Medical Nutrition Therapy for Neurologic Disorders
Neurologic diseases arise from causes that vary in complexity. Some neurologic diseases result from a simple deficiency or excess of a nutrient, whereas others have more complex causes like diabetic neuropathy, stroke, or trauma. More complicated conditions arise with the interaction of genetics with other factors, as is the case with multiple sclerosis (MS), Parkinson’s disease (PD), Alzheimer’s disease (AD), and alcoholism. The medical and health history is often the most important part of a neurologic evaluation.
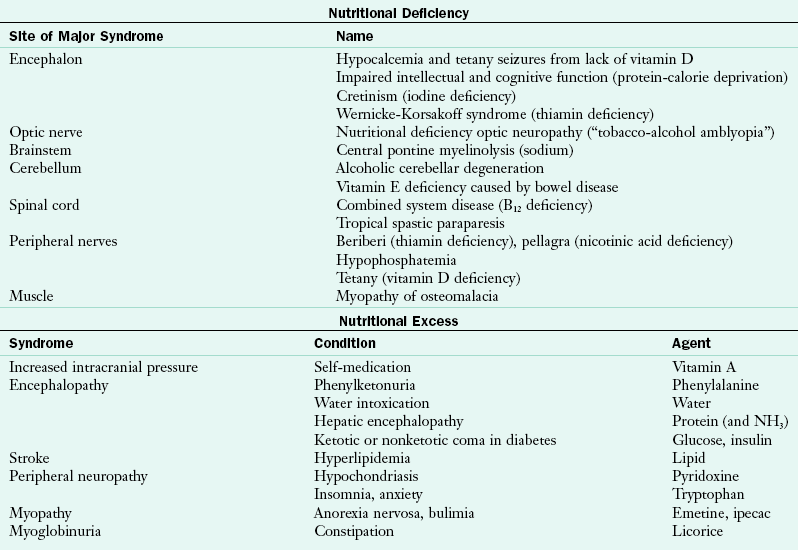
Numerous symptoms and malnutrition often accompany neurologic diseases. Complaints of even minor symptoms such as headaches, dizziness, insomnia, fatigue, weakness, pain, or discomfort must be skillfully evaluated for the presence of a nutrition component in their cause and treatment. Although not all neurologic diseases have a nutritional cause, nutritional considerations are integral to effective medical and clinical management (Table 41-1). Some neurologic dysfunctions such as peripheral neuropathy occur secondary to disease or a deficiency of a single or several vitamins, whereas other diseases of the nervous system may be attributed to dietary deficiency or excess (Table 41-2). This chapter emphasizes conditions with nutritionally significant dysfunction.
TABLE 41-1
Nutritional Considerations for Neurologic Conditions
| Medical Condition | Relevant Nutrition Therapy |
| Adrenoleukodystrophy | Dietary avoidance of VLCFAs has not been proven. |
| Lorenzo’s oil may lower VLCFA levels. | |
| Alzheimer’s disease (see Table 41-7) | Recommend antioxidants and antiinflammatory diet. |
| Minimize distractions at mealtime. | |
| Initiate smell or touch of food. | |
| Guide hand to initiate eating. | |
| Provide nutrient-dense foods and ω-3 fatty acids. | |
| Amyotrophic lateral sclerosis (see Table 41-9) | Intervene to prevent malnutrition and dehydration. |
| Monitor dysphagia. | |
| Antioxidant use ( vitamins C, E, selenium, methionine) is well tolerated, but not proven. | |
| Epilepsy | Provide ketogenic diet (see Table 41-10). |
| Guillain-Barré | Attain positive energy balance with high-energy, high-protein tube feedings. |
| Assess dysphagia. | |
| Migraine headache | Follow general recommendations for food avoidance. |
| Maintain adequate dietary and fluid intake. | |
| Keep extensive records of symptoms and foods. | |
| Myasthenia gravis | Provide nutritionally dense foods at beginning of meal. |
| Small, frequent meals are recommended. | |
| Limit physical activity before meals. | |
| Place temporary feeding tube. | |
| Multiple sclerosis | Recommend antioxidants and antiinflammatory diet. |
| Possibly recommend linoleic acid supplement. | |
| Evaluate health and especially vitamin D status of patient. | |
| Nutrition support may be needed in advanced stages. | |
| Distribute fluids throughout waking hours; limit before bed. | |
| Neurotrauma | Enteral or parenteral nutrition support needed. |
| Parkinson’s disease (see Table 41-11) | Focus on drug-nutrient interactions. |
| Minimize dietary protein at breakfast and lunch. | |
| Recommend antioxidants and antiinflammatory diet. | |
| Pernicious anemia | Administer vitamin B12 injections. |
| Provide diet liberal in HBV protein. | |
| Provide diet supplemented with Fe+, vitamin C, and B complex vitamins. | |
| Spinal trauma | Provide enteral or parenteral nutrition support. |
| Provide high-fiber diet, adequate hydration to minimize constipation. | |
| Provide dietary intake to maintain nutrition health and adequate weight. | |
| Stroke | Dietary alterations for primary prevention. |
| Maintain good nutrition status. | |
| Assess possible dysphagia. | |
| Enteral or parenteral nutrition support may be needed. | |
| Wernicke-Korsakoff syndrome | Provide thiamin supplementation. |
| Provide adequate hydration. | |
| Provide diet liberal in high-thiamin foods. | |
| Eliminate alcohol. | |
| Dietary protein may need to be restricted. |
Fe+, Iron; HBV, high biologic value; VLCFA, very-long-chain fatty acid.
In those neurologic diseases with a nonnutritional cause, nutrition therapies are important adjuncts to effective medical management. Traumatic head, brain, and spinal cord injuries are of increasing interest to some researchers secondary to athletic or sports trauma as well as injuries to military personnel, especially during war. Many elements of nutrition care for neurologic diseases and conditions are similar, regardless of the origin of the disease process.
The Central Nervous System
The central nervous system (CNS) in mammals is differentiated functionally into three components so that lesions in the nervous system leave a unique “calling card” for localized dysfunction. Localizing the defect (lesion) to muscle, nerve, spinal cord, or brain is part of the medical diagnosis. Nerve tracts coming to and from the brain cross to opposite sides in the CNS (Figure 41-1). Therefore a lesion in the brain that affects the right arm is found on the left side of the brain. Figure 41-2 shows the segments of the brain.
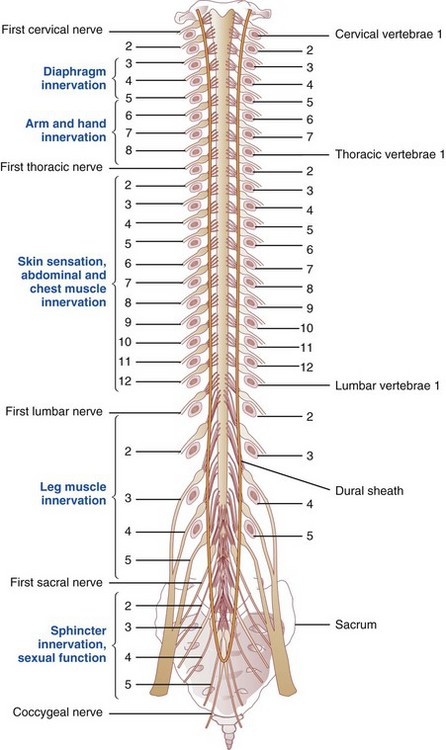
FIGURE 41-1 Spinal cord lying within the vertebral canal. Spinal nerves are numbered on the left side; vertebrae are numbered on the right side; body areas supplied by various levels are in blue.
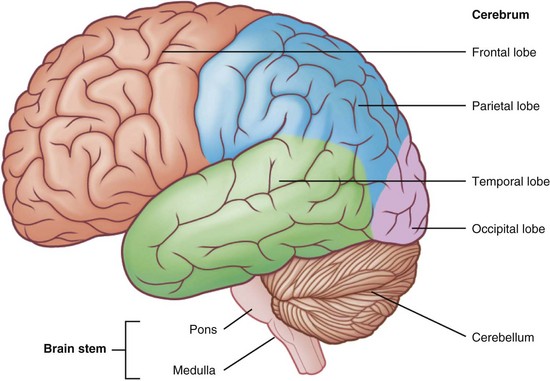
FIGURE 41-2 Parts of the brain. Trauma or disease in one area may affect speech, vision, movement or eating ability. (From http://projectflexner.sites.medinfo.ufl.edu/files/2009/04/brain-regions.jpg. From Scully C: Medical problems in dentistry, ed 6, 2010, Churchill Livingstone.)
Signs of weakness are the most quantifiable clinical signs of nervous system disease. The neurons in the motor strip (upper motor neurons) receive input from all parts of the brain and project their axons all the way to their destinations in the spinal cord. Axons connect to the spinal cord motor neurons (lower motor neurons). These neurons extend from the spinal cord to muscles without interruption. The location of a lesion in the nervous system can often be deduced clinically by observing stereotypical abnormalities and function of either upper or lower motor neurons (Table 41-3).
TABLE 41-3
Basic Functions of Cranial Nerves
| Number | Nerve Function |
| Olfactory (I) | Smell |
| Optic (II) | Vision |
| Oculomotor (III) | 1. Eye movement |
| 2. Pupil constriction | |
| Trochlear (IV) | Eye movement |
| Trigeminal (V) | 1. Mastication |
| 2. Facial heat, cold, touch | |
| 3. Noxious odors | |
| 4. Input for corneal reflex | |
| Abducens (VI) | Eye movement |
| Facial (VII) | 1. All muscles of facial expression |
| 2. Corneal reflex | |
| 3. Facial pain | |
| 4. Taste on anterior two thirds of tongue | |
| Vestibulocochlear (VIII) | Hearing and head acceleration and input for oculocephalic reflex |
| Glossopharyngeal (IX) | 1. Swallowing |
| 2 Gag reflex | |
| 3. Palatal, glossal, and oral sensation | |
| Vagus (X) | 1. Heart rate, gastrointestinal activity, sexual function |
| 2. Cough reflex | |
| 3. Taste on posterior third of tongue | |
| Spinal accessory (XI) | 1. Trapezius |
| 2. Sternocleidomastoid muscle | |
| Hypoglossal (XII) | Tongue movement |
Pathophysiology and Signs of Mass Lesions
The frontal lobes in the brain are the source of the most complex activities and commonly offer the most complex presentations. Psychiatric manifestations such as depression, mania, or personality change may herald a tumor or other frontal lobe mass, either right or left. With a lesion or tumor near the skull base, one may lose the sense of smell or have visual changes because olfactory and optic nerves track along the bottom of these frontal lobes. Chemosensory losses of smell have been described as anosmia (absence of smell), hyperosmia (increased sensitivity of smell), or dysomia (distortion of normal smell).
Frontal lobes are larger, and the posterior portions of the frontal lobes contain the motor strips. Lesions in the central portion of the frontal lobes may present as a motor apraxia. A person with apraxia cannot properly execute a complex activity, although he or she may understand a request to perform the activity.
Temporal lobes control memory and speech; lesions there may affect these abilities, as seen with AD and stroke. Although any lesion of cerebral gray matter may produce seizures, the temporal lobes are particularly prone to seizures. A right parietal lobe mass may result in chronic inability to focus attention, thus completely ignoring the body’s left side. Because speech centers are located near the junction of the left temporal, parietal, and frontal lobes, pathologic conditions in this region may cause speech problems.
The occipital lobes are reserved for vision, and dysfunction here may bring about cortical blindness of varying degrees. In this condition the person is unaware that he or she cannot see. Lesions at other points along the visual pathway can cause several different types of visual field deficits.
Lesions of the cerebellum and brainstem may obstruct the ventricular system where it is the narrowest. This obstruction may precipitate life-threatening hydrocephalus, a condition of increased intracranial pressure (ICP) that may quickly result in death. Other signs of hydrocephalus include trouble with balance, walking and coordination abnormalities, marked sleepiness, and complaints of a headache that is worse on awakening. Lesions in the brainstem may infiltrate any of the cranial nerves that enervate structures of the face and head, including the eyes, ears, jaw, tongue, pharynx, and facial muscles (see Table 41-3). These lesions have consequences for nutrition because the patient is often unable to eat without risking aspiration of food or liquids into the lung. Tumors or other lesions in the medulla may infiltrate respiratory and cardiac centers, and dysregulation of these centers has grim consequences.
Lesions in the spinal cord are much less common than brain tumors and ordinarily cause lower motor neuron signs at the level of the lesion and upper motor signs in segments below the level of the lesion. Spinal cord injury (SCI) is the most common pathologic condition in this region. Other examples of spinal cord abnormalities are MS, amyotrophic lateral sclerosis (ALS), tumor, syrinx (fluid-filled neurologic cavity), chronic meningitis, vascular insufficiency, and mass lesions of the epidural space.
Lesions of the pituitary gland and hypothalamus are often heralded by systemic manifestations that may include electrolyte and metabolic abnormalities secondary to adrenocortical, thyroid, and antidiuretic hormone dysregulation. Because of the proximity to the visual pathways, changes may occur in visual field or acuity. The syndrome of inappropriate antidiuretic hormone secretion (SIADH) is often a complication; volume status and hyponatremia are part of the medical diagnosis (see Chapter 7). Because the hypothalamus is the regulatory center for hunger and satiety, lesions here may present as anorexia or overeating.
