Clinical
Food-Drug Interactions
The management of many diseases requires drug therapy, frequently involving the use of multiple drugs. Food-drug interactions can change the effects of drugs, and the therapeutic effects or side effects of medications can affect the nutrition status of an individual. Alternatively, the diet and use of supplements, genetic makeup, or the nutritional status of the patient can decrease the efficacy of a drug or increase its toxicity.
The terms drug-nutrient interaction and food-drug interaction are often used interchangeably. In actuality, drug-nutrient interactions are some of the many possible food-drug interactions. Drug-nutrient interactions include specific changes to the pharmacokinetics of a drug caused by a nutrient or nutrients or changes to the kinetics of a nutrient caused by a drug. Food-drug interaction is a broader term that also includes the effects of a medication on nutritional status. Nutritional status may be affected by the side effects of a medication, which could include an effect on appetite or the ability to eat.
For clinical, economic, and legal reasons, it is important to recognize food-drug interactions. Food-drug interactions that reduce the efficacy of a drug can result in longer or repeated stays in health care facilities, the use of multiple drugs, and deterioration of the patient because of the effects of the disease. Additional health problems can occur because of long-term drug-nutrient interactions. An example of this type of interaction is the long-term effects of corticosteroids on calcium metabolism and the resulting osteoporosis. Medical team members should be aware that therapeutically important food-drug interactions can do the following:
Awareness of these interactions enables the health care professional and patient to work together to avoid or minimize problems (Box 9-1).
Pharmacologic Aspects Of Food-Drug Interactions
Medication is administered to produce a pharmacologic effect in the body or, more specifically, in a target organ or tissue. To achieve this goal, the drug must move from the site of administration into the bloodstream and eventually to the site of drug action. In due course the drug may be changed to active or inactive metabolites and is ultimately eliminated from the body. An interaction between the drug and food, a food component, or a nutrient can alter this process at any point. Food-drug interactions may be divided into two broad types: (1) pharmacodynamic interactions, which affect the pharmacologic action of the drug; and (2) pharmacokinetic interactions, which affect the movement of the drug into, around, or out of the body.
Pharmacodynamics
Pharmacodynamics is the study of the biochemical and physiologic effects of a drug. The mechanism of action of a drug might include the binding of the drug molecule to a receptor, enzyme, or ion channel, resulting in the observable physiologic response. Ultimately this response may be enhanced or attenuated by the addition of other substances with similar or opposing actions. Pharmacokinetics is the study of the time course of a drug in the body involving the absorption, distribution, metabolism (biotransformation), and excretion of the drug. Absorption is the process of the movement of the drug from the site of administration to the bloodstream. This process depends on (1) the route of administration, (2) the chemistry of the drug and its ability to cross biologic membranes, (3) the rate of gastric emptying (for orally administered drugs) and gastrointestinal (GI) movement, and (4) the quality of the product formulation. Food, food components, and nutrition supplements can interfere with the absorption process, especially when the drug is administered orally.
Distribution occurs when the drug leaves the systemic circulation and travels to various regions of the body. Body areas of distribution vary with different drugs, depending on the drug’s chemistry and ability to cross biologic membranes. The rate and extent of blood flow to an organ or tissue strongly affect the amount of drug that reaches the area. Many drugs are highly bound to plasma proteins such as albumin. The bound fraction of drug does not leave the vasculature and therefore does not produce a pharmacologic effect. Only the unbound fraction is able to produce an effect at a target organ.
A drug is eliminated from the body as either an unchanged drug or a metabolite of the original compound. The major organ of metabolism, or biotransformation, in the body is the liver, although other sites, such as the intestinal membrane, contribute to variable degrees. One of the more important enzyme systems that facilitate drug metabolism is the cytochrome P-450 enzyme system. This is a multienzyme system in the smooth endoplasmic reticulum of numerous tissues that is involved in phase I of liver detoxification (see Chapter 20). Food or dietary supplements may either increase or inhibit the activity of this enzyme system, which can significantly change the rate or extent of drug metabolism. The general tendency of the process of metabolism is to transform a drug from a lipid-soluble to a more water-soluble compound that can be handled more easily by the kidneys and excreted in the urine.
Renal excretion is the major route of elimination for drugs and drug metabolites either by glomerular filtration or tubular secretion. To a lesser extent drugs may be eliminated in feces, bile, and other body fluids. Under certain circumstances, such as a change in urinary pH, drugs that have reached the renal tubule may pass back into the bloodstream. This process is known as tubular resorption. The recommended dose of a drug generally assumes normal liver and kidney function. The dose and dosing interval of an excreted drug or active metabolite must be adjusted to meet the degree of renal dysfunction in patients with kidney disease (see Chapter 36).
Risk Factors For Food-Drug Interactions
Patients must be assessed individually for the effect of food on drug action and the effect of drugs on nutrition status. Interactions can be caused or complicated by polypharmacy, nutrition status, genetics, underlying illness, special diets, nutrition supplements, tube feeding, herbal or phytonutrient products, alcohol intake, drugs of abuse, nonnutrients in food, excipients in drugs or food, allergies, or intolerances. Poor patient compliance and physicians’ prescribing patterns further complicate the risk. Drug-induced malnutrition occurs most commonly during long-term treatment for chronic disease, and older patients are at a particularly high risk for many reasons (see Focus On: Polypharmacy in Older Adults).
 Focus on
Focus on
Polypharmacy in Older Adults
Older patients are more likely to take multiple drugs, both prescription and over-the-counter, than are younger patients. They have a higher risk of food-drug interactions because of physical changes related to aging, such as the increase in the ratio of fat tissue to lean body mass, a decrease in liver mass and blood flow, and impairment of kidney function. Illness, cognitive or endocrine dysfunction, and ingestion of restricted diets also increase this risk. Malnutrition and dehydration affect drug kinetics. The use of herbal or phytonutrient products has increased significantly in all developed countries, including use by older adults. Drugs of abuse or excessive alcohol intake are often missed in the older patient.
Central nervous system side effects of drugs can interfere with the ability or desire to eat. Drugs that cause drowsiness, dizziness, ataxia, confusion, headache, weakness, tremor, or peripheral neuropathy can lead to nutritional compromise, particularly in older patients. Recognition of these problems as a drug side effect rather than a consequence of disease or aging can be overlooked. An old list known as the Beers criteria lists some medications that can cause cardiac, gastrointestinal or urinary effects, although the usefulness of the Beers criteria is now controversial (Steinman et al., 2009).
Care must be taken to evaluate intake of interacting nutrients (in the oral diet, supplements, or tube feedings) when specific drugs are used. Examples are vitamin K with warfarin (Coumadin); calcium and vitamin D with tetracycline; and potassium, sodium, and magnesium with loop diuretics such as furosemide (Lasix). Patients with Parkinson disease may be concerned with the amount and timing of protein intake because of interaction with levodopa (Sinemet, Dopar). The interdisciplinary team, which includes the physician, pharmacist, nurse, and dietitian, must work together to plan and coordinate the medication regimen and diet and nutritional supplements to preserve optimal nutrition status and minimize food-drug interactions (Figure 9-1).

Existing malnutrition places patients at greater risk for drug-nutrient interactions. Protein alterations—specifically low albumin levels—and changes in body composition secondary to malnutrition can affect drug disposition by altering protein binding and drug distribution. Patients with active cancer or human immunodeficiency virus (HIV) infection who have significant anorexia and wasting are at special risk because of the high prevalence of malnutrition and reduced intakes. Treatment modalities such as chemotherapy and radiation may also exacerbate nutritional disturbances. For example, cisplatin (Platinol-AQ) and other cytotoxic agents commonly cause mouth sores, nausea, vomiting, diarrhea, anorexia, and reduced food intake.
Drug disposition can be affected by alterations in the GI tract, such as vomiting, diarrhea, hypochlorhydria, mucosal atrophy, and motility changes. Malabsorption caused by intestinal damage from disease such as cancer, celiac disease, or inflammatory bowel disease creates greater potential for food-drug interactions. Body composition is another important consideration in determining drug response. In obese or older patients, the proportion of adipose tissue to lean body mass is increased. In theory, accumulation of fat-soluble drugs such as the long-acting benzodiazepines (e.g., diazepam [Valium]) is more likely to occur. Accumulation of a drug and its metabolites in adipose tissue may result in prolonged clearance and increased toxicity (Spriet et al., 2009). In older patients this interaction may be complicated by decreased hepatic clearance of the drug.
The developing fetus, infant, and pregnant woman are also at high risk for drug-nutrient interactions. Many drugs have not been tested on these populations, making it difficult to assess the risks of negative drug effects, including food-drug interactions.
