Medical Nutrition Therapy for Anemia
It is important for nutrition professionals to understand the myriad terms related to blood disorders. Hemoglobin is a conjugated protein containing four heme groups and globin; it is the oxygen-carrying pigment of the erythrocytes. The hematocrit is the volume percentage of erythrocytes in the blood. Plasma is the liquid portion of whole blood containing coagulation factors; serum is the liquid portion of whole blood without coagulation factors.
Anemia is a deficiency in the size or number of red blood cells (RBCs) or the amount of hemoglobin they contain. This deficiency limits the exchange of oxygen and carbon dioxide between the blood and the tissue cells. Anemia classification is based on cell size—macrocytic (larger than normal), normocytic (normal), and microcytic (small)—and on hemoglobin content—hypochromic (pale color from deficiency of hemoglobin) and normochromic (normal color; (Table 33-1). Macrocytic anemia presents with larger-than-normal RBCs, plus increased mean corpuscular volume (MCV) and mean corpuscular hemoglobin concentration (MCHC). Microcytic anemia is characterized by smaller-than-normal erythrocytes and less circulating hemoglobin, as in iron deficiency anemia and thalassemia.
TABLE 33-1
Morphologic Classification of Anemia
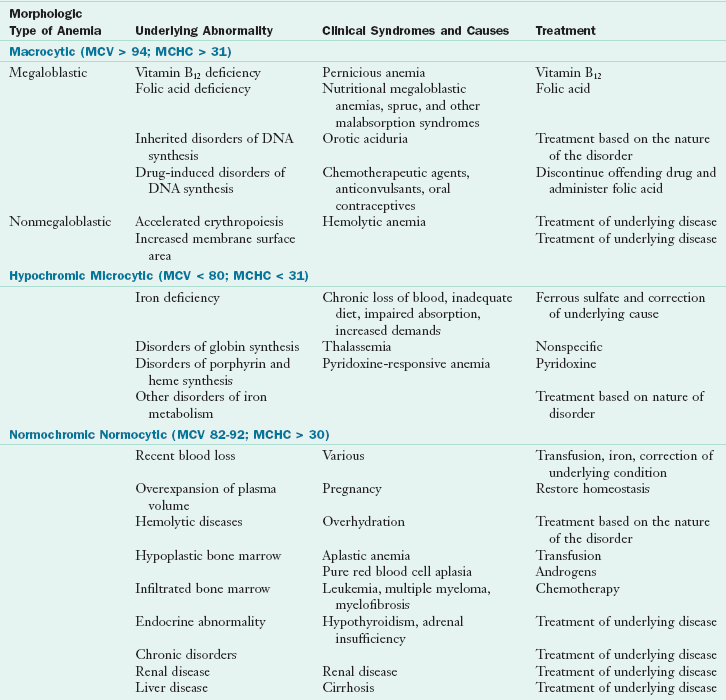
DNA, Deoxyribonucleic acid; MCHC, Mean corpuscular hemoglobin concentration: concentration of hemoglobin expressed in grams per deciliter (g/dL); MCV, mean corpuscular volume: volume of one red blood cell expressed in femtoliters (fL).
Modified from Wintrobe MM et al: Clinical hematology, ed 8, Philadelphia, 1981, Lea & Febiger.
Most anemias are caused by a lack of nutrients required for normal erythrocyte synthesis, principally iron, vitamin B12, and folic acid. These anemias that result from an inadequate intake of iron, protein, certain vitamins (B12, folic acid, pyridoxine, and ascorbic acid), copper, and other heavy metals are called nutritional anemias. Other anemias result from conditions such as hemorrhage, genetic abnormalities, chronic disease states, or drug toxicity, and have varying degrees of nutritional consequence.
Iron-Related Blood Disorders
Iron status can range from overload to deficiency and anemia. Routine measurement of iron status is necessary because approximately 6% of Americans have a negative iron balance, approximately 10% have a gene for positive balance, and approximately 1% have iron overload. Deviations from normal iron status are summarized in Box 33-1. Iron overload disease develops in persons with stage II positive balance after years of iron overload have caused progressive damage to tissues and organs (Figure 33-1).
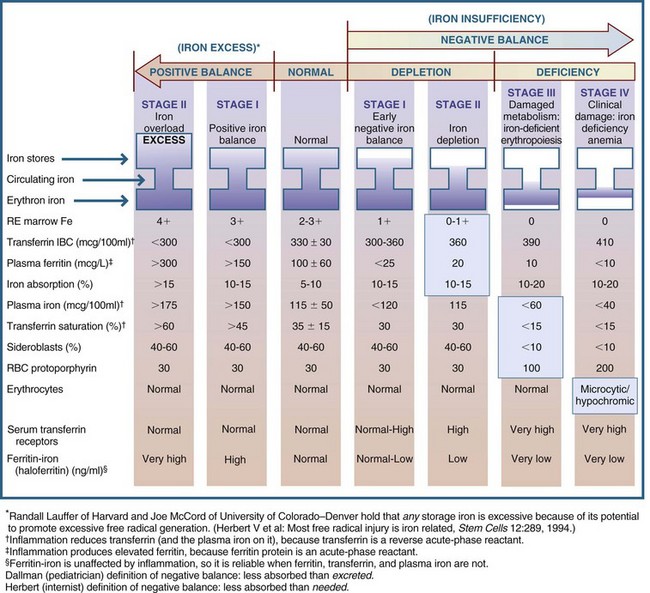
FIGURE 33-1 Sequential stages of iron status. IBC, Iron-binding capacity; RBC, red blood cell; RE, reticuloendothelial cells. (Copyright Victor Herbert, 1995.)
Iron status has a variety of indicators. Serum ferritin is an iron apoferritin complex, one of the chief storage forms of iron. Serum ferritin levels are in equilibrium with body iron stores. Very early (stage I) positive iron balance may best be recognized by measuring total iron-binding capacity (TIBC), the capacity of transferrin to take on or become saturated with iron. Conversely, measurement of serum or plasma ferritin levels may best reveal early (stage I or II) negative iron balance, although serum TIBC may be as good an indicator (see Chapter 8). Transferrin saturation is the measure of the amount of iron bound to transferrin and is a gauge of iron supply to the tissues; the percent saturation = serum iron/TIBC × 100.
Iron-Deficiency Anemia
Iron-deficiency anemia is characterized by the production of small (microcytic) erythrocytes and a diminished level of circulating hemoglobin. This microcytic anemia is actually the last stage of iron deficiency, and it represents the end point of a long period of iron deprivation.
Pathophysiology
There are many possible causes of iron-deficiency anemia (see Pathophysiology and Care Management Algorithm: Iron Deficiency Anemia). The condition can arise from:
1. Inadequate dietary intake secondary to a poor diet without supplementation
2. Inadequate absorption resulting from diarrhea, achlorhydria, intestinal disease such as celiac disease, atrophic gastritis, partial or total gastrectomy, or drug interference
3. Inadequate utilization secondary to chronic gastrointestinal disturbances
4. Increased iron requirement for growth of blood volume, which occurs during infancy, adolescence, pregnancy, and lactation
5. Increased excretion because of excessive menstrual blood (in females); hemorrhage from injury; or chronic blood loss from a bleeding ulcer, bleeding hemorrhoids, esophageal varices, regional enteritis, ulcerative colitis, parasitic or malignant disease
6. Defective release of iron from iron stores into the plasma and defective iron use caused by a chronic inflammation or other chronic disorder
With few exceptions, iron-deficiency anemia in adult men is the result of blood loss. Large losses of menstrual blood can cause iron deficiency in women, many of whom are unaware that their menses are unusually heavy.