Finally, disorders of peripheral nerves and the neuromuscular junction affect one’s ability to maintain proper nutrition. Disorders such as Guillain-Barré syndrome (GBS) or myasthenia gravis (MG) may counteract the efforts to maintain nutritional balance. To eat and drink effectively, many parts of the nervous system are required. A problem at any step along the way can result in an inability to meet metabolic demands.
Issues Complicating Nutrition Therapy
The nutritional management of patients with neurologic disease is complex. Severe neurologic impairments often compromise the mechanisms and cognitive abilities needed for adequate nourishment. Many patients have dysphagia (difficulty swallowing), and the ability to obtain, prepare, and present food to the mouth can be compromised. As a result, all patients with neurologic disease are at risk for malnutrition. Early recognition of signs and symptoms, implementation of an appropriate care plan to meet the nutritional requirements of the individual, and counseling for the patient and family members on dietary choices are essential. Regular evaluation of the patient’s nutrition status and disease management are priorities, with the ultimate goal of improving outcomes and the patient’s nutritional quality of life.
Nutrition assessment requires detailed histories. The diet history and mealtime observations are used to assess patterns of normal chewing, swallowing, and rate of ingestion. Weight loss history establishes a baseline weight; a weight loss of 10% or more is indicative of nutritional risk. Assessment for nutrients involved in neurotransmitter synthesis is particularly important in these patients (Table 41-4). Nutrition diagnoses common in the neurologic patient population include:

• Increased energy expenditure
• Inadequate oral food and beverage intake
Meal Preparation
Confusion, dementia, impaired vision, or poor ambulation may contribute to difficulty with meal preparation, thus hindering oral food and beverage intake. Assistance with shopping and meal planning are frequently necessary.
Self-Feeding Difficulties and Inadequate Access to Food or Fluid
With chronic neurologic diseases, a decline in function may hinder the ability for self-care and nourishment. Access to food and satisfying basic needs may depend on the involvement of family, friends, or professionals. With acute neurologic situations such as trauma, stroke, or GBS, the entire process of eating can be interrupted abruptly. The patient may require enteral nutrition for a time until overall function improves and adequate oral intake is resumed.
Feeding Issues: Presentation of Food to the Mouth
The patient with neurologic disease may be unable to feed himself or herself because of limb weakness, poor body positioning, hemianopsia, apraxia, confusion, or neglect. Tremors in PD, spastic movements, or involuntary movements that occur with cerebral palsy, Huntington’s disease, or tardive dyskinesia may further restrict dietary intake. The affected region of the CNS determines the resulting disability (Table 41-5).
TABLE 41-5
Common Impairments with Neurologic Diseases
| Site in the Brain | Impairment | Results |
| Cortical lesions of the parietal lobe (perception of sensory stimuli) | Sensory deficits | Fine regulation of muscle activities impossible if the patient is unable to perceive joint position and motion and tension of contracting muscles. |
| Lesions of the nondominant hemisphere | Hemi inattention syndrome (neglect) | Patient neglects that side of the body. |
| Optic tract lesions (usually of the middle cerebral artery or the artery near the internal capsule) | Visual field cuts | Patient reads one half of a page, eats from only half of the plate, etc. (see Figure 41-3). |
| Loss of subcortically stored pattern of motor skills | Apraxia | Inability to perform a previously learned task (e.g., walking, rising from a chair), but paralysis, sensory loss, spasticity, and incoordination are not present. |
| No identification with a particular brain disorder or a specifically located lesion | Language apraxia | Inability to produce meaningful speech, even though oral muscle function is intact and language production has not been affected. |
| Lesion of Broca area | Nonfluent aphasia | Thought and language formulation are intact, but the patient is unable to connect them into fluent speech production. |
| Lesion of Wernicke area | Fluent aphasia | Flow of speech and articulation seem normal, but language output makes little or no sense. |
| Extensive brain damage | Global aphasia | Both expression and speech perception are severely impaired. |
| Brainstem lesions, bilateral hemispheric lesions, cerebellar disorders | Dysarthria | Inability to produce intelligible words with proper articulation. |
From Steinberg FU: Rehabilitating the older stroke patient: what’s possible? Geriatrics 41:85, 1986.
If limb weakness or paralysis occurs on the dominant side of the body, poor coordination resulting from a new reliance on the nondominant side may make eating difficult and unpleasant. The patient may have to adjust to eating with one hand and also to using the nondominant hand. Hemiparesis is weakness on one side of the body that causes the body to slump toward the affected side; it may increase a patient’s risk of aspiration.
Hemianopsia is blindness for one half of the field of vision. The patient must learn to recognize that he or she no longer has a normal field of vision and must compensate by turning the head. Neglect is inattention to a weakened or paralyzed side of the body; this occurs when the nondominant (right) parietal side of the brain is affected. The patient ignores the affected body part, and his or her perception of the body’s midline is shifted. Hemianopsia and neglect can occur together and severely impair the patient’s function. A patient may eat only half of the contents of a meal because he or she recognizes only half of it (Figure 41-3).

Another potential interference with self-feeding is apraxia because the person is unable to carry out an action and follow directions. Demonstration may make it possible to do the action; however, judgment may be affected as well and can result in the performance of dangerous tasks. This makes it unsafe to leave the patient alone.
Dysphagia
Dysphagia often leads to malnutrition because of inadequate intake. Symptoms of dysphagia may include drooling, choking, or coughing during or following meals; inability to suck from a straw; a gurgly voice quality; holding pockets of food in the buccal recesses (of which the patient may be unaware); absent gag reflex; and chronic upper respiratory infections. Patients with intermediate or late-stage PD, MS, ALS, dementia, or stroke are likely to have dysphagia.
A swallowing evaluation by a speech-language pathologist (SLP) is important in assessing and treating swallowing disorders. The SLP is often consulted for individual patients following traumatic brain injury (TBI), stroke, or cancers of the head and neck, and for those at risk of aspiration or with other conditions that result in a lack of coordination in swallowing. Many registered dietitians (RDs) have acquired additional training in swallowing therapies to help coordinate this evaluative process.
Phases of Swallowing
Proper position for effective swallowing should be encouraged (i.e., sitting bolt upright with the head in a chin-down position). Concentrating on the swallowing process can also help reduce choking. Initiation of the swallow begins voluntarily but is completed reflexively. Normal swallowing allows for safe and easy passage of food from the oral cavity through the pharynx and esophagus into the stomach by propulsive muscular force, with some benefit from gravity. The process of swallowing can be organized into three phases, as shown in Figure 41-4.
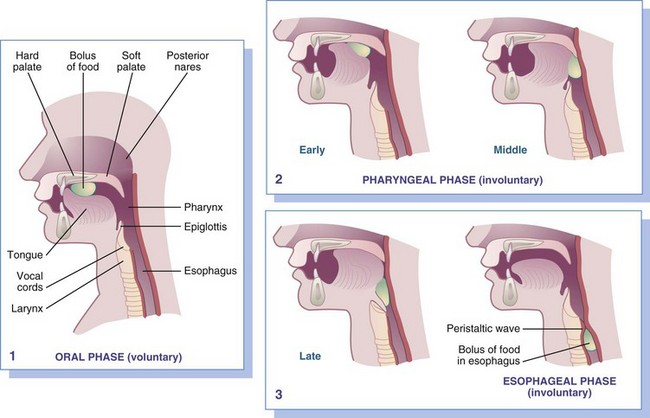
FIGURE 41-4 Swallowing occurs in three phases:
1.
Voluntary or oral phase: Tongue presses food against the hard palate, forcing it toward the pharynx.
2.
Involuntary, pharyngeal phase: Early: wave of peristalsis forces a bolus between the tonsillar pillars. Middle: soft palate draws upward to close posterior nares, and respirations cease momentarily. Late: vocal cords approximate, and the larynx pulls upward, covering the airway and stretching the esophagus open.
3.
Involuntary, esophageal phase: Relaxation of the upper esophageal (hypopharyngeal) sphincter allows the peristaltic wave to move the bolus down the esophagus.
Oral Phase
During the preparatory and oral phases of swallowing, food is placed in the mouth, where it is combined with saliva, chewed if necessary, and formed into a bolus by the tongue. The tongue pushes the food to the rear of the oral cavity by gradually squeezing it backward against the hard and soft palate. Increased ICP or intracranial nerve damage may result in weakened or poorly coordinated tongue movements and lead to problems in completing the oral phase of swallowing. Weakened lip muscles result in the inability to completely seal the lips, form a seal around a cup, or suck through a straw. Patients are often embarrassed by drooling and may not want to eat in front of others. The patient may have difficulty forming a cohesive bolus and moving it through the oral cavity. Food can become pocketed in the buccal recesses, especially if sensation in the cheek is lost or facial weakness exists.
Pharyngeal Phase
The pharyngeal phase is initiated when the bolus is propelled past the faucial arches. Four events must occur in rapid succession during this phase. The soft palate elevates to close off the nasopharynx and prevent oropharyngeal regurgitation. The hyoid and larynx elevate, and the vocal cords adduct to protect the airway. The pharynx sequentially contracts while the cricopharyngeal sphincter relaxes, allowing the food to pass into the esophagus. Breathing resumes at the end of the pharyngeal phase. Symptoms of poor coordination during this phase include gagging, choking, and nasopharyngeal regurgitation.
Esophageal Phase
The final or esophageal phase, during which the bolus continues through the esophagus into the stomach, is completely involuntary. Difficulties that occur during this phase are generally the result of a mechanical obstruction, but neurologic disease cannot be ruled out. For example, impaired peristalsis can arise from a brainstem infarct.
Medical Nutrition Therapy
Weight loss and anorexia are key concerns with dysphagia. Observation during meals allows the nurse or RD to screen informally for signs of dysphagia and bring them to the attention of the health care team. Environmental distractions and conversations during mealtime increase the risk for aspiration and should be curtailed. Reports of coughing and unusually long mealtimes are associated with tongue, facial, and masticator muscle weakness. Changing the consistency of foods served may be beneficial. Keep the diet palatable and nutritionally adequate by recommending changes in food consistency. A mechanically soft or pureed consistency can reduce the need for oral manipulation and to conserve energy while eating (Box 41-1). Appendix 35 provides more detail for the National Dysphagia Diet.
Liquids
Swallowing liquids of thin consistency such as juice or water requires the most coordination and control. Liquids are easily aspirated into the lungs and may pose a life-threatening event because aspiration pneumonia may ensue, even from sterile water in the lungs. Sterile water is no longer sterile once it is introduced to the bacterial load of the oral cavity.
If a patient has difficulty consuming thin liquids, fluid requirements must still be met. Liquids of all types can be thickened with nonfat dry milk powder, cornstarch, modular carbohydrate supplements, or commercial thickeners that contain a modified cornstarch thickener. Thick liquids that contain a high percentage of water are needed to maintain fluid balance. Fatigue and malaise are often associated with a “mild chronic dehydration” that results from decreased fluid intake. Fresh fruit is an additional source of free water.
Some long-term care facilities use the Frazier Water Protocol, which allows for drinking water in those who otherwise require thickened liquids. This protocol is based on the following assumptions:
1. Aspiration of water poses little risk to the patient if oral bacteria associated with the development of aspiration pneumonia can be minimized.
2. Allowing free water decreases the risk of dehydration.
3. Allowing free water increases patient compliance with swallowing precautions and improves quality of life.
4. Good oral hygiene is a key ingredient of the water protocol and offers other benefits to swallowing.
Liquid intake is a concern in those with neurogenic bladder and urinary retention, a common management issue in patients with a myelopathy (a pathologic condition of the spinal cord) or an SCI. This predisposes the individual to urinary tract infections (UTIs) and miscalculation of fluid balance. Alternately, myelopathy and SCI may result in urinary urgency, frequency, or incontinence. To minimize these problems, distributing fluids evenly throughout the waking hours and limiting them before bedtime may help. Some patients limit fluid intake severely to decrease urgency or frequent urination. This practice increases the risk of UTI and is not recommended.
One nontraumatic cause of myelopathy and neurogenic bladder is MS, an unpredictable and severe, progressive disease of the CNS. Individuals with MS have a higher incidence of UTIs. Increased intake of cranberry juice may reduce the frequency of UTIs (see Chapter 36).
Milk is considered a liquid with unique properties. Some people associate consumption of milk with symptoms of excess mucus production; however, no evidence shows an increase in mucus production. When the dysphagic patient reports increased phlegm after milk consumption, it may actually be a consequence of poor swallowing ability rather than mucus production. Patients are encouraged to “chase” the milk products with appropriately thickened liquids to help flush the throat rather than eliminate nutrient-rich dairy products.
Textures
As chronic neurologic disease progresses, cranial nerves become damaged, leading to neurologic deficits often manifested by dysphagia or elimination of entire food groups. Nutrition intervention should be individualized according to the type and extent of dysfunction. Vitamin and mineral supplementation may be necessary. If chewable supplements are not handled safely, liquid forms may be added to acceptable foods.
Presented with small, frequent meals, the patient may eat more. Swallowing can also be improved by carefully selecting various tastes, textures, and temperatures of foods. Juices can be substituted for water and provide flavor, nutrients, and calories. A cool temperature facilitates swallowing; therefore cold food items may be better tolerated. Carbonation may also be better tolerated because there is the beneficial effect of texture. Sauces and gravies lubricate foods for ease in swallowing and can help prevent fragmentation of foods in the oral cavity. Moist pastas, casseroles, and egg dishes are generally well tolerated. Avoid foods that crumble easily in the mouth, because they can increase choking risk.
