Chapter 22 Neurological disease
The impact of neurological disease
Neurology is a large and diverse subject which covers many conditions that require long-term coordinated care and have serious effects on the daily lives of patients and their families. Neurology includes conditions as diverse as cognitive disorders involving higher level mental functioning through to disorders of peripheral nerve and skeletal muscle. It is a specialty requiring good clinical skills and examination technique which cannot be replaced with investigations or imaging techniques alone (Table 22.1).
Table 22.1 UK incidence of common neurological conditions
| Conditions | Events per 100 000/year |
|---|---|
Cerebrovascular events |
210 |
Shingles (herpes zoster) and postherpetic neuralgia |
150 |
Diabetic and other neuropathies |
105 |
Epilepsy |
46 |
Parkinson’s disease |
19 |
Severe brain injury and subdural haematoma |
13 |
All CNS tumours |
9 |
Trigeminal neuralgia |
8 |
Meningitis |
7 |
Multiple sclerosis |
7 |
Presenile dementia (below 65 years) |
4 |
Myasthenia, all muscle and motor neurone disease |
5 |
Common symptoms and signs
Pattern recognition in neurology – interpretation of history, symptoms and examination – is very reliable. Practical experience is vital. There are three critical questions in formulating a clinical diagnosis:
Difficulty walking and falls
Change in walking pattern is a common complaint (Box 22.1). Arthritis and muscle pain make walking painful and slow (antalgic gait). The pattern of gait is valuable diagnostically.
Spasticity and hemiparesis
Spasticity (p. 1082), more pronounced in extensor muscles, with or without weakness, causes stiff and jerky walking. Toes of shoes become scuffed, catching level ground. Pace shortens; a narrow base is maintained. Clonus – involuntary extensor rhythmic leg jerking – may occur.
In a hemiparesis when spasticity is unilateral and weakness marked, the stiff, weak leg is circumducted and drags.
Parkinson’s disease: shuffling gait
There is muscular rigidity (p. 1118) throughout extensors and flexors. Power is preserved; the pace shortens, and slows to a shuffle; its base remains narrow. A stoop and diminished arm swinging become apparent. Gait becomes festinant (hurried) with short rapid steps. There is difficulty turning quickly and initiating movement, sometimes with falls. Retropulsion means small backward steps, taken involuntarily when a patient is halted.
Cerebellar ataxia: broad-based gait
In lateral cerebellar lobe disease (p. 1083) stance becomes broad-based, unstable and tremulous. Ataxia describes this incoordination. When walking, the person tends to veer to the side of the affected cerebellar lobe.
In disease of midline structures (cerebellar vermis), the trunk becomes unsteady without limb ataxia, with a tendency to fall backwards or sideways – truncal ataxia.
Sensory ataxia: stamping gait
Peripheral sensory lesions (e.g. polyneuropathy, p. 1145) cause ataxia because of loss of proprioception (position sense). Broad-based, high-stepping, stamping gait develops. This form of ataxia is exacerbated by removal of sensory input (e.g. vision) and worse in the dark. Romberg’s test, first described in sensory ataxia of tabes dorsalis (p. 1129), becomes positive.
Lower limb weakness: slapping and waddling gaits
When weakness is distal, each foot must be lifted over obstacles. When ankle dorsiflexors are weak, e.g. in a common peroneal nerve palsy (p. 1144), the sole returns to the ground with an audible slap.
Weakness of proximal lower limb muscles (e.g. polymyositis, muscular dystrophy) causes difficulty rising from sitting. Walking becomes a waddle, the pelvis being poorly supported by each leg.
Gait apraxia
With frontal lobe disease (e.g. tumour, hydrocephalus, infarction), acquired walking skills become disorganized. Leg movement is normal when sitting or lying but initiation and organization of walking fail. Shuffling small steps (marche à petits pas), gait ignition failure or undue hesitancy may predominate. Urinary incontinence and dementia are often present.
Dizziness, vertigo and blackouts
Dizziness covers many complaints, from a vague feeling of unsteadiness to severe, acute vertigo. It is frequently used to describe light-headedness, panic, anxiety, palpitations and chronic ill-health. The real nature of this symptom must be determined.
Vertigo (p. 1078) means the illusion of movement, a sensation of rotation or tipping. The patient feels the surroundings are spinning or moving. This is distressing and often accompanied by nausea or vomiting.
Blackout, like dizziness, is simply descriptive, implying either altered consciousness, visual disturbance or falling. Epilepsy (p. 1112) and syncope are mentioned in detail (p. 1116); hypoglycaemia and anaemia must be considered. Commonly no sinister cause is found. A careful history is essential.
Collapse is a vague term, but often used. Avoid it.
No serious disease is found in many patients (>20%) referred with symptoms suggestive of possible neurological conditions.
Fatigue is common: when it is an isolated symptom, neurological disease is rarely discovered. There is a borderland (sometimes contentious) between neurology and psychiatry.
Examination and formulation
Following a short or detailed examination, relevant findings are summarized in a brief formulation – the basis for investigation, transfer of information and management (Practical Boxes 22.1, 22.2 and Table 22.2).
 Practical Box 22.2
Practical Box 22.2
10-part neurological examination
1. State of consciousness, arousal, appearance
2. Mental state, attitude, insight (see Box 22.5, p. 1069)
3. Cognitive function – orientation, recall, level of intellect, language, other cortical problems, e.g. apraxia
6. Neck – stiffness, palpation and auscultation of carotids
7. Cranial nerves (Table 22.3)
Table 22.2 Six grades of muscle power
| Grade | Definition |
|---|---|
5 |
Normal power |
4 |
Active movement against gravity and resistance |
3 |
Active movement against gravity |
2 |
Active movement with gravity eliminated |
1 |
Flicker of contraction |
0 |
No contraction |
Functional neuroanatomy
The neurone and synapse
The neurone is the functional unit of the entire nervous system (Fig. 22.1). Its cell body and axon terminate in a synapse. Size and type of each group of neurones vary. A thoracic spinal cord α-motor neurone has an axonal length of >1 metre and innervates between several hundred and 2000 muscle fibres in one leg – a motor unit. By contrast, some spinal or intracerebral interneurones have axons under 100 µm long, terminating on one neuronal cell body.
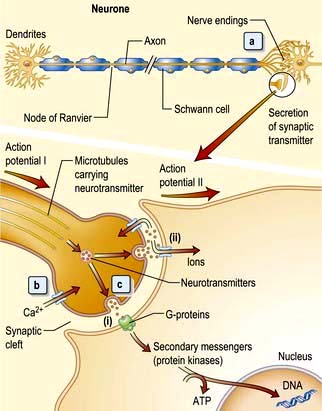
Figure 22.1 The functional unit: neurone and neurotransmitters. (a) The action potential, i.e. nerve impulse, travels down the axon. Microtubules carry neurotransmitters to nerve endings. (b) Action potential I depolarizes the synaptic membrane, opening voltage-gated calcium channels. (c) Influx of calcium ions cause vesicles to fuse with the membrane allowing neurotransmitter binding to receptors (i) and activation of secondary messengers that modulate gene transcription and also open ligand-gated channels (ii). This allows ions to enter, depolarize the membrane and initiate action potential II.
Neurotransmitters
Neurotransmitters are excitatory (acetylcholine, noradrenaline, adrenaline, 5-hydroxytryptamine, dopamine, glutamate and aspartate) or inhibitory (γ-aminobutyric acid (GABA), histamine and glycine). Neuropeptides, e.g. vasopressin, ACTH, substance P and opioid peptides, as well as the purines (ATP and AMP) are both excitatory and inhibitory.
Synaptic transmission is mediated by neurotransmitters released by action potentials passing down an axon. Neurotransmitters activate postsynaptic receptors and are removed by transporter proteins. The neurotransmitter-receptor reaction increases ionic permeability and propagates a further action potential. Axonal electrical activity and synaptic chemical release is the basis of neurological function.
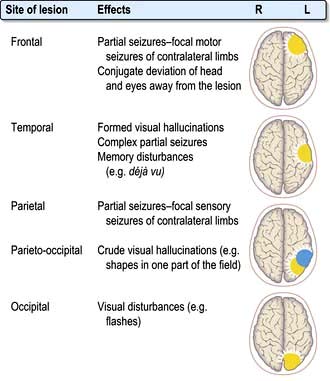
Clinical features of focal brain lesions: general mechanisms
The symptoms and signs suggest the area of the brain that is malfunctioning (e.g. aphasia – the left frontal lobe, hemiparesis – internal capsule or a Bell’s palsy – Vllth cranial (facial) nerve).
Focal lesions of the cortex, and lesions throughout the nervous system, cause symptoms and signs by two processes:
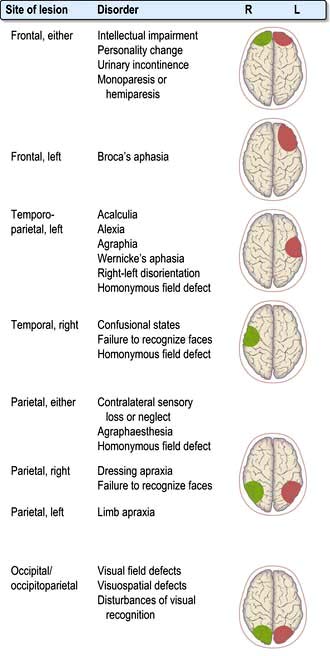
Localization within the cerebral cortex
This subject causes unnecessary difficulty. Work on neuronal networks, functional imaging and plasticity questions the traditional views of highly specific localization of cortical function. The following paragraphs summarize areas of clinical relevance.
The dominant hemisphere (usually left)
The concept of cerebral dominance arose from a simple observation: right-handed stroke patients with acquired language disorders had destructive lesions within the left hemisphere. Right-handed (and 70% of left-handed) people have language function on the left.
More specifically, destructive lesions within the left fronto-temporo-parietal region cause disorders of communication:
Developmental dyslexia describes delayed, disorganized reading and writing ability in children, usually with normal intelligence.
Aphasia
Aphasia is loss of or defective language from damage to the speech centres within the left hemisphere. Numerous varieties have been described.
Broca’s (expressive, anterior) aphasia
Damage in the left frontal lobe causes reduced speech fluency with relatively preserved comprehension. The patient makes great efforts to initiate language, which becomes reduced to a few disjointed words with failure to construct sentences. Patients who recover say they knew what they wanted to say, but could not get the words out.
Wernicke’s (receptive, posterior) aphasia
Left temporo-parietal damage leaves fluency of language but words are muddled. This varies from insertion of a few incorrect or non-existent words into speech to a profuse outpouring of jargon (i.e. rubbish with wholly non-existent words). Severe jargon aphasia is bizarre and often mistaken for psychotic behaviour.
Patients who recover from Wernicke’s aphasia say that they found speech, both their own and others’, like an unintelligible foreign language, i.e. incomprehensible, but they could neither stop speaking nor understand speech.
Nominal (anomic, amnestic) aphasia
This means difficulty naming familiar objects. Naming difficulty is an early feature in all types of aphasia.
Global (central) aphasia
This means the combination of the expressive problems of Broca’s aphasia and the loss of comprehension of Wernicke’s with loss of both language production and understanding. This is due to widespread damage to speech areas and is the commonest aphasia after a severe left hemisphere infarct. Writing and reading are also affected.
Dysarthria
Dysarthria is disordered articulation – slurred speech. Language is intact. Paralysis, slowing or incoordination of muscles of articulation or local discomfort causes various patterns of dysarthria. Examples are the gravelly speech of pseudobulbar palsy (p. 1080), the jerky, ataxic speech of cerebellar lesions, the monotone of Parkinson’s, and speech in myasthenia that fatigues and dies away. Many aphasic patients are also dysarthric.
The non-dominant hemisphere
Disorders in right-handed patients with right hemisphere lesions are often difficult to recognize. There are abnormalities of perception of internal and external space. Examples are losing the way in familiar surroundings, failing to put on clothing correctly (dressing apraxia), or failure to draw simple shapes – constructional apraxia.
Memory and its disorders
Disorders of memory follow damage to the medial surfaces of both temporal lobes and their brainstem connections – the hippocampi, fornices and mammillary bodies. Bilateral lesions are necessary to cause amnesia. In all organic memory disorders recent events are recalled poorly, in contrast to the relative preservation of distant memories.
