Medical Nutrition Therapy for Hepatobiliary and Pancreatic Disorders
The liver is of primary importance; one cannot survive without a liver. The pancreas and liver are essential to digestion and metabolism. Although it is important, the gallbladder can be removed, and the body will adapt comfortably to its absence. Knowledge of the structure and functions of these organs is vital. When they are diseased, the necessary medical nutrition therapy (MNT) is complex.
Physiology and Functions of the Liver
The liver is the largest gland in the body, weighing approximately 1500 g. The liver has two main lobes: the right and left. The right lobe is further divided into the anterior and posterior segments; the right segmental fissure, which cannot be seen externally, separates the segments. The externally visible falciform ligament divides the left lobe into the medial and lateral segments. The liver is supplied with blood from two sources: the hepatic artery, which supplies approximately one third of the blood from the aorta; and the portal vein, which supplies the other two thirds and collects blood drained from the digestive tract.
Approximately 1500 mL of blood per minute circulates through the liver and exits via the right and left hepatic veins into the inferior vena cava. Just as there is a system of blood vessels throughout the liver, there also exists a series of bile ducts. Bile, which is formed in the liver cells, exits the liver through a series of bile ducts that increase in size as they approach the common bile duct. It is a thick, viscous fluid secreted from the liver, stored in the gallbladder, and released into the duodenum when fatty foods enter the duodenum. It emulsifies fats in the intestine and forms compounds with fatty acids to facilitate their absorption.
Functions
The liver has the ability to regenerate itself. Only 10% to 20% of functioning liver is required to sustain life, although removal of the liver results in death, usually within 24 hours. The liver is integral to most metabolic functions of the body and performs more than 500 tasks. The main functions of the liver include metabolism of carbohydrate, protein, and fat; storage and activation of vitamins and minerals; formation and excretion of bile; conversion of ammonia to urea; metabolism of steroids; and action as a filter and flood chamber.
The liver plays a major role in carbohydrate metabolism. Galactose and fructose, products of carbohydrate digestion, are converted into glucose in the hepatocyte or liver cell. The liver stores glucose as glycogen (glycogenesis) and then returns it to the blood when glucose levels become low (glycogenolysis). The liver also produces “new” glucose (gluconeogenesis) from precursors such as lactic acid, glycogenic amino acids, and intermediates of the tricarboxylic acid cycle (see Chapter 3).
Important protein metabolic pathways occur in the liver. Transamination and oxidative deamination are two such pathways that convert amino acids to substrates that are used in energy and glucose production as well as in the synthesis of nonessential amino acids. Blood-clotting factors such as fibrinogen; prothrombin; and serum proteins, including albumin, α-globulin, β-globulin, transferrin, ceruloplasmin, and lipoproteins are formed by the liver.
Fatty acids from the diet and adipose tissue are converted in the liver to acetyl-coenzyme A by the process of β-oxidation to produce energy. Ketones are also produced. The liver synthesizes and hydrolyzes triglycerides, phospholipids, cholesterol, and lipoproteins as well.
The liver is involved in the storage, activation, and transport of many vitamins and minerals. It stores all the fat-soluble vitamins in addition to vitamin B12 and the minerals zinc, iron, copper, and magnesium. Hepatically synthesized proteins transport vitamin A, iron, zinc, and copper in the bloodstream. Carotene is converted to vitamin A, folate to 5-methyl tetrahydrofolic acid, and vitamin D to an active form (25-hydroxycholecalciferol) by the liver.
In addition to functions of nutrient metabolism and storage, the liver forms and excretes bile. Bile salts are metabolized and used for the digestion and absorption of fats and fat-soluble vitamins. Bilirubin is a metabolic end product from red blood cell destruction; it is conjugated and excreted in the bile.
Hepatocytes detoxify ammonia by converting it to urea, 75% of which is excreted by the kidneys. The remaining urea finds its way back to the gastrointestinal tract (GIT). The liver also metabolizes steroids. It inactivates and excretes aldosterone, glucocorticoids, estrogen, progesterone, and testosterone. It is responsible for the detoxification of substances, including drugs and alcohol. Finally, the liver acts as a filter and flood chamber by removing bacteria and debris from blood through the phagocytic action of Kupffer cells located in the sinusoids and by storing blood backed up from the vena cava as in right heart failure.
Laboratory Assessment of Liver Function
Biochemical markers are used to evaluate and monitor patients having or suspected of having liver disease. Enzyme assays measure the release of liver enzymes, and other tests measure liver function. Screening tests for hepatobiliary disease include serum levels of bilirubin, alkaline phosphatase, aspartate amino transferase, and alanine aminotransferase. Table 30-1 elaborates common laboratory tests for liver disorders (see also Appendix 30).
TABLE 30-1
Common Laboratory Tests Used to Test Liver Function



Anti-HBe, Antibody to HBeAg; HbeAg, hepatitis B e-antigen; Anti-HBs, antibody to HBsAg; HAV, hepatic A virus; HBc, hepatitis B core; HBsAg, hepatitis B surface antigen; HCV, hepatitis C virus; HDV, hepatitis D virus; HEV, hepatitis E virus; IgG, immunoglobin G; IgM, immunoglobulin M; PBC, primary biliary cirrhosis; RNA, ribonucleic acid; SGOT, serum glutamic oxaloacetic transaminase; SGPT, serum glutamic pyruvic transaminase.
Data from Baker AL: Liver chemistry tests. In Kaplowitz N, editor: Liver and biliary diseases, ed 2, Baltimore, 1996, Williams & Wilkins; Hoofnagle JH, Lindsay KL: Acute viral hepatitis. In Goldman L, Bennett JC, editors: Cecil textbook of medicine, ed 21, Philadelphia, 2000, Saunders; Kamath PS: Clinical approach to the patient with abnormal liver test results, Mayo Clin Proc 71:1089, 1996; Lindsay KL, Hoofnagle JH: Serologic tests for viral hepatitis. In Kaplowitz N, editor: Liver and biliary diseases, ed 2, Baltimore, 1996, Williams & Wilkins; Weisiger RA: Laboratory tests in liver disease. In Goldman L, Bennett JC, editors: Cecil textbook of medicine, ed 21, Philadelphia, 2000, Saunders.
Diseases of the Liver
Diseases of the liver can be acute or chronic, inherited or acquired. Liver disease is classified in various ways: acute viral hepatitis, fulminant hepatitis, chronic hepatitis, nonalcoholic steatohepatitis (NASH), alcoholic hepatitis and cirrhosis, cholestatic liver diseases, inherited disorders, and other liver diseases.
Acute Viral Hepatitis
Acute viral hepatitis is a widespread inflammation of the liver and is caused by hepatitis viruses A, B, C, D, and E (Figure 30-1, Table 30-2). Hepatitis A and E are the infectious forms, mainly spread by fecal-oral route. Hepatitis B, C, and D are the serum forms that are spread by blood and body fluids (Hoofnagle, 2007). Minor agents such as Epstein-Barr virus, cytomegalovirus, herpes simplex, yellow fever, and rubella can also cause an acute hepatitis.
TABLE 30-2
| Virus | Transmission | Comments |
| Hepatitis A | Fecal-oral route; is contracted through contaminated drinking water, food, and sewage. | Anorexia is the most frequent symptom, and it can be severe. Other common symptoms include nausea, vomiting, right upper quadrant abdominal pain, dark urine, and jaundice (icterus). Recovery is usually complete, and long-term consequences are rare. Serious complications may occur in high-risk patients; subsequently, great attention must be given to adequate nutritional intake. |
| Hepatitis B & C | HBV and HCV are transmitted via blood, blood products, semen, and saliva. For example, they can be spread from contaminated needles, blood transfusions, open cuts or wounds, splashes of blood into the mouth or eyes, or sexual contact. | HBV and HCV can lead to chronic and carrier states. Chronic active hepatitis can also develop, leading to cirrhosis and liver failure. |
| Hepatitis D | HDV is rare in the United States and depends on the HBV for survival and propagation in humans. | HDV may be a coinfection (occurring at the same time as HBV) or a superinfection (superimposing itself on the HBV carrier state). This form of hepatitis usually becomes chronic. |
| Hepatitis E | HEV is transmitted via the oral-fecal route. | HEV is rare in the United States (typically only occurs when imported), but it is reported more frequently in many countries of southern, eastern, and central Asia; northern, eastern, and western Africa; and Mexico. Contaminated water appears to be the source of infection, which usually afflicts people living in crowded and unsanitary conditions. HEV is generally acute rather than chronic. |
| Hepatitis G/GB | HGV and a virus labeled GBV-C appear to be variants of the same virus. | Although HGV infection is present in a significant proportion of blood donors and is transmitted through blood transfusions, it does not appear to cause liver disease. |
HBV, Hepatitis B virus; HCV, hepatitis C virus; HDV, hepatitis D virus; HEV, hepatitis E virus; HGV, hepatitis G virus.
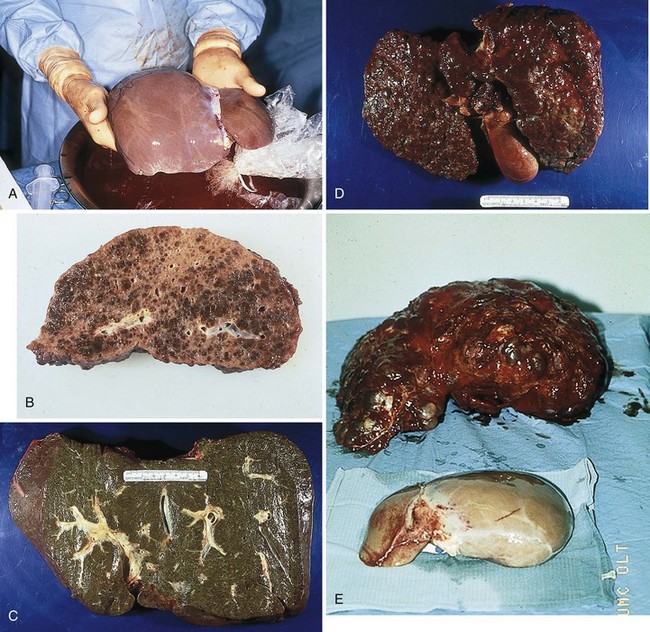
FIGURE 30-1 A, Normal liver. B, Liver with damage from chronic active hepatitis. C, Liver with damage from sclerosing cholangitis. D, Liver with damage from primary biliary cirrhosis. E, Liver with damage from polycystic liver disease (background) and normal liver (foreground). (Courtesy Baylor Transplant Institute, Baylor University Medical Center, Dallas, TX.)
The general symptoms of acute viral hepatitis are divided into four phases. The first phase, the early prodromal phase, affects approximately 25% of patients, causing fever, arthralgia, arthritis, rash, and angioedema. This is followed by the preicteric phase, in which malaise, fatigue, myalgia, anorexia, nausea, and vomiting occur. Some patients complain of epigastric or right upper quadrant pain. The third phase is the icteric phase, in which jaundice appears. Finally, during the convalescent phase, jaundice and other symptoms begin to subside.
Complete recovery is expected in more than 95% of hepatitis A cases, in 90% of acute hepatitis B cases, but in only 15% to 50% of acute hepatitis C cases. Chronic hepatitis does not usually develop with hepatitis E, and symptoms and liver function tests usually normalize within 6 weeks (Hoofnagle, 2007).