Pharmacogenomics
Gene-nutrient interactions reflect the genetic heterogeneity among humans, environmental factors and dietary chemicals, and diverse physiologies (Wise and Kaput, 2009). Because the efficacy and safety disparity of drugs varies according to race and genetic variants, pharmacogenetic knowledge is important for the interpretation and prediction of drug interaction-induced adverse events (Bai, 2010). Pharmacogenomics involves genetically determined variations that are revealed solely by the effects of drugs and can be a driver for nutrigenomics, as discussed in Chapter 5 (Ghosh et al., 2007). Food-drug interaction ramifications are seen in glucose-6-phosphate dehydrogenase (G6PD) enzyme deficiency, slow inactivation of isoniazid (INH) or phenelzine (Nardil), and warfarin (Coumadin) resistance. Warfarin resistance affects individual requirements for and response to warfarin.
Slow inactivation of INH used in tuberculosis (TB) represents the effect of slow acetylation, a conjugation reaction that metabolizes and inactivates amines, hydrazines, and sulfonamides. “Slow acetylators” are persons who metabolize these drugs more slowly than average because of inherited lower levels of the hepatic enzyme acetyl transferase. Therefore unacetylated drug levels remain higher for longer periods in these persons than in those who are “rapid acetylators.” For example, the half-life of INH for fast acetylators is approximately 70 minutes, whereas the half-life is more than 3 hours for slow acetylators. A dose of drug prescribed normally for fast acetylators can be toxic for slow acetylators. Elevated blood levels of affected drugs in slow acetylators increase the potential for food-drug interactions. Slow inactivation of INH increases the risk of pyridoxine deficiency and peripheral neuropathy. Slow inactivation of phenelzine, a monoamine oxidase (MAO) inhibitor, increases the risk for hypertensive crisis if foods high in tyramine are consumed. Dapsone (DDS) and hydralazine (Apresoline) are also metabolized by acetylation and are affected by inherited differences in acetylase enzymes.
Deficiency of G6PD is an X-chromosome–linked deficiency of G6PD enzyme in red blood cells that can lead to neonatal jaundice, hemolytic anemia, or acute hemolysis. Most common in African, Middle Eastern, and Southeast Asian populations, it is also called favism. Intake of fava beans, aspirin, sulfonamides, and antimalarial drugs can cause hemolysis and acute anemia in G6PD-deficient persons. The potential exists for food-drug interactions in G6PD deficiency resulting from the ingestion of fava beans (broad beans), as well as vitamin C or vitamin K.
Another factor that affects drug metabolism is genetically different activity of cytochrome P450 (CYP) enzymes. Therapeutic proteins affect the disposition of drugs that are metabolized by these enzymes (Lee et al., 2010). “Slow metabolizers” may have less of a specific enzyme or their enzymes may be less active. Such individuals have a higher risk of adverse drug effects. Slow CYP2D6 metabolizers make up approximately 5% to 10% of whites, whereas approximately 20% of Asians are CYP2C19 poor metabolizers. Tests are now available to analyze deoxyribonucleic acid (DNA) to determine variations in the activity of these two enzymes. CYP2D6 and CYP2C19 metabolize approximately 25% of all drugs, including many antipsychotics, antidepressants, and narcotics. Slow metabolizers achieve a higher blood level with usual doses of such drugs, whereas fast metabolizers may have an unpredictable response as a result of rapid metabolism of the drug (Medical Letter, 2005).
Drug response genotyping helps to determine which drugs will be effective, depending on an individual’s genetic makeup (see Chapter 5). The ability to predict response to specific drugs determines more effective treatments for cancer, mental illness, and even pain management. Genotyping will help reduce adverse drug reactions, including food-medication interactions.
Effects Of Food On Drug Therapy
The presence of food and nutrients in the stomach or intestinal lumen may reduce the absorption of a drug. Bioavailability describes the fraction of an administered drug that reaches the systemic circulation. If a medication is administered intravenously, its bioavailability is 100%, but bioavailability decreases because absorption and metabolism are incomplete when taken orally. Examples of a critically significant reduction in drug absorption are the antiosteoporosis drugs alendronate (Fosamax), risedronate (Actonel), or ibandronate (Boniva). Absorption is negligible if these drugs are given with food and reduced by 60% if taken with coffee or orange juice. The manufacturer’s instructions for alendronate or risedronate are to take the drug on an empty stomach with plain water at least 30 minutes before any other food, drink, or medication. Ibandronate must be taken at least 60 minutes before any other food, drink, or medication. The absorption of the iron from supplements can be decreased by 50% if taken with food. Iron is best absorbed when taken with 8 oz of water on an empty stomach. If iron must be taken with food to avoid GI distress, it should not be taken with bran, eggs, high-phytate foods, fiber supplements, tea, coffee, dairy products, or calcium supplements, because each of these can decrease iron absorption (see Chapter 3).
Various mechanisms may contribute to the reduction in the rate or extent of drug absorption in the presence of food or nutrients. The presence and type of meal or food ingested influence the rate of gastric emptying. Gastric emptying may be delayed by the consumption of high-fiber meals and meals with high fat content. In general, a delay in drug absorption is not clinically significant as long as the extent of absorption is not affected. However, delayed absorption of antibiotics or analgesics may be clinically significant. Chelation reactions occur between certain medications and divalent or trivalent cations, such as iron, calcium, magnesium, zinc, or aluminum, and the absorption of drugs may be reduced by chelation with one of these metal ions.
The Parkinson disease drug entacapone (Comtan) chelates with iron; therefore the iron must be taken 1 hour before or 2 hours after taking the drug. The antibiotics ciprofloxacin (Cipro) and tetracycline (Achromycin-V or Sumycin) form insoluble complexes with calcium in dairy products or calcium-fortified foods and beverages; calcium, magnesium, zinc, or iron supplements; or aluminum in antacids, thus preventing or reducing the absorption of both drug and nutrient (Neuhofel et al., 2002). The optimal approach to avoid this interaction is to stop noncritical supplements for the duration of the antibiotic prescription. If this is not possible, particularly with magnesium or with long-term antibiotic use, it is advisable to give the drug at least 2 hours before or 6 hours after the mineral.
Adsorption, or the adhesion to food or a food component, is another mechanism by which drug absorption is slowed or reduced. A high-fiber diet may decrease the absorption of tricyclic antidepressants such as amitriptyline (Elavil), leading to loss of therapeutic effect of the antidepressant because of the adsorption of the drug to the fiber. Likewise, the cardiovascular drug digoxin (Lanoxin) should not be taken with high-phytate foods such as wheat bran or oatmeal.
Gastrointestinal pH is another important factor in the absorption of drugs. Any situations resulting in changes in gastric acid pH, such as achlorhydria or hypochlorhydria, may reduce drug absorption. An example of such an interaction is the failure of ketoconazole (Nizoral) to clear a Candida infection in patients with HIV infection or in persons taking potent acid-reducing agents for gastroesophageal reflux disease (GERD). Ketoconazole achieves optimal absorption in an acid medium. Because of the high prevalence of achlorhydria in HIV infected patients, dissolution of ketoconazole tablets in the stomach is reduced, leading to impaired drug absorption. This is also a concern with hypochlorhydria in persons receiving chronic acid suppression therapy, such as antacids, histamine 2 (H2) receptor antagonists (e.g., famotidine [Pepcid]), or proton-pump inhibitors (e.g., omeprazole [Prilosec]). Ingestion of ketoconazole with an acidic liquid such as cola or a dilute hydrochloric acid (HCl) solution may improve bioavailability in these patients.
The presence of food in the stomach enhances the absorption of some medications, such as the antibiotic cefuroxime axetil (Ceftin) or the antiretroviral drug saquinavir (Invirase). These drugs are prescribed to be taken after a meal to reduce the dose that must be taken to reach an effective level. The bioavailability of cefuroxime axetil is substantially greater when taken with food, compared with taking it in the fasting state.
Medication and Enteral Nutrition Interactions
Continuous enteral feeding is an effective method of providing nutrients to patients who are unable to swallow or eat adequately. However, use of the feeding tube to administer medication can be a problem. When liquid medications are mixed with enteral formulas, incompatibilities may occur. Types of physical incompatibility include granulation, gel formation, and separation of the enteral product; these frequently clog feeding tubes and interrupt delivery of nutrition to the patient. Examples of drugs that can cause granulation and gel formation are ciprofloxacin suspension (Cipro), chlorpromazine (Thorazine) concentrate, ferrous sulfate elixir, guaifenesin (Robitussin expectorant), and metoclopramide (Reglan) syrup (Wohlt et al., 2009). Emulsion breakage occurs when acidic pharmaceutical syrups are added to enteral formulas, more commonly in enteral formulas with intact protein and less so with hydrolyzed protein or free amino acids.
