42 Local anaesthetics and other drugs affecting sodium channels
Overview
As described in Chapter 4, the property of electrical excitability is what enables the membranes of nerve and muscle cells to generate propagated action potentials, which are essential for communication in the nervous system and for the initiation of mechanical activity in striated and cardiac muscle. Initiation of the action potential depends on voltage-gated sodium channels, which open transiently when the membrane is depolarised. Here we discuss local anaesthetics, which act mainly by blocking sodium channels, and mention briefly other drugs that affect sodium channel function.
There are, broadly speaking, two ways in which channel function may be modified, namely block of the channels and modification of gating behaviour. Blocking sodium channels reduces excitability. On the other hand, different types of drugs can either facilitate channel opening and thus increase excitability, or inhibit channel opening and reduce excitability.
Local Anaesthetics
Although many drugs can, at high concentrations, block voltage-sensitive sodium channels and inhibit the generation of the action potential, the only drugs used clinically for this effect are the local anaesthetics, various antiepileptic and analgesic drugs (see Chs 41 and 44) and class I antidysrhythmic drugs (see Ch. 21).
History
Coca leaves have been chewed for their psychotropic effects for thousands of years (see Ch. 47) by South American Indians, who knew about the numbing effect they produced on the mouth and tongue. Cocaine was isolated in 1860 and proposed as a local anaesthetic for surgical procedures. Sigmund Freud, who tried unsuccessfully to make use of its ‘psychic energising’ power, gave some cocaine to his ophthalmologist friend in Vienna, Carl Köller, who reported in 1884 that reversible corneal anaesthesia could be produced by dropping cocaine into the eye. The idea was rapidly taken up, and within a few years cocaine anaesthesia was introduced into dentistry and general surgery. A synthetic substitute, procaine, was discovered in 1905, and many other useful compounds were later developed.
Chemical aspects
Local anaesthetic molecules consist of an aromatic part linked by an ester or amide bond to a basic side-chain (Fig. 42.1). They are weak bases, with pKa values mainly in the range 8–9, so that they are mainly, but not completely, ionised at physiological pH (see Ch. 8 for an explanation of how pH influences the ionisation of weak bases). This is important in relation to their ability to penetrate the nerve sheath and axon membrane; quaternary derivatives such as QX-314, which are fully ionised irrespective of pH, are ineffective as local anaesthetics but have important experimental uses (see below). Benzocaine, an atypical local anaesthetic, has no basic group.
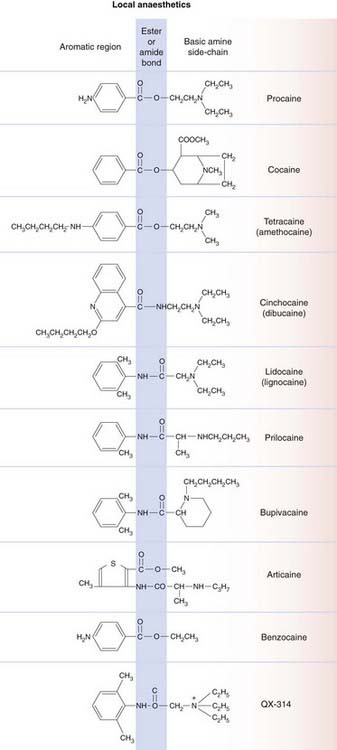
Fig. 42.1 Structures of local anaesthetics.
The general structure of local anaesthetic molecules consists of an aromatic group (left), ester or amide group (shaded blue) and amine group (right).
The presence of the ester or amide bond in local anaesthetic molecules is important because of its susceptibility to metabolic hydrolysis. The ester-containing compounds are fairly rapidly inactivated in the plasma and tissues (mainly liver) by non-specific esterases. Amides are more stable, and these anaesthetics generally have longer plasma half-lives.