Fulminant Hepatitis
Fulminant hepatitis is a syndrome in which severe liver dysfunction is accompanied by hepatic encephalopathy, a clinical syndrome characterized by impaired mentation, neuromuscular disturbances, and altered consciousness. Fulminant liver disease is defined by the absence of preexisting liver disease and the development of hepatic encephalopathy within 2 to 8 weeks of the onset of illness. The causes of fulminant hepatitis include viral hepatitis in approximately 75% of cases, chemical toxicity (e.g., acetaminophen, drug reactions, poisonous mushrooms, industrial poisons), and other causes (e.g., Wilson’s disease, fatty liver of pregnancy, Reye syndrome, hepatic ischemia, hepatic vein obstruction, and disseminated malignancies). Extrahepatic complications of fulminant hepatitis are cerebral edema, coagulopathy and bleeding, cardiovascular abnormalities, renal failure, pulmonary complications, acid-base disturbances, electrolyte imbalances, sepsis, and pancreatitis.
Chronic Hepatitis
To be diagnosed with chronic hepatitis, a patient must have at least a 6-month course of hepatitis or biochemical and clinical evidence of liver disease with confirmatory biopsy findings of unresolving hepatic inflammation (Hoofnagle, 2007). Chronic hepatitis can have autoimmune, viral, metabolic, or medicine or toxin causes. The most common causes of chronic hepatitis are hepatitis B, hepatitis C, and autoimmune hepatitis. Other causes are drug-induced liver disease, metabolic diseases, and NASH. Cryptogenic cirrhosis is cirrhosis of an unknown cause.
Clinical symptoms of chronic hepatitis are usually nonspecific, occur intermittently, and are mild. Common symptoms include fatigue, sleep disorders, difficulty concentrating, and mild right upper quadrant pain. Severe advanced disease can lead to jaundice, muscle wasting, tea-colored urine, ascites, edema, hepatic encephalopathy, gastrointestinal bleeding, splenomegaly, palmar erythema, and spider angiomata.
Nonalcoholic Fatty Liver Disease
Nonalcoholic fatty liver disease (NAFLD) is a spectrum of liver disease ranging from steatosis to steatohepatitis. It involves the accumulation of fat droplets in the hepatocytes and can lead to fibrosis, cirrhosis, and even hepatocellular carcinoma. Steatosis is the simple accumulation of fat within the liver. Causes of NAFLD include drugs, inborn errors of metabolism, and acquired metabolic disorders (type 2 diabetes mellitus, lipodystrophy, jejunal ileal bypass, obesity, and malnutrition) (Diehl, 2007). It is commonly associated with obesity, diabetes mellitus, dyslipidemia, and insulin resistance.
Nonalcoholic steatohepatitis (NASH) is associated with accumulation of fibrous tissue in the liver. A two-hit hypothesis has been proposed to explain why some patients who develop NAFLD do not progress to NASH, and others do. Insulin resistance may lead to steatosis, but some type of oxidative stress is theorized to cause the disease to progress to NASH.
Patients with NASH may be asymptomatic but can experience malaise, weakness, or hepatomegaly. The treatment is often gradual weight loss, insulin-sensitizing drugs such as thiazolidinediones or possibly metformin, and treatment of dyslipidemia. Extreme, rapid weight loss can accelerate NASH developing into cirrhosis and increase the chance of gallstone development.
Chronic liver disease and cirrhosis can develop in patients with NASH. The progression to cirrhosis is variable, depending on age and the presence of obesity and type 2 diabetes, which contribute to a worsening prognosis (Diehl, 2007). Some studies suggest that vitamin E, betaine, and S-adenosylmethionine may be beneficial in reducing NASH by reducing tumor necrosis factor–α activity.
Alcoholic Liver Disease
Alcoholic liver disease is the most common liver disease in the United States, with age-adjusted death rates of 4.2 per 100,000 people (National Institute on Alcohol Abuse and Alcoholism, 2005). Acetaldehyde is a toxic byproduct of alcohol metabolism that causes damage to mitochondrial membrane structure and function. Acetaldehyde is produced by multiple metabolic pathways, one of which involves alcohol dehydrogenase (see Focus On: Metabolic Consequences of Alcohol Consumption).
 Focus On
Focus On
Metabolic Consequences of Alcohol Consumption
Ethanol is metabolized primarily in the liver by alcohol dehydrogenase. This results in acetaldehyde production with the transfer of hydrogen to nicotinamide adenine dinucleotide (NAD), reducing it to NADH. The acetaldehyde then loses hydrogen and is converted to acetate, most of which is released into the blood.
Many metabolic disturbances occur because of the excess of NADH, which overrides the ability of the cell to maintain a normal redox state. These include hyperlacticacidemia, acidosis, hyperuricemia, ketonemia, and hyperlipemia. The tricarboxylic acid (TCA) cycle is depressed because it requires NAD. The mitochondria, in turn, use hydrogen from ethanol rather than from the oxidation of fatty acids to produce energy via the TCA cycle, which leads to a decreased fatty acid oxidation and accumulation of triglycerides. In addition, NADH may actually promote fatty acid synthesis. Hypoglycemia can also occur in early alcoholic liver disease secondary to the suppression of the TCA cycle, coupled with decreased gluconeogenesis due to ethanol.
Several variables predispose some people to alcoholic liver disease. These include genetic polymorphisms of alcohol-metabolizing enzymes, gender (women more than men), simultaneous exposure to other drugs, infections with hepatotropic viruses, immunologic factors, and poor nutrition status. The pathogenesis of alcoholic liver disease progresses in three stages (Figure 30-2): hepatic steatosis (Figure 30-3), alcoholic hepatitis, and finally cirrhosis.
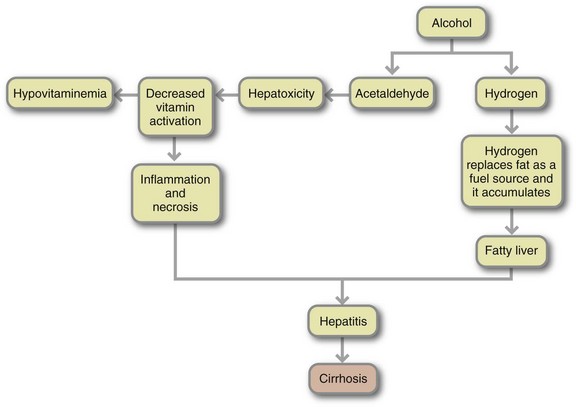
FIGURE 30-2 Complications of excessive alcohol consumption stem largely from excess hydrogen and from acetaldehyde. Hydrogen produces fatty liver and hyperlipemia, high blood lactic acid, and low blood sugar. The accumulation of fat, the effect of acetaldehyde on liver cells, and other factors as yet unknown lead to alcoholic hepatitis. The next step is cirrhosis. The consequent impairment of liver function disturbs blood chemistry, notably causing a high ammonia level that can lead to coma and death. Cirrhosis also distorts liver structure, inhibiting blood flow. High pressure in vessels supplying the liver may cause ruptured varices and accumulation of fluid in the abdominal cavity. Response to alcohol differs among individuals; in particular, not all heavy drinkers develop hepatitis and cirrhosis.
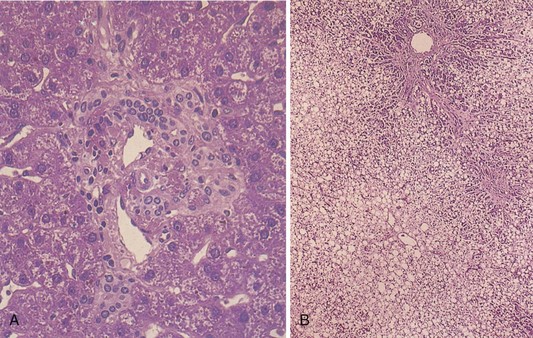
FIGURE 30-3 A, Microscopic appearance of a normal liver. A normal portal tract consists of the portal vein, hepatic arteriole, one to two interlobular bile ducts, and occasional peripherally located ductules. B, Acute fatty liver. This photomicrograph on low power exhibits fatty change involving virtually all the hepatocytes, with slight sparing of the liver cells immediately adjacent to the portal tract (top). (From Kanel G, Korula J: Atlas of liver pathology, Philadelphia, 1992, Saunders.)
Hepatic Steatosis
Fatty infiltration, known as hepatic steatosis or fatty liver, is caused by a culmination of these metabolic disturbances: (1) an increase in the mobilization of fatty acids from adipose tissue; (2) an increase in hepatic synthesis of fatty acids; (3) a decrease in fatty acid oxidation; (4) an increase in triglyceride production; and (5) a trapping of triglycerides in the liver. Hepatic steatosis is reversible with abstinence from alcohol. Conversely, if alcohol abuse continues, cirrhosis can develop.
Alcoholic Hepatitis
Alcoholic hepatitis is generally characterized by hepatomegaly, modest elevation of transaminase levels, increased serum bilirubin concentrations, normal or depressed serum albumin concentrations, or anemia. Patients may also have abdominal pain, anorexia, nausea, vomiting, weakness, diarrhea, weight loss, or fever. If patients discontinue alcohol intake, hepatitis may resolve; however, the condition often progresses to the third stage. Nutrition support is the main treatment in addition to counseling or support to continue avoidance of alcohol. Molecular genetics may lead to new therapies in the future (Willner and Reuben, 2005).
Alcoholic Cirrhosis
Clinical features of alcoholic cirrhosis, the third stage, vary. Symptoms can mimic those of alcoholic hepatitis; or patients can develop gastrointestinal bleeding, hepatic encephalopathy, or portal hypertension (elevated blood pressure in the portal venous system caused by the obstruction of blood flow through the liver). They often develop ascites, the accumulation of fluid, serum protein, and electrolytes within the peritoneal cavity caused by increased pressure from portal hypertension and decreased production of albumin (which maintains serum colloidal osmotic pressure). A liver biopsy usually reveals micronodular cirrhosis, but it can be macronodular or mixed. Prognosis depends on abstinence from alcohol and the degree of complications already developed. Ethanol ingestion creates specific and severe nutritional abnormalities (see Clinical Insight: Malnutrition in the Alcoholic).
 Clinical Insight
Clinical Insight
Malnutrition in the Alcoholic
Several factors contribute to the malnutrition that is common in chronic alcoholics with liver disease:
1. Alcohol can replace food in the diet of moderate and heavy drinkers, displacing the intake of adequate calories and nutrients. In light drinkers, it is usually an additional energy source, or empty calories. Although alcohol yields 7.1 kcal/g, when it is consumed in large amounts it is not used efficiently as a fuel source. When individuals consume alcohol on a regular basis but do not fulfill criteria for alcohol abuse, they are often overweight because of the increased calories (alcohol addition). This is different from the heavy drinker who replaces energy-rich nutrients with alcohol (alcohol substitution).
2. In the alcoholic, impaired digestion and absorption are related to pancreatic insufficiency, as well as morphologic and functional alterations of the intestinal mucosa. Acute and chronic alcohol intake impairs hepatic amino acid uptake and synthesis into proteins, reduces protein synthesis and secretion from the liver, and increases catabolism in the gut.
3. Use of lipids and carbohydrates is compromised. An excess of reduction equivalents (e.g., nicotinamide adenine dinucleotide phosphate) and impaired oxidation of triglycerides result in fat deposition in the hepatocytes and an increase in circulating triglycerides. Insulin resistance is also common.
4. Vitamin and mineral deficiencies occur in alcoholic liver disease as a result of reduced intake and alterations in absorption, storage, and ability to convert the nutrients to their active forms (Leevy and Moroianu, 2005). Steatorrhea resulting from bile acid deficiency is also common in alcoholic liver disease affecting fat-soluble vitamin absorption. Vitamin A deficiency can lead to night blindness (Leevy and Moroianu, 2005). Thiamin deficiency is the most common vitamin deficiency in alcoholics and is responsible for Wernicke encephalopathy (Leevy and Moroianu, 2005). Folate deficiency can occur as a result of poor intake, impaired absorption, accelerated excretion, and altered storage and metabolism. Inadequate dietary intake and interactions between pyridoxal-5′-phosphate (active coenzyme of vitamin B6) and alcohol reduce vitamin B6 status. Deficiency of all B vitamins and vitamins C, D, E, and K is also common (Leevy and Moroianu, 2005). Hypocalcemia, hypomagnesemia, and hypophosphatemia are not uncommon among alcoholics; furthermore, zinc deficiency and alterations in other micronutrients can accompany chronic alcohol intake (Leevy and Moroianu, 2005).