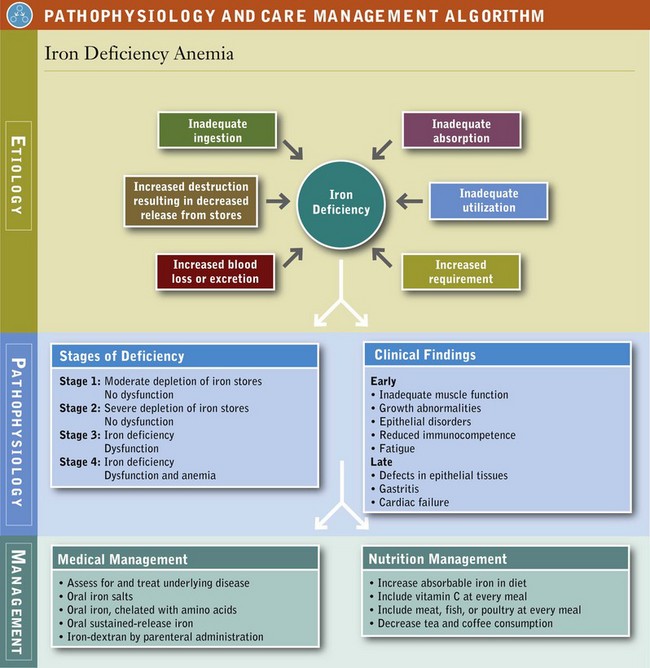
Because anemia is the last manifestation of chronic, long-term iron deficiency, the symptoms reflect a malfunction of a variety of body systems. Inadequate muscle function is reflected in decreased work performance and exercise tolerance. Neurologic involvement is manifested by behavioral changes such as fatigue, anorexia, and pica, especially pagophagia (ice eating). Abnormal cognitive development in children suggests iron deficiency before it has developed into overt anemia.
Growth abnormalities, epithelial disorders, and a reduction in gastric acidity are also common. A possible sign of early iron deficiency is reduced immunocompetence, particularly defects in cell-mediated immunity and the phagocytic activity of neutrophils, which may lead to frequent infections. Restless legs syndrome (RLS) with leg pain or discomfort may result from a lack of iron in the brain; this alters dopamine production and movement. Besides iron deficiency, kidney failure, Parkinson disease, diabetes, rheumatoid arthritis, or pregnancy can aggravate RLS (National Heart, Blood and Lung Institute, 2010).
As iron-deficiency anemia becomes more severe, defects arise in the structure and function of the epithelial tissues, especially of the tongue, nails, mouth, and stomach. The skin may appear pale, and the inside of the lower eyelid may be light pink instead of red. Mouth changes include atrophy of the lingual papillae, burning, redness, and in severe cases a completely smooth, waxy, and glistening appearance of the tongue (glossitis). Angular stomatitis may also occur, as may a form of dysphagia. Gastritis occurs frequently and may result in achlorhydria. Fingernails can become thin and flat, and eventually koilonychia (spoon-shaped nails) may be noted (Figure 33-2).
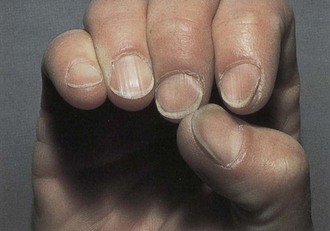
FIGURE 33-2 Fingernails with cuplike depressions (koilonychia) are a sign of iron deficiency in adults. (From Callen JP et al: Color atlas of dermatology, Philadelphia, 1993, Saunders.)
Progressive, untreated anemia results in cardiovascular and respiratory changes that can eventually lead to cardiac failure. Some behavioral symptoms respond to iron therapy before the anemia is cured, suggesting they may be the result of tissue depletion of iron-containing enzymes rather than from a decreased level of hemoglobin.
Diagnosis
A definitive diagnosis of iron-deficiency anemia requires more than one method of iron evaluation; ferritin, iron and transferrin are the most useful. The evaluation should also include an assessment of cell morphology. By itself, hemoglobin concentration is unsuitable as a diagnostic tool in cases of suspected iron-deficiency anemia for three reasons: (1) it is affected only late in the disease, (2) it cannot distinguish iron deficiency from other anemias, and (3) hemoglobin values in normal individuals vary widely.
After absorption, iron is transported by plasma transferrin—a β1-globulin (protein) that binds iron derived from the gastrointestinal tract, iron storage sites, or hemoglobin breakdown—to the bone marrow (hemoglobin synthesis), endothelial cells (storage), or placenta (fetal needs). Transferrin molecules are generated on the surface of RBCs in response to the need for iron. With iron deficiency, so many transferrin receptors are on the cell surface looking for iron that some of them break off and float in the serum. Their presence is an early measurement of developing iron deficiency; a higher quantity of soluble serum transferrin receptors (STFRs) means greater deficiency of iron. Progressive stages of iron deficiency can be evaluated by measurements, as shown in Table 33-2.
TABLE 33-2
Biochemical Evaluation of Iron Deficiency
| Measure | Comment |
| Quantity of serum or plasma ferritin | The most sensitive indicator of negative iron balance |
| Quantity of serum or plasma iron | |
| Quantity of total circulating transferrin | |
| Percent saturation of circulating transferrin | This measures the iron supply to the tissues. It is calculated by dividing serum iron by the TIBC; levels less than 16% are considered inadequate for erythropoiesis. |
| Percent saturation of ferritin with iron | |
| Quantity of STFR | STFR measures early iron deficiency. |
STFR, Soluble serum transferrin receptor; TIBC, total iron-binding capacity.
Protoporphyrin is the iron-containing portion of the respiratory pigments that combines with protein to form hemoglobin or myoglobin. The zinc protoporphyrin (ZnPP)/heme ratio is measured to assess iron deficiency. However, this ZnPP/heme ratio and hemoglobin levels are affected by chronic infection and other factors that can produce a condition that mimics iron-deficiency anemia when, in fact, iron is adequate.
Medical Management
Treatment of iron-deficiency anemia should focus primarily on the underlying cause, although this is often difficult to determine. The goal is repletion of iron stores.
Oral Supplementation: The chief treatment for iron-deficiency anemia involves oral administration of inorganic iron in the ferrous form. Although the body uses both ferric and ferrous iron, the reduced ferrous is easier on the gut and better absorbed. At a dose of 30 mg, absorption of ferrous iron is three times greater than if the same amount were given in the ferric form. Chelated forms of iron (combined with amino acids) are more bioavailable than nonchelated iron. Chelated iron is less affected by phytate, oxalate, phosphate, and calcium (all iron absorption inhibitors). Chelated iron causes less gastrointestinal disturbances than elemental iron because it is needed in lower doses when it is absorbed into mucosal cells (Ashmead, 2001).
Iron is best absorbed when the stomach is empty; however, under these conditions it tends to cause gastric irritation. Gastrointestinal side effects can include nausea, epigastric discomfort and distention, heartburn, diarrhea, or constipation. If these side effects occur, the patient is told to take the iron with meals instead of on an empty stomach; however, this sharply reduces the absorbability of the iron. Gastric irritation is a direct result of the high quantity of free ferrous iron in the stomach.
Health professionals usually prescribe oral iron for 3 months (three times daily) to treat iron deficiency. Depending on the severity of the anemia and the patient’s tolerance, the daily dose of elemental iron should be 50 to 200 mg for adults and 6 mg/kg of body weight for children. Ascorbic acid greatly increases both iron absorption and gastric irritation through its capacity to maintain iron in the reduced state.
Absorption of 10 to 20 mg of iron per day permits RBC production to increase to approximately three times the normal rate and, in the absence of blood loss, hemoglobin concentration to rise at a rate of 0.2 g/dL daily. Increased reticulocytosis (an increase in the number of young RBCs) is seen within 2 to 3 days after iron administration, but affected persons may report subjective improvements in mood and appetite sooner. The hemoglobin level will begin to increase by day 4. Iron therapy should be continued for 4 to 5 months, even after restoration of normal hemoglobin levels, to allow for repletion of body iron reserves.