Enteral Nutrition
Patients with acute and chronic neurologic diseases may benefit from nutrition support. In acute disease it may be required initially until a degree of function is regained, whereas in chronic neurologic disease it may be required in the late stages to meet changing metabolic demands. Well-managed nutrition support helps to prevent pneumonia and sepsis, which can complicate these diseases. Enteral tube feedings may be necessary if the risk of aspiration from oral intake is high, or if the patient cannot eat enough to meet his or her nutritional needs. In the latter case nocturnal tube feedings can bridge the gap between oral intake and actual nutritional requirements. This should allow the normal sensation of hunger to be generated and provide freedom from tube feeding during the day.
In most instances the gastrointestinal tract function remains intact, and enteral nutrition is the preferred method of administering nutrition support. One noted exception occurs after SCI, in which ileus is common for 7 to 10 days after the insult, and parenteral nutrition may be necessary. Although a nasogastric (NG) tube can be a short-term option, a percutaneous endoscopic gastrostomy (PEG) or gastrostomy-jejunostomy (PEG/J) tube is preferred for long-term management. These should be considered for patients whose swallowing is inadequate to ensure their nutritional health (see Chapter 14).
Malnutrition itself can produce neuromuscular weakness that negatively affects quality of life; it is a prognostic factor for poor survival. In the acutely ill but previously well-nourished individual who is unable to resume oral nourishment within 7 days, nutrition support is used to prevent decline in nutritional health and aid in recovery until oral intake can be resumed. Conversely, in the chronically ill, nutrition support is an issue that each patient must eventually address because it may result in prolonged therapy. However, adequate nutrition can promote health and may be a welcome relief to an overburdened patient.
Some patients may decline early placement of a feeding tube because of the emotional, economic, or physical effects of this choice. In advanced stages of disease the patient may refuse tube feedings, choosing not to prolong life. Nutrition support should be used when it can enhance quality of life. The health care team should alleviate patient and family concerns and support informed decisions. The patient needs to be fully informed about the effects of tube feeding on daily life. Discussion of both the advantages and disadvantages of nutrition support should be initiated with the patient and family well ahead of need. Options should include a description of feeding schedules, tube placement procedures, and appropriate training for on-going care.
Neurologic Diseases of Nutritional Origin
Dietary deficiencies of thiamin and niacin can directly result in neurologic symptoms. With Wernicke-Korsakoff syndrome (WKS), the neurologic effect occurs secondary to alcoholism. Most neurologic symptoms arising from nutritional deficiencies can be corrected with increased food intake or supplements. See Table 41-2 through Table 41-6.

Neurologic Disorders from Trauma
Cerebrovascular Accident (Stroke)
Stroke (cerebrovascular accident) is defined as an acute onset of focal or global neurologic deficit lasting more than 24 hours; it is attributable to diseases of the intracranial or extracranial neurovasculature. Severe strokes are often preceded by transient ischemic attacks (TIAs), brief attacks of cerebral dysfunction of vascular origin with no persistent neurologic defect. Stroke is the third most common cause of death in the United States and the most common cause of disability in the United States (National Institutes of Health [NIH], 2006). Old age is the most significant risk factor for stroke. Among modifiable risk factors, hypertension and smoking contribute most often. (See Chapter 33.) Other factors include obesity, coronary heart disease, diabetes, physical inactivity, and genetics (Goldstein et al., 2006). The high costs of stroke are attributed to the degree of disability imparted by these events.
Pathophysiology
Embolic stroke occurs when a cholesterol plaque is dislodged from a proximal vessel, travels to the brain, and blocks an artery, most commonly the middle cerebral artery (MCA). In patients with dysfunctional cardiac atria, clots may be dislodged from there and embolize. In thrombotic stroke a cholesterol plaque within an artery ruptures, and platelets subsequently aggregate to clog an already narrowed artery. Most strokes are incited by a thromboembolic event, which may be aggravated by atherosclerosis, hypertension, diabetes, and gout (Pathophysiology and Care Management Algorithm: Stroke).
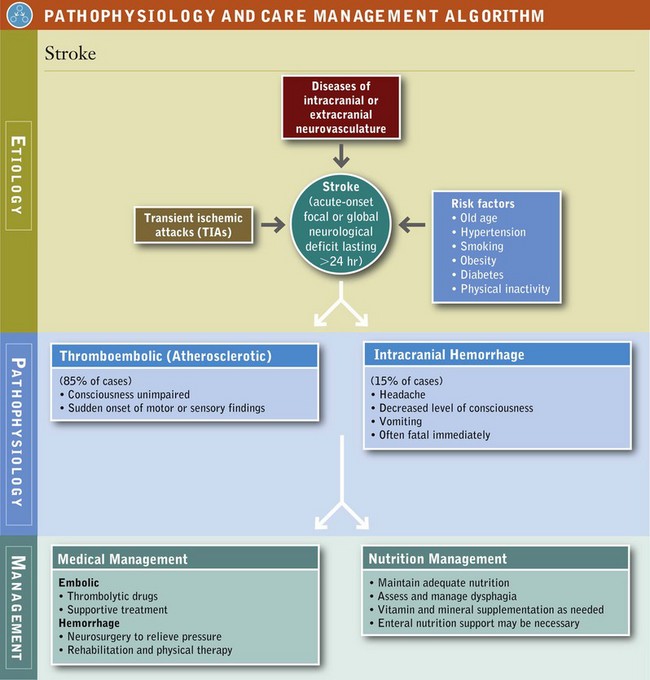
Intracranial hemorrhage occurs in only 15% of strokes but is often fatal immediately. Intracranial hemorrhage occurs more commonly in individuals with hypertension. In intraparenchymal hemorrhage, a vessel inside the brain ruptures. A variation of intraparenchymal hemorrhage is a lacunar (deep pool) infarct. These smaller infarcts occur in the deep structures of the brain such as the internal capsule, basal ganglia, pons, thalamus, and cerebellum. Even a small lacunar infarct can produce significant disability because the brain tissue in the deep structures is so densely functional. A second type of intracranial hemorrhage is subarachnoid hemorrhage (SAH). SAH occurs commonly as a result of head trauma but more often as a result of a ruptured aneurysm of a vessel in the subarachnoid space.
Medical Management
Hemorrhage is suspected when the patient presents with headache, decreased level of consciousness, and vomiting, all of which occur within minutes to hours. A thromboembolic stroke is more likely to occur when the patient is fully conscious, but onset of motor or sensory changes occurs suddenly. As with all neurologic disease, the clinical presentation depends on the location of the abnormality. An infarct of a particular cerebrovascular territory can be suspected by seeking out various neurologic deficits. An MCA occlusion produces paresis, with sensory deficits of limbs on the opposite side of the body because this artery supplies the motor and sensory strips. If the left MCA is occluded, aphasia, or loss of speech or expression may be present.
In the past, treatment for embolic stroke was supportive; it focused on prevention of further brain infarction and rehabilitation. Use of thrombolytic, “clot-busting” drugs reverses brain ischemia by lysing the clots. Initiation of therapy needs to occur within 6 hours of the onset of symptoms. Use of aspirin may be of some value in preventing further cerebrovascular events, but its effectiveness varies from one patient to another.
Controlling ICP while maintaining sufficient perfusion of the brain is the treatment for intracranial hemorrhage. This may include surgical evacuation of large volumes of intracranial blood, ventricular drainage, or other neurosurgical interventions. Rehabilitation is a key component. Hemorrhage, particularly SAH, has severe functional consequences and therefore has a longer period of convalescence than ischemic stroke.
Medical Nutrition Therapy
Primary prevention is the cornerstone for preventing stroke, in part by diet as well as by other lifestyle behaviors (Goldstein et al., 2006). Nutrition-related factors have been compiled from various large population-based prospective studies (Box 41-2).
Given the prevalence of stroke and its associated burden of disease, treatment for those afflicted with this disease cannot be ignored. In 2003 alone, strokes cost the United States an estimated $51 billion in lost productivity and health, including $12 billion in nursing home expenses (Centers for Disease Control and Prevention, 2006). Efforts should be directed toward maintaining the overall health of the patient. Omega-3 fatty acids may prevent some types of stroke, but should be avoided by anyone taking a blood thinner like warfarin or aspirin.
Feeding difficulties are determined by the extent of the stroke and the area of the brain affected. Dysphagia, an independent predictor of mortality, commonly accompanies stroke and contributes to complications and poor outcome from malnutrition, pulmonary infections, disability, increased length of hospital stay, and institutional care. In some instances nutrition support is required to maintain nutritional health until oral alimentation can be resumed. As motor functions improve, eating and other activities of daily living are part of the patient’s rehabilitation process and necessary for resuming independence. Malnutrition predicts a poor outcome and should be prevented.
Head Trauma or Neurotrauma
Traumatic brain injury (TBI) refers to any of the following, alone or in combination: brain injury, skull fractures, extraparenchymal hemorrhage—epidural, subdural, subarachnoid—or hemorrhage into the brain tissue itself, including intraparenchymal or intraventricular hemorrhage. In the United States trauma is the leading cause of death in persons up to 44 years of age, and more than one half of these deaths are the result of head injuries (Victor and Ropper, 2005). The annual incidence is estimated to be 200 per 100,000 people, with a peak frequency between 15 and 24 years of age. Motor vehicle collisions are the major source of injury.
Morbidity is high and headache is one of the most common complaints. It is difficult to accurately predict neurologic recovery. Despite intensive intervention, long-term disability occurs in a large portion of the survivors of severe head injury.
Pathophysiology
Brain injury can be categorized as three types: concussion, contusion, and diffuse axonal injury. A concussion is a brief loss of consciousness, less than 6 hours, with no damage found on computed tomography (CT) or magnetic resonance imaging (MRI) scans. Microscopic studies have failed to find any evidence of structural damage in areas of known concussion, although evidence of change in cellular metabolism exists. A contusion is characterized by damaged capillaries and swelling, followed by resolution of the damage. Large contusions may dramatically increase ICP and may lead to ischemia or herniation. Contusions can be detected by CT or MRI scans. Diffuse axonal injury results from the shearing of axons by a rotational acceleration of the brain inside the skull. Damaged areas are often found in the corpus callosum (the bridge between the two hemispheres) and the upper, outer portion of the brainstem.
Skull fractures of the calvarium and the base are described in the same manner as other fractures. Comminution refers to splintering of bone into many fragments. Displacement refers to a condition in which bones are displaced from their original positions. Open or closed describes whether a fracture is exposed to air. Open fractures dramatically increase the risk of infection (osteomyelitis), and open skull fractures in particular carry an increased risk for meningitis because the dura mater is often violated.
Epidural and subdural hematomas are often corrected by surgical intervention. The volume of these lesions often displaces the brain tissue and may cause diffuse axonal injury and swelling. When the lesion becomes large enough, it may cause herniation of brain contents through various openings of the skull base. Consequent compression and ischemia of vital brain structures often rapidly lead to death.
Medical Management
The body’s response to stress from TBI results in production of cytokines (interleukin-1, interleukin-6, interleukin-8, and tumor necrosis factor). These are elevated in the body after head injury and are associated with the hormonal milieu that negatively affects metabolism and organ function. Some of the metabolic events include fever, neutrophilia, muscle breakdown, altered amino acid metabolism, production of hepatic acute-phase reactants, increased endothelial permeability, and expression of endothelial adhesion molecules. Specific cytokines tend to cause organ demise; tissue damage has been observed in the gut, liver, lung, and brain. Overall, the molecular basis of functional recovery is poorly understood. (See Chapter 39.)
Clinical findings of brain injury often include a transient decrease in level of consciousness. Headache and dizziness are relatively common and not worrisome unless they become more intense or are accompanied by vomiting. Focal neurologic deficits, progressively decreasing level of consciousness, and penetrating brain injury demand prompt neurosurgical evaluation.
Skull fractures underneath lacerations can often be felt as a “drop off” or discontinuity on the surface of the skull, and are readily identifiable by CT scan. Basilar skull fractures are manifested by otorrhea (fluid leaking from the ear) or rhinorrhea (salty fluid dripping from the nose or down the pharynx). Other signs include raccoon eyes and Battle’s sign—blood behind the mastoid process. Basilar skull fractures may precipitate injuries to cranial nerves, which are essential for chewing, swallowing, taste, and smell.
Hematomas are neurosurgical emergencies because they may rapidly progress to herniation of brain contents through the skull base and to subsequent death. These lesions may present similarly, with decreased level of consciousness, contralateral hemiparesis, and pupillary dilation. These lesions damage brain tissue by gross displacement and traction. Classically the epidural hematoma presents with progressively decreasing consciousness after an interval of several hours during which the patient had only a brief loss of consciousness. Subdural hematoma usually features progressively decreasing consciousness from the time of injury.
Sequelae most often include epilepsy and the postconcussive syndrome, a constellation of headache, vertigo, fatigue, and memory difficulties. Treatment for these patients can be highly complex, but the two goals of any therapeutic intervention are to maintain cerebral perfusion and to regulate ICP. Perfusion and pressure control have implications for nutrition therapy.
Medical Nutrition Therapy
The goal of nutrition therapy is to oppose the hypercatabolism and hypermetabolism associated with inflammation. Hypercatabolism is manifested by protein degradation, evidenced by profound urinary urea nitrogen excretion. Nitrogen catabolism in a fasting normal human is only 3 to 5 g of nitrogen per day, whereas nitrogen excretion is 14 to 25 g of nitrogen per day in the fasting patient with severe head injury. In the absence of nutritional intake, this degree of nitrogen loss can result in a 10% decrease in lean mass within 7 days. A 30% weight loss increases mortality rate (Brain Trauma Foundation, 2007).
