The small intestine
Structure
The small intestine extends from the duodenum to the ileocaecal valve. It is approximately 3–6 m in length, and 300 m2 in surface area. The upper 40% is the duodenum and jejunum, the remainder is the ileum. Its surface area is enormously increased by circumferential mucosal folds that have on them multiple finger-like projections called villi. On the villi the surface area is further increased by microvilli on the luminal side of the epithelial cells (enterocytes) (Fig. 6.23).
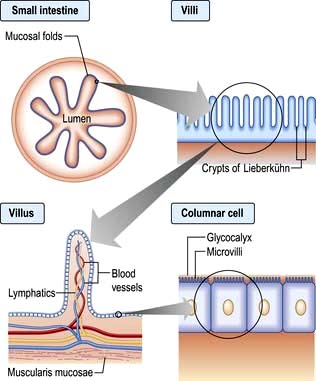
Each villus consists of a core containing blood vessels, lacteals (lymphatics) and cells (e.g. plasma cells and lymphocytes). The lamina propria contains plasma cells, lymphocytes, macrophages, eosinophils and mast cells. The crypts of Lieberkühn are the spaces between the bases of the villi.
Enterocytes are formed at the bottom of the crypts and migrate toward the tops of the villi, where they are shed. This process takes 3–4 days. On its luminal side, the enterocyte is covered by microvilli and a gelatinous layer called the glycocalyx. Scattered between the epithelial cells are mucin-secreting goblet cells and occasional intraepithelial lymphocytes and Paneth cells. Most of the blood supply to the small intestine is via branches of the superior mesenteric artery. The terminal branches are end arteries – there are no local anastomotic connections.
Enteric nervous system (ENS)
This controls the functioning of the small bowel; it is an independent system that coordinates absorption, secretion, blood flow and motility. It is estimated to contain 108 neurones (as many as the spinal cord) contained in two major ganglionated plexuses: the myenteric plexus between the muscular layers of the intestinal wall, and the submucosal plexuses associated with the mucosa. The ENS communicates with the central nervous system via autonomic afferent and efferent pathways but can operate autonomously.
Coordination of small intestinal function involves a complex and poorly understood interplay between many neuroactive mediators and their receptors, ion channels, GI hormones, nitric oxide and other transmitters. Acetylcholine, adrenaline, ATP, vasoactive intestinal peptide (VIP) and other hormones and opioids have been shown to have actions in the small bowel, but the exact role for each is far from being understood.
Gut motility
The contractile patterns of the small intestinal muscular layers are primarily determined by the enteric nervous system. The CNS and gut hormones also have a modulatory role on motility. The interstitial cells of Cajal which lie within the smooth muscle appear to govern rhythmic contractions.
During fasting, a distally migrating sequence of motor events termed the migrating motor complex (MMC) occurs in a cyclical fashion. The MMC consists of
 A period of motor quiescence (phase I),
A period of motor quiescence (phase I),
followed by a period of irregular contractile activity (phase II),
culminating in a short (5–10 min) burst of regular phasic contractions (phase III).
Each MMC cycle lasts for approximately 90 min. In the duodenum, phase III is associated with increased gastric, pancreatic and biliary secretions. The role of the MMC is unclear, but the strong phase III contractions propel secretions, residual food and desquamated cells towards the colon. It is named the ‘intestinal housekeeper’.
After a meal, the MMC pattern is disrupted and replaced by irregular contractions. This seemingly chaotic pattern lasts typically for 2–5 hours after feeding, depending on the size and nutrient content of the meal. The irregular contractions of the fed pattern have a mixing function, moving intraluminal contents to and fro, aiding the digestive process.
Neuroendocrine peptide production
The hormone-producing cells of the gut are scattered diffusely throughout its length and also occur in the pancreas. The cells that synthesize hormones are derived from neural ectoderm and are known as APUD (amine precursor uptake and decarboxylation) cells. Many of these hormones have very similar structures and their action is probably local.
Gut hormones play a part in the regulation and integration of the functions of the small bowel and other metabolic activities. Their actions are complex and interacting, both with each other and with the ENS (Table 6.8).
Table 6.8 Gut regulatory peptides
| Peptide | Localization | Main actions |
|---|---|---|
Gastrin/cholecystokinin family |
|
|
Cholecystokinin (CCK) |
|
|
Multiple forms from CCK8 (8 amino acids) to CCK83; 8, 33 and 58 are predominant. Terminal 5 amino acids same as gastrin |
Duodenum and jejunum (I cells) |
Causes gall bladder contraction and sphincter of Oddi relaxation. Trophic effects on duodenum and pancreas. Pancreatic secretion (minor role). Role in satiety – acting in CNS |
Gastrin |
G cells in gastric antrum and duodenum |
Stimulates acid secretion. |
Secretin-glucagon family |
|
|
Secretin |
Duodenum and jejunum (S cells) |
Stimulates pancreatic bicarbonate secretion |
Glucagon |
Alpha cells of pancreas |
Opposes insulin in blood glucose control |
Vasoactive polypeptide (VIP) |
Enteric nerves |
Intestinal secretion of water and electrolytes. |
Glucose-dependent insulinotropic peptide (GIP) |
Duodenum (K cells) |
Release by intraduodenal glucose causes greater insulin release by islets than i.v. glucose (incretin effect) |
Glucagon-like peptide-1 (GLP-1) |
Ileum and colon (L cells) |
Incretin. Stimulates insulin synthesis. Trophic to islet cells. Inhibits glucagon secretion and gastric emptying |
Glicentin |
L cells |
Stimulates insulin secretion and gut growth, inhibits gastric secretion |
Growth hormone-releasing factor (GHRF) |
Small intestine |
Unclear |
Pancreatic polypeptide family |
|
|
Pancreatic polypeptide (PP) |
Pancreas (PP cells) |
Inhibits pancreatic and biliary secretion |
Peptide YY (PYY) |
Ileum and colon (L cells) |
Inhibits pancreatic exocrine secretion. Slows gastric and small bowel transit (‘ileal brake’). Reduces food intake and appetite |
Neuropeptide Y (NPY) |
Enteric nerves |
Stimulates feeding. Regulates intestinal blood flow |
Other |
|
|
Motilin |
Whole gut |
Increases gastric emptying and small bowel contraction |
Ghrelin |
Stomach |
Stimulates appetite, increases gastric emptying |
Obestatin |
Stomach and small intestine |
Opposes ghrelin |
Oxyntomodulin |
Colon |
Inhibits appetite |
Gastrin releasing-polypeptide (bombesin) |
Whole gut and pancreas |
Stimulates pancreatic exocrine secretion and gastric acid secretion |
Somatostatin |
Stomach and pancreas (D cells) |
Inhibits secretion and action of most hormones |
Substance P |
Enteric nerves |
Enhances gastric acid secretion, smooth muscle contraction |
Neurotensin |
Ileum |
Affects gut motility. Increases jejunal and ileal fluid secretion |
Insulin |
Pancreatic β cells |
Increases glucose utilization |
Chromogranins |
Neuroendocrine cells |
Precursor for other regulatory peptides that inhibit neuroendocrine secretion |
Physiology
In the small bowel digestion and absorption of nutrients and ions takes place, as does the regulation of fluid absorption and secretion. The epithelial cells of the small bowel form a physical barrier that is selectively permeable to ions, small molecules and macromolecules. Digestive enzymes such as proteases and disaccharidases are produced by intestinal cells and expressed on the surface of microvilli; others such as lipases produced by the pancreas are associated with the glycocalyx. Some nutrients are absorbed most actively in specific parts of the small intestine; iron and folate in the duodenum and jejunum, vitamin B12 and bile salts in the terminal ileum where they have specific receptors.
General principles of absorption
Simple diffusion
This process is nonspecific, requires no carrier molecule or energy and takes place if there is a concentration gradient from the intestinal lumen (high concentration) to the bloodstream (low concentration). Vitamin B12 can be absorbed from the jejunum by this means.
Facilitated diffusion
Absorption takes place down a concentration gradient, but a membrane carrier protein is involved, conferring specificity on the process. Fructose is an example.
Active transport
Absorption occurs via a specific carrier protein, powered by cellular energy, and thus a substance can be transported against a concentration gradient. Many carrier proteins are powered by ion gradients across the enterocyte wall. For example, glucose crosses the enterocyte microvillous membrane from the lumen into the cell against a concentration gradient by using a co-transporter carrier molecule. This is the sodium/glucose co-transporter, SGLT1 (Fig. 6.24). The process is powered by the energy derived from the flow of Na+ ions from a high concentration outside the cell to a low concentration inside. The sodium gradient across the cell wall is maintained by a separate ATP-consuming Na+/K+ exchanger in the basolateral membrane. Glucose leaves the cell on the serosal side by facilitated diffusion via a sodium-independent carrier (GLUT-2) in the basolateral membrane.