Parenteral Iron-Dextran: If iron supplementation fails to correct the anemia, (1) the patient may not be taking the medication as prescribed; (2) bleeding may be continuing at a rate faster than the erythroid marrow can replace blood cells; or (3) the supplemental iron is not being absorbed, possibly as a result of malabsorption secondary to steatorrhea, celiac disease, or hemodialysis. In these circumstances parenteral administration of iron in the form of iron-dextran may be necessary. Although replenishment of iron stores by this route is faster, it is more expensive than, and not as safe as oral administration.
Medical Nutrition Therapy
In addition to iron supplementation, attention should be given to the amount of absorbable dietary iron consumed. A good source of iron contains a substantial amount of iron in relation to its calorie content and contributes at least 10% of the recommended dietary allowance (RDA) for iron. Liver; kidney; beef; dried fruits; dried peas and beans; nuts; green leafy vegetables; and fortified whole-grain breads, muffins, cereals, and nutrition bars are among the foods that rank highest in iron content (see Appendix 54). It is estimated that 1.8 mg of iron must be absorbed daily to meet the needs of 80% to 90% of adult women and adolescent boys and girls.
Bioavailability of Dietary Iron: Because typical Western diets generally contain 6 mg/1000 kcal of iron, the bioavailability of iron in the diet is more important in correcting or preventing iron deficiency than the total amount of dietary iron consumed. The rate of absorption depends on the iron status of the individual, as reflected in the level of iron stores. The lower the iron stores, the greater the rate of iron absorption. Individuals with iron-deficiency anemia absorb approximately 20% to 30% of dietary iron compared with the 5% to 10% absorbed by those without iron deficiency.
Form of Iron: Heme iron (approximately 15% of which is absorbable) is the organic form in meat, fish, and poultry, and is known as the meat-fish-poultry (MFP) factor. It is much better absorbed than nonheme iron. Nonheme iron can be found in MFP, as well as in eggs, grains, vegetables, and fruits, but it is not part of the heme molecule. The absorption rate of nonheme iron varies between 3% and 8%, depending on the presence of dietary enhancing factors, specifically ascorbic acid and meat, fish, and poultry. Ascorbic acid is not only a powerful reducing agent, but it also binds iron to form a readily absorbed complex. The mechanism by which the MFP factor potentiates the absorption of nonheme iron in other foodstuffs is unknown.
Inhibitors: Iron absorption can be inhibited to varying degrees by factors that chelate iron, including carbonates, oxalates, phosphates, and phytates (unleavened bread, unrefined cereals, and soybeans). Factors in vegetable fiber may inhibit nonheme iron absorption. If taken with meals, tea and coffee can reduce iron absorption by 50% through the formation of insoluble iron compounds with tannin. Iron in egg yolk is poorly absorbed because of the presence of phosvitin.
Iron Overload
Excess iron is stored as ferritin and hemosiderin in the macrophages of the liver, spleen, and bone marrow. The body has a limited capacity to excrete iron. Approximately 1 mg of iron is excreted daily through the gastrointestinal tract, urinary tract, and skin. To maintain a normal iron balance, the daily obligatory loss must be replaced by the absorption of heme and nonheme food iron. Persons with iron overload excrete increased amounts of iron, especially in the feces, to compensate partially for the increased absorption and higher stores.
Excessive iron intake usually stems from accidental incorporation of iron into the diet from environmental sources. In developing countries the iron overload can result from eating foods cooked in cast-iron cooking vessels or contaminated by iron-containing soils. In developed countries it likely results from excessive intake of iron-supplemented foods or multivitamin and mineral supplementation.
Uncommon disorders associated with iron overload or toxicity include thalassemias, sideroblastic anemia, chronic hemolytic anemia, ineffective erythropoiesis, transfusional iron overload (secondary to multiple blood transfusions), porphyria cutanea tarda, aplastic anemia, and alcoholic cirrhosis. Aplastic anemia is a normochromic-normocytic anemia accompanied by a deficiency of all the formed elements in the blood; it can be caused by exposure to toxic chemicals, ionizing radiation, and medications, although the cause is often unknown.
Brain iron increases with age, is higher in men, and is abnormally elevated in neurodegenerative diseases, including Alzheimer disease and Parkinson disease (Bartzokis et al., 2010). Several gene variants affect iron metabolism and may contribute to early onset of these conditions.
Hemochromatosis
Hemochromatosis is the most common form of iron overload that causes progressive hepatic, pancreatic, cardiac, and other organ damage. People with this condition absorb three times more iron from their food than those without hemochromatosis. This disease, associated with the HFE gene, is often underdiagnosed. Persons who have two affected genes (homozygotes) will die of iron overload unless they donate blood frequently. Otherwise, the excessive iron absorption continues unabated.
Asians and Pacific Islanders have the highest levels of iron in their blood of all racial and ethnic groups, but they have the lowest prevalence of the gene mutation found with the typical form of hemochromatosis (Adams et al., 2005). The Hemochromatosis and Iron Overload Screening Study notes that non-Hispanic whites have the highest prevalence of the C282Y mutation of the hereditary iron (HFE) gene, followed by Native Americans, Hispanics, blacks, Pacific Islanders, and Asians.
In women, monthly menses slow the associated organ damage until after menopause (Adams et al., 2005). Men are particularly susceptible to hemochromatosis because they have no physiologic mechanisms for losing iron such as menstruation, pregnancy, or lactation.
Pathophysiology
Hepcidin is a peptide synthesized in the liver that functions as the principal regulator of systemic iron homeostasis. It regulates iron transport from iron-exporting tissues into plasma. Hepcidin deficiency underlies most known forms of hereditary hemochromatosis (Nemeth and Ganz, 2006). Hepcidin inhibits the cellular efflux of iron by binding to and inducing the degradation of ferroprotein, the sole iron exporter in iron-transporting cells. Hepcidin controls plasma iron concentration and tissue distribution of iron by inhibiting intestinal iron absorption, iron recycling by macrophages, and iron mobilization from hepatic stores.
Hepcidin synthesis is increased by iron loading and decreased by anemia and hypoxia. Its synthesis is also greatly increased during inflammation, trapping iron in macrophages, decreasing plasma iron concentrations, and causing iron-restricted erythropoiesis that is characteristic of anemia of chronic disease. There is evidence that the mutation of the HFE gene leading to hemochromatosis is also associated with increased levels of gastrin in the stomach, leading to increased levels of gastric acid and thus increased absorption of iron (Smith et al., 2006).
In hemochromatosis iron absorption is enhanced, resulting in a gradual, progressive accumulation of iron. Most affected persons do not know they have it. In its early stages iron overload may result in symptoms similar to iron deficiency such as fatigue and weakness; later it can cause chronic abdominal pain, aching joints, impotence, and menstrual irregularities.
A progressive, positive iron balance may result in a variety of serious problems, including hepatomegaly, skin pigmentation, arthritis, heart disease, hypogonadism, diabetes mellitus, and cancer. Individuals with abnormally high iron levels are more likely to develop cancer of the colon. Iron is a prooxidant that can be used for tumor cell growth and proliferation. There also seems to be increased risk for age-related macular degeneration and Alzheimer disease because of the oxidant effect of iron overload (Connor and Lee, 2006; Dunaief, 2006).
Assessment
If an iron overload is suspected, the following screening tests should be performed: serum ferritin level (storage iron), serum iron concentration, TIBC, and percent of transferrin saturation ([serum iron/TIBC] × 100). Iron overload may be present if the percent of transferrin saturation is greater than 50 in women and 60 in men, and if the serum iron level is greater than 180 mg/dL. Deoxyribonucleic acid (DNA) testing, using blood or cheek cell samples, is also available for early detection of hemochromatosis. Liver biopsy is the gold standard for the diagnosis of iron overload.