Mechanism of action
Local anaesthetics block the initiation and propagation of action potentials by preventing the voltage-dependent increase in Na+ conductance (see Ch. 4) (see Strichartz & Ritchie, 1987; Hille, 2001). At low concentrations they decrease the rate of rise of the action potential, increasing its duration and reducing the firing rate. At higher concentrations they prevent action potential firing. Currently available local anaesthetic agents do not distinguish between different sodium channel subtypes (see Ch. 4; Lai et al., 2004). They block sodium channels, by physically plugging the transmembrane pore, interacting with various amino acid residues of the S6 transmembrane helical domain of the channel protein (see Ragsdale et al., 1994).
 Local anaesthetic activity is strongly pH dependent, being increased at alkaline extracellular pH (i.e. when the proportion of ionised molecules is low) and reduced at acid pH. This is because the compound needs to penetrate the nerve sheath and the axon membrane to reach the inner end of the sodium channel (where the local anaesthetic-binding site resides). Because the ionised form is not membrane permeant, penetration is very poor at acid pH. Once inside the axon, it is primarily the ionised form of the local anaesthetic molecule that binds to the channel and blocks it (Fig. 42.2), the unionised form having only weak channel-blocking activity. This pH dependence can be clinically important, because inflamed tissues are often acidic and thus somewhat resistant to local anaesthetic agents.
Local anaesthetic activity is strongly pH dependent, being increased at alkaline extracellular pH (i.e. when the proportion of ionised molecules is low) and reduced at acid pH. This is because the compound needs to penetrate the nerve sheath and the axon membrane to reach the inner end of the sodium channel (where the local anaesthetic-binding site resides). Because the ionised form is not membrane permeant, penetration is very poor at acid pH. Once inside the axon, it is primarily the ionised form of the local anaesthetic molecule that binds to the channel and blocks it (Fig. 42.2), the unionised form having only weak channel-blocking activity. This pH dependence can be clinically important, because inflamed tissues are often acidic and thus somewhat resistant to local anaesthetic agents.
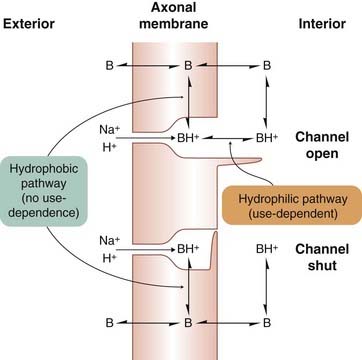
Fig. 42.2 Interaction of local anaesthetics with sodium channels.
The blocking site within the channel can be reached via the open channel gate on the inner surface of the membrane by the charged species BH+ (hydrophilic pathway), or directly from the membrane by the uncharged species B (hydrophilic pathway).
Further analysis of local anaesthetic action (see Strichartz & Ritchie, 1987) has shown that many drugs exhibit the property of ‘use-dependent’ block of sodium channels, as well as affecting, to some extent, the gating of the channels. Use-dependence means that the more the channels are opened, the greater the block becomes. It is a prominent feature of the action of many class I antidysrhythmic drugs (Ch. 21) and antiepileptic drugs (Ch. 44), and occurs because the blocking molecule enters the channel much more readily when the channel is open than when it is closed. For local anaesthetics that rapidly dissociate from the channel, block only occurs at high frequencies of action potential firing when the time between action potentials is too short for drug dissociation from the channel to occur. The channel can exist in three functional states: resting, open and inactivated (see Ch. 4). Many local anaesthetics bind most strongly to the inactivated state of the channel. Therefore, at any given membrane potential, the equilibrium between resting and inactivated channels will, in the presence of a local anaesthetic, be shifted in favour of the inactivated state, and this factor contributes to the overall blocking effect. The passage of a train of action potentials causes the channels to cycle through the open and inactivated states, both of which are more likely to bind local anaesthetic molecules than the resting state; thus both mechanisms contribute to use-dependence.
Quaternary amine local anaesthetics only work when applied to the inside of the membrane and the channels must be cycled through their open state a few times before the blocking effect appears. With tertiary amine local anaesthetics, block can develop even if the channels are not open, and it is likely that the blocking molecule (uncharged) can reach the channel either directly from the membrane phase or via the open gate (Fig. 42.2). The relative importance of these two blocking pathways—the hydrophobic pathway via the membrane and the hydrophilic pathway via the inner mouth of the channel—varies according to the lipid solubility of the drug.