Hypermetabolism contributes to increased energy expenditure. Correlations between the severity of brain injury as measured by the Glasgow Coma Scale and energy requirements have been shown. Replacement of 100% to 140% resting metabolism expenditure with 15% to 20% nitrogen calories reduces nitrogen loss (Brain Trauma Foundation, 2007). In patients medicated with barbiturates, metabolic expenditure may be decreased to 100% to 120% of basal metabolic rate. This decreased metabolic rate in pharmacologically paralyzed patients suggests that maintaining muscle tone is an important part of metabolic expenditure.
Nourishing the neurologically critically ill patient is accomplished by administering either enteral or parenteral nutrition support. Nutrition support is usually begun within 72 hours after injury and is necessary to achieve nutritional replacement by 7 days after injury (Brain Trauma Foundation, 2007). Both modes of therapy must be initiated at levels below actual requirements and increased gradually to meet nutritional requirements. For more guidelines, refer to Chapter 39.
Research in this area is exciting. An experimental dietary intervention based on a pyrazole curcumin derivative reestablishes membrane integrity and homeostasis following TBI (Sharma et al., 2010). The ω-3 fatty acids docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA) have antioxidative, anti-inflammatory, and antiapoptosis effects, leading to neuron protection in the damaged brain (Su, 2010). Clearly, more nutrient trials are needed in this population.
Spine Trauma and Spinal Cord Injury
Spine trauma encompasses many types of injuries, ranging from stable fractures of the spinal column to catastrophic transsection of the spinal cord. A complete spinal cord injury (SCI) is defined as a lesion in which there is no preservation of motor or sensory function more than three segments below the level of the injury. With an incomplete injury there is some degree of residual motor or sensory function more than three segments below the lesion. SCI is somewhat less common than head injury; both are most often seen in the young. Motor vehicle collisions account for one third to one half of SCIs; athletic injuries and domestic and industrial accidents account for the remainder.
Pathophysiology
The spinal cord responds to insult in a manner similar to the brain. Bleeding, contusion, and shorn axons appear first, followed by a several-year remodeling process consisting of gliosis and fibrosis. Liquefactive necrosis may predispose to the formation of a syrinx, fluid collection in the center of the spinal cord; effects manifest as a slow but progressive neurologic deficit. SCIs are usually associated with spinal column fractures and ligament instability. Such processes may be amenable to either surgical or nonsurgical reduction and stabilization.
The location of the SCI and the disruption of the descending axons determines the extent of paralysis. Tetraplegia (formerly known as quadraplegia) exists when the injury to the spinal cord affects all 4 extremities. When the SCI location results in only lower extremity involvement it is called paraplegia.
Medical Management
Spinal cord injuries have numerous clinical manifestations, depending on the level of the injury. Complete transsection results in complete loss of function below the level of the lesion, including the bladder and sphincters. After the patient is stabilized hemodynamically, the doctor evaluates the degree of neurologic deficit. Patients with suspected SCI are usually immobilized promptly in the field. Complete radiographic evaluation of the spinal column is obligatory in multitrauma and unconscious patients.
In the awake patient, clinical evidence of spine compromise is usually sufficient to determine the need for further workup. CT and MRI are used to more accurately delineate bony damage and spinal cord compromise. A dismal 3% of patients with complete spinal cord insults recover some function after 24 hours. Failure to regain function after 24 hours predicts a 0% chance of reestablishment of function in the future. Incomplete spinal cord syndromes may have somewhat improved outcomes.
Morbidity and mortality rates associated with SCI have improved dramatically, particularly in the last two decades. Advances in acute-phase care have reduced early mortality and prevented complications frequently associated with early death, such as respiratory failure and pulmonary emboli. Today, fewer than 10% of patients with SCI die from the acute injury.
Medical Nutrition Therapy
Technologic advances in enteral and parenteral feeding techniques and formulas have played a role in maintaining nutrition status of these patients. Although the metabolic response to neurotrauma has been studied extensively, the acute metabolic response to SCI has not; but it is similar to other forms of neurotrauma during the acute phase. Initially paralytic ileus may occur but often resolves within 72 hours after injury. Because DHA and EPA have antioxidative, anti-inflammatory, antiapoptosis effects, patients may benefit from fish oil supplementation (Su, 2010).
For those who survive the injury but are disabled for life, there are significant alterations in lifestyle as well as the possibility of secondary complications. In general the number and frequency of complications, and the presence of constipation, pressure ulcers, obesity, and pain vary but will involve nutrition. Box 41-3 describes the rehabilitation potential based on the level of injury. Evidence-based practice guidelines for SCI were released in 2010 by the American Dietetic Association.
Individuals with SCI have significantly high fat mass and low lean mass. Loss of muscle tone caused by skeletal muscle paralysis below the level of injury contributes to decreased metabolic activity, initial weight loss, and predisposition to osteoporosis. Guidelines for accepted weights adjusted for paraplegia and tetraplegia are as follows: the paraplegic should weigh 10 to 15 lb less than the ideal body mass index (BMI) would indicate; the tetraplegic should weigh 15 to 20 lb less than ideal weight dictated by the BMI. The higher the injury, the lower the metabolic rate; a lower energy requirement occurs.
Tetraplegic patients have lower metabolic rates than paraplegic patients, proportional to the amount of denervated muscle in their arms and legs, caused in part by the loss of residual motor function. In the rehabilitation phase, tetraplegics may require approximately 25% to 50% fewer calories than conventional equations predict. Thus these patients have the potential to become overweight. It has been proposed that obesity may slow the eventual rehabilitation process by limiting functional outcome.
As a consequence of bone loss resulting from the loss of mineralization caused by immobilization, SCI is associated with osteopenia and osteoporosis, and the prevalence of long-bone fractures increases. Adequate intake of vitamin D and calcium should be planned without excessive daily intakes.
Neurologic Diseases
Pathophysiology
Adrenomyeloleukodystrophy (ALD) is a rare congenital enzyme deficiency that affects the metabolism of very-long-chain fatty acids (VLCFAs) in young men. This leads to accumulation of VLCFAs, particularly hexacosanoic acid (C26:0) and tetracosanoic acid (C24:0) in the brain and adrenal glands (Deon et al., 2006). The incidence is 1 in 21,000 male births and 1 in 14,000 female (Moser, 2006). It is an X-linked recessive disorder characterized by myelopathy, peripheral neuropathy, and cerebral demyelination. The adult variant, adrenomyeloneuropathy, has chronic distal axonopathy of spinal cord and peripheral nerves marked by cerebral inflammatory demyelination; head trauma is an environmental factor that is detrimental in those people genetically at risk (Raymond et al., 2010). The mental and physical deterioration progresses to dementia, aphasia, apraxia, dysarthria, and blindness.
Medical Management
Clinical manifestations usually occur before age 7 and may manifest as adrenal insufficiency or cerebral decompensation.
Dysarthria (impairment of the tongue or other muscles needed for speech) and dysphagia may interfere with oral alimentation. Bronzing of the skin is a late clinical sign. With adrenal insufficiency, replacement of steroids is indicated, which may improve neurologic symptoms and prolong life. Numerous therapies have been directed at the root of the disorder but have been disappointing. The selective use of bone marrow transplant is one current therapy; gene therapy holds promise for the future.
Medical Nutrition Therapy
Nutritional therapy by dietary avoidance of VLCFAs does not lead to biochemical change because of endogenous synthesis. A specialty altered fatty acid product, Lorenzo’s oil (C18:1 oleic acid and C22:1 erucic acid), lowers the VLCFA level. Although the clinical course is not significantly improved, a slower decline in function may result.
Alzheimer’s Disease
Alzheimer’s Disease (AD) is the most common form of dementia, with varying patterns and rates of cognitive decline (Soto et al., 2005). It is named after Alois Alzheimer, who first described the clinical features and pathologic changes of this degenerative brain disease in 1907. The incidence rate of new cases of AD is similar for both sexes and throughout the world, but prevalence is three times higher in women (who tend to live longer than men). Age is the most important risk factor for AD; the number of people with the disease doubles every 5 years beyond age 65. More than 35 million people worldwide are estimated to have this disease (Querfurth, 2010). Given the increase in the number of adults living longer, the personal, familial, financial, and clinical effects of AD are staggering.
Etiology: At least three genes have been discovered that cause early-onset, familial AD; other genetic mutations have been associated with age-related, sporadic AD. Apolipoprotein-E4 (Apo-E4) is a protein located on chromosome 19; it binds β-amyloid and is involved in the transport of cholesterol. Thus, Apo-E4 has both cardiovascular and nutritional implications. Damage to key mitochondrial components (Kidd, 2005), oxidative stress, impaired insulin signaling, elevated homocysteine (Hcy), low folate, and high serum cholesterol may be causal factors.
Lead, iron, aluminum, copper and zinc have been implicated in AD pathogenesis because they catalyze the production of free radicals and induce dementia (Ramesh et al., 2010). With aging, changes occur in neurogenesis and Reelin signaling (Shetty, 2010). Inflammatory pathways have been implicated as well. Beclin 1 is a protein involved in the regulation of autophagy, a cellular degradation and maintenance pathway that is reduced in patients with AD (Jaeger and Wyss-Coray, 2010).
Pathophysiology: Alzheimer’s disease (AD) is a progressive, neurodegenerative disease characterized in the brain by abnormal clumps of amyloid beta (Aβ) peptide and neurofibrillary tangles (NIH, 2006). The early symptoms of AD, forgetfulness and loss of concentration, are often missed because they are misinterpreted as natural signs of aging (see Pathophysiology and Care Management Algorithm: Alzheimer’s Disease).
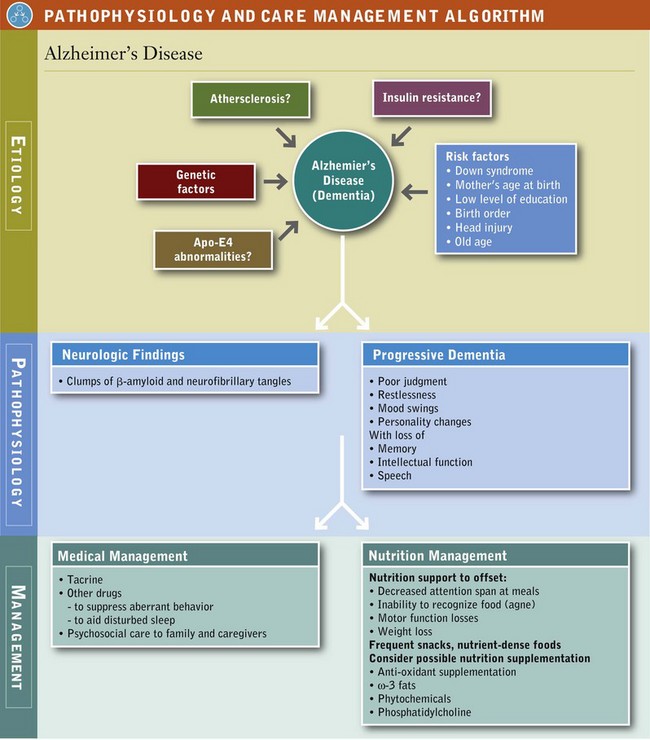
AD begins gradually; advances; and eventually leads to confusion, personality and behavior changes, and impaired judgment. Manifestations of AD result in a progressive dementia, with increasing loss of memory, intellectual function, disturbances in speech, loss of independence, disordered eating behavior, and weight change.
Persons with poor physical function seem to have a high risk for developing AD (Wang, 2006). Initially, day-to-day events are forgotten, possessions are misplaced, and appointments are missed but memories are retained. Cerebral function declines, mostly evident after pronounced memory loss. Names of objects are forgotten (anomia), words spoken by others are repeated (echolalia), and comprehension is mostly lost (agnosia). Over time, motor skills deteriorate, evidenced by changes in reflexes and a shuffling gait. When AD reaches the terminal stage, bowel and bladder controls are lost; limb weakness and contractures occur; intellectual activity ceases. The patient becomes completely incapacitated in a vegetative state as death approaches.
Medical Management: AD is diagnosed by histopathologic examination. The disease includes a preclinical disease stage, a mild cognitive impairment stage, and finally dementia. For the first time in decades, new diagnostic and treatment guidelines are being developed by the National Institute on Aging. Nonsteroidal antiinflammatory drugs (NSAIDs) or aspirin in combination with intake of food sources of vitamin E, other antioxidants, and ω-3 fatty acids are currently most effective (Shetty, 2010).
Cerebral vasodilators, stimulants, levodopa, and megadoses of vitamins remain unproven and drug treatment is experimental. Tacrine, the first cholinesterase inhibitor approved by the Food and Drug Administration (FDA) for treatment of AD, gives only modest improvement in function and cognition. Some other medications are used to suppress aberrant behavior, disturbed sleep, anxiety, or agitation. Collaborative interdisciplinary care may improve behavioral and psychological symptoms (Callahan et al., 2006). Stem cell transplantation may be tested for improving memory (Shetty, 2010). Other therapies are under development; see New Directions: Supplementation of Cell Membrane Phospholipids in Alzheimer’s Disease?
 New Directions
New Directions
Supplementation of Cell Membrane Phospholipids in Alzheimer’s Disease?
