41 Analgesic drugs
Overview
Pain is a disabling accompaniment of many medical conditions, and pain control is one of the most important therapeutic priorities.
In this chapter, we discuss the neural mechanisms responsible for different types of pain, and the various drugs that are used to reduce it. The ‘classic’ analgesic drugs, notably opioids and non-steroidal anti-inflammatory drugs (NSAIDs; described in Ch. 26), have their origins in natural products that have been used for centuries. The original compounds, typified by morphine and aspirin, are still in widespread use, but many synthetic compounds that act by the same mechanisms have been developed. Opioid analgesics are described in this chapter. Next, we consider various other drug classes, such as antidepressants and antiepileptic drugs, which clinical experience has shown to be effective in certain types of pain. Finally, looking into the future, many potential new drug targets have emerged over the past decade or so as our knowledge of the neural mechanisms underlying pain has advanced. We describe briefly some of these new approaches at the end of the chapter.
Neural Mechanisms of Pain
Pain is a subjective experience, hard to define exactly, even though we all know what we mean by it. Typically, it is a direct response to an untoward event associated with tissue damage, such as injury, inflammation or cancer, but severe pain can arise independently of any obvious predisposing cause (e.g. trigeminal neuralgia), or persist long after the precipitating injury has healed (e.g. phantom limb pain). It can also occur as a consequence of brain or nerve injury (e.g. following a stroke or herpes infection). Painful conditions of the latter kind, not directly linked to tissue injury, are often described as ‘neuropathic pains’. They are very common and a major cause of disability and distress, and in general they respond less well to conventional analgesic drugs than do conditions where the immediate cause is clear. In these cases, we need to think of pain in terms of disordered neural function rather than simply as a ‘normal’ response to tissue injury.
Good accounts of the neural basis of pain can be found in McMahon & Koltzenburg (2006).
Nociceptive Afferent Neurons
Under normal conditions, pain is associated with impulse activity in small-diameter (C and Aδ) primary afferent fibres of peripheral nerves. These nerves have sensory endings in peripheral tissues and are activated by stimuli of various kinds (mechanical, thermal, chemical; Julius & Basbaum, 2001; Julius & McCleskey, 2006). The majority of umyelinated (C) fibres are associated with polymodal nociceptive endings and convey a dull, diffuse, burning pain, whereas myelinated (Aδ) fibres convey a sensation of sharp, well-localised pain. C and Aδ fibres convey nociceptive information from muscle and viscera as well as from the skin.
With many pathological conditions, tissue injury is the immediate cause of the pain and results in the local release of a variety of chemicals that act on the nerve terminals, either activating them directly or enhancing their sensitivity to other forms of stimulation (Fig. 41.1). The pharmacological properties of nociceptive nerve terminals are discussed in more detail below.
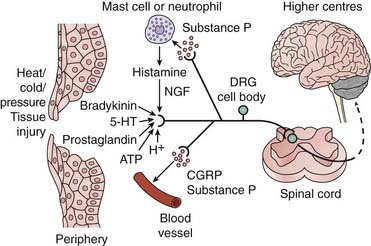
Fig. 41.1 Activation of nociceptive neurons.
Various stimuli (physical and chemical) can initiate or enhance the rate of action potential firing in nociceptive primary afferent neurons (i.e. induce pain). These afferent fibres project to the dorsal horn of the spinal cord where they synapse on neurons projecting to higher centres. 5-HT, 5-hydroxytryptamine; ATP, adenosine triphosphate; CGRP, calcitonin gene-related peptide; DRG, dorsal root ganglion; NGF, nerve growth factor.
(Adapted from Julius D, Basbaum A I 2001 Nature 413: 203–210.)
The cell bodies of spinal nociceptive afferent fibres lie in dorsal root ganglia; fibres enter the spinal cord via the dorsal roots, ending in the grey matter of the dorsal horn. Most of the nociceptive afferents terminate in the superficial region of the dorsal horn, the C fibres and some Aδ fibres innervating cell bodies in laminae I and II (also known as the substantia gelatinosa), while other A fibres penetrate deeper into the dorsal horn (lamina V). The substantia gelatinosa is rich in both endogenous opioid peptides and opioid receptors, and may be an important site of action for morphine-like drugs (see below).
Cells in laminae I and V give rise to the main projection pathways from the dorsal horn to the thalamus. For a more detailed account of dorsal horn circuitry, see Fields et al. (2006).
The nociceptive afferent neurons release glutamate and possibly ATP as the fast neurotransmitters at their central synapses in the dorsal horn. They also contain several neuropeptides (see Ch. 19), particularly substance P and calcitonin gene-related peptide (CGRP). These are released as mediators at both the central and the peripheral terminals, and play an important role in the pathology of pain. For a detailed description of synaptic transmission in the dorsal horn, see McMahon & Koltzenburg (2006).
Modulation in The Nociceptive Pathway
Acute pain is generally well accounted for in terms of nociception—an excessive noxious stimulus giving rise to an intense and unpleasant sensation. In contrast, most chronic pain states1 are associated with aberrations of the normal physiological pathway, giving rise to hyperalgesia (an increased amount of pain associated with a mild noxious stimulus), allodynia (pain evoked by a non-noxious stimulus) or spontaneous pain without any precipitating stimulus. Some of the main mechanisms are summarised in Figure 41.2.