Cholestatic Liver Diseases
Primary biliary cirrhosis (PBC) is a chronic cholestatic disease caused by progressive destruction of small and intermediate-size intrahepatic bile ducts. The extrahepatic biliary tree and larger intrahepatic ducts are normal. Ninety-five percent of patients with PBC are women. This disease progresses slowly, eventually resulting in cirrhosis, portal hypertension, liver transplantation, or death (Afdhal, 2007).
PBC is an immune-mediated disease in which serum autoantibodies, elevated immunoglobulin levels, circulating immune complexes, and depressed cell-mediated immune response are present. PBC typically presents with a mild elevation of liver enzymes with physical symptoms of pruritus and fatigue. Treatment with ursodeoxycholic acid can slow progression of the disease (Afdhal, 2007). Several nutritional complications from cholestasis can occur with PBC, including osteopenia, hypercholesterolemia, and fat-soluble vitamin deficiencies.
Sclerosing Cholangitis
Sclerosing cholangitis shows fibrosing inflammation of segments of extrahepatic bile ducts, with or without involvement of intrahepatic ducts. Progression of the disease leads to complications of portal hypertension, hepatic failure (liver function diminished to 25% or less), and cholangiocarcinoma. Primary sclerosing cholangitis (PSC) is the most common type. Like PBC, PSC may be an immune disorder because of its strong association with human leukocyte antigen haplotypes, autoantibodies, and multiple immunologic abnormalities. Of patients with PSC, 70% to 90% also have inflammatory bowel disease (especially ulcerative colitis), and men are more likely than women (2.3 : 1) to have PSC (Afdhal, 2007). Patients with PSC are also at increased risk of fat-soluble vitamin deficiencies resulting from steatorrhea associated with this disease. Hepatic osteodystrophy may occur from vitamin D and calcium malabsorption, resulting in secondary hyperparathyroidism, osteomalacia, or rickets. No treatment slows progression of the disease or improves survival. Ursodeoxycholic acid may improve laboratory values (serum bilirubin, alkaline phosphatase, and albumin), but has no effect on survival (Afdhal, 2007).
Inherited Disorders
Inherited disorders of the liver include hemochromatosis, Wilson disease, α1-antitrypsin deficiency, protoporphyria, cystic fibrosis, glycogen storage disease, amyloidosis, and sarcoidosis. The first three disorders most commonly result in liver failure.
Hemochromatosis
Hemochromatosis is an inherited disease of iron overload associated with the gene HFE. Patients with hereditary hemochromatosis absorb excessive iron from the gut and may store 20 to 40 g of iron compared with 0.3 to 0.8 g in normal persons (see Chapter 33). Increased transferrin saturation (≥45%) and ferritin (more than two times normal) suggest hemochromatosis. Hepatomegaly, esophageal bleeding, ascites, impaired hepatic synthetic function, abnormal skin pigmentation, glucose intolerance, cardiac involvement, hypogonadism, arthropathy, and hepatocellular carcinoma may develop. Early diagnosis includes clinical, laboratory, and pathologic testing, including elevated serum transferrin levels. Life expectancy is normal if phlebotomy is initiated before the development of cirrhosis or diabetes mellitus.
Wilson’s Disease
Wilson’s disease is an autosomal-recessive disorder associated with impaired biliary copper excretion. Copper accumulates in various tissues, including the liver, brain, cornea, and kidneys. Kayser-Fleischer rings are greenish-yellow pigmented rings encircling the cornea just within the corneoscleral margin, formed by copper deposits. Patients can present with acute, fulminant, or chronic active hepatitis and with neuropsychiatric symptoms. Low serum ceruloplasmin levels, elevated copper concentration in a liver biopsy, and high urinary copper excretion confirm the diagnosis (Kowdley, 2007).
Copper-chelating agents and zinc supplementation (to inhibit intestinal copper absorption and binding in the liver) are used to treat Wilson disease once it is diagnosed. Copper chelation improves survival but does not prevent cirrhosis; transplantation corrects the metabolic defect (Medici, 2006). A low-copper diet is no longer required, but could be implemented if other therapies are unsuccessful (see Table 30-3). If this disease is not diagnosed until onset of fulminant failure, survival is not possible without transplantation.
TABLE 30-3
Copper Content of Commonly Used Foods*


*Copper content of the usual American diet varies, with estimates from 1 mg of copper a day to 5 mg/day. The concentration of copper in foods is affected by soil conditions, geographic location, species, diet, processing method, and contamination in processing. The exact copper content of foods is difficult to verify. It is estimated that avoiding high-copper foods and restricting moderate-copper foods result in a diet of approximately 1 mg/day. For practical purposes, diets are designed to limit foods with higher copper content rather than to achieve a specific level of copper in the diet.
†Portions commonly used are those generally accepted as typical portion sizes in various nutrient data source manuals.
‡A water sample from the patient’s home water supply should be analyzed for copper content. Demineralized water should be used if the water contains more than 100 mcg/L.
§Although not necessarily high in copper, alcohol is discouraged because of its action as a hepatotoxin.
From Pemberton CM et al: Mayo Clinic diet manual: a handbook of nutrition practices, ed 7, St Louis, 1994, Mosby.
α1-Antitrypsin Deficiency
α1-Antitrypsin deficiency is an inherited disorder that can cause both liver and lung disease. α1-Antitrypsin is a glycoprotein found in serum and body fluids; it inhibits neutrophil proteinases. Cholestasis or cirrhosis is caused by this deficiency and there is no treatment except liver transplantation.
Other Liver Diseases
Liver disease has several other causes. Liver tumors can be primary or metastatic, benign or malignant. Hepatocellular carcinoma usually develops in cirrhotic livers. The highest risk occurs in those with hepatitis B, hepatitis C, and hereditary hemochromatosis. The liver can also be affected by systemic diseases such as rheumatoid arthritis, systemic lupus erythematosus, polymyalgia, temporal arteritis, polyarteritis nodosa, systemic sclerosis, and Sjögren’s syndrome. When hepatic blood flow is altered, as in acute ischemic and chronic congestive hepatopathy, Budd-Chiari syndrome, and hepatic venoocclusive disease, dysfunction occurs. Individuals with hepatic or portal vein thromboses should be evaluated for a myeloproliferative disorder. Parasitic, bacterial, fungal, and granulomatous liver diseases also occur. Finally, cryptogenic cirrhosis is any cirrhosis for which the cause is unknown.
Treatment of Cirrhosis and Its Complications
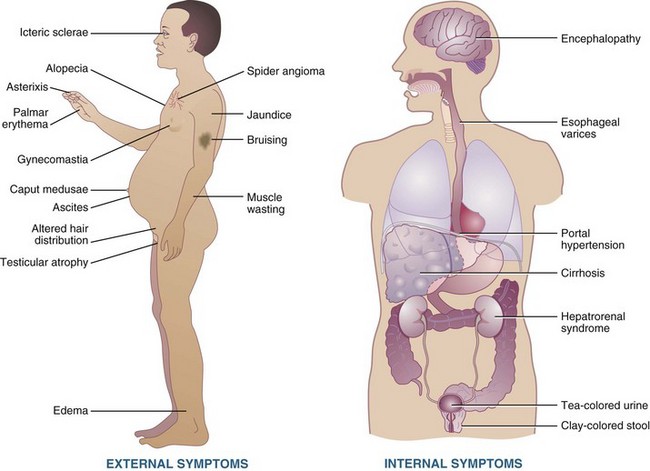
Cirrhosis has many clinical manifestations, as illustrated in Figure 30-4. Several major complications of cirrhosis and end-stage liver disease (ESLD), including malnutrition, ascites, hyponatremia, hepatic encephalopathy, glucose alterations, fat malabsorption, hepatorenal syndrome, and osteopenia, have nutritional implications. When appropriate nutrition therapy is provided to patients with liver disease, malnutrition can be reversed, and clinical outcomes improved. Studies to date have shown positive outcomes with oral and enteral nutrition (EN) in malnourished patients with cirrhosis, including improvement in nutrition status and clinical complications of cirrhosis such as ascites, encephalopathy, and infection (Campillo et al., 2005).
Nutrition Assessment
A specific nutrition assessment must be performed to determine the extent and cause of malnutrition. Many traditional markers of nutrition status are affected by liver disease and its consequences, making traditional assessment difficult. Table 30-4 summarizes the factors that affect interpretation of nutrition assessment parameters in patients with liver dysfunction.
TABLE 30-4
Factors That Affect Interpretation of Nutrition Assessment in Patients With End-Stage Liver Disease

Modified from Hasse J: Nutritional aspects of adult liver transplantation. In Busuttil RW, Klintmalm GB, editors: Transplantation of the liver, ed 2, Philadelphia, 2005, Saunders.
Objective parameters that are helpful when monitored serially include anthropometric measurements and dietary intake evaluation (see Chapter 9). The best way to perform a nutrition assessment may be to combine these parameters with the subjective global assessment (SGA) approach, which has demonstrated an acceptable level of reliability and validity. This method uses a few readily available parameters obtained by an experienced clinician. The SGA gives a broad perspective, but it is not sensitive to changes in nutrition status. Other available parameters should also be reviewed. The SGA approach is summarized in Box 30-1.
Malnutrition
Moderate to severe malnutrition is a common finding in patients with advanced liver disease (Figure 30-5). This is extremely significant, considering that malnutrition plays a major role in the pathogenesis of liver injury and has a profound negative effect on prognosis. The prevalence of malnutrition depends on nutrition assessment parameters used, type and degree of liver disease, and socioeconomic status.
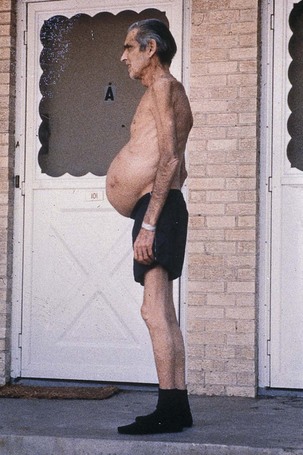
Numerous coexisting factors are involved in the development of malnutrition in liver disease (see Pathophysiology and Care Management Algorithm: Malnutrition in Liver Disease). Inadequate oral intake, a major contributor, is caused by anorexia, dysgeusia, early satiety, nausea, or vomiting associated with liver disease and the drugs used to treat it. Another cause of inadequate intake is dietary restriction.
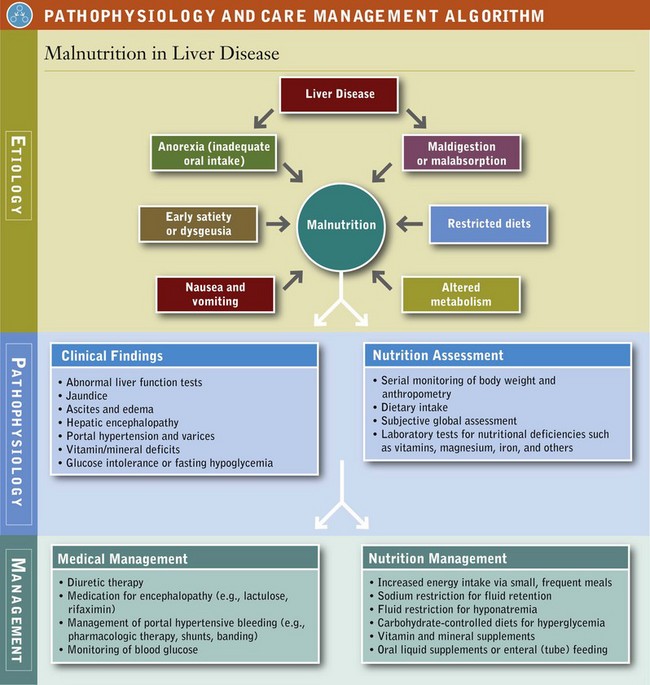
Maldigestion and malabsorption also play a role. Steatorrhea, presence of fat in the stool, is common in cirrhosis, especially if there is disease involving bile duct injury and obstruction. Medications may also cause specific malabsorptive losses. In addition, altered metabolism secondary to liver dysfunction causes malnutrition in various ways. Micronutrient function is affected by altered storage in the liver, decreased transport by liver-synthesized proteins, and renal losses associated with alcoholic and advanced liver disease. Abnormal macronutrient metabolism and increased energy expenditure can also contribute to malnutrition. Finally, protein losses can occur from large-volume paracentesis when fluid from the abdomen (ascites) is removed through a needle.
