Medical Nutrition Therapy for Intellectual and Developmental Disabilities
Individuals with developmental disabilities were generally housed in institutions for the first half of the 20th century. Little attention was paid to their education or medical or nutritional care. In 1963 the Developmental Disabilities Assistance and Bill of Rights Act was passed. Through this Act federal funds supported the development and operation of state councils, protection and advocacy systems, university centers, and projects of national significance. This Act provided the structure to assist people with developmental disabilities to pursue meaningful and productive lives. The institutions that housed these individuals were gradually closed or reduced in size. By 1975 these individuals were cared for at home, in schools, or in small residential facilities. In 1975 Public Law (P.L.) 94-142 was passed, opening public schools to children with developmental disabilities. In 1985 P.L. 99-487 (102-119 in 1992), the Early Intervention Act, was passed, bringing services to children from birth to school age.
A developmental disability is defined as a severe chronic disability that is attributable to a mental or physical impairment or combination of mental and physical impairments. It is manifested before the person attains age 22; is likely to continue indefinitely; results in substantial functional limitations in three or more areas of major life activity (self-care, receptive and expressive language, learning, mobility, self-direction, capacity for independent living, and economic self-sufficiency); and reflects the person’s need for a combination of generic or specialized interdisciplinary care, treatments, or other services that are lifelong or of extended duration and individually planned and coordinated. Developmental disabilities affect individuals of all ages and are not a disease state. They are conditions that are caused by fetal abnormalities, birth defects, and metabolic and chromosomal disorders.
Intellectual disability replaced the term mental retardation in the 11th edition of the Definition Manual of the American Association on Intellectual and Developmental Disabilities (AAIDD, 2011). Intellectual disability is the most common developmental disability, characterized by significantly below-average intellectual functioning along with related limitations in areas such as communication, self-care, functional academics, home living, self-direction, health and safety, leisure, or work and social skills. It is estimated that 1% to 3% of the population have this diagnosis.
Developmental disabilities have been traced to many causes: chromosomal aberrations, congenital anomalies, specific syndromes, neuromuscular dysfunction, neurologic disorders, prematurity, cerebral palsy (CP), untreated inborn errors of metabolism, toxins in the environment, and nutrient deficiencies. The Centers for Disease Control and Prevention (CDC) reports that 3% of all live births have a birth defect (CDC, 2010) and there are 4.5 million Americans living with developmental disabilities (American Dietetic Association [ADA], 2010).
Medical Nutrition Therapy
Medical nutrition therapy (MNT) services vary depending on the individual’s physical or mental problem, and much has been learned about the role of nutrition in both the prevention of disabilities and intervention when a nutrition problem exists. The role of the registered dietitian (RD) is essential. Because there is an abundance of information that parents and caretakers use from support groups and websites that are untested scientifically, RDs are often providing evidence-based counseling to counteract misinformation.
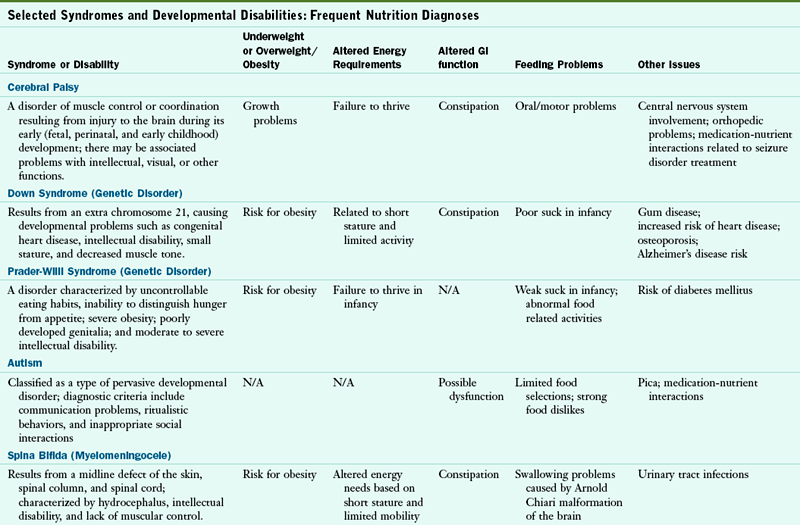
Numerous nutrition problems have been identified in the individual with developmental disabilities. Growth retardation, obesity, failure to thrive, feeding problems, metabolic disorders, medication-nutrient interactions, constipation, and renal problems may be present. Other health problems exist, depending on the disorder. Table 45-1 lists the most common developmental disabilities and their associated nutrition problems.
TABLE 45-1
Selected Syndromes and Developmental Disabilities: Frequent Nutrition Diagnoses
GI, Gastrointestinal; N/A, not applicable.
Data from American Dietetic Association: Position of the American Dietetic Association: providing nutrition services for people with developmental disabilities and special health care needs, J Am Diet Assoc 110:297, 2010.
The ADA confirmed that nutrition services provided by RDs are essential components of comprehensive care for infants, children, and adults with intellectual and developmental disabilities. Nutrition services should be provided throughout the life cycle. Educational and vocational programs should provide MNT in a manner that is interdisciplinary, family centered, community based, and culturally competent (ADA, 2010).
Nutrition Assessment
Anthropometric measures are altered when an individual is unable to stand, suffers from contractions, or has other gross motor problems. Measuring body weight may require special equipment such as chair scales or bucket scales. Wheelchair scales are used in some clinics but require that the wheelchair weight be known. Obtaining height for the nonambulatory individual requires either a recumbent board that can be purchased or constructed. Other measures of height include arm span, knee-to-ankle height, or sitting height (Figure 45-1; see Appendixes 19 and 20).
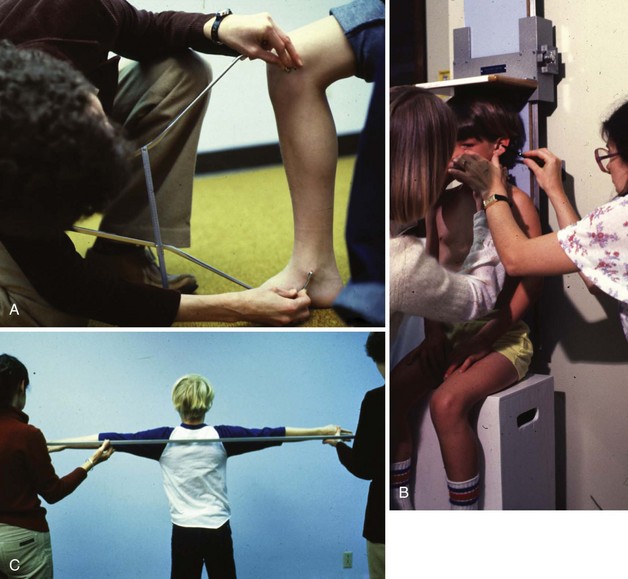
FIGURE 45-1 A, Knee height measure. B, Sitting height measure. C, Arm span measure. (Courtesy Cristine M. Trahms, 2002.)
Although growth charts for children with various syndromes do exist, most clinicians recommend using the general CDC charts (Appendixes 9 to 16) because the information in the specialized charts is often based on small numbers, mixed populations, and old data (CDC, 2010). See Chapter 6.
Other measures that can be used to explore weight issues include arm circumference, triceps skinfold measures, and body mass index (BMI). BMI is a part of the CDC growth charts and can also be found in Appendixes 12, 16, and 23. Using the BMI for age can be controversial. For example, BMI is limited for identifying overweight in children who are overly fat because of decreased muscle mass and short stature.
Biochemical Measures
Laboratory assessment for the child and adult with developmental disabilities is generally the same as that discussed in Chapter 8 and Appendix 30. Additional tests may be indicated for the individual with epilepsy or seizures who is receiving an anticonvulsant medication such as phenytoin, divalproex sodium, topiramate, or carbamazepine. Use of these medications can lead to low blood levels of folic acid, carnitine, ascorbic acid, calcium, vitamin D, alkaline phosphatase, phosphorus, and pyridoxine. Assessment of thyroid status is part of the protocol for children with Down syndrome (DS), and a glucose tolerance test is recommended for evaluation of the child with Prader-Willi syndrome (PWS). As appropriate, genetic testing may be encouraged for both affected individuals and their biological family members.
Dietary Intake and Feeding Problems
Dietary information should be obtained for the child with a developmental disability through a diet history. However, there are difficulties in obtaining an accurate recall when the child is in a day-care center. Written diaries are also difficult to obtain when the child has multiple caretakers or when he or she is in school. When working with an adult with developmental disabilities, it is often difficult to obtain accurate information unless the individual has supervision, such as in special residential housing. Use of pictures and food models can often help in obtaining an estimate of the individual’s intake.
Many children and adults with developmental disabilities display feeding problems that seriously decrease their ability to eat an adequate diet. Feeding problems are defined as the inability or refusal to eat certain foods because of neuromotor dysfunction, obstructive lesions such as strictures, and psychosocial factors (Cloud et al., 2005). Other causes of feeding problems in this population include oral-motor difficulties, positioning problems, conflict in parent-child relationships, sensory issues, and tactile resistance from previous intubation (Tobin et al., 2005). The nutritional consequences of feeding problems include inadequate weight gain, poor growth in length, lowered immunity, anemia, vitamin and mineral deficiencies, dental caries, and psychosocial problems. Feeding problems should be assessed with an understanding of the normal development of feeding and the physical makeup of the mouth and pharynx (see Chapters 17-19 and Chapter 41).
Estimates are that feeding problems are found in 40% to 70% of children with special health care needs and 80% of children with developmental delays. Feeding problems are classified as oral motor, positioning, behavioral, and self-feeding. Oral-motor problems include difficulty with suckling, sucking, swallowing, and chewing. They also include sensory motor integration and problems with self-feeding and are described as exaggerations of normal neuromotor mechanisms that disrupt the rhythm and organization of oral-motor function and interfere with the feeding process (Box 45-1).