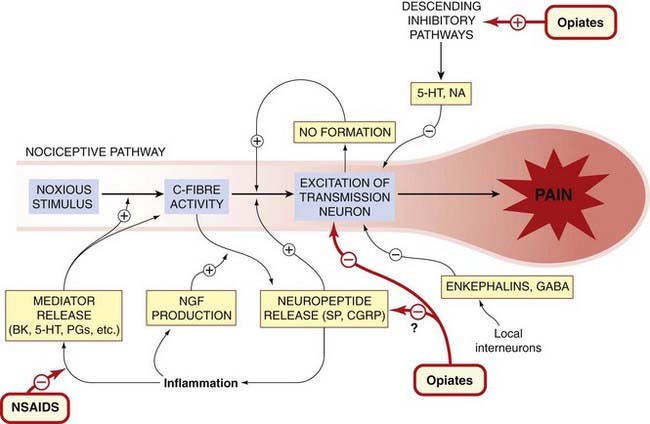
Fig. 41.2 Summary of modulatory mechanisms in the nociceptive pathway.
5-HT, 5-hydroxytryptamine; BK, bradykinin; CGRP, calcitonin gene-related peptide; NA, noradrenaline; NGF, nerve growth factor; NO, nitric oxide; NSAID, non-steroidal anti-inflammatory drug; PG, prostaglandin; SP, substance P.
Hyperalgesia and Allodynia
 Anyone who has suffered a burn or sprained ankle has experienced hyperalgesia and allodynia. Hyperalgesia involves both sensitisation of peripheral nociceptive nerve terminals and central facilitation of transmission at the level of the dorsal horn and thalamus—changes defined by the term neuroplasticity. The peripheral component is due to the action of mediators such as bradykinin and prostaglandins acting on the nerve terminals. The central component reflects facilitation of synaptic transmission in the dorsal horn of the spinal cord (see Yaksh, 1999). The synaptic responses of dorsal horn neurons to nociceptive inputs display the phenomenon of ‘wind-up’—i.e. the synaptic potentials steadily increase in amplitude with each stimulus—when repeated stimuli are delivered at physiological frequencies. This activity-dependent facilitation of transmission has features in common with the phenomenon of long-term potentiation in the hippocampus, described in Chapter 37, and the chemical mechanisms underlying it may also be similar (see Ji et al., 2003). In the dorsal horn, the facilitation is blocked by NMDA receptor antagonists and also in part by antagonists of substance P and by inhibitors of nitric oxide (NO) synthesis (see Figs 41.2 and 41.3).
Anyone who has suffered a burn or sprained ankle has experienced hyperalgesia and allodynia. Hyperalgesia involves both sensitisation of peripheral nociceptive nerve terminals and central facilitation of transmission at the level of the dorsal horn and thalamus—changes defined by the term neuroplasticity. The peripheral component is due to the action of mediators such as bradykinin and prostaglandins acting on the nerve terminals. The central component reflects facilitation of synaptic transmission in the dorsal horn of the spinal cord (see Yaksh, 1999). The synaptic responses of dorsal horn neurons to nociceptive inputs display the phenomenon of ‘wind-up’—i.e. the synaptic potentials steadily increase in amplitude with each stimulus—when repeated stimuli are delivered at physiological frequencies. This activity-dependent facilitation of transmission has features in common with the phenomenon of long-term potentiation in the hippocampus, described in Chapter 37, and the chemical mechanisms underlying it may also be similar (see Ji et al., 2003). In the dorsal horn, the facilitation is blocked by NMDA receptor antagonists and also in part by antagonists of substance P and by inhibitors of nitric oxide (NO) synthesis (see Figs 41.2 and 41.3).
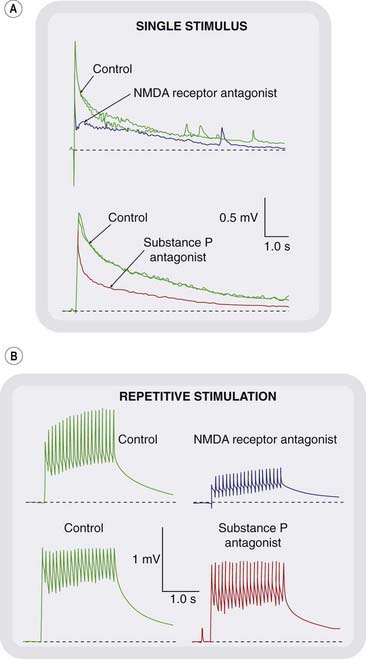
Fig. 41.3 Effect of glutamate and substance P antagonists on nociceptive transmission in the rat spinal cord.
The rat paw was inflamed by ultraviolet irradiation 2 days before the experiment, a procedure that induces hyperalgesia and spinal cord facilitation. The synaptic response was recorded from the ventral root, in response to stimulation of C fibres in the dorsal root with A single stimuli or B repetitive stimuli. The effects of the NMDA receptor antagonist D-AP-5 (see Ch. 37) and the substance P antagonist RP 67580 (selective for neurokinin type 2, (NK2) receptors) are shown. The slow component of the synaptic response is reduced by both antagonists (A), as is the ‘wind-up’ in response to repetitive stimulation (B). These effects are much less pronounced in the normal animal. Thus both glutamate, acting on NMDA receptors, and substance P, acting on NK2 receptors, are involved in nociceptive transmission, and their contribution increases as a result of inflammatory hyperalgesia.
(Records kindly provided by L Urban and S W Thompson.)
Substance P and CGRP released from primary afferent neurons (see Fig. 41.1) also act in the periphery, promoting inflammation by their effects on blood vessels and cells of the immune system (Ch. 17). This mechanism, known as neurogenic inflammation, amplifies and sustains the inflammatory reaction and the accompanying activation of nociceptive afferent fibres.
