressive cases. When jaundice appears, hepatomegaly is usually found. In the later stages, patients are pigmented and jaundiced with severe pruritus. Pigmented xanthelasma on eyelids or other deposits of cholesterol in the creases of the hands are seen.
Associated disorders
Autoimmune disorders (e.g. Sjögren’s syndrome, scleroderma, thyroid disease) occur with increased frequency. Keratoconjunctivitis sicca (dry eyes and mouth) is seen in 70% of cases. Renal tubular acidosis, membranous glomerulonephritis, coeliac disease and interstitial pneumonitis are also associated with PBC.
Investigations
Mitochondrial antibodies – measured routinely by ELISA (in titres >1 : 160) – are present in over 95% of patients; M2 antibody is specific. Other nonspecific antibodies (e.g. anti-nuclear factor and smooth muscle) may also be present.
High serum alkaline phosphatase is often the only abnormality in the liver biochemistry.
Ultrasound can show a diffuse alteration in liver architecture.
Liver biopsy shows characteristic histological features of a portal tract infiltrate, mainly of lymphocytes and plasma cells: approximately 40% have granulomas. Most of the early changes are in zone 1. Later, there is damage to and loss of small bile ducts with ductular proliferation. Portal tract fibrosis and, eventually, cirrhosis is seen.
Hepatic granulomas are not specific and are also seen in sarcoidosis, tuberculosis, schistosomiasis, drug reactions, brucellosis, parasitic infestation (e.g. strongyloidiasis) and other conditions.
Differential diagnosis
The classical picture presents little difficulty with diagnosis (high serum alkaline phosphatase and the presence of AMA); this can be confirmed by the characteristic histological features although this is not necessary except in doubtful cases. There is a group of patients with the histological changes of PBC but the serology of autoimmune hepatitis. This has been given the name of autoimmune cholangitis and responds to steroids and azathioprine.
In the jaundiced patient, extrahepatic biliary obstruction should be excluded by ultrasound and, if there is doubt about the diagnosis, MRCP (or ERCP) should be performed to make sure that the bile ducts are normal.
Treatment
Ursodeoxycholic acid (10–15 mg/kg) improves bilirubin and aminotransferase levels. It should be given early in the asymptomatic phase. It is not clear if prognosis is altered. Symptoms are not improved. Steroids improve biochemical and histological disease but may lead to increased osteoporosis and other side-effects and should not be used.
Malabsorption of fat-soluble vitamins (A, D and K) occurs and supplementation is required when deficiency is detected and prophylactically in the jaundiced patient. Bisphosphonates are required for osteoporosis. Despite raised serum lipid concentrations there is no increased risk from cardiovascular disease, although this has been disputed by one group.
Pruritus is difficult to control, but cholestyramine, one 4 g sachet three times daily, can be helpful, although it is unpalatable. Rifampicin, naloxone hydrochloride and naltrexone (opioid antagonists) have been shown to be of benefit. Intractable pruritus can be relieved by plasmapheresis or a molecular absorbent recirculating system (MARS).
The lack of effective medical therapy has made PBC a major indication for liver transplantation (p. 331).
Complications
The complications are those of cirrhosis. In addition, osteoporosis, and rarely osteomalacia and a polyneuropathy can also occur.
Course and prognosis
This is very variable. Asymptomatic patients and those presenting with pruritus will survive for more than 20 years. Symptomatic patients with jaundice have a more rapidly progressive course and die of liver failure or bleeding varices in approximately 5 years. Liver transplantation should therefore be offered when the serum bilirubin is persistently above 100 µmol/L. Transplantation has a 5-year survival of at least 80%.