Local anaesthetics exert a number of effects on other ion channels as well as on membrane and intracellular signalling proteins. The importance of these actions to local anaesthetic action is as yet unclear (see Yanagidate & Stricharz, 2007).
In general, local anaesthetics block conduction in small-diameter nerve fibres more readily than in large fibres. Because nociceptive impulses are carried by Aδ and C fibres (Ch. 41), pain sensation is blocked more readily than other sensory modalities (touch, proprioception, etc.). Motor axons, being large in diameter, are also relatively resistant. The differences in sensitivity among different nerve fibres, although easily measured experimentally, are not of much practical importance, and it is not possible to block pain sensation without affecting other sensory modalities.
Local anaesthetics, as their name implies, are mainly used to produce local nerve block. At low concentrations, they are also able to suppress the spontaneous action potential discharge in sensory neurons that occurs in neuropathic pain. Lidocaine (lignocaine) can be used intravenously to control neuropathic pain (see Ch. 41).
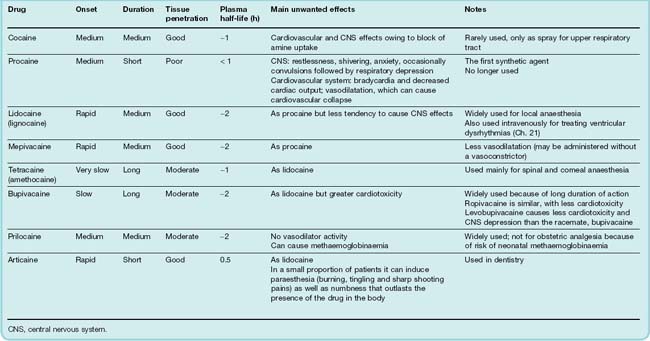
The properties of individual local anaesthetic drugs are summarised in Table 42.1.
Action of local anaesthetics 
Unwanted effects
When used clinically as local anaesthetics, the main unwanted effects involve the central nervous system (CNS) and the cardiovascular system (Table 42.1). Their action on the heart can also be therapeutic in cardiac arrhythmias (see Ch. 21). Although local anaesthetics are usually administered in such a way as to minimise their spread to other parts of the body, they are ultimately absorbed into the systemic circulation. They may also be injected into veins or arterioles by accident.
Most local anaesthetics produce a mixture of depressant and stimulant effects on the CNS. Depressant effects predominate at low plasma concentrations, giving way to stimulation at higher concentrations, resulting in restlessness, tremor and sometimes convulsions, accompanied by subjective effects ranging from confusion to extreme agitation. Further increasing the dose produces profound CNS depression and death due to respiratory depression. The only local anaesthetic with markedly different CNS effects is cocaine (see Ch. 47), which produces euphoria at doses well below those that cause other CNS effects. This relates to its specific effect on monoamine uptake (see Ch. 47), an effect not shared by other local anaesthetics. Procaine is particularly liable to produce unwanted central effects, and has been superseded in clinical use by agents such as lidocaine and prilocaine. Studies with bupivacaine, a widely used long-acting local anaesthetic prepared as a racemic mixture of two optical isomers, suggested that its CNS and cardiac effects were mainly due to the S(+) isomer. The R(−) isomer (levobupivacaine) has a better margin of safety.
The adverse cardiovascular effects of local anaesthetics are due mainly to myocardial depression, conduction block and vasodilatation. Reduction of myocardial contractility probably results indirectly from an inhibition of the Na+ current in cardiac muscle (see Ch. 21). The resulting decrease of [Na+]i in turn reduces intracellular Ca2+ stores (see Ch. 4), and this reduces the force of contraction. Interference with atrioventricular conduction can result in partial or complete heart block, as well as other types of dysrhythmia. Ropivacaine has less cardiotoxicity than bupivacaine.
Vasodilatation, mainly affecting arterioles, is due partly to a direct effect on vascular smooth muscle, and partly to inhibition of the sympathetic nervous system. This leads to a fall in blood pressure, which may be sudden and life-threatening. Cocaine is an exception in respect of its cardiovascular effects, because of its ability to inhibit noradrenaline reuptake (see Chs 14 and 47). This enhances sympathetic activity, leading to tachycardia, increased cardiac output, vasoconstriction and increased arterial pressure.