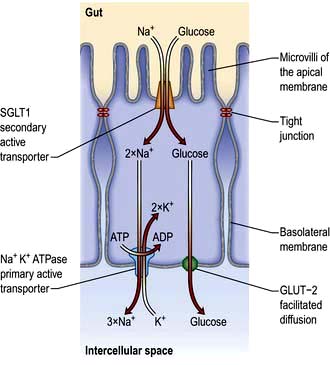
Figure 6.24 Transcellular uptake of glucose across the intestinal epithelia. Glucose is co-transported across the apical membrane with sodium ions by the sodium-dependent glucose transporter (SGLT). This is secondary active transport as the sodium is travelling down its electrochemical gradient. The sodium gradient is maintained by the primary active transport of sodium across the basolateral membrane by the Na+K+ ATPase (thus intracellular Na+ is kept low). Transcellular transport of glucose is achieved by facilitated diffusion across the basolateral membrane as glucose is moved down its concentration gradient by GLUT-2.
Another active transport mechanism operates for Na+ absorption in the ileum using an Na+/H+ exchange mechanism powered by the outwardly directed gradient of H+ across the cell membrane.
Absorption of nutrients in the small intestine
Carbohydrate
Dietary carbohydrate consists mainly of starch, with some sucrose and a small amount of lactose. Starch is a polysaccharide made up of numerous glucose units. In order to have a nutrient value starch must be digested into smaller oligo-, di- and finally monosaccharides which may then be absorbed. Polysaccharide hydrolysis begins in the mouth catalysed by salivary amylase, though the majority takes place under the action of pancreatic amylase in the upper intestine. The breakdown products of starch digestion are maltose and maltotriose, together with sucrose and lactose. These are further hydrolysed on the microvillous membrane by specific oligo- and disaccharidases to form glucose, galactose and fructose. These monosaccharides are then able to be transported across the enterocytes into the blood (Fig. 6.24).
Protein
Dietary protein is digested by pancreatic proteolytic enzymes to amino acids and peptides prior to absorption. These enzymes are secreted by the pancreas as pro-enzymes and transformed to active forms in the lumen. Protein in the duodenal lumen stimulates the enzymatic conversion of trypsinogen to trypsin, and this in turn activates the other pro-enzymes, chymotrypsin and elastase.
These enzymes break down protein into oligopeptides. Some di- and tri-peptides are absorbed intact by carrier-mediated processes, while the remainder are broken down into free amino acids by peptidases on the microvillous membranes of the enterocytes, prior to absorption into the cell by a variety of amino acid and peptide carrier systems.
Fat
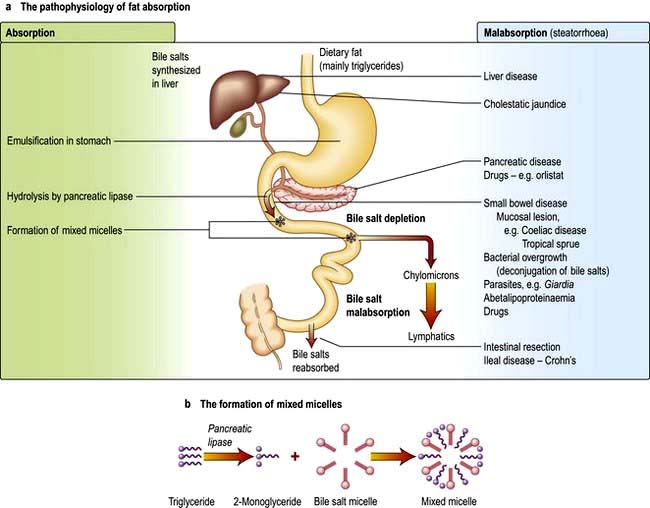
Dietary fat consists mainly of triglycerides with some cholesterol and fat-soluble vitamins. Fat is emulsified by mechanical action in the stomach. Bile containing the amphipathic detergents, bile acids and phospholipids enters the duodenum following gall bladder contraction. They act to solubilize fat and promote hydrolysis of triglycerides in the duodenum by pancreatic lipase to yield fatty acids and monoglycerides. Bile acids, phospholipids and the products of fat digestion cluster together with their hydrophilic ends on the outside to form aggregations called mixed micelles. Trapped in the centre of the micelles are the hydrophobic monoglycerides, fatty acids and cholesterol. At the cell membrane the lipid contents of the micelles are absorbed, while the bile salts remain in the lumen. Inside the cell the monoglycerides and fatty acids are re-esterified to triglycerides. The triglycerides and other fat-soluble molecules (e.g. cholesterol, phospholipids) are then incorporated into chylomicrons to be transported into the lymph.
By contrast, another mechanism exists for medium-chain triglycerides (MCT; fatty acids of chain length 6–12) which are transported via the portal vein with a small amount of long-chain fatty acid. Patients with pancreatic exocrine or bile salt insufficiency can therefore supplement their fat absorption with MCT.
Bile salts are not absorbed in the jejunum, so the intraluminal concentration in the upper gut is high. They pass down the intestine to be absorbed in the terminal ileum and are transported back to the liver. This enterohepatic circulation prevents excess loss of bile salts (see p. 306).
The pathophysiology of fat absorption is shown in Figure 6.25. Interference with absorption can occur at all stages, as indicated, giving rise to steatorrhoea (>17 mmol or 6 g of faecal fat per day).
Water and electrolytes
Large amounts of water and electrolytes, partly dietary, but mainly from intestinal secretions, are absorbed coupled with absorption of monosaccharides, amino acids and bicarbonate in the upper jejunum. Water and electrolytes are also absorbed paracellularly (between the enterocytes) down electrochemical and osmotic gradients. Additional water and electrolytes are absorbed in the ileum and colon, where active sodium transport is not coupled to solute absorption. Secretion of fluid and electrolytes occur together to maintain the normal functioning of the gut. Secretory diarrhoea (see p. 292) can occur because of defects in intestinal secretory mechanisms.
Water-soluble vitamins, essential metals and trace elements
These are all absorbed in the small intestine. Vitamin B12 (see p. 382) and bile salts are absorbed by specific transport mechanisms in the terminal ileum; malabsorption of both these substances often occurs following ileal resection.
Response of the small bowel to antigens and pathogens
The small bowel has a number of mechanisms to prevent colonization and invasion by pathogens while simultaneously preventing inappropriate responses to foreign antigens or the indigenous bacterial population. At the same time commensal bacteria maintain the integrity of the small bowel and play a major role in host physiology.
Mechanisms
Continuous shedding of surface epithelial cells
The physical movement of the luminal contents
Colonization resistance – the ability of the indigenous microbiota to outcompete pathogens for a survival niche in the gut.
Enzymes such as lysozyme and phospholipase A2 secreted by Paneth cells at the base of the crypts help ensure an infection-free environment in the gut, even in the presence of commensal bacteria.
Antimicrobial peptides are secreted from enterocytes and Paneth cells in response to pathogenic bacteria. These include defensins, which are 15–20 amino acid peptides with potent activity against a broad range of pathogens including Gram-positive and Gram-negative bacteria, fungi and viruses.
Trefoil peptides are a family of small proteins secreted by goblet cells. They consist of a three-loop structure with intra-chain disulphide bonds which makes the molecules highly resistant to digestion. Their actions include stabilization of mucus, promotion of cell migration over injured areas, and promotion of repair. Three trefoil factors (TFF) are found in humans (TFF1, TFF2 and TFF3) all of which have been implicated in the response to gastrointestinal injury in experimental models. Their molecular mode of action is not yet known.
Innate immunological defence
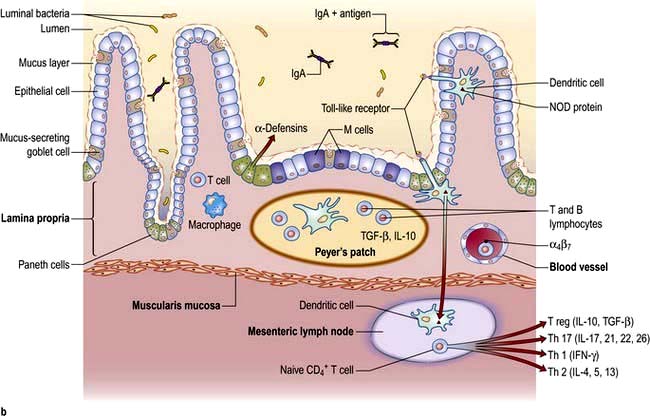
Humoral. IgA is the principal mucosal antibody. IgA mediates mucosal immunity by agglutinating and neutralizing pathogens in the lumen and preventing colonization of the epithelial surface (Fig. 6.26). IgA is secreted from immunocytes in the lamina propria as dimers joined by a protein called the ‘Joining chain’ (J-chain); in this form it is known as polymeric IgA (pIgA). This pIgA is internalized by endocytosis at the basolateral membrane of enterocytes. It crosses the cell as a complex of pIgA/pIgAR and is secreted onto the mucosal surface.