Primary sclerosing cholangitis
Primary sclerosing cholangitis (PSC) is a chronic cholestatic liver disease characterized by fibrosing inflammatory destruction of both the intra- and extrahepatic bile ducts. In 75% of patients, PSC is associated with inflammatory bowel disease (usually ulcerative colitis) and it is not unusual for the PSC to predate the onset of the inflammatory bowel disease. The causes are unknown but genetic susceptibility to PSC is associated with the HLA A1-B8-DR3 haplotype. The autoantibody pANCA (anti-neutrophil cytoplasmic antibody) is found in the serum of 60% of cases. Seventy per cent of patients are men and the average age of onset is approximately 40 years. Secondary PSC is seen in patients with HIV and cryptosporidium (p. 189).
Clinical features
With increasing screening of patients with inflammatory bowel disease PSC is detected at an asymptomatic phase with abnormal liver biochemistry, usually a raised serum alkaline phosphatase. Symptomatic presentation is usually with fluctuating pruritus, jaundice and cholangitis.
Diagnosis
The typical biliary changes associated with PSC may be identified by MRCP. This technique may fail to identify minor, but still clinically significant, intrahepatic duct abnormalities and this may require endoscopic retrograde cholangiography (ERC). The cholangiogram characteristically shows irregularity of calibre of both intra- and extrahepatic ducts, although either may be involved alone (Fig. 7.25).
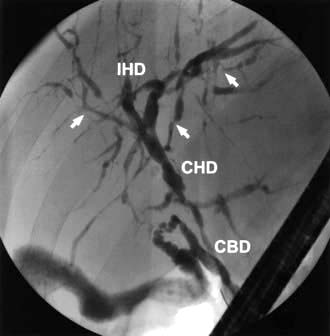
Figure 7.25 Primary sclerosing cholangitis. An endoscopic cholangiogram showing the typical features of primary sclerosing cholangitis. There are calibre irregularities of the intrahepatic ducts (IHD). There is also minor stricturing of the extrahepatic ducts at the confluence between the common bile duct (CBD) and the common hepatic duct (CHD).
Pathology
Histology can be contributory: it shows inflammation of the intrahepatic biliary radicles with associated scar tissue classically described as being onion skin in appearance. These changes range from minor inflammatory infiltrates to the level of established cirrhosis. The presence of cirrhosis has prognostic implications.
Management
PSC is a slowly progressive lesion (symptoms and biochemical tests may fluctuate), ultimately leading to liver cirrhosis and associated decompensation. Recurrent cholangitis may be a feature before the onset of cirrhosis. Cholangiocarcinoma occurs in up to 15% of patients.
The only proven treatment is liver transplantation. The bile acid ursodeoxycholic acid has been evaluated extensively in the treatment of PSC, but there is no evidence of long-term symptomatic, histological or survival benefit. High-dose therapy (30 mg/kg) may be deleterious. In a small minority of patients with PSC the dominant lesion is of the extrahepatic ducts. Such lesions may be amenable to endoscopic biliary intervention with balloon dilatation and temporary stent placement (p. 357).
Secondary biliary cirrhosis
Cirrhosis can result from prolonged (for months) large duct biliary obstruction. Causes include bile duct strictures, gallstones and sclerosing cholangitis. An ultrasound examination, followed by ERCP or PTC, is performed to outline the ducts and any remedial cause is treated.
Hereditary haemochromatosis (see also p. 310)
Hereditary haemochromatosis (HH) is an inherited disease characterized by excess iron deposition in various organs leading to eventual fibrosis and functional organ failure. There are four main types of inherited disorders:
Type 1 HFE. The HFE gene (mutation C282Y): commonest on chromosome 6
Type 2A. Juvenile HJV gene (mutation G320V)
Type 2B. Juvenile HAMP gene (mutation 93delG)
All are transmitted by an autosomal recessive gene, apart from the ferroportin iron overload which is dominantly transmitted.
Prevalence and aetiology
HH has a prevalence in Caucasians of homozygotes (affected) of 1 in 400, but very variable phenotypic expression and a heterozygote (carrier) frequency of 1 in 10. It is the most common single gene disorder in Caucasians.