Children with developmental disabilities such as DS, CP, or cleft lip and palate often have oral-motor feeding problems that may be related to the cleft, muscle tone, and inability to accept texture changes. The oral-motor problem can also be related to the developmental level, which may be delayed.
Positioning a child for feeding relates to his or her motor development, head control, trunk stability, and ability to have hips and legs at a right angle (Figures 45-2 and 45-3). This is frequently a problem for individuals with CP, spina bifida, and DS. However, without proper positioning, oral-motor problems are difficult to correct. The ability to self-feed may be delayed in the child with developmental disabilities and requires training by a feeding specialist. A feeding evaluation is best completed with actual observation by a team composed of a speech therapist, a dentist, a physical therapist, an occupational therapist, and an RD. An excellent interdisciplinary feeding evaluation form is the Developmental Feeding Tool shown in Figure 45-4. Frequently, adaptive feeding equipment is needed.
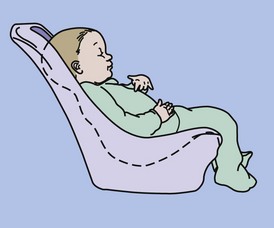
FIGURE 45-2 Proper feeding position for the infant. (From Cloud H: Team approach to pediatric feeding problems, Chicago, 1987, American Dietetic Association. © American Dietetic Association. Reprinted with permission.)

FIGURE 45-3 Good feeding position for a child ages 6 to 24 months, showing hip flexion, trunk in midline, and head in midline. Good foot support with a stool should continue throughout childhood. (From Cloud H: Team approach to pediatric feeding problems, Chicago, 1987, American Dietetic Association. © American Dietetic Association. Reprinted with permission.)

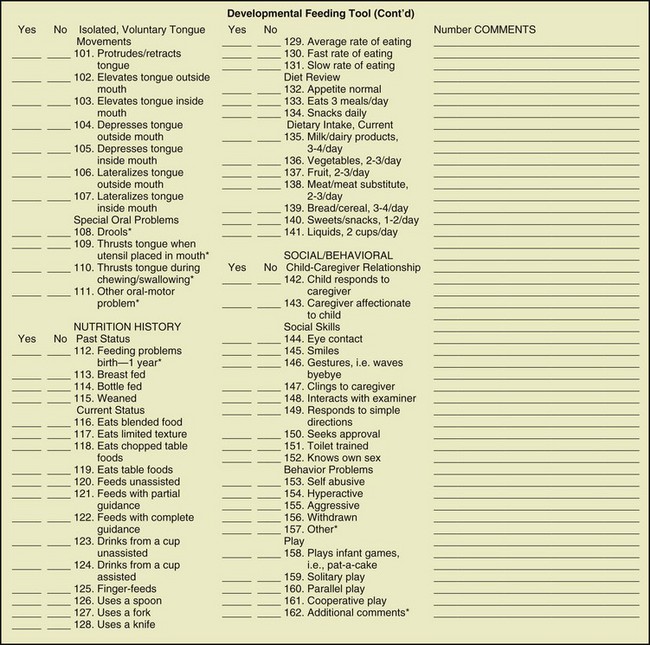
FIGURE 45-4 Developmental feeding tool. (Smith MAH et al: Feeding management for a child with a handicap: a guide for professionals, Memphis, 1982, University of Tennessee: The Boling Child Development Center, University of Tennessee Center for Health Sciences).
Behavioral issues may result from oral-motor or sensory problems, medical problems, certain medications, and the amount of emphasis placed on feeding. Issues such as control of the feeding process along with lack of autonomy in the child may create negative behavior. Environmental factors also influence the eating behavior of the child. Examples include where the child is fed, distractibility, serving sizes, delayed weaning, and frequency of feeding.
Nutrition Diagnosis
Once the nutrition assessment has been completed, problems should be identified related to growth. Excessive or inadequate weights; inadequate dietary intake; excessive or inadequate fluid intake; altered gastrointestinal problems such as constipation, vomiting, and diarrhea; intake of foods that are unsafe because of contamination or food allergies; food-medication interactions; chewing and swallowing difficulties; and problems with self-feeding may be issues. The nutrition diagnoses should be listed and priorities established. When possible, this information is shared with the parent, caregiver, or the adult client before the intervention process begins.
Interventions
Once the nutrition diagnoses are identified and prioritized, short- and long-term goals should be set. Consideration must be given to the motivational level of the parent or client, his or her cultural background, and how the therapy can be community based and family centered. This means that consideration must be given to where the client will be served so that it becomes a part of the individualized education plan (IEP) or the individualized family plan.
Intervention plans should include all aspects of an individual’s treatment program to avoid issuing an isolated set of instructions relevant to only one treatment goal. In some cases MNT may not be the family’s first priority in the care of the child or adult, making it important for the RD to recognize the family’s cues (see Chapter 15). Even when the family is ready for an intervention, such as weight management for a child with spina bifida, many factors require consideration. The parent or caregiver’s educational level and income, language barriers, access to safe and appropriate food, and family coping strategies should always be identified (see Chapter 15).
Monitoring and Evaluation
Once MNT has been initiated, the need for follow-up evaluation and monitoring either by the RD or another health care professional is important. Giving information in writing, followed by a phone call, gives the chance to repeat some of the discussion and to answer any questions not asked during the initial session. Clarification of suggestions is often needed when monitoring nutrition changes that affect growth and development; a follow-up visit may also be needed.
A case manager may be involved who communicates with the adult with the disability. The RD may need to find appropriate resources to pay for supplemental nutrition products, tube feedings, and special food products as a part of the follow-up process. Community and agency resources will be discussed.
Chromosomal Aberrations
Down syndrome (DS) is a chromosomal aberration of chromosome 21 (trisomy 21). It has an incidence of 1 in 700 live births and results from the presence of an extra chromosome in each cell of the body. This anomaly causes the physical and developmental features of short stature; congenital heart disease; mental retardation; decreased muscle tone; hyperflexibility of joints; speckling of the iris (Brushfield spots); upward slant of the eyes; epicanthal folds; small oral cavity; short, broad hands with the single palmar crease; and a wide gap between the first and second toes (Capone et al., 2005).
Pathophysiology
Normally every cell of the human body except for the gametes (sperm or ova) contains 46 chromosomes, which are arranged in pairs (see Chapter 5). With DS there is one extra chromosome for a total of 47. There are three processes by which this anomaly can occur: nondysjunction, translocation, and mosaicism. In nondysjunction, chromosome 21 fails to separate before conception and the abnormal gamete joins with a normal gamete at conception to form a fertilized egg with three of chromosome 21. This may also occur during the first cell division after conception. This type of DS is usually sporadic and has a recurrence rate of 0.5% to 1%. In translocation, the extra chromosome is attached to another chromosome (usually 14, 15, or 22). Approximately half the time, this type of DS is inherited from a parent who is a carrier; it has a much higher risk of recurrence in another pregnancy. In mosaicism the abnormal separation of chromosome 21 occurs sometime after conception. All future divisions of the affected cell result in cells with an extra chromosome. Therefore the child has some cells with the normal number of chromosomes and other cells with an extra chromosome. Frequently the child with this type of DS lacks some of the more distinctive features of the syndrome (Capone et al., 2005) (see Pathophysiology and Care Management Algorithm: Down Syndrome).
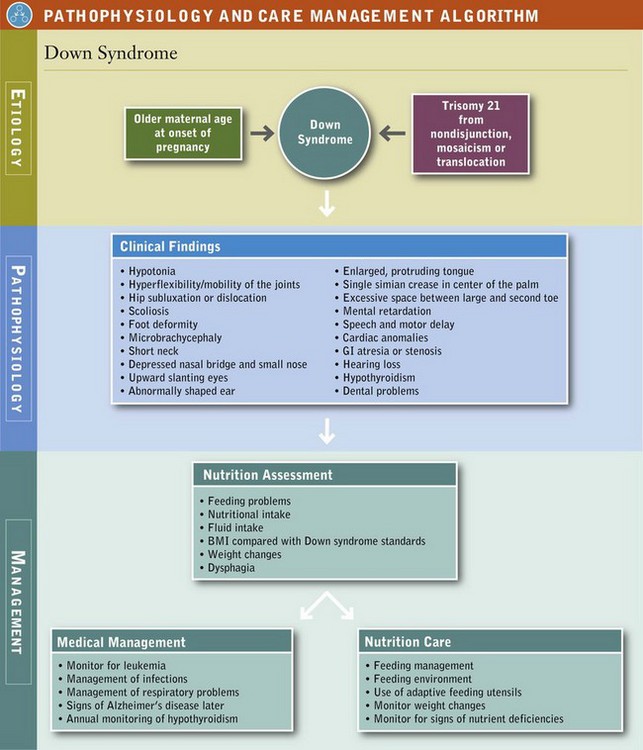
Medical Treatment
The National Down Syndrome Congress has published a listing of the health concerns for individuals with DS; many have nutrition implications (Table 45-2).
TABLE 45-2
Health Concerns of Children with Down Syndrome
| Health Concern | Implications | Treatment |
| Congenital heart disease | 40%-50% of population | Medication or surgical repair |
| Hypotonia | Reduced muscle tone, increased range of joints | |
| Motor function problem | Poor physical function | Early intervention for physical therapy, occupational therapy |
| Delayed growth | Short stature | In some cases growth hormone |
| Developmental delays | Poor physical, emotional function | Early intervention |
| Hearing concerns | Small ear canals, otitis media, conductive impairment | Early intervention |
| Dental problems | Decreased saliva, reflux, and vomiting | Low sucrose intake |
| Ocular problems | Refractive errors, strabismus, cataracts | Corrective glasses |
| Cervical spine abnormality | None | |
| Thyroid disease | Hypothyroidism | Thyroid supplement, tests repeated annually |
| Overweight | Excessive weight gain, inactivity | Decrease energy, increase activity |
| Seizure disorders | Variable nutrient intake | Medications |
| Emotional disorders | May occur late in childhood | Medication, counseling |
Updated from Saenz RB: Primary care of infants and young children with Down syndrome, Am Fam Phys 59:381, 1999.