Nutrition Intake Problems
Because anorexia, nausea, dysgeusia, and other gastrointestinal symptoms are common, adequate nutrition intake is difficult to achieve. With ascites, early satiety is also a frequent complaint. Smaller, more frequent meals are better tolerated than three traditional meals. In addition, frequent feedings may also improve nitrogen balance and prevent hypoglycemia. Oral liquid supplements should be encouraged, and, when necessary, enteral tube feedings used. Adjunctive nutrition support should be given to malnourished patients with liver disease if their intake is less than dietary reference intake (DRI) levels of 0.8 g of protein and 30 calories/kg of body weight daily and if they are at risk for fatal complications from the disease. Esophageal varices are usually not a contraindication for tube feeding (Crippin, 2006).
Nutrient Requirements
Energy requirements vary among patients with cirrhosis. Several studies have measured resting energy expenditure (REE) in patients with liver disease to determine energy requirements. Some found that patients with ESLD had normal metabolism and that others had hypometabolism or hypermetabolism. Ascites or shunt placement may increase energy expenditure slightly.
In general, energy requirements for patients with ESLD and without ascites are approximately 120% to 140% of the REE. Requirements increase to 150% to 175% of REE if ascites, infection, and malabsorption are present or if nutritional repletion is necessary. This equates to approximately 25 to 35 calories per kilogram body weight. Estimated dry body weight should be used in calculations to prevent overfeeding. Oral nutritional supplements or tube feeding can increase or ensure optimal intake in malnourished patients, thus reducing complications and prolonging survival (Plauth et al., 2006).
Carbohydrates
Determining carbohydrate needs is challenging in liver failure because of the primary role of the liver in carbohydrate metabolism. Liver failure reduces glucose production and peripheral glucose use. The rate of gluconeogenesis is decreased, with preference for lipids and amino acids for energy. Alterations in the hormones insulin, glucagon, cortisol, and epinephrine are responsible in part for the preference for alternative fuels. In addition, insulin resistance can be present with liver dysfunction.
Lipid
In cirrhosis, plasma free fatty acids, glycerol, and ketone bodies are increased in the fasting state. The body prefers lipids as an energy substrate. Lipolysis is increased with active mobilization of lipid deposits, but the net capacity to store exogenous lipid is not impaired. A range of 25% to 40% of calories as fat is generally recommended.
Protein
Protein is by far the most controversial nutrient in liver failure, and its management is also the most complex. Cirrhosis has long been thought of as a catabolic disease with increased protein breakdown, inadequate resynthesis, depleted status, and muscle wasting. However, protein kinetic studies demonstrate increased nitrogen losses only in patients with fulminant hepatic failure or decompensated disease but not in stable cirrhosis.
Patients with cirrhosis also have increased protein use. Studies suggest that 0.8 g of protein/kg/day is the mean protein requirement to achieve nitrogen balance in stable cirrhosis. Therefore in uncomplicated hepatitis or cirrhosis without encephalopathy, protein requirements range from 0.8 to 1 g/kg of dry weight per day to achieve nitrogen balance.
To promote nitrogen accumulation or positive balance, at least 1.2 to 1.3 g/kg daily is needed. In situations of stress such as alcoholic hepatitis or decompensated disease (sepsis, infection, gastrointestinal bleeding, or severe ascites), at least 1.5 g of protein per kilogram per day should be provided.
Vitamins and Minerals
Vitamin and mineral supplementation is needed in all patients with ESLD because of the intimate role of the liver in nutrient transport, storage, and metabolism, in addition to the side effects of drugs (Table 30-5). Vitamin deficiencies can contribute to complications. For example, folate and vitamin B12 deficiencies can lead to macrocytic anemia. Deficiency of pyridoxine, thiamin, or vitamin B12 can result in neuropathy. Confusion, ataxia, and ocular disturbances can result from a thiamin deficiency.
TABLE 30-5
Vitamin and Mineral Deficits in Severe Hepatic Failure
| Vitamin or Mineral | Predisposing Factors | Signs of Deficiency |
| Vitamin A | Steatorrhea, neomycin, cholestyramine, alcoholism | Night-blindness, increased infection risk |
| Vitamin B1 (thiamine) | Alcoholism, high CHO diet | Neuropathy, ascites, edema, CNS dysfunction |
| Vitamin B3 (niacin) | Alcoholism | Dermatitis, dementia, diarrhea, inflammation of mucous membranes |
| Vitamin B6 (pyridoxine) |
Alcoholism | Mucous membrane lesions, seborrheic dermatitis, glossitis, angular stomatitis, blepharitis, peripheral neuropathy, microcytic anemia, depression |
| Vitamin B12 (Cyanocobalamin) |
Alcoholism, cholestyramine | Megaloblastic anemia, glossitis, CNS dysfunction |
| Folate | Alcoholism | Megaloblastic anemia, glossitis, irritability |
| Vitamin D | Steatorrhea, glucocorticoids, cholestyramine | Osteomalacia, rickets (in children), possible link to cancer or autoimmune disorders |
| Vitamin E | Steatorrhea, cholestyramine | Peripheral neuropathy, ataxia, skeletal myopathy, retinopathy, immune system impairment |
| Vitamin K | Steatorrhea, antibiotics, cholestyramine | Excessive bleeding; bruising |
| Iron | Chronic bleeding | Stomatitis, microcytic anemia, malaise |
| Magnesium | Alcoholism, diuretics | Neuromuscular irritability, hypokalemia, hypocalcemia |
| Phosphorus | Anabolism, alcoholism | Anorexia, weakness, cardiac failure, glucose intolerance |
| Zinc | Diarrhea, diuretics, alcoholism | Immunodeficiency, impaired taste acuity, wound healing, protein synthesis |
Deficiencies of fat-soluble vitamins have been found in all types of liver failure, especially in cholestatic diseases in which malabsorption and steatorrhea occur. Impaired dark adaptation can occur from vitamin A deficiency. Hepatic osteodystrophy or osteopenia can develop from vitamin D deficiency. Therefore supplementation is necessary, and may require using water-soluble forms. Intravenous or intramuscular vitamin K is often given for 3 days to rule out vitamin K deficiency as the cause of a prolonged prothrombin times. Water-soluble vitamin deficiencies associated with liver disease include thiamin (which can lead to Wernicke encephalopathy), pyridoxine (B6), cyanocobalamin (B12), folate, and niacin (B3). Large doses (100 mg) of thiamin are given daily for a limited time if deficiency is suspected.
Mineral nutriture is also altered in liver disease. Iron stores may be depleted in patients experiencing gastrointestinal bleeding; however, iron supplementation should be avoided by persons with hemochromatosis or hemosiderosis (see Chapter 33). Elevated serum copper levels are found in cholestatic liver diseases (i.e., PBC and PSC). Because copper and manganese are excreted primarily via bile, supplements should not contain these minerals. Manganese deposition has been noted to accumulate in brains of patients with cirrhosis leading to impaired motor function (Garcia-Tsao, 2007).
In Wilson’s disease, excess copper in various organs causes severe damage. Oral chelating agents such as zinc acetate or d-penicillamine are the primary treatment. Dietary copper restriction (see Table 30-3) is not routinely prescribed unless other therapies are unsuccessful. A vegetarian diet may be useful as adjunctive therapy; copper is less available.
Zinc and magnesium levels are low in liver disease related to alcoholism, in part because of diuretic therapy. Calcium, as well as magnesium and zinc, may be malabsorbed with steatorrhea. Therefore the patient should take supplements of these minerals at least at the level of the DRI.
Herbal Supplements
There are multiple case reports of different herbal supplements resulting in liver failure. Terpenoid-containing dietary supplements have been implicated in causing severe and sometimes fatal hepatotoxicity, including teucrium polium (germander), Sho-saiko-to, Centella asiatica, and black cohosh (Chitturi and Farrell, 2008). Liver injury has also been caused by N-nitrosofenfluramine, ephedra alkaloids, Boh-Gol-Zhee, Kava, and pyrrolizidine alkaloids (Chitturi and Farrell, 2008).
Two herbal supplements have become popular in the treatment of liver disease. Milk thistle is popular among those suffering from viral hepatitis or alcoholic liver disease. The active component in milk thistle is silymarin. It is proposed to reduce free radical production and lipid peroxidation associated with hepatotoxicity. S-adenosyl-L-methionine (SAMe) is another popular complementary medicine product purported to act as a methyl donor for methylation reactions and participate in glutathione (an antioxidant) synthesis. A Cochrane review did not show any evidence to support or refute a beneficial effect of either milk thistle or SAMe in patients with alcoholic liver disease (Rambaldi et al., 2006, 2007).
Portal Hypertension
Pathophysiology and Medical Treatment
Portal hypertension increases collateral blood flow and can result in swollen veins (varices) in the GIT. These varices often bleed, causing a medical emergency. Treatment includes administration of α-adrenergic blockers to decrease heart rate, endoscopic banding or variceal ligation, and radiologic or surgical placement of shunts. During an acute bleeding episode, somatostatin analog may be administered to decrease bleeding, or a nasogastric tube equipped with an inflatable balloon is placed to tamponade bleeding vessels.
Medical Nutrition Therapy
During acute bleeding episodes, nutrition cannot be administered enterally. Parenteral nutrition (PN) is indicated if a patient will be taking nothing orally for at least 5 days. Repeated endoscopic therapies may cause esophageal strictures or impair a patient’s swallowing. Finally, surgically or radiologically placed shunts may increase the incidence of encephalopathy and reduce nutrient metabolism because blood is shunted past the liver cells.
Ascites
Pathophysiology and Medical Treatment
Fluid retention is common, and ascites (accumulation of fluid in the abdominal cavity) is a serious consequence of liver disease. Portal hypertension, hypoalbuminemia, lymphatic obstruction, and renal retention of sodium and fluid contribute to fluid retention. Increased release of catecholamines, renin, angiotensin, aldosterone, and antidiuretic hormone secondary to peripheral arterial vasodilation causes renal retention of sodium and water.
Large-volume paracentesis may be used to relieve ascites. Diuretic therapy is often used and includes spironolactone and furosemide. These drugs are often used in combination for best effect. Major side effects of loop diuretics such as furosemide include hyponatremia, hypokalemia, hypomagnesemia, hypocalcemia, and hypochloremic acidosis. Conversely, spironolactone is potassium sparing. Therefore serum potassium levels must be monitored carefully and supplemented or restricted if necessary because deficiency or excess can contribute to metabolic abnormalities. Weight; abdominal girth; urinary sodium concentration; and serum levels of urea nitrogen, creatinine, albumin, uric acid, and electrolytes should be monitored during diuretic therapy.