Medical Management
The patient with iron overload may simultaneously be anemic as a result of damage to the bone marrow, an inflammatory disorder, cancer, internal bleeding, or chronic infection. Iron supplements should not be taken until the cause is known.
For patients with significant iron overload, weekly phlebotomy for 2 to 3 years may be required to eliminate all excess iron. Treatment for iron overload may also involve iron depletion with intravenous desferrioxamine-B, a chelating agent that is excreted by the kidneys, or with calcium disodium ethylenediaminetetraacetic acid (EDTA). Mortality is preventable if excess body iron is removed by phlebotomy therapy before hepatic cirrhosis develops. Patients diagnosed as having hemochromatosis should inform all blood relatives so they can be evaluated.
Medical Nutrition Therapy
Individuals with iron overload should ingest less heme iron from meat, fish, and poultry compared with nonheme iron from plant foods. Persons with iron overload should also avoid alcohol and vitamin C supplements because both enhance iron absorption. In addition, vitamin C supplements may cause release of harmful free radical–generating excess iron from body stores.
Affected persons should avoid foods that are highly fortified with iron (i.e., many breakfast cereals, fortified “energy” or sports bars, and many meal-replacement drinks or shakes that are fortified with vitamins and minerals). They should also avoid iron supplements or multiple vitamin and mineral supplements that contain iron. The dietary requirement for iron should not be exceeded, and perhaps the intake of iron should be less in some persons. The dietary reference intakes (DRIs) for iron are summarized on the inside front cover. The RDA for women in their childbearing years is 18 mg; for pregnant women, 27 mg; and the RDA for adult men and women 51 years of age and older is 8 mg.
Megaloblastic Anemias
Megaloblastic anemia reflects a disturbed synthesis of DNA, which results in morphologic and functional changes in erythrocytes, leukocytes, platelets, and their precursors in the blood and bone marrow. This anemia is characterized by the presence of large, immature, abnormal, RBC progenitors in the bone marrow; 95% of cases are attributable to folic acid or vitamin B12 deficiency. Two disorders of cobalamin metabolism arise from mutations of the methionine synthase and methionine synthase reductase genes; these disorders feature both megaloblastic anemia and neurologic manifestations.
Both vitamins are essential to the synthesis of nucleoproteins. Hematologic changes are the same for both; however, the folic acid deficiency is the first to appear. Normal body folate stores are depleted within 2 to 4 months in individuals consuming folate-deficient diets. By contrast, vitamin B12 stores are depleted only after several years of a vitamin B12–deficient diet. In persons with vitamin B12 deficiency, folic acid supplementation can mask B12 deficiency (Figure 33-3). In correcting the anemia, the vitamin B12 deficiency may remain undetected, leading to the irreversible neuropsychiatric damage that is only corrected with B12 supplementation (see Chapters 3 and 8).
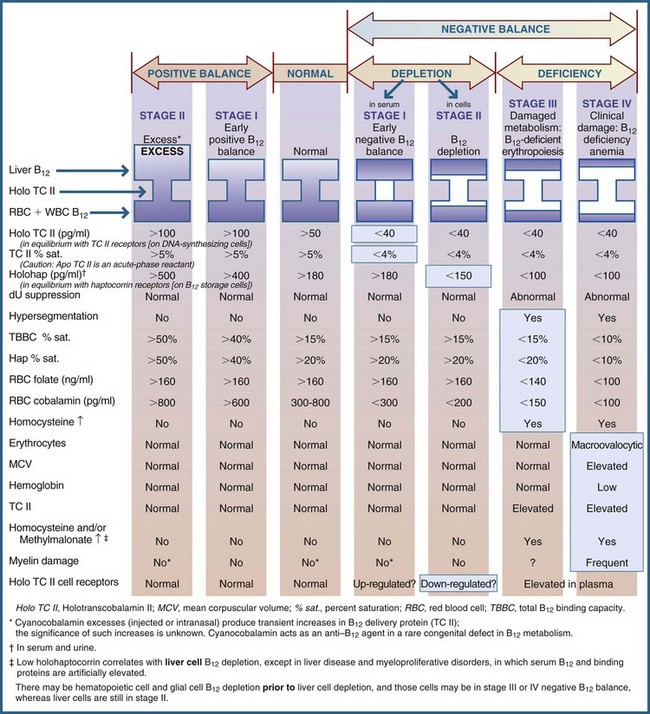
FIGURE 33-3 Sequential stages of vitamin B12 status. (From Herbert V: Staging vitamin B12. In Ziegler EE, Filer LJ, editors: Present knowledge in nutrition, ed 7, Washington, DC, 1996, International Life Sciences Institute Press.)
Folic Acid Deficiency Anemia
Folic acid deficiency anemia is associated with tropical sprue, can affect pregnant women, and occurs in infants born to mothers with folic acid deficiency. Folic acid deficiency in early pregnancy can also result in an infant with a neural tube defect (see Chapter 16). Prolonged inadequate diets, faulty absorption and use of folic acid, and increased requirements resulting from growth are believed to be the most frequent causes. Other causes include gluten-induced enteropathy (childhood and adult celiac disease), idiopathic steatorrhea, nontropical sprue and drugs (anticonvulsants, barbiturates, cycloserine, ethanol, sulfasalazine, cholestyramine and metformin), and amino acid excess (glycine and methionine).
Because alcohol interferes with the folate enterohepatic cycle, most alcoholics have a negative folate balance or a folate deficiency. Alcoholics constitute the only group that generally has all six causes of folic acid deficiency simultaneously: inadequate ingestion, absorption, and use with increased excretion, requirement, and destruction of folic acid. Box 33-2 describes the causes of folate deficiency.
Folate absorption takes place in the small intestine. Enzyme conjugases (e.g., pteroylpolyglutamate hydrolase, the folate conjugase), found in the brush border of the small intestine, hydrolyze the polyglutamates to monoglutamates and reduce them to dihydrofolate and tetrahydrofolic acid (THFA) in the small intestinal epithelial cells (enterocytes). From the enterocytes these forms are transported to the circulation, where they are bound to protein and transported as methyl THFA into the cells of the body.
In the absence of vitamin B12, 5-methyl THFA, the major circulating and storage form of folic acid, is metabolically inactive. To be activated the 5-methyl group is removed, and THFA is cycled back into the folate pool, where it functions as the main 1-carbon-unit acceptor in mammalian biochemical reactions. THFA may then be converted to the coenzyme form of folate required to convert deoxyuridylate to thymidylate, which is necessary for DNA synthesis.
MTHFR Allele: A genetic defect found in 10% of whites is the methylenetetrahydrofolate reductase (MTHFR) deficiency. (See Chapters 5 and 8.) The allele is problematic in pregnancy, and may contribute to miscarriages, anencephaly, or neural defects. Because MTHFR irreversibly reduces 5,10-methylenetetrahydrofolate to 5-methyltetrahydrofolate, its deficiency may result in developmental delay, motor and gait dysfunction, seizures, neurologic impairment, extremely high levels of homocysteine, clotting disorders, and other conditions.
Methylfolate Trap: Vitamin B12 deficiency can result in a folic acid deficiency by causing folate entrapment in the metabolically useless form of 5-methyl THFA (Figure 33-4). The lack of vitamin B12 to remove the 5-methyl unit means that metabolically inactive methyl THFA is trapped. It cannot release its 1-carbon methyl group to become THFA, the basic 1-carbon carrier that picks up 1-carbon units from one molecule and delivers them to another. Hence a functional folic acid deficiency results.