Medical Nutrition Therapy
Anthropometric Measures: Height, weight, head circumference, triceps skinfold, and arm circumference are obtained for the child with DS with the usual measurements. See Chapter 6. BMI can be taken but may be higher than normal because of short stature. Growth measures are an important part of the assessment and ongoing nutrition therapy because these individuals tend to be short. Muscle tone is low and gross motor ability is often delayed, leading to the possibility of the individual becoming overweight. Monitoring should be frequent, and growth plotted on the CDC charts (see Appendixes 9 through 16).
Biochemical Measures: Numerous studies have shown biochemical and metabolic abnormalities in individuals with DS; however, many have involved small samples and were difficult to interpret (Capone et al., 2005). Although serum concentrations of albumin have been found to be low, the guidelines from the Down Syndrome Medical Congress do not list serum albumin assessment as routine. Increased glucose levels have been reported, with an increased incidence of diabetes mellitus.
Current guidelines for the treatment of infants and children with DS include evaluation of thyroid function at birth and thereafter annually.
Dietary Intake: During infancy the food intake of the infant with DS may differ from that of the normal infant. Although human breast milk is recommended, many infants with DS are formula fed. Infant illnesses, admission to the neonatal unit, frustration, depression, perceived milk insufficiency, and difficulty in suckling by the infant are reasons why formula feeding is used.
Progression to solid food has been found to be delayed in children with DS, mostly as a result of delays in feeding and motor development. Introduction of solid food may not be offered at 6 months if the infant has poor head control or is not yet sitting. Low tone and sucking problems also delay weaning from the breast or bottle to the cup. IEPs include feeding and feeding progression instruction and practice.
An important part of evaluating the dietary intake is determining energy and fluid needs, because children with DS have a high prevalence of obesity. Studies have indicated that the resting energy expenditure (REE) of the child with DS is lower than for controls without DS—and may be as much as 10% lower than the dietary reference intake (DRI) for energy. For the child older than the age of 5, calculations for energy requirements may need to be based on height rather than weight (Table 45-3) (see Chapter 2).
TABLE 45-3
Estimated Caloric Needs for Special Conditions
| Condition | kcal/cm | Comments |
| Normal child | Average 16 | |
| Prader-Willi | Maintain growth: 10-11 Promote weight loss: 8.5 |
For all children and adolescents |
| Cerebral palsy | ||
| Mild | 14 | Reliable for ages 5-11 yr |
| Severe, limited mobility | 11 | Reliable for ages 5-11 yr |
| Down syndrome | Girls: 14.3 Boys: 16.1 |
Reliable for ages 5-11 yr |
| Motor dysfunction | ||
| Nonambulatory | 7-11 | Reliable for ages 5-12 yr |
| Ambulatory | 14 | Reliable for ages 5-12 yr |
| Spina bifida | Maintain weight: 9-11 Promote weight loss: 7 |
For all children older than 8 years of age and minimally active |
Modified from Rokusek C, Heindicles E: Nutrition and feeding for persons with special needs, with permission of the South Dakota University Affiliated Program, Interdisciplinary Center for Disabilities, 1992.
Feeding Skills: Feeding skills are delayed in the infant and child with DS. Some parents find difficulty in initiating oral motor skills such as suckling and sucking. The infant with DS often has difficulty in coordinating sucking, swallowing, and breathing, which are the foundations for early feeding. When the infant has a congenital heart defect, which occurs in 40% to 60% of the DS infants, sucking is weakened, and fatigue interferes with the feeding process. Gastrointestinal anomalies are found in 8% to 12% of infants with DS, and these infants often require nasogastric or gastrostomy feedings.
Other physical factors that make feeding difficult in the first years of life include a midfacial hypoplasia (a craniofacial deformity common in cleft palate), a small oral cavity, a small mandible, delayed or abnormal dentition, malocclusion, nasal congestion, small hands, and short fingers. Weaning and self-feeding are usually late when compared with the normal infant, and frequently do not emerge until 15 to 18 months of age. The DS infant strives for independence and autonomy approximately 6 months later than the child without DS.
Intervention Strategies
Overweight: The most effective intervention for the overweight child with DS is to design a calorie-controlled eating plan based on kilocalories per centimeter of height (see Table 45-3). Dietary management includes assessing the feeding developmental level of the child, working with a physical therapist related to gross motor skills to determine possible activity levels, and making environmental changes. Environmental changes should include following a regular eating schedule that includes three meals at regular times with the child sitting either in a high chair or at the table. Planned snacks should be low in fat and sugar. Soft drinks should be eliminated, and milk should be low fat (after age 2). Physical activity should be encouraged. Counseling in which the parent helps determine a realistic plan should focus on serving sizes and food preparation and decreasing the number of times meals are purchased in fast-food restaurants. If the child or adolescent is school age, a prescription for a special meal at school can be obtained by using the school food service prescription (to be discussed later in the chapter).
Feeding Skills: Often parents wrongly expect different feeding development for the child with DS. Behavioral problems related to feeding usually develop based on what happens between the parent and child at mealtime. An example of this is the unnecessary delay of weaning to a cup or progression of food textures because of inadequate effort or education. During intervention programs the feeding team can guide the parent in positioning the child and working toward attainable feeding skills related to the developmental level of the child.
Constipation: This is a frequent problem for the child with DS because of overall low tone followed by lack of fiber and fluid in the diet. Treatment should involve increasing fiber and fluid, with water consumption emphasized. Fiber content of the diet for children after age 3, is 5 to 6 g per year of age per day. For adults the recommendation is for 25 to 30 g of dietary fiber daily.
Prader-Willi Syndrome
Prader-Willi syndrome (PWS) was first described in 1956 by Doctors Prader, Willi, and Lambert. It is a genetic condition caused by the absence of chromosomal material. PWS occurs with a frequency of 1 in 10,000 to 1 in 25,000 live births. Characteristics of the syndrome include developmental delays, poor muscle tone, short stature, small hands and feet, incomplete sexual development, and unique facial features. Insatiable appetite leading to obesity is the classic feature of PWS; however, in infancy the problem of hypotonia (low muscle tone) interferes with feeding and leads to failure to thrive (McCune and Driscoll, 2005). Developmental delays (affecting 50% of the population), learning disabilities, and mental retardation (affecting 10%) are associated with PWS.
The genetic basis of PWS is complex. Individuals with PWS have a portion of genetic material deleted from chromosome 15 received from the father. Of the cases of PWS, 70% are caused from the paternal deletion, occurring in a specific region on the q arm of the chromosome. PWS can also develop if a child receives both chromosome 15s from the mother. This is seen in approximately 25% of the cases of PWS and is called maternal uniparental disomy. Early detection of PWS is now possible because of the use of deoxyribonucleic acid methylation analysis, which correctly diagnoses 99% of the cases (McCune and Driscoll, 2005). This is an important development in the early identification and subsequent treatment of these children to prevent obesity and growth retardation and is used to identify the infant born with features and characteristics described previously
Pathophysiology
Metabolic Abnormalities: Short stature in the individual with PWS has been attributed to growth hormone deficiency. In addition to decreased growth hormone release, children have low serum insulin-like growth factor (IGF)-1, low IGF-binding protein-1, and low insulin compared with normal obese children. Growth hormone therapy was approved by the Food and Drug Administration in 2000, and in one 5-year study in Japan 37 patients from age 3 to 21 years experienced significant increase in height gain velocity when given growth hormone (Obata et al., 2003). A more recent study (Carrel et al., 2010) found that GH therapy of infants and toddlers for 12 months significantly improved body composition and mobility skill acquisition.
In addition to the growth hormone deficiency, individuals have a deficiency in the hypothalamic-pituitary-gonadal axis, causing delayed and incomplete sexual development. Finally there is a decreased insulin response to a glucose load in children with PWS compared with age-matched non-PWS obese children (Talebizadeh and Butler, 2005).
Appetite and Obesity: Appetite control and obesity are common problems for individuals with PWS. After the initial period of failure to thrive, children begin to gain excessively between the ages of 1 and 4, and appetite slowly becomes excessive. Based on longitudinal study, Miller et al describe this gradual and complex progression in terms of 7 nutritional phases based on levels of appetite, metabolic changes and growth. In fact, some adults with PWS may progress to the last stage - one, with no insatiable appetite and the person is able to feel full (Miller et al, 2011).
This uncontrollable appetite, a classic feature of PWS, when combined with overeating, a low basal metabolic rate, and decreased activity, leads to the characteristic obesity. The cause of the uncontrollable appetite is suspected to involve the hypothalamus and altered levels of satiety hormones and peptides such as ghrelin (Scerif et al, 2011).
Body composition is an important consideration in the evaluation of individuals with PWS. They have decreased lean body mass and increased body fat, even in infancy (Reus et al, 2011). Body fat is generally deposited in the thighs, buttocks, and abdominal area. The lowered energy expenditure is found in young children, adolescents, and adults with PWS, with one study showing adolescents with PWS having a total energy expenditure (TEE) 53% of that of normal obese adolescents (McCune and Driscoll, 2005). The low muscle tone contributes greatly to the lack of interest in physical activity.
Nutrition Assessment
Anthropometric Measures: Height measurements tend to be lower in PWS infants and young children, with the rate of height gain tapering off between the ages of 1 and 4. The usual measurements of length or height, weight, and head circumference should be taken and plotted on the CDC growth curves. Other measures of interest include arm circumference and triceps skinfold measures. BMI may be distorted for the individual with PWS because of the short stature; however, plotting the BMI over time is useful in determining unusual changes (see Appendixes 12 and 16). It is important that anthropometric measures be taken frequently and reported to the parents or caregiver.
Biochemical Measures: Biochemical studies are generally the same for the PWS individual, with the exception of either fasting blood glucose tests or glucose tolerance tests. These are added because of the risk for diabetes mellitus, possibly related to the decreased insulin response and obesity that usually accompanies PWS.