Central facilitation is an important component of pathological hyperalgesia (e.g. that associated with inflammatory responses). The mediators responsible for central facilitation include substance P, CGRP, nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF) and NO as well as many others (see Ji et al., 2003). For example, NGF, a cytokine-like mediator produced by peripheral tissues, particularly in inflammation, acts on a kinase-linked receptor (known as TrkA) on nociceptive afferent neurons, increasing their electrical excitability, chemosensitivity and peptide content, and also promoting the formation of synaptic contacts. Increased NGF production may be an important mechanism by which nociceptive transmission becomes facilitated by tissue damage, leading to hyperalgesia (see Pezet & McMahon, 2006). Increased gene expression in sensory neurons is induced by NGF and other inflammatory mediators; the upregulated genes include those for neuropeptides and neuromodulators (e.g. CGRP, substance P and BDNF) as well as for receptors (e.g. transient receptor potential TRPV1 and P2X3) and sodium channels, and have the overall effect of facilitating transmission at the first synaptic relay in the dorsal horn. BDNF released from primary afferent nerve terminals activates the kinase-linked TrkB receptor on postsynaptic dorsal horn neurons leading to phosphorylation of the NMDA subunit GluN1 (NR1) and thus sensitisation of these glutamate receptors, resulting in synaptic facilitation, in the dorsal horn.
Excitation of nociceptive sensory neurons depends, as in other neurons (see Ch. 4), on voltage-gated sodium channels. Individuals who express non-functional mutations of Nav1.7 are unable to experience pain (see Cox et al., 2006). The expression of certain sodium channel subtypes (e.g. Nav1.3, Nav1.8 and Nav 1.7 channels) is increased in sensory neurons in various pathological pain states and their enhanced activity underlies the sensitisation to external stimuli that occurs in inflammatory pain and hyperalgesia (see Ch. 4 for a detailed description of voltage-activated sodium channels). Consistent with this hypothesis is the fact that many antiepileptic and antidysrhythmic drugs, which act by blocking sodium channels (see Chs 21 and 44) also find clinical application as analgesics (see below).
Transmission of Pain to Higher Centres
From the dorsal horn, ascending nerve axons travel in the contralateral spinothalamic tracts, and synapse on neurons in the ventral and medial parts of the thalamus, from which there are further projections to the somatosensory cortex. In the medial thalamus in particular, many cells respond specifically to noxious stimuli in the periphery, and lesions in this area cause analgesia. Functional brain imaging studies in conscious subjects have been performed to localise regions involved in pain processing. These include sensory, discriminatory areas such as primary and secondary somatosensory cortex, thalamus and posterior parts of insula as well as affective, cognitive areas such as the anterior parts of insula, anterior cingulate cortex and prefrontal cortex (see Tracey, 2008).
Descending Inhibitory Controls
Descending pathways (Fig. 41.4) control impulse transmission in the dorsal horn (see Millan, 2002). A key part of this descending system is the periaqueductal grey (PAG) area of the midbrain, a small area of grey matter surrounding the central canal. In 1969, Reynolds found that electrical stimulation of this brain area in the rat caused analgesia sufficiently intense that abdominal surgery could be performed without anaesthesia and without eliciting any marked response. Non-painful sensations were unaffected. The PAG receives inputs from many other brain regions, including the hypothalamus, amygdala and cortex, and is the main pathway through which cortical and other inputs act to control the nociceptive ‘gate’ in the dorsal horn.
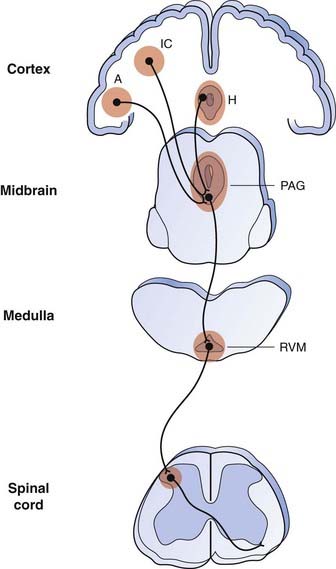
Fig. 41.4 The descending pain control system and sites of action of opioids to relieve pain.
Opioids induce analgesia when microinjected into the insular cortex (IC), amygdala (A), hypothalamus (H), periaqueductal grey (PAG) region and rostroventral medulla (RVM) as well as into the dorsal horn of the spinal cord. The PAG receives input from higher centres and is the main output centre of the limbic system. It projects to the rostral ventromedial medulla (RVM). From the RVM, descending inhibitory fibres, some of which contain 5-hydroxytryptamine, project to the dorsal horn of the spinal cord. Shaded areas indicate regions expressing µ-opioid receptors. The pathways shown in this diagram represent a considerable oversimplification.
(Adapted from Fields 2001 Prog Brain Res 122: 245–253. For a fuller account of the descending pain modulating pathways, see Fields, 2004.)
The PAG projects first to the rostroventral medulla (RVM) and thence via the dorsolateral funiculus of the spinal cord to the dorsal horn. Two important transmitters in this pathway are 5-hydroxytryptamine and the enkephalins, which act directly or via interneurons to inhibit the discharge of spinothalamic neurons (Fig. 41.4).
The descending inhibitory pathway is probably an important site of action for opioid analgesics. Both PAG and substantia gelatinosa (SG) are particularly rich in enkephalin-containing neurons, and opioid antagonists such as naloxone (see later section) can prevent electrically induced analgesia, which would suggest that endogenous opioid peptides may function as transmitters in this system. The physiological role of opioid peptides in regulating pain transmission has been controversial, mainly because under normal conditions naloxone has relatively little effect on pain threshold. Under pathological conditions, however, when stress is present, naloxone causes hyperalgesia, implying that the opioid system is active.