Memory loss (the amnestic syndrome) is part of dementia (p. 1137) but also occurs as an isolated entity (Box 22.2).
Cranial nerves (Table 22.3)
I: Olfactory nerve
This sensory nerve arises from olfactory (smell) receptors within nasal mucosa. Branches pierce the cribriform plate and synapse in the olfactory bulb. The olfactory tract passes to the olfactory cortex.
| Number | Name | Main clinical action |
|---|---|---|
I |
Olfactory |
Smell |
II |
Optic |
Vision, fields, afferent light reflex |
III |
Oculomotor |
Eyelid elevation, eye elevation, ADduction, depression in ABduction, efferent (pupil) |
IV |
Trochlear |
Eye intorsion, depression in ADduction |
V |
Trigeminal |
Facial (and corneal) sensation, mastication muscles |
VI |
Abducens |
Eye ABduction |
VII |
Facial |
Facial movement, taste fibres |
VIII |
Vestibular |
Balance and hearing |
|
Cochlear |
|
IX |
Glossopharyngeal |
Sensation – soft palate, taste fibres |
X |
Vagus |
Cough, palatal and vocal cord movements |
XI |
Accessory |
Head turning, shoulder shrugging |
XII |
Hypoglossal |
Tongue movement |
Anosmia (loss of sense of smell) is caused by head injury (shearing of olfactory neurones as they pass through the cribriform plate at the skull base) or tumours of the olfactory groove (e.g. meningioma). Olfaction is temporarily (occasionally permanently) lost or diminished after upper respiratory infections and with local disorders of the nose. Many patients with gradual onset anosmia over many years may be unaware of the deficit, e.g. in Parkinson’s disease where anosmia precedes motor symptoms by many years but is often not noticed by the patient.
Detailed smell testing is difficult in routine clinical practice and rarely performed. Adequate testing requires use of commercially available kits such as scratch and sniff cards or odour filled pens with forced multiple choice identification.
II: Optic nerve and visual system (Fig. 22.4)
Light regulated by the pupillary aperture is converted into action potentials by retinal rod, cone and ganglion cells (see page 1055). The lens, under control of the ciliary muscle, produces the image (inverted) on the retina. Axons in the optic nerve (1) decussate at the optic chiasm (2), fibres from the nasal retina cross and join with uncrossed fibres originating in the temporal retina to form the optic tract (3). Each optic tract thus carries information from the contralateral visual hemifield.
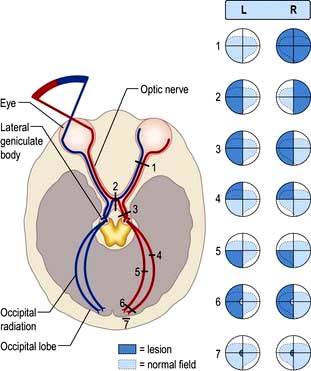
Figure 22.4 The visual pathway. 1. Mononuclear field loss – complete optic nerve lesion. 2. Bitemporal hemianopia – chiasmal lesion. 3. Homonymous hemianopia – optic tract lesion. 4. Homonymous quadrantanopia – temporal lesion. 5. Homonymous quadrantanopia – parietal lesion. 6. Homonymous hemianopia with macular sparing – occipital cortex or optic radiation. 7. Homonymous hemianopia (hemiscotoma) – occipital pole lesion.
From the lateral geniculate body, fibres pass in the optic radiation through the parietal and temporal lobes (4 and 5) to reach the visual cortex of the occipital lobe (6 and 7), which is somatotopically organized with macular vision located at the occipital pole (see Fig. 22.4).
Beyond the visual cortex visual information is further processed by neighbouring visual association areas to detect lines, orientation, shapes, movement, colour and depth; there is even a distinct area responsible for face recognition.
Visual acuity
This is assessed in each eye with a Snellen chart and/or Near Vision Reading Types, corrected for refractive errors with lenses or a pinhole. The patient should stand 6 metres from a well-lit chart. Acuity is recorded as distance in metres from the chart over distance at which the line should be legible, e.g. 6/6 indicates ‘normal’ acuity and 6/60 very poor acuity.
Visual field defects
Visual fields are assessed at the bedside by confrontation – comparing the examiner’s and patient’s fields, one eye at a time and quadrant by quadrant. Patience and good technique are required to get reliable results. White and red targets (traditionally hatpins) are used to assess peripheral and central fields respectively although in practice a fingertip is often substituted as a cruder screening test. More detailed quantification of fields may be obtained using Goldmann (manual) or Humphrey (automated) perimetry testing.
Field defects are described as hemianopic when half the field is affected and quadrantanopic when a quadrant is affected. Lesions posterior to the optic chiasm produce homonymous field defects, indicating involvement of the same part of the visual field in both eyes as information from the two visual hemifields is separated beyond this point. Lesions damaging decussating nasal fibres at the optic chiasm cause bitemporal defects.
Optic nerve lesions
Unilateral visual loss, commencing with a central or paracentral (off-centre) scotoma, is the hallmark of an optic nerve lesion. Because most fibres in the optic nerve subserve macular vision, lesions within the nerve disproportionately affect central vision and colour vision. A total optic nerve lesion causes unilateral blindness with loss of pupillary light reflex. Examination findings in optic neuropathy:
 Reduced acuity in affected eye
Reduced acuity in affected eye
Impaired colour vision (assess with Ishihara plates)
Causes are listed in Box 22.3.
 Box 22.3
Box 22.3
Causes of optic neuropathy
Inflammatory (optic neuritis), e.g. demyelination, sarcoidosis, vasculitis
Optic nerve trauma or compression, e.g. glioma, meningioma, aneurysm, bone disorders affecting orbit
Toxic, e.g. tobacco-alcohol, ethambutol, methyl alcohol, quinine, hydroxy chloroquine, radiation
Ischaemic optic neuropathy, e.g. giant cell arteritis
Hereditary optic neuropathies, e.g. Leber’s
Nutritional deficiency, e.g. vitamin B1 and B12
Infection, e.g. orbital cellulitis, syphilis, TB
Neurodegenerative disorders, e.g. leucodystrophies
Papilloedema and its causes (Box 22.4)
Papilloedema
Papilloedema means swelling of the optic disc. Causes are shown in Box 22.4. The earliest signs of swelling are disc pinkness, with blurring and heaping up of disc margins, nasal first. There is loss of spontaneous pulsation of retinal veins within the disc. The physiological cup becomes obliterated, the disc engorged with dilated vessels. Small haemorrhages often surround the disc.
Box 22.4
Causes of optic disc swelling
Raised intracranial pressure (papilloedema)
Brain tumour, abscess, or haemorrhage. Idiopathic intracranial hypertension, hydrocephalus
Optic neuritis, e.g. multiple sclerosis
Ischaemic optic neuropathy, e.g. giant cell arteritis
Toxic optic neuropathy, e.g. methanol
Central retinal vein thrombosis
Vasculitis, e.g. systemic lupus erythematosus
Various conditions simulate true disc swelling. Marked hypermetropic (long-sighted) refractive errors make a disc appear pink, distant and ill-defined. Myelinated nerve fibres at disc margins and hyaline bodies (drusen, p. 1064) can be mistaken for disc swelling.
Disc infiltration also causes a swollen disc with raised margins (e.g. in leukaemia).
When there is doubt about disc oedema, i.v. fluorescein angiography is diagnostic; retinal leakage is seen with papilloedema.
Papilloedema produces few if any visual symptoms other than momentary visual obscurations with changes in posture. The underlying disease is the source of the patient’s symptoms. The blind spot is enlarged but this is not noticed by the patient. However, over time progressive and permanent constriction of visual fields occurs, ultimately culminating in optic atrophy.
Inflammatory optic neuropathy (optic neuritis)
Optic neuritis is one of the most common causes of subacute visual loss. Symptoms may vary from a mild fogging of central vision with colour desaturation to a dense central scotoma, but very rarely complete blindness. Pain on eye movements is almost universal. The optic disc usually appears normal despite severe visual loss (unless the inflammation is at the optic nerve head in which case the disc may appear swollen in the acute phase).
A plaque of demyelination within the optic nerve is the most common cause in Western populations. Dedicated MRI imaging of the optic nerves may show the inflammatory plaque and imaging of the brain may show additional inflammatory lesions which confer a higher risk of developing multiple sclerosis (MS). Approximately 50% of patients go on to develop MS with prolonged follow-up (see p. 1123). Recovery of visual acuity to 6/9 or better occurs in 95% of cases over months, with recovery time improved by high-dose i.v. steroids given acutely.
Optic neuritis may be caused by infective or other inflammatory disorders, e.g. sarcoidosis or vasculitides (see Box 22.3).
Anterior ischaemic optic neuropathy
The anterior part of the optic nerve is supplied by the posterior ciliary arteries, occlusion or hypoperfusion of which leads to infarction of all or part of the optic nerve head. There is sudden or stuttering altitudinal visual loss (typically the lower half of the visual field) with disc swelling, later replaced by optic atrophy. The other eye is later affected in one-third of cases.
Individuals with small hypermetropic discs seem to be predisposed, and often there are relatively few vascular risk factors. Less commonly arteritis is the cause (see p. 536).
Optic chiasm
Bitemporal hemianopia or quadrantanopia occurs with compression of the chiasm from above or below. Common causes are:
Optic tract and optic radiation
Optic tract lesions (rare) cause a homonymous hemianopia (loss of the contralateral visual field in both eyes). Optic radiation lesions cause homonymous quadrantanopic defects. Temporal lobe lesions (e.g. tumour, infarction) cause upper quadrantic defects, and parietal lobe, lower.
Occipital cortex
Homonymous hemianopic defects are produced by unilateral posterior cerebral artery infarction (see p. 1101 stroke). The macular cortex (at each occipital pole) may be spared.
Widespread bilateral occipital lobe damage by infarction (‘top of the basilar’ syndrome), trauma or coning causes cortical blindness (Anton’s syndrome). The patient cannot see but characteristically lacks insight into this; he or she may even deny it. Pupillary responses remain normal (p. 1101).
The pupils
A slight difference between the size of each pupil (up to 1 mm) is common (physiological anisocoria) and does not vary with differing light levels. The pupil tends to become smaller and irregular in old age (senile miosis); anisocoria is more pronounced. Convergence becomes sluggish with ageing.
Pupillary reactions to light and accommodation may be tested (Fig. 22.5). A bright torch (not an ophthalmoscope light!) should be used to test the pupillary light reaction.
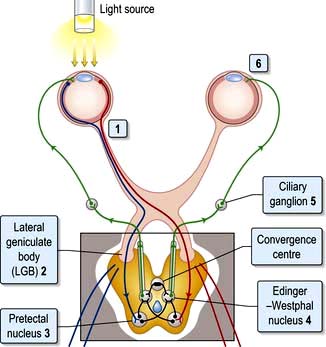
Figure 22.5 Pupillary light reflex. Afferent pathway: (1) Light activates optic nerve axons. (2) Axons (some decussating at the chiasm) pass through each lateral geniculate body, and (3) synapse at pretectal nuclei. Efferent pathway: (4) Action potentials pass to Edinger–Westphal nuclei of IIIrd nerve, then, (5) via parasympathetic neurones in IIIrd nerves to cause (6) pupil constriction.
Afferent pupillary defect. A complete optic nerve lesion causes a dilated pupil and an afferent pupillary defect (APD). For a left APD:
The pupil is unreactive to light (i.e. the direct reflex is absent).
The consensual reflex (constriction of the right pupil when the left is illuminated) is absent. Conversely, the left pupil constricts when light is shone in the intact right eye, i.e. the consensual reflex of the right eye remains intact.
Relative afferent pupillary defect (RAPD). This occurs with incomplete damage to one optic nerve relative to the other. An RAPD is a sensitive sign of optic nerve pathology and can provide evidence of an optic nerve lesion even after recovery of vision. For a left RAPD:
Direct and indirect reflexes are intact in each eye but differ in relative strength.
When the light is swung from one eye to the other, the left pupil dilates slightly when illuminated and constricts slightly when the right eye is illuminated (the consensual reflex is stronger than the direct).
Horner’s syndrome (see Box 22.5)
The sympathetic nervous supply to the eye is a three neurone pathway originating in the hypothalamus and descending by way of the brainstem and cervical cord to T1 nerve root, paravertebral sympathetic chain and, on via the carotid artery wall, to the eye. Damage to any part of the pathway results in Horner’s syndrome. This is significant not only because it affects vision but also because it may indicate a serious underlying pathology.