Most compatibility studies of medication and enteral products have focused on the effect of the drug on the integrity of the enteral product. More important is the effect of the enteral product on the bioavailability of the drug. This area requires much more research as feeding tube placement becomes a more common practice. Bioavailability problems are common with phenytoin (Dilantin) suspension and tube feeding. Because blood levels of phenytoin are routinely performed to monitor the drug, much information exists about the reduction of phenytoin bioavailability when given with enteral feedings. Stopping the tube feeding before and after the phenytoin dose is generally suggested; a 2-hour feeding-free interval before and after the dose of phenytoin is administered can safely be recommended.
Information may not be readily available concerning a drug and enteral product interactions even though the manufacturer may have unpublished information about their drug’s interaction with enteral products. Checking with the manufacturer’s medical information department may yield more information for the clinician.
Drug Distribution
Albumin is the most important drug-binding protein in the blood. Low serum albumin levels, often the result of inadequate protein intake and poor nutrition, provide fewer binding sites for highly protein-bound drugs. Fewer binding sites mean that a larger free fraction of drug will be present in the serum. Only the free fraction (unbound fraction) of a drug is able to leave the vasculature and exert a pharmacologic effect at the target organ. Patients with albumin levels below 3 g/dL are at increased risk for adverse effects from highly protein-bound drugs. Usual adult doses of highly protein-bound drugs in such persons may produce more pronounced pharmacologic effects than the same dose in persons with normal serum albumin levels. A lower dose of such drugs is often recommended for patients with low albumin levels. In addition, the risk for displacement of one drug from albumin-binding sites by another drug is greater when albumin levels are less than 3 g/dL.
The anticoagulant warfarin, which is 99.9% serum protein–bound, and the anticonvulsant phenytoin, which is greater than 90% protein-bound, are common drugs used in older patients. Low albumin levels tend to be more common in older patients and in critically ill patients. In the case of warfarin, higher levels of free drug lead to risk of excessive anticoagulation and bleeding. Phenytoin toxicity can result from higher serum levels of free phenytoin.
Drug Metabolism
Enzyme systems in the intestinal tract and the liver, although not the only sites of drug metabolism, account for a large portion of the drug metabolizing activity in the body. Food can both inhibit and enhance the metabolism of medication by altering the activity of these enzyme systems. A diet high in protein and low in carbohydrates can increase the hepatic metabolism of the antiasthma drug theophylline (Theo-Dur).
Conversely, a substance found in grapefruit and grapefruit juice can inhibit the intestinal metabolism of drugs such as calcium channel blockers that are dihydropyridine derivatives (felodipine [Plendil]) (Sica, 2006) and some 3-hydroxy-3-methylglutaryl (HMG)–coenzyme A (CoA) reductase inhibitors such as simvastatin (Zocor). Grapefruit inhibits the cytochrome P-450 3A4 enzyme system responsible for the oxidative metabolism of many orally administered drugs. This interaction appears to be clinically significant for drugs with low oral bioavailability, which are substantially metabolized and inactivated in the intestinal tract by the cytochrome P-450 3A4 enzyme in the intestinal wall. When grapefruit or grapefruit juice is ingested, the metabolizing enzyme is irreversibly inhibited, which reduces the normal metabolism of the drug. This reduction in metabolism allows more of the drug to reach the systemic circulation; the increase in blood levels of unmetabolized drug results in a greater pharmacologic effect and possible toxicity. Unfortunately, the effects of grapefruit on intestinal cytochrome P-4503A4 last up to 72 hours, until the body can reproduce the enzyme. Therefore separating the ingestion of the grapefruit and the drug does not appear to alleviate this interaction.
Seville oranges (used in some marmalades but not in commercial orange juice), pomelos, and tangelos may also cause similar reactions (Egashira et al., 2003). Even a small amount of these foods may be dangerous and should be totally avoided with some drugs such as the immunosuppressant tacrolimus (Prograf) or simvastatin (Zocor). These foods may be taken in small amounts with other drugs such as fluvoxamine (Luvox). The interaction is not significant in drugs that are not metabolized by cytochrome P-450 3A4 in the intestinal wall, such as the HMG-CoA reductase inhibitors pravastatin (Pravachol) or fluvastatin (Lescol).
Competition between food and drugs such as propranolol (Inderal) and metoprolol (Lopressor) for metabolizing enzymes in the liver may alter the first-pass metabolism of these medications. Drugs absorbed from the intestinal tract by the portal circulation are first transported to the liver before they reach the systemic circulation. Because many drugs are highly metabolized during this first pass through the liver, only a small percentage of the original dose is actually available to the systemic circulation and the target organ. In some cases, however, this percentage can be increased by concurrent ingestion of food with the drug. When food and drug compete for the same metabolizing enzymes in the liver, more of the drug is likely to reach the systemic circulation, which can lead to a toxic effect if the dose of the drug is titrated to an optimal level in the fasting state.
Drug Excretion
Food and nutrients can alter the resorption of drugs from the renal tubule. Resorption of the antimanic agent lithium (Lithobid or Eskalith) is closely associated with the resorption of sodium. When sodium intake is low or when a patient is dehydrated, the kidneys resorb more sodium. In the person treated with lithium, the kidney resorbs lithium as well as sodium under these conditions. Higher lithium levels and possible toxicity result. When excess sodium is ingested, the kidneys eliminate more sodium in the urine and likewise more lithium. This produces lower lithium levels and possible therapeutic failure.
Drugs that are weak acids or bases are resorbed from the renal tubule into the systemic circulation only in the nonionic state. An acidic drug is largely in the nonionic state in urine with an acidic pH, whereas a basic drug is largely in a nonionic state in urine with an alkaline pH. A change in urinary pH by food may change the amount of drug existing in the nonionic state, thus increasing or decreasing the amount of drug available for tubular resorption. Foods such as milk, most fruits (including citrus fruits), and most vegetables are urinary alkalinizers (see Clinical Insight: Urinary pH—How Does Diet Affect It? in Chapter 36). This change can affect the ionic state of a basic drug such as the antiarrhythmic agent quinidine gluconate (Quinaglute Dura-Tabs). In alkaline urine the drug will be predominately in the nonionic state and available for resorption from the urine into the systemic circulation, which may lead to higher blood quinidine levels. The excretion of memantine (Namenda), a drug used to treat Alzheimer dementia, is also decreased by alkaline pH, thus raising the drug blood levels. Higher drug levels increase the risk of toxicity. This interaction is most likely to be clinically significant when the diet is composed exclusively of a single food or food group. Patients should be cautioned against initiating major diet changes without consulting their physician or dietitian.
Licorice, or glycyrrhizic acid, is an extract of glycyrrhiza root used in “natural” licorice candy. Approximately 100 g of licorice (the amount in two or more twists of natural licorice) can increase cortisol concentration, resulting in pseudohyperaldosteronism with increased sodium resorption, water retention, increased blood pressure, and greater excretion of potassium. The action of diuretics and antihypertensive drugs may be antagonized. The resultant hypokalemia may alter the action of some drugs (Pronsky and Crowe, 2010).
Effects Of Drugs On Food And Nutrition
Many of the interactions discussed in this section are the opposite of those discussed previously under the Effects of Food on Drug Therapy. For instance, the chelation of a mineral with a medication not only decreases the absorption and therefore the action of the drug, but also decreases the absorption and availability of the nutrient.
Nutrient Absorption
Medication can decrease or prevent nutrient absorption. Chelation reactions between medications and minerals (metal ions) reduce the amount of mineral available for absorption. An example is tetracycline (Achromycin-V or Sumycin) and ciprofloxacin, which chelates calcium found in supplements or in dairy products such as milk or yogurt. This is also true for other divalent or trivalent cations such as iron, magnesium, and zinc found in individual mineral supplements or multivitamin-mineral supplements. Standard advice is to take the minerals at least 2 to 6 hours apart from the drug.
Adsorption also can decrease nutrient absorption. Antihyperlipidemic, bile acid sequestrant cholestyramine (Questran) is also used to treat diarrhea. It adsorbs fat-soluble vitamins A, D, E, and K. Vitamin supplementation is recommended with long-term use of this drug, especially when it is taken more than once a day. More than 2 tbsp (30 ml) of mineral oil per day decreases absorption of fat-soluble vitamins A, D, E, and K. It is advised to take the mineral oil in the morning and the vitamins at least 2 hours later, primarily with chronic mineral oil use.
Drugs can reduce nutrient absorption by influencing the transit time of food and nutrients in the gut. Cathartic agents and laxatives reduce transit time and may cause diarrhea, leading to losses of calcium and potassium. Diarrhea may be induced by drugs containing sorbitol, such as syrup or solution forms of furosemide (Lasix), valproic acid (Depakene), carbamazepine (Tegretol), trimethoprim/sulfamethoxazole (Septra), or by drugs that increase peristalsis such as the gastric mucosa protectant misoprostol (Cytotec).