There is also a noradrenergic pathway from the locus coeruleus (LC; see Ch. 38) which has a similar inhibitory effect on transmission in the dorsal horn. Surprisingly, opioids inhibit rather than activate this pathway. The use of tricyclic antidepressants to control pain probably depends on potentiating this pathway.
Modulation of pain transmission 
Neuropathic Pain
Neurological disease affecting the sensory pathway can produce severe chronic pain—termed neuropathic pain—unrelated to any peripheral tissue injury. This occurs with central nervous system disorders such as stroke and multiple sclerosis, or with conditions associated with peripheral nerve damage, such as mechanical injury, diabetic neuropathy or herpes zoster infection (shingles). The pathophysiological mechanisms underlying this kind of pain are poorly understood, although spontaneous activity in damaged sensory neurons, due to overexpression or redistribution of voltage-gated sodium channels, is thought to be a factor (see Lai et al., 2004; Chahine et al., 2005). The sympathetic nervous system also plays a part, because damaged sensory neurons can express α adrenoceptors and develop a sensitivity to noradrenaline that they do not possess under normal conditions. Thus, physiological stimuli that evoke sympathetic responses can produce severe pain, a phenomenon described clinically as sympathetically mediated pain. Neuropathic pain, which appears to be a component of many types of clinical pain (including common conditions such as back pain and cancer pain, as well as amputation pain), responds poorly to conventional analgesic drugs but can be relieved by some antidepressant and antiepileptic agents (see later section). Potential new targets are discussed at the end of this chapter.
Pain and Nociception
The perception of noxious stimuli (termed nociception by Sherrington) is not the same thing as pain, which is a subjective experience and includes a strong emotional (affective) component. The amount of pain that a particular stimulus produces depends on many factors other than the stimulus itself. It is recognised clinically that many analgesics, particularly those of the morphine type, can greatly reduce the distress associated with pain even though the patient reports no great change in the intensity of the actual sensation. The affective component may be at least as significant as the antinociceptive component in the action of these drugs. There is thus often a poor correlation between the activity of analgesic drugs in animal tests (which mainly assess antinociceptive activity) and their clinical effectiveness.
Chemical Signalling in The Nociceptive Pathway
Chemosensitivity of Nociceptive Nerve Endings
In most cases, stimulation of nociceptive endings in the periphery is chemical in origin. Excessive mechanical or thermal stimuli can obviously cause acute pain, but the persistence of such pain after the stimulus has been removed, or the pain resulting from inflammatory or ischaemic changes in tissues, generally reflects an altered chemical environment of the pain afferents. The current state of knowledge is reviewed by McMahon et al. (2006) and summarised in Figure 41.5.
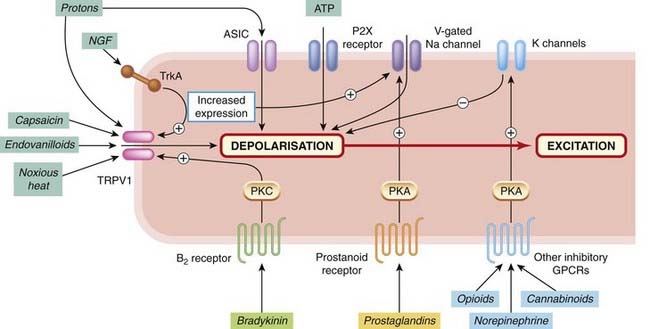
Fig. 41.5 Channels, receptors and transduction mechanisms of nociceptive afferent terminals.
Only the main channels and receptors are shown. Ligand-gated channels include acid-sensitive ion channels (ASICs), ATP-sensitive channels (P2X receptors) and the capsaicin-sensitive channel (TRPV1), which is also sensitive to protons and to temperature. Various facilitatory and inhibitory G-protein-coupled receptors (GPCRs) are shown, which regulate channel function through various second messenger systems. Growth factors such as nerve growth factor (NGF) act via kinase-linked receptors (TrkA) to control ion channel function and gene expression. B2 receptor, bradykinin type 2 receptor; PKA, protein kinase A; PKC, protein kinase C.
TRP channels—thermal sensation and pain
The transient receptor potential (TRP) channel family comprises some 27 or more structurally related ion channels that serve a wide variety of physiological functions (for review, see Flockerzi & Nilius, 2007). Within this family are a group of channels present on sensory neurons that are activated both by thermal stimuli across a wide range of temperatures and by chemical agents (Table 41.1). With respect to pain, the most important channels are TRPV1, TRPM8 and TRPA1 (see Patapoutian et al., 2009).
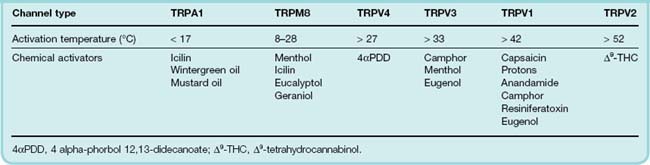
Capsaicin, the substance in chilli peppers that gives them their pungency, selectively excites nociceptive nerve terminals, causing intense pain if injected into the skin or applied to sensitive structures such as the cornea.2 It produces this effect by activating TRPV1.3 Agonists such as capsaicin open the channel, which is permeable to Na+, Ca2+ and other cations, causing depolarisation and initiation of action potentials. The large influx of Ca2+ into peripheral nerve terminals also results in peptide release (mainly substance P and CGRP), causing intense vascular and other physiological responses. The Ca2+ influx may be enough to cause nerve terminal degeneration, which takes days or weeks to recover. Attempts to use topically applied capsaicin to relieve painful skin conditions have had some success, but the initial strong irritant effect is a major disadvantage. Capsaicin applied to the bladder causes degeneration of primary afferent nerve terminals, and has been used to treat incontinence associated with bladder hyper-reactivity in stroke or spinal injury patients. C-fibre afferents in the bladder serve a local reflex function, which promotes emptying when the bladder is distended, the reflex being exaggerated when central control is lost.