Box 22.5
Causes of Horner’s syndrome
Myotonic pupil (Holmes–Adie pupil)
This is a dilated, often irregular, pupil, more frequent in women; it is common and usually unilateral. There is no (or very slow) reaction to bright light and also incomplete constriction to convergence. This is due to denervation in the ciliary ganglion, of unknown cause, and has no other pathological significance. A myotonic pupil is sometimes associated with diminished or absent tendon reflexes.
III, IV, VI: Oculomotor, trochlear and abducens nerves
These cranial nerves supply the extraocular muscles and disorders commonly result in abnormal eye movements and diplopia (double vision) due to breakdown of conjugate (yoked) eye movements. Diplopia may also occur with local orbital lesions or myasthenia gravis.
Examining eye movements
Pursuit (slow) eye movements and saccadic eye movements are tested separately. The examiner assesses the range of eye movements in all directions and asks the patient to report double vision. Jerky pursuit movements with saccadic intrusion (i.e. brief fast saccades interspersed with slower pursuit movements), overshoot on saccadic movements and nystagmus may indicate cerebellar or brainstem pathology.
Control of eye movements
Fast voluntary eye movements originate in the frontal lobes. Fibres descend and cross in the pons to end in the centre for lateral gaze (paramedian pontine reticular formation – PPRF), close to the VIth nerve nucleus. Each PPRF also receives input from:
the ipsilateral occipital cortex – pathway concerned with tracking objects
the vestibular nuclei – pathways linking eye movements with position of the head and neck (vestibulo-ocular reflex, p. 1072).
Conjugate lateral eye movements are coordinated from each PPRF via the medial longitudinal fasciculus (MLF, Fig. 22.6). Fibres from the PPRF pass both to the ipsilateral VIth nerve nucleus (lateral rectus) and, having crossed the midline, to the opposite IIIrd nerve nucleus (medial rectus and other muscles) via the MLF, thus linking the eyes for lateral gaze.
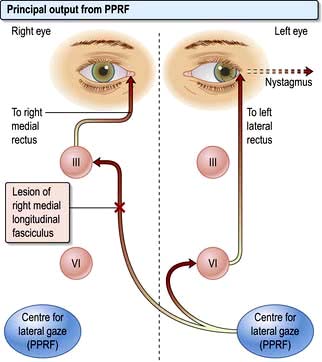
Figure 22.6 PPRF and INO. Impulses from PPRF pass via ipsilateral VIth nerve nucleus to lateral rectus muscle (ABduction) and via medial longitudinal fasciculus to opposite IIIrd nerve nucleus and thus to opposite medial rectus muscle (ADduction). A lesion of the MLF (X) causes failure of or slow ADduction in the right eye and nystagmus in the left eye with left lateral gaze. PPRF, para-median pontine reticular formation; INO, internuclear ophthalmoplegia; MLF, medial longitudinal fasciculus.
Abnormalities of conjugate lateral gaze
A destructive lesion on one side allows the eyes to be driven by the intact opposite pathway. A left frontal destructive lesion (e.g. an infarct) leads to failure of conjugate lateral gaze to the right. In an acute lesion the eyes are often deviated to the side of the lesion, past the midline and therefore look towards the left (normal) limbs; there is usually a contralateral (i.e. right) hemiparesis.
In the brainstem a unilateral destructive lesion involving the PPRF leads to failure of conjugate lateral gaze towards that side. There is usually a contralateral hemiparesis and lateral gaze is deviated towards the side of the paralysed limbs.
Internuclear ophthalmoplegia (INO)
Damage to one MLF causes internuclear ophthalmoplegia (INO), a common complex brainstem eye movement disorder seen frequently in MS. In a right INO there is a lesion of the right MLF (Fig. 22.6). On attempted left lateral gaze the right eye fails to ADduct. The left eye develops nystagmus in ABduction. The side of the lesion is on the side of impaired ADduction, not on the side of the (obvious, unilateral) nystagmus. When present bilaterally, INO is almost pathognomonic of MS.
One and a half syndrome
Pontine infarction involving the PPRF, VIth nerve nucleus and MLF on one side results in an ipsilateral horizontal gaze palsy and an INO so that abduction of the opposite eye (with nystagmus) is the only horizontal eye movement possible. Vertical gaze and convergence are preserved as they have distinct neural control mechanisms.
Abnormalities of vertical gaze
Failure of up-gaze may be caused by dorsal midbrain lesions, e.g. pinealoma or infarcts. When the pupillary light reflex fails in addition, this is called Parinaud’s syndrome. Defective up-gaze also develops in certain degenerative disorders (e.g. progressive supranuclear palsy). Some impairment of up-gaze occurs as part of normal ageing.
Nystagmus
Nystagmus is rhythmic oscillation of eye movement, and a sign of disease of the retina, cerebellum and/or vestibular systems and their connections. Nystagmus is either jerk or pendular. Nystagmus must be sustained within binocular gaze to be of diagnostic value – a few beats at the extremes of gaze are normal.
Jerk nystagmus
Jerk nystagmus (usual in neurological disease) is a fast/slow oscillation. This is seen in vestibular, VIIIth nerve, brainstem, cerebellar lesions. Direction of nystagmus is decided by the fast component, a reflex attempt to correct the slower, primary movement.
Horizontal or rotary jerk nystagmus may be either of peripheral (vestibular) or central origin (VIIIth nerve, brainstem, cerebellum and connections).
Vertical jerk nystagmus is caused typically by central lesions.
Down-beat jerk nystagmus is a rarity caused by lesions around the foramen magnum (e.g. meningioma, cerebellar ectopia).
III: Oculomotor nerve
The nucleus of the IIIrd nerve lies ventral to the aqueduct in the midbrain. It supplies four external ocular muscles (superior, inferior and medial recti, and inferior oblique), levator palpebrae superioris (which lifts the eyelid) and parasympathetic constriction of the pupil. Causes of a IIIrd nerve lesion are listed in Box 22.6.
Signs of a complete IIIrd nerve palsy:
Unilateral complete ptosis (levator weakness)
Eye deviated down and out (unopposed lateral rectus and superior oblique)
Patients do not complain of diplopia as the ptosis effectively covers the eye. Sparing of the pupil indicates parasympathetic fibres are undamaged; these run in a discrete bundle on the surface of the nerve, thus the pupil is of normal size and reacts normally. Diabetic IIIrd nerve infarction is usually painless and pupil sparing, unlike compression by a posterior communicating artery aneurysm.
IV: Trochlear nerve
This supplies the superior oblique muscle. The patient complains of torsional diplopia (two objects at an angle) when attempting to look down (e.g. descending stairs); the head is tilted away from that side. The most common cause is head injury, often with bilateral trochlear nerve palsies occurring.
VI: Abducens nerve
This supplies the lateral rectus muscle (ABduction). Lesions cause horizontal diplopia when looking into the distance, maximal when looking to the side of the lesion. The eye cannot be fully abducted and an esotropia (inwards eye deviation) may be visible in the primary position.
The VIth nerve has a long intracranial course. It can be damaged in the brainstem (e.g. by MS or infarction). In raised intracranial pressure, it is compressed against the tip of the petrous temporal bone (may be bilateral). The nerve sheath may be infiltrated by tumours, particularly nasopharyngeal carcinoma. Microvascular ischaemia of the nerve may occur in diabetes with acute onset followed by recovery within 3 months in most cases.
Complete external ophthalmoplegia
Complete external ophthalmoplegia describes an immobile eye when IIIrd, IVth and VIth nerves are paralysed at the orbital apex (e.g. by metastasis) or within the cavernous sinus (e.g. by sinus thrombosis or meningioma).
Wernicke’s encephalopathy due to thiamine deficiency (p. 1147) may cause a complex eye movement disorder or complete ophthalmoplegia, as may neuromuscular junction disorders such as myasthenia, botulism and some myopathies or metabolic disorders.
V: Trigeminal nerve
The largest cranial nerve; mainly sensory with a motor component to the muscles of mastication.
Sensory fibres (Fig. 22.7; see also Figs 22.11 and 22.12) of the three divisions – ophthalmic (V1), maxillary (V2) and mandibular (V3) – pass to the trigeminal (Gasserian) ganglion at the apex of the petrous temporal bone. Ascending fibres transmitting light touch enter the Vth nucleus in the pons. Descending central fibres carrying pain and temperature form the spinal tract of V, to end in the spinal Vth nucleus that extends from the medulla into the cervical cord.
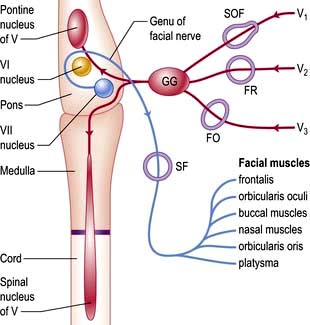
Figure 22.7 Sensory input of Vth nerve (red) and motor output of VIIth nerve (blue). GG, Gasserian ganglion; SOF, superior orbital fissure; FR, foramen rotundum; FO, foramen ovale; SF, stylomastoid foramen.
Signs of a Vth nerve lesion
A complete Vth nerve lesion causes unilateral sensory loss on the face, scalp anterior to the vertex, anterior two-thirds of the tongue and buccal mucosa; the jaw deviates to that side as the mouth opens (motor fibres). Diminution of the corneal reflex is an early and sometimes isolated sign of a Vth nerve lesion.
Causes
Brainstem pathology (infarction, demyelination or tumour) may damage the nucleus, with light touch and spinothalamic pathways sometimes being differentially involved.
Cerebellopontine angle tumours (acoustic neuroma or meningioma) may compress the nerve and also affect the VIIth and VIIIth nerves producing facial weakness and deafness.
Cavernous sinus and skull base pathology (tumour or infection) may affect the ganglion and proximal branches.
Peripheral branches may be picked off individually, e.g. the ‘numb chin syndrome’ seen with a breast cancer metastasis in the mandible.
Trigeminal sensory neuropathy
Causes gradually progressive unilateral facial sensory loss and tingling with normal imaging. The condition is probably heterogeneous in aetiology but may have an autoimmune basis, with inflammation of the trigeminal ganglion, occurring mainly in association with mixed and undifferentiated connective tissue disease and primary Sjögren’s syndrome.
VII: Facial nerve
The VIIth nerve is largely motor, supplying muscles of facial expression. VII carries sensory taste fibres from the anterior two-thirds of the tongue via the chorda tympani and supplies motor fibres to the stapedius muscle. The VIIth nerve (Fig. 22.7) arises from its nucleus in the pons and leaves the skull through the stylomastoid foramen. Neurones in each VIIth nucleus supplying the upper face (principally frontalis) receive bilateral supranuclear innervation.
Unilateral facial weakness
Upper motor neurone (UMN lesions) cause weakness of the lower part of the face on the opposite side. Frontalis is spared: normal furrowing of the brow is preserved; eye closure and blinking are largely unaffected. The earliest sign is slowing of one side of the face, e.g. on baring teeth. There is sometimes relative preservation of spontaneous emotional movement (e.g. smiling) compared with voluntary movement.
Lower motor neurone (LMN) lesions. A complete unilateral LMN VIIth lesion causes weakness (ipsilateral) of all facial expression muscles. The angle of the mouth falls; unilateral dribbling develops. Frowning (frontalis) and eye closure are weak. Corneal exposure and ulceration occur if the eye does not close during sleep. Taste sensation is frequently also impaired.
Causes of facial weakness
The commonest cause of a UMN lesion is hemispheric stroke with hemiparesis on the opposite side. At lower levels, lesion sites are recognized by LMN weakness with additional signs.
Pons. Here the VIIth nerve loops around the VIth (abducens) nucleus (Fig. 22.7), leading to a lateral rectus palsy (see p. 1076) with unilateral LMN facial weakness. When the neighbouring PPRF and corticospinal tract are involved, there is the combination of:
Causes include pontine tumours (e.g. glioma), MS and infarction.
Cerebellopontine angle (CPA). The neighbouring Vth, VIth and VIIIth nerves are compressed with VII in the CPA, e.g. by acoustic neuroma, meningioma or metastasis.