B-cell sensitization. Antigens from the lumen of the bowel are transported by M cells and dendritic cells in the follicle-associated epithelium (FAE). This covers Peyer’s patches in the ‘dome’ region which contain abundant virgin B cells, helper T cells and antigen-presenting cells. Activated B cells then produce IgA locally and are programmed to home back to the lamina propria. They travel through mesenteric lymph nodes and then via the thoracic duct to the blood and back to the small bowel and other mucosal surfaces (such as the airways) where they undergo terminal differentiation into plasma cells. Homing back to the gut is facilitated by the α4β7-integrin on gut-derived lymphocytes binding to MAdCAM-1, uniquely expressed on blood vessels in the gut.
Cellular defence. T lymphocytes also provide host defence and initiate, activate and regulate adaptive immune responses. Intestinal T lymphocytes occur principally in three major compartments:
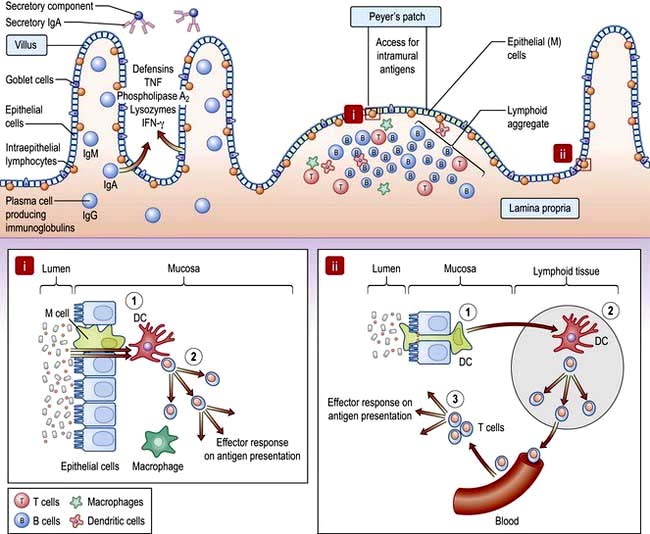
Figure 6.26 Small intestinal mucosa with a Peyer’s patch, showing the gut-associated lymphoid tissue (GALT). Inset i: (1) Specialized ‘M’ cells within a lymphoid follicle pass antigen to underlying APC such as DC. (2) DC present antigen to T cells resulting in activation and effector responses. Inset ii: (1) DC pass dendrites through epithelial tight junctions to sample luminal antigen. (2) DC traffic to the mesenteric lymph node and present antigen to naive T cells which differentiate into effector or regulatory subtypes and are imprinted with a gut homing integrin. (3) On subsequent antigen presentation, activated T cells home to lamina propria and exert regulatory or inflammatory effect depending on their phenotype. DC, dendritic cell.
Commensal bacteria
The relationship between the hundred thousand billion microbes in the human gut and the host are only beginning to be appreciated. Germ-free mice have essentially no mucosal immune system showing that the abundant and activated immune system seen in healthy individuals is driven by the flora, without adverse effects. Bacteria also release chemical signals such as LPS and lipoteichoic acid that are recognized by Toll-like receptors (TLRs) (see p. 55) present on a variety of intestinal cells, priming repair processes and enhancing the ability of the epithelium to respond to injury.
Oral tolerance
The immune system must guard against pathogens and toxins while avoiding an excessive response to the multiplicity of food antigens and commensal bacteria. The mechanisms by which tolerance occurs are undoubtedly multiple, including maintaining barrier function to prevent excess antigen uptake, active inhibition via regulatory T cells, and dendritic cells which promote tolerogenic rather than immunogenic T cell responses. All of these are likely to play a role in diseases such as coeliac disease, caused by an excessive T cell response to gluten, or Crohn’s disease, where tolerance to the indigenous bacterial population is defective.
Presenting features of small bowel disease
Regardless of the cause, the common presenting features of small bowel disease are listed below. However, 10–20% of patients will have no diarrhoea or any other gastrointestinal symptoms.
Diarrhoea is common and may be watery.
Steatorrhoea occurs when the stool fat is >17 mmol/day (or 6 g/day). The stools are pale, bulky, offensive, float (because of their increased air content), leave a fatty film on the water in the pan and are difficult to flush away.
Abdominal pain and discomfort. Abdominal distension can cause discomfort and flatulence. The pain has no specific character or periodicity and is not usually severe.
Weight loss. Weight loss is largely due to the anorexia that invariably accompanies small bowel disease. The calorie deficit due to malabsorption is small relative to the reduction in intake.
Nutritional deficiencies. Deficiencies of iron, B12, folate or all of these, leading to anaemia, are the only common deficiencies. Occasionally malabsorption of other vitamins or minerals occurs, causing bruising (vitamin K deficiency), tetany (calcium deficiency), osteomalacia (vitamin D deficiency), or stomatitis, sore tongue and aphthous ulceration (multiple vitamin deficiencies). Oedema due to hypoproteinaemia is due to low intake and intestinal loss of albumin (protein-losing enteropathy).
Physical signs are few and nonspecific. If present, they are usually associated with anaemia and the nutritional deficiencies described above.
Abdominal examination is often normal, but sometimes distension and, rarely, hepatomegaly or an abdominal mass are found. Visible peristalsis and high pitched bowel sounds can indicate chronic subacute obstruction of the small intestine, e.g. due to stricturing Crohn’s disease. Gross weight loss, oedema and muscle wasting is seen only in severe cases. A neuropathy, not always due to B12 deficiency, can be present.
Investigation of small bowel disease (Fig. 6.27)
The emphasis in the investigation of malabsorption is on the structural features of the underlying disorder, rather than on the documentation of malabsorption itself.
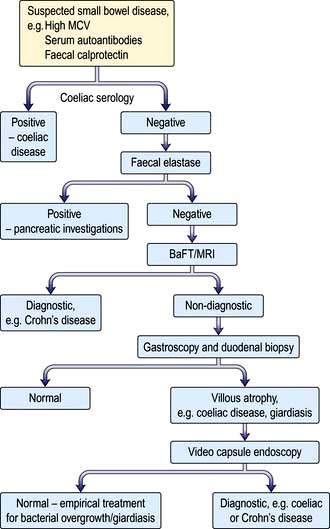
Figure 6.27 Flow diagram for investigation of patients with suspected small bowel disease. BaFT, barium follow-through; MRI, magnetic resonance imaging.
Blood tests
Full blood count and film. Anaemia can be microcytic, macrocytic or normocytic and the blood film may be dimorphic. Other abnormal cells (e.g. Howell–Jolly bodies, p. 406) may be seen in splenic atrophy associated with coeliac disease.
Low serum calcium and raised alkaline phosphatase may indicate the presence of osteomalacia due to vitamin D deficiency.
Liver biochemistry and serum albumin and prothrombin time.
Immunological tests. Measurement of serum antibodies to endomysium and tissue transglutaminase is useful for the diagnosis of coeliac disease. These should always be accompanied by an assessment of total immunoglobulin levels.
Small bowel anatomy
MRI enteroclysis. This is cross-sectional imaging which does not involve radiation. Using oral loading with water or a hyperonic solution to distend the small bowel lumen.
Small bowel barium follow-through (see p. 234). This detects gross anatomical defects such as diverticula, strictures and Crohn’s disease. Dilatation of the bowel and a changed fold pattern may suggest malabsorption but these are nonspecific findings. Gross dilatation is seen in myopathic pseudo-obstruction.
Small bowel biopsy. This is used to assess the microanatomy of the small bowel mucosa. Biopsies are usually obtained via an endoscope passed into the duodenum and should be well-orientated for correct evaluation. The histological appearances are described in the sections on individual diseases. A smear of the jejunal juice or a mucosal impression should also be made when Giardia intestinalis is suspected.
Ultrasound is a useful preliminary investigation which can show thickened small bowel or distended loops.
CT scanning is used to look for small bowel wall thickening, diverticula and for extraintestinal features such as abscesses (e.g. in Crohn’s disease).
Video-capsule enteroscopy is increasingly widely used to directly visualize the small bowel lumen and mucosa along its entire length. It is particularly useful in the diagnosis of occult GI bleeding.
Tests of absorption
These are required only in complicated cases.