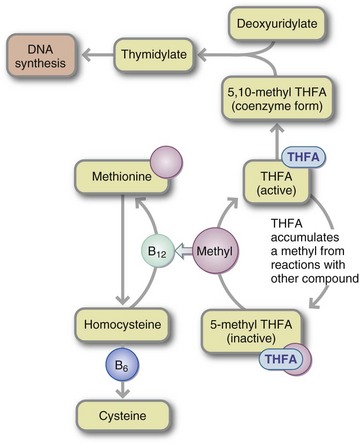
FIGURE 33-4 Methylfolate trap. A deficiency of vitamin B12 can result in a deficiency of folic acid because folate is trapped in the form of 5-methyltetrahydrofolate (5-methyl THFA), which cannot be converted to THFA and methyl groups donated by the vitamin B12–dependent pathway. DNA, Deoxyribonucleic acid.
Pathophysiology
Folate deficiency develops in four stages: two that involve depletion, followed by two marked by deficiency (Figure 33-5):
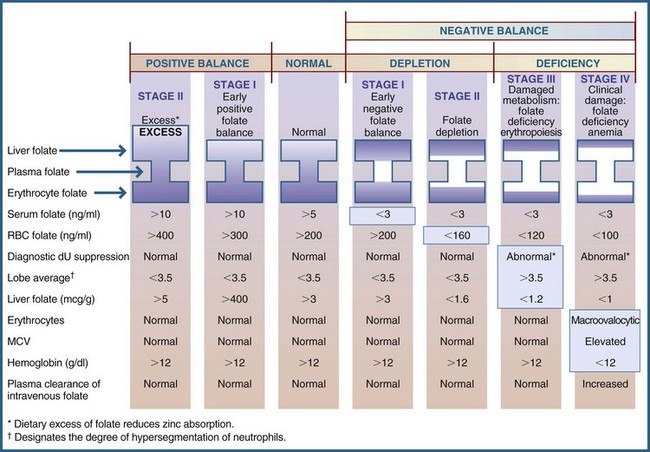
FIGURE 33-5 Sequential stages of folate status. dU, Deoxyuridine; MCV, mean corpuscular volume; RBC, red blood cell. (From Herbert V: Folic acid. In Shils ME, Olson JA, Shike M, editors: Modern nutrition in health and disease, ed 9, Philadelphia 1998, Lea & Febiger.)
Stage 1: Characterized by early negative folate balance (serum depletion to less than 3 ng/mL).
Stage 2: Characterized by negative folate balance (cell depletion), with a decrease in erythrocyte folate levels to less than 160 ng/mL.
Stage 3: Characterized by damaged folate metabolism, with folate-deficient erythropoiesis. This stage is characterized by slowed DNA synthesis, manifested by an abnormal diagnostic deoxyuridine (dU) suppression test correctable in vitro by folates, granulocyte nuclear hypersegmentation, and macroovalocytic red cells.
Stage 4: Characterized by clinical folate-deficiency anemia, with an elevated MCV and anemia.
Because of their interrelated roles in the synthesis of thymidylate in DNA formation, a deficiency of either vitamin B12 or folic acid results in a megaloblastic anemia. The immature nuclei do not mature properly in the deficient state; and large (macrocytic), immature (megaloblastic) RBCs are the result. The common clinical signs of folic acid deficiency include fatigue, dyspnea, sore tongue, diarrhea, irritability, forgetfulness, anorexia, glossitis, and weight loss.
Assessment
Normal body folate stores are depleted within 2 to 4 months on a folate-deficient diet, resulting in a macrocytic, megaloblastic anemia with a decreased number of erythrocytes, leukocytes, and platelets. Folate-deficiency anemia is manifested by very low serum folate (<3 ng/mL) and RBC folate levels of less than 140-160 ng/mL. Whereas a low serum folate level merely diagnoses a negative balance at the time the blood is drawn, an red cell folate (RCF) level measures actual body folate stores and thus is the superior measurement for determining folate nutriture. To differentiate folate deficiency from vitamin B12 deficiency, levels of serum folate, RCF, serum vitamin B12, and vitamin B12 bound to transcobalamin II (TCII) can be measured simultaneously using a radioassay kit. Also diagnostic for folate deficiency is an elevated level of formiminoglutamic acid in the urine, as well as the dU suppression test in bone marrow cells or peripheral blood lymphocytes. See Chapter 8.
Medical Management
Before treatment is initiated, it is important to diagnose the cause of the megaloblastosis correctly. Administration of folate will correct megaloblastosis from either folate or vitamin B12 deficiency, but it can mask the neurologic damage of vitamin B12 deficiency, allowing the nerve damage to progress to the point of irreversibility.
A dosage of 1 mg of folate taken orally every day for 2 to 3 weeks replenishes folate stores. Maintaining repleted stores requires an absolute minimum oral intake of 50 to 100 mcg of folic acid daily. When folate deficiency is complicated by alcoholism or other conditions that suppress hematopoiesis, increase folate requirements, or reduce folate absorption, therapy should remain at 500 to 1000 mcg daily. Symptomatic improvement, as evidenced by increased alertness, cooperation, and appetite, may be apparent within 24 to 48 hours, long before hematologic values revert to normal, a gradual process that takes approximately a month.
Medical Nutrition Therapy
After the anemia is corrected, the patient should be instructed to eat at least one fresh, uncooked fruit or vegetable or to drink a glass of fruit juice daily. One cup of orange juice supplies approximately 135 mcg of folic acid (see Appendix 46 for a list of foods). Fresh, uncooked fruits and vegetables are good sources of folate because folate can easily be destroyed by heat. In 1998 the Food and Drug Administration required that grains be fortified with folic acid. The DRIs for folate are RDAs and are summarized on the inside front cover of this text. The RDA for adults is 400 mcg daily. The Dietary Guidelines for Americans recommend that women of childbearing age who may become pregnant and those in their first trimester of pregnancy consume adequate synthetic folic acid (600 mcg/day) from fortified foods and supplements in addition to consuming a variety of foods containing folate.
Vitamin B12 Deficiency and Pernicious Anemias
Intrinsic factor (IF) is a glycoprotein in the gastric juice that is necessary for the absorption of dietary vitamin B12. Secreted by parietal cells of the gastric mucosa, IF is necessary for the absorption of exogenous vitamin B12. Ingested vitamin B12 is freed from protein by gastric acid and gastric and intestinal enzymes. The free vitamin B12 attaches to salivary R-binder, which has a higher affinity for the vitamin than does IF. An acid pH (2.3) is needed, such as that found in the healthy stomach.
The release of pancreatic trypsin into the proximal small intestine destroys R-binder and releases vitamin B12 from its complex with R-protein. With an alkaline pH (6.8) in the intestine, IF binds the vitamin B12. The vitamin B12–IF complex is then carried to the ileum. In the ileum, with the presence of ionic calcium (Ca2+) and a pH (>6), the complex attaches to the surface vitamin B12–IF receptors on the ileal cell brush border. Here, the vitamin B12 is released and attaches to holotranscobalamin II (holo TCII). Holo TCII is vitamin B12 attached to the β-globulin, the major circulating vitamin B12 delivery protein. Like IF, holo TCII plays an active role in binding and transporting vitamin B12. The TCII–vitamin B12 complex then enters the portal venous blood.
Other binding proteins in the blood include haptocorrin, also known as transcobalamin I (TCI) and transcobalamin III (TCIII). These are α-globulins, larger macromolecular-weight glycoproteins that make up the R-binder component of the blood. Unlike IF, the R-proteins are capable of binding not only vitamin B12 itself but also to many of its biologically inactive analogs. Although approximately 75% of the vitamin B12 in human serum is bound to haptocorrin and roughly 25% is bound to TCII, only TCII is important in delivering vitamin B12 to all the cells that need it. After transport through the bloodstream, TCII is recognized by receptors on cell surfaces. Patients with haptocorrin abnormalities have no symptoms of vitamin B12 deficiency. Those lacking TCII rapidly develop megaloblastic anemia. Vitamin B12 is excreted in urine.