Petrous temporal bone. The nerve may be damaged within the bony facial canal, within which lies the sensory geniculate ganglion (receiving taste fibres from the anterior two-thirds of the tongue via the chorda tympani). As well as LMN facial weakness, lesions in this region cause:
Loss of taste on the anterior two-thirds of the tongue
Hyperacusis (loud noise distortion – paralysis of stapedius).
Skull base, parotid gland and within the face. The facial nerve can be compressed by skull base tumours and in Paget’s disease of bone. Branches of VII may be damaged by parotid gland tumours as the nerve traverses the parotid, sarcoidosis (p. 845) and trauma.
Bell’s palsy
This common (1 per 5000 incidence), acute facial palsy is thought to be due to viral infection (often herpes simplex) causing swelling of nerve within the tight petrous bone facial canal. There is unilateral LMN facial weakness developing over 24–48 hours, sometimes with lost or altered taste on the tongue, and hyperacusis. Pain behind the ear is common at onset. Patients often suspect a stroke and may be very distressed. Vague altered facial sensation is often reported although examination of facial sensation is normal.
Diagnosis is made on clinical grounds and tests are usually not required. The ear (and palate) should be examined for vesicles (see Ramsay Hunt syndrome below), hearing loss or evidence of local pathology such as cholesteatoma or malignant otitis externa and parotid tumours should be excluded. Involvement of other cranial nerves means facial weakness is not due to Bell’s palsy. Lyme disease may account for one-quarter of cases of facial palsy in endemic areas and HIV seroconversion is the commonest cause in parts of Africa.
(Bell’s phenomenon is the upward conjugate eye movement that occurs when the eyes are closed.)
Management and outcome
Complete, or almost complete, recovery over 3–8 weeks occurs in at least 85% of patients, even without specific treatment. Patients should be reassured that the prognosis is good and it is unlikely to recur.
Inability to blink in severe facial weakness may lead to exposure keratitis. Use of lubricating eye drops is often required and patients should be advised to carefully tape the eye closed at night. For more severe facial weakness with complete inability to close the eye, early ophthalmological assessment is required and lateral tarsorrhaphy and/or insertion of a gold weight into the upper lid may be required until recovery occurs.
Early treatment with corticosteroids (prednisolone 1 mg/kg for 7 days) improves outcome. Evidence to support use of antiviral agents is limited but aciclovir or valaciclovir is often given in combination with steroids.
Recovery sometimes takes up to a year if axons have to regrow rather than just remyelinate, in which case aberrant reinnervation of facial muscles (e.g. mouth twitching with blinking) is a frequent late complication. Cosmetic surgery may be helpful where recovery is not complete. Bell’s palsy rarely recurs and recurrence should prompt a search for an alternative cause.
Ramsay hunt syndrome
This is herpes zoster (shingles) of the geniculate ganglion. There is a facial palsy (identical to Bell’s) with vesicles around the external auditory meatus and/or the soft palate (sensory twigs from VII). Deafness and unsteadiness may occur. Complete recovery is less likely than in Bell’s. Antiviral treatment for shingles (above) should be given with steroids.
Bilateral facial weakness
Bilateral facial palsy is rare, accounting for fewer than 1% of cases of facial palsy, but is more likely than unilateral palsies to have an identifiable underlying cause. Paradoxically, bilateral weakness is often less obviously apparent than unilateral weakness as there is no facial asymmetry.
Hemifacial spasm (HFS)
This is an irregular, painless unilateral spasm of facial muscles, usually occurring after middle age. It starts in the orbicularis oculi and usually progresses gradually over the years to involve other facial muscles on the same side. It varies from a mild to a severe, disfiguring spasm.
HFS is usually caused by compression of the root entry zone of the facial nerve, generally by vascular structures such as the vertebral or basilar arteries or their branches (a mechanism similar to that of trigeminal neuralgia see p. 1110). Other mass lesions in the cerebellopontine angle, including tumours, are the cause in approximately 1% of cases.
Occasionally HFS may occur with ipsilateral trigeminal neuralgia, one symptom usually preceding the other, a combination called tic convulsif. The paroxysms of pain and spasm occur independently. A compressive cause such as a vascular loop or other structural lesion is usually identified.
Management
Mild cases require no treatment. Botulinum toxin injection into affected muscles every 3–4 months is now the standard treatment. Drugs (e.g. carbamazepine) are of little value. Decompression of the VIIth nerve in the CPA is sometimes helpful. Surgical decompression of the facial nerve in the posterior fossa involves interposing a non-resorbable sponge between the nerve and any adjacent vascular loop identified at operation. The procedure results in complete resolution of symptoms in up to 90% of cases but is associated with a risk of facial weakness or deafness.
Other involuntary facial movements
Myokymia of orbicularis oculi is an irritating twitch, usually of the lower eyelid. It is a normal phenomenon, but sometimes a cause of anxiety. More extensive facial myokymia may result from intrinsic brainstem pathology.
Tics and tardive dyskinesia frequently involve facial or perioral muscles (see p. 1122).
Blepharospasm is a form of focal dystonia affecting orbicularis oculi (see p. 1122)
VIII: Vestibulo-cochlear nerve; cochlear nerve
Auditory fibres from the spiral organ of Corti within the cochlea pass to the cochlear nuclei in the pons. Fibres from these nuclei cross the midline and pass upwards via the medial lemnisci to the medial geniculate bodies and then to the temporal cortex.
Symptoms of a cochlear nerve lesion are deafness and tinnitus (p. 1050). Sensorineural and conductive deafness can be distinguished with tuning fork tests, e.g. Rinne’s and Weber’s (256 Hz, not 128 Hz tuning fork) (p. 1048).
Basic investigations of cochlear lesions
Pure tone audiometry and auditory thresholds.
Auditory evoked potentials (recording responses from repetitive clicks via scalp electrodes; lesion levels are determined from the response pattern).
Causes of deafness are listed in Table 22.4 and Table 21.1 (see p. 1048).
Table 22.4 Some causes of vertigo and hearing loss
| Conditions | Symptom(s) |
|---|---|
Benign paroxysmal positional vertigo |
V |
Vestibular neuritis |
V |
Ménière’s disease |
V, D |
Alcohol, antiepileptic drug intoxication |
V |
Cerebellar lesions |
V |
Partial (temporal lobe) seizures |
V |
Migraine |
V+ phonophobia |
Brainstem ischaemia |
V, occasionally D |
Multiple sclerosis |
V, occasionally D |
Mumps, intrauterine rubella and congenital syphilis (D) |
D |
Advancing age (presbyacusis) and otosclerosis (D) |
D |
Acoustic trauma (D) |
D |
Congenital, e.g. Pendred’s syndrome |
D |
Gentamicin, furosemide |
V, D |
Middle and external ear disease |
V, D |
Cerebellopontine angle lesions, e.g. acoustic neuroma |
V, D |
Carcinomatous meningitis, sarcoid and tuberculous meningitis |
V, D |
V, vertigo; D, hearing loss.
Vertigo and the vestibular system
The vestibular system of the inner ear detects head movements and has three primary functions:
To stabilize gaze during head movements, e.g. looking ahead while running (the vestibulo-ocular reflex)
Nerve impulses generated by movement of hair cells within the three semicircular canals detect head motion in the three planes (yaw/pitch/roll). Balance is maintained by integrating information from:
The main symptoms of vestibular lesions are vertigo and loss of balance.
Vertigo
Vertigo, is the illusion of movement of the subject or surroundings, typically rotatory, and should be distinguished from other causes of nonspecific dizziness. It can be described to patients as being similar to the sensation one feels after getting off a child’s roundabout or after spinning on the spot and suddenly stopping.
Vomiting frequently accompanies acute vertigo of any cause. Vertigo is always made worse by head movements and patients prefer to remain still and maintain visual fixation. Nystagmus (p. 1075) is the principal sign.
Causes of vertigo
Vertigo indicates a disturbance of the vestibular apparatus or brainstem and associated neural pathways (Table 22.4). Causes:
Peripheral (vestibular system) causes are common. Deafness and tinnitus accompanying vertigo indicate involvement of the ear or cochlear nerve (p. 1048).
Central causes (brainstem and connections) are rarer. Other clinical features such as diplopia, weakness, cerebellar signs or cranial nerve palsies may help localize the lesion.
Vestibular disorders
Vestibular disorders are fully discussed elsewhere (p. 1049). Attack duration and frequency and trigger factors help the clinician distinguish on history between different pathological causes. The ability to perform a Hallpike test, head impulse test and Epley particle repositioning manoeuvre are invaluable skills for all clinicians (p. 1049; see Fig. 22.8 and Fig. 21.3).
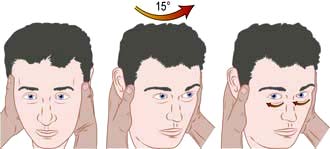
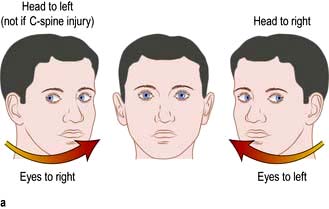
Figure 22.8 The head impulse test. When tested on the normal side, a patient can maintain fixation on a distant object as the examiner rapidly turns the patient’s head sideways through about 15°. If there is unilateral vestibulopathy, the patient cannot maintain fixation on the target and can be observed to make a voluntary eye movement (a saccade) back to the target after the head impulse, as shown on the right. It is essential that the head is turned as rapidly as possible otherwise smooth pursuit eye movements will compensate for the head turn.
The most commonly encountered vestibular disorders presenting with vertigo are:
Benign positional vertigo (BPPV) – frequent attacks lasting seconds only. Triggered by specific movements – lying down, sitting up or rolling over in bed, neck flexion or extension.
Vestibular neuronitis – acute onset lasting days or weeks. Non-recurrent.
Migrainous vertigo (p. 1108), really a central cause – lasting hours, occurring every few weeks or months
Meniere’s disease – recurrent attacks lasting minutes or hours, usually accompanied by hearing loss, tinnitus and a feeling of fullness in the ear.
Central causes of vertigo
Vertigo may be a manifestation of brainstem pathology including:
Infarcts involving the vestibular nuclei in the medulla (e.g. the lateral medullary syndrome)
Demyelination involving the brainstem
Posterior fossa mass lesions, e.g. tumours, haemorrhage or vascular malformations
CPA mass lesions and tumours compressing the vestibular nerve (technically these should be classified as ‘peripheral’ disorders, but are distinct from disorders of the vestibular apparatus)
Although vertigo occasionally occurs in isolation with brainstem pathology, it is more typically a single component of a more complex clinical picture, associated with other symptoms or examination findings. The brainstem nuclei and tracts are tightly packed into a small space and most pathological processes affect multiple contiguous neural pathways, resulting, e.g. in diplopia, eye movement disorders, cranial nerve palsies, cerebellar signs or hemiparesis.
Slowly growing CPA tumours such as vestibular nerve schwannomas may cause vertigo but rarely do so in the absence of unilateral deafness and tinnitus.
Basic investigations for vestibular problems
Bedside assessment is usually sufficient to make a diagnosis in the majority of patients:
Examination of eye movements for nystagmus (p. 1074)
Assessment of hearing and otoscopic examination of the ear (p. 1047)
Head impulse (thrust) test – to assess the vestibulo-ocular reflex (VOR) and identify a unilateral vestibulopathy
Hallpike manoeuvre – a positioning test to stimulate the posterior semicircular canal and trigger an attack in BPPV (see Fig. 21.3)
Specialist testing is occasionally required to assess vestibular function and hearing. This includes:
Caloric testing – irrigation of the external auditory, meatus with cold and then warm water to stimulate the horizontal semicircular canal and induce nystagmus. Test labyrinthine function in each ear separately.
Electro-nystagmography – to quantify and characterize nystagmus under different conditions, e.g. in a rotating chair.
Posturography – assesses body sway on a moving platform.
High-definition MRI provides the best structural imaging of the brainstem and CPA and is useful where a central cause of vertigo is suspected.
Vestibular neuronitis
Vestibular neuronitis is a common, poorly understood problem. It is an acute attack of isolated vertigo with nystagmus, often with vomiting, believed to follow viral infections. The disturbance lasts for several days or weeks, is self-limiting and rarely recurs. Vestibular neuronitis is sometimes followed by benign positional vertigo (p. 1071). Deafness is absent. Acute treatment is with vestibular sedatives. Similar symptoms can be caused by MS or brainstem vascular lesions. Other signs are usually apparent.