A drug also can prevent nutrient absorption by changing the GI environment. H2-receptor antagonists, such as famotidine (Pepcid) or ranitidine (Zantac), and proton-pump inhibitors, such as omeprazole (Prilosec) or esomeprazole (Nexium), are antisecretory drugs used to treat ulcer disease and GERD. They inhibit gastric acid secretion and raise gastric pH. These effects may impair absorption of vitamin B12 by reducing cleavage from its dietary sources. Cimetidine (Tagamet) is an antagonist that also reduces intrinsic factor secretion; this can be a problem for vitamin B12 absorption and can result in vitamin B12 deficiency with long-term use. Because of the hypothesized effect on calcium absorption, protein pump inhibitors were thought to raise the risk of osteoporosis (Fourniet et al., 2009) but recent information refutes this hypothesis (Targownik et al., 2010).
Drugs with the greatest effect on nutrient absorption are those that damage the intestinal mucosa. Damage to the structure of the villi and microvilli inhibits the brush-border enzymes and intestinal transport systems involved in nutrient absorption. The result is general or varying degrees of specific malabsorption, which can alter the ability of the GI tract to absorb minerals, especially iron and calcium. Damage to the gut mucosa commonly results from chemotherapeutic agents, nonsteroidal antiinflammatory drugs (NSAIDs), and long-term antibiotic therapy. NSAIDs may adversely affect the colon by causing a nonspecific colitis or by exacerbating a preexisting colonic disease (Valley et al., 2006). Patients with NSAID-induced colitis present with bloody diarrhea, weight loss, and iron deficiency anemia; the pathogenesis of this colitis is still controversial.
Drugs that affect intestinal transport mechanisms include (1) colchicine, an antiinflammatory agent used to treat gout; (2) paraaminosalicylic acid, an anti-TB drug; (3) sulfasalazine (Azulfidine), used to treat ulcerative colitis; and (4) trimethoprim (antibiotic in sulfamethoxazole-trimethoprim [Bactrim]) and antiprotozoal agent pyrimethamine (Daraprim). The first two agents impair absorption of vitamin B12; the others are competitive inhibitors of folate transport mechanisms.
Nutrient Metabolism
A drug may increase the metabolism of a nutrient, causing it to pass through the body faster, resulting in higher requirements; or a drug may cause vitamin antagonism by blocking conversion of a vitamin to the active form. Anticonvulsants phenobarbital and phenytoin induce hepatic enzymes and increase the metabolism of vitamins D and K, and folic acid (Crawford, 2005; Nicolaidou et al., 2006). Supplements of these vitamins are often prescribed with these drugs. Carbamazepine (Tegretol) has been reported to affect the metabolism of biotin, vitamin D, and folic acid, leading to possible depletion. Measurement of vitamin D levels and supplementation if indicated are recommended with these anticonvulsants (Holick, 2007).
The anti-TB drug INH blocks the conversion of pyridoxine (vitamin B6) to its active form, pyridoxal 5-phosphate. Particularly in patients with low pyridoxine intake, this interaction may cause pyridoxine deficiency and peripheral neuropathy. Pyridoxine supplementation (25 to 50 mg/day) is generally recommended with the prescription of INH because it is prescribed for at least 6 months at a time. Some other drugs that function as pyridoxine antagonists are hydralazine (Apresoline), penicillamine, levodopa (Dopar), and cycloserine (Seromycin).
Methotrexate (MTX; Rheumatrex) is a folic acid antagonist used to treat cancer and rheumatoid arthritis. Without folic acid, DNA synthesis is inhibited, cell replication stops, and the cell dies. Pyrimethamine (Daraprim), used to treat malaria and ocular toxoplasmosis, is also a folic acid antagonist. These drugs bind to and inhibit the enzyme dihydrofolate reductase, preventing conversion of folate to its active form (see Chapter 3), which could lead to megaloblastic anemia from folate deficiency (see Chapter 33). Leucovorin (folinic acid, the reduced form of folic acid) is used with folic acid antagonists to prevent anemia and GI damage, especially with chemotherapy such as high-dose MTX. Leucovorin does not require reduction by dihydrofolate reductase; thus, unlike folic acid, it is not affected by folic acid antagonists. Therefore leucovorin may “rescue” normal cells from MTX damage by competing for the same transport mechanisms into the cells. Administration of daily folic acid supplements or folinic acid can lower toxicity without affecting efficacy of the drug. See also Chapter 8, “Clinical: Biochemical Assessment,” for more information on the assessment for folic acid.
Statin drugs (HMG-CoA reductase inhibitors) such as atorvastatin (Lipitor) affect the formation of coenzyme Q10 (CoQ10; ubiquinone) See Box 9-2 on the mechanism of this effect. When HMG-CoA reductase is inhibited by statins, the production of cholesterol is significantly decreased. It is reasonable to conclude that the production of CoQ10 is also decreased (Ghirlanda, 1993). Studies have shown that circulatory, platelet, and lymphocyte levels of CoQ10 are also diminished. Although reports and small studies suggest that muscle pain and weakness can be relieved by CoQ10 supplementation (Littarru 2007), further large-scale studies are still needed. It may be worthwhile to supplement patients taking HMG-CoA reductase inhibitors with at least 100 mg CoQ10 daily for the preventive effect.
Nutrient Excretion
Some drugs can either increase or decrease the urinary excretion of nutrients. Drugs can increase the excretion of a nutrient by interfering with nutrient resorption by the kidneys. For instance, most clinicians know that loop diuretics such as furosemide (Lasix) or bumetanide (Bumex) increase the excretion of potassium; but these diuretics also increase the excretion of magnesium, sodium, chloride, and calcium. Potassium supplements are routinely prescribed with loop diuretics. In addition, clinicians need to consider supplements of magnesium and calcium, especially with long-term drug use, high doses of the diuretics, or poor dietary intake. Electrolyte and magnesium blood levels should be monitored. Prolonged use of high-dose diuretics, particularly by older patients on low-sodium diets, can cause sodium depletion. Hyponatremia may be overlooked in older patients because the mental confusion that is symptomatic of sodium depletion may be misdiagnosed as organic brain syndrome or dementia. Thiazide diuretics such as hydrochlorothiazide (HCTZ) increase the excretion of potassium and magnesium but reduce the excretion of calcium by enhancing renal resorption of calcium. High-dose HCTZ plus calcium supplementation may result in hypercalcemia.
Potassium-sparing diuretics such as spironolactone (Aldactone) or triamterene (Dyrenium) increase excretion of sodium, chloride, and calcium. Blood levels of potassium can rise to dangerous levels if patients also take potassium supplements or suffer from renal insufficiency. Antihypertensive angiotensin-converting enzyme (ACE) inhibitors such as enalapril (Vasotec) or fosinopril (Monopril) decrease potassium excretion, leading to increased serum potassium levels. The combination of a potassium-sparing diuretic and an ACE inhibitor increases the danger of hyperkalemia.
Corticosteroids such as prednisone decrease sodium excretion, resulting in sodium and water retention. Conversely, enhanced excretion of potassium and calcium is caused by these drugs; so a low-sodium, high-potassium diet is recommended. Calcium and vitamin D supplements are generally recommended with long-term corticosteroid use to prevent osteoporosis, such as might be the case for a person with asthma, lupus, or rheumatoid arthritis. With corticosteroid use this risk is important because it appears that not only is calcium lost in the urine, but corticosteroids may impair intestinal calcium absorption.
Phenothiazine-class antipsychotic drugs such as chlorpromazine (Thorazine) increase excretion of riboflavin and can lead to riboflavin deficiency in those with poor dietary intake. A complication associated with the use of another drug, cisplatin, is the development of acute hypomagnesemia resulting from nephrotoxicity; hypocalcemia, hypokalemia, and hypophosphatemia are also common. Both intravenous magnesium supplementation via rectal treatment or posttreatment hydration and oral magnesium supplements taken between chemotherapeutic courses have been used to prevent magnesium depletion. Hypomagnesemia can result from cisplatin use even with high-dose magnesium replacement therapy. Hypomagnesemia can persist for months or even years after the final course. When any drugs known to cause hypomagnesemia are administered, preventive treatment is warranted (Atsmon and Dolev, 2005).