Choline, a B-vitamin, makes several metabolic contributions to cell membrane phospholipids. Because of this, it is gaining interest for its potential role in neurologic conditions. It is a precursor for phosphatidylserine and phosphatidylcholine, phospholipids located in the cell membrane of cells, including nerve cells (Tan, 1998; Zeisel, 2010).
Phosphatidylserine is located on the cytosol side of the lipid membrane of cells. It has been studied in brain health, and may offer neuro-protection by inhibiting the formation of beta amyloid fragments in those with AD. Early studies using bovine phosphatidylserine showed positive results in those with cognitive decline. Due to fears of mad cow disease, the compound was replaced in studies with a soy-based phosphatidylserine which is not exactly the same. Weak evidence supporting improvement in age-related memory impairment has been associated with the soy-based compound, but overall results are inconclusive and more research is needed (Wollen, 2010).
Because phosphatidylcholine may offer protection, it is noted that precursor CDP-choline shows promise. There may be positive effects on memory and behavior in patients with various forms of dementia, including AD. Longer term studies are needed to investigate full benefits (Fioravanti et al, 2010). Nonetheless, nutritional intake can be enhanced. Choline is found in eggs (especially yolks), beef (especially brains and liver), fish, pork, poultry, beans, nuts, peas, and soy products.
Medical Nutrition Therapy: Diets rich in saturated fatty acids and alcohol, and deficient in antioxidants and vitamins promote the onset of the disease; diets rich in unsaturated fatty acids, vitamins, antioxidants, wine, curcumin and some spices suppress its onset by scavenging free radicals and preventing oxidative damage (Ramesh et al., 2010). Proper inclusion of antioxidants and specific nutrients may protect the AD patient. A 2010 report of a 4 year prospective cohort study of 2148 community-based adults (65+ years of age) found that persons with a diet characterized by higher intakes of salad dressing, nuts, fish, tomatoes, poultry, cruciferous vegetables, fruits, and dark and green leafy vegetables were less likely to acquire AD than those eating diets of high-fat dairy products, red meat, organ meats, and butter (Gu et al., 2010). Because several dietary polyphenols are known to chelate metals, their routine use may also be protective (Ramesh et al., 2010). Garlic may be neuroprotective (Chauhan, 2005) and resveratrol prevents neuronal decline with aging (Anekonda, 2006; Ramesh et al., 2010). See also Figure 41-5.
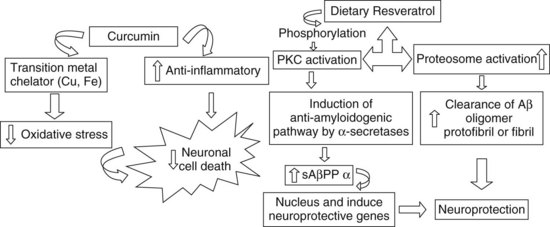
FIGURE 41-5 The role of curcumin and resveratrol in neuroprotection.
Left: Curcumin has multiple biologic effects: it chelates transition metals (iron and copper) and acts as an antioxidant and antiinflammatory molecule, protecting from oxidative stress.
Right: Resveratrol favors phosphorylation in protein kinase C, activating the nonamyloidogenic pathway of AβPP cleavage, and this leads to reduction in Aβ formation. sAβPPβ, a product of AβPP cleavage, gets translocated to the nucleus and modulates the genes. All these events favor neuronal cell survival.
Aβ, Amyloid beta; PKC, protein kinase C; sAPPβ, secreted fragment of amyloid protein precursor beta. (From Ramesh BN et al: Neuronutrition and Alzheimer’s disease, J Alzheimer’s Dis 19:1123, 2010. With permission from IOS Press.)
Problems with Self-Feeding, Oral Intake, and Weight Management: A wide range of neurologic functions are impaired that interfere with eating. Cognitive losses impair attention span, reasoning, and judgment; the ability to recognize feelings of hunger, thirst, and satiety decline. As the disease progresses, attention span decreases, and meals may be forgotten as soon as they are eaten or may not be eaten at all. Dehydration is also a problem; recognizing thirst and then seeking water is neglected.
Although a gluttonous appetite and weight gain may develop in some individuals with AD, generally weight loss is the norm. Whether increased resting metabolic rate or increased energy expenditure from constant pacing causes weight loss is unclear. For others, eating is neglected and inadequate oral intake results from impaired self-feeding. In still other cases weight loss may be secondary to frequent infections. Weight loss in turn increases the risk of skin ulcers and consequently decreases quality of life.
Mealtimes should be kept simple. Noise can be distracting; the radio or television should be turned off during mealtime. Food may need to be placed on small plates or bowls and served one at a time so as not to offer too many choices. As social inhibitions decrease, the patient may take another person’s food, consume inedible items or spoiled foods, or drink hazardous fluids. AD patients should be closely supervised during meals.
With sensory losses, perception of the surrounding world and related auditory, visual, or tactile recognition are distorted; this is called agnosia. Visual agnosia, the inability to recognize food, is manifested by not eating. The touch or smell of food is needed to initiate eating responses. Another sensory loss is the inability to recognize food when it is served in a bowl the same color as the food item. Use of colored bowls and plates that are in contrast to the color of the food may be necessary so that food can be distinguished from the place setting. Patients may have difficulty using eating utensils, but may model behaviors when demonstrated by staff or caregivers.
Motor losses occur during the course of the illness. Some clients may need hand guidance to initiate eating, then verbal cues to continue. As motor skills decline, the patient may be able to use only a spoon. However, eating utensils should not be removed prematurely because this may contribute to agitation, excessive disability, inadequate oral intake, and eventual weight loss. In addition, motor skills should be assessed routinely. Finger foods may be used, but only if the patient has no difficulty with chewing or swallowing. If the patient is inclined to swallow large boluses of food, finger foods are not appropriate. Although adaptive equipment is sometimes useful, it may be unfamiliar to the patient. As end-stage disease approaches, swallowing often becomes impossible. Dysphagia should be managed to prevent aspiration.
Frequent snacks, nutrient-dense foods, and nutrition supplements are needed to combat weight loss. Behavior modification and the use of altered food choices can improve quality of life. Evaluation of nutrition status is needed throughout the stages of AD to ensure that objectives of nutrition therapy continue to be met. Table 41-7 lists additional interventions.
TABLE 41-7
Practical Interventions for Eating-Related Behavioral Problems Common in Individuals with Dementia


Modified from National Institute on Aging (NIA) at http://www.nia.nih.gov/nia.nih.gov/templates/ADEAR Accessed 3/22/2011
Further, a succinct listing of drugs commonly used in the treatment of various neurologic diseases is shown in Table 41-8.
TABLE 41-8
Drugs Commonly Used in the Treatment of Neurologic Diseases



COMT (catechol-O-methyltransferase); GI, gastrointestinal; IV, intravenous; MAO-B (monoamine oxidase B); MS, multiple sclerosis; NSAID, nonsteroidal antiinflammatory drug; N/V, nausea and vomiting.
Data from National Institute of Neurologic Disorders and Stroke: Disorders index. Accessed 15 July 2010 from http://www.ninds.nih.gov/disorders; Accessed 4 November 2010 from http://www.mayoclinic.com/health-information.
Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS) is the most common motor system disease. ALS is also called Lou Gehrig disease, after the famous baseball player afflicted with the disease. The incidence is 2 in 100,000 (International Alliance of ALS, 2010). ALS involves a progressive denervation, atrophy, and weakness of muscles; hence the name amyotrophy. The prevalence is constant throughout the world, but men are affected more than women. The average age of onset is the mid-50s, and it is usually found in the 40- to 70-year age group.
Etiology: The cause of ALS is not clear. Genetic analysis of patients with familial, chromosome 21–linked ALS suggests that misfolding mutations in the copper-zinc superoxide dismutase (SOD1) gene may be involved (Nordlund and Oliveberg, 2006). Risk factors related to occupation, trauma, diet, or socioeconomic status are not consistent. However, it has been noted in several studies that elevated Hcy has neurotoxic effects. Because high Hcy levels induce oxidative stress and stimulate excitotoxic receptors, they damage motor neurons (Zoccolella et al., 2010).
Pathophysiology: The pathologic basis of weakness in ALS is the selective death of motor neurons in the ventral gray matter of the spinal cord, brainstem, and in the motor cortex. Clinical manifestations are characterized by generalized skeletal muscular weakness, atrophy, and hyperreflexia. The natural course for ALS is unpleasant. Decline is relentless and without remissions, relapses, or plateaus; it finally progresses to death usually in 2 to 6 years (Czaplinski, 2006).
The typical presentation is evidenced with both lower motor neuron (weakness, wasting, fasciculation) and upper motor neuron deficits (hyperactive tendon reflexes, Hoffman signs, Babinski signs, or clonus). Muscle weakness begins in the legs and hands and progresses to the proximal arms and oropharynx. As these motor nerves deteriorate, almost all of the voluntary skeletal muscles are at risk for atrophy and complete loss of function. The loss of spinal motor neurons causes the denervation of voluntary skeletal muscles of the neck, trunk, and limbs, resulting in muscle wasting, flaccid weakness, involuntary twitching (fasciculations) and loss of mobility.
Progressive loss of function in cortical motor neurons can lead to spasticity of jaw muscles, resulting in slurred speech and dysphagia. The onset of dysphagia is usually insidious. Swallowing difficulties usually follow speech difficulties. Although some weight loss is inevitable given the muscle atrophy, consistent or dramatic loss may be an indicator of chewing difficulties or dysphagia (Bulat and Orlando, 2005). Eye movement and eye blink are spared, as are the sphincter muscles of the bowel and bladder; thus incontinence is rare. Sensation remains intact and mental acuity is maintained.
Medical Management: No currently known therapy cures the disease. To lower high serum Hcy levels, short-term treatment with a high dose of methylcobalamin (B12) shows promise (Zoccolella et al., 2010). Use of folate and antioxidant treatments should also be investigated. Although mechanical ventilation can extend the life of patients, most decline this option. Quality of life is poor in advanced ALS and supportive comfort measures are primarily used.
Medical Nutrition Therapy: There are decreases in body fat, lean body mass, muscle power, and nitrogen balance and an increase in resting energy expenditure as death approaches. Some of the nutritional changes during the different stages of ALS are noted in Table 41-9.
TABLE 41-9
Nutritional and Metabolic Changes During the Progression of Amyotrophic Lateral Sclerosis

From Kasarskis EJ et al: Nutritional status of patients with amyotrophic lateral sclerosis: relation to the proximity of death, Am J Clin Nutr 63:130, 1996.
Hypermetabolic status and increased resting energy expenditure measurements have been noted. The relationship between dysphagia and respiratory status is important. As ALS progresses, a progressive loss of function in bulbar and respiratory muscles contributes to oral and pharyngeal dysphagia. In late stages, when the respiratory status is impaired, alternative tube placement besides PEG may be required.
The clinician should become familiar with common clinical findings in ALS to prevent secondary complications of malnutrition and dehydration. The functional status of each patient should be monitored closely so that timely intervention with the appropriate management techniques can be started. In particular, dysphagia should be monitored closely. Oropharyngeal weakness affects survival in ALS by placing the patient at continuous risk of aspiration, pneumonia, and sepsis and by curtailing the adequate intake of energy and protein. These problems can compound the deteriorating effects of the disease. The Amyotrophic Lateral Sclerosis Severity Scale is often used to assess the functional level of swallowing, speech, upper and lower extremities. Once the severity of deficits has been identified, appropriate interventions can be implemented (see Focus On: Dysphagia Intervention for Amyotrophic Lateral Sclerosis).
 Focus On
Focus On
Dysphagia Intervention for Amyotrophic Lateral Sclerosis
Strand and colleagues (1996) outlined dysphagia intervention on a continuum of five stages that correlate to the amyotrophic lateral sclerosis (ALS) severity scale. They include:
1. Normal Eating Habits (ALS Severity Scale Rating 10-9). Early assessment and intervention are critical for maintaining nutritional health in ALS. This is the appropriate time to begin educating the patient, before the development of speech or swallowing symptoms. Hydration and maintenance of nutritional health are critical at this stage. Fluid intake of at least 2 q/day is important. Dehydration contributes to fatigue and thickens saliva. For patients with spinal ALS, emphasis on fluids is important because they may intentionally limit fluid intake because of difficulties with toileting. The diet history is helpful to assess patterns of normal chewing, swallowing, and the rate of ingestion. Weight loss history establishes a baseline weight. A weight loss of 10% or more is indicative of nutritional risk.
2. Early Eating Problems (Severity Scale Rating 8-7). At this point patients begin to report difficulties eating; reports of coughing and unusually long mealtimes are associated with tongue, facial, and masticator muscle weakness. Dietary intervention begins to focus on modification of consistency, avoidance of thin liquids, and use of foods that are easier to chew and swallow.