Hypersensitivity reactions sometimes occur with local anaesthetics, usually in the form of allergic dermatitis but rarely as an acute anaphylactic reaction. Other unwanted effects that are specific to particular drugs include mucosal irritation (cocaine) and methaemoglobinaemia (which occurs after large doses of prilocaine, because of the production of a toxic metabolite). Articaine, used in dentistry, can induce prolonged numbness (paraesthesia) that outlasts the presence of the drug in the body.
Pharmacokinetic aspects
Local anaesthetics vary a good deal in the rapidity with which they penetrate tissues, and this affects the rate at which they cause nerve block when injected into tissues, and the rate of onset of, and recovery from, anaesthesia (Table 42.1). It also affects their usefulness as surface anaesthetics for application to mucous membranes.
Most of the ester-linked local anaesthetics (e.g. tetracaine) are rapidly hydrolysed by plasma cholinesterase, so their plasma half-life is short. Procaine—now rarely used—is hydrolysed to p-aminobenzoic acid, a folate precursor that interferes with the antibacterial effect of sulfonamides (see Ch. 50). The amide-linked drugs (e.g. lidocaine and prilocaine) are metabolised mainly in the liver, usually by N-dealkylation rather than cleavage of the amide bond, and the metabolites are often pharmacologically active.
Benzocaine is an unusual local anaesthetic of very low solubility, which is used as a dry powder to dress painful skin ulcers, or as throat lozenges. The drug is slowly released and produces long-lasting surface anaesthesia.1
The routes of administration, uses and main adverse effects of local anaesthetics are summarised in Table 42.2.
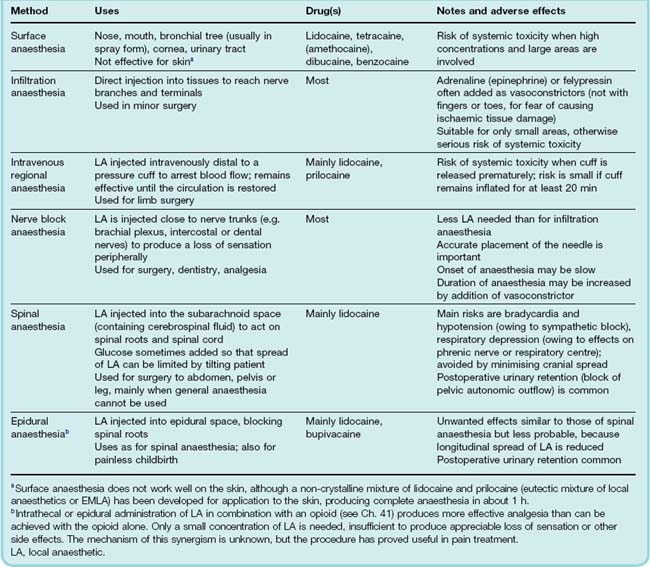
Most local anaesthetics have a direct vasodilator action, which increases the rate at which they are absorbed into the systemic circulation, thus increasing their potential toxicity and reducing their local anaesthetic action. Adrenaline (epinephrine) or felypressin, a short-acting vasopressin analogue (see Ch. 32), is often added to local anaesthetic solutions injected locally in order to cause vasoconstriction.
Unwanted effects and pharmacokinetics of local anaesthetics
Other therapeutic uses
Blocking specific sodium channel subtypes is seen as a promising therapeutic strategy for a variety of clinical conditions, including epilepsy (see Ch. 44), neurodegenerative diseases and stroke (see Ch. 39), neuropathic pain (see Ch. 41) and myopathies. As our understanding of the role of specific sodium channel subtypes in different pathophysiological situations has increased, it is likely that selective blocking agents will be developed for use in different clinical situations.