Dietary Intake: Dietary information varies for individuals with PWS, depending on their age. In infancy the dietary information should be obtained with a careful dietary history and analyzed for energy and nutrient intake. Infants are commonly difficult to feed because of their hypotonia, poor suck, and delayed motor skills. Generally their feeding development is slower than in the normal infant, and transitioning to food at 4 to 6 months of age may be difficult. Many of these infants have gastroesophageal reflux requiring medication or thickening of their formula.
During the toddler years weight gain may increase rapidly as dietary intake increases. This requires careful assessment of portion sizes, frequency of feeding, and types of foods served. Although some parents may report that the child with PWS does not eat more than other children in the family, they need to be educated that the energy needs of the child are lower because of the reduced lean muscle mass and slow development of motor skills and activity. As the child gets older, interest in food increases; and, starting around ages 5 through 12, the child may be hungry all the time and display difficult behaviors such as tantrums, stubbornness, and food stealing. Many parents have found it necessary to lock cabinets, refrigerators, and the kitchen door to control food intake. Information gathered during the dietary interview should include asking about environmental control techniques.
Determination of energy needs for the infant with PWS is the same as for a normal infant. However, as the child enters the toddler years, he or she will need fewer calories to maintain weight gain along the growth curve. This will apply in adulthood when fewer calories are needed to maintain weight. Energy needs have been calculated according to centimeters of height from 2 years on. It has been recommended that the macronutrient intake of the diet be 25% protein, 50% carbohydrate, and 25% fat (see Table 45-3).
Feeding Skills: The infant with PWS often presents with weak oral skills and poor sucking skills in the first year of life. As the child matures, feeding skills are not a problem, but they may be delayed. Chewing and swallowing problems are not usually seen, although they may be associated with the low muscle tone. Behavioral feeding issues are associated with an insatiable appetite and not being provided with food. This can bring about tantrums.
Intervention Strategies
Intervention for PWS should occur at each developmental stage: infancy, toddler, preschool age, school age, and adult.
Infancy: Providing adequate nutrition as established by the American Academy of Pediatrics (AAP) related to breast-feeding or formula feeding is recommended. Because feeding may be difficult related to sucking, concentrating the formula or breast milk may be necessary to promote adequate weight gain. Feeding intervention will assist in improving the sucking problems caused by hypotonia. As the infant matures, a concentrated formula is not necessary, and foods can be added when head control and trunk stability are achieved, usually at approximately ages 4 to 6 months.
Toddler and Preschool Age: Most children begin to gain excessive weight between 1 and 4 years of age. Beginning a structured dietary protocol for the child and the family is important so that the toddler learns that meals are provided at specified times so that a pattern of grazing doesn’t develop. Parents should be taught to provide small servings of meats, vegetables, grains, and fruits and limited amounts of sweets. Early intervention for these children in the preschool years is very important in working with feeding issues and intake control as they grow older. Weight, height, and nutrient intake should be monitored monthly, and energy needs adjusted if weight gain becomes excessive. Concurrently physical activity must be encouraged as a part of the IEP, and physical therapy services made available if necessary.
School Age: For the school-age child, collaboration with the school food service program becomes important. Energy needs should be calculated per centimeter of height (see Table 45-3) and are generally 50% to 75% of the energy needs of unaffected children. This may require using the prescription for special meals through the school food service program. At home environmental controls may be required, with cupboards and refrigerators being locked, because the child and adolescent have limited satiety, and will search for food away from mealtime. Some parents say that growth hormone therapy for their child helps, but it doesn’t seem to change the child’s lack of satiety. Appetite-suppressing medications have been used but are largely unsuccessful.
Adulthood: Prevention of obesity is truly the key for successful treatment of PWS; however, many adults who are not identified early become very obese. Weight management programs providing a very low 6 to 8 kcal per centimeter of height may be required. Nutrient values should be calculated, and vitamin-mineral supplements added, as well as essential fatty acids (EFAs) if indicated. Many dietary treatments have been tried such as the ketogenic diet and the protein-sparing modified-fast diets. However, with any approach strict supervision is usually required, and great emphasis must be placed on physical activity. A behavior management approach has also been recommended to implement both the dietary management and physical activity plans. In many states there are group homes for adults with PWS where supervised independent living is possible and meals can be very structured and exercise programs implemented.
MNT of children and adults with PWS requires follow-up with many health care providers and schools. Fortunately parents of the individual with PWS now have access to a number of support groups and organizations dedicated to education, research, and establishing treatment programs.
Neurologic Disorders
Spina bifida is a neurologic tube defect that presents in a number of ways: meningocele, myelomeningocele (MM), and spina bifida occulta. MM is the most common derangement in the formation of the spinal cord and generally occurs between 26 to 30 days of gestation, with the date of occurrence affecting the location of the lesion. The lesion may occur in the thoracic, lumbar, or sacral area and will influence the amount of paralysis. The higher the lesion, the greater is the paralysis. Manifestations range from weakness in the lower extremities to complete paralysis and loss of sensation. Other manifestations include incontinence and hydrocephalus. The incidence of spina bifida is about 1 in 1430 births in the United States (CDC, 2010).
Prevention of spina bifida is now possible. In the 1980s, studies reported a positive effect from supplementation of mothers with folic acid plus multivitamins (Smithells et al, 1983). This reduced the risk of a second pregnancy with spina bifida as an outcome. As a result of numerous studies showing folic acid supplementation before conception to be effective, the national recommendation is 400 mcg/day for all women of childbearing age. Folic acid has been added to many flours and other cereal and grain products in the food supply since 1996 (CDC, 2010). These public health measures have resulted in increased folic acid blood levels in U.S. women of childbearing age and a decrease of 20% in the national rate of spina bifida (Robbins et al., 2006) (see Chapter 16).
Pathophysiology
The spinal lesion may be open and can be surgically repaired shortly after birth, usually within 24 hours to prevent infection. Although the spinal opening can be surgically repaired, the nerve damage is permanent, resulting in the varying degrees of paralysis of the lower limbs. In addition to physical and mobility issues, most individuals have some form of learning disability.
The spinal lesion affects many systems of the body and can result in weakness in the lower extremities, paralysis, and nonambulation; poor skin condition caused by pressure sores; loss of sensation and bladder incontinence; hydrocephalus; urinary tract infections; constipation; and obesity. Seizures also occur in approximately 20% of children with MM and require medication. Chronic medication is required for prevention and treatment of urinary tract infections and for bladder control. The resultant nutrition problems include obesity, feeding problems, constipation, and drug-nutrient interaction problems. Children with spina bifida may be allergic to latex. It has been recommended that they avoid certain foods such as bananas, kiwi, and avocados. Mild reactions can occur from apples, carrots, celery, tomatoes, papaya, and melons (Cloud et al., 2005) (see Box 27-3 in Chapter 27).
Nutrition Assessment
Anthropometric Measures: Infants and children with neural tube defects are usually shorter because of reduced length and atrophy of the lower extremities, although other problems such as hydrocephalus, scoliosis, renal disease, and malnutrition may contribute. The level of the lesions can also affect the length and height of the individual.
Obtaining accurate length and height measures can be difficult, especially as the child grows older. An alternate measure for determining height, the arm span/height ratio, is used and modified, depending on leg muscle mass. Arm span can be used directly as a height measure (arm span × 1) if there is no leg muscle mass loss, as in a sacral lesion. Arm span × 0.95 can be used to determine height if there is partial leg muscle loss, and arm span × 0.90 is used for a height measurement when there is complete leg muscle loss such as with a thoracic spinal lesion (Ekvall and Cerniglia, 2005). See Fig. 45-1 and Appendix 20.
Weight measures can be obtained for the child unable to stand by using chair scales, bucket scales, and wheelchair scales. To monitor the weight accurately, it should be obtained in a consistent manner, with the person in light clothing or undressed. Triceps skinfold measures can also be used, along with subscapular measures and abdominal and thorax measures, to determine the amount of body fat. See Chapter 6.
Head circumference should be measured in infants and toddlers up to age 3. A high percentage of children with spina bifida have head shunts as a result of their hydrocephalus. Unusual changes in the size of the head may indicate a problem with the shunt.
Biochemical Measures: Most protocols in the treatment of spina bifida include iron status tests, measurements of vitamin C and zinc levels, and other tests related to the nutritional consequences of medications needed for seizures and urinary tract infection control (see Chapter 9 and Appendix 31).
Dietary Intake: Many children with spina bifida eat a limited variety of foods, and they are frequently described as a “picky eaters” by the parents. When doing a dietary history, it is important to ask about the variety of foods, particularly of high-fiber foods. The school-age child may be prone to skipping breakfast because early morning preparations for school require more time than for the nonaffected child.
Energy needs are lower for the child with spina bifida (see Table 45-3), and calorie requirements must be determined carefully to prevent the obesity to which many are prone. Ekvall and Cerniglia (2005) found that for MM children 8 years or older, the caloric need is 7 cal/cm of height for weight loss and 9 to 11 cal/cm of height to maintain weight. It is important to evaluate how the mother or caretaker perceives food for the child since it represents sympathy and love for many parents.
It is important to evaluate fluid intake because so many children have urinary tract infections and may be drinking inadequate amounts of water and excessive amounts of soft drinks or tea. Cranberry juice can be offered. Physical activity must also be evaluated and may be found to be very limited, particularly when the child is nonambulatory. Ambulatory individuals with a shunt may be restricted from contact sports but can be involved in walking and running.
Feeding skills need to be evaluated, along with oral motor function in particular. Many children with spina bifida are born with Arnold Chiari malformation of the brain, which affects the brainstem and swallowing. See Appendix 35 for dietary recommendations for dysphagia. Difficulty in swallowing may contribute to the child avoiding certain foods later in life. Because of this there may be delays in weaning from the breast or bottle to the cup, but there should be no delays in gaining self-feeding skills.
Clinical Evaluation: Evaluation should include looking for pressure sores and signs of dehydration, along with asking about the amount and type of fluids consumed. Constipation may be caused by the neurogenic bowel combined with a diet low in fiber and fluids. The evaluation should include a review of food intake, fiber content, and fluids.