3. Dietary Consistency Changes (Severity Scale Rating 6-5). As symptoms progress, the oral transport of food becomes difficult as dry, crumbly foods tend to break apart and cause choking. Foods that require more chewing (e.g., raw vegetables or steak) are typically avoided. As dysphagia progresses, ingestion of thin liquids, especially water, may become more problematic. Often the patient has fatigue and malaise, which may be associated with a mild chronic dehydration resulting from a decreased fluid intake. Dietary intervention should change food consistency to mechanically soft or pureed (see Appendix 35) to reduce the need for oral manipulation and to conserve energy. Small, frequent meals may also increase intake. Thick liquids that contain a high percentage of water, as well as attempts to increase fluid intake, need to be emphasized to maintain fluid balance. Popsicles, gelatin, ice, and fresh fruit are additional sources of free water. Liquids can be thickened with a modified cornstarch thickener. Swallowing can be improved by emphasizing taste, texture, and temperature. Juices can be substituted for water to provide taste, nutrients, and calories. A cool temperature facilitates the swallowing mechanism; therefore cold food items may be better tolerated; heat does not provide the same advantage. Carbonation may also be better tolerated because of the beneficial effect of texture. Instructions for preventing aspiration should be addressed: safe swallowing includes sitting bolt upright with the head in a chin-down position. Concentrating on the swallowing process can also help reduce choking. Avoid environmental distractions and conversation during mealtime; however, families should be encouraged to maintain a normal mealtime routine. As dysphagia progresses, the limitation of food consistencies may result in the exclusion of entire food groups. Vitamin and mineral supplementation may be necessary. If chewable supplements are not handled safely, liquid forms may be added to acceptable foods. Fiber may also need to be added along with fluids for constipation problems.
4. Tube Feeding (Severity Scale Rating 4-3). Dehydration will occur acutely before malnutrition, a more chronic state, is exhibited. This may be an early indication of the need for nutrition support. Weight loss from muscle wasting and dysphagia will eventually lead to placement of a percutaneous endoscopic gastrostomy (PEG) tube for nutrition and protection against aspiration caused by dysphagia. Enteral nutrition support is preferred because the gastrointestinal tract should be functioning properly. Given the progressive nature of ALS, placing feeding tubes with signs of dysphagia and dehydration is better than initiating this therapy later, after the patient has become overtly malnourished or when respiratory status is marginal. The decision of whether to place a feeding tube for nutrition support is part of the decision-making process each patient must face. Adequate nutriture can maintain health of the individual longer and may be a welcome relief for the patient. The purpose of nutrition support should be to enhance the quality of life. Long-term access should be considered via a PEG or percutaneous endoscope jejunostomy tube (see Chapter 14).
5. Nothing by Mouth (Severity Scale Rating 2-1). The final level of dysphagia is reached when the patient can neither eat orally nor manage his or her own oral secretions. Although saliva production is not increased, it tends to pool in the front of the mouth as a result of a declining swallow response. Once the swallowing mechanism is absent, mechanical ventilation is required to manage saliva flow. Tube feeding is permanent at this stage.
Data from Strand EA et al: Management of oral-pharyngeal dysphagia symptoms in amyotrophic lateral sclerosis, Dysphagia 11:129, 1996.
Epilepsy
Epilepsy is a chronic condition characterized by unprovoked, recurring seizures that disrupt the nervous system. Seizures (or convulsions) are intermittent derangements in brain function caused by abnormal electrical activity of a group of brain cells, and present with apparent clinical symptoms and findings. It is estimated that 2.3 million individuals in the United States have epilepsy; 45,000 children younger than age 15 develop epilepsy each year according to the Epilepsy Foundation (2006).
Pathophysiology: Most seizures begin in early life, but a resurgence of epileptic events occurs after age 60. The first occurrence of a seizure in adults should prompt investigation into a cause. A clinical workup usually reveals no anatomic abnormalities, and the cause of the seizure may remain unknown (idiopathic). Seizures before age 2 are usually caused by developmental defects, birth injuries, or a metabolic disease (see Chapters 44 and 45). The medical history is the key component for suggesting further avenues of diagnostic investigation and potential treatments, especially in children. An electroencephalogram can help to delineate seizure activity. It is most helpful in localizing partial complex seizures.
Medical Management: The dramatic tonic-clonic (grand mal) seizure is the most common image of a seizure (lasting 1 to 2 minutes), yet numerous classifications of seizures, each with a different and often less dramatic clinical presentation, exist. A generalized seizure involves or appears to involve the entire brain cortex from its beginning phases. The tonic-clonic seizure comes under this heading. After such a seizure the patient wakes up slowly after a time; he or she will be groggy and disoriented for minutes to hours after the event. This is termed the postictal phase and is characterized by deep sleep, headache, confusion, and muscle soreness.
The absence seizure (petit mal) is also generalized in nature. A patient with absence seizures may appear to be daydreaming during an episode, but he or she recovers consciousness within a few seconds and has no postictal fatigue or disorientation. Partial seizures occur when there is a discrete focus of epileptogenic brain tissue. A simple partial seizure involves no loss of consciousness, whereas a complex partial seizure is characterized by a change in consciousness. Failure of partial seizure control may prompt consideration of seizure surgery. A localized focus resected from nonessential brain renders a patient seizure free in 75% of cases.
Determining the seizure type is key to implementing effective therapy. Generalized seizures are ordinarily managed with valproate or phenytoin. Phenytoin metabolism has unusual kinetics; thus toxic levels may be attained with very small dosage adjustments. These drugs interact with other drugs metabolized in the liver and may cause liver damage. Liver enzymes and serum drug levels must be monitored periodically. Gabapentin is rapidly gaining popularity because of its safety and ease of use. Carbamazepine or phenytoin can usually control partial seizures. Use of just one antiseizure medication is recommended initially, resorting to combination therapies only when needed.
Medications used in anticonvulsant therapy may alter the nutrition status of the patient (see Chapter 9 and Appendix 31). Phenobarbital has been associated with decreased intelligence quotient when used in children. It is occasionally considered for use after failure of other antiepileptic drugs. Phenobarbital, phenytoin, and primidone interfere with intestinal absorption of calcium by increasing vitamin D metabolism in the liver. Long-term therapy with these drugs may lead to osteomalacia in adults or rickets in children, and vitamin D supplementation is recommended. Folic acid supplementation interferes with phenytoin metabolism; thus it contributes to difficulties in achieving therapeutic levels. If folic acid supplementation is necessary, it should be consistent, and medication adjusted accordingly.
Phenytoin and phenobarbital are bound primarily to albumin in the bloodstream. Decreased serum albumin levels in malnutrition or advanced cirrhosis limits the amount of drug that can be bound. This results in an increased free drug concentration and possible drug toxicity even with a standard dose.
Absorption of phenobarbital is delayed by the consumption of food; therefore administration of the drug must be staggered around mealtimes if it is used. Continuous enteral feeding slows the absorption of phenytoin, thus necessitating an increase in the dose to achieve a therapeutic level. Stopping the tube feeding 1 hour before and 1 hour after the phenytoin dose is generally recommended. Whenever tube feedings are stopped, the dose of phenytoin needs to be adjusted to avoid toxicity.
Medical Nutrition Therapy: A ketogenic diet can be used for treatment of all types of seizures in children with intractable epilepsy in whom drug therapies have failed. The ketogenic diet has minimal side effects, and risks of the diet are low blood sugar, upset stomach at first caused by the high amounts of fat, and constipation. The long-term risk of kidney stones is rare; elevated serum cholesterol is usually is temporary and disappears with discontinuation of the diet; and growth, which is sometimes slowed while on the diet, resumes at the child’s normal rate (Patel et al., 2010). Although the diet is very demanding and requires continued effort even after the first phase, it completely controls epilepsy in one third of the children whose seizures are otherwise uncontrollable (Groomes et al., 2011). Practical guidelines for implementing the diet have been released by the American Academy of Neurology and the American Epilepsy Society and can be retrieved from the Epilepsy Foundation website (Kossoff et al., 2009).
The diet is designed to create and maintain a state of ketosis (Bough and Rho, 2007). The beneficial effect in epilepsy may be caused by a change in neuronal metabolism; ketones may inhibit neurotransmitters, thus producing an anticonvulsant effect in the body. To begin the diet the individual usually fasts in the hospital for 24 to 72 hours until ketonemia is produced as measured by the level of beta-hydroxybutyrate in the blood. For many patients, if the diet is going to work, it usually works during this initial period with a reduction in seizures; however, sometimes the child needs to follow the subsequent ketogenic diet for 3 months before there are any reductions in seizure activity. It should also be noted that antiepileptic drugs are not stopped when administering the ketogenic diet.
In the traditional approach, once ketosis is established, caloric intake is resumed in a ratio of 3 : 1 or 4 : 1, meaning that there are 3 or 4 g of fat for every 1 g of protein and carbohydrate combined in the diet. With a 4 : 1 ratio, the diet is calculated so that at least 80% of the kilocalories are from fat. Protein is calculated to provide appropriate intake for growth (approximately 1 g/kg/day). Carbohydrates are added to make up the remaining small portion of calories.
The majority of the diet is composed of fresh meats, eggs, cheese, fish, heavy whipping cream, butter, oils, nuts, and seeds. Vegetables and fruits are added in small amounts, within the current diet prescription (Table 41-10). Ketosis is monitored by regular measurements of serum β-hydroxybutyrate. Even though goal levels are patient specific, most individuals need to be in the range of 35-60 mg/L (4-7mmol/L) for seizure control. A carbohydrate-free multiple vitamin and mineral supplement is necessary to ensure that the diet is nutritionally complete. However, additional vitamins and minerals are often necessary, including calcium, vitamin D, and selenium.
TABLE 41-10
Typical Ketogenic Diet Pattern
Created by Marta Mazzanti, RD, Seattle Children’s Hospital, Seattle, WA.
Other nonfood items such as sugar-containing toothpaste, shampoo, and lotions need to be avoided. It is important that the diet be strictly followed; the smallest amount of extra carbohydrate can cause a breakthrough seizure. The child should be monitored, because too rapid a rate of weight gain can decrease ketosis and reduce effectiveness.
A variation of the ketogenic diet is the modified Atkins diet in which calories are not restricted, but carbohydrates are drastically limited to 10 to 20 g/day. The ratio of fat to protein and carbohydrate is usually 1 : 1. It appears that the best results are achieved when the diet begins with 10 g/day of carbohydrate and is then adjusted upward, always maintaining ketosis (Porta et al., 2009; Weber et al., 2009).
Another modification of the traditional ketogenic diet is the medium-chain triglyceride (MCT) oil–based ketogenic diet, used frequently in the United Kingdom and Canada, in which some of the long-chain fats of the traditional diet are replaced with MCTs. MCT oil is an odorless, colorless, tasteless oil and was originally used as a means of improving the palatability of the diet. A greater amount of nonketogenic foods such as fruits and vegetables and small amounts of bread and other starches can be allowed because ketosis from MCT can be more readily achieved with a lower percentage of fat in the diet. MCT can be added in to the classic ketogenic diet and modified Atkins diet to increase ketosis. Coconut oil can also be used as a source of MCT to increase palatability, but it contains only 45% to 50% MCT so more may be needed. MCT oil or coconut oils are also added if the serum triglycerides are too high with the use of the traditional ketogenic diet.
Initiating the ketogenic diet is intense. Further, the diet may be unpalatable as well as complex, requiring weighing and measuring of all foods, thus making compliance difficult to achieve. To be successful, children may benefit from behavioral techniques, whereas parents most often require substantial psychosocial support. Attention required during the fine-tuning phase varies and is affected by the patient’s health status, growth, and development. For the child whose epilepsy is controlled by the diet, complying with the diet is much easier than dealing with devastating seizures and injuries.
Discontinuation of the diet can be considered after 2 to 3 years, especially if the child has been seizure free for a year, although seizure control medication may still be necessary. The person is “weaned” off the ketogenic diet over a period of several months to a year as small amounts of carbohydrates are added with observation for seizure reoccurrence.
Guillain-Barré Syndrome
Guillain-Barré syndrome (GBS) is an acute-onset, inflammatory, demyelinating polyneuropathy of the proximal motor nerves, including the cranial nerves and the diaphragm. The incidence is approximately 2 in 100,000.
Etiology: In 60% of cases the disorder follows an infection, surgery, or an immunization. Some of the more common organisms are Campylobacter jejuni and Mycoplasma. Several pathologic varieties exist, and the nature of the distinction is related to the segment of the immune system that is inflicting nerve damage. The clinical course of GBS is similar regardless of subtype, although GBS after a Campylobacter infection tends to be more severe.
Pathophysiology: Relatively symmetric weakness with paresthesia usually begins in the legs and progresses to the arms. The loss of function in affected nerves occurs because of demyelination. Myelin is the specialized fatty insulation that envelops the conducting part of the nerve, the axon. In GBS the immune system recognizes myelin and mounts an attack against it. Presumably myelin shares a common characteristic with the pathogen from the antecedent infection; thus the immune system cannot differentiate what is foreign (the pathogen) from what is native (myelin). When the nerve is demyelinated, its ability to conduct signals is severely impaired, resulting in neuropathy.
Medical Management: GBS reveals itself in a matter of days. The most common sequence of symptoms is areflexia (absence of reflexes), followed by proximal limb weakness, cranial nerve weakness and respiratory insufficiency. These symptoms normally peak by 2 weeks but may progress up to 1 month. Medical diagnosis is ordinarily made on clinical grounds, but nerve conduction studies are also beneficial. Before the clinical course is apparent, myelopathic disorders need to be considered.
Because of the precipitous progression of GBS, hospitalization is usually necessary. Vital capacity and swallowing function may rapidly deteriorate such that intensive care is sometimes necessary. Intubation and respiratory support should be instituted early in respiratory decline to avoid the need for resuscitation. Plasmapheresis, the exchange of the patient’s plasma for albumin, is often helpful to reduce the load of circulating antibodies. Intravenous immunoglobulin or steroids have been shown to be of benefit.