Pathophysiology
Pernicious anemia is a megaloblastic, macrocytic anemia caused by a deficiency of vitamin B12, most commonly from a lack of IF. Rarely, vitamin B12 deficiency anemia occurs in strict vegetarians whose diet contains no vitamin B12 except for traces found in plants contaminated by microorganisms capable of synthesizing vitamin B12. Other causes include antibody to IF in saliva or gastric juice; small intestinal disorders affecting the ileum such as celiac disease, idiopathic steatorrhea, tropical sprue, cancers involving the small intestine; drugs (paraaminosalicylic acid, colchicine, neomycin, metformin, antiretrovirals); and long-term ingestion of alcohol or calcium-chelating agents (Table 33-3).
TABLE 33-3
Causes of Vitamin B12 Deficiency
| Inadequate ingestion Inadequate absorption Inadequate utilization Increased requirement Increased excretion Increased destruction |
Poor diet resulting from a vegan diet and lack of supplementation, chronic alcoholism, poverty Gastric disorders, small intestinal disorders, competition for absorption sites, pancreatic disease, HIV or AIDS Vitamin B12 antagonists, congenital or acquired enzyme deficiency, abnormal binding proteins Hyperthyroidism, increased hematopoiesis Inadequate vitamin B12 binding protein, liver disease, renal disease Pharmacologic doses of ascorbic acid by antioxidants |
AIDS, Acquired immune deficiency syndrome; HIV, human immunodeficiency virus.
Stages of Deficiency
As a result of normal enterohepatic circulation (i.e., excretion of vitamin B12 and analogs in bile and resorption of vitamin B12 in the ileum), it generally takes decades for strict vegetarians who are not receiving vitamin B12 supplementation to develop a vitamin B12 deficiency. Serum B12, homocysteine, and methylmalonic acid levels are not as effective as predictors of B12-responsive neurologic disorders; patients with unexplained leukoencephalopathy should be treated proactively because even long-standing deficits may be reversible (Graber et al., 2010).
Stage 1: Early negative vitamin B12 balance begins when vitamin B12 intake is low or absorption is poor, depleting the primary delivery protein, TCII. A low TCII (<40 pg/mL) may be the earliest detectable sign of a vitamin B12 deficiency (Serefhanoglu et al., 2008). This is a vitamin B12 predeficiency stage.
Stage 2: Vitamin B12 depletion shows a low B12 on TCII and a gradual lowering of B12 in haptocorrin (holohap <150 pg/mL), the storage protein.
Stage 3: Damaged metabolism and vitamin B12–deficient erythropoiesis includes an abnormal dU suppression, hypersegmentation, a decreased TIBC and holohap percent saturation, a low RCF level (<140 ng/mL), and subtle neuropsychiatric damage (impaired short-term and recent memory).
Stage 4: Clinical damage occurs, including vitamin B12 deficiency anemia; includes all preceding parameters, including macroovalocytic erythrocytes, elevated MCV, elevated TCII levels, increased homocysteine (see Chapter 34) and methylmalonic acid levels, and myelin damage. Leukoencephalopathy and autonomic dysfunction occur with very low serum B12 levels (<200 pg/mL); psychiatric changes, neuropathy, and dementia may also occur (Graber et al., 2010).
Clinical Findings
Pernicious anemia affects not only the blood but also the gastrointestinal tract and the peripheral and central nervous systems. This distinguishes it from folic acid deficiency anemia. The overt symptoms, which are caused by inadequate myelinization of the nerves, include paresthesia (especially numbness and tingling in the hands and feet), diminution of the senses of vibration and position, poor muscular coordination, poor memory, and hallucinations. If the deficiency is prolonged, the nervous system damage may be irreversible, even with initiation of vitamin B12 treatment.
Helicobacter pylori causes peptic ulcer disease and chronic gastritis. Both conditions are associated with hypochlorhydria, reduced production of IF by epithelial cells in the stomach, vitamin B12 malabsorption, and pernicious anemia. There is also a correlation between autoimmune gastritis and pernicious anemia. More than 90% of patients with pernicious anemia have parietal cell antibodies (PCAs) and 50% to 70% have elevated IF antibodies. Serum vitamin B12 levels of the H. pylori–infected patients are significantly lower than that of uninfected patients (Sarari et al., 2008).
A study on H. pylori infection and autoimmune type atrophic gastritis examined serum markers for gastric atrophy (pepsinogen I, pepsinogen I/II, and gastrin) and autoimmunity. Positive serum autoimmune markers (IF antibodies and PCA) suggest that H. pylori contributes to autoimmune gastritis and pernicious anemia (Veijola et al., 2010).
Vitamin B12 deficiency is an important modifiable risk factor for osteoporosis in both men and women. Adults with vitamin B12 levels below 148 pg/mL have a lower average bone mineral density and greater risk for osteoporosis (Tucker and Mayer, 2005).
Reduced vitamin B12 status and elevated homocysteine concentrations are common. These alterations are problematic among vegans (Elmadfa and Singer, 2009). B12-folate-homocysteine interactions aggravate heart disease and may lead to adverse pregnancy outcomes (Moreiras et al., 2009).
Diagnosis
Vitamin B12 stores are depleted after several years without vitamin B12 intake. A low holo-TCII value (<40 pg/mL) is a sign of early B12 deficiency. Radioassays measure more than one component within the same biologic medium. The Becton-Dickinson SimulTRAC Radioassay Kit measures the levels of serum vitamin B12 and serum folate simultaneously in a single test tube.
Other laboratory tests that may be helpful in diagnosing a vitamin B12 deficiency and determining its cause include measurements of unsaturated B12 binding capacity, IF antibody (IFAB), the Schilling test, the dU suppression test, and tests to determine serum homocysteine and serum methionine levels (see Chapter 8). The IFAB and Schilling urinary excretion tests can determine whether the deficiency is caused by a lack of IF. The IFAB assay is performed on a patient’s serum, whereas the Schilling test requires that the patient first swallow radioactive B12 alone and then a second time with IF.
The vitamin B12 assay is performed on the patient’s urine after both steps of the Schilling test are completed. Patients with pernicious anemia excrete very little vitamin B12 during the first step because little or no vitamin B12 is absorbed. However, during the second step the urinary excretion becomes almost normal because more vitamin B12 is absorbed with the addition of the IF. Vitamin B12 deficiency secondary to malabsorption syndrome is manifested by a decrease in urinary excretion of B12 that remains unchanged with IF administration.
Medical Management
Treatment usually consists of an intramuscular or subcutaneous injection of 100 mcg or more of vitamin B12 once per week. After an initial response is elicited, the frequency of administration is reduced until remission can be maintained indefinitely with monthly injections of 100 mcg. Very large oral doses of vitamin B12 (1000 mcg daily) are also effective, even in the absence of IF, because approximately 1% of vitamin B12 will be absorbed by diffusion. Initial doses should be increased when vitamin B12 deficiency is complicated by debilitating illness such as infection, hepatic disease, uremia, coma, severe disorientation, or marked neurologic damage. A response to treatment is evidenced by improved appetite, alertness, and cooperation, followed by improved hematologic results, as manifested by marked reticulocytosis within hours of an injection.
Medical Nutrition Therapy
A high-protein diet (1.5 g/kg of body weight) is desirable both for liver function and for blood regeneration. Because green leafy vegetables contain both iron and folic acid, the diet should contain increased amounts of these foods. Meats (especially beef and pork), eggs, milk, and milk products are particularly rich in vitamin B12 (see Appendix 46).
For those individuals prescribed metformin for treatment of diabetes, 10% to 30% have reduced vitamin B12 absorption. Metformin negatively affects the calcium-dependent membrane and the B12-IF complex by decreasing the absorbability by the ileal cell surface receptors. Increased intake of calcium reverses the vitamin B12 malabsorption (see Chapter 9).