Fat malabsorption. The confirmation of the presence of steatorrhoea is only occasionally necessary. Three-day faecal fat analysis, triglyceride breath tests and serum β carotene are now rarely performed. In rare cases when it is essential to confirm steatorrhoea, Sudan III staining of a faecal sample can be used.
Lactose tolerance test. Testing is of little use in adults because lactose intolerance is rarely a clinical problem; patients who are upset by milk usually avoid it. Formal testing involves giving an oral dose of 50 g of lactose and serial measurement of blood glucose over 2 hours. (Note: 500 mL of milk contain 20 g of lactose). There is a high incidence of lactase deficiency in many parts of the world (e.g. the Mediterranean countries, and parts of Africa and Asia).
Other tests
Hydrogen breath test. This is frequently used as a screening test to measure transit time and to detect small bowel bacterial overgrowth. Bacteria are present in the oral cavity so the mouth should be rinsed out with an antiseptic mouthwash beforehand. The appearance of a breath hydrogen peak after oral lactulose is used to estimate mouth to caecum transit time. An earlier rise in the breath hydrogen after lactulose indicates bacterial breakdown in the small intestine. This test is simple to perform and it does not involve radioisotopes. However, interpretation is often difficult with a low sensitivity and specificity.
Tests for pancreatic insufficiency are used in the differential diagnosis of steatorrhoea. Human pancreatic elastase 1 (E1) remains undegraded during intestinal transit so its concentration in faeces reflects exocrine pancreatic function. The faecal elastase test quantifies E1 in stool, allowing the diagnosis or exclusion of severe pancreatic exocrine insufficiency (see p. 360).
Other blood tests. Serum immunoglobulins are measured to exclude immune deficiencies in particular IgA deficiency which may lead to false-negative coeliac antibody tests. Gut peptides (e.g. VIP) are measured in high-volume secretory diarrhoea, and chromogranins A and B are raised in endocrine tumours.
Tests for protein-losing enteropathy (PLE) (see p. 234). These tests are rarely required unless a low serum albumin is a major clinical feature.
Measurement of α1 antitrypsin clearance does not require an isotope. α1 antitrypsin is a large molecule (>50 000 daltons), which is resistant to proteolysis. Simultaneous measurements of serum and stool concentration (24-hour collection) are made.
Bile salt loss. This can be demonstrated by giving oral 75Se-homocholyl taurine (SeHCAT – a synthetic taurine conjugate) and measuring the retention of the bile acid by whole-body counting at 7 days.
Stool tests. Faecal calprotectin is 93% sensitive and 96% specific for IBD. Faecal lactoferrin is also an inflammatory marker.
Malabsorption
In many small bowel diseases, malabsorption of specific substances occurs, but these deficiencies do not usually dominate the clinical picture. An example is Crohn’s disease, in which malabsorption of vitamin B12 can be demonstrated, but this is not usually the major problem; diarrhoea and general ill-health are the major features.
The major disorders of the small intestine that cause malabsorption are shown in Table 6.9.
Table 6.9 Disorders of the small intestine causing malabsorption
Coeliac disease (gluten-sensitive enteropathy)
Coeliac disease (CD) is a condition in which there is inflammation of the mucosa of the upper small bowel that improves when gluten is withdrawn from the diet and relapses when gluten is reintroduced. Up to 1% of many populations are affected, though most have clinically silent disease.
Aetiology
Gluten is the entire protein content of the cereals wheat, barley and rye. Prolamins (gliadin from wheat, hordeins from barley, secalins from rye) are damaging factors. These proteins are resistant to digestion by pepsin and chymotrypsin because of their high glutamine and proline content and remain in the intestinal lumen triggering immune responses.
Immunology. Gliadin peptides pass through the epithelium (para- and/or intracellularly) and are deaminated by tissue transglutaminase which increases their immunogenicity. Gliadin peptides then bind to antigen-presenting cells which interact with CD4+ T cells in the lamina propria via HLA class II molecules DQ2 or DQ8. These T cells produce pro-inflammatory cytokines, particularly interferon-γ. CD4+ T cells also interact with B cells to produce endomysial and tissue transglutaminase antibodies. Gliadin peptides also cause release of interleukin-15 from enterocytes, activating intraepithelial lymphocytes with a natural killer cell marker. This inflammatory cascade releases metalloproteinases and other mediators that contribute to the villous atrophy and crypt hyperplasia which are typical of the disease.
The mucosa of the proximal small bowel is predominantly affected, the mucosal damage decreasing in severity towards the ileum as gluten is digested into smaller ‘non-toxic’ fragments.
Genetic factors. There is an increased incidence of coeliac disease within families but the exact mode of inheritance is unknown; 10–15% of 1st-degree relatives will have the condition, although it may be asymptomatic. The concordance rate in identical twins is about 70%.
HLA-DQ2 (DQAI*0501, DQBI*0201) and HLA-DQ8 (DQAI*0301, DQBI*0302) are associated with CD. Over 90% of patients will have HLA-DQ2, compared with 20–30% of the general population. Studies in twins and siblings indicate that HLA genes are responsible for <50% of the genetic cause of the disease. Many unaffected people also carry these genes, so other factors must also be involved. Non-HLA genes may also contribute to coeliac disease, e.g. chromosome regions 19p13.1, 11q, 5q31-33 and 6q21-22. The CD28/CTLA4/ILO5 gene cluster has also shown linkage with coeliac disease.
Environmental factors. Breast-feeding and the age of introduction of gluten into the diet are significant.
Rotavirus infection in infancy also increases the risk, and adenovirus-12 which has sequence homology with α-gliadin has been suspected as a causative agent but this is now thought to be unlikely.
Clinical features
Coeliac disease can present at any age. In infancy it sometimes appears after weaning onto gluten-containing foods. The peak period for diagnosis in adults is in the 5th decade, with a female preponderance. Many patients are asymptomatic (silent) and come to attention because of routine blood tests, e.g. a raised MCV, or iron deficiency in pregnancy. The symptoms are very variable and often nonspecific, e.g. tiredness and malaise often associated with anaemia.
GI symptoms may be absent or mild. Coeliac disease should be tested for in all patients with symptoms suggestive of IBS. Diarrhoea or steatorrhoea, abdominal pain and weight loss suggest more severe disease. Mouth ulcers and angular stomatitis are frequent and can be intermittent. Infertility and neuropsychiatric symptoms of anxiety and depression occur.
Rare complications include tetany, osteomalacia or gross malnutrition with peripheral oedema. Neurological symptoms such as paraesthesia, ataxia (due to cerebellar calcification), muscle weakness or a polyneuropathy occur; the prognosis for these symptoms is variable. There is an increased incidence of atopy and autoimmune disease, including thyroid disease, type 1 diabetes and Sjögren’s syndrome. Other associated diseases include inflammatory bowel disease, primary biliary cirrhosis, chronic liver disease, interstitial lung disease and epilepsy. IgA deficiency is more common than in the general population. Long-term problems include osteoporosis which occurs even in patients on long-term gluten-free diets.
Physical signs are usually few and nonspecific and are related to anaemia and malnutrition.
Diagnosis
Small bowel biopsy is still considered ‘gold standard’ for positive diagnosis, and is therefore desirable in all but the most clear-cut cases, because treatment involves a life-long diet that is both expensive and socially limiting. However with the increasing accuracy of serological tests, it is no longer necessary to take duodenal biopsies for suspected coeliac disease in patients without antibodies. For example in patients being endoscoped for iron deficiency anaemia with negative coeliac serology, the pretest value of small bowel histology is <0.03%.
If biopsies are to be taken, because the disease is sometimes patchy and it can be difficult to orientate endoscopic biopsies for histological section, four to six forceps biopsies should be taken from the second part of the duodenum. Endoscopic signs including absence of mucosal folds, mosaic pattern of the surface and scalloping of mucosal folds are often present; however, their absence is not conclusive because they are markers of relatively severe disease.
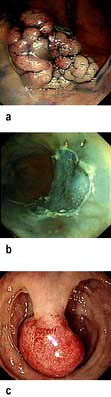
Histology (Fig. 6.28). Histological changes are of variable severity and, though characteristic, are not specific. Villous atrophy can be caused by many other conditions, but coeliac disease is the commonest cause of subtotal villous atrophy.