Medical Nutrition Therapy: Guillain-Barré syndrome evolves quickly; during the acute stage, the metabolic response of GBS is similar to the stress response that occurs in neurotrauma. Energy needs assessed by indirect calorimetry may be as high as 40 to 45 kcal/kg and protein needs twice the usual amount. Supportive nutritional care should be offered to attenuate muscle wasting.
For a small percentage of patients, oropharyngeal muscles may be affected, leading to dysphagia and dysarthria. In this situation, a visit by the dietitian at mealtime can be a valuable way to observe difficulties the patient may have with chewing or swallowing. Specific difficulties warrant evaluation by a swallowing specialist. The speech therapist can evaluate the degree of dysphagia and make appropriate dietary recommendations pertaining to texture. As the patient recovers, it is important to discuss safe food handling and future prevention of C. jejuni infection.
Migraine Syndrome
The migraine syndrome is defined clinically as an episodic intense, throbbing head pain that lasts from 4 to 72 hours. It is usually on one side of the head and becomes worse with exertion. It may be accompanied by nausea and classically is associated with a prodrome of visual disturbances or unusual olfactory and gustatory perception. Most persons report an associated transient visual aura, including flashing lights. Migraines are more common between the ages 15 and 55 and affect women three times more often than men.
Pathophysiology: Although the cause is unknown, migraine headache is thought to be vascular in origin and follows a family history of migraines or of visual prodromata. The leading theory proposes that dural blood vessels become dilated and the pulsatile blood flow through these vessels distends and irritates the highly pain-sensitive dura mater. This would explain the throbbing quality of the headache. An inflammatory component to migraine headache is also present along with mitochondrial dysfunction (Gardner and Boles, 2010).
Medical Management: Treatment depends on the frequency of attacks and the presence of comorbid illness. A thorough history is the key to medical diagnosis. To qualify for a diagnosis of migraine headache, the headache must be throbbing, episodic, and supremely intense. A history of intercurrent nausea, vomiting, photophobia, and visual or olfactory auras should be present.
Numerous medicines are used to prevent or abort migraine, indicating a less than clear understanding of its pathophysiologic characteristics. NSAIDs are often the first line, followed by sympathomimetics and serotonin agonists such as sumatriptan. Prophylaxis can include calcium channel antagonists, β-adrenergic blockers, and serotonin antagonists.
Medical Nutrition Therapy: Migraine attacks are triggered by a variety of factors, including food, drugs, odors, and changes in sleep habits; they respond to a variety of treatments (Dowson et al., 2006). Because foods implicated in one individual may not trigger attacks in another and food intolerance thresholds vary over time, generalized recommendations about food avoidance are ill advised. There has been attention to biogenic amines such as tyramine or phenylethylamine in foods as triggers of migraine headaches. A trial of a tyramine- or phenylethylamine-restricted diet may be recommended for some clients (see Box 9-3 in Chapter 9 for biogenic amine–containing foods), but evidence is not clear on its success (Sun-Edelstein and Mauskop, 2009a). Because suspect food items or “trigger foods” can only be correctly identified if eliminated and then reintroduced into the diet, the dietitian can strategize on this approach. See Chapter 27 for a food diary form to record intake and timing of symptoms, and to determine which foods may be a problem. In some patients dehydration may be the instigator; one simple measure is to drink more water (at least 2-3 cups) to rehydrate (Blau, 2005; Spigt et al., 2005).
Many patients try herbs or botanical products or nutritional supplementation to manage their headaches. Feverfew is used by many migraine sufferers, even though a Cochrane Database Review has not proven its efficacy. Because of their roles in energy metabolism, two nutrients, riboflavin and coenzyme Q10, have been suggested (Hershey et al., 2007; Schiapparelli et al., 2010). Magnesium has also been shown effective in treating migraine headaches (Sun-Edelstein and Mauskop, 2009b).
Myasthenia Gravis
Myasthenia gravis (MG) is the most well-known disorder of the neuromuscular junction. The neuromuscular junction is the site on the striated muscle membrane where a spinal motor neuron connects. Here the signal from the nerve is carried to the muscle via a submicron-size gap, a synapse. The molecule that carries the signal from the nerve ending to the muscle membrane is acetylcholine (Ach), and acetylcholine receptors (AchRs) populate the muscle membrane. These receptors translate the chemical signal of Ach into an electrical signal that is required for contraction of muscle fibers. MG is one of the most well-characterized autoimmune diseases, a class of disorders in which the body’s immune system raises a response to AchRs. The incidence of MG is low, approximately 14 in 100,000 people.
Pathophysiology: In MG the body unwittingly makes antibodies to AchR. These antibodies are the same that fight off colds and give immunity. The AchR antibodies bind to AchR and make them unresponsive to Ach. There is no disorder of nerve conduction and no intrinsic disorder of muscle. The characteristic weakness in MG occurs because the signal of the nervous system to the muscle is garbled at the neuromuscular junction. Patients with MG commonly have an overactive thymus gland. This gland resides in the anterior thorax and plays a role in the maturation of B lymphocytes, the cells that are charged with synthesizing antibodies.
Medical Management: Relapsing and remitting weakness and fatigue, varying from minutes to days, characterize MG. The most common presentation is diplopia (double vision) caused by extraocular muscle weakness, followed by dysarthria, facial muscle weakness, and dysphagia. Dysphagia or swallowing disorders (resulting from fatigue following mastication) may cause malnutrition. Less commonly, proximal limb weakness in the hips and shoulders may be present. Severe diaphragmatic weakness can result in respiratory difficulty. No involvement of sensory nerves occurs.
Anticholinesterases are medicines that inhibit acetylcholinesterase, thus serving to increase the amount of Ach in the neuromuscular junction. Corticosteroids are immunosuppressive. Removal of the thymus results in symptomatic improvement in most patients.
Medical Nutrition Therapy: Chewing and swallowing are often compromised in MG. Because this compromise occurs with fatigue, it is important to provide nutritionally dense foods at the beginning of meals before the patient tires. Small, frequent meals that are easy to chew and swallow are helpful. Difficulties holding a bolus on the tongue have also been observed, suggesting that foods that do not fall apart easily may be better tolerated. For patients treated with anticholinesterase drugs, it is crucial to time medication with feeding to facilitate optimal swallowing.
Physical activity should be limited before mealtime to ensure maximum strength to eat a meal. It is also important not to encourage food consumption once the patient begins to tire because this may contribute to aspiration. If and when respiratory crisis occurs, it is usually temporary. Nutrition support via NG tube may be implemented in the interim to assist in maintaining vital functions of the patient until the crisis subsides. Once extubated, a swallow evaluation using cinefluoroscopy is appropriate to assess the degree of deglutitory dysfunction (swallowing irregularity) or risk of aspiration associated with an oral diet.
Multiple Sclerosis
Multiple sclerosis (MS) is a chronic disease that affects the CNS and is characterized by destruction of the myelin sheath, the function of which is transmission of electrical nerve impulses. Multiple areas of optic nerves, spinal cord, and brain undergo “sclerosis” whereby myelin is replaced with sclera or scar tissue. No single test can ascertain whether a patient has MS; however, diagnostic criteria (McDonald criteria) were developed for use by practicing clinicians (Polman et al., 2005).
The signs and symptoms of MS are easily distinguished, and they recur over the natural history of this disease. In the worst scenario MS can render a person unable to write, speak, or walk. Fortunately the majority of patients are only mildly affected. The prevalence is lower in equatorial areas, southern United States, and southern Europe; it is higher in Canada, northern Europe, and the northern United States. MS is the most common demyelinating disorder of the CNS, affecting 2.5 million people worldwide (Freedman, 2006).
Pathophysiology: The precise cause of MS remains undetermined. A familial predisposition to MS has been noted in a minority of cases. However, familial tendency is not well established and no consistent pattern of Mendelian inheritance has emerged (Victor and Ropper, 2005). Geographic latitude and diet are therefore implicated. Epidemiologic studies have linked the incidence of MS to geographic location and sunshine exposure. The degree of sunlight exposure catalyzes the production of vitamin D3 in the skin. This form of vitamin D3 is a selective immune system regulator and may inhibit MS progression (Mark and Carson, 2006). Further, in cross-sectional evaluations of MS and vitamin D, an increased prevalence of clinical vitamin D deficiency was associated with decreased bone density (Mark and Carson, 2006). Given the current evidence of the potential benefits of vitamin D, it appears to be reasonable and safe to consider vitamin D supplementation adequate to achieve normal levels in patients with MS (Solomon and Whitham, 2010).
Medical Management: Fluctuating symptoms and spontaneous remissions make treatments difficult to evaluate. Currently no proven treatment for changing the course of MS, preventing future attacks, or preventing deterioration exists. Initially recovery from relapses is nearly complete, but over time, neurologic deficits remain. Therefore measures to maximize recovery from initial attacks or exacerbations, prevent fatigue and infection, and use all of the available rehabilitative measures to postpone the bedridden stage of disease are imperative. Physical and occupational therapies are standard for weakness, spasticity, tremor, uncoordination, and other symptoms.
Drugs for spasticity can be initiated at a low dose and cautiously increased until the patient responds. Physical therapy for gait training and range-of-motion exercises helps. Steroid therapy is used in treating exacerbations; adrenocorticotropic hormone (ACTH) and prednisolone are the drugs of choice. However, treatment is not consistently effective and tends to be more useful in cases of less than 5 years’ duration. Side effects of short-term steroid treatment include increased appetite, weight gain, fluid retention, nervousness, and insomnia. Reduced cerebrospinal fluid and serum levels of vitamin B12 and folate have been noted in MS patients who receive high-dose steroids. Methotrexate may also be used with ACTH, causing anorexia and nausea. Drug therapies may be a challenge (see Chapter 9 and Appendix 31).
Medical Nutrition Therapy: Several dietary regimens for managing MS have been studied, all of which have yielded equivocal results. Various diets such as allergen-free, gluten-free, pectin-free, fructose-restricted, raw food diet, megadoses of micronutrients, zinc phosphates with calcium, and other combinations have been ruled ineffective. The RD’s evaluation of the patient to maximize nutritional intake is imperative. Vitamin D status should be assessed by measuring 25-hydroxy vitamin D and supplementation may be warranted; total intake should be monitored from multivitamin and mineral supplements and fortified foods (Brown, 2006).
As the disease progresses, neurologic deficits and dysphasia may occur as the result of damaged cranial nerves. Thus diet consistency may need to be modified from solids to mechanically soft or pureed items, even progressing to thick liquids to prevent aspiration. Impaired vision, dysarthria, and poor ambulation make meal preparation into a difficult task. In this situation reliance on comfort foods or prepackaged, single-serving, or convenience foods often permits independent preparation of meals. Given the chronic nature of this debilitating disease, patients may require enteral nutrition support.
Neurogenic bladder is common, causing urinary incontinence, urgency, and frequency. To minimize these problems, distributing fluids evenly throughout the waking hours and limiting them before bed is helpful. Some patients limit fluid intake severely to decrease frequency of urination but thereby increase the risk of UTIs. UTIs are common in patients with MS, and some patients increase their intake of cranberry juice as a form of self-treatment (see Chapter 36).
Neurogenic bowel can cause either constipation or diarrhea, and incidence of fecal impaction is increased in MS. A diet that is high in fiber with additional prunes and adequate fluid can moderate both problems.
Parkinson’s Disease
Parkinson’s disease (PD) is a progressive, disabling, neurodegenerative disease, first described by James Parkinson in 1817. PD is characterized by slow and decreased movement, muscular rigidity, resting tremor, postural instability, and decreased dopamine transmission to the basal ganglia. Although the natural history of this disease can be remarkably benign in some cases, approximately 66% of patients are disabled within 5 years, and 80% are disabled after 10 years (Victor and Ropper, 2005).
PD is one of the most common neurologic diseases in North America; it affects approximately 1% of the population older than 65 years of age. The incidence is similar across socioeconomic groups, although PD is less common in blacks and Asians in comparison with whites. It most commonly occurs between the ages of 40 and 70.
Pathophysiology: Although the cause of PD remains unclear, it is multifactorial and the pathogenesis is well described. It involves an interaction of inheritance with environmental factors. There is a marked loss of dopaminergic neurons (pigmented cells) in the substantia nigra, as well as tyrosine hydroxylase, the rate-limiting enzyme for dopamine.
In studying the brains of people with PD after they die, accumulations of protein called Lewy bodies (named after the doctor who first found them) are found. More than 10 genes causing familial PD have been identified. Genetic testing for PD is not warranted at this time, but continued investigations hold promise.
The role of endogenous toxins from cellular oxidative reactions has emerged because aging has been associated with a loss of neurons containing dopamine and an increase in monoamine oxidase. When metabolized (enzymatic oxidation and autooxidation), dopamine produces endogenous toxins (hydrogen peroxide and free radicals), causing peroxidation of membrane lipids and cell death. In the presence of an inherited or acquired predisposition, severe oxidative injury can lead to substantial loss of dopaminergic neurons similar to that observed in PD.
Several other environmental factors have also been implicated as causal factors of PD. The connection between smoking and a lowered risk for PD has been evaluated, but results are inconsistent (Zhang et al., 2006). In older patients, drug-induced PD may occur as a side effect of neuroleptics or metoclopramide (see Chapter 9 and Appendix 31).