TRPV1 responds not only to capsaicin-like agonists but also to other stimuli (see Table 41.1), including temperatures in excess of about 42°C (the threshold for pain) and proton concentrations in the micromolar range (pH 5.5 and below), which also cause pain. The receptor thus has unusual ‘polymodal’ characteristics that closely match those of nociceptive neurons, and it is believed to play a central role in nociception. TRPV1 is, like many other ionotropic receptors, modulated by phosphorylation, and several of the pain-producing substances that act through G-protein-coupled receptors (e.g. bradykinin) work by sensitising TRPV1. A search for endogenous ligands for TRPV1 revealed, surprisingly, that anandamide (a lipid mediator previously identified as an agonist at cannabinoid receptors; see Ch. 18) is also a TRPV1 agonist, although less potent than capsaicin. Confirming the role of TRPV1 in nociception, it has been found that TRPV1 knockout mice show reduced responsiveness to noxious heat and also fail to show thermal hyperalgesia in response to inflammation. The latter observation is interesting, because TRPV1 expression is known to be increased by inflammation and this may be a key mechanism by which hyperalgesia is produced. A number of pharmaceutical companies are actively developing TRPV1 antagonists as analgesic agents.
TRPM8 and TRPA1 respond to cold rather than heat (Table 41.1). TRPM8 is important in cold hypersensitivity in neuropathy. It may also be capable of eliciting a novel inhibitory, analgesic control over noxious inputs in chronic pain states (see Fleetwood-Walker et al., 2007). TRPA1 is activated in some experimental settings by noxious cold temperatures, calcium, pain-producing substances and inflammatory mediators (see Patapoutian et al., 2009); it can therefore also be considered to be a polymodal sensor.
Kinins
The most active pain-producing substances are bradykinin and kallidin (see Ch. 17), two closely related peptides produced under conditions of tissue injury by the proteolytic cleavage of the active kinins from a precursor protein contained in the plasma. Bradykinin is a potent pain-producing substance, acting partly by release of prostaglandins, which strongly enhance the direct action of bradykinin on the nerve terminals (Fig. 41.6). Bradykinin acts on B2 receptors (see Ch. 17) on nociceptive neurons. B2 receptors are coupled to activation of a specific isoform of protein kinase C (PKCε), which phosphorylates TRPV1 and facilitates opening of the TRPV1 channel.
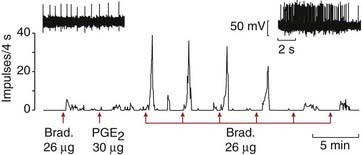
Fig. 41.6 Response of a nociceptive afferent neuron to bradykinin (Brad.) and prostaglandin.
Recordings were made from a nociceptive afferent fibre supplying a muscle, and drugs were injected into the arterial supply. Upper records: single-fibre recordings showing discharge caused by bradykinin alone (left), and by bradykinin following injection of prostaglandin (right). Lower trace: ratemeter recording of single-fibre discharge, showing long-lasting enhancement of response to bradykinin after an injection of prostaglandin E2 (PGE2). Prostaglandin itself did not evoke a discharge.
(From Mense S 1981 Brain Res 225: 95.)
Bradykinin is converted in tissues by removal of a terminal arginine residue to des-Arg9 bradykinin, which acts selectively on B1 receptors. B1 receptors are normally expressed at very low levels, but their expression is strongly upregulated in inflamed tissues (see Calixto et al., 2004). Transgenic knockout animals lacking either type of receptor show reduced inflammatory hyperalgesia. Specific competitive antagonists for both B1 and B2 receptors are known, including peptides such as the B2 antagonist icatibant (Ch. 17), as well as non-peptides. These show analgesic and anti-inflammatory properties, and may prove suitable for clinical use as analgesics (see Marceau & Regoli, 2004).
Prostaglandins
Prostaglandins do not themselves cause pain, but they strongly enhance the pain-producing effect of other agents such as 5-hydroxytryptamine or bradykinin (Fig. 41.6). Prostaglandins of the E and F series are released in inflammation (Ch. 17) and also during tissue ischaemia. Antagonists at EP1 receptors decrease inflammatory hyperalgesia in animal models (Hall et al., 2007). Prostaglandins sensitise nerve terminals to other agents partly by inhibiting potassium channels and partly by facilitating—through second messenger-mediated phosphorylation reactions (see Ch. 3)—the cation channels opened by noxious agents. It is of interest that bradykinin itself causes prostaglandin release, and thus has a powerful ‘self-sensitising’ effect on nociceptive afferents. Other eicosanoids, including prostacyclin, leukotrienes and the unstable hydroxyeicosatetraenoic acid (HETE) derivatives (Ch. 17), may also be important (see Samad et al., 2002). The analgesic effects of NSAIDs (Ch. 26) result from inhibition of prostaglandin synthesis.
Other peripheral mediators
Various metabolites and substances are released from damaged or ischaemic cells, or inflamed tissues, including ATP, protons (produced by lactic acid), 5-hydroxytryptamine, histamine and K+, many of which affect nociceptive nerve terminals.