Lower cranial nerves IX, X, XI, XII
The glossopharyngeal (IX), vagus (X) and accessory (XI) nerves arise in the medulla and leave the skull through the jugular foramen. The hypoglossal (XII) arises in the medulla, to leave the skull base via the hypoglossal foramen. Outside the skull, the four cranial nerves lie together, close to the carotid artery and sympathetic trunk.
Glossopharyngeal (IX)
This nerve is largely sensory, supplying sensation and taste from the posterior third of the tongue and the pharynx (afferent pathway of gag reflex). Motor fibres supply some pharyngeal muscles and parasympathetic fibres the parotid.
Vagus (X)
The vagus is a mixed nerve, largely motor, which supplies striated muscle of the pharynx (efferent gag reflex pathway), larynx (including vocal cords via recurrent laryngeal nerves) and upper oesophagus. There are sensory fibres from the larynx. Parasympathetic fibres supply the heart and abdominal viscera.
IXth and Xth nerve lesions
Principal causes of IXth, Xth, XIth and XIIth nerve lesions are listed in Box 22.7.
Isolated lesions of IXth and Xth nerves are unusual, since disease at the jugular foramen affects both nerves and sometimes XI.
A unilateral IXth nerve lesion causes diminished sensation on the same side of the pharynx, and is hard to recognize in isolation. A Xth nerve palsy produces ipsilateral failure of voluntary and reflex elevation of the soft palate (which is drawn to the opposite side) and ipsilateral vocal cords.
Bilateral lesions of IXth and Xth nerves cause palatal weakness, reduced palatal sensation, an absent gag reflex, dysphonia and choking with nasal regurgitation. Bulbar palsy is a general term describing palatal, pharyngeal and tongue weakness of LMN or muscle origin.
Recurrent laryngeal nerve lesions. Paralysis of this branch of each vagus causes hoarseness (dysphonia) and failure of the forceful, explosive part of coughing (‘bovine cough’). There is no visible palatal weakness; vocal cord paralysis is seen endoscopically. Bilateral acute lesions (e.g. postoperatively) cause respiratory obstruction – an emergency.
The left recurrent laryngeal nerve (looping beneath the aorta) is damaged more commonly than the right.
XIth nerve lesions
XIth nerve lesions cause weakness of sternomastoid (rotation of the head and neck to the opposite side) and trapezius (shoulder shrugging). Nerve section (e.g. following lymph node biopsy in the neck posterior triangle) is followed by persistent neuralgic pain.
XIIth nerve lesions
LMN lesions of XII lead to unilateral tongue weakness, wasting and fasciculation. The protruded tongue deviates towards the weaker side. Bilateral supranuclear (UMN) lesions produce slow, limited tongue movements and a stiff tongue that cannot be protruded far. Fasciculation is absent.
Bulbar and pseudobulbar palsy
Bulbar palsy
This is LMN weakness of muscles whose cranial nerve nuclei lie in the medulla (the bulb). Paralysis of bulbar muscles is caused by disease of lower cranial nerve nuclei, lesions of IXth, Xth and XIIth nerves (Box 22.7), malfunction of their neuromuscular junctions (e.g. myasthenia gravis, botulism) or disease of muscles themselves (e.g. dystrophies).
Pseudobulbar palsy
Describes bilateral supranuclear (UMN) lesions of lower cranial nerves producing weakness of the tongue and pharyngeal muscles. This resembles, superficially, a bulbar palsy, hence pseudobulbar. Findings are a stiff, slow, spastic tongue (not wasted), dysarthria and dysphagia. Gag and palatal reflexes are preserved and the jaw jerk exaggerated. Emotional lability (inappropriate laughing or crying) often accompanies pseudobulbar palsy. Principal causes:
Motor neurone disease – often both UMN and LMN lesions (i.e. elements of both pseudobulbar and bulbar palsy)
Cerebrovascular disease, typically following multiple infarcts
Neurodegenerative disorders such as progressive supranuclear palsy (p. 1120)
Difficulty swallowing, dysarthria and drooling also develop in later stages of Parkinson’s disease.
Dropped head syndrome
As the name suggests, weakness of neck extensors cause neck flexion and inability to hold the head up. Seen mainly in the elderly it is often due to isolated neck extensor myopathy of uncertain cause, but may be a presenting feature of motor neurone disease, myasthenia or various myopathic disorders.
Motor control systems
There are three systems, each of which interacts by feedback loops with the other two, with sensory input and the reticular formation:
The corticospinal (or pyramidal) system enables purposive, skilled, intricate, strong and organized movements. Defective function is recognized by a distinct pattern of signs – loss of skilled voluntary movement, spasticity and reflex change – seen, e.g. in a hemiparesis, hemiplegia or paraparesis.
The extrapyramidal system facilitates fast, fluid movements that the corticospinal system has generated. Defective function produces slowness (bradykinesia), stiffness (rigidity) and/or disorders of movement (rest tremor, chorea and other dyskinesias). One feature (e.g. stiffness, tremor or chorea) will often predominate.
The cerebellum and its connections have a role coordinating smooth and learned movement, initiated by the pyramidal system, and in posture and balance control. Cerebellar disease leads to unsteady and jerky movements (ataxia), with characteristic limb signs of past pointing, action tremor and incoordination, gait ataxia and/or truncal ataxia.
Corticospinal (pyramidal) system
The corticospinal tracts originate in neurones of the cortex and terminate at motor nuclei of cranial nerves and spinal cord anterior horn cells. The pathways of particular clinical significance (Fig. 22.9) congregate in the internal capsule and cross in the medulla (decussation of the pyramids), passing to the contralateral cord as the lateral corticospinal tracts. This is the pyramidal system, disease of which causes upper motor neurone (UMN) lesions. ‘Pyramidal’ is simply a descriptive term that draws together anatomy and characteristic physical signs, and is used interchangeably with the term UMN.
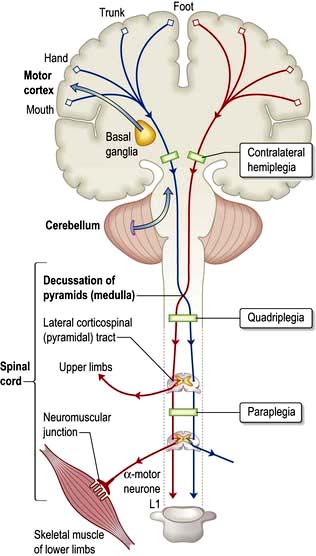
A proportion of the corticospinal outflow is uncrossed (anterior corticospinal tracts). This is of no relevance in practice.
Characteristics of pyramidal lesions (Box 22.8)
Signs of an early pyramidal lesion may be minimal. Weakness, spasticity or changes in superficial reflexes can predominate, or be present in isolation.
Pyramidal drift of an upper limb
Normally, the outstretched upper limbs are held symmetrically, when the eyes are closed. With a pyramidal lesion, when both upper limbs are held outstretched, palms uppermost, the affected limb drifts downwards and medially. The forearm tends to pronate and the fingers flex slightly. This sign is often first to emerge, sometimes before weakness and/or reflex changes become apparent.
Weakness and loss of skilled movement
A unilateral pyramidal lesion above the decussation in the medulla causes weakness of the opposite limbs. When acute and complete, this weakness will be immediate and total, a hemiplegia, e.g. following an internal capsule infarct. With slowly progressive lesions (e.g. a hemisphere glioma) a characteristic pattern of weakness emerges – a hemiparesis.
In the upper limb, flexors remain stronger than extensors, whereas in the lower limb, extensors remain stronger than flexors. In the upper arm, weaker movements are thus shoulder abduction and elbow extension; in the forearm and hand, wrist and finger extensors and abductors are weaker than their antagonists. In the lower limb, weaker movements are hip flexion and abduction, knee flexion, ankle dorsiflexion and eversion. There is also loss of skilled movement – fine finger and toe control diminishes. Wasting (except from disuse) is not a feature. Muscles remain normally excitable electrically.
When a UMN lesion is below the decussation of the pyramids, e.g. in the cervical cord, hemiparesis is on the same side as the lesion, an unusual situation.
Changes in tone and tendon reflexes
An acute lesion of one pyramidal tract (e.g. internal capsule stroke) causes initially flaccid paralysis with loss of tendon reflexes. Increase in tone follows, usually within several days due to loss of inhibitory effects of the corticospinal pathways and an increase in spinal reflex activity. This increase in tone (spasticity) is detectable most easily in stronger muscles. Spasticity is characterized by sudden changing resistance to passive movement – the clasp-knife effect. Relevant tendon reflexes become exaggerated; clonus may emerge.
Changes in superficial reflexes
The normal flexor plantar response becomes extensor. In a severe acute lesion, this extensor response can be elicited from a wide area of the foot. As recovery progresses, the receptive area diminishes until the lateral posterior third of the sole remains receptive to an orange-stick stimulus (an appropriate instrument). An extensor plantar is certain when great toe dorsiflexion is accompanied by abduction of adjacent toes. Abdominal (and cremasteric) reflexes are abolished on the side affected.
Patterns of UMN disorders
There are three main patterns:
Hemiparesis means weakness of the limbs on one side; it is usually caused by a lesion in the brain and occasionally in the cord.
Paraparesis means weakness of both lower limbs and is usually diagnostic of a cord lesion; bilateral brain lesions occasionally cause paraparesis.
Tetraparesis (syn. quadriparesis) means weakness of four limbs.
Hemiplegia, paraplegia and tetraplegia indicate (strictly) total paralysis, but are often used to describe severe weakness.
Hemiparesis
The level within the corticospinal system is recognized by particular features.
Motor cortex. Weakness and/or loss of skilled movement confined to one contralateral limb (an arm or a leg – mono-paresis) or part of a limb (e.g. a clumsy hand) is typical of an isolated motor cortex lesion (e.g. a secondary neoplasm). A defect in cognitive function (e.g. aphasia) and focal epilepsy may occur.
Internal capsule. Corticospinal fibres are tightly packed in the internal capsule (about 1 cm2), thus a small lesion causes a large deficit. A middle cerebral artery branch infarction (p. 1100) produces a sudden, dense, contralateral hemiplegia.
Pons. A pontine lesion (e.g. an MS plaque) is rarely confined to the corticospinal tract. Adjacent structures, e.g. VIth and VIIth nuclei, MLF and PPRF (p. 1075) are involved – diplopia, facial weakness, internuclear ophthalmoplegia (INO) and/or a lateral gaze palsy occur with contralateral hemiparesis.
Spinal cord. An isolated lesion of one lateral corticospinal tract (e.g. a cervical cord injury) causes an ipsilateral UMN lesion, the level indicated by changes in reflexes (e.g. absent biceps, C5/6), features of a Brown–Séquard syndrome (p. 1086) and muscle wasting at the level of the lesion (p. 1087).
Spastic paraparesis
Paraparesis indicates bilateral damage to corticospinal pathways causing weakness and spasticity (or flaccid weakness in the initial phase of spinal shock after an acute cord insult). Cord compression (p. 1135) or cord diseases are the usual causes; cerebral lesions occasionally produce paraparesis. Paraparesis is a feature of many neurological conditions; finding the cause is crucial (p. 1135).
Extrapyramidal system
The extrapyramidal system is a general term for basal ganglia motor systems, i.e. corpus striatum (caudate nucleus + globus pallidus + putamen), subthalamic nucleus, substantia nigra and parts of the thalamus. In basal ganglia/extrapyramidal disorders, two features (either or both) become apparent, in limbs and axial muscles:
Reduction in speed (bradykinesia, meaning slow movement) or akinesia (no movement), with muscle rigidity
Involuntary movements (e.g. tremor, chorea, hemiballismus, athetosis, dystonia).
Extrapyramidal disorders are classified broadly into akinetic-rigid syndromes (p. 1130) where poverty of movement predominates, and dyskinesias where there are involuntary movements (p. 1121).
The most common extrapyramidal disorder is Parkinson’s disease.
Essential anatomy
The corpus striatum lies close to the substantia nigra, thalami and subthalamic nuclei, lateral to the internal capsule (Figs 22.9, 22.10).