Nutrient-related findings are biologically plausible and support the hypothesis that oxidative stress may contribute to the pathogenesis of PD (Czlonkowska et al, 2010). The relationships of folate, elevated plasma homocysteine levels, fiber and caloric deficits are being evaluated. Vitamin B6 intake may also decrease the risk of PD (De Lau et al., 2006). The final environmental factor associated with the incidence of PD is exposure to chemicals and toxins, as in farming communities where pesticides are used. However, no one chemical toxin or heavy metal is known as the cause (Victor and Ropper, 2005).
Medical Management: The “classic triad” signs—tremor at rest, rigidity, and bradykinesia—remain the criterion for medical diagnosis. However, it was well over a century before l-dopa (a precursor to dopamine) was introduced for controlling symptoms. Exelon, a cholinesterase inhibitor, was approved by the FDA in 2006 for use in mild to moderate PD dementia. Pharmacotherapy agents, surgical interventions, and physical therapy are the best adjunctive therapies.
Medical Nutrition Therapy: The primary nutrition intervention should be to focus on drug-nutrient interactions, especially between dietary protein and l-dopa. Side effects of medications for PD include anorexia, nausea, reduced sense of smell, constipation, and dry mouth. To diminish the gastrointestinal side effects of l-dopa, it should be taken with meals. Avoid foods that contain natural l-dopa such as broad beans (fava beans). For some patients dyskinesia may be reduced by limiting dietary protein at breakfast and lunch and including it in the evening meal. Table 41-11 presents a sample menu for this diet.
{kind=link}
Fiber and fluid adequacy lessen constipation, a common concern for persons with PD. Interactions between pyridoxine and aspartame should be considered as well. Pyridoxine has a possible interaction with l-dopa. Decarboxylase, the enzyme required to convert l-dopa to dopamine, depends on pyridoxine. If excessive amounts of the vitamin are present, l-dopa may be metabolized in the periphery and not in the CNS where its therapeutic activity occurs. Therefore vitamin preparations containing pyridoxine should not be taken with doses of l-dopa. In addition, manganese should be carefully monitored to avoid excesses above DRI levels.
The high demand for molecular oxygen, the enrichment of polyunsaturated fatty acids in membrane phospholipids, and the relatively low abundance of antioxidant defense enzymes are all relevant factors (Sun et al., 2008). Antiinflammatory and neuroprotective effects come from phenolic compounds, such as resveratrol from grapes and red wine, curcumin from turmeric, apocynin from Picrorhiza kurroa, and epigallocatechin from green tea (Sun et al., 2008). Sufficient intake of vitamin D3 and ω-3 fatty acids can be suggested.
As the disease progresses, rigidity of the extremities can interfere with the patient’s ability to perform self-feeding. Rigidity interferes with the ability to control the position of the head and trunk, necessary for eating. Eating is slowed; mealtimes can take up to an hour. Simultaneous movements such as those required to handle both a knife and fork become difficult. Tremor in the arms and hands may make self-feeding of liquids impossible without spilling. Perception and spatial organization can become impaired.
Dysphagia is often a late complication. A large number of patients may be silent aspirators, which affects nutrition status.
Experimental treatment procedures are being tested and reported with increasing frequency. Deep brain penetration, other surgical interventions, and efforts with stem cell research continue in hopes that a “cure” is possible.
 Clinical Scenario
Clinical Scenario
Clarence is a 74-year-old white man hospitalized for evaluation of dementia progression and recent hearing difficulty. He was diagnosed with Alzheimer’s disease 5 years ago and has been taking donepezil (Aricept) and furosemide (Lasix). He lives at home with his 68-year-old Italian wife who is his primary caregiver. Clarence is 5′10″ and currently weighs 155 lb. His medical chart indicates that his weight was 170 lb at his last hospital visit 6 months ago, following a fall. Clarence’s wife notes that his appetite and hearing seem to have changed and he shows little interest in food at meal times. They don’t talk much during home meals and nursing staff have observed his wife helping him eat and suspect some problems with feeding. His wife describes their meals as follows:
Breakfast:  C oatmeal,
C oatmeal,  C 2% milk, 1 orange ; food is usually consumed while watching the morning news
C 2% milk, 1 orange ; food is usually consumed while watching the morning news
Lunch:  C vegetable beef soup,
C vegetable beef soup,  turkey sandwich with white bread, light mayonnaise, lettuce, tomato slice, 10 baby carrots, water
turkey sandwich with white bread, light mayonnaise, lettuce, tomato slice, 10 baby carrots, water
Supper: frozen TV dinner (generally small meat serving, vegetable, and starch item;) TV is on in the background
Alzheimer’s Disease Education and Referral Center
http://www.nia.nih.gov/Alzheimer’s
http://www.strokeassociation.org
http://www.epilepsyfoundation.org
National Human Genome Research Institute
National Institute of Neurological Disorders and Stroke
National Institutes of Health: Swallowing Disorders
http://www.ninds.nih.gov/disorders/swallowing_disorders/swallowing_disorders.htm
National Institute of Neurologic Disorders and Stroke: Stroke Page
http://www.ninds.nih.gov/disorders/stroke/stroke.htm
References
American Dietetic Association. Spinal cord injury and nutrition guideline. Accessed 30 October 2010 from http://www.adaevidencelibrary.com/category.cfm?cid=14&cat=0.
Anekonda, TS. Resveratrol—a boon for treating Alzheimer’s disease? Brain Res Rev. 2006;52:316.
Blau, JN. Water deprivation: a new migraine precipitant? Headache. 2005;45:757.
Brain Trauma Foundation, Guidelines for the management of severe traumatic brain injury. ed 3. New York: Brain Trauma Foundation; 2007. Accessed 30 October 2010 from. http://www.braintrauma.org/pdf/protected/Guidelines_Management_2007w_bookmarks.pdf
Bough, KJ, Rho, JM. Anticonvulsant mechanisms of the ketogenic diet. Epilepsia. 2007;48:43.
Brown, SJ. The role of vitamin D in multiple sclerosis. Ann Pharmacother. 2006;40:1158.
Bulat, RD, Orlando, RC. Oropharyngeal dysphagia. Curr Treat Options Gastroenterol. 2005;8:269.
Callahan, CM, et al. Effectiveness of collaborative care for older adults with Alzheimer’s disease in primary care. JAMA. 2006;295:2148.
Centers for Disease Control and Prevention. Heart disease and stroke. http://www.cdc.gov/nccdphp/scientific.htm, 2006. [Accessed 30 June 2006 from].
Chauhan, NB. Multiplicity of garlic health effects and Alzheimer’s disease. J Nutr Health Aging. 2005;9:421.
Czaplinski, A, et al. Amyotrophic lateral sclerosis: early predictors of prolonged survival. J Neurol. 2006;13 June:2226.
Czlonkowska, A, Kurkowska-Jastrzebska, I. Inflammation and gliosis in neurological diseases—clinical implications. J Neuroimmunol. 2011;231:78.
De Lau, LM, et al. Dietary folate, vitamin B12, and vitamin B6 and the risk of Parkinson’s disease. Neurology. 2006;67:315.
Deon, M, et al. The effect of Lorenzo’s oil on oxidative stress in X-linked adrenoleukodystrophy. J Neurol Sci. 2006;247(2):157.
Dowson, AJ, et al. Review of clinical trials using acute intervention with oral triptans for migraine management. Int J Clin Pract. 2006;60:698.
Epilepsy Foundation. Statistics. http://www.epilepsyfoundation.org/answerplace/statistics.cfm, 2006. [Accessed 30 June 2006 from].
Freedman, MS. Disease-modifying drugs for multiple sclerosis: current and future aspects. Expert Opin Pharmacother. 2006;7:S1.
Fioravanti, M, Yangi, M. Cytidinediphophocholine (CDP-choline) for cognitive and behavioral disturbances associated with chronic cerebral disorders in the elderly, Cochrane Database of Systematic Reviews 2010. Accessed 28 March 2011 from http://onlinelibrary.wiley.com/o/cochrane/clsysrev/articles/CD000269/pdf_fs.html.
Gardner, A, Boles, RG. Beyond the serotonin hypothesis: mitochondria, inflammation and neurodegeneration in major depression and affective spectrum disorders. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:730.
Goldstein, LB, et al. Primary prevention of ischemic stroke: a guideline from the American Heart Association/American Stroke Association Stroke Council. Stroke. 2006;37:1583.
Groomes, LB, et al. Do patients with absence epilepsy respond to ketogenic diets. J Child Neurol. 2011;26:160.
Gu, Y, et al. Food combination and Alzheimer’s disease risk. Arch Neurol. 2010;67:699.
Hershey, AD, et al. Coenzyme Q10 deficiency and response to supplementation in pediatrics and adolescent migraine. Headache. 2007;47:73.
International Alliance of ALS. What is ALS/MND? Accessed 22 March 2010 from www.alsmndalliance.org/whatis.html.
Jaeger, PA, Wyss-Coray, T. Beclin 1 complex in autophagy and Alzheimer’s disease. Arch Neurol. 2010;67:1181.
Kidd, PM. Neurodegeneration from mitochondrial insufficiency: nutrients, stem cells, growth factors, and prospects for brain rebuilding using integrative management. Altern Med Rev. 2005;10:268.
Kossoff, EH, et al. Optimal clinical management of children receiving the ketogenic diet: recommendations of the International Ketogenic Diet Study Group. Epilepsia. 2009;50:304.
Mark, BL, Carson, JA. Vitamin D and autoimmune disease—implications for practice from the multiple sclerosis literature. J Am Diet Assoc. 2006;106:418.
Moser, HW. Therapy of X-linked adrenoleukodystrophy. NeuroRx. 2006;3:246.
National Institutes of Health. Stroke. http://www.ninds.nih.gov/, 2006. [Accessed 22 March 2011 from].
Nordlund, A, Oliveberg, M. Folding of Cu/Zn superoxide dismutase suggests structural hotspots for gain of neurotoxic function in ALS: parallels to precursors in amyloid disease. Proc Natl Acad Sci USA. 2006;103:10218.
Patel, A, et al. Long-term outcomes of children treated with the ketogenic diet in the past. Epilepsia. 2010;51:1277.
Polman, CH, et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald” criteria. Ann Neurol. 2005;58:840.
Porta, N, et al. The ketogenic diet and its variants: state of the art. Rev Neurol. 2009;165:430.
Querfurth, HW, LaFerla, FM. Alzheimer’s disease. N Engl J Med. 2010;362:329.
Ramesh, BN, et al. Neuronutrition and Alzheimer’s disease. J Alzheimer’s Dis. 2010;19:1123.
Raymond, GV, et al. Head trauma can initiate the onset of adrenoleukodystrophy. J Neurol Sci. 2010;290:70.
Schiapparelli, P, et al. Non-pharmacological approach to migraine prophylaxis: part II. Neurol Sci. 2010;31(Suppl 1):S137.
Sharma, S, et al. A pyrazole curcumin derivative restores membrane homeostasis disrupted after brain trauma. Exp Neurol. 2010;226:191.
Shetty, AK. Reelin signaling, hippocampal neurogenesis, and efficacy of aspirin intake & stem cell transplantation in aging and Alzheimer’s disease. Aging Dis. 2010;1:2.
Solomon, AJ, Whitham, RH. Multiple sclerosis and vitamin D: a review and recommendations. Curr Neurol Neurosci Rep. 2010;10:389.
Soto, ME, et al. Rapid cognitive decline: searching for a definition and predictive factors among elderly with Alzheimer’s disease. J Nutr Health Aging. 2005;9:158.
Spigt, MG, et al. Increasing the daily water intake for the prophylactic treatment of headache: a pilot trial. Europ J Neurol. 2005;12:715.
Strand, EA, et al. Management of oral-pharyngeal dysphagia symptoms in amyotrophic lateral sclerosis. Dysphagia. 1996;11:129.
Su, HM. Mechanisms of n-3 fatty acid-mediated development and maintenance of learning memory performance. J Nutr Biochem. 2010;21:364.
Sun, AY, et al. Botanical phenolics and brain health. Neuromolecular Med. 2008;10:259.
Sun-Edelstein, C, Mauskop, A. Foods and supplements in the management of migraine headaches. Clin J Pain. 2009;25:446.
Sun-Edelstein, C, Mauskop, A. Role of magnesium in the pathogenesis and treatment of migraine. Expert Rev Neurother. 2009;9:369.
Tan, J, et al. Lack of effect of oral choline supplement on the concentrations of choline metabolites in human brain. Magn Reson Med. 1998;39:1005.
Victor, M, Ropper, AH. Adams and Victor’s principles of neurology, ed 8. New York: McGraw-Hill, Health Professions Division; 2005.
Wang, L, et al. Performance-based physical function and future dementia in older people. Arch Intern Med. 2006;166:1115.
Weber, S, et al. Modified Atkins diet to children and adolescents with medical intractable epilepsy. Seizure. 2009;18:237.
Wollen, KA. Alzheimer’s Disease: the pros and cons of pharmaceutical, nutritional, botanical, and stimulatory therapies, with a discussion of treatment strategies from the perspective of patients and practitioners. Altern Med Rev. 2010;15:223.
Zeisel, SH, Caudill, MA. Choline. Advances in Nutrition. 2010;1:46.
Zhang, ML, et al. Dietary factors and smoking as risk factors for PD in a rural population in China: a nested case-control study. Acta Neurol Scand. 2006;113:278.
Zoccolella, S, et al. Homocysteine levels and amyotrophic lateral sclerosis: a possible link. Amytroph Lateral Scler. 2010;11:140.