Medical Nutrition Therapy
Dietary treatment for ascites includes sodium restriction in addition to diuretic therapy. Sodium is commonly restricted to 2 g/day (see Chapter 34 and Appendix 37 for discussion of low-sodium diets). More severe limitations may be imposed; however, caution is warranted because of limited palatability and the risk of overrestricting sodium. Adequate protein intake is also important when a patient undergoes frequent paracentesis.
Hyponatremia
Hepatic Encephalopathy
Pathophysiology and Medical Treatment
Hepatic encephalopathy is a syndrome characterized by impaired mentation, neuromuscular disturbances, and altered consciousness. Gastrointestinal bleeding, fluid and electrolyte abnormalities, uremia, infection, use of sedatives, hyperglycemia or hypoglycemia, alcohol withdrawal, constipation, azotemia, dehydration, portosystemic shunts, and acidosis can precipitate hepatic encephalopathy. Subclinical or minimal hepatic encephalopathy also affects patients with chronic hepatic failure. Hepatic or portal systemic encephalopathy results in neuromuscular and behavioral alterations. Box 30-2 describes the four stages of hepatic encephalopathy.
There are three main theories regarding the mechanism by which hepatic encephalopathy occurs. Ammonia accumulation is considered an important causal factor in the development of encephalopathy. When the liver fails, it is unable to detoxify ammonia to urea, and ammonia is a direct cerebral toxin. Ammonia levels are elevated in the brain and bloodstream, leading to impaired neural function through cytotoxicity, cell swelling, and depletion of glutamate (Fitz, 2006). The main source of ammonia is its endogenous production by the GIT from the metabolism of protein and from the degradation of bacteria and blood from gastrointestinal bleeding. Exogenous protein is also a source of ammonia. Some clinicians suggest that dietary protein causes an increase in ammonia levels and subsequently hepatic encephalopathy, but this has not been proven in studies.
Drugs such as lactulose and rifaximin are given. Lactulose is a nonabsorbable disaccharide. It acidifies the colonic contents, retaining ammonia as the ammonium ion. It also acts as an osmotic laxative to remove the ammonia. Rifaximin is a nonabsorbable antibiotic that helps decrease colonic ammonia production.
Another suggested mechanism involves the γ-aminobutyric acid (GABA) receptor complex in contributing to neuronal inhibition in hepatic encephalopathy. Flumazenil or other benzodiazepine receptor antagonists may help reduce hepatic encephalopathy.
A final hypothesis is the “altered neurotransmitter theory.” A plasma amino acid imbalance exists in ESLD in which the branched-chain amino acids (BCAAs) valine, leucine, and isoleucine are decreased. The BCAAs furnish as much as 30% of energy requirements for skeletal muscle, heart, and brain when gluconeogenesis and ketogenesis are depressed causing serum BCAA levels to fall. Aromatic amino acids (AAAs) tryptophan, phenylalanine, and tyrosine, plus methionine, glutamine, asparagine, and histidine are increased, Plasma AAAs and methionine are released into circulation by muscle proteolysis, but the synthesis into protein and liver clearance of AAAs is depressed. This changes the plasma molar ratio of BCAAs to AAAs and may contribute to the development of hepatic encephalopathy. High levels of AAAs may limit the cerebral uptake of BCAAs because they compete for carrier-mediated transport at the blood-brain barrier.
Medical Nutrition Therapy
The practice of protein restriction in patients with low-grade hepatic encephalopathy is based on the premise that protein intolerance causes hepatic encephalopathy, but it has never been proven in a study. True dietary protein intolerance is rare except in fulminant hepatic failure, or in a rare patient with chronic endogenous hepatic encephalopathy. Unnecessary protein restriction may worsen body protein losses and must be avoided.
Patients with encephalopathy often do not receive adequate protein. More than 95% of patients with cirrhosis can tolerate mixed-protein diets up to 1.5 g/kg of body weight. Studies evaluating the benefit of supplements enriched with BCAAs and restricted in AAAs have varied in study design, sample size, composition of formulas, level of encephalopathy, type of liver disease, duration of therapy, and control groups. When high methodologic quality studies are evaluated, there are no significant improvements or survival benefits associated with giving extra BCAAs to patients.
Other theories postulate that vegetable proteins and casein may improve mental status compared with meat protein. Casein-based diets are lower in AAAs and higher in BCAAs than meat-based diets. Vegetable protein is low in methionine and ammoniagenic amino acids, but BCAA-rich. The high-fiber content of a vegetable-protein diet may also play a role in the excretion of nitrogenous compounds.
Finally, it has been proposed that probiotics and synbiotics (sources of gut-friendly bacteria and fermentable fibers) can be used to treat hepatic encephalopathy. Probiotics (see Chapter 29) may improve hepatic encephalopathy by reducing ammonia portal blood (Pereg et al., 2010) or by preventing production or uptake of lipopolysaccharides in the gut (Gratz et al., 2010). Thus they decrease inflammation and oxidative stress in the hepatocyte (thus increasing hepatic clearance of toxins including ammonia), and minimizing uptake of other toxins.
Glucose Alterations
Glucose intolerance occurs in almost two thirds of patients with cirrhosis, and 10% to 37% of patients develop overt diabetes. Glucose intolerance in patients with liver disease occurs because of insulin resistance in peripheral tissues. Hyperinsulinism also occurs in patients with cirrhosis, possibly because insulin production is increased, hepatic clearance is decreased, portal systemic shunting occurs, there is a defect in the insulin-binding action at the receptor site, or there is a postreceptor defect.
Fasting hypoglycemia, or low blood glucose, can occur because of the decreased availability of glucose from glycogen in addition to the failing gluconeogenic capacity of the liver when the patient has ESLD. Hypoglycemia occurs more often in acute or fulminant liver failure than in chronic liver disease. Hypoglycemia may also occur after alcohol consumption in patients whose glycogen stores are depleted by starvation because of the block of hepatic gluconeogenesis by ethanol.
Medical Nutrition Therapy
Patients with diabetes should receive standard medical and nutrition therapy to achieve normoglycemia (see Chapter 31). Patients with hypoglycemia should eat frequently to prevent this condition (see Clinical Insight: Fasting Hypoglycemia).
Clinical Insight
Fasting Hypoglycemia
Two-thirds of the glucose requirement in an adult is used by the central nervous system. During fasting, plasma glucose concentrations are maintained for use by the nervous system and the brain because liver glycogen is broken down, or new glucose is made from nonglucose precursors such as alanine. Fasting hypoglycemia occurs when there is reduced synthesis of new glucose or reduced liver glycogen breakdown.
Causes of fasting hypoglycemia include cirrhosis, consumption of alcohol, extensive intrahepatic cancer, deficiency of cortisol and growth hormone, or non–β cell tumors of the pancreas. The method for detecting it involves measuring plasma insulin when plasma glucose is low. The diagnostic hallmark of an insulinoma is altered insulin secretion in the presence of hypoglycemia. Fasting hypoglycemia may also be caused by spontaneously produced antibodies. All patients with liver or pancreatic disease should be monitored for fasting hypoglycemia. Nutrition therapy involves balanced meals with small, frequent snacks to avoid periods of fasting. Monitoring of blood glucose and insulin levels is required.
Fat Malabsorption
Fat absorption may be impaired in liver disease. Possible causes include decreased bile salt secretion (as in PBC, sclerosing cholangitis, and biliary strictures), administration of neomycin or cholestyramine, and pancreatic enzyme insufficiency. Stools may be greasy, floating, or light- or clay-colored, signifying malabsorption, which can be verified by a 72-hour fecal fat study (see Chapter 29 and Appendix 30).
Medical Nutrition Therapy
If significant steatorrhea is present, replacement of some of the long-chain triglycerides or dietary fat with medium-chain triglycerides (MCTs) may be useful. Because MCTs do not require bile salts and micelle formation for absorption, they are readily taken up via the portal route (see Chapter 29). Some nutrition supplements contain MCTs, which can be used in addition to liquid MCT oil (see Chapter 14).
Significant stool fat losses may warrant a trial of a low-fat (40 g/day) diet. If diarrhea does not resolve, fat restriction should be discontinued because it decreases the palatability of the diet and severely hampers adequate calorie intake.
Renal Insufficiency and Hepatorenal Syndrome
Pathophysiology, Medical and Nutrition Therapies
Hepatorenal syndrome is renal failure associated with severe liver disease without intrinsic kidney abnormalities. Hepatorenal syndrome is diagnosed when the urine sodium level is less than 10 mEq/L and oliguria persists in the absence of intravascular volume depletion. If conservative therapies, including discontinuation of nephrotoxic drugs, optimization of intravascular volume status, treatment of underlying infection, and monitoring of fluid intake and output fail, dialysis may be required. In any case, renal insufficiency and failure may necessitate alteration in fluid, sodium, potassium, and phosphorus intake (see Chapter 36).
Osteopenia
Osteopenia often exists in patients with PBC, sclerosing cholangitis, and alcoholic liver disease. Depressed osteoblastic function and osteoporosis also can occur in patients with hemochromatosis, and osteoporosis is prevalent in patients who have had long-term treatment with corticosteroids. Corticosteroids increase bone resorption; suppress osteoblastic function; and affect sex hormone secretion, intestinal absorption of dietary calcium, renal excretion of calcium and phosphorus, and the vitamin D system.
Medical Nutrition Therapy
Prevention or treatment options for osteopenia include weight maintenance, ingestion of a well-balanced diet, adequate protein to maintain muscle mass, 1500 mg of calcium per day, adequate vitamin D from the diet or supplements, avoidance of alcohol, and monitoring for steatorrhea, with diet adjustments as needed to minimize nutrient losses.
Liver Resection and Transplantation
Liver resection and ablation are fairly common now that problem areas can be located by means of tomography and arteriography. As with any major surgery, protein and energy needs increase after liver resection. Needs are also increased for liver cell regeneration. EN is vital because of the role of portal hepatotropic factors necessary for liver cell proliferation. Optimal nutrition is most important for patients with poor nutrition status before hepatectomy (e.g., patients with hepatocellular carcinoma or cholangiocarcinoma).
Liver transplantation has become an established treatment for ESLD. Malnutrition is common in liver transplant candidates. Dietary intake can often be enhanced if patients eat small, frequent, nutrient-dense meals; oral nutritional supplements may also be well tolerated. Enteral tube feeding is indicated when oral intake is inadequate or contraindicated. Varices are not an absolute contraindication for placement of a feeding tube. Because PN can adversely affect liver function, EN is preferred. PN is reserved for patients without adequate gut function. (See Chapter 14.)
In the acute posttransplant phase, nutrient needs are increased to promote healing, deter infection, provide energy for recovery, and replenish depleted body stores. Nitrogen requirements are elevated in the acute posttransplant phase and can be met with early postoperative tube feeding. Probiotics and fiber added to tube feeding may reduce postoperative infection rate better than tube feeding or fiber alone (Rayes et al, 2005).
Multiple medications used after transplant have nutritional side effects such as anorexia, gastrointestinal upset, hypercatabolism, diarrhea, hyperglycemia, hyperlipidemia, sodium retention, hypertension, hyperkalemia, and hypercalciuria. Therefore dietary modification is based on the specific side effects of drug therapy (Table 30-6). During the posttransplant phase, nutrient requirements are adjusted to prevent or treat problems of obesity, hyperlipidemia, hypertension, diabetes mellitus, and osteopenia. Table 30-7 summarizes nutrient needs following liver transplantation.

TABLE 30-7
Nutrition Care Guidelines for Liver Transplant Patient
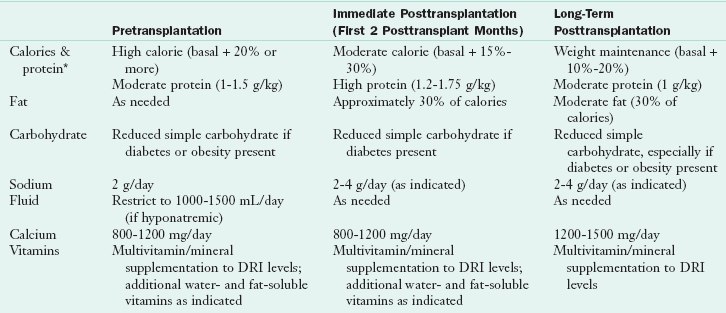
DRI, Dietary reference intake.