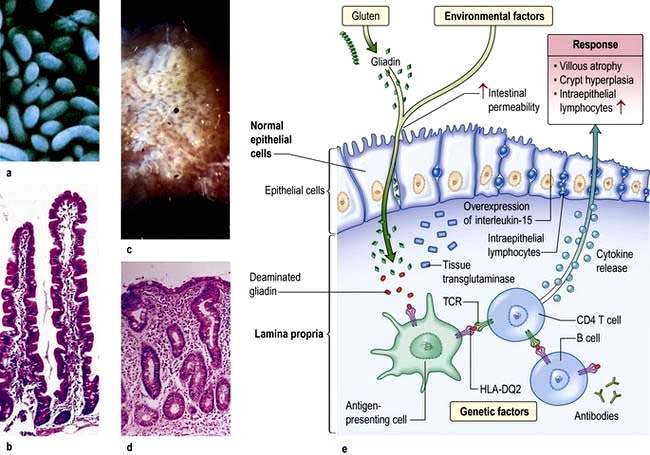
Figure 6.28 Small bowel mucosal appearances – macroscopic and microscopic. (a) Normal mucosa under the dissecting microscope (DM). (b) Normal mucosal histology. (c) Coeliac disease – flattened mucosa. (d) Coeliac disease – showing subtotal villous atrophy. (e) Immune and genetic mechanisms in coeliac disease. TCR, T cell receptor.
(After Green PH, Cellier C. Coeliac disease. New England Journal of Medicine 2007; 357:1731–1743.)
Histological examination shows crypt hyperplasia with chronic inflammatory cells in the lamina propria, and villous atrophy. The enterocytes become cuboidal with an increase in the number of intraepithelial lymphocytes. In the lamina propria there is an increase in lymphocytes and plasma cells. The most severe histological change with mucosal atrophy and hypoplasia is seen in patients who do not respond to a gluten-free diet.
In mild cases, the villous architecture is almost normal but there are increased numbers of intraepithelial lymphocytes.
Serology. Persistent diarrhoea, folate or iron deficiency, a family history of coeliac disease and associated autoimmune disease are indications for serological testing.
The most sensitive tests are for endomysial and anti-tissue transglutaminase antibodies. The sensitivity of these tests is >90% though both are not always positive in the same subject. Titres of either correlate with the severity of mucosal damage so they can be used for dietary monitoring. Standard tests use IgA class antibodies. Selective IgA deficiency occurs in 2.5% of coeliac disease patients but only 0.25% of normals and renders these tests falsely negative. All patients should have concomitant IgA levels tested and if deficient, IgG-based tests should then be used.
HLA typing. HLA-DQ2 is present in 90–95% of CD patients and HLA-DQ8 in about 8%, i.e. most of the rest. The absence of both alleles has a high negative predictive value for coeliac disease. HLA typing may occasionally be useful for risk assessment, e.g. in patients already on a gluten free diet.
Other investigations
Haematology. Mild or moderate anaemia is present in 50% of cases. Folate deficiency is common, often causing macrocytosis. B12 deficiency is rare. Iron deficiency due to malabsorption of iron and increased loss of desquamated cells is common. A blood film may therefore show microcytes and macrocytes as well as hypersegmented polymorphonuclear leucocytes and Howell–Jolly bodies (see p. 406) due to splenic atrophy.
Biochemistry, liver biochemistry and function. In severe cases, biochemical evidence of osteomalacia may be seen (low calcium and high phosphate) and hypoalbuminaemia.
Radiology. A small bowel follow-through may show dilatation of the small bowel with slow transit. Folds become thicker and in severe disease total effacement is seen. Radiology is mainly used when a complication, e.g. lymphoma, is suspected.
Bone densitometry (DXA) should be performed on all patients because of the risk of osteoporosis.
Capsule endoscopy (see p. 233) is used to look for gut abnormalities when a complication is suspected.
Treatment and management
Replacement minerals and vitamins, e.g. iron, folic acid, calcium, vitamin D, may be needed initially to replace body stores.
Treatment is with a gluten-free diet for life. Dietary elimination of wheat, barley and rye usually produces a clinical improvement within days or weeks. Morphological improvement often takes months, especially in adults. Oats are tolerated by most coeliacs, but must not be contaminated with flour during their production. Meat, dairy products, fruits and vegetables are naturally gluten free and are all safe.
Gluten-free products can be expensive, unless subsidized by national health services. Patient support organizations such as The Coeliac Society are valuable as information sources and for advice about diet, recipes and gluten-free processed foods. Despite advice, many patients do not keep to a strict diet but maintain good health. The long-term effects of this low gluten intake are uncertain but osteoporosis can occur even in treated cases.
The usual cause for failure to respond to the diet is poor compliance. Dietary adherence can be monitored by serial tests for endomysial antibody (EMA) and tissue transglutaminase (tTG). If clinical progress is suboptimal then a repeat intestinal biopsy should be taken. If the diagnosis is equivocal on the diagnostic mucosal biopsy, or if the patient has already started on a gluten-free diet, then a gluten challenge, i.e. reintroduction of oral gluten, with evidence of jejunal morphological change, can confirm the diagnosis.
Patients should have pneumococcal vaccinations (because of splenic atrophy) once every 5 years (see p. 406).
Complications
A few patients do not improve on a strict diet and are said to have non-responsive coeliac disease. Many of these patients are still ingesting gluten (see above). A few of the others may have concomitant problems, e.g. microscopic colitis, IBD, small bowel bacterial overgrowth or lactase deficiency.
A very small percentage will have the rare complication of refractory coeliac disease (RCD). In type 1 RCD, the lymphocytes are normal and the T cell receptors are polyclonal, whilst in type 2 there are abnormal clonal lymphocytes with loss of CD8 and CD3 surface markers. The 5-year survival rates are 93% and 40–60% respectively.
Very rarely, enteropathy-associated T cell lymphoma (EATCL) (8–20% 5-year survival), or ulcerative jejunitis can occur as part of a spectrum of neoplastic T cell disorders. Small bowel adenocarcinoma is also increased in coeliac disease.
Ulcerative jejunitis presents with fever, abdominal pain, perforation and bleeding.
Diagnosis for these conditions is with MRI or barium studies but laparoscopy with full-thickness small bowel biopsies is often required. Steroids and immunosuppressive agents, e.g. azathioprine, are used in ulcerative jejunitis
Carcinoma of the oesophagus as well as extragastrointestinal cancers are also increased in incidence. Malignancy seems to be unrelated to the duration of the disease but the incidence is reduced by a gluten-free diet.
Dermatitis herpetiformis
This is an uncommon blistering subepidermal eruption of the skin associated with a gluten-sensitive enteropathy (see also p. 1222). Rarely gross malabsorption occurs, but usually the jejunal morphological abnormalities are not as severe as in coeliac disease. The inheritance and immunological abnormalities are the same as for coeliac disease. The skin condition responds to dapsone but a gluten-free diet improves both the enteropathy and the skin lesion, and is recommended for long-term benefit.
Tropical sprue
This is a condition presenting with chronic diarrhoea and malabsorption that occurs in residents or visitors to affected tropical areas. The disease is endemic in most of Asia, some Caribbean islands, Puerto Rico and parts of South America. Epidemics occur, lasting up to 2 years, and in some areas repeated epidemics occur at varying intervals of up to 10 years.
The term tropical sprue is reserved for severe malabsorption (of two or more substances) accompanied by diarrhoea and malnutrition. Malabsorption of a mild degree, sometimes following an enteric infection, is quite common in the tropics; it is usually asymptomatic and is sometimes called tropical malabsorption.
Aetiology
The aetiology is unknown, but is likely to be infective because the disease occurs in epidemics and patients improve on antibiotics. A number of agents have been suggested but none has been unequivocally shown to be responsible. Different agents could be involved in different parts of the world.
Clinical features
These vary in intensity and consist of diarrhoea, anorexia, abdominal distension and weight loss. The onset is sometimes acute and occurs either a few days or many years after being in the tropics. Epidemics can break out in villages, affecting thousands of people at the same time. The onset can also be insidious, with chronic diarrhoea and evidence of nutritional deficiency. The clinical features of tropical sprue vary in different parts of the world, particularly as different criteria are used for diagnosis.
Diagnosis
Acute infective causes of diarrhoea must be excluded (see p. 293), particularly Giardia, which can produce a syndrome very similar to tropical sprue. Malabsorption should be demonstrated, particularly of fat and B12. The jejunal mucosa is abnormal, showing some villous atrophy (partial villous atrophy). In most cases, the lesion is less severe than that found in coeliac disease, although it affects the whole small bowel. Mild mucosal changes can be seen in asymptomatic individuals in the tropics.
Treatment and prognosis
Many patients improve when they leave the sprue area and take folic acid (5 mg daily). Most patients also require an antibiotic to ensure a complete recovery (usually tetracycline 1 g daily for up to 6 months).
Severely ill patients require resuscitation with fluids and electrolytes for dehydration, and nutritional deficiencies should be corrected. Vitamin B12 (1000 µg) is also given to all acute cases.
The prognosis is excellent. Mortality is usually associated with water and electrolyte depletion, particularly in epidemics.