Intervention Strategies
Many children with spina bifida are overweight. It usually occurs when ambulation is a problem leading to decreased energy needs. Refusal to accept a wide variety of foods is common. Frequent feeding is both an oral-motor and a behavioral problem. Counseling should include introducing foods around age 6 months, limiting the intake of high-sucrose infant jar foods, and training the child to accept a wide variety of flavors and textures.
Obesity prevention should include addressing the problems of limited physical activity, increasing fluids and fiber and calculating the appropriate amount of calories. Once the child is in school, the food service manager should be provided with a prescription for a low-calorie breakfast and lunch, and weight management program should be listed as a part of the IEP. Enrollment in a group weight management program has been used successfully with modification of the accompanying physical exercise. The ideal program uses a team approach with involvement of the RD, nurse, occupational therapist, physical therapist, educator, and psychologist.
In many clinics the child or adult with spina bifida is seen on a semiannual or annual basis. This frequent follow-up is necessary and should include monitoring of growth, particularly weight; food and fluid intake; and medication use. School programs and IEPs are excellent follow-up tools; however, the school often lacks appropriate scales for weighing a nonambulatory student. In this situation the parent should be encouraged to bring the child to the clinic for weight checks or, if distance is a problem, find a long-term care facility that will permit use of its scales. Follow-up by phone contact or e-mail can be done for evaluating dietary intake and fluid management.
Cerebral Palsy
Cerebral palsy (CP) is a disorder of motor control or coordination resulting from injury to the brain during its early development. Among the causative agents of CP are prematurity; blood-type incompatibility; placental insufficiency; maternal infection that includes German measles; other viral diseases; neonatal jaundice; anoxia at birth; and other bacterial infections of the mother, fetus, or infant that affect the central nervous system.
The problem in CP lies in the inability of the brain to control the muscles, even though the muscles themselves and the nerves connecting them to the spinal cord are normal. The extent and location of the brain injury determine the type and distribution of CP. The incidence of CP varies with different studies, but the most commonly used rate is 2 to 3 in 1000 live births. The prevalence of premature births has contributed to maintenance of this figure despite electro-fetal monitoring.
Pathophysiology
There are various types of CP, which are classified according to the neurologic signs involving muscle tone and abnormal motor patterns and postures. The diagnosis of CP is generally made between 9 to 12 months of age and as late as 2 years with some types (Box 45-2).
Poor nutrition status and growth failure, often related to feeding problems, are common in children with CP. Meeting energy and nutrient needs is particularly difficult in children and adults with more severe forms of CP such as spastic quadriplegia and athetoid CP. For example, bone mineral density of children and adolescents with moderate to severe CP is reduced in those with gross motor function and feeding difficulties (Henderson et al., 2005).
Other health problems include constipation, usually caused by inactivity and lack of fiber and fluids, often connected to feeding problems. Dental problems occur and are often related to malocclusion, dental irregularities, and fractured teeth. Lengthy and prolonged bottle-feedings of milk and juice promote the decay of the primary upper front teeth and molars (see Chapter 26). Hearing problems and especially visual impairments, mental retardation, respiratory problems, and seizures affect nutrition status. Seizures are controlled with anticonvulsants, and a number of drug-nutrient interaction problems occur (see Chapter 9 and Appendix 31).
Nutrition Assessment
Anthropometric Measures: This is an important area of assessment because of the growth failure of the more severely involved child or adult with CP. Children with CP are often shorter, and, depending on the level of severity, some children with CP may need to be measured for length using recumbent length boards or standing boards even as they grow older. See Appendix 20. However, some of the measuring devices are inappropriate for the child with contractures and inability to be stretched out full length. Arm span can be used when the individual’s arms are stretchable, as well as upper arm and lower leg length. Stevenson (2005) has recommended lower leg length or knee height as a possible measure for determining height for both children and adults with lower-leg CP (Fig. 45-1). The CDC recommends using the CDC curves designed for nonaffected children, plotting sequentially for indications of malnutrition rather than using the disease-specific curves.
Weight measures should be collected over time. Scales may require modifications, with positioning devices for the individual with CP who has developed scoliosis, contractures, and spasticity. Working with a physical therapist to find a positioning device that can be placed in a chair scale or using a bucket scale often works well. Mid–upper arm circumference and triceps skinfold measures are recommended reliable ways to screen for fat stores in children with CP. Head circumference should be measured regularly from birth to 36 months and plotted on the CDC growth curves.
Biochemical Measures: Although there are no specific laboratory tests indicated for the child with CP, a complete blood count, including hemoglobin and hematocrit, should be done when food intake is limited and malnutrition is a possibility. Because bone fractures are a significant problem for many children and adults with spastic quadriplegia, bone mineral density may need evaluating. Medications for seizures may be given; many have nutrition interaction problems (see Appendix 31). Evaluation of vitamin D, calcium, carnitine, and vitamin K levels is recommended.
Dietary Intake: Feeding methods can result in limiting the intake of food and fluid; caretakers may not provide sufficient food to meet nutritional needs. The energy needs of the individual with CP vary according to the type of CP. Studies show that the REE and TEE are lower in those with spastic quadriplegic CP than in normal controls (see Table 45-3).
Intervention Strategies
A high percentage of children with CP have feeding problems that are largely the result of oral-motor, positioning, and behavioral factors. As infants they have difficulty swallowing and coordinating swallowing and chewing, so that the normal progression to solid foods is later than usual. All this may lead to inadequate intake and growth limitations. For those infants and children with IEPs, the team of dietitian, speech therapist, occupational therapist, and physical therapist should evaluate the problem and work together in planning therapy.
Gastroesophageal reflux is frequently seen in these infants and toddlers. A tube feeding may be required if a modified barium swallow reveals aspiration. Alternative techniques in feeding should be considered, which could include thickening all beverages or placing a gastrostomy tube (Sullivan, 2005). RDs should evaluate gastrostomy feedings for caloric and nutritional value, volume required, and osmolality; and offer directions for inclusion of solid foods in addition to the formula if necessary.
Usual problems identified in the evaluation will be altered growth, inadequate energy or fluid intakes, drug-nutrient interaction problems, constipation, and feeding problems. Working out an intervention plan is most successful when it involves the parent as part of the team, addresses cultural issues, and recognizes the importance of the feeding problem. Children with CP have complex problems that require continuing follow-up with the family in the community and will take time to correct. There are state agencies that provide tube-feeding formulas and special wheelchairs and equipment to assist with feeding problems. These agencies vary from state to state.
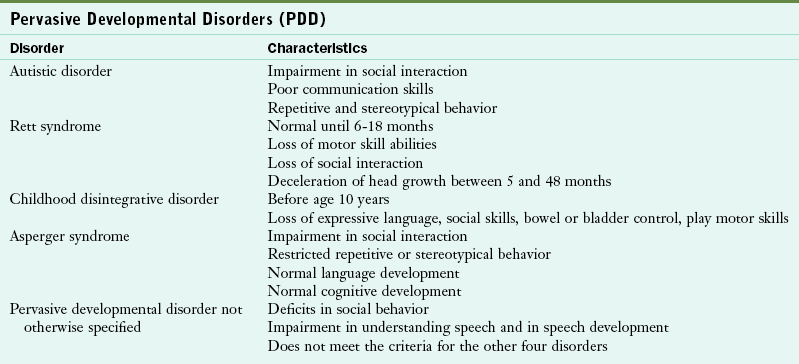
Autism
Autism is one of five disorders under the category pervasive developmental disorder (PDD). PDD was first used in the 1980s to describe a class of disorders as shown in Table 45-4. All types of PDD are neurologic disorders that are usually evident by age 3. In general, children who have a type of PDD have difficulty in talking, playing with other children, and relating to others, including their families.
Pathophysiology
Autism spectrum disorders (ASDs) are diagnosed by the presence of qualitatively impaired reciprocal social interaction; impaired communication skills; and restricted, repetitive, stereotypical interests and behaviors. Many children with autism also have intellectual compromise. ASD is four times more common in boys than in girls.
Asperger syndrome describes children with the problems of ASD but who have normal to high cognitive levels. These children have a difficult time socializing, but may otherwise be able to attend school successfully.
ASDs may occur with other developmental or physical disabilities. They have been associated with tuberous sclerosis and maternal rubella. Macrocephaly has been a common finding in large surveys of individuals with autism and also among their relatives. Overall growth is usually normal, and medical problems nonexistent. However, with the limited variety of foods usually eaten by these children, vitamin and mineral intake could be inadequate.
Efforts to find the cause of ASD have led to many studies looking at a possible toxic environment or food, a nutritionally deficient diet, immune system problems, oxidative stress, and pesticide exposure as important factors. Other studies have studied neurotransmitters such as elevated serotonin levels and disturbances in gamma-amino butyric acid (GABA) receptors, glutamate transmitters, and cholinergic activity. A number of prenatal causes have been evaluated, including pesticides. In one California study (Roberts et al., 2007) women in the first 8 weeks of pregnancy who lived near farm fields sprayed with dicofol and endosulfan were several times more likely to give birth to children with autism. More research is needed with greater numbers of mothers included.
Some treatment and research programs are using genomic panels to identify specific intervention protocols. The genomic panel identifies single-nucleotide polymorphisms, which are identified from blood samples or cell cultures (see Chapter 5). This work has revealed that the child with autism may need additional ω-3 or EFAs; nutrients with antioxidants qualities such as vitamins A, C, E, and selenium; mineral supplementation with zinc, calcium, and magnesium; a mercury-free diet; or an allergy-elimination diet (see Chapter 27).
Interest in a neurochemical cause of ASD has noted gluten and casein as the suspected sources. Intestinal inflammation has been reported in children with ASD and has improved with dietary restriction of gluten and casein (Reichelt and Knivsberg, 2003). Antibodies to casein, gluten, and soy have been noted in some children with ASD.