Modification Of Drug Action By Food And Nutrients
Food or nutrients can alter the intended pharmacologic action of a medication by enhancing the medication effects or by opposing it. The classic example of an enhanced drug effect is the interaction between monoamine oxidase inhibitors (MAOIs) such as phenelzine sulfate (Nardil) or tranylcypromine (Parnate) and pressor agents such as dopamine, histamine, and especially tyramine. These biologically active amines are normally present in many foods (Box 9-3), but they rarely constitute a hazard because they are deaminated rapidly by MAO and diamine oxidases. Inhibition of MAO by medication prevents the breakdown of tyramine and other pressor agents. Tyramine is a vasoconstrictor that raises blood pressure. Significant ingestion of high-tyramine foods such as aged cheeses and cured meats while being treated with an MAOI antidepressant can cause a hypertensive crisis with increased heart rate, flushing, headache, stroke, and even death. This reaction may be avoided with use of a transdermal administration method that bypasses the GI tract and omits contact with the indicated foods (Blob et al., 2007).
BOX 9-3
Pressor Agents in Foods and Beverages (Tyramine, Dopamine, Histamine, Phenylethylamine)
Avoid with MAOI medications: phenelzine (Nardil), tranylcypromine (Parnate), isocarboxazid (Marplan), selegiline (Eldepryl) in doses >10 mg/day, and the antibiotic linezolid (Zyvox).
Aged cheeses (e.g., cheddar, blue, Gorgonzola, Stilton)
Aged meats (e.g., dry sausage such as salami, mortadella, Chinese dried duck)
Fermented soya beans, soya bean paste, teriyaki sauce
Tofu/fermented bean curd, tempeh
Fava (broad) beans or pods, snow pea pods (contain dopamine)
Concentrated yeast extracts (Marmite)
All casseroles made with aged cheese
Meats, fish or poultry stored longer than 3-4 days in the refrigerator
Foods That May Be Used with Caution
Red or white wine 2-4 oz per day
Coffee, cola*
Pizza (homemade or gourmet pizzas may have higher content)
Bottled beer, two 12-oz bottles, maximum
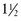 -oz servings per day)
-oz servings per day)Foods Not Limited (based on current analyses)
Unfermented cheeses (cream, cottage, ricotta, mozzarella, processed American if refrigerated less than 2-3 weeks)
Smoked white fish, salmon, carp, or anchovies
Baked raised products, English cookies
Boiled egg, yogurt, junket, ice cream
Avocado, figs, banana, raspberries
Brewer’s yeast (vitamin supplements)
Packaged or processed meats (e.g., hot dogs, bologna, liverwurst), although they should be stored in refrigerator immediately and eaten as soon as possible; histamine content is highest in improperly stored or spoiled fish, tuna
*Contains caffeine, a weak pressor agent; in quantities >500 mg/day may exacerbate reactions.
From Pronsky ZM & Crowe JP: Food medication-interactions, ed 16, Birchrunville, Pa, 2010, Food-Medication Interactions.
Caffeine in foods or beverages (see Appendix 39) increases the adverse effects of stimulant drugs such as amphetamines, methylphenidate (Ritalin, Concerta), or theophylline, causing nervousness, tremor, and insomnia. Conversely, the central nervous system (CNS) stimulatory properties of caffeine can oppose or counteract the antianxiety effect of tranquilizers such as lorazepam (Ativan).
Warfarin (Coumadin) is an oral anticoagulant that reduces the hepatic production of four vitamin K–dependent clotting factors by inhibiting the conversion of vitamin K to a usable form. Because this is a competitive interaction, the ingestion of vitamin K in the usable form will oppose the action of warfarin and allow the production of more clotting factors. To achieve an optimal level of anticoagulation, a balance must be maintained between the dose of the drug and the ingestion of vitamin K. Counseling of a person taking oral anticoagulation therapy should include nutrition therapy to maintain a consistent dietary vitamin K intake rather than prohibiting all high–vitamin K foods, such as dark green leafy vegetables (Johnson, 2005). CoQ10, St. John’s wort, or avocado may also counteract the effect of warfarin.
Ingestion of other substances may enhance the anticoagulant effect of warfarin. These substances include onions, garlic, quinine, papaya, mango, or vitamin E supplements in doses greater than 400 IU. Certain herbal products such as dong quai, which contain coumarin-like substances, or ginseng, which is a platelet inhibitor, also enhance the effect of the warfarin. Enhancement of the anticoagulation effects of warfarin may lead to serious bleeding events (Greenblatt and von Moltke, 2005). Recently there has been concern about an interaction with cranberry and warfarin (Coumadin) and the Food and Drug Administration (FDA) required a label warning change because of anecdotal reports. However, several studies found no evidence-based information to support this warning (Ansell, 2009).
Alcohol
Ethanol combined with certain medications produces additive toxicity, affecting various body organs and systems. Ethanol combined with CNS-depressant medications such as a benzodiazepine (e.g., diazepam [Valium]) or a barbiturate (e.g., phenobarbital) may produce excessive drowsiness, incoordination, and other signs of CNS depression.
In the GI tract ethanol acts as a stomach mucosal irritant. Combining ethanol with drugs that cause the same effect such as aspirin or other NSAIDs (ibuprofen [Advil or Motrin]) may increase the risk of GI ulceration and bleeding. Because of the hepatotoxic potential of ethanol, it should not be combined with medications that also exhibit a risk of hepatotoxicity such as acetaminophen (Tylenol), amiodarone (Cordarone), or methotrexate (Rheumatrex).
Ethanol can inhibit gluconeogenesis, particularly when consumed in a fasting state. Inhibition of gluconeogenesis will prolong a hypoglycemic episode caused by insulin or an oral hypoglycemic agent such as glyburide (Diabeta, Micronase).
The combination of disulfiram (Antabuse) and ethanol produces a potentially life-threatening reaction characterized by flushing, rapid heartbeat, palpitations, and elevation of blood pressure. Disulfiram inhibits aldehyde dehydrogenase, an enzyme necessary for the normal catabolism of ethanol by the liver. As a result of this enzyme inhibition, high levels of acetaldehyde accumulate in the blood. Symptoms such as flushing, headache, and nausea appear within 15 minutes of alcohol ingestion. Because these symptoms are unpleasant, the drug is sometimes used as an aid to prevent alcoholics from returning to drinking. However, because these symptoms may also be life threatening, candidates for this drug must be chosen carefully. Other medications, when ingested concurrently with ethanol, may produce disulfiram-like reactions. Some of these medications are the antibiotics metronidazole (Flagyl) and cefoperazone (Cefobid), the oral hypoglycemic agent chlorpropamide (Diabinese), and the antineoplastic agent procarbazine (Matulane).
Ethanol can also affect the physical characteristics of a medication. The FDA recently required a change in the labeling of the extended-release capsules of morphine sulfate (Avinza, Kadian). The label now includes a black box warning that patients must not consume alcoholic beverages or take morphine sulfate with medications containing alcohol. If taken with alcohol, the extended-release beads of morphine can dissolve rapidly, delivering a potentially fatal dose of morphine.
Effects Of Drugs On Nutrition Status
The desired effects of medications often are accompanied by effects that are considered undesirable, or side effects. Side effects are often an extension of the desired effects, such as bacterial overgrowth as a result of use of an antibiotic. Overgrowth of Clostridium difficile causes pseudomembranous colitis. Suppression of natural oral bacteria may lead to oral yeast overgrowth, or candidiasis (see Chapter 26).
Oral, Taste, and Smell
Many drugs affect the ability to taste or smell foods (Box 9-4). Drugs can cause an alteration in taste sensation (dysgeusia), reduced acuity of taste sensation (hypogeusia), or an unpleasant aftertaste, any of which may affect food intake. The mechanisms by which drugs alter the chemical senses are not well understood. They may alter the turnover of taste cells or interfere with transduction mechanisms inside taste cells; or they may alter neurotransmitters that process chemosensory information. Common drugs that cause dysgeusia include the antihypertensive drug captopril (Capoten), the antineoplastic cisplatin (Platinol-AQ), and the anticonvulsant phenytoin. When exploring taste changes related to medication use it is always important to consider changes in zinc absorption related to the medication. An underlying zinc deficiency may affect the sense of taste (Heckmann and Lang, 2006).
Captopril (Capoten) may cause a metallic or salty taste and the loss of taste perception. The antibiotic clarithromycin (Biaxin) enters the saliva. The drug itself has a bitter taste that stays in the mouth as long as the drug is present in the body. An unpleasant or metallic taste has been reported by up to 34% of patients taking the sleep aid eszopiclone (Lunesta).
Antineoplastic drugs, used in chemotherapy for cancer, affect cells that reproduce rapidly, including the mucous membranes. Inflammation of the mucous membranes, or mucositis, occurs and is manifest as stomatitis (mouth inflammation), glossitis (tongue inflammation), or cheilitis (lip inflammation and cracking). Mucositis can be extremely painful to the point that patients are not able to eat or even drink (see Chapter 38.) Aldesleukin, also called interleukin-2 (Proleukin), paclitaxel (Taxol), and carboplatin (Paraplatin), are examples of antineoplastic agents that commonly cause severe mucositis.