Bacterial overgrowth
The gut contains many resident bacteria in the terminal ileum and colon. Anaerobic bacteria, e.g. Bacteroides, bifidobacteria, are 100–1000 times more abundant than aerobic (facultative anaerobes), e.g. Escherichia, Enterobacter, Enterococcus. This gut microflora has major functions including metabolic, e.g. fermentation of non-digestible dietary residues into short-chain fatty acids as an energy source in the colon.
The microflora which influences epithelial cell proliferation, is involved in the development and maintenance of the immune system and protects the gut mucosa from colonization by pathogenic bacteria. Bacteria also initiate vitamin K production.
The upper part of the small intestine is almost sterile, containing only a few organisms derived from the mouth. Gastric acid kills some ingested organisms and intestinal motility keeps bacterial counts in the jejunum low. The normal terminal ileum contains faecal-type organisms, mainly Escherichia coli and anaerobes, and the colon has abundant bacteria.
Bacterial overgrowth is normally found associated with a structural abnormality of the small intestine such as a stricture or diverticulum, although it can occur occasionally in the elderly without. E. coli and/or Bacteroides, both in concentrations greater than 106/mL, are found as part of a mixed flora. These bacteria are capable of deconjugating and dehydroxylating bile salts, so that unconjugated and dehydroxylated bile salts can be detected in small bowel aspirates.
Clinical features
The clinical features of overgrowth are chiefly diarrhoea and steatorrhoea. There may also be symptoms due to the underlying small bowel pathology. Steatorrhoea (see p. 263) occurs because of conjugated bile salt deficiency. Some bacteria can metabolize vitamin B12 and interfere with its binding to intrinsic factor, leading to mild B12 deficiency (see Chapter 8) rarely severe enough to produce a neurological deficit. Some bacteria produce folic acid giving a high serum folate. Bacterial overgrowth has only minimal effects on the absorption of other substances. Confirmation of bacterial overgrowth is with the hydrogen breath test (see p. 264).
Intestinal resection
Small intestinal resection is usually well tolerated, but massive resection leaving <1 m of small bowel in continuity is followed by the short-bowel syndrome. The effects of resection depend on the amount and location of the resection and the presence or absence of the colon. Resection of the jejunum is better tolerated than ileal resection, where there is less adaptation, probably due to low levels of glucagon-like peptide 2 (GLP-2), which is a specific growth hormone for the enterocyte.
Ileal resection
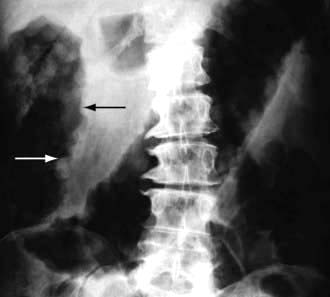
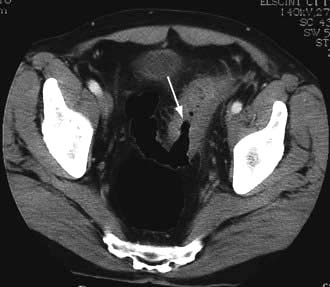
The ileum is the site of specific mechanisms for the absorption of bile salts and vitamin B12. Relatively small resections lead to malabsorption of these substances. Removal of the ileocaecal valve increases the incidence of diarrhoea (Fig. 6.29).
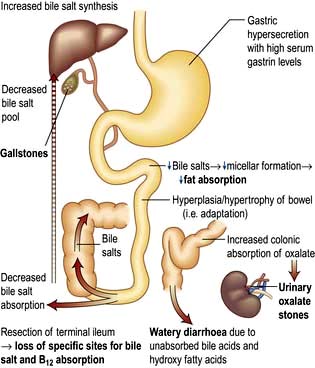
The following occur after ileal resection:
Bile-salt induced diarrhoea: bile salts and fatty acids enter the colon and cause malabsorption of water and electrolytes (see p. 293).
Steatorrhoea and gallstone formation: increased bile salt synthesis can compensate for loss of approximately one-third of the bile salts in the faeces. Greater loss than this results in decreased micelle formation and steatorrhoea, and lithogenic bile and gallstone formation.
Oxaluria and oxalate stones: bile salts in the colon cause increased oxalate absorption with oxaluria leading to urinary stone formation.
B12 deficiency: low serum B12, macrocytosis and other effects of B12 deficiency.
Investigations include a small bowel follow-through, measurement of B12, bile salt retention (SeHCAT) test (see p. 236). A hydrogen breath test may show rapid transit (see p. 264). Many patients require B12 replacement and some need a low-fat diet if there is steatorrhoea. Diarrhoea is often improved by cholestyramine which binds bile salts and reduces the level of diarrhoeogenic bile salts in the colon.
Jejunal resection
The ileum can compensate for loss of jejunal absorptive function. Jejunal resection may lead to gastric hypersecretion with high gastrin levels; the exact mechanism of this is unclear. Structural and functional intestinal adaptation takes place over the course of a year, with an increase in the absorption per unit length of bowel.
Massive intestinal resection (short-bowel syndrome)
Intestinal failure results from obstruction, dysmotility, surgical resection, congenital defect, or disease-associated loss of absorption and is characterized by the inability to maintain protein-energy, fluid, electrolyte, or micronutrient balance. This most often occurs following resection for Crohn’s disease, mesenteric vessel occlusion (see p. 270), radiation enteritis (see p. 268) or trauma. There are two common situations:
Shortened small intestine ending at a terminal small bowel stoma
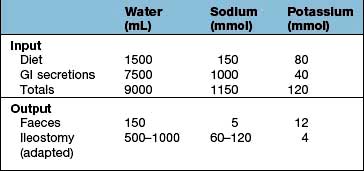
The major problem is of sodium and fluid depletion, and the majority of patients with ≤100 cm of jejunum remaining will require parenteral supplements of fluid and electrolytes, often with nutrients. Sodium losses can be minimized by increasing salt intake, restricting hypotonic fluids between meals and administering oral glucose-electrolyte mixture with a sodium concentration 90 mmol/L. Jejunal transit time can be increased and stomal effluent loss reduced by treatment with the somatostatin analogue octreotide, often used in combination with a proton pump inhibitor, loperamide and codeine phosphate. There is no benefit from a low-fat diet, but fat assimilation can be increased on treatment with cholestyramine and synthetic bile acids.
Shortened small intestine in continuity with colon
Because of the absorptive capacity of the colon for fluid and electrolytes, only a small proportion of these patients require parenteral supplementation. Unabsorbed fat results in impairment of colonic fluid and electrolyte absorption so patients should be on a low-fat diet. A high carbohydrate intake is advised as unabsorbed carbohydrate is metabolized anaerobically to short-chain fatty acids (SCFAs) which are absorbed; they also stimulate fluid and electrolyte absorption in the colon and act as an energy source (1.6 kcal/g). Patients are often treated with cholestyramine to reduce diarrhoea and colonic oxalate absorption.
Whipple’s disease
Whipple’s disease is a rare infectious bacterial disease caused by Tropheryma whipplei. About 1000 cases have been described; 87% are males, usually white and middle-aged. It presents with arthritis and arthralgia, progressing over years to weight loss and diarrhoea with abdominal pain, systemic symptoms of fever and weight loss. Peripheral lymphadenopathy and involvement of the heart, lung, joints and brain occur, simulating many neurological conditions.
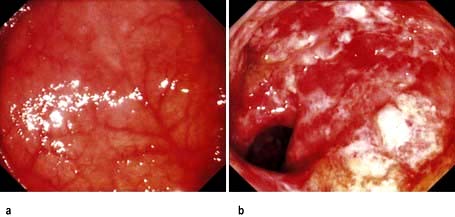
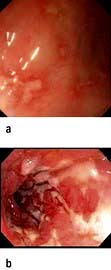
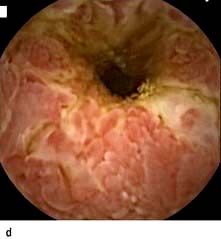
Blood tests show features of chronic inflammation and malabsorption. Endoscopy typically shows pale, shaggy duodenal mucosa with eroded, red, friable patches.
Diagnosis is made by small bowel biopsy. Periodic acid–Schiff (PAS)-positive macrophages are present but are nonspecific. On electron microscopy, the characteristic trilaminar cell wall of T. whipplei can be seen within macrophages. T. whipplei antibodies can be identified by immunohistochemistry. A confirmatory PCR-based assay is available.
Treatment is with antibiotics which cross the blood-brain barrier, such as 160 mg trimethoprim and 800 mg sulphamethoxazole (co-trimoxazole) daily for 1 year. This is preceded by a 2-week course of streptomycin and penicillin or ceftriaxone. Treatment periods of less than a year are associated with relapse in about 40%.
Radiation enteritis
Radiation of >40 Gy will damage the intestine. The chronic effects of radiation are muscle fibre atrophy, ulcerative changes due to ischaemia and obstruction due to radiation-induced fibrotic strictures.