*Use estimated dry or ideal weight.
Modified from Porayko MK et al: Impact of malnutrition and its therapy on liver transplantation, Semin Liv Dis 11(4):305, 1991.
Physiology and Functions of the Gallbladder
The gallbladder lies on the undersurface of the right lobe of the liver (Figure 30-6). The main function of the gallbladder is to concentrate, store, and excrete bile, which is produced by the liver. During the concentration process, water and electrolytes are resorbed by the gallbladder mucosa. The chief constituents of bile are cholesterol, bilirubin, and bile salts. Bilirubin, the main bile pigment, is derived from the release of hemoglobin from red blood cell destruction. It is transported to the liver, where it is conjugated and excreted via bile. Bile salts are made by liver cells from cholesterol and are essential for the digestion and absorption of fats, fat-soluble vitamins, and some minerals (see Chapter 1).
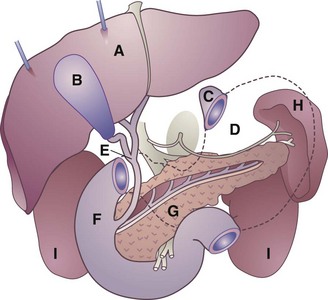
FIGURE 30-6 Schematic drawing showing relationship of organs of the upper abdomen. A, Liver (retracted upward); B, gallbladder; C, esophageal opening of stomach; D, stomach (shown in dotted outline); E, common bile duct; F, duodenum; G, pancreas and pancreatic duct; H, spleen; I, kidneys. (Courtesy Cleveland Clinic, Cleveland, Ohio, 2002.)
The primary transporter responsible for bile salt secretion is the bile salt export pump (BSEP). Overall, bile salts play a key role in a wide range of physiologic and pathophysiologic processes (Lam et al., 2010). Excreted into the small intestine via bile, bile salts are later resorbed into the portal system (enterohepatic circulation). This is the primary excretory pathway for the minerals copper and manganese.
Bile contains immunoglobulins that support the integrity of the intestinal mucosa. Fibroblast growth factor receptor (FGFR) 4 controls bile acid metabolism and protects the liver from fibrosis; FGFR1 and FGFR2 assist in regeneration of the liver (Böhm et al., 2010). Molecular crosstalk between bile acid–activated nuclear receptors and proinflammatory nuclear mediators provides new understanding of inflammation-induced cholestasis (Kosters and Karpen, 2010).
Bile is removed by the liver via bile canaliculi that drain into intrahepatic bile ducts. The ducts lead to the left and right hepatic ducts, which leave the liver and join to become the common hepatic duct. The bile is directed to the gallbladder via the cystic duct for concentration and storage. The cystic duct joins the common hepatic duct to form the common bile duct. The bile duct then joins the pancreatic duct, which carries digestive enzymes.
During the course of digestion, food reaches the duodenum, causing the release of intestinal hormones such as cholecystokinin (CCK) and secretin. This stimulates the gallbladder and pancreas and causes the sphincter of Oddi to relax, allowing pancreatic juice and bile to flow into the duodenum at the ampulla of Vater to assist in fat digestion. For this reason diseases of the gallbladder, liver, and pancreas are often interrelated.
Diseases of the Gallbladder
Disorders of the biliary tract affect millions of people each year, causing significant suffering and even death by precipitating pancreatitis and sepsis. A diverse spectrum of disease affects the biliary system, often presenting with similar clinical signs and symptoms. Treatment may involve diet, medication, or surgery.
Cholestasis
Pathophysiology and Medical Management
Cholestasis is a condition in which little or no bile is secreted or the flow of bile into the digestive tract is obstructed. This can occur in patients without oral or enteral feeding for a prolonged period, such as those requiring PN, and can predispose to acalculous cholecystitis. BSEP deficiency results in several different genetic forms of cholestasis and acquired forms of cholestasis such as drug-induced cholestasis and intrahepatic cholestasis of pregnancy (Lam et al., 2010). Prevention of cholestasis requires stimulation of biliary motility and secretions by at least minimum enteral feedings. If this is not possible, drug therapy is used.
Cholelithiasis
The formation of gallstones (calculi) is cholelithiasis. Virtually all gallstones form within the gallbladder. Gallstone disease affects millions of Americans each year and causes significant morbidity. In most cases gallstones are asymptomatic. Gallstones that pass from the gallbladder into the common bile duct may remain there indefinitely without causing symptoms, or they may pass into the duodenum with or without symptoms.
Choledocholithiasis develops when stones slip into the bile ducts, producing obstruction, pain, and cramps. If passage of bile into the duodenum is interrupted, cholecystitis can develop. In the absence of bile in the intestine, lipid absorption is impaired, and without bile pigments, stools become light in color (acholic). If uncorrected, bile backup can result in jaundice and liver damage (secondary biliary cirrhosis). Obstruction of the distal common bile duct can lead to pancreatitis if the pancreatic duct is blocked.
Most gallstones are unpigmented cholesterol stones composed primarily of cholesterol, bilirubin, and calcium salts. Bacteria also play a role in gallstone formation. Low-grade chronic infections produce changes in the gallbladder mucosa, which affect its absorptive capabilities. Excess water or bile acid may be absorbed as a result. Cholesterol may then precipitate out and cause gallstones (Volzke et al., 2005).
High dietary fat intake over a prolonged period may predispose a person to gallstone formation because of the constant stimulus to produce more cholesterol for bile synthesis required in fat digestion. Rapid weight loss (as with jejunoileal and gastric bypass and fasting or severe calorie restriction) is associated with a high incidence of biliary sludge and gallstone formation.
Indeed, cholelithiasis and fatty liver disease share risk factors including central obesity, insulin resistance, and diabetes (Weikert et al., 2010).
Risk factors for cholesterol stone formation include female gender, pregnancy, older age, family history, obesity and truncal body fat distribution, diabetes mellitus, inflammatory bowel disease, and drugs (lipid-lowering medications, oral contraceptives, and estrogens). Certain ethnic groups are at greater risk of stone formation, including Pima Indians, Scandinavians, and Mexican-Americans.
Pigmented stones typically consist of bilirubin polymers or calcium salts. They are associated with chronic hemolysis. Risk factors associated with these stones are age, sickle cell anemia and thalassemia, biliary tract infection, cirrhosis, alcoholism, and long-term PN (Abayli et al., 2005).
Medical & Surgical Management
Cholecystectomy is surgical removal of the gallbladder, especially if the stones are numerous, large, or calcified. The cholecystectomy may be done as a traditional open laparotomy or as a less invasive laparoscopic procedure. Recently, it has been noted that cholecystectomy is a predictor of the development of cirrhosis and is associated with elevated serum liver enzymes (Ioannou, 2010).
Chemical dissolution with the administration of bile salts, chenodeoxycholic acid, and ursodeoxycholic acid (litholytic therapy) or dissolution by extracorporeal shockwave lithotripsy may also be used less often than surgical techniques. Patients with gallstones that have migrated into the bile ducts may be candidates for endoscopic retrograde cholangiopancreatography techniques (Adler et al., 2005).
Medical Nutrition Therapy
No specific dietary treatment is available to prevent cholelithiasis in susceptible persons. Gallstones are more prevalent in low-fiber, high-fat, Westernized diets. Consumption of large amounts of animal protein and animal fat, especially saturated fat, and a lack of dietary fiber, promote gallstone development.
There may also be some benefit in replacing simple sugars and refined starches with high-fiber carbohydrates. Individuals consuming refined carbohydrates have a 60% greater risk for developing gallstones, compared with those who consumed the most fiber, in particular insoluble fiber (Tsai, 2005). Thus plant-based diets may reduce the risk of cholelithiasis. Vegetarian diets are high in fiber and low in fat, consisting primarily of unsaturated fat. Vitamin C, which is generally high in vegetarian diets, affects the rate-limiting step in the catabolism of cholesterol to bile acids and is inversely related to the risk of gallstones in women.
Weight cycling (repeatedly losing and regaining weight), fasting, and very-low-calorie diets increase the likelihood of cholelithiasis. Along with weight reduction, there is some evidence that physical activity will reduce the risk of cholecystitis. In cholecystitis, MNT includes a high-fiber, low-fat, plant-based diet to prevent gallbladder contractions. Data are conflicting as to whether intravenous lipids stimulate gallbladder contraction.
After surgical removal of the gallbladder, oral feedings can be advanced to a regular diet as tolerated. In the absence of the gallbladder, bile is secreted directly by the liver into the intestine. The biliary tract dilates, forming a “simulated pouch” over time, to allow bile to be held in a manner similar to the original gallbladder.
Cholecystitis
Inflammation of the gallbladder is known as cholecystitis, and it may be chronic or acute. It is usually caused by gallstones obstructing the bile ducts (calculous cholecystitis), leading to the backup of bile. Bilirubin, the main bile pigment, gives bile its greenish color. When biliary tract obstruction prevents bile from reaching the intestine, it backs up and returns to the circulation. Bilirubin has an affinity for elastic tissues; therefore when it overflows into the general circulation, it causes the yellow skin pigmentation and eye discoloration typical of jaundice.
Acute cholecystitis without stones (acalculous cholecystitis) may occur in critically ill patients or when the gallbladder and its bile are stagnant. Impaired gallbladder emptying in chronic acalculous cholecystitis appears to be due to diminished spontaneous contractile activity and decreased contractile responsiveness to CCK. The walls of the gallbladder become inflamed and distended, and infection can occur. During such episodes, the patient experiences upper-quadrant abdominal pain accompanied by nausea, vomiting, and flatulence.
Chronic cholecystitis is long-standing inflammation of the gallbladder. It is caused by repeated, mild attacks of acute cholecystitis. This leads to thickening of the walls of the gallbladder. The gallbladder begins to shrink and eventually loses the ability to perform its function: concentrating and storing bile. Eating foods that are high in fat may aggravate the symptoms of cholecystitis, because bile is needed to digest such foods. Chronic cholecystitis occurs more often in women than in men, and the incidence increases after the age of 40. Risk factors include the presence of gallstones and a history of acute cholecystitis.
Surgical Management
Acute cholecystitis requires surgical intervention unless medically contraindicated. Without surgery, the condition may either subside or progress to gangrene.
Medical Nutrition Therapy
Acute Cholecystitis: In an acute attack, oral feedings are discontinued. PN may be indicated if the patient is malnourished and it is anticipated that he or she will not be taking anything orally for a prolonged period. When feedings are resumed, a low-fat diet is recommended to decrease gallbladder stimulation. A hydrolyzed low-fat formula or an oral low-fat diet consisting of 30 to 45 g of fat per day can be given. Table 30-8 shows a fat-restricted diet.