Anticholinergic drugs (Box 9-5) compete with the neurotransmitter acetylcholine for its receptor sites, thereby inhibiting transmission of parasympathetic nerve impulses. This results in decreased secretions, including salivary secretions, causing dry mouth (xerostomia). Tricyclic antidepressants such as amitriptyline (Elavil), antihistamines such as diphenhydramine (Benadryl), and antispasmodic bladder control agents such as oxybutynin (Ditropan) are particularly problematic. Dry mouth immediately causes loss of taste sensation. Long-term dry mouth can cause dental caries and loss of teeth, gum disease, stomatitis, and glossitis, as well as nutritional imbalance and undesired weight loss (Friedlander et al., 2003) (see Chapter 26).
Gastrointestinal Effects
GI irritation and ulceration are serious problems with many drugs. The antiosteoporosis drug alendronate is contraindicated in patients who are unable to sit upright for at least 30 minutes after taking it because of the danger of esophagitis. NSAIDs such as ibuprofen or aspirin can cause stomach irritation, dyspepsia, gastritis, ulceration, and sudden serious gastric bleeding, sometimes leading to fatalities. Fluoxetine (Prozac) and other selective serotonin reuptake inhibitors can also cause serious gastric irritation, leading to hemorrhage, especially when aspirin or NSAIDs are also used (Yuan et al., 2006) (Box 9-6).
Antineoplastic drugs, used to treat cancer, often cause severe nausea and vomiting. Severe, prolonged nausea and vomiting, lasting as long as a week, have been reported with cisplatin (Platinol-AQ). Dehydration and electrolyte imbalances are of immediate concern. Weight loss and malnutrition are common long-term effects of these drugs, although it is often difficult to distinguish these effects from the complications of the disease itself (see Chapter 37). Serotonin antagonists such as ondansetron (Zofran) help to reduce these GI side effects.
Drugs can cause changes in bowel function that can lead to constipation or diarrhea. Narcotic agents such as codeine and morphine (MS Contin, MSIR, Avinza) cause a nonproductive increase in smooth muscle tone of the intestinal muscle wall, thereby decreasing peristalsis and causing constipation. A new parenteral drug methylnaltrexone (Relistor) is a laxative, administered subcutaneously, specifically indicated for severe opioid-induced constipation.
Drugs with anticholinergic effects decrease intestinal secretions, slow peristalsis, and cause constipation. The atypical antipsychotic clozapine (Clozaril), tricyclic antidepressant amitriptyline (Elavil), and antihistamine diphenhydramine (Benadryl) cause constipation and possibly impaction. Patients should be closely monitored and kept adequately hydrated.
Some drugs are used to inhibit intestinal enzymes, such as the diabetic drugs acarbose (Precose) and miglitol (Glyset), which are α-glucosidase inhibitors. Such action leads to a delayed and reduced rise in postprandial blood glucose levels and plasma insulin responses. The major adverse effect is GI intolerance, specifically diarrhea, flatulence, and cramping secondary to both the osmotic effect and bacterial fermentation of undigested carbohydrates in the distal bowel.
Prescription orlistat (Xenical, or over-the-counter [OTC] Alli), is a lipase inhibitor for weight loss that reduces the absorption of fat by binding to lipase in the intestine, thereby inhibiting its action. Consequently, fecal fat excretion is increased, a factor that contributes to the GI complaints associated with the drug, specifically oily spotting, increased fecal urgency, and possible fecal incontinence. A low-fat diet of no more than 30% of calories from fat is essential. Fat intake should be distributed among all three meals. Orlistat is not an appetite suppressant, and some persons may find it difficult to maintain a low-fat diet. Sufficient counseling and support is needed for success with this medication. Attention should also be given to potential malabsorption of fat soluble vitamins A, D, E, and K and carotenoids requiring the presence of fat for optimal absorption. Obviously any of these problems, from dry mouth, to GI irritation, to constipation or diarrhea, can negatively affect food intake and nutrient absorption and nutrition status (see Chapter 22).
The use of antibiotics, particularly broad-spectrum antibiotics (Box 9-7) for long periods, destroy all sensitive bacteria of the gut flora. Intestinal flora that are not sensitive to the antibiotic will continue to grow because they are no longer inhibited by the bacteria that have been destroyed. An example of this situation is the overgrowth of C. difficile, causing pseudomembranous colitis with very strong-smelling yellow diarrhea (see also Chapter 29).
Appetite Changes
Drugs can suppress appetite (Box 9-8), leading to undesired weight changes, nutritional imbalance, and growth retardation in children. In the past the stimulant drug dextroamphetamine (Dexedrine) was used as an appetite suppressant. Because of the potential for abuse, the use of amphetamines for appetite suppression is no longer legal. Dextroamphetamine (part of Adderall) is now only indicated for treatment of attention-deficit hyperactivity disorder (ADHD) or narcolepsy.
In general, most CNS stimulants, including the amphetamine mixture (Adderall) and methylphenidate (Ritalin, Concerta, Metadate, Daytrana), suppress appetite or cause frank anorexia. These drugs are used extensively to treat ADHD in children and may cause weight loss and inhibit growth (see Chapter 18).
Sibutramine (Meridia) and phentermine (Adipex-P, Ionamin), structurally related to amphetamines, are used as appetite suppressants. These drugs are indicated for short-term use, along with a reduced-calorie diet and exercise, in obese patients (i.e., patients with a body mass index [BMI] greater than 30) or in overweight patients (BMI greater than 27) with additional risk factors such as hypertension, diabetes, or hyperlipidemia.
A major side effect of stimulant drugs is hypertension. Thus they are often contraindicated for hypertensive patients or those who have seizures or cardiac disease. Because hypertension is common among obese persons, these contraindications may limit the use of stimulants in obese or overweight hypertensive patients.
CNS side effects can interfere with the ability or desire to eat. Drugs that cause drowsiness, dizziness, ataxia, confusion, headache, weakness, tremor, or peripheral neuropathy can lead to nutritional compromise, particularly in older or chronically ill patients. Recognition of these problems as a drug side effect rather than a consequence of disease or aging is often overlooked.
Many medications stimulate appetite and lead to weight gain (Box 9-9). Antipsychotic drugs such as clozapine (Clozaril), olanzapine (Zyprexa), tricyclic antidepressant drugs such as amitriptyline (Elavil), and the anticonvulsant divalproex (Depakote) often lead to weight gain. Patients complain of a ravenous appetite and the inability to “feel full.” Weight gains of 40 to 60 lb in a few months are not uncommon. Corticosteroid use is associated with dose-dependent weight gain in many patients. Sodium and water retention, as well as appetite stimulation, causes weight increases with corticosteroids. Medical nutrition therapy (MNT) is essential, as is routine exercise.
Appetite stimulation is desirable for patients suffering from wasting (cachexia) resulting from disease states such as cancer or HIV or the acquired immunodeficiency (AIDS) virus (Tisdale, 2006). Drugs indicated as appetite stimulants or antiwasting agents are the hormone megestrol acetate (Megace, Megace ES), human growth hormone somatropin (Serostim), the anabolic steroid oxandrolone (Oxandrin), and the marijuana derivative dronabinol (Marinol). Drugs also used as appetite stimulants, although not FDA-indicated as such, are the anabolic steroids oxymetholone (Anadrol-50) and nandrolone (Deca-Durabolin), the antihistamine cyproheptadine (Periactin), and the hormone testosterone (Androderm, Virilon). The ω-3 fatty acid, eicosapentaenoic acid has been suggested as an appetite stimulant. Although some studies have not shown improvement in appetite or weight gain (Fearon et al., 2006), one has shown improvement in cachexia (Stehr and Heller, 2006). Obviously this is an area of further study. With the successful advent of highly active antiretroviral therapy (HAART), lipodystrophy is often a problem for patients with HIV/AIDS. Debate about an accurate definition of lipodystrophy is ongoing. Redistribution of body fat, fat wasting, glucose intolerance, hypertension, and hyperlipidemia are common aspects of this syndrome. Antidiabetic drugs such as metformin (Glucophage) and rosiglitazone (Avandia) are used to normalize glucose and insulin levels. Antihyperlipidemic drugs such as atorvastatin (Lipitor), pravastatin (Pravachol), or fenofibrate (Tricor) are used to control elevated triglycerides and cholesterol.