Pelvic irradiation is frequently used for gynaecological and urinary tract malignancies, so the ileum and rectum are the areas most often involved.
At the time of the irradiation, there may be nausea, vomiting, diarrhoea and abdominal pain, usually improving within 6 weeks of completion of therapy.
Chronic radiation enteritis is diagnosed if symptoms persist for ≥3 months. The prevalence is >15%. Abdominal pain due to obstruction is the main symptom. Malabsorption can be due to bacterial overgrowth in dilated segments and mucosal damage.
Many patients suffer from increased bowel frequency.
Treatment is symptomatic, although often unsuccessful in chronic radiation enteritis. Surgery should be avoided if possible, being reserved for obstruction or perforation.
Acute radiation damage to the rectum produces a radiation proctitis with diarrhoea and tenesmus, with or without blood. Local steroids sometimes help initially. When the acute phase heals, mucosal telangiectases form and may cause persistent bleeding. They can be treated with argon plasma coagulation or, under a light anaesthetic, by packing the rectum with a formalin-soaked swab for 2 min, both of which destroy the telangiectases.
Parasite infestation
Giardia intestinalis (see p. 151) not only produces diarrhoea but can produce malabsorption with steatorrhoea. Minor changes are seen in the jejunal mucosa and the organism can be found in the jejunal fluid or mucosa.
Cryptosporidiosis (see p. 151) can also produce malabsorption.
Patients with HIV infection are particularly prone to parasitic infestation (see Table 6.23).
Other causes of malabsorption
Drugs that bind bile salts (e.g. cholestyramine) and some antibiotics (e.g. neomycin) produce steatorrhoea.
Orlistat (see p. 220) is used in obesity to reduce fat absorption by inhibiting gastric and pancreatic lipase causing diarrhoea and steatorrhoea.
Thyrotoxicosis: diarrhoea, rarely with steatorrhoea, occurs in thyrotoxicosis owing to increased gastric emptying and increased motility.
Zollinger–Ellison syndrome (see p. 370).
Intestinal lymphangiectasia produces diarrhoea and rarely steatorrhoea (see p. 270).
Lymphoma that has infiltrated the small bowel mucosa causes malabsorption.
Diabetes mellitus: diarrhoea, malabsorption and steatorrhoea occur, sometimes due to bacterial overgrowth from autonomic neuropathy causing small bowel stasis.
Hypogammaglobulinaemia, which is seen in a number of conditions including lymphoid nodular hyperplasia, causes steatorrhoea due either to an abnormal jejunal mucosa or to secondary infestation with Giardia intestinalis.
Miscellaneous intestinal diseases
Protein-losing enteropathy
Protein-losing enteropathy refers to intestinal conditions causing protein loss, usually manifest by hypoalbuminaemia. The causes include Crohn’s disease, tumours, Ménétrier’s disease, coeliac disease and lymphatic disorders (e.g. lymphangiectasia).
Usually protein-losing enteropathy forms a minor part of the generalized disorder, but occasionally hepatic synthesis of albumin cannot compensate for the protein loss, and peripheral oedema dominates the clinical picture. The investigations are described on page 264 and treatment is that of the underlying disorder.
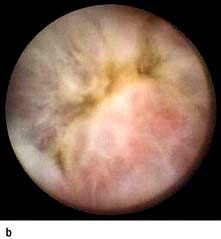
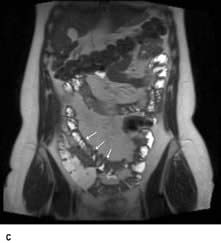
Meckel’s diverticulum
This is the most common congenital abnormality of the GI tract, affecting 2–3% of the population. The diverticulum projects from the wall of the ileum approximately 60 cm from the ileocaecal valve. It is usually symptomless, but 50% contain gastric mucosa that secretes hydrochloric acid. Peptic ulcers can occur and may bleed (see p. 254) or perforate.
Acute inflammation of the diverticulum also occurs and is indistinguishable clinically from acute appendicitis. Obstruction from an associated band rarely occurs.
Tuberculosis
Tuberculosis (TB) (see also p. 839) can affect the intestine as well as the peritoneum (see p. 302). In developed countries, most patients are from ethnic minority groups, or are immunocompromised due to HIV or drugs. Intestinal tuberculosis is due to reactivation of primary disease caused by Mycobacterium tuberculosis. Bovine TB occurs in areas where milk is unpasteurized and is rare in western countries.
Clinical features are abdominal pain, weight loss, anaemia, fever with night sweats, obstruction, right iliac fossa pain or a palpable mass. The ileocaecal area is most commonly affected, but the colon, and rarely other parts of the gastrointestinal tract, can be involved. One-third of patients present acutely with intestinal obstruction or generalized peritonitis; 50% have X-ray evidence of pulmonary tuberculosis.
Diagnosis
Differential diagnosis includes Crohn’s disease and caecal carcinoma.
A small bowel follow-through may show transverse ulceration, diffuse narrowing of the bowel with shortening of the caecal pole.
Ultrasound or CT shows additional mesenteric thickening and lymph node enlargement.
Histology and culture of tissue is desirable, but it is not always possible. Specimens can be obtained by colonoscopy or laparoscopy but laparotomy is required in some cases.
Treatment
Drug treatment is similar to that for pulmonary TB (see page 842). Treatment should be started if there is a high degree of suspicion.
Amyloid
Systemic amyloidosis may affect any part of the GI tract (see also p. 1042). Rectal biopsy may be diagnostic. Occasionally, amyloid deposits occur as polypoid lesions. The symptoms depend on the site of involvement; amyloidosis in the small intestine gives rise to diarrhoea.
Rheumatic autoimmune disorders
Systemic sclerosis (see p. 538) most commonly affects the oesophagus (see p. 243), although the small bowel and colon are often found to be involved if investigated. There may be no symptoms of this involvement, but diarrhoea and steatorrhoea can occur due to bacterial overgrowth as a result of reduced motility, dilatation and the presence of diverticula.
Intestinal ischaemia
Intestinal ischaemia results from occlusion of arterial inflow, occlusion of venous outflow or failure of perfusion; these factors may act alone or in combination and usually occur in the elderly.
Arterial inflow occlusion can be caused by atheroma, thrombosis, and embolism (cardiac arrhythmia), including cholesterol emboli (see p. 599), aortic disease (occluding ostia of mesenteric vessels) or vasculitis (see p. 542), thromboangiitis and Takayasu’s syndrome (see p. 745)
Venous outflow occlusion occurs in 5–15% of cases and usually in sick patients with circulatory failure.
Infarction without occlusion can occur due to reduced cardiac output, hypotension and shock causing reduced intestinal blood flow.
Acute small intestinal ischaemia
An embolus from the heart in a patient with atrial fibrillation is the commonest cause, usually occluding the superior mesenteric artery. Patients present with sudden abdominal pain and vomiting with a distended and tender abdomen, and absent bowel sounds. The patient is hypotensive and ill. Surgery is necessary to resect the gangrenous bowel. Mortality is high (up to 90%) and is related to co-existing disease, the development of multiorgan failure (MOF) (see p. 882) and massive fluid and electrolyte losses in the postoperative period. Survivors may go on to develop nutritionally inadequate short-bowel syndrome (see p. 268).
Chronic small intestinal ischaemia
This is due to atheromatous occlusion or cholesterol emboli of the mesenteric vessels, particularly in the elderly. Good collateral circulation can minimize clinical effects. The characteristic symptom is post-prandial abdominal pain and weight loss. Loud bruits may be heard but, as these are heard in normal subjects, they are of doubtful significance. The diagnosis is made using angiography.
Eosinophilic gastroenteritis
In this condition of unknown aetiology there is eosinophilic infiltration and oedema of any part of the gastrointestinal mucosa. The gastric antrum and proximal small intestine are usually involved either as a localized lesion (eosinophilic granuloma) or diffusely with sheets of eosinophils seen in the serosal and submucosal layers. There is an association with asthma, eczema and urticaria.
The condition occurs mainly in the third decade. The clinical presentation depends on the site of gut involvement. Abdominal pain, nausea and vomiting and upper GI bleeding occur. Peripheral eosinophilia occurs in only 20% of patients. Endoscopic biopsy is useful for making the diagnosis histologically. Radiology may demonstrate mass lesions.
Treatment is with corticosteroids for the widespread infiltration, particularly if peripheral eosinophilia is present.
In some adults, the condition appears to be allergic (allergic gastroenteritis) and is associated with peripheral eosinophilia and high levels of plasma and tissue IgE. Eosinophilic oesophagitis’s relationship to eosinophilic gastroenteritis is unclear.