Nutrition Assessment
Anthropometric Measures: Height and weight are determined for the child and adult with ASD using the equipment and growth charts for nonaffected individuals. Head circumference has been found to be larger than that of the non-ASD individual.
Biochemical Measures: There is no standard pattern of tests that should be given other than the regular blood work for health monitoring. However, amino acid screening shortly after birth, thyroid testing, and allergy testing may be indicated (see Chapter 27).
Dietary Intake: Evaluations are sometimes difficult to complete for the child with a very limited intake. An effective measure may be provided by having the parents and caregivers keep a food diary for several days to determine the macronutrient intake in addition to the vitamin and mineral intake. See Chapter 4. Obtaining information related to when food is presented and the amount eaten is important, along with fluid consumption. Often excessive fluids are provided to compensate for limited food consumption.
Evaluations should include an observation of the child during mealtime. Some children are slow in arriving at developmental milestones for self-feeding and require feeding. Others finger feed or insist on self-feeding. The texture of the food presented should be recorded because sensory integration is difficult for children with ASD, and they may be very resistant to texture progression or variety. This is reflected in their fixation on one food (e.g., crackers, dry cereal, or chips). Food jags and picky eating are common. Evaluation should include a description of the feeding environment, whether there is a high chair or age-appropriate toddler chair, the timing of meals, and the location for meals.
Intervention Strategies
No one therapy or method will work for all individuals with ASD. Many professionals and families use a range of treatments simultaneously, including behavior modification, structured educational approaches, medication, speech therapy, occupational therapy, and counseling. Popular nutrition interventions include mineral and vitamin therapy and elimination diets such as a gluten-free (see Chapter 29), casein-free diet (see Chapter 27); allergy diets (see Chapter 27); supplementation with essential fatty acids (EFAs) and megavitamins.
There are anecdotal reports of success. The exclusion diets are now used in some treatment centers and are publicized on various websites (http://www.autismndi.com). Table 45-5 describes some exclusion diets. It is important for the RD to understand these various forms of therapy in order to counsel the parent effectively. Because of the increasing prevalence of ASD, research on potential MNT should be promoted. One of the problems with the gluten-, casein-free diet is cost, because special foods needed to provide sufficient food choices are expensive and sometimes difficult to find (see Chapter 27).
TABLE 45-5
Comparison of Foods Allowed in the Gluten-Free and Casein-Free Diet, Specific Carbohydrate Diet, and Body Ecology Diet
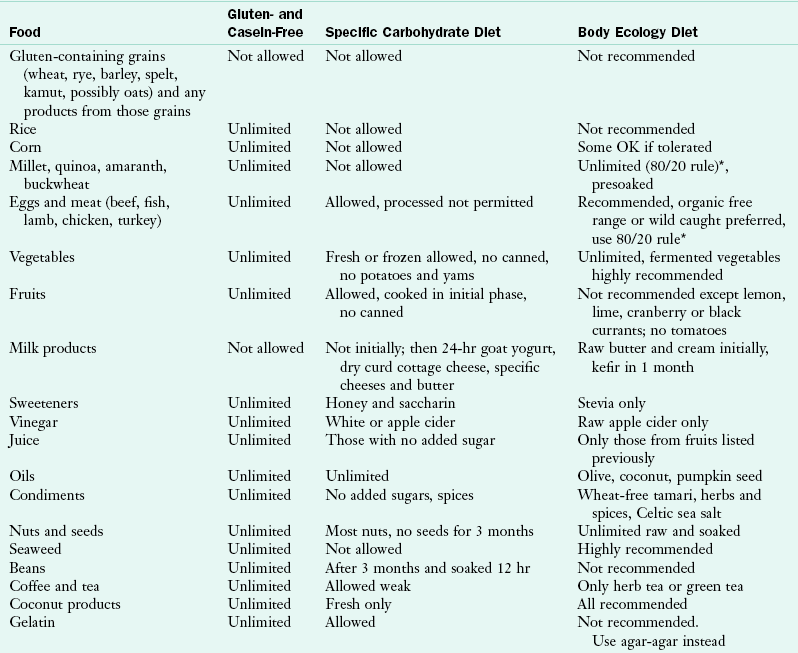
*80/20 = meal contains: 1) 80% land and ocean vegetables and 20% either protein or a grain and 2) 80% alkaline-forming foods and 20% acid forming foods.
Developed by G. A. Houston-Ludlam. Reprinted with permission from The ANDI News, Autism Network for Dietary Intervention, 2005.
When MNT is used, taking a team approach and working with the occupational therapist, speech therapist, and others is important for success. Parents also should be members of the team and counseled that changes will take time. Unfortunately, there have been no double-blind, randomized, controlled studies.
Follow-up is an important component of all therapy. From a nutritional standpoint, routine measures of height and weight should be scheduled, and there should be regular evaluation of eating and feeding behavior related to increasing ability to self-feed and accept new and different foods.
Attention-Deficit/Hyperactivity Disorder
Attention-deficit/hyperactivity disorder (ADHD) is a neurobehavioral problem seen in children with increasing frequency. It has been associated with learning disorders, inappropriate degrees of impulsiveness, hyperactivity, and attention deficit. Diagnostic criteria were developed by the American Psychiatric Association and have designated three types: (1) combined type of hyperactivity and attention deficit, (2) predominately inattentive type, and (3) predominately hyperactive-impulse type. ADHD affects the child at home, in school, and in social situations.
Nutrition Assessment
Many factors should be considered, along with the usual anthropometric measures, particularly when the individual is on medication.
Anthropometric Measures: Measurements of height and weight should be taken and recorded on a regular basis because the medications used in treatment may cause anorexia if given at inappropriate times, resulting in inadequate energy intake and potential slowing of growth. A 10 year prospective study of over 250 children with and without ADHD, treated or not treated with medication, found no evidence of limited growth in height over time.(Biederman J et al, 2010).
Biochemical Measures: These measurements should include a complete blood count and blood and tissue levels of vitamin and minerals if megavitamin therapy is used.
Dietary Intake: A detailed dietary history would include infant feeding history, food likes and dislikes, behavior at mealtimes, snacking behavior, presence of food allergies or food intolerances, or use of special diets. If the individual is on medications, the time of administration in relation to mealtime is important. Information should be obtained regarding any specific diet for the child or individual and how closely it is being followed.
Feeding evaluations should include observing the individual at mealtime. Generally the problems around feeding will be behavioral and will not include oral-motor or positioning peculiarities. Evaluating the environment around mealtime is important because distractions can be problematic.
Intervention Strategies
Current treatment may include psychotropic medications and the use of consistent behavioral management techniques. The timing and type of medication must be adjusted so that there is minimum influence on the child’s dietary intake.
Specific diets have been used for many years, but they are not based on scientific research. For example, parents have been advised to use the Feingold diet, which states that foods containing synthetic food colors and naturally occurring salicylates be removed from the diet because of their neurologic effect.
Recently there has been renewed interest in the role of artificial food colorings (previously advised by Feingold) in exacerbating hyperactivity in selective children. Eight dyes are included: FD&C Blue 1 and 2, FD&C green 3, Orange B, FD&C Red 3, Red 40, FD&C Yellow 5 and 6. The outcome of current interest involves the Food and Drug Administration and committee discussion related to removing food coloring from the food supply (Pelsser LM et al, 2011).
Other recommendations have included the elimination of sugar, the elimination of caffeine, or the addition of large doses of vitamins (megavitamin therapy). A series of well-designed studies to evaluate the effectiveness of these recommendations have generally had negative results, and successful outcomes are largely anecdotal (see Chapter 18).
For the child or adult who is up and down throughout the meal, behavior modification may be indicated, and it should be a part of the overall behavioral management program. Distractions should be eliminated.
The most effective treatment for the individual with ADHD is a diet based on wholesome foods as outlined in the Dietary Guidelines or MyPlate and in Chapter 12. The food should be served at regular times, with small servings followed by refills. This is an important concept because of the tendency of the child or individual to eat very small amounts and leave the table, planning to return or graze throughout the day. Some programs recommend removing the food and returning it only once after explaining why this is being done. The intervention requires that the child or individual sit at the table in the high chair away from television or other distractions. These suggestions are most applicable to children in preschool settings and in the school cafeteria or classroom.
It has been suggested that a lack of EFAs is a possible cause of hyperactivity in children. It is more likely the result of varying biochemical influences. These children have a deficiency of EFAs because they cannot metabolize linoleic acid normally, they cannot absorb EFAs effectively from the intestine, or their EFA requirements are higher than normal. A recent study in Germany of 810 children ages 5-12 provided a food supplement containing ω-3 and ω-6 fatty acids plus zinc and magnesium resulting in considerable reduction in attention deficit and hyperactivity after 12 weeks of supplementation (Huss et al., 2010). This type of supplementation can be considered.
Cleft Lip and Palate
Cleft lip and cleft palate (CL/CP) are the most commonly occurring craniofacial birth defects (Merritt, 2005). Cleft lip is a condition that creates an opening of the upper lip. It can range from a slight notch to complete separation in one or both sides of the lips, and extending upward. If it occurs on one side of the lip, it is called a unilateral cleft lip; if it occurs on both sides, it is called a bilateral cleft lip. The cleft palate occurs when the roof of the mouth has not joined completely; it can be either unilateral or bilateral. Cleft palate can range from just an opening at the back of the soft palate or separation of the roof of the mouth with both soft and hard palate involved. CL/CP result from incomplete merging and fusion of embryonic processes during formation of the face. There is also a condition called submucous cleft palate in which there is incomplete fusion of the muscular layers of the soft palate with fusion of the overlying mucosa (Figures 45-5 and 45-6).
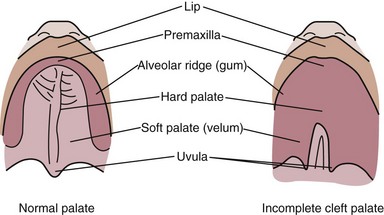
FIGURE 45-5 Cleft palate. (From the Cleft Palate Foundation. Accessed 28 December 2006 from www.cleftline.org.)