The Dietary Guidelines for Americans recommend that people older than age 50 consume vitamin B12 in its crystalline form (i.e., fortified cereals or supplements) to overcome the effects of atrophic gastritis. The DRIs for B12 are RDAs and are summarized on the inside front cover. The RDA for adult men and women is 2.4 mcg daily.
Other Nutritional Anemias
Anemia of Protein-Energy Malnutrition
Protein is essential for the proper production of hemoglobin and RBCs. Because of the reduction in cell mass and thus oxygen requirements in protein-energy malnutrition (PEM), fewer RBCs are required to oxygenate the tissue. Because blood volume remains the same, this reduced number of RBCs with a low hemoglobin level (hypochromic, normocytic anemia), which can mimic an iron-deficiency anemia, is actually a physiologic (nonharmful) rather than harmful anemia. In acute PEM, the loss of active tissue mass may be greater than the reduction in the number of RBCs, leading to polycythemia. The body responds to this RBC production, which is not a reflection of protein and amino acid deficiency but of an oversupply of RBCs. Iron released from normal RBC destruction is not reused in RBC production but is stored, so that iron stores are often adequate. Iron-deficiency anemia can reappear with rehabilitation when RBC mass expands rapidly.
The anemia of PEM may be complicated by deficiencies of iron and other nutrients and by associated infections, parasitic infestation, and malabsorption. A diet lacking in protein is usually deficient in iron, folic acid, and, less frequently, vitamin B12. The nutrition counselor plays an important role in assessing recent and typical dietary intake of these nutrients.
Copper-Deficiency Anemia
Copper and other heavy metals are essential for the proper formation of hemoglobin. Ceruloplasmin, a copper-containing protein, is required for normal mobilization of iron from its storage sites to the plasma. In a copper-deficient state, iron cannot be released; this leads to low serum iron and hemoglobin levels, even in the presence of normal iron stores. Other consequences of copper deficiency suggest that copper proteins are needed for use of iron by the developing erythrocyte and for optimal functions of the erythrocyte membrane (see Chapter 3). The amounts of copper needed for normal hemoglobin synthesis are so minute that they are usually amply supplied by an adequate diet; however, copper deficiency may occur in infants who are fed cow’s milk or a copper-deficient infant formula. It may also be seen in children or adults who have a malabsorption syndrome or who are receiving long-term total parenteral nutrition that does not supply copper.
Sideroblastic (Pyridoxine-Responsive) Anemia
Sideroblastic anemia is characterized by a derangement in the final pathway of heme synthesis, leading to a buildup of iron-containing immature RBCs. It has four primary characteristics: (1) microcytic and hypochromic RBCs; (2) high serum and tissue iron levels (causing increased transferrin saturation); (3) the presence of an inherited defect in the formation of δ-aminolevulinic acid synthetase, an enzyme involved in heme synthesis (pyridoxal-5-phosphate is necessary in this reaction); and (4) a buildup of iron-containing immature RBCs (sideroblasts, for which the anemia is named). The iron that cannot be used for heme synthesis is stored in the mitochondria of immature RBCs. These iron-laden mitochondria do not function normally, and the development and production of RBCs become ineffective. The symptoms are those of both anemia and iron overload. The neurologic and cutaneous manifestations of vitamin B6 deficiency are not observed. The anemia responds to the administration of pharmacologic doses of pyridoxine and thus is referred to as vitamin B6 (pyridoxine)–responsive anemia, to distinguish it from anemia caused by a dietary vitamin B6 deficiency.
Treatment consists of a therapeutic trial dose of 50 to 200 mg daily of pyridoxine or pyridoxal phosphate, which is 25 to 100 times the RDA. If the anemia responds to one or the other, pyridoxine therapy is continued for life. However, the anemia is only partially corrected; a normal hematocrit value is never regained. Patients respond to this treatment to varying degrees, and some may achieve near-normal hemoglobin levels.
Acquired sideroblastic anemias such as those attributable to drug therapy (isoniazid, chloramphenicol), copper deficiency, hypothermia, and alcoholism are not responsive to vitamin B6 (pyridoxine) administration.
Vitamin E–Responsive Hemolytic Anemia
Hemolytic anemia occurs when defects in RBC membranes lead to oxidative damage and eventually to cell lysis.
This anemia is caused by shortened survival of mature RBCs. Vitamin E, an antioxidant, is involved in protecting the membrane against oxidative damage, and one of the few signs noted in vitamin E deficiency is early hemolysis of RBCs (see Chapter 3). Vitamin E–responsive hemolytic anemia in neonates is discussed in Chapter 43.
Nonnutritional Anemias
A physiologic anemia is the anemia of pregnancy, which is related to increased blood volume and usually resolves with the end of the pregnancy; however, demands for iron during pregnancy are also increased so that inadequate iron intake may also play a role (see Chapter 16 for further discussion).
Anemia of Chronic Disease
Anemia of chronic disease occurs from inflammation, infection, or malignancy because there is decreased RBC production, possibly as a result of disordered iron metabolism. Ferritin levels are normal or increased, but serum iron levels and TIBC are low. It is important that this form of anemia, which is mild and normocytic, not be mistaken for iron-deficiency anemia; iron supplements should not be given. Recombinant erythropoietin therapy usually corrects this anemia. See Chapters 6 and 39.
Sickle Cell Anemia
Sickle cell anemia (SCA), a chronic hemolytic anemia also known as hemoglobin S disease, affects 1 of 600 African Americans in the United States as a result of homozygous inheritance of hemoglobin S. This results in defective hemoglobin synthesis, which produces sickle-shaped RBCs that get caught in capillaries and do not carry oxygen well. The disease is usually diagnosed toward the end of the first year of life.
In addition to the usual symptoms of anemia, SCA is characterized by episodes of pain resulting from the occlusion of small blood vessels by the abnormally shaped erythrocytes. The occlusions frequently occur in the abdomen, causing acute, severe abdominal pain. The hemolytic anemia and vasoocclusive disease result in impaired liver function, jaundice, gallstones, and deteriorating renal function. The constant hemolysis of erythrocytes increases iron stores in the liver; however, iron-deficiency anemia and SCA can coexist. Iron overload is less common and is usually a problem only in those who have received multiple blood transfusions.
Typically serum homocysteine levels are elevated, which may be due to low concentrations of vitamin B6. Children with SCA were found to have these lower vitamin B6 levels despite B6 intakes comparable to those of unaffected children.
Medical Management
No specific treatment exists for SCA other than relieving pain during a crisis, keeping the body oxygenated, and possibly administering an exchange transfusion. It is important that SCA not be mistaken for iron-deficiency anemia, which can be treated with iron supplements, because iron stores in the patient with SCA secondary to transfusions are frequently excessive.
Zinc can increase the oxygen affinity of both normal and sickle-shaped erythrocytes. Thus zinc supplements may be beneficial in managing sickle cell disease, especially because decreased plasma zinc is common in children with the SS genotype sickle cell disease and is associated with decreased linear and skeletal growth, muscle mass, and sexual maturation. Zinc supplementation (as little as 10 mg daily) may also prevent the deficit in growth that appears in these children (Zemel et al., 2007). Because zinc competes with copper for binding sites on proteins, the use of high doses of zinc may precipitate copper deficiency.
Medical Nutrition Therapy
Children with SCA and their families should receive instruction about how they can develop a well-balanced food plan providing enough calories and protein for growth and development. Their dietary intake may be low because of the abdominal pain characteristic of the disease. They also have increased metabolic rates, leading to a need for a higher caloric intake. This hypermetabolism is probably due to a constant inflammation and oxidative stress (Akohoue et al., 2007; Hibbert et al., 2005). Therefore their diets must be high enough in calories to meet these needs and must provide foods high in folate and the trace minerals zinc and copper (see Appendix 58 for sources of these minerals). In addition, they may be low in vitamins A, C, D, and E, folate, calcium and fiber. The diet should be high in folate (400 to 600 mcg daily) because the increased production of erythrocytes needed to replace the cells being continuously destroyed also increases folic acid requirements.