Between 85% and 90% of patients with overt HH are homozygous for the Cys 282 Tyr (C282Y mutation), i.e. type 1 HFE. A second mutation (His 63 Asp, H63D) occurs in about 25% of the population and is in complete linkage disequilibrium with Cys 282 Tyr.
Another form of haemochromatosis (type 3) occurs in Southern Europe and is associated with TfR2, a transferrin receptor isoform. The other types, ferroportin related (type 4) and juvenile forms (types 2A and 2B), are much rarer.
Dietary intakes of iron and chelating agents (ascorbic acid) may be relevant. Iron overload may be present in alcoholics, but alcohol excess per se does not cause HH although there is a history of excess alcohol intake in 25% of patients.
Mechanism of damage. This is still unclear. The HFE gene protein interacts with the transferrin receptor 1, which is a mediator in intestinal iron absorption (see Fig. 8.8). Iron is taken up by the mucosal cells inappropriately, exceeding the binding capacity of transferrin.
Hepcidin, a protein synthesized in the liver (Fig. 7.26), is central to the control of iron absorption; it is increased in iron deficiency states and decreased with iron overload. The mutations described above disrupt hepcidin expression, thereby internalizing ferroportin leading to uninhibited iron overload.
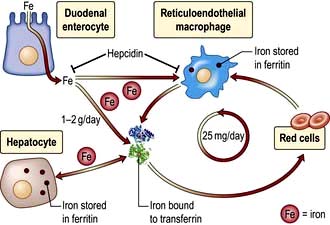
Figure 7.26 The circulation of iron from the duodenal enterocyte to and from the liver, red cells and the reticuloendothelial macrophages.
1–2 g of iron is absorbed from the intestine (Fig. 8.8, p. 378) and circulates bound to transferrin, The reticuloendothelial cells clear old erythrocytes and release iron to circulate and be stored as ferritin in the liver. The liver is the major site of production of the peptide hormone hepcidin. Hepcidin blocks release of iron from the erythrocytes and macrophages by degrading the iron exporter transferrin. Fe, iron.
(Modified from Fleming RE, Ponka P. Iron overload in human disease. N Engl J Med 2012; 366:348–359, with permission.)
Hepatic expression of the hepcidin gene is decreased in HFE haemochromatosis, facilitating liver iron overload. Excess iron is then taken up by the liver and other tissues gradually over a long period. It seems likely that it is the iron itself that precipitates fibrosis.
Pathology
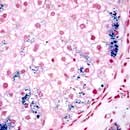
In symptomatic patients the total body iron content is 20–40 g, compared with 3–4 g in a normal person. The iron content is particularly increased in the liver and pancreas (50–100 times normal) but is also increased in other organs (e.g. the endocrine glands, heart and skin).
In established cases the liver shows extensive iron deposition and fibrosis. Early in the disease, iron is deposited in the periportal hepatocytes (in pericanalicular lysosomes). Later it is distributed widely throughout all acinar zones, biliary duct epithelium, Kupffer cells and connective tissue. Cirrhosis is a late feature.
Clinical features
The course of the disease depends on a number of factors, including gender, dietary iron intake, presence of associated hepatotoxins (especially alcohol) and genotypes. Overt clinical manifestations occur more frequently in men; the reduced incidence in women is probably explained by physiological blood loss and a smaller dietary intake of iron. Most affected individuals present in the 5th decade. The classic triad of bronze skin pigmentation (due to melanin deposition), hepatomegaly and diabetes mellitus is only present in cases of gross iron overload.
Hypogonadism secondary to pituitary dysfunction is the most common endocrine feature. Deficiency of other pituitary hormones is also found, but symptomatic endocrine deficiencies, such as loss of libido, are very rare. Cardiac manifestations, particularly heart failure and arrhythmias, are common, especially in younger patients. Calcium pyrophosphate is deposited asymmetrically in both large and small joints (chondrocalcinosis) leading to an arthropathy. The exact relationship of chondrocalcinosis to iron deposition is uncertain.