Organ System Toxicity
Drugs can cause specific organ system toxicity such as hepatotoxicity, nephrotoxicity, pulmonary toxicity, neurotoxicity, ototoxicity, ocular toxicity, pancreatitis, or cardiotoxicity. MNT may be indicated as part of the treatment of these toxicities. Although all toxicities are of concern, hepatotoxicity and nephrotoxicity are addressed here because drugs are eliminated from the body predominately through the liver and kidney.
Examples of drugs that cause hepatotoxicity (liver damage) leading to hepatitis, jaundice, hepatomegaly, or even liver failure are amiodarone (Cordarone), amitriptyline (Elavil), lovastatin (Mevacor) and other “statin” antihyperlipidemic drugs, divalproex (Depakote), carbamazepine (Tegretol), methotrexate, kava, niacin, and sulfasalazine (Azulfidine). Monitoring of hepatic function through routine blood work for liver enzyme levels is generally prescribed with use of these drugs (see Table 8-1).
Nephrotoxicity (kidney damage) may change the excretion of specific nutrients or cause acute or chronic renal insufficiency, which may not resolve with cessation of drug use. Examples of drugs that often cause nephrotoxicity are antiinfectives amphotericin B (especially with intravenous desoxycholate form [Fungizone]) and cidofovir (Vistide), as well as antineoplastics cisplatin (Plaquenil-AQ), gentamicin (Garamycin), ifosfamide (Ifex), methotrexate, and pentamidine (Pentam 300). Adequate or extra prehydration, often administered intravenously, is prescribed to reduce renal toxicity. For example, with cidofovir, 1 L of intravenous normal saline (0.9% sodium chloride [NaCl]) is infused 1 to 2 hours before infusion of the drug. If tolerated, up to an additional liter may be infused after the drug infusion. Oral probenecid (Benemid) is also prescribed with cidofovir to reduce nephrotoxicity.
Glucose Levels
Many drugs affect glucose metabolism, causing hypoglycemia or hyperglycemia and in some cases frank diabetes (Box 9-10). The mechanisms of these effects vary from drug to drug and from individual to individual. Drugs may stimulate glucose production or impair glucose uptake. They may inhibit insulin secretion, decrease insulin sensitivity, or increase insulin clearance.
Glucose levels may be affected by changes in parameters, such as hypokalemia induced by thiazide diuretics or weight gain induced by antipsychotic medications (Izzedine et al., 2005). Corticosteroids, particularly prednisone, prednisolone, and hydrocortisone, are diabetogenic because of increased gluconeogenesis, but they also cause insulin resistance and therefore inhibit glucose uptake. Second-generation antipsychotics, particularly clozapine (Clozaril) or olanzapine (Zyprexa), have been reported to cause treatment-emergent hyperglycemia. Recently the FDA added a labeling requirement on all second-generation antipsychotics to warn of the possibility of developing hyperglycemia and diabetes.
Excipients And Food-Drug Interactions
An excipient is added to drug formulations for its action as a buffer, binder, filler, diluent, disintegrant, glidant, flavoring, dye, preservative, suspending agent, or coating. Excipients are also called inactive ingredients (Box 9-11). Hundreds of excipients are approved by the FDA for use in pharmaceuticals. Several common excipients have potential for interactions in persons with an allergy or enzyme deficiency. Often just one brand of a drug or one formulation or strength of a particular brand may contain the excipient of concern. For example, tartrazine, listed as yellow dye No. 5, is used in a brand of clindamycin (Cleocin) capsules in the 75- and 150-mg strengths but not in the 300-mg strength. A brand of metoclopramide (Reglan) 5-mg tablets contain lactose, but the 10-mg tablets do not. Micronized progesterone (Prometrium) capsules contain peanut oil and lecithin, whereas other progesterone forms do not. Micronized progesterone labeling includes a warning that anyone allergic to peanuts should not use the drug (see Chapter 27).
Lactose is commonly used as a filler in many pills and capsules. The amount of lactose may be significant enough to cause GI problems for lactase-deficient patients, particularly those on multiple drugs throughout the day (see Chapter 29). Product information on prescription drugs and labeling on OTC drugs contain information on excipients, usually called “inactive ingredients,” including lactose.
Patients with celiac disease have gluten sensitivity and must practice lifelong abstinence from wheat, barley, rye, and oats (which may be contaminated with gluten; see Chapter 29). They are particularly concerned with the composition and source of excipients such as wheat starch or flour, which might contain gluten. Only a few pharmaceutical companies guarantee their products to be gluten-free. Excipients such as dextrin and sodium starch glycolate are usually made from corn and potato, respectively, but can be made from wheat or barley. For example, the excipient dextrimaltose, a mixture of maltose and dextrin, is produced by the enzymatic action of barley malt on corn flour (Crowe and Falini, 2001; Kibbe, 2000). The source of each drug ingredient, if not specified, should be checked with the manufacturer.
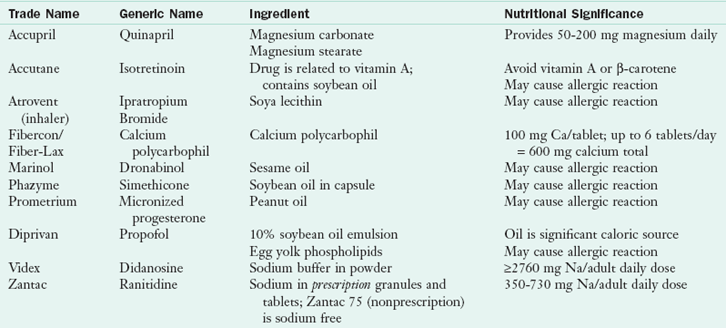
Finally, some drug brands may contain enough excipient to be nutritionally significant (see Table 9-1), magnesium in quinapril (Accupril), calcium in calcium polycarbophil (Fibercon, Fiber-Lax), and soybean oil lipid emulsion in propofol (Diprivan). Propofol is commonly used in the long term for sedation of patients in the intensive care unit. Its formulation includes 10% emulsion, which contributes 1.1 kcal/mL. When infused at doses up to 9 mg/kg/hr in a patient weighing 70 kg, for instance, it may contribute an additional 1663 kcal/day from the emulsion. For a patient receiving total parenteral nutrition, limiting the use of long-chain fatty acids and using medium-chain triglyceride (MCT) oil may be recommended while he or she is taking propofol (Dubey and Kumar, 2005). Specific brands or formulations of a specific brand provide significant amounts of sodium and therefore may be contraindicated for patients who need to limit sodium.
Medical Nutrition Therapy
MNT can be divided into prospective and retrospective care. Prospective MNT occurs when the patient first starts a drug. A diet history must be obtained, including information about the use of OTC (nonprescription) drugs, alcohol, vitamin and mineral supplements, and herbal or phytonutrient supplements. The patient should be evaluated for genetic characteristics, weight and appetite changes, altered taste, and GI problems (see Chapter 6).
Prospective drug MNT provides basic information about the drug: the name, purpose, and duration of prescription of the drug plus when and how to take the drug. This information includes whether to take the drug with or without food. Specific foods and beverages to avoid while taking the drug and potential interactions between drug and vitamin or mineral supplements need to be emphasized. For instance, the patient taking tetracycline (Achromycin-V or Sumycin) or ciprofloxacin (Cipro) should be warned not to combine the drug with milk, yogurt, or supplements containing divalent cations, calcium, iron, magnesium, zinc, or vitamin-minerals containing any of these cations.
Potential significant side effects must be delineated, and possible dietary suggestions to relieve the side effects should be described. For instance, information about a high-fiber diet with adequate fluids should be part of MNT about an anticholinergic drug such as oxybutynin (Ditropan), which often causes constipation. Conversely, diarrhea can be controlled by the use of psyllium (Metamucil) or probiotics, such as Lactobacillus acid-ophilus (Lactinex), particularly for antibiotic-associated diarrhea even in children (Szajewska et al., 2006). Probiotics may be contraindicated for some individuals and should be prescribed and monitored by the physician.
Patients should be warned about potential nutritional problems, particularly when dietary intake is inadequate, such as hypokalemia with a potassium-depleting diuretic. Dietary changes that may alter drug action should be included, such as the effect of an increase in foods high in vitamin K on warfarin action. Special diet information, such as a low-cholesterol, low-fat, limited-sugar diet with atorvastatin (Lipitor) or other antihyperlipidemic drugs, is essential information. Written information should list medication ingredients such as nonnutrient excipients in the medication. Examples include lactose, starch, tartrazine, aspartame, and alcohol. Patients with lactose intolerance, celiac disease, allergies, phenylketonuria, or alcoholism need to avoid or limit one or more of these ingredients.
Prospective MNT should also cover potential concerns with OTC drugs and herbal and natural products (Herr, 2005). It is important to emphasize that the pharmacokinetic and pharmacodynamic interactions explained in this chapter occur with all medications, whether obtained by prescription, OTC, or as natural or herbal products.