ATP excites nociceptive nerve terminals by acting on homomeric P2X3 receptors or heteromeric P2X2/P2X3 receptors (see Ch. 16), ligand-gated ion channels that are selectively expressed by these neurons. Downregulation of P2X3 receptors, by antisense DNA technology, reduces inflammatory pain.4 Antagonists at this receptor are analgesic in animal models (see Jarvis, 2003) and may be developed for clinical use. Other P2X receptors (P2X4 and P2X7) are expressed on microglia in the spinal cord; activation results in the release of cytokines and chemokines that then act on neighbouring neurons to promote hypersensitivity. ATP and other purine mediators, such as adenosine, also play a role in the dorsal horn, and other types of purinoceptor may also be targeted by analgesic drugs in the future (see Sawynok, 2007).
Low pH excites nociceptive afferent neurons partly by opening proton-activated cation channels (acid-sensitive ion channels) and partly by facilitation of TRPV1 (see above).
5-Hydroxytryptamine causes excitation, but studies with antagonists suggest that it plays at most a minor role. Histamine is also active but causes itching rather than actual pain. Both these substances are released locally in inflammation (see Ch. 17).
In summary, pain endings can be activated or sensitised by a wide variety of endogenous mediators, the receptors for which are often up- or downregulated under pathophysiological conditions. Neuroplasticity plays an important role in persistent pain states, irrespective of their primary cause; not surprisingly, the signalling pathways have much in common with, and are at least as complex as, those involved in other neuroplasticity-based CNS pathologies discussed in later chapters. The strategies for developing the next wave of analgesic drugs therefore follow similar lines.5
Mechanisms of pain and nociception
Transmitters and Modulators in The Nociceptive Pathway
The family of endogenous opioid peptides (see Ch. 19) plays a key role in modulating nociceptive transmission. Opioid analgesics act on the various receptors for these peptides.
Several neuropeptides are thought to play key roles in the transmission of nociceptive information in the dorsal horn of the spinal cord. These include substance P, CGRP and galanin, each of which is expressed by nociceptive afferent neurons and, it should be noted, can be released at their peripheral as well as their central terminals. In the periphery, substance P and CGRP produce some of the features of neurogenic inflammation whereas galanin is anti-inflammatory. CGRP antagonists have potential for the treatment of migraine (see Ch. 15) but not as analgesics for other pain states. In the dorsal horn, substance P may be involved in wind-up and central sensitisation. In animal models, antagonists of substance P at the NK1 receptor were shown to be effective analgesic drugs, but clinical trials have failed to confirm this in humans, so the high hopes for developing a new type of analgesic for clinical use have so far not come to fruition (see Hill & Oliver, 2007). The reason for this failure is not clear, but it may imply that substance P is less important as a pain mediator in humans than in rats.
Other mediators include the following:
Analgesic Drugs
Opioid Drugs
Opium is an extract of the juice of the poppy Papaver somniferum that contains morphine and other related alkaloids. It has been used for social and medicinal purposes for thousands of years as an agent to produce euphoria, analgesia and sleep, and to prevent diarrhoea. It was introduced in Britain at the end of the 17th century, usually taken orally as ‘tincture of laudanum’, addiction to which acquired a certain social cachet during the next 200 years. The situation changed when the hypodermic syringe and needle were invented in the mid-19th century, and opioid dependence began to take on a more sinister significance (see Ch. 48).
The opioid field is reviewed thoroughly by Corbett et al. (2006).
Chemical Aspects
The structure of morphine (Fig. 41.7) was determined in 1902, and since then many semisynthetic compounds (produced by chemical modification of morphine) and fully synthetic opioids have been studied.
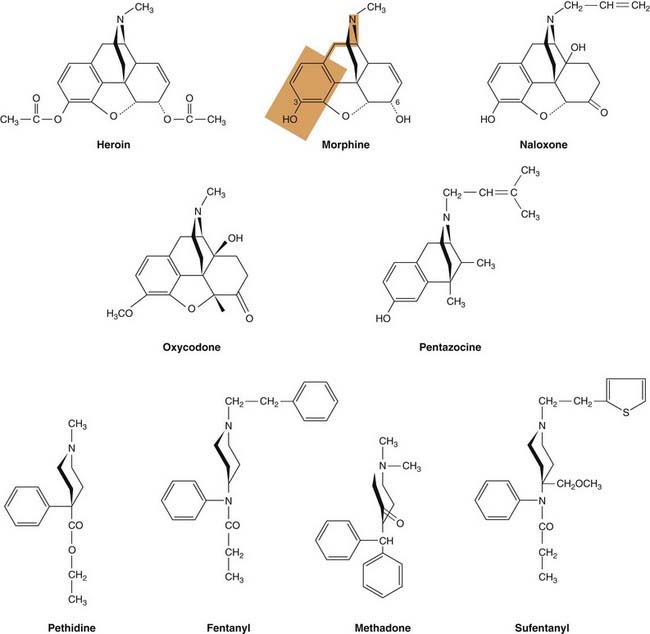
Fig. 41.7 Structures of some opioid analgesics.
The red shaded area indicates the part of the morphine molecule that is structurally similar to tyrosine, the N-terminal amino acid in the endorphins. Carbon atoms 3 and 6 in the morphine structure are indicated. Diamorphine (heroin) is 3,6-diacetylmorphine and morphine is metabolised by addition of a glucuronide moiety at either position 3 or position 6.
Morphine analogues
Morphine is a phenanthrene derivative with two planar rings and two aliphatic ring structures, which occupy a plane roughly at right angles to the rest of the molecule (Fig. 41.7). The most important parts of the molecule for opioid activity are the free hydroxyl on the benzene ring that is linked by two carbon atoms to a nitrogen atom. Variants of the morphine molecule have been produced by substitution at one or both of the hydroxyls (e.g. diamorphine6 3,6-diacetylmorphine, codeine 3-methoxymorphine and oxycodone). Substitution of a bulky substituent on the nitrogen atom introduces antagonist activity to the molecule (e.g. naloxone).