Complications
Of people with cirrhosis, 30% will develop primary hepatocellular carcinoma (HCC). HCC has only very rarely been described in non-cirrhotic patients in whom the excess iron stores have been removed. Early diagnosis is vital.
Investigations
Heterozygotes may have normal biochemical tests or modest increases in serum iron transferrin saturation (>45%) or serum ferritin (usually >400 µg/L).
This is not required for diagnosis, but is useful to assess the extent of tissue damage, assess tissue iron, and measure the hepatic iron concentration (>180 µmol/g dry weight of liver indicates haemochromatosis).
Mild degrees of parenchymal iron deposition in patients with other forms of cirrhosis, particularly due to alcohol, can often cause confusion with true homozygous HH.
MRI shows dramatic reduction in the signal intensity of the liver and pancreas owing to the paramagnetic effect of ferritin and haemosiderin. A highly T2-weighted gradient recalled echo (GRE) technique detects all clinically relevant liver iron overload (>60 µmol/g of liver). In secondary iron overload (haemosiderosis), which involves the reticuloendothelial cells, the pancreas is spared, enabling distinction between these two conditions.
Treatment and management
This prolongs life and may reverse tissue damage; the risk of malignancy still remains if cirrhosis is present. All patients should have excess iron removed as rapidly as possible. This is achieved using venesection of 500 mL performed twice-weekly for up to 2 years, i.e. 160 units with 250 mg of iron per unit, equals 40 g removed. During venesection, serum iron and ferritin and the mean corpuscular volume (MCV) should be monitored. These fall only when available iron is depleted. Three or four venesections per year are required to prevent reaccumulation of iron. Serum ferritin should remain within the normal range.
Manifestations of the disease usually improve or disappear, except for diabetes, testicular atrophy and chondrocalcinosis. The requirements for insulin often diminish in diabetic patients. Testosterone replacement is often helpful.
In the rare patient who cannot tolerate venesection (because of severe cardiac disease or anaemia), chelation therapy with desferrioxamine, either intermittently or continuously by infusion, has been successful in removing iron.
Screening
In all cases of HH, all first-degree family members must be screened to detect early and asymptomatic disease. HFE mutation analysis is performed with measurement of transferrin saturation and serum ferritin.
In the general population, the serum iron and transferrin saturation are the best and cheapest tests available.
Wilson’s disease (progressive hepatolenticular degeneration)
Dietary copper is normally absorbed from the stomach and upper small intestine. Copper is transported to the liver loosely bound to albumin where it is incorporated into apocaeruloplasmin forming caeruloplasmin, a glycoprotein synthesized in the liver, and secreted into the blood. The remaining copper is normally excreted in the bile and excreted in faeces.
Wilson’s disease is a very rare inborn error of copper metabolism that results in copper deposition in various organs, including the liver, the basal ganglia of the brain and the cornea. It is potentially treatable and all young patients with liver disease must be screened for this condition.
Aetiology
It is an autosomal recessive disorder with a molecular defect within a copper-transporting ATPase encoded by a gene (designated ATP7B) located on chromosome 13, affecting between 1 in 30 000 and 1 in 100 000 individuals. Over 300 mutations have been identified, the most frequent being His 1069 Gly (H1069Q) found in approximately 50% of Caucasian patients, with compound heterozygotes being frequent. This mutation is rare in India and Asia. Wilson’s disease occurs worldwide, particularly in countries where consanguinity is common. There is a failure of both incorporation of copper into procaeruloplasmin, which leads to low serum caeruloplasmin, and biliary excretion of copper. There is a low serum caeruloplasmin in over 80% of patients but this is not the cause of the copper deposition. The precise mechanism for the failure of copper excretion is not known.
Pathology
The liver histology is not diagnostic and varies from that of chronic hepatitis to macronodular cirrhosis. Stains for copper show a periportal distribution but this can be unreliable (see below). The basal ganglia are damaged and show cavitation, the kidneys show tubular degeneration, and erosions are seen in bones.