TABLE 30-8
Fat-Restricted Diet*
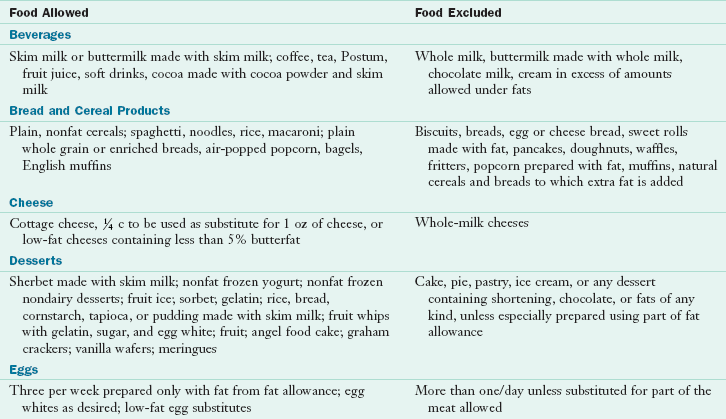

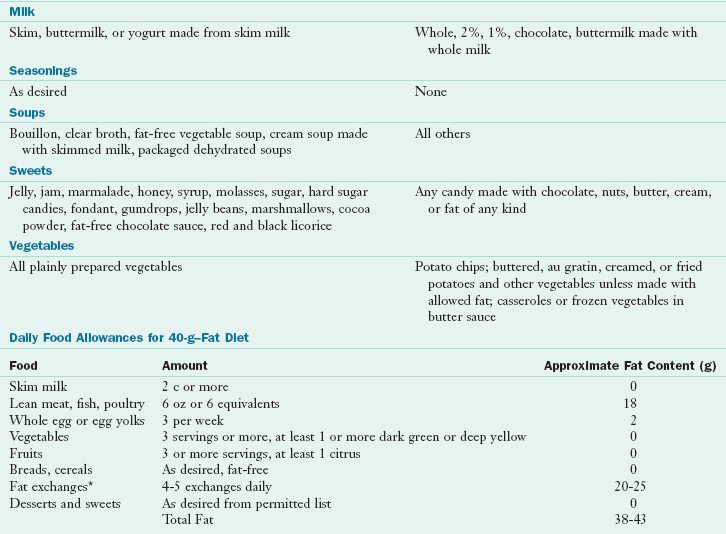
*Fat content can be reduced further by reducing the fat exchanges. 1 Fat exchange = 5 g of fat.
Following cholecystectomy, patients may experience symptoms of gastritis secondary to duodenogastric reflux of bile acids. The reflux may also be responsible for symptoms in this postcholecystectomy syndrome. At present, there are no well-established pharmacologic approaches in the management of postcholecystectomy gastritis. The symptoms are not caused, but exacerbated, by the cholecystectomy. It has been proposed that the addition of soluble fiber to the diet will act as a sequestering agent and bind the bile in the stomach between meals to avoid gastritis.
Chronic Cholecystitis: Patients with chronic conditions may require a long-term, low-fat diet that contains 25% to 30% of total kilocalories as fat. Stricter limitation is undesirable because fat in the intestine is important for some stimulation and drainage of the biliary tract. The degree of food intolerance varies widely among persons with gallbladder disorders; many complain of foods that cause flatulence and bloating. For this reason it is best to determine with the patient which foods should be eliminated. See Chapter 29 for a discussion of potential gas-forming foods. Administration of water-soluble forms of fat-soluble vitamins may be of benefit in patients with chronic gallbladder conditions or in those in whom fat malabsorption is suspected.
Cholangitis
Pathophysiology and Medical Management
Inflammation of the bile ducts is known as cholangitis. Patients with acute cholangitis need resuscitation with fluids and broad-spectrum antibiotics. If the patient does not improve with conservative treatment, placement of a percutaneous biliary stent or cholecystectomy may be needed.
Sclerosing cholangitis can result in sepsis and liver failure. Most patients have multiple intrahepatic strictures, which makes surgical intervention difficult, if not impossible. Patients are generally on broad-spectrum antibiotics. Percutaneous ductal dilation may provide short-term bile duct patency in some patients. When sepsis is recurrent, patients may require chronic antibiotic therapy. See the section on sclerosing cholangitis in the liver disease section.
Physiology and Functions of the Exocrine Pancreas
The pancreas is an elongated, flattened gland that lies in the upper abdomen behind the stomach. The head of the pancreas is in the right upper quadrant below the liver within the curvature of the duodenum, and the tapering tail slants upward to the hilum of the spleen. This glandular organ has both an endocrine and exocrine function. Pancreatic cells manufacture glucagon, insulin, and somatostatin for absorption into the bloodstream (endocrine function) for regulation of glucose homeostasis (see Chapter 31). Other cells secrete enzymes and other substances directly into the intestinal lumen, where they aid in digesting proteins, fats, and carbohydrates (exocrine function).
In most people the pancreatic duct, which carries the exocrine pancreatic secretions, merges with the common bile duct into a unified opening through which bile and pancreatic juices drain into the duodenum at the ampulla of Vater. Many factors regulate exocrine secretion from the pancreas. Neural and hormonal responses play a role, with the presence and composition of ingested foods being a large contributor. The two primary hormonal stimuli for pancreatic secretion are secretin and CCK (see Chapter 1).
Factors that influence pancreatic secretions during a meal can be divided into three phases: (1) the cephalic phase, mediated through the vagus nerve and initiated by the sight, smell, taste, and anticipation of food that leads to the secretion of bicarbonate and pancreatic enzymes; (2) gastric distention with food initiates the gastric phase of pancreatic secretion, which stimulates enzyme secretion; and (3) the intestinal phase, mediated by the release of CCK, with the most potent effect.
Diseases of the Exocrine Pancreas
Pathophysiology and Medical Management
Pancreatitis is an inflammation of the pancreas and is characterized by edema, cellular exudate, and fat necrosis. The disease can range from mild and self-limiting to severe, with autodigestion, necrosis, and hemorrhage of pancreatic tissue. Ranson and colleagues (1974) identified 11 signs that could be measured during the first 48 hours of admission and that have prognostic significance (Box 30-3). By using these observations, one can determine the likely outcome of hospitalization. Surgical intervention may be necessary. Pancreatitis is classified as either acute or chronic, the latter with pancreatic destruction so extensive that exocrine and endocrine function are severely diminished, and maldigestion and diabetes may result.
The symptoms of pancreatitis can range from continuous or intermittent pain of varying intensity to severe upper abdominal pain, which may radiate to the back. Symptoms may worsen with the ingestion of food. Clinical presentation may also include nausea, vomiting, abdominal distention, and steatorrhea. Severe cases are complicated by hypotension, oliguria, and dyspnea. There is extensive destruction of pancreatic tissue with subsequent fibrosis, enzyme production is diminished, and serum amylase and lipase may appear normal. However, absence of enzymes to aid in the digestion of food leads to steatorrhea and malabsorption. Table 30-9 describes several tests used to determine the extent of pancreatic destruction.
TABLE 30-9
Some Tests of Pancreatic Function
| Test | Significance |
| Secretin stimulation test | Measures pancreatic secretion, particularly bicarbonate, in response to secretin stimulation |
| Glucose tolerance test | Assesses endocrine function of the pancreas by measuring insulin response to a glucose load |
| 72-hr stool fat test | Assesses exocrine function of the pancreas by measuring fat absorption that reflects pancreatic lipase secretion |
Medical Nutrition Therapy
Alcohol use, smoking, body weight, diet, genetic factors, and medications all affect the risk of developing pancreatitis. Thus diet modification has an important role after diagnosis. Dietary recommendations differ, depending on whether the condition is acute or chronic. Obesity appears to be a risk factor for the development of pancreatitis and an increased severity.
Depressed serum calcium levels are common. Hypoalbuminemia occurs, with subsequent third spacing of fluid. The calcium, which is bound to albumin, is thus affected and may appear artificially low. Another occurrence is “soap” formation in the gut by calcium and fatty acids, created by the fat necrosis that results in less calcium absorption. Checking an ionized calcium level is a method of determining available calcium.
Acute Pancreatitis: Pain associated with acute pancreatitis (AP) is partially related to the secretory mechanisms of pancreatic enzymes and bile. Therefore nutrition therapy is adjusted to provide minimum stimulation of these systems (see Pathophysiology and Care Management Algorithm: Pancreatitis). In the past the pancreas was put “at rest.” During acute attacks all oral feeding is withheld, and hydration maintained intravenously. In less severe attacks, a clear liquid diet with negligible fat may be given in a few days. The patient should be monitored for any symptoms of pain, nausea, or vomiting. The diet should be progressed as tolerated to easily digested foods with a low fat content and then advanced as tolerated. Foods may be better tolerated if they are divided into six small meals (see Table 30-8).
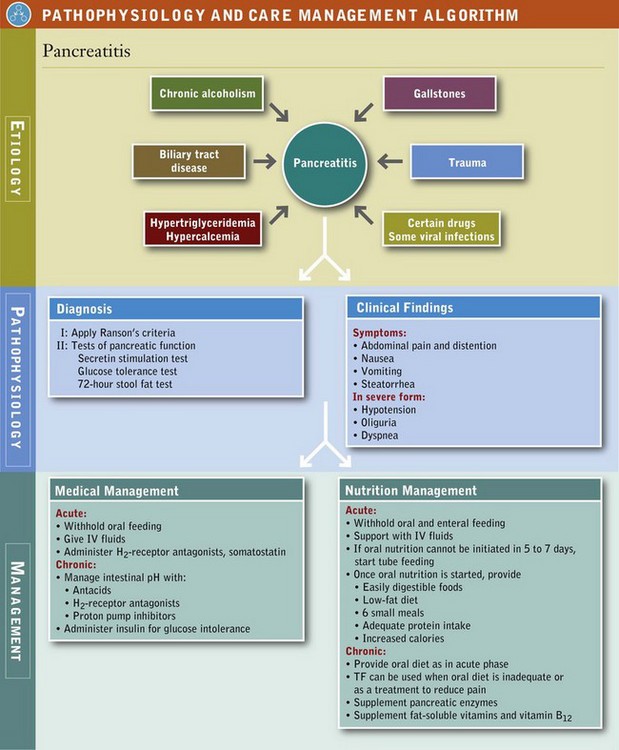
Severe AP results in a hypermetabolic, catabolic state with immediate metabolic alterations in the pancreas and also in remote organs. Metabolic demands are similar to those of sepsis. Amino acids are released from muscle and used for gluconeogenesis. These patients often exhibit signs of malnutrition such as decreased serum levels of albumin, transferrin, and lymphocytes. Attention should be given to a nutrition regimen with adequate protein in an effort to achieve positive nitrogen balance.
Oral nutrition must be further delayed when the acute illness persists longer than a few days as evidenced by persistent or recurrent elevation of serum amylase, continued abdominal pain, and ileus; or when cessation of nasogastric suction is followed by return of symptoms, the presence of a complication such as pancreatic abscess or pseudocyst, or a suspected obstruction to the main pancreatic ducts.
The optimal route of nutrition in AP has been the subject of much controversy over the years. Failure to use the GIT in patients with AP may exacerbate the stress response and disease severity, leading to more complications and prolonged hospitalization; thus EN is preferred for nutrition therapy (McClave et al., 2006; Louie et al., 2005). There is a substantial cost savings with EN and fewer septic complications. Most patients with AP have a return of bowel function within 2 to 3 days following an attack and can advance quickly from intravenous fluids to diet.
Both PN and EN are equally effective in terms of days to normalization of serum amylase, days to resumption of oral feeding, serum albumin levels, nosocomial infections, and clinical outcome in patients with mild to moderate pancreatitis (Petrov, 2009). The favorable effect of either EN or PN on patient outcome may be enhanced by supplementation with modulators of inflammation and systemic immunity (McClave et al., 2006).
Aggressive nutrition support may include attempts to use the GIT. The location of the feeding and the composition of the formula determine the degree of pancreatic stimulation. Infusion into the jejunum eliminates the cephalic and gastric phases of exocrine pancreatic stimulation (McClave et al., 2006; Stanga et al., 2005). Although various formulations have been used in pancreatitis, no studies have determined the relative merits of standard, partially digested, elemental, or “immune-enhanced” formulations. Polymeric formulas infused at various sections of the gut stimulate the pancreas more than elemental and hydrolyzed formulas. Close observation for patient tolerance is important. When the patient is allowed to eat, supplemental pancreatic enzymes may be needed to treat steatorrhea. See Chapter 14) for jejunal feeding details.