Synthetic derivatives
Phenylpiperidine series.
Pethidine (known as meperidine in the USA), the first fully synthetic morphine-like drug (Fig. 41.7), was discovered accidentally when new atropine-like drugs were being sought. It is chemically unlike morphine, although its pharmacological actions are very similar. Fentanyl and alfentanyl as well as sufentanil (not used in the UK) are more potent and shorter-acting derivatives. Remifentanyl was designed as a potent analogue of fentanyl that is rapidly broken down by esterases in both blood and tissues, resulting in rapid elimination.
Methadone series.
Methadone, although its structural formula bears no obvious chemical relationship to that of morphine, assumes a similar conformation in solution and was designed by reference to the common three-dimensional structural features of morphine and pethidine (Fig. 41.7).
Benzomorphan series.
Therapeutically the most important member of this class is pentazocine (Fig. 41.7). Benzomorphans differ from morphine in their receptor-binding profile (see below), and so have somewhat different actions and side effects. Cyclazocine was an important pharmacological tool in the original description of the putative σ receptor (see below); it is not used in the UK.
Thebaine derivatives.
Buprenorphine resembles morphine but is a partial agonist that binds very tightly to opioid receptors. Because it is a partial agonist, it induces less respiratory depression than other opioids. It is a very potent drug that can also antagonise the effect of other opioids. Etorphine is a highly potent full agonist used in veterinary practice.
Opioid analgesics
Opioid Receptors
The proposal that opioids produce analgesia and their other behavioural effects by interacting with specific receptors first arose in the 1950s, based on the strict structural and stereochemical requirements essential for activity. It was, however, only with the development of molecules with antagonist activity (first nalorphine and then naloxone) that the notion of a specific receptor became accepted. Martin and co-workers then provided evidence for multiple types of opioid receptors. They proposed three different types of receptor, called µ, κ and σ7 for which the prototypical agonists were morphine, ketocyclazocine and N-allylnormetazocine (SKF 10047), respectively. Subsequently, in the early 1970s three research groups led by Simon, Snyder and Terenius simultaneously described the use of radioligand binding to demonstrate the presence of µ receptors in the brain.
Why are there specific receptors in the brain for morphine, a drug that is present in the opium poppy? Hughes and Kosterlitz rationalised that there must be an endogenous substance or substances in the brain that activated these receptors.8 In 1975 they reported the isolation and characterisation of the first endogenous ligands, the enkephalins. We now know that the enkephalins are only two members of a larger family of endogenous opioid peptides known collectively as the endorphins, all of which possess a tyrosine residue at their N-terminus. The chemical structure of tyrosine includes an amine group separated from a phenol ring by two carbon atoms. This same structure (phenol-2 carbon atom chain-amine) is also contained within the morphine structure (Fig. 41.7). It is probably just serendipity that the opium poppy synthesises a semirigid alkaloid molecule, morphine, part of which structurally resembles the tyrosine residue in the endogenous opioid peptides.
Following on from the discovery of the enkephalins, another receptor, δ, was discovered using a combination of classical pharmacological and radioligand binding approaches. Later, another opioid receptor (ORL1) that had a high a degree of amino acid sequence homology (> 60%) towards the µ, δ and κ opioid receptors was identified by cloning techniques, although the antagonist, naloxone, did not bind to this new opioid receptor. The terminology used for opioid receptors has in recent years been through several revisions; in this chapter we shall use the classical terminology. The four opioid receptors, µ, δ, κ and ORL1 are all G-protein-coupled receptors (see Ch 3).9 The main behavioural effects resulting from their activation are summarised in Table 41.2. The interaction of various endogenous opioid peptides with the various receptor types is summarised in Table 41.3. Some agents that are used as experimental tools for distinguishing the different receptor types are also shown.
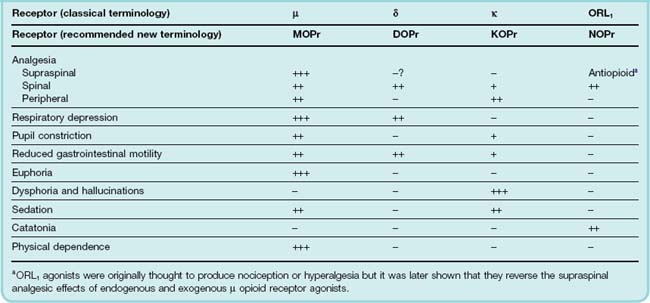

The development of transgenic mouse strains lacking each of the three main opioid receptor types (see Kieffer, 1999) has revealed that the major pharmacological effects of morphine, including analgesia, are mediated by the µ receptor.
All four opioid receptors apear to form homomeric as well as heteromeric receptor complexes (see Milligan, 2004). Opioid receptors are, in fact, quite promiscuous and can form heterodimers with non-opioid receptors. Heterodimerisation between opioid receptors has been shown to result in changes in the pharmacology of the receptors from that observed with the monomeric receptors and may explain some of the subtypes of each receptor that have been proposed. Another level of complexity may reflect ‘protean agonism’ (see Ch. 3), whereby different ligands acting on the same opioid receptor can elicit different cellular responses and differential receptor trafficking (see Kelly et al., 2008).