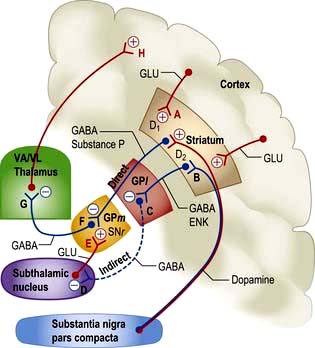
Figure 22.10 Extrapyramidal system: connections and neurotransmitters. The inhibitory pathways are in blue (B, C, D, F, G) and excitory in red (A, E, H). VA/VL, ventral anterior and ventrolateral thalamic nuclei. GPl, lateral globus pallidus. GPm: medial globus pallidus. SNr, substantia nigra pars reticulata. GLU, glutamate; ENK, enkephalin; GABA, γ-aminobutyric acid.
Function and dysfunction
Overall function of this system is modulation of cortical motor activity by a series of servo loops between cortex and basal ganglia (Fig. 22.10). In involuntary movement disorders there are specific changes in neurotransmitters (Table 22.5) rather than focal lesions seen on imaging or at autopsy.
Table 22.5 Changes in neurotransmitters in Parkinson’s and Huntington’s diseases
| Condition | Site | Neurotransmitter |
|---|---|---|
Parkinson’s |
Putamen Substantia nigra Cerebral cortex |
Dopamine ↓ 90%Norepinephrine (noradrenaline) ↓ 60%5-HT ↓ 60%Dopamine ↓ 90%GAD + GABA ↓↓GAD + GABA ↓↓ |
Huntington’s |
Corpus striatum |
Acetylcholine ↓↓GABA ↓↓Dopamine: normalGAD + GABA ↓↓ |
GABA, γ-aminobutyric acid; GAD, glutamic acid decarboxylase, the enzyme responsible for synthesizing GABA; 5-HT, 5-hydroxytryptamine.
Proposed model of principal pathways
1. Direct pathway from striatum to medial globus pallidus (GPm) and substantia nigra pars reticulata (SNr). Inhibitory synapse F, GABA and substance P.
2. Indirect pathway from striatum to globus pallidus; via lateral globus pallidus (GPl; inhibitory synapse C, GABA, enkephalin) and subthalamic nucleus (inhibitory synapse D, GABA). Terminates in GPm-SNr (in excitatory synapse E, glutamate).
3. Direct pathways, both inhibitory and excitatory from substantia nigra pars compacta (SNc) to striatum. Synapse A, dopamine, D1, excitatory; and synapse B, D2, inhibitory.
4. GPm and SNr to thalamus. Inhibitory synapse G, GABA.
The model helps explain how basal ganglia disease can either reduce excitatory thalamo-cortical activity at synapse H, i.e. movement – causing bradykinesia, or increase it, causing dyskinesias.
Parkinson’s disease (PD). This is characterized by slowness, stiffness and rest tremor (p. 1118). Degeneration in SNc causes loss of dopamine activity in the striatum. Dopamine is excitatory for synapse A and inhibitory for synapse B. Through the direct pathway there is reduced activity at synapse F, leading to increased inhibitory output (G) and decreased cortical activity (H).
Also in PD, in the indirect pathway, dopamine deficiency results in disinhibition of neurones synapsing at C. This leads to reduced activity at D, and to increased activity of neurones in the subthalamic nucleus. There is excess stimulation at synapse E, enhancing further inhibitory output of GPm-SNr.
The net effect via both pathways is to inhibit the ventral anterior (VA) and ventrolateral (VL) nuclei of the thalamus at synapse G. Cortical (motor) activity at H is thus reduced.
Levodopa helps slowness and tremor in PD (p. 1119) but induces unwanted dyskinesias by increasing dopamine activity at synapses A and B, it is thought by reversing sequences in both direct and indirect pathways.
Hemiballismus (p. 1121). Wild, flinging (ballistic) limb movements are caused by a lesion in the subthalamic nucleus, typically an infarct. This reduces excitatory activity at synapse E, reduces inhibition at G, with increased thalamo-cortical neuronal activity, and increases activity at H.
Cerebellum
The third system of motor control modulates coordination and learned movement patterns, rather than speed. Ataxia, i.e. unsteadiness, is characteristic.
The cerebellum receives afferents from:
Efferents pass from the cerebellum to:
Each lateral cerebellar lobe coordinates movement of the ipsilateral limbs. The vermis (a midline structure) is concerned with maintenance of axial (midline) posture and balance.
Cerebellar lesions (Box 22.9)
Expanding lesions obstruct the aqueduct to cause hydrocephalus, with severe pressure headaches, vomiting and papilloedema. Coning of the cerebellar tonsils (p. 1133) through the foramen magnum leads to respiratory arrest, sometimes within minutes/hours. Rarely, tonic seizures (attacks of limb stiffness) occur.
Lateral cerebellar hemisphere lesions
A lesion within one cerebellar lobe (e.g. tumour or infarction) causes disruption of the normal sequence of movements (dyssynergia) on the same side.
Posture and gait. The outstretched arm is held still in the early stages of a cerebellar lobar lesion (cf. the drift of pyramidal lesions) but there is rebound upward overshoot when the limb is pressed downwards and released. Gait becomes broad and ataxic, faltering towards the side of the lesion.
Tremor and ataxia. Movement is imprecise in direction, force and distance (dysmetria). Rapid alternating movements (tapping, clapping or rotary hand movements) become disorganized (dysdiadochokinesis). Intention tremor (action tremor with past-pointing) is seen, but speed of fine movement is preserved, cf. extrapyramidal and pyramidal lesions.
Nystagmus. Coarse horizontal nystagmus (p. 1075) develops with a lateral cerebellar lobe lesion. The fast component is always towards the side of the lesion.
Dysarthria. Halting, jerking speech develops – scanning speech.
Other signs. Titubation – rhythmic head tremor as either forward and back (yes-yes) movements or rotary (no-no) movements – can occur, mainly when cerebellar connections are involved (e.g. in essential tremor and MS, pp. 1080 and 1124). Hypotonia (floppy limbs) and depression of reflexes (with slow, pendular reflexes) are also sometimes seen.
Midline cerebellar lesions
Cerebellar vermis lesions have dramatic effects on trunk and axial muscles. There is difficulty standing and sitting unsupported (truncal ataxia), with a rolling, broad, ataxic gait. Lesions of the flocculonodular region cause vertigo and vomiting with gait ataxia if they extend to the roof of the IVth ventricle. Box 22.9 summarizes the main causes of cerebellar disease.
Tremor
Tremor means a regular and sinusoidal oscillation of the limbs, head or trunk.
Postural tremor
Everyone has a physiological tremor (often barely perceptible) of the outstretched hands at 8–12 Hz. This is increased with anxiety, caffeine, hyperthyroidism and drugs (e.g. sympathomimetics, sodium valproate, lithium) and occurs in mercury poisoning. A coarser, postural tremor is seen in benign essential tremor (usually at 5–8 Hz) and in chronic alcohol use.
Intention tremor
Tremor exacerbated by action, with past-pointing and accompanying incoordination of rapid alternating movement (dysdiadochokinesis), occurs in cerebellar lobe disease and with lesions of cerebellar connections. Titubation and nystagmus may be present.
Lower motor neurone (LMN) lesions
The LMN is the pathway from anterior horn cell (or cranial nerve nucleus) via a peripheral nerve to muscle motor endplates. The motor unit consists of one anterior horn cell, its single fast-conducting axon that leaves the cord via the anterior root, and the group of muscle fibres (100–2000) supplied via the nerve. Anterior horn cell activity is modulated by impulses from:
Signs of lower motor neurone lesions
These are seen in voluntary muscles that depend upon an intact nerve supply both for contraction and metabolic integrity. Signs follow rapidly if the LMN is interrupted (Box 22.10).
Spinal reflex arc
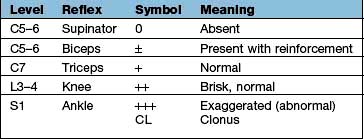
Components are illustrated in Figure 22.11. The stretch reflex is the physiological basis for all tendon reflexes. In the knee jerk, a tap on the patellar tendon activates stretch receptors in the quadriceps. Impulses in first-order sensory neurones pass directly to LMNs (L3 and L4) that contract quadriceps. Loss of a tendon reflex is caused by a lesion anywhere along the spinal reflex path. The reflex lost indicates its level (Table 22.6).
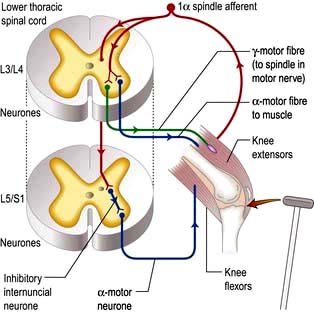
Figure 22.11 Knee jerk: a spinal reflex arc. Sudden patellar tendon stretching generates sensory action potentials in 1α muscle spindle afferents that synapse with γ-motor fibres to spindles and α-motor fibres. Motor action potentials cause brisk extensor muscle contraction; there is also inhibition of knee flexors.
Reinforcement. Distraction of the patient’s attention, clenching teeth or pulling interlocked fingers enhances reflex activity. Such reinforcement manoeuvres should be done before a reflex is recorded as absent.
Sensory pathways and pain
Peripheral nerves and spinal roots
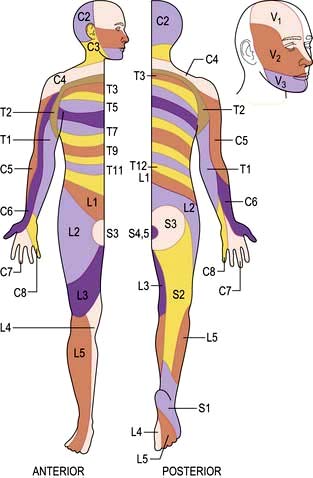
Peripheral nerves carry all modalities of sensation from either free or specialized nerve endings to dorsal roots and thence to the cord. Sensory distribution of spinal roots (dermatomes) is shown in Figure 22.12.
Spinal cord
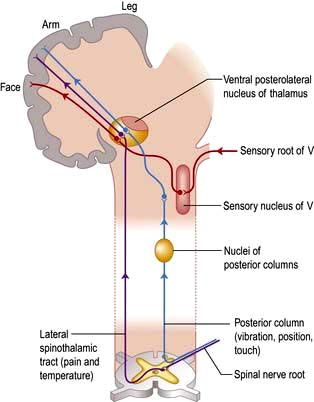
Posterior columns
Axons in the posterior columns whose cell bodies are in the ipsilateral gracile and cuneate nuclei in the medulla carry sensory modalities of vibration, joint position (proprioception), light touch and two-point discrimination. Axons from second-order neurones then cross in the brainstem to form the medial lemniscus, passing to the thalamus (Fig. 22.13).
Sensory cortex
Fibres from the thalamus pass to the parietal region sensory cortex (Fig. 22.13). Connections exist between the thalamus, sensory cortex and motor cortex.
Lesions of the sensory pathways
Altered sensation (paraesthesia), tingling, clumsiness, numbness and pain are the principal symptoms of sensory lesions. The pattern and distribution point to the site of pathology (Fig. 22.14).
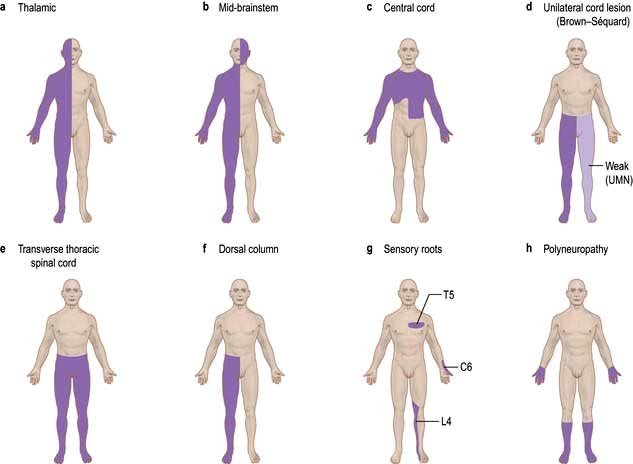
Figure 22.14 Principal patterns of loss of sensation. (a) Thalamic lesion: sensory loss throughout opposite side (rare). (b) Brainstem lesion: contralateral sensory loss below face and ipsilateral loss on face. (c) Central cord lesion, e.g. syrinx: ‘suspended’ areas of loss, often asymmetrical and ‘dissociated’, i.e. pain and temperature loss but light touch intact. (d) Hemisection of cord/unilateral cord lesion = Brown-Séquard syndrome: contralateral spinothalamic (pain and temperature) loss with ipsilateral weakness and dorsal column loss below lesion. (e) Transverse cord lesion: loss of all modalities, including motor, below lesion. (f) Dorsal column lesion, e.g. MS: loss of proprioception, vibration and light touch. (g) Individual sensory root lesions, e.g. C6, T5, L4. (h) Polyneuropathy: distal sensory loss.