Intestinal lymphangiectasia
Dilatation of the lymphatics may be primary or secondary to lymphatic obstruction, such as occurs in malignancy or constrictive pericarditis. Hypoproteinaemia with ankle oedema is the main feature. The rare primary form may be detected incidentally as dilated lacteals on a jejunal biopsy or it can produce steatorrhoea of varying degrees. White-tipped villi are seen on capsule endoscopy. Serum immunoglobulin levels are reduced, with low circulating lymphocytes. Treatment is with a low-fat diet, mid-chain triglycerides and fat-soluble vitamin supplements as required. Octreotide has a dramatic effect in a few primary cases, although the mechanism of action is unknown.
Abetalipoproteinaemia
This rare congenital disorder is due to a failure of apo B-100 synthesis in the liver and apo B-48 in the intestinal cell, so that chylomicrons are not formed. This leads to fat accumulation in the intestinal cells, giving a characteristic histological appearance to the jejunal mucosa. Clinical features include acanthocytosis (spiky red cells owing to membrane abnormalities), a form of retinitis pigmentosa, and mental and neurological abnormalities. The latter can be prevented by vitamin E injections.
Tumours of the small intestine
The small intestine is relatively resistant to the development of neoplasia and only 3–6% of all GI tumours and fewer than 1% of all malignant lesions occur here. The reason for the rarity of tumours is unknown. Explanations include the fluidity and relative sterility of small bowel contents and the rapid transit time, reducing the time of exposure to potential carcinogens. It is also possible that the high population of lymphoid tissue and secretion of IgA in the small intestine protect against malignancy.
Adenocarcinoma of the small intestine is rare and found most frequently in the duodenum (in the periampullary region) and in the jejunum. It is the most common tumour of the small intestine, accounting for up to 50% of primary tumours.
Lymphomas are most frequently found in the ileum. These are of the non-Hodgkin’s type and must be distinguished from peripheral or nodal lymphomas involving the gut secondarily.
In developed countries, the most common type of lymphoma is the B cell type arising from MALT (see p. 468). These lymphomas tend to be annular or polypoid masses in the distal or terminal ileum, whereas most T cell lymphomas are ulcerated plaques or strictures in the proximal small bowel.
A tumour similar to Burkitt’s lymphoma also occurs and commonly affects the terminal ileum of children in North Africa and the Middle East.
Predisposing factors for adenocarcinoma and lymphoma
Coeliac disease
There is an increased incidence of lymphoma of the T cell type and adenocarcinoma of the small bowel, as well as an unexplained increase in all malignancies both in the GI tract and elsewhere. The reason for the local development of malignancy is unknown. It is now accepted that coeliac disease is a premalignant condition, but there is no association with the length of the symptoms. Treatment with a gluten-free diet reduces the risk of both lymphoma and carcinoma.
Crohn’s disease
There is a small increase in the incidence of adenocarcinoma of the small bowel in Crohn’s disease.
Immunoproliferative small intestinal disease (IPSID)
IPSID is a rare B cell disorder in which there is proliferation of plasma cells in the lamina propria of the upper small bowel producing truncated monoclonal heavy chains, without associated light chains. The α heavy chains are found in the gut mucosa on immunofluorescence and can also be detected in the serum. It occurs usually in countries surrounding the Mediterranean, but it has also been found in other developing countries in South America and the Far East. IPSID predominantly affects people in lower socioeconomic groups in areas with poor hygiene and a high incidence of bacterial and parasitic infection of the gut. IPSID presents as a malabsorptive syndrome associated with diffuse lymphoid infiltration of the small bowel and neighbouring lymph nodes, progressing in some cases to a lymphoma. The condition has also been documented in the developed world.
Clinically, patients present with abdominal pain, diarrhoea, anorexia, weight loss and symptoms of anaemia. There may be a palpable mass, and a small bowel follow-through may detect a mass lesion. Endoscopic biopsy is useful where lesions are within reach. Ultrasound and CT may show bowel wall thickening and the involvement of lymph nodes, which is common with lymphoma. Wireless capsule endoscopy can be used where obstruction by the capsule is not likely, but cannot deliver histology.
Treatment of small intestinal tumours
Adenocarcinoma. Most patients are treated surgically with a segmental resection. The overall 5-year survival rate is 20–35%; this varies with the histological grade and the presence or absence of lymph node involvement. Radiotherapy and chemotherapy are used in addition.
IPSID. If there is no evidence of lymphoma, antibiotics, e.g. tetracycline, should be tried initially. In the presence of lymphoma, combination chemotherapy is used; in one series the 3–5-year survival rate was 58%.
Lymphoma. Most patients require surgery and radiotherapy with chemotherapy for more extensive disease. The prognosis varies with the type. The 5-year survival rate for T cell lymphomas is 25%, but is better for B cell lymphomas, varying from 50% to 75%, depending on the grade of lymphoma.
Carcinoid tumours
These originate from the enterochromaffin cells (APUD cells) of the intestine. They make up 10% of all small bowel neoplasms, the most common sites being in the appendix and terminal ileum. It is often difficult to be certain histologically whether a particular tumour is benign or malignant. A total of 10% of carcinoid tumours in the appendix present as acute appendicitis, secondary to obstruction. Surgical resection of the tumour is usually performed.
Most carcinoids do not secrete hormones or vasoactive compounds, and may present with liver enlargement due to metastases.
Carcinoid syndrome occurs in only 5% of patients with carcinoid tumours and only when there are liver metastases. Patients complain of spontaneous or induced bluish-red flushing, predominantly on the face and neck, sometimes leading to permanent changes with telangiectases.
Gastrointestinal symptoms consist of abdominal pain and recurrent watery diarrhoea. Cardiac abnormalities are found in 50% of patients and consist of pulmonary stenosis or tricuspid incompetence. Examination of the abdomen reveals hepatomegaly. The tumours secrete a variety of biologically active amines and peptides, including serotonin (5-hydroxy-tryptamine – 5-HT), bradykinin, histamine, tachykinins and prostaglandins. The diarrhoea and cardiac complications are probably caused by 5-HT itself, but the cutaneous flushing is thought to be produced by one of the kinins, such as bradykinin. This is known to cause vasodilatation, bronchospasm and increased intestinal motility.
Diagnosis and treatment of carcinoid syndrome
Ultrasound examination confirms the presence of liver secondary deposits.
Urine shows a high concentration of 5-hydroxyindoleacetic acid (5-HIAA) which is the major metabolite of 5-HT.
Serum chromogranin A is raised in nearly all hindgut tumours and 80–90% of patients with symptomatic foregut and midgut tumours.
Treatment is with octreotide and lanreotide; both are octapeptide somatostatin analogues that inhibit the release of many gut hormones. They alleviate the flushing and diarrhoea and can control a carcinoid crisis. Octreotide is given subcutaneously in doses up to 200 µg three times daily initially; a depot preparation 30 mg every 4 weeks can then be used. Lanreotide 30 mg is given every 7–10 days or as a gel 60 mg every 28 days. Long-acting octreotide also sometimes inhibits tumour growth. Interferon and other chemotherapeutic regimens also occasionally reduce tumour growth, but have not been shown to increase survival.
Peutz–Jeghers syndrome
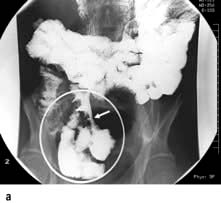
This consists of mucocutaneous pigmentation (circumoral; 95% of patients), hands (70%) and feet (60%) and gastrointestinal polyps. It has an autosomal dominant inheritance. The gene STK11 (also known as LKB1) responsible for Peutz–Jeghers codes for a serine protein kinase and can be used for genetic analysis. The brown buccal pigment is characteristic of the condition. The polyps, which are hamartomas, can occur anywhere in the GI tract but are most frequent in the small bowel. They may bleed or cause small bowel obstruction or intussusception (50% of patients).
Treatment is by endoscopic polypectomy. Balloon enteroscopy may be necessary to reach all the small bowel polyps. Bowel resection should be avoided if possible, but may be necessary in patients presenting with gangrenous bowel due to intussusception. Follow-up is with yearly pan-endoscopy. There is an increased incidence of GI cancers. Non-GI cancers also occur with increased frequency, so yearly screening for uterine, ovarian and cervical cancer should start in the teens, and breast and testicular screening by the age of 20.
Other tumours
Adenomas, lipomas and stromal tumours (see p. 253) are rarely found and are usually asymptomatic and picked up incidentally. They occasionally present with iron deficiency anaemia. In familial adenomatous polyposis (FAP) duodenal adenomas form in one-third of patients and may progress to adenocarcinoma. This is the commonest cause of death in FAP patients who have been treated by prophylactic colectomy.
 Box 6.6
Box 6.6