In severe, prolonged cases, PN may be necessary. Patients with mild to moderate stress can tolerate dextrose-based solutions, whereas patients with more severe stress require a mixed fuel system of dextrose and lipid to avoid complications of glucose intolerance. Lipid emulsion should not be included in a PN regimen if hypertriglyceridemia is the cause of the pancreatitis. A serum triglyceride level should be obtained before PN with lipids is initiated. Lipids may be given to patients with triglyceride values less than 400 mg/dL. Because of the possibility of pancreatic endocrine abnormalities and a relative insulin resistance, close glucose monitoring is also warranted. H2-receptor antagonists may be prescribed to decrease hydrochloric acid production, which will reduce stimulation of the pancreas. Somatostatin is considered the best inhibitor of pancreatic secretion and may be added to the PN solution.
Chronic Pancreatitis: In contrast to AP, chronic pancreatitis (CP) evolves insidiously over many years. CP is characterized by recurrent attacks of epigastric pain of long duration that may radiate into the back. The pain can be precipitated by meals. Associated nausea, vomiting, or diarrhea makes it difficult to maintain adequate nutrition status. Patients with CP are at increased risk of developing protein-calorie malnutrition because of pancreatic insufficiency and inadequate oral intake. Patients with CP admitted to a tertiary care center usually have malnutrition, increased energy requirements, weight loss, deficits of lean muscle and adipose tissue, visceral protein depletion, and impaired immune function.
The objective of therapy for patients is to prevent further damage to the pancreas, decrease the number of attacks of acute inflammation, alleviate pain, decrease steatorrhea, and correct malnutrition. Dietary intake should be as liberal as possible, but modifications may be necessary to minimize symptoms.
The first goal of MNT is to provide optimal nutrition support, and the second is to decrease pain by minimizing stimulation of the exocrine pancreas. Because CCK stimulates secretion from the exocrine pancreas, one approach is to decrease CCK levels. If postprandial pain is a limiting factor, alternative enteral therapies that minimally stimulate the pancreas are warranted. Nutrition counseling, antioxidants, and pancreatic enzymes may play a role in effective management of CP as well.
Idiopathic CP is often associated with a cystic fibrosis gene mutation, and therapies directed toward cystic fibrosis may benefit these patients. When pancreatic function is diminished by approximately 90%, enzyme production and secretion are insufficient; maldigestion and malabsorption of protein and fat thus become a problem. Large meals with high-fat foods and alcohol should be avoided.
The patient may present with weight loss despite adequate energy intake and will complain of bulky, greasy stools. Pancreatic enzyme replacement is mandatory at this time. Pancreatic enzyme replacements are given orally with meals; the dosage should be at least 30,000 units of lipase with each meal. To promote weight gain, the level of fat in the diet should be the maximum a patient can tolerate without increased steatorrhea or pain. Additional therapies that may be tried to maintain nutrition status and minimize symptoms in patients with maximum enzyme supplementation include a lower-fat diet (40 to 60 g/day) or substitution of some dietary fat with MCT oil to improve fat absorption and weight gain. Meals should be small and frequent.
The diet should be low fat, primarily from vegetable-based oils such as olive oil. Reduce significantly or eliminate trans-fatty acids, found in commercially baked goods. Substitution of dietary fat with MCT oil may relieve steatorrhea and lead to weight gain. Malabsorption of the fat-soluble vitamins may occur in patients with significant steatorrhea. Also, deficiency of pancreatic protease, necessary to cleave vitamin B12 from its carrier protein, could potentially lead to vitamin B12 deficiency. With appropriate supplemental enzyme therapy, vitamin absorption should be improved; however, the patient should still be monitored periodically for vitamin deficiencies. Water-soluble forms of the fat-soluble vitamins or parenteral administration of vitamin B12 may be necessary (see Chapter 33). There is some evidence that increasing intake of antioxidants (found in fruits and vegetables) may help protect against pancreatitis or alleviate symptoms of the condition.
Because pancreatic bicarbonate secretion is frequently defective, medical management may also include maintenance of an optimal intestinal pH to facilitate enzyme activation. Antacids, H2-receptor antagonists, or proton pump inhibitors that reduce gastric acid secretion may be used to achieve this effect.
In chronic cases with extensive pancreatic destruction, the insulin-secreting capacity of the pancreas decreases, and glucose intolerance develops. Treatment with insulin and nutrition therapy is then required (see Chapter 31). Management is delicate and should focus on control of symptoms rather than normoglycemia.
Effort should be made to cater to the patient’s tolerances and preferences for nutritional management; however, alcohol is prohibited because of the possibility of exacerbating the pancreatic disease. There is evidence that the progressive destruction of the pancreas will be slowed in the alcoholic patient who abstains from alcohol.
Pancreatic Surgery
A surgical procedure often used for pancreatic carcinoma is a pancreaticoduodenectomy (Whipple procedure). A cholecystectomy, vagotomy, or a partial gastrectomy may also be done during the surgery. The pancreatic duct is reanastomosed to the jejunum. Partial or complete pancreatic insufficiency can result, depending on the extent of the pancreatic resection. Most patients who have undergone pancreatic resection are at risk for vitamin and mineral deficiencies and will benefit from vitamin and mineral supplementation. Nutrition care is similar to that for CP.
 Clinical Scenario
Clinical Scenario
A 40-year-old man is admitted to the hospital with chief complaints of right upper quadrant pain, anorexia, nausea, dysgeusia, and frequent loose stools. On physical examination, he has mild peripheral edema with a slightly jaundiced appearance. No asterixis is noted. The patient’s mental status is clear, but he appears lethargic. He reports no history of portal hypertension, ascites, or gastrointestinal bleeding. Muscle wasting is noted along with stomatitis. A liver biopsy shows steatosis and fibrosis. The patient has a significant alcohol abuse history spanning 15 years, suggesting alcoholic hepatitis. Abnormal laboratory values include elevated liver enzymes and total bilirubin; serum albumin, 2.5 g/dL; transferrin, 150 mg/dL; megaloblastic anemia profile; ammonia, 75 mmol/L. Nutritional data include height, 177.8 cm; weight, 67 kg; ideal body weight, 75 kg ± 10%; usual body weight, 82 kg (5 years ago), 73 kg (6 months ago).
References
Abayli, B, et al. Helicobacter pylori in the etiology of cholesterol gallstones. J Clin Gastroenterol. 2005;39:134.
Adler, DG, et al. Standards of Practice Committee of American Society for Gastrointestinal Endoscopy: ASGE guideline: the role of ERCP in diseases of the biliary tract and the pancreas. Gastrointest Endosc. 2005;62:1.
Afdhal, NH. Diseases of the gallbladder and bile ducts. In Goldman L, et al, eds.: Cecil textbook of medicine, ed 23, Philadelphia: Saunders, 2007.
Böhm, F, et al. FGF receptors 1 and 2 control chemically-induced injury and compound detoxification in regenerating livers of mice. Gastroenterology. 2010;139:1385.
Campillo, B, et al. Enteral nutrition in severely malnourished and anorectic cirrhotic patients in clinical practice. Gastroenterol Clin Biol. 2005;29:645.
Chitturi, S, Farrell, GC. Hepatotoxic slimming aids and other herbal hepatotoxins. J Gastroenterol Hepatol. 2008;23:366.
Crippin, JS. Is tube feeding an option in patients with liver disease? Nutr Clin Pract. 2006;21:296.
Diehl, AM. Alcoholic and nonalcoholic steatohepatitis. In Goldman L, et al, eds.: Cecil textbook of medicine, ed 23, Philadelphia: Saunders, 2007.
Fitz, JG. Hepatic encephalopathy, hepatopulmonary syndromes, hepatorenal syndrome, and other complications of liver disease. In Feldman M, ed.: Sleisenger and Fordtran’s gastrointestinal and liver disease, ed 8, Philadelphia: Saunders, 2006.
Garcia-Tsao, G. Cirrhosis and its sequelae. In Goldman L, et al, eds.: Cecil textbook of medicine, ed 23, Philadelphia: Saunders, 2007.
Gratz, SW, et al. Probiotics and gut health: a special focus on liver diseases. World J Gastroenterol. 2010;16:403.
Hoofnagle, JH. Hepatitis. In Goldman L, et al, eds.: Cecil textbook of medicine, ed 23, Philadelphia: Saunders, 2007.
Ioannou, GN. Cholelithiasis, cholecystectomy, and liver disease. Am J Gastroenterol. 2010;105:1364.
Kosters, A, Karpen, SJ. The role of inflammation in cholestasis: clinical and basic aspects. Semin Liver Dis. 2010;30:186.
Kowdley, KV. Inherited and metabolic hepatic disorders. In Goldman L, et al, eds.: Cecil textbook of medicine, ed 23, Philadelphia: Saunders, 2007.
Lam, P, et al. The bile salt export pump: clinical and experimental aspects of genetic and acquired cholestatic liver disease. Semin Liver Dis. 2010;30:125.
Leevy, CM, Moroianu, SA. Nutritional aspects of alcoholic liver disease. Clin Liver Dis. 2005;9:67.
Louie, BE, et al. 2004 MacLean-Mueller prize enteral or parenteral nutrition for severe pancreatitis: a randomized controlled trial and health technology assessment. Can J Surg. 2005;48:298.
McClave, S, et al. Nutrition support in acute pancreatitis: a systematic review of the literature. JPEN J Parenter Enteral Nutr. 2006;30:143.
Medici, V, et al. Diagnosis and management of Wilson’s disease: results of a single center experience. J Clin Gastroenterol. 2006;40:936.
National Institute on Alcohol Abuse and Alcoholism. Age-specific and age-adjusted death rates for cirrhosis with and without mention of alcohol, United States, 1970-2005. October 2008. Available at http://www.niaaa.nih.gov/Resources/DatabaseResources/QuickFacts/Liver/Pages/cirmrt3b.aspx. [Accessed February 21, 2011].
Pereg, D, et al. Probiotics for patients with compensated liver cirrhosis: a double-blind placebo-controlled study. Nutrition. 6 May 2010. [[Epub ahead of print.]].
Petrov, MS, et al. Systemic review: nutrition support in acute pancreatitis. Alimen Pharmacol Ther. 2009;28:704.
Plauth, M, et al. ESPEN Guidelines on enteral nutrition: liver disease. Clin Nutr. 2006;25:285.
Rambaldi A, Gluud C: S-adenosyl-L-methionine for alcoholic liver diseases, Cochrane Database Syst Rev 2006, Issue 2. Art. No.: CD002235.
Rambaldi A, et al: Milk thistle for alcoholic and/or hepatitis B or C virus liver diseases, Cochrane Database Syst Rev 2007, Issue 4. Art. No.: CD003620.
Ranson, JH, et al. Prognostic signs and the role of operative management in acute pancreatitis. Surg Gynecol Obstet. 1974;139:69.
Rayes, N, et al. Supply of pre- and probiotics reduces bacterial infection rates after liver transplantation—a randomized, double-blind trial. Am J Transplant. 2005;5:125.
Stanga, Z, et al. Effect of jejunal long-term feeding in chronic pancreatitis. JPEN J Parenter Enteral Nutr. 2005;29:12.
Tsai, CJ, et al. Dietary carbohydrates and glycaemic load and the incidence of symptomatic gall stone disease in men. Gut. 2005;54:823.
Volzke, H, et al. Independent risk factors for gallstone formation in a region with high cholelithiasis prevalence. Digestion. 2005;71:97.
Weikert, C, et al. Presence of gallstones or kidney stones and risk of type 2 diabetes. Am J Epidemiol. 2010;171:447.
Willner, IR, Reuben, A. Alcohol and the liver. Curr Opin Gastroenterol. 2005;21:323.