Clinical features
Children usually present with hepatic problems, whereas young adults have more neurological problems, such as tremor, dysarthria, involuntary movements and eventually dementia. The liver disease varies from episodes of acute hepatitis, especially in children, which can go on to fulminant hepatic failure, to chronic hepatitis or cirrhosis.
Typical signs are of chronic liver disease with neurological signs of basal ganglia involvement (p. 1082). A specific sign is the Kayser–Fleischer ring, which is due to copper deposition in Descemet’s membrane in the cornea. It appears as a greenish brown pigment at the corneoscleral junction and frequently requires slit-lamp examination for identification. It may be absent in young children.
Investigations
Serum copper and caeruloplasmin are usually reduced but can be normal.
Urinary copper is usually increased 100–1000 µg in 24 h (1.6–16 µmol); normal levels <40 µg (0.6 µmol).
Liver biopsy. The diagnosis depends on measurement of the amount of copper in the liver (>250 µg/g dry weight), although high levels of copper are also found in the liver in chronic cholestasis.
Haemolysis and anaemia may be present.
Genetic analysis is limited but selected exons are screened according to population group.
Treatment
Lifetime treatment with penicillamine, 1–1.5 g daily, is effective in chelating copper. If treatment is started early, clinical and biochemical improvement can occur. Urine copper levels should be monitored and the drug dose adjusted downwards after 2–3 years. Serious side-effects of the drug occur in 10% and include skin rashes, leucopenia, skin changes and renal damage. Trientine (1.2–1.8 g/day) and zinc acetate (150 mg/day) have been used as maintenance therapy and for asymptomatic cases. All siblings and children of patients should be screened (ATP7B mutation analysis is useful) and treatment given even in the asymptomatic if there is evidence of copper accumulation.
α1-Antitrypsin deficiency
A deficiency of α1-antitrypsin (α1-AT) (see also p. 793) is sometimes associated with liver disease and pulmonary emphysema (particularly in smokers). α1-AT is a glycoprotein, part of a family of serine protease inhibitors, or serpin superfamily. α1-AT deficiency is a genetic disorder and 1 in 10 northern Europeans carries an abnormal gene.
The protein is a 394-amino acid 52 kDa acute-phase protein that is synthesized in the liver and constitutes 90% of the serum α1-globulin seen on electrophoresis. Its main role is to inhibit the proteolytic enzyme, neutrophil elastase.
The gene is located on chromosome 14. The genetic variants of α1-ATare characterized by their electrophoretic mobilities as medium (M), slow (S) or very slow (Z). The normal genotype is protease inhibitor MM (PiMM), the homozygote for Z is PiZZ, and the heterozygotes are PiMZ and PiSZ. S and Z variants are due to a single amino acid replacement of glutamic acid at positions 264 and 342 of the polypeptide, respectively. This results in decreased synthesis and secretion of the protein by the liver as protein-protein interactions occur between the reactive centre loop of one molecule and the β-pleated sheet of a second (loop sheet polymerization).
How this causes liver disease is uncertain. It is postulated that the failure of secretion of the abnormal protein leads to an accumulation in the liver, causing liver damage.
Clinical features
The majority of patients with clinical disease are homozygotes with a PiZZ phenotype. Some may present in childhood and a few require transplantation. Approximately 10–15% of adult patients will develop cirrhosis, usually over the age of 50 years, and 75% will have respiratory problems. Approximately 5% of patients die of their liver disease. Heterozygotes (e.g. PiSZ or PiMZ) may develop liver disease, but the risk is small.
Investigations
Serum α1-antritrypsin is low, at 10% of the normal level in the PiZZ phenotypes, and 60% of normal in the S variant.
Histologically, periodic acid-Schiff (PAS)-positive, diastase-resistant globules which contain α1-AT are seen in periportal hepatocytes. Fibrosis and cirrhosis can be present.
Treatment
There is no treatment apart from dealing with the complications of liver disease. Patients with hepatic decompensation should be assessed for liver transplantation. Patients should stop smoking (see p. 317).