Other Drugs that Affect Sodium Channels
Tetrodotoxin and Saxitoxin
Tetrodotoxin (TTX) is produced by a marine bacterium and accumulates in the tissues of a poisonous Pacific fish, the puffer fish. The puffer fish is regarded in Japan as a special delicacy partly because of the mild tingling sensation that follows eating its flesh. To serve it in public restaurants, however, the chef must be registered as sufficiently skilled in removing the toxic organs (especially liver and ovaries) so as to make the flesh safe to eat. Accidental TTX poisoning is quite common, nonetheless. Historical records of long sea voyages often contained reference to attacks of severe weakness, progressing to complete paralysis and death, caused by eating puffer fish. It was suggested that the powders used by voodoo practitioners to induce zombification may contain TTX but this has subsequently been disputed.
Saxitoxin (STX) is produced by a marine microorganism that sometimes proliferates in very large numbers and even colours the sea, giving the ‘red tide’ phenomenon. At such times, marine shellfish can accumulate the toxin and become poisonous to humans.
These toxins, unlike conventional local anaesthetics, act exclusively from the outside of the membrane. Both are complex molecules, bearing a positively charged guanidinium moiety. The guanidinium ion is able to permeate voltage-sensitive sodium channels, and this part of the TTX or STX molecule lodges in the channel, while the rest of the molecule blocks its outer mouth. In the manner of its blockade of sodium channels, TTX can be likened to a champagne cork. In contrast to the local anaesthetics, there is no interaction between the gating and blocking reactions with TTX or STX—their association and dissociation are independent of whether the channel is open or closed. Some voltage-sensitive sodium channels are insensitive to TTX, notably those of cardiac muscle and those upregulated in sensory neurons in neuropathic pain (see Ch. 41).
Both TTX and STX are unsuitable for clinical use as local anaesthetics, being expensive to obtain from their exotic sources and poor at penetrating tissues because of their very low lipid solubility. They have, however, been important as experimental tools for the isolation and cloning of sodium channels (see Ch. 4).
Agents that Affect Sodium Channel Gating
Various substances are known that modify sodium channel gating in such a way as to increase the probability of opening of the channels (see Hille, 2001). They include various toxins, mainly from frog skin (e.g. batrachotoxin), scorpion or sea anemone venoms; plant alkaloids such as veratridine; and insecticides such as DDT and the pyrethrins. They facilitate sodium channel activation so that sodium channels open at more negative potentials close to the normal resting potential; they also inhibit inactivation, so that the channels fail to close if the membrane remains depolarised. The membrane thus becomes hyperexcitable, and the action potential is prolonged. Spontaneous discharges occur at first, but the cells eventually become permanently depolarised and inexcitable. All these substances affect the heart, producing extrasystoles and other dysrhythmias, culminating in fibrillation; they also cause spontaneous discharges in nerve and muscle, leading to twitching and convulsions. The very high lipid solubility of substances like DDT makes them effective as insecticides, for they are readily absorbed through the integument. Drugs in this class are useful as experimental tools for studying sodium channels but have no clinical uses.

References and Further Reading
Hille B. Ionic channels of excitable membranes. Sunderland: Sinauer; 2001. (Excellent, clearly written textbook for those wanting more than the basic minimum)
Lai J., Porreca F., Hunter J.C., Gold M.S. Voltage-gated sodium channels and hyperalgesia. Annu. Rev. Pharmacol.. 2004;44:371-397. (Review summarising the role of particular sodium channel subtypes in the pathogenesis of pain, and the use of local anaesthetic and antidysrhythmic drugs in controlling neuropathic pain)
Ragsdale D.R., McPhee J.C., Scheuer T., Catterall W.A. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science. 1994;265:1724-1728. (Use of site-directed mutations of the sodium channel to show that local anaesthetics bind to residues in the S6 transmembrane domain)
Strichartz G.R., Ritchie J.M. The action of local anaesthetics on ion channels of excitable tissues. Handb. Exp. Pharmacol.. 1987;81:21-52. (Excellent review of actions of local anaesthetics—other articles in the same volume cover more clinical aspects)
Yanagidate F., Stricharz G.R. Local anesthetics. Handb. Exp. Pharmacol.. 2007;177:95-127. (Review of sodium channel block by local anaesthetics and description of other actions of these drugs that might also be important)
1Benzocaine is also used in condoms to delay ejaculation.