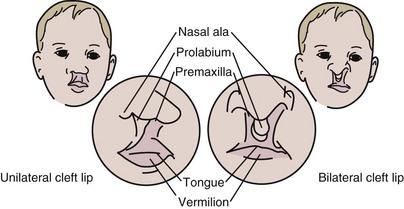
FIGURE 45-6 Cleft lip. (From the Cleft Palate Foundation. Accessed 28 December 2006 from www.cleftline.org.)
Lip and palate development occur between 5 and 12 weeks of gestation. Lip development begins first, usually at 5 weeks of gestation, followed by the development of the maxilla prominences and the primary palate. Fusion of the hard palate is completed by 10 weeks of gestation and the soft palate by 12 weeks.
The incidence of CL/CP varies but is generally 1 in 700 live births. CL/CP have multiple causes and are often associated with underlying syndromes such as Pierre Robin sequence. The Pierre Robin syndrome or complex is a birth condition that involves the lower jaw being either small in size (micrognathia) or set back from the upper jaw (retrognathia) (Cleft Palate Foundation, 2006). As a result, the tongue tends to be displaced back toward the throat, where it can fall back and obstruct the airway. Most infants will have a cleft palate, but none will have a cleft lip. The incidence of Pierre Robin ranges from 1 in 2000 to 30,000 births, based on how strictly the diagnosis is made. The basic cause appears to be the failure of the lower jaw to develop normally before birth.
Approximately 50% of children with cleft palate have an underlying syndrome or multiple anomalies. Wide ranges of studies in developmental biology have shown that both genetics and environmental factors are involved in causing oral clefts. Some of the risk factors from the environment include maternal folic acid deficiency, smoking, alcohol use, anticonvulsant use, and some maternal illnesses. Genetic counseling can now identify high-risk families.
Nutrition Assessment
Nutrition assessment for CL/CP includes the usual anthropometric measures for all infants and children. Biochemical measures are also those used with nonaffected children, and dietary intake information depends on the feeding problems that exist. Other problems include dental abnormalities and missing teeth, speech difficulties, and increased incidence of middle ear infections. The feeding evaluation is a major part of the assessment and is best accomplished with a team approach, including the parents. Because the major problem in CL/CP is feeding and providing adequate intake, growth can be jeopardized and needs to be assessed regularly.
Intervention Strategies
Surgical repair of the cleft lip is generally done at 2 to 3 months of age, and cleft palate repair at 9 months. Other operations may involve minor improvements to the lip or nose and are usually completed before the child starts school.
Breast-feeding is difficult for these infants because of problems with sucking, although those infants with just the cleft lip may be successful. It is generally recommended that the mother who wishes to breast-feed express her milk and give it to her baby from a specialized bottle and nipple. Parents and caregivers need to be educated in the positioning of the child for feeding, nipple selection, bottle selection, and monitoring of intake.
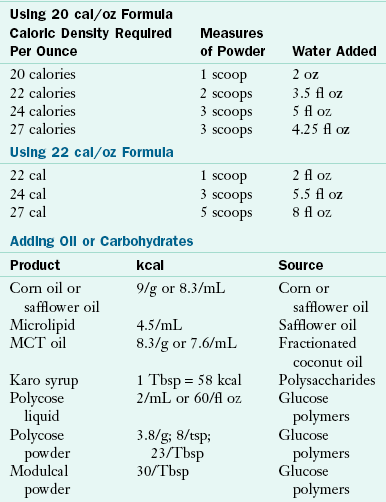
Energy needs are generally the same as for a nonaffected infant or child, but, if the feeding process is too difficult, the energy needs may not be met. Strategies for solving that problem vary, with some professionals advising tube feeding, whereas others recommend continuing with appropriate bottles and nipples but using more concentrated formula or breast milk; see Chapter 43 and Table 45-6.
TABLE 45-6
Increasing Calories through Concentration of Formulas and Addition of Oils and Carbohydrates
Effective feeding requires that the infant be able to form a vacuum inside the mouth and form a seal around the nipple with the lips. This is achieved through the proper bottle, nipple, and position for feeding. Some of the acceptable nipples and bottles include a regular newborn nipple with enlarged holes, a lamb’s nipple, a Ross cleft palate assembly, an obturator nipple, and the orthodontic vented nipple. Bottles can vary from a very soft bottle with a cross-cut nipple to a Haberman feeder, squeeze bottle, or an Asepto feeder.
Individuals with CL/CP are different; thus it is extremely important that the feeding team evaluate various types of equipment and carefully educate the parent in their use. Palate obturators have been used to cover the cleft palate until the child can have surgery to close it; their use results in improved intake, better feeding skills, increased weight gain, and growth of the dental arches. Disadvantages include cost and the inconvenience of refabricating the devices as the infant grows to maintain effectiveness. One study (Prahl et al., 2005) measured growth and the length of feeding between two groups, one using the obturator and one without, and found no significant difference in growth, leading the researchers to conclude that the obturator could be abandoned. Positioning in an upright position, choosing the appropriate nipple, and directing the liquid flow to the side or back of the mouth seem to be just as effective in promoting optimal feeding and are recommended. The baby should be given ample opportunities for frequent burping in an upright position.
Introduction of solid foods for the CL/CP infant can follow the usual protocol at 4 to 6 months of age. By this time the cleft lip should have been repaired, and the child has achieved good head control and trunk stability. Care needs to be taken that the food is presented slowly, allowing the infant to control each bite while gradually learning how to direct the food around the cleft until it is repaired. Following the repair and healing of the cleft palate, feeding along the developmental pathway should progress slowly but normally. See Chapter 17.
Fetal Alcohol Syndrome
Fetal alcohol syndrome (FAS) is a pattern of mental and physical defects that can develop in the fetus of a woman who drinks alcohol during pregnancy. The effects of the alcohol consumption include growth retardation and usually characteristic facial stigmata, damaged neurons and brain structures which can result in psychological or behavioral problems and other physical problems. The prevalence rate in the US and Europe is estimated to be between 0.2-1.5 in every 1000 live births. FAS was named in 1973 by Drs. Kenneth Jones and David Smith at the University of Washington School of Medicine after identifying a pattern of craniofacial, limb and cardiovascular defects in eight unrelated children of three ethnic groups all born to women who were alcoholics (Jones, KL, et al, 1973).
Diagnosis of fetal alcohol syndrome requires that the following criteria are fully met: 1) growth deficiency, 2) FAS facial features, 3) central nervous system damage, and 4) prenatal alcohol exposure. The three facial features include smooth filtrum (the groove between the upper lip and the nose; thin upper lip; small palpebral (between the upper and lower eyelid) fissures. See Fig. 16-4 on p. 361.
Nutrition Assessment
Anthropometric measures are very important in the assessment of the FAS child since growth deficiency is part of the diagnosis. In severe growth deficiency both the height and weight are below the 3rd percentile, while in moderate cases of growth deficiency, either the height or weight is below the 3rd percentile, but not both. Growth should be evaluated frequently and plotted on the CDC and WHO growth curves. Studies have shown that the prenatal growth retardation of the infant may persist postnatally and some infants have failure to thrive. (Huber A, Ekvall S. 2005). Feeding problems have also been associated with FAS, including having a week suck and oral motor problems, making these babies difficult to feed. This would contribute to failure to thrive.
Intervention Strategies
Nutritional intervention for the child with FAS is focused on the specific nutrition problem that exists for that child. Working with growth deficiency, failure to thrive and feeding problems requires the usual evaluation and intervention used in other infants and children with developmental disabilities. Energy and nutrient needs are the same as for non-affected children, although strategies for increasing calories would be required for those who have failure to thrive. For some children ADHD has been reported, and the treatment should be the same as already described.
Controversial Nutrition Therapy
An important factor in providing MNT for children and adults with developmental disabilities is realizing that counseling may have been inadequate in helping the parent accept the limitations of the disorder, limitations such as growth, feeding problems, and cognitive ability. As a result, many parents look for unusual medical or nutrition therapies. Major sources of information are often the Internet and parent support groups. Recent media coverage has promoted the use of antioxidant vitamins (A, C, and E) and minerals (zinc, copper, manganese, and selenium) along with the amino acids glucosamine, tyrosine, and tryptophan. The expected outcomes are improved growth; increased cognition, alertness, and attention span; and changed facial features.
There is little scientific information to back up these therapies. Research studies have addressed the vitamin needs of children with DS, spina bifida, fragile X syndrome, and autism, and findings do not indicate that the vitamin and mineral needs of these children with developmental disabilities are higher than normal. Numerous historical studies have searched for nutritional deficiencies as causative factors in DS. Traditionally, the studies have included numerous vitamins, minerals, fatty acids, digestive enzymes, lipotropic nutrients, and numerous drugs, with no definitive results.
The key concept in the proposed nutrition interventions for DS is metabolic correction of genetic overexpression. It is postulated that presence of the third chromosome 21 causes overproduction of superoxide dismutase and cystathionine b-synthase, which disrupts active methylation pathways. Vitamin supplements of folic acid and antioxidants counteract this and are considered key to the treatment. However, these are just theories, and at this point nutrition supplements are considered an expensive, questionable approach.
Parents of children with ADHD report that omitting sugar from the children’s diets decreases hyperactivity, but there is no scientific evidence to support this. However, it probably is a good idea to eliminate or at least reduce the sugar intake in any child’s diet to promote better nutritional intake. Blue-green algae has been promoted for children with DS and other developmental disabilities, purportedly to increase attention span and concentration. High-dose supplementation of vitamin B6 and magnesium has been proposed for autism to diminish tantrums and self-stimulation activities and improve attention and speech. Another proposed treatment is dimethyl glycine. Limited research is available.
Community Resources
For many types of nutrition problems and MNT, the school system is an excellent resource through the school lunch and school breakfast programs. Children and adolescents may receive modified meals at school. Child and adult care food programs must provide meals at no extra cost for children and adolescents with special needs and developmental disabilities. School food service is required to offer special meals at no additional cost to children whose disabilities restrict their diets as defined in the U.S. Department of Agriculture’s nondiscrimination regulations.