Retrospective MNT evaluates symptoms to determine if medical problems might be the result of food-drug interactions. To determine whether a patient’s symptoms are the result of a food-drug interaction, a complete medical and nutrition history is essential, including prescription and nonprescription drugs, vitamin-mineral supplements, and herbal or phytonutrient products. The date of beginning to take the drugs versus the date of symptom onset is significant information. It is important to identify the use of nutrition supplements such as enteral products or significant dietary changes such as fad diets during the course of drug prescription. Finally it is important to investigate the reported incidence of side effects (by percentage as compared with a placebo). For example, vomiting occurs in 1.5% of those taking omeprazole (Prilosec) compared with 4.7% of those taking a placebo. Therefore in a patient treated with omeprazole, it would be appropriate to consider other causes for vomiting. A rare drug effect is less likely to be the reason for a negative symptom than an effect that is common.
In summary, although food provides energy for sustenance and physiological benefits for good health, and drugs prevent or treat many diseases, together the synergistic effects can be very positive (MacDonald et al., 2009). The nutrition therapist must assess, intervene, and evaluate the mixtures with care.
 Clinical Scenario
Clinical Scenario
Henry is a 31-year-old man who began to suffer seizures after a head trauma injury from a motorcycle accident at the age of 18. For the first 2 years after the accident, he was prescribed various anticonvulsant regimens. The combination of phenytoin (Dilantin), 300 mg daily, and phenobarbital, 120 mg daily, has proven to be the most effective therapy to control his seizures. Henry has been stabilized on this regimen for the last 11 years.
Henry is a senior computer programmer for a large corporation. He is 6 feet 2 inches tall and weighs 182 lb. Henry admits to having an aversion for exercise and athletics. In his free time, he enjoys reading, playing computer games, and watching television. During the past year, Henry has broken his left femur and tibia on two separate occasions. He broke his femur when he missed the bottom step on the stairway in his office building. Several months later he broke his tibia when he tripped over a broken branch in his yard. Henry recently complained to his orthopedic surgeon about hip and pelvic pain of several weeks’ duration. An orthopedic examination with x-ray examination, bone scan, and Dexa scan revealed that Henry is suffering from osteomalacia. A review of Henry’s typical diet reveals a nutritionally marginal diet that commonly includes fast foods and frozen dinners. His diet is generally deficient in fresh fruits, vegetables, and dairy products.
Nutrition Diagnostic Statement
Food-medication interaction related to inadequate calcium and vitamin D intake while taking anticonvulsant medications as evidenced by osteomalacia.
1. Is osteomalacia common in young men?
2. How does Henry’s lifestyle contribute to the development of osteomalacia?
3. What vitamin or mineral deficiency may have contributed to the current state of Henry’s bones?
4. Describe the food-drug interaction that has contributed to Henry’s osteomalacia.
5. What medical nutritional therapy would you recommend for Henry?
Food and Drug Administration Center for Drug Evaluation and Research
Food and Nutrition Information Center
www.powernetdesign.com/grapefruit
National Institutes of Health Patient Handouts
www.cc.nih.gov/ccc/patient_education/
References
Ansell, J, et al. The absence of an interaction between warfarin and cranberry juice: a randomized, double-blind trial. J Clin Pharmacol. 2009;49:824.
Atsmon, J, Dolev, E. Drug-induced hypomagnesemia: Scope and management. Drug Saf. 2005;28:763.
Bai, JP. Ongoing challenges in drug interaction safety: from exposure to pharmacogenomics. Drug Metab Pharmacokinet. 2010;25:62.
Blob, LF, et al. Effects of a tyramine-enriched meal on blood pressure response in healthy male volunteers treated with selegiline transdermal system 6 mg/24 hr. CNS Spectr. 2007;12:25.
Crawford, P. Best practice guidelines for the management of women with epilepsy. Epilepsia. 2005;46:117.
Crowe, JP, Falini, NP. Gluten in pharmaceutical products. Am J Health Syst Pharmacol. 2001;58:396.
Dubey, PK, Kumar, A. Pain on injection of lipid-free propofol and propofol emulsion containing medium-chain triglyceride: a comparative study. Anesth Analg. 2005;101:1060.
Egashira, K, et al. Pomelo-induced increase in the blood level of tacrolimus in a renal transplant patient. Transplantation. 2003;75:1057.
Fearon, KC, et al. Double-blind, placebo-controlled, randomized study of eicosapentaenoic acid diester in patients with cancer cachexia. J Clin Oncol. 2006;24:3401.
Fourniet, MR, et al. Proton pump inhibitors, osteoporosis, and osteoporosis-related fractures. Maturitas. 2009;64:9.
Friedlander, AH, et al. Late-life depression: its oral health significance. Int Dent J. 2003;53:41.
Ghosh, D, et al. Pharmacogenomics and nutrigenomics: synergies and differences. Eur J Clin Nutr. 2007;61:567.
Ghirlanda, G, et al. Evidence of plasma CoQ10-lowering effect by HMG-CoA reductase inhibitors: a double blind, placebo-controlled study. J Clin Pharmacol. 1993;33:226.
Greenblatt, DJ, Von Moltke, LL. Interaction of warfarin with drugs, natural substances and foods. J Clin Pharmacol. 2005;45:127.
Heckmann, JG, Lang, CJ. Neurological causes of taste disorders. Adv Otorhinolaryngol. 2006;63:255.
Herr, SM. Herb-drug interaction handbook, ed 3. Nassau, NY: Church Street Books; 2005.
Holick, MF. Vitamin D deficiency. N Engl J Med. 2007;357:266.
Izzedine, H, et al. Drug-induced diabetes mellitus. Expert Opin Surg Saf. 2005;4:1097.
Johnson, MA. Influence of vitamin K on anticoagulant therapy depends on vitamin K status and the source and forms of vitamin K. Nutr Rev. 2005;63:91.
Kibbe AH, ed. Handbook of pharmaceutical excipients, ed 3, Washington, DC: American Pharmaceutical Association, 2000.
Lee, JI, et al. CYP-mediated therapeutic protein-drug interactions: clinical findings, proposed mechanisms and regulatory implications. Clin Pharmacokinet. 2010;49:295.
Littarru, GP, Langsjoen, P. Coenzyme Q10 and statins: biochemical and clinical implications. Mitochondrion. 2007;7:S168.
MacDonald, L, et al. Food and therapeutic product interactions—a therapeutic perspective. J Pharm Pharm Sci. 2009;12:367.
Medical Letter. AmpliChip CYP450 tes. Med Lett Drugs Ther. 2005;47:71.
Neuhofel, AL, et al. Lack of bioequivalence of ciprofloxacin when administered with calcium-fortified orange juice: a new twist on an old interaction. J Clin Pharmacol. 2002;42:461.
Nicolaidou, P, et al. Effects of anticonvulsant therapy on vitamin D status in children: prospective monitoring study. J Child Neurol. 2006;21:2005.
Pronsky, ZM, Crowe, JP. Food medication interactions,, ed 16. Birchrunville, Pa: Food Medication Interactions; 2010.
Sica, DA. Interaction of grapefruit juice and calcium channel blockers. Am J Hyperts. 2006;19:768.
Spriet, I, et al. Mini-series: II. Clinical aspects. Clinically relevant CYP450-mediated drug interactions in the ICU. Intensive Care Med. 2009;35:603.
Stehr, SN, Heller, AR. ω-3 fatty acid effects on biochemical indices following cancer surgery. Clin Chim Acta. 2006;373:1.
Steinman, MA, et al. Agreement between drugs-to-avoid criteria and expert assessments of problematic prescribing. Arch Int Med. 2009;169:1326.
Szajewska, H, et al. Probiotics in the prevention of antibiotic associated diarrhea in children: a meta-analysis of randomized controlled trials. J Pediatr. 2006;149:367.
Targownik, LE, et al. Proton-Pump inhibitor use is not associated with osteoporosis of accelerated bone miner density loss. Gastroenterology. 2010;138:896.
Tisdale, MJ. Clinical anticachexia treatments. Nutr Clin Pract. 2006;21:168.
Valley, M, et al. Emerging peptide therapeutics for inflammatory diseases. Curr Pharm Biotechnol. 2006;7:241.
Wise, C, Kaput, J. A strategy for analyzing gene-nutrient interactions in type 2 diabetes. J Diabetes Sci Technol. 2009;3:710.
Wohlt, PD, et al. Recommendations for use of medications with continuous enteral nutrition. Am J Health-Syst Pharm. 2009;66:1458.
Yuan, Y, et al. Selective serotonin reuptake inhibitors and risk of upper GI bleeding: confusion or confounding? Am J Med. 2006;119:719.