Peripheral nerve lesions
Symptoms are felt within the distribution of a peripheral nerve (p. 1143). Section of a sensory nerve is followed by complete sensory loss. Nerve entrapment (p. 1144) causes numbness, pain and tingling. Tapping the site of compression sometimes causes a sharp, electric-shock-like pain in the distribution of the nerve, known as Tinel’s sign, e.g. in carpal tunnel syndrome (p. 1144).
Neuralgia
Neuralgia refers to pain, usually of great severity, in the distribution of a damaged nerve. Examples are:
Spinal root lesions
Root pain
Pain of root compression is felt in the myotome supplied by the root, and there is also a tingling discomfort in the dermatome. The pain is worsened by manoeuvres that either stretch the root (e.g. straight leg raising in lumbar disc prolapse) or increase pressure in the spinal subarachnoid space (coughing and straining). Cervical and lumbar disc protrusions (p. 1148) are common causes of root lesions.
Dorsal spinal root lesions
Section of a dorsal root causes loss of all modalities of sensation within a dermatome (see Fig. 22.12). However, overlap between adjacent dermatomes makes it difficult to detect anaesthesia when a single root is destroyed.
Lightning pains. Tabes dorsalis (rare in the UK) is a form of neurosyphilis that causes low-grade inflammation of dorsal roots and spinal cord root entry zones. Irregular, sharp, stabbing pains (like lightning) involve one or two spots, typically in a calf, thigh or ankle.
Spinal cord lesions
Posterior column lesions
These symptoms, though lateralized, are often felt vaguely without a clear sensory level. Position sense, vibration sense, light touch and two-point discrimination are diminished below the lesion. Position sense loss produces a stamping gait (sensory ataxia, p. 1068).
Lhermitte’s phenomenon
Electric-shock-like sensations radiate down the trunk and limbs on neck flexion. This points to a cervical cord lesion. Lhermitte’s is common in acute exacerbations of MS (p. 1124), and also occurs in cervical myelopathy (p. 1149), subacute combined degeneration of the cord (p. 1147), radiation myelopathy (p. 1149) and cord compression.
Spinothalamic tract lesions
Pure spinothalamic spinal lesions cause contralateral loss of pain and temperature sensation with a clear level below the lesion. This is called dissociated sensory loss – pain and temperature are dissociated from light touch, which remains preserved. This is seen typically in syringomyelia where a cavity occupies the central cord (p. 1137).
The spinal level is modified by lamination of fibres within the spinothalamic tracts. Fibres from lower spinal roots lie superficially and are damaged first by compressive lesions from outside the cord. As an external compressive lesion (e.g. a midthoracic extradural meningioma; Fig. 22.15) enlarges, the spinal sensory level ascends as deeper fibres become involved. Conversely, a central cord lesion (e.g. a syrinx, p. 1137) affects deeper fibres first. Spinothalamic tract lesions cause loss of pain and temperature perception (e.g. painless burns). Perforating ulcers and neuropathic (Charcot) joints develop.
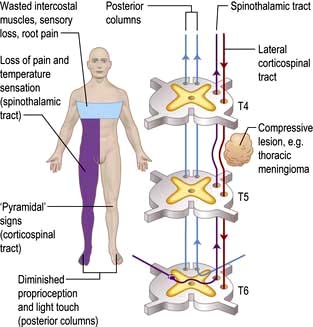
Spinal cord compression (Fig. 22.15)
Cord compression causes progressive spastic paraparesis (or tetraparesis/quadriparesis) with sensory loss below the level of compression. Sphincter disturbance is common. Root pain is frequent but not invariable, felt characteristically at the level of compression. With thoracic cord compression (e.g. an extradural meningioma), pain radiates around the chest, exacerbated by coughing and straining, as meningeal root sheaths are stretched.
Damage to one spinothalamic tract (contralateral loss of pain and temperature) with the ipsilateral corticospinal tract is known as the Brown-Séquard syndrome (originally, cord hemisection). The patient complains of numbness on one side and weakness on the other. Paraparesis/spinal cord lesions are discussed on page 1135.
Pontine lesions
Since lesions (e.g. an MS plaque) lie above the decussation of the posterior columns, and both medial lemniscus and spinothalamic tracts are close together, there is loss of all forms of sensation on the side opposite the lesion. Combinations of III, IV, V, VI and VII cranial nerve nuclei are seen, and may indicate a level (Fig. 22.13).
Thalamic lesions
Thalamic pain (also called central post-stroke pain or thalamic syndrome) follows a small thalamic infarct. The patient has a stroke (hemiparesis and sensory loss). Weakness improves, but deep-seated constant pain in the paretic limbs develops. Choreo-athetotic movements occur. Secondary depression may lead to self-harm. Thalamic lesions can also cause diminished sensation alone, on the opposite side; this is less usual.
Parietal cortex lesions
Sensory loss, neglect of one side, apraxia (p. 1068) and subtle disorders of sensation occur. Pain is not a feature of destructive cortical lesions. Irritative phenomena (e.g. partial sensory seizures from a parietal cortex glioma) cause tingling sensations in a limb, or elsewhere.
Pain
Pain is an unpleasant, unique physical and psychological experience. Acute pain serves a biological purpose (e.g. withdrawal) and is typically self-limiting, ceasing as healing ensues. Some forms of chronic pain (e.g. causalgia) outlast the period required for healing, and may be permanent.
Essential physiology of pain
Pain perception is mediated by free nerve endings, terminations of finely myelinated A-delta and of non-myelinated C fibres. Chemicals released following injury produce pain either by direct stimulation or by sensitizing nerve endings. A-delta fibres give rise to perception of sharp, immediate pain, then slower-onset, more diffuse and prolonged pain is mediated by slower-conducting C fibres.
Sensory impulses enter the cord via dorsal spinal roots. Impulses ascend either in each dorsal (posterior) column or in each spinothalamic tract. Grey matter neurones in the cord are arranged in laminae labelled I–X (dorsal to ventral). A fibres terminate in laminae I and V and excite second-order neurones that project to the contralateral side via the anterior commissure and via the anterolateral column of the direct spinothalamic tract. C fibres mostly terminate in the substantia gelatinosa (laminae II and III); axons then pass through the anterior commissure to the contralateral side and rostrally, up the spino-reticulo-thalamic tract.
The spinothalamic tracts carry impulses that localize pain. Thalamic pathways to and from the cortex mediate emotional components. Sympathetic activity increases pain, e.g. hyperaemia in a painful limb.
Gate theory of pain
Gate theory, a useful way of thinking about pain, proposes that entry of afferent impulses is monitored first by the substantia gelatinosa. This gate determines whether or not sufficient activity penetrates to fire secondary neurones in the dorsal horn. This, and subsequent gates, are influenced by brain and cord regulators that alter how far a gate remains open.
Animal studies suggest that downstream regulators binding to calcium-regulated transcription factors control CNS endogenous opiate precursors (e.g. pro-dynorphin). These modulate pain.
Endogenous opiates
Endorphin peptides have opioid activity and probably account for placebo phenomena, effects of stress and acupuncture, and may explain in part why some patients develop chronic pain syndromes. Endorphins are CNS neurotransmitters acting at inhibitory synapses via δ, κ and µ-receptors. Dynorphin (from the precursor, pro-dynorphin) is thought to modulate nociceptive (pain) impulses entering the cord.
FURTHER READING
American Society of Anesthesiologists Task Force on Chronic Pain Management and the American Society of Regional Anesthesia and Pain Medicine. Practice guidelines for chronic pain management. Anaesthesiology 2010; 112:810–833.
Turk DC, Wilson HD, Cahana A. Treatment of chronic non-cancer pain. Lancet 2011; 377:2226–2235.
Management of chronic pain
Chronic pain is gravely disabling, distressing, and taxing to treat (p. 509). Multidisciplinary pain-relief clinics provide specific and supportive therapy.
Management plans for intractable pain have seven components:
Diagnostic
Rigorous attention must be paid to the diagnosis, reviewing the entire history and investigations. A specific surgical approach may become apparent (e.g. spinal stenosis, trigeminal neuralgia, glossopharyngeal neuralgia, or syringomyelia).
Psychological
Chronic pain influences quality of life. Depression (p. 1170) is commonly associated with pain when the pathology is benign; antidepressants can help. Of patients suffering pain from secondary cancer about one-third are clinically depressed.
Analgesics
Perseverance and compliance with therapy is a common problem. The WHO analgesic ladder (p. 487) is useful.
Co-analgesics
Co-analgesics have a primary use other than for pain but help either alone or when added to analgesics. Examples are:
Tricyclic antidepressants, e.g. amitriptyline
Duloxetine – a serotonin-norepinephrine reuptake blocker
Anticonvulsants, e.g. carbamazepine, gabapentin and pregabalin (p. 486)
Calcium-channel blockers, e.g. ziconotide used intrathecally in chronic refractory pain
Capsaicin topical preparations – extracts from capsicum plants (chilli peppers) which bind to vanilloid receptors on pain neurones and deplete them of neurotransmitters such as substance P.
Stimulation
Acupuncture, ice, heat, ultrasound, massage, transcutaneous electrical nerve stimulation (TENS) and spinal cord stimulation all achieve analgesia by gating effects on large myelinated nerve fibres.
Bladder control and sexual dysfunction
Changes in micturition and failure of normal sexual activity due to neurological conditions are seen in sacral, spinal cord and cortical disease.
Essential functions and anatomy
The bladder has two functions: storage and voiding. Afferent pathways (T12–S4) respond to pressure within the bladder and sensation from the genitalia. As the bladder distends, continence is maintained by suppression of parasympathetic and reciprocal activation of sympathetic outflow. Both are under some voluntary control. Voiding takes place by para-sympathetic activation of the detrusor, and relaxation of the internal sphincter (Table 22.7).
Table 22.7 Efferents to bladder and genitalia
| Nerve supply | Function |
|---|---|
Parasympathetic |
Bladder wall: contraction |
S2–4 |
Internal sphincter: relaxationPenis/clitoris: engorgement |
Sympathetic |
Bladder wall: relaxation |
T12–L2 |
Internal sphincter: contractionOrgasm, ejaculation |
Pudendal nerves |
External sphincter (skeletal muscle) |
Cortical awareness of bladder fullness is located in the post-central gyrus, parasagittally, while initiation of micturition is in the pre-central gyrus. Voluntary control of micturition is located in the frontal cortex, parasagittally.
Neurological disorders of micturition
Urogenital tract disease is dealt with largely by urologists. Incontinence is common and easy to recognize; neurological causes are sometimes not obvious. These are:
Post-central lesions cause loss of sense of bladder fullness
Spinal cord. Bilateral UMN lesions (pyramidal tracts) cause urinary frequency and incontinence. The bladder is small and hypertonic, i.e. sensitive to small changes in intravesical pressure. Frontal lesions can also cause a hypertonic bladder.
LMN. Sacral lesions (conus medullaris, sacral root and pelvic nerve – bilateral) cause a flaccid, atonic bladder that overflows (cauda equina, p. 1149), often unexpectedly.
Management. Assessment of both urological causes (e.g. calculi, prostatism, gynaecological problems) and potential neurological causes of incontinence is necessary. Intermittent self-catheterization is used by many patients, with for example spinal cord lesions.
Male erectile dysfunction
Failure of penile erection often has mixed organic and psychological causes. Depression is common. Endocrine aspects of sexual dysfunction are described on page 977. Erectile dysfunction is sometimes helped by phosphodiesterase type 5 inhibitors, e.g. sildenafil.
Neurological tests
Neuro-imaging
Skull and spinal X-rays
Fractures of the vault or base
Vault and skull base disease (e.g. metastases, osteomyelitis, Paget’s disease, abnormal skull foramina, fibrous dysplasia)
Enlargement or destruction of the pituitary fossa-intrasellar tumour, raised intracranial pressure
Intracranial calcification – tuberculoma, oligodendroglioma, wall of an aneurysm, cysticercosis.