When assessing the nutrition status of patients with SCA, the questions related to the use of vitamin and mineral supplements, the consumption of alcohol (which increases iron absorption), and sources of protein (animal sources being high in both zinc and iron) in the diet must be given special attention. A multivitamin and mineral supplement containing 50% to 150% of the RDA for folate, zinc, and copper (not iron) is recommended.
Dietary fluid and sodium intake influence the risk for vasoocclusive events in SCA; increasing fluid intake and limiting high-sodium foods should be discussed (Fowler et al., 2010). Intake of 2 to 3 quarts of water daily is recommended. Finally, it is important to remember that patients with sickle cell disease may require higher than RDA amounts of protein.
If it is necessary for the diet to be low in absorbable iron, the diet should emphasize vegetable proteins. Iron-rich foods, such as liver, iron-fortified formula, iron-fortified cereals, and iron-fortified energy bars are excluded. Substances such as alcohol and ascorbic acid supplements, both of which enhance iron absorption should be avoided. However, it is important to remember that iron deficiency may be present in some patients with SCA owing to repeated phlebotomies, excessive transfusions, or hematuria secondary to renal papillary necrosis. This should be assessed, and the diet adjusted appropriately.
Hypochromic Microcytic Transient Anemia (Sports Anemia)
Increased RBC destruction, along with decreased hemoglobin, serum iron, and ferritin concentrations, may occur at the initiation and early stages of a vigorous training program. Once called march hemoglobinuria, this anemia was believed to arise in soldiers as a result of mechanical trauma incurred by erythrocytes (RBCs) during long marches. The RBCs in the capillaries are compressed every time the foot lands until they burst, releasing hemoglobin. It was thought that a similar situation existed in runners, especially long-distance runners; however, it is now thought that it is a physiologic anemia (i.e., a transient problem of blood volume and dilution) (see Chapter 24 for further discussion).
Athletes who have hemoglobin concentrations below those needed for optimal oxygen delivery may benefit from consuming nutrient and iron-rich foods; ensuring that their diets contain adequate protein; and avoiding tea, coffee, antacids, H2-blockers, and tetracycline, all of which inhibit iron absorption. No athlete should take iron supplements unless true iron deficiency is diagnosed based on a complete blood cell count with differential, serum ferritin level, serum iron level, TIBC, and percent saturation of iron-binding capacity. Athletes who are female, vegetarian, involved in endurance sports, or entering a growth spurt are at risk for iron-deficiency anemia and therefore should undergo periodic monitoring.
Thalassemias
Thalassemias (α and β) are severe inherited anemias characterized by microcytic, hypochromic, and short-lived RBCs resulting from defective hemoglobin synthesis, which affects mostly persons in the Mediterranean region. The ineffective erythropoiesis leads to an increase in plasma volume, progressive splenomegaly, and bone marrow expansion with the result of facial deformities, osteomalacia, and bone changes. Ultimately there is increased iron absorption and progressive iron deposition in tissues, resulting in oxidative damage. The accumulation of iron causes dysfunction of the heart, liver, and endocrine glands. Because these patients require transfusions to stay alive, they must also have regular chelation therapy to prevent the damaging buildup of iron that can occur. Impaired growth in children accompanying thalassemia major can be partially corrected by increasing caloric intake.
 Clinical Scenario
Clinical Scenario
Dana is a 30-year-old mother of a 2-year-old and is now planning to become pregnant with her second child. Struggling to lose the last 10 pounds from her first pregnancy, her diet of choice over this past year has been a version of the low-carbohydrate diet. Dana’s food intake lacks variety and balance. She is low on fruits, vegetables, and grains. She complains of diarrhea, loss of appetite, weakness, and irritability. Her blood work reveals a normal hemoglobin level but a low serum folate level. She has scheduled an appointment to see you.
Nutrition Diagnostic Statement
Inadequate B vitamin intake related to consumption of very low-carbohydrate diet as evidenced by low serum folate level.
1. What are the risks of following a low-carbohydrate diet, especially before pregnancy?
2. What folate-containing, nutrient-dense foods could be included in her diet that would be beneficial to her pending pregnancy?
3. What supplements, if any, and in what amounts, would you recommend to Dana?
4. Which websites can you refer Dana to for her to learn more about the role of folate and neural tube defects?
5. What information do you need to gather before developing a plan for Dana? Of what would this plan consist?
Anemia Institute for Research and Education
http://www.anemiainstitute.org
References
Adams, PC, et al. Hemochromatosis and iron-overload screening in a racially diverse population. N Engl J Med. 2005;352:1769.
Akohoue, SA, et al. Energy expenditure, inflammation and oxidative stress in steady-state adolescents with sickle cell anemia. Pediatr Res. 2007;61:233.
Ashmead, D. The absorption and metabolism of iron amino acid chelate. Archivos Latino Americano De Nutricion. 2001;51:1.
Bartzokis, G, et al. Prevalent iron metabolism gene variants associated with increased brain ferritin iron in healthy older men. J Alzheimers Dis. 2010;20:333.
Connor, JR, Lee, SY. HFE mutations and Alzheimer’s disease. J Alzheimers Dis. 2006;10:267.
Dunaief, JL. Iron induced oxidative damage as a potential factor in age-related macular degeneration: the Cogan Lecture. Invest Opthamol Vis Sci. 2006;47:4660.
Elmadfa, I, Singer, I. Vitamin B12 and homocysteine status among vegetarians: a global perspective. AM J Clin Nutr. 2009;89:1693S.
Fowler, JT, et al. Dietary water and sodium intake of children and adolescents with sickle cell anemia. J Pediatr Hematol Oncol. 2010;32:350.
Graber, JJ, et al. Vitamin B12-responsive severe leukoencephalopathy and autonomic dysfunction in a patient with “normal” serum B12 levels. J Neurol Neurosurg Psychiatry. 2010;81:1369.
Hibbert, JM, et al. Proinflammatory cytokines and the hypermetabolism of children with sickle cell disease. Exp Biol Med (Maywood). 2005;230:68.
Moreiras, GV, et al. Cobalamin, folic acid, and homocysteine. Nutr Rev. 2009;67:69S.
Nemeth, E, Ganz, T. Regulation of iron metabolism by hepcidin. Ann Rev Nutr. 2006;26:323.
National Heart, Blood and Lung Institute (NHBLI). What is restless legs syndrome. Accessed 9 August 2010 from http://www.nhlbi.nih.gov/health/dci/Diseases/rls/rls_WhatIs.html.
Serefhanoglu, S, et al. Measuring holotranscobalamin II, an early indicator of negative B12 balance, by radioimmunoassay in patients with ischemic cerebrovascular disease. Ann Hematol. 2008;87:391.
Sarari, A, et al. Helicobacter pylori, a causative agent of vitamin B12 deficiency. J Infect Dev Ctries. 2008;2:346.
Smith, KA, et al. Circulating gastrin is increased in hemo-chromatosis. FEBS Lett. 2006;580:6195.
Tucker, K, Mayer, M. Low plasma vitamin B12 is associated with lower bone mineral density: the Framingham Osteoporosis Study. J Bone Miner Res. 2005;20:152.
Veijola, L. Association of autoimmune type atrophic corpus gastritis with Helicobacter pylori infection. World J Gastroenterol. 2010;16.1:83.
Zemel, BS, et al. Effects of delayed pubertal development, nutritional status, and disease severity on longitudinal patterns of growth failure in children with sickle cell disease. Pediatr Res. 2007;61:607.