The term “child with a disability” under Part B of the Individuals with Disabilities Education Act (IDEA) refers to a child evaluated in accordance with IDEA as having one of the 13 recognized disability categories: (1) autism; (2) deaf-blindness; (3) deafness; (4) mental retardation; (5) orthopedic impairments; (6) other health impairments caused by chronic or acute health problems such as asthma, nephritis, diabetes, sickle cell anemia, a heart condition, epilepsy, rheumatic fever, hemophilia, leukemia, lead poisoning, or tuberculosis; (7) emotional disturbances; (8) specific learning disabilities; (9) speech or language impairment; (10) traumatic brain injury; (11) visual impairment; (12) multiple disabilities; and (13) developmental delays. Attention deficit disorder may fall under 1 of the 13 categories.
When a referral is made to the school system for a special meal related to a developmental disability, it must be accompanied by a medical statement for a child with special dietary needs (Figure 45-7). The request requires an identification of the medical or other special dietary condition, the food or foods to be omitted, and the food or choice of foods to be substituted. The statement requires the signature of the physician or recognized medical authority. The school food service may make food substitutions for individual children who do not have a disability but who are medically certified as having a special medical or dietary need. An example is the child with severe allergies or an inborn error of metabolism. The availability of school food service for children with developmental disabilities is an important resource in the long-term implementation of MNT.

FIGURE 45-7 Diet prescription for meals at school. (Reprinted with permission from CARE: special nutrition for kids, Birmingham, AL, 1999, Alabama Department of Education.)
 Clinical Scenario 1
Clinical Scenario 1
Mitchell is a 2-month-old boy with Down syndrome. He was born prematurely (30 weeks of gestation) and was started on gastrostomy tube feeding at 10 days of age because of his poor weight gain and severe gastroesophageal reflux. The poor weight gain was caused by a poor suck, although swallowing was not a problem. He was first seen by a nutritionist in an early intervention program at 4 months of age when he was 22.5 in long and weighed 10 lb 7 oz.
At 16 months he started eating table foods, and his tube-feeding formula was PediaSure. By 21 months his height was 28 inches, and his weight 18.5 lb. He was at the 5th percentile for length and weight using the CDC growth curves, but at the 25th percentile when the Down syndrome curves were used. He had been taking oral feeds since 7 months of age, but his total oral consumption was one jar of baby food a day in addition to his gastrostomy feeds. He was crawling but not yet walking, and he had very limited self-feeding skills. Now at age 21 months his mother’s highest priority is to stop the tube feeding and have Mitchell continue to grow well; she is concerned about his rate of weight gain. She is also concerned that constipation has become a problem requiring medication.
Nutrition Diagnostic Statement
Self-feeding difficulty related to developmental delays as evidenced by inability to feed self most foods offered.
1. What would be your approach in working with this mother and the other team members?
2. What do you think would be his nutritional needs, starting with energy?
3. How many ounces of a 30 cal/oz tube-feeding formula would you recommend for Mitchell to promote weight gain?
4. What steps should be taken to increase his oral intake and decrease the tube feeding?
5. What would you recommend for management of his constipation?
Clinical Scenario 2
Luke is a 2-year-old boy with Prader-Willi syndrome. He was born weighing 7 lb and was 18 in long. A test done in the nursery determined that he had Prader-Willi syndrome. Typical of Prader-Willi syndrome, Luke was a very hypotonic infant with a very poor suck. Mom wanted to breast-feed, but Luke was unable to latch on at the breast, so she pumped her milk. Also it was recommended that human milk fortifier (HMF) be added to the breast milk. She was very concerned about adding the HMF because she had read about potential obesity as Luke grew older. After discharge from the hospital Luke entered an early intervention program with nutrition services. Eventually the mother changed Luke to an infant formula, but Luke continued to have a weak suck, and feeding services were needed.
Nutrition Diagnostic Statement
Inability to feed self related to weak suck and hypotonia as evidenced by need for feeding services.
1. What would be a good plan for continuing nutrition care for Luke?
2. What kind of information should the nutritionist provide to the mother related to her fear of eventual obesity for Luke?
3. As Luke grows older, what would be a good way to determine the number of calories he should receive to prevent obesity?
Centers for Disease Control and Prevention Birth Defects Research
http://www.cdc.gov/ncbddd/birthdefects/research.html
National Center for Education in Maternal and Child Health
National Dissemination Center for Children with Disabilities
References
American Dietetic Association (ADA). Position of the American Dietetic Association: providing nutrition services for people with developmental disabilities and special health care needs. J Am Diet Assoc. 2010;110:297.
AAIDD. American Association on Intellectual and Developmental Disabilities. Definitions. Accessed April 10 http://www.aaidd.org/content_100.cfm?navID=21, 2011. [from].
Biederman, J, et al. A naturalistic 10-year prospective study of height and weight in children with attention-deficit hyperactivity disorder grown up: sex and treatment effects. J Pediatr. 2010;157:635.
Carrel, AL, et al. Long-term growth hormone therapy changes the natural history of body composition and motor function in children with Prader-Willi syndrome. J Clin Endo Met. 2010;95:1131.
Capone, G, et al. Down syndrome. In: Ekvall SW, Ekvall VK, eds. Pediatric nutrition in chronic disease and developmental disorders. New York: Oxford University Press, 2005.
Centers for Disease Control and Prevention. CDC Grand Rounds: additional opportunities to prevent neural tube detects. MMWR Morb Mortal Wkly Rep. 2010;59(31):980.
Centers for Disease Control and Prevention. Updated national birth prevalence estimates for selected birth defects in the United States, 2004-2006. http://www.cdc.gov/ncbdddfeatures/birthdefects, 2010. [Atlanta, Ga. Accessed 28 October 2010 from].
Cleft Palate Foundation. Information about Pierre Robin sequence/complex. http://www.cleftline.org/publications/pierre_robin, 2006. [Accessed 21 April 2011 from].
Cloud, HH, et al. Feeding problems of the child with special health care needs. In Ekvall SV, Ekvall VK, eds.: Pediatric nutrition in chronic disease and developmental disorders, ed 2, New York: Oxford University Press, 2005.
Ekvall, SW, Cerniglia, F. Myelomeningocele. In Ekvall SW, Ekvall VK, eds.: Pediatric nutrition in developmental disabilities and chronic disorders, ed 2, New York: Oxford University Press, 2005.
Henderson, RC, et al. Longitudinal changes in bone density in children and adolescents with moderate to severe cerebral palsy. J Pediatr. 2005;146:769.
Huber, A, Ekvall, SW. Fetal Alcohol Syndrome. In Ekvall SV, Ekvall VK, eds.: Pediatric nutrition in chronic disease and developmental disorders, ed 2, New York: Oxford University Press, 2005.
Huss, M, et al. Supplementation of polyunsaturated fatty acids, magnesium and zinc in children seeking medical advice for attention-deficit/hyperactivity problems-an observational cohort study. Lipids Health Dis. 2010;9:105.
Jones, KL, et al. Pattern of malformation in offspring of chronic alcoholic mothers. Lancet. 1973;1(7815):1267.
McCune, H, Driscoll, D. Prader-Willi syndrome. In Ekvall SW, Ekvall VK, eds.: Pediatric nutrition in chronic disease and developmental disorders, ed 2, New York: Oxford University Press, 2005.
Merritt, L. Physical assessment of the infant with cleft lip and/or palate. Adv Neonatal Care. 2005;5:125.
Miller, JL, et al. Nutritional phases in Prader-Willi syndrome. Am J Med Genet A. 2011;155:1040.
Obata, K, et al. Effects of 5 years growth hormone treatment in patients with Prader-Willi syndrome. J Pediatr Endocrinol Metab. 2003;16:155.
Pelsser, LM, et al. Effects of a restricted elimination diet on the behavior of children with attention-deficit hyperactivity disorder (INCA study): a randomised controlled trial. Lancet. 2011;377(9764):494.
Prahl, C, et al. Infant orthopedics in UCLP: effect on feeding, weight, and length: a randomized clinical trial (Dutch cleft). Cleft Palate Craniofac J. 2005;42:171.
Reichelt, K, Knivsberg, AM. Why use the gluten free and casein-free diet? What the results have shown so far. Autism Research Institute www.autismwebsite.com/ARI/fsn/reicvhelt.htm, 2003. [accessed April 14, 2007].
Reus, L, et al. Motor problems in Prader-Willi syndrome: a systematic review on body composition and neuromuscular functioning. Neurosci Biobehav Rev. 2011;35:956.
Robbins, JM, et al. Hospitalizations of newborns with folate-sensitive birth defects before and after fortification of foods with folic acid. Pediatrics. 2006;118:906.
Roberts, EM, et al. Maternal residence near agricultural pesticide applications and autism spectrum disorders among children in the Californiat Central valley. Environ Health Perspect. 2007;115:1482.
Scerif M, et al: Ghrelin in obesity and endocrine diseases, Mol Cell Endocrinol [Feb 21, 2011, e-pub ahead of print].
Smithells, RN, et al. Further experience of vitamin supplementation for prevention of neural tube defect recurrences. Lancet. 1983;1:1027.
Stevenson, RD. Use of segmental measures to estimate stature in children with cerebral palsy. Arch Paediatr Adolesc Med. 2005;149:658.
Sullivan, PB, et al. Gastrostomy tube feeding in children with cerebral palsy: a prospective longitudinal study. Dev Med Child Neurol. 2005;47:77.
Talebizadeh, Z, Butler, MG. Insulin resistance and obesity-related factors in Prader-Willi syndrome: comparison with obese subjects. Clin Genet. 2005;67:230.
Tobin, SP, et al. The role of an interdisciplinary feeding team in the assessment and treatment of feeding problems: building blocks for life: Pediatric Nutrition Practice Group. J Am Diet Assoc. 2005;28:3.