Spinal X-rays show fractures, congenital and destructive lesions (bone cysts, infection, metastases) and degenerative spondylosis.
Imaging brain and spinal cord
Brain CT and MRI are widely available worldwide. Myelography (contrast imaging of the cord and ventricles; ventriculography) is obsolete.
Brain computed tomography (CT)
A collimated X-ray beam moves synchronously across a brain slice 2–13 mm thick. Transmitted X-irradiation from a pixel, an element <1 mm2, is computer-processed to assign a Hounsfield number to its density (air = −1000 units; water = 0; bone = +1000 units). The digital data are converted to cross-sectional images to reconstruct brain anatomy. Helical CT (spiral or volumetric CT) provides greater definition in a shorter time. It is used for two-dimensional reconstruction.
Differences in attenuation (density) between bone, brain and CSF enable recognition of normal and infarcted tissue, tumour, blood, intracerebral haemorrhage, free subarachnoid blood, subdural and extradural haematoma and oedema.
Enhancement with i.v. contrast delineates areas of altered blood supply (CT angiography).
Safety. The irradiation involved is relatively small. There are occasional reactions to contrast.
Limitations of brain CT
Lesions under 1 cm in diameter may be missed.
Lesions with attenuation close to bone may be missed, if near the skull.
Lesions with attenuation similar to brain are poorly displayed (e.g. MS plaques, isodense subdural haematoma).
CT is not good at detecting posterior fossa lesions because of surrounding bone.
Patient cooperation: an anaesthetic is very occasionally needed.
Magnetic resonance imaging: MRI
A hydrogen nucleus is a proton whose electrical charge creates a local electrical field. Protons are aligned by sudden strong magnetic impulses and then imaged with radiofrequency waves at right angles to their alignment. The protons resonate and spin, then revert to their normal alignment. As they do so, images are made at different phases of relaxation, known as T1, T2, T2 STIR, FLAIR, diffusion-weighted imaging (DWI) and other sequences. From these sequences, often referred to as different weightings, recorded images are compared. Gadolinium is used as i.v. contrast to show areas of increased vascularity.
Advantages of MRI
MR distinguishes between white and grey matter in the brain and cord.
Cord and nerve roots are imaged directly.
MRI has resolution superior to CT (around 0.5 cm).
MR angiography (MRA) images blood vessels without contrast.
Tumours, infarction, haemorrhage, MS plaques, posterior fossa, foramen magnum and cord are demonstrated well by MRI.
Limitations are principally time and cost. Patients need to keep still within a narrow tube: claustrophobia is an issue; open machines are available that are less claustrophobic. Patients with pacemakers or metallic fragments in the brain cannot be imaged. MR imaging frequently shows diffuse meningeal enhancement with gadolinium for some days after lumbar puncture (p. 1091).
Digital cerebral and spinal angiography
Where advanced MRI and CT are available, these techniques are little used. Contrast is injected intra-arterially or intravenously. Angiography carries a mortality and stroke risk (<1%). Images of aorta, carotid, vertebral and brain arteries demonstrate occlusion, stenoses, atheroma, aneurysms and arteriovenous malformations (AVMs). Spinal angiography images cord AVMs.
Positron emission tomography (PET), single proton emission computed tomography (SPECT), dopamine transporter imaging (DAT) and functional MRI (fMRI)
These functional imaging techniques track uptake of radioisotopes and/or metabolites. PET is used principally in the detection of occult neoplasms, outside the CNS. SPECT is little used in cerebrovascular disease and traumatic brain injury – there are issues of reliability. DAT is used in basal ganglia disease. fMRI is largely a research tool for mapping brain function, in health and disease.
Isotope bone scanning
The radioisotope [99mTc]-pertechnetate is given intravenously. The technique is used principally in detection of vertebral, skull and bone metastases.
Electroencephalography (EEG)
EEG (Fig. 22.16) recorded from scalp electrodes (16 channels simultaneously) is of value in epilepsy and diffuse brain diseases. Videotelemetry, combining EEG with video, is invaluable in attacks difficult to diagnose.
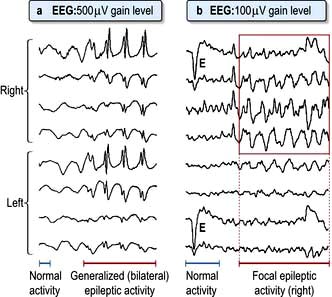
Figure 22.16 Examples of EEG traces. (a) Normal activity followed by generalized epileptic spikes in all leads. (b) Normal activity followed by focal spikes in right leads. E, eye-movement artefact.
Epilepsy
Spikes, or spike-and-wave abnormalities, are hallmarks of epilepsy, but it should be emphasized that patients with epilepsy often have a normal EEG between seizures (p. 1092).
Diffuse brain disorders
Slow-wave EEG abnormalities appear in encephalitis, prion (Creutzfeldt–Jakob) diseases and metabolic states (e.g. hypoglycaemia, hepatic coma).
Brain death
The EEG is isoelectric (flat); EEG is no longer necessary to confirm brain death (p. 898).
Electromyography and conduction studies
Electromyography
A concentric needle electrode is inserted into voluntary muscle. Amplified recordings, on an oscilloscope, are also heard through a speaker. The main EMG features:
Denervation and reinnervation changes
Myopathic, myotonic and myasthenic changes (p. 1090).
Peripheral nerve conduction (Fig. 22.17)
Four measurements are of principal value in neuropathies and nerve entrapment:
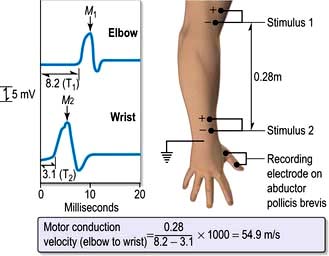
Figure 22.17 Measurement of motor conduction velocity (MCV) in ulnar nerve. The recording electrode on abductor pollicis brevis measures muscle action potential (M) from ulnar nerve stimulation at elbow (Stimulus 1) and at wrist (Stimulus 2).
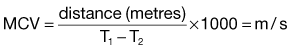
Measurements differentiate between axonal and demyelinating damage and determine whether pathology is focal or diffuse.
Cerebral-evoked potentials
Visual-evoked potentials (VEPs) record the interval visual stimuli take to reach the occipital cortex, and the amplitude of response. VEPs are used to confirm previous retrobulbar neuritis (p. 1073); this leaves a delayed latency despite clinical recovery.
Similar techniques for auditory and somatosensory potentials (from a limb) are also used.
Lumbar puncture and CSF examination
See Table 22.8 and Practical Box 22.3.
| Appearance | Crystal clear, colourless |
|---|---|
Pressure |
60–150 mm of CSF, recumbent |
Cell count |
<5/mm3No polymorphsMononuclear cells only |
Protein |
0.2–0.4 g/L |
Glucose |
 – –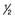 of blood glucose of blood glucose |
IgG |
<15% of total CSF protein |
Oligoclonal bands |
Absent |
Practical Box 22.3
Lumbar puncture
The procedure should be explained to the patient, and consent obtained. LP should not be performed in the presence of raised intracranial pressure or when an intracranial mass lesion is a possibility.
Technique
The patient is placed on the edge of the bed in the left lateral position with knees and chin as close together as possible.
The third and fourth lumbar spines are marked. The fourth lumbar spine usually lies on a line joining the iliac crests.
Using sterile precautions, 2% lidocaine is injected into the dermis by raising a bleb in either the third or fourth lumbar interspace.
The LP needle is pushed through the skin in the midline, steadily forwards and slightly towards the head, with the head and spine bolstered horizontally with pillows.
When the needle is felt to penetrate the dura, the stylet is withdrawn and a few drops of CSF allowed to escape.
The CSF pressure can then be measured with a manometer connected to the needle. The patient’s head must be on the same level as the sacrum. Normal CSF pressure is 60–150 mm of CSF. The level rises and falls with respiration and heart beat, and rises on coughing.
CSF specimens are collected in three sterile bottles and a sample for CSF glucose, together with a simultaneous blood glucose sample.
Record CSF naked-eye appearance: clear, cloudy, yellow (xanthochromic), red.
The patient is asked to lie flat after the procedure to avoid subsequent headaches, but this manoeuvre is probably of little value.
Contraindications
Suspicion of a mass lesion in the brain or cord. Caudal herniation of the cerebellar tonsils (coning) may occur if an intracranial mass is present and the pressure below is reduced by removal of CSF
Any cause of raised intracranial pressure
Local infection near the LP site
Congenital lesions in the lumbosacral region (e.g. meningomyelocele)
Platelet count <40 × 109/L and other clotting abnormalities, including anticoagulant drugs
Unconscious patients and those with papilloedema must have a CT scan before lumbar puncture.
Notes
Contraindications are relative; there are circumstances when LP is carried out despite them.
Composition of normal CSF is shown in Table 22.8.
Indications for lumbar puncture (LP):
Diagnosis of meningitis and encephalitis
Diagnosis of subarachnoid haemorrhage (sometimes)
Measurement of CSF pressure, e.g. idiopathic intracranial hypertension (p. 1110)
Removal of CSF therapeutically, e.g. idiopathic intracranial hypertension
Diagnosis of various conditions, e.g. MS, neurosyphilis, sarcoidosis, Behçet’s, neoplastic involvement, polyneuropathies
Meticulous attention should focus on microbiology in suspected CNS infection. Close liaison between clinician and microbiologist is essential. Specific techniques (e.g. polymerase chain reaction to identify bacteria) are invaluable. Repeated CSF examination is often necessary in chronic infection such as tuberculosis. Post-LP headaches, worse on standing, are a common complaint for several days (or more). Prolonged headaches can be treated by an ‘autologous intrathecal blood patch’ – injection of 20 mL of the patient’s venous blood into the CSF.
Biopsy
Interpretation of brain, tonsillar, muscle and nerve histology requires specialist neuropathology services.
Brain and meninges
Brain biopsy (e.g. of a non-dominant frontal lobe) is sometimes used to diagnose inflammatory and degenerative brain diseases. CT and MR stereotactic biopsies of intracranial lesions are standard procedures.
Muscle
Biopsy, with light and electron microscopy and biochemical analysis, elucidates diagnosis of inflammatory, metabolic and dystrophic disorders (p. 1152).
Psychometric assessment
Psychometric testing assesses cognitive function. Preservation of verbal IQ (a measure of past attainments) with deterioration of performance IQ (a measure of present abilities) indicates decline of cognitive function, seen for example following brain injury or in dementia. Low subtest scores (e.g. block design, various aspects of memory, visual, speech and constructional skills) indicate impaired function of specific brain regions.
Depression and lack of attention also reduce scores – a substantial problem. Opinions sometimes vary between psychologists about interpretation of tests, particularly after brain injury, limiting the value of the tests.
Routine tests
See Table 22.9.
Table 22.9 Value of routine investigations neurology
| Test | Yield | Condition |
|---|---|---|
Urinalysis |
Glycosuria |
Polyneuropathy |
Ketones |
Coma |
|
Bence Jones protein |
Cord compression |
|
Blood picture |
↑ T MCV↑↑ T T ESR |
B12 deficiencyGiant cell arteritis |
Blood glucose |
HypoglycaemiaHyperglycaemia |
ComaComa |
Serum electrolytes |
Hyponatraemia |
Coma |
Hypokalaemia |
Weakness |
|
Serum calcium |
Hypocalcaemia |
Tetany, spasms |
Serum CPK |
Raised |
Muscle disease |
Chest X-ray |
Lytic bone or mass lesion |
Bronchial cancer, thymoma |
Specialized tests in specific diseases
Various tests are employed to diagnose individual (sometimes rare) diseases. Examples are:
Anti-cardiolipin and lupus anticoagulant antibody and detailed clotting studies in stroke (p. 538)
Antibody to acetylcholine receptor protein and anti-Mu SK antibodies in myasthenia gravis (p. 1151)
Serum copper and caeruloplasmin in Wilson’s disease (p. 1098)
Blood lactate studies (failure to rise on exercise) in McArdle’s syndrome (p. 1069)
Anti-neuronal antibodies (paraneoplastic syndromes)
Serum phytanic acid (elevated) in Refsum’s disease
Serum long-chain fatty acid (present) in adrenoleucodystrophy
Genetic studies, e.g. Huntington’s disease, hereditary sensorimotor neuropathies and ataxias (p. 1121 and p. 1147).