Opioid receptors
Agonists and Antagonists
Opioids vary not only in their receptor specificity but also in their efficacy at the different types of receptor. Thus, some agents act as agonists or partial agonists on one type of receptor, and antagonists or partial agonists at another, producing a very complicated pharmacological picture.
Four main pharmacological categories are recognised:
Mechanism of Action of Opioids
The opioids have probably been studied more intensively than any other group of drugs in the effort to understand their powerful effects in molecular, cellular and physiological terms, and to use this understanding to develop new drugs as analgesics with significant advantages over morphine. Even so, morphine—described by Osler as ‘God’s own medicine’—remains the standard against which any new analgesic is assessed.
Cellular actions
All four types of opioid receptor belong to the family of Gi/Go-protein-coupled receptors. Opioids thus exert powerful effects on ion channels on neuronal membranes through a direct G-protein coupling to the channel. Opioids promote the opening of a specific type of potassium channel (the inwardly rectifying potassium channel) and inhibit the opening of voltage-gated calcium channels (mainly the N type of calcium channel). These membrane effects decrease neuronal excitability (because the increased K+ conductance causes hyperpolarisation of the membrane making the cell less likely to fire action potentials) and reduce transmitter release (due to inhibition of Ca2+ entry). The overall effect is therefore inhibitory at the cellular level. Nonetheless, opioids do increase activity in some neuronal pathways (see below). They do this by a process of disinhibition whereby they cause excitation of projection neurons by suppressing the firing of inhibitory interneurons that tonically inhibit the projection neurons (see Ch. 36, Fig. 36.2).
At the biochemical level, all four receptor types inhibit adenylyl cyclase and cause MAP kinase (ERK) activation (see Ch. 3). These cellular responses are likely to be important in mediating the long-term adaptive changes that occur in response to prolonged receptor activation and which, for µ-receptor agonists, may underlie the phenomenon of physical dependence (see Ch. 48).
At the cellular level, therefore, all four types of opioid receptor mediate very similar effects. It is their heterogeneous anatomical distributions across the CNS that give rise to the different behavioural responses seen with selective agonists for each type of receptor.
Sites of action of opioids to produce analgesia
Opioid receptors are widely distributed in the brain and spinal cord. Opioids are effective as analgesics when injected in minute doses into a number of specific brain nuclei (such as the insular cortex, amygdala, hypothalamus, PAG region and RVM) as well as into the dorsal horn of the spinal cord see Fig. 41.4 and (for a fuller description, see Fields, 2004). There is evidence to suggest that supraspinal opioid analgesia involves endogenous opioid peptide release both at supraspinal and spinal sites and that at the spinal level there is also a component of the analgesia that results from the release of serotonin (5-HT) from descending inhibitory fibres. Surgical interruption of the descending pathway from the RVM to the spinal cord reduces analgesia induced by morphine that has been given systemically or microinjected into supraspinal sites, implying that in man a combination of effects at supraspinal and spinal sites contribute to the analgesic response.
At the spinal level, morphine inhibits transmission of nociceptive impulses through the dorsal horn and suppresses nociceptive spinal reflexes, even in patients with spinal cord transection. It can act presynaptically to inhibit release of various neurotransmitters from primary afferent terminals in the dorsal horn as well as acting postsynaptically to reduce the excitability of dorsal horn neurons.
There is also evidence (see Sawynok, 2003) that opioids inhibit the discharge of nociceptive afferent terminals in the periphery, particularly under conditions of inflammation, in which the expression of opioid receptors by sensory neurons is increased. Injection of morphine into the knee joint following surgery to the joint provides effective analgesia, undermining the age-old belief that opioid analgesia is exclusively a central phenomenon.
Pharmacological Actions
Morphine is typical of many opioid analgesics and will be taken as the reference compound.
The most important effects of morphine are on the CNS and the gastrointestinal tract, although numerous effects of lesser significance on many other systems have been described.
Effects on the central nervous system
Analgesia
Morphine is effective in most kinds of acute and chronic pain, although opioids in general are less effective in neuropathic pain syndromes (such as phantom limb and other types of deafferentation pain, and trigeminal neuralgia) than in pain associated with tissue injury, inflammation or tumour growth.
As well as being antinociceptive, morphine also reduces the affective component of pain. This reflects its supraspinal action, possibly at the level of the limbic system, which is probably involved in the euphoria-producing effect. Drugs such as pentazocine share the antinociceptive actions of morphine but have much less effect on the psychological response to pain.
In both animal studies and in patients receiving opioids for pain relief, prolonged exposure to opioids may sometimes paradoxically induce a state of hyperalgesia in which pain sensitisation or allodynia occurs (see Chu et al., 2008). This can appear as a reduced analgesic response to a given dose of opioid but should not be confused with tolerance which is a reduced responsiveness due in large part to µ-receptor desensitisation (see below) and occurs with other opioid-induced behaviours in addition to analgesia. Hyperalgesia appears to have peripheral, spinal and supraspinal components. At the cellular level, the mechanisms underlying this phenomenon are still unclear but appear to involve PKC and NMDA receptor activation. Opioid-induced hyperalgesia can be reduced by ketamine, an NMDA antagonist, propofol, α2 adrenoceptor agonists and COX-2 inhibitors. Switching to another opioid can also reduce hyperalgesia; in this regard, methadone may be a good choice as it is a weak NMDA receptor antagonist.
Euphoria
Morphine causes a powerful sense of contentment and well-being (see also Ch. 48). This is an important component of its analgesic effect, because the agitation and anxiety associated with a painful illness or injury are thereby reduced. If morphine or diamorphine (heroin) is given intravenously, the result is a sudden ‘rush’ likened to an ‘abdominal orgasm’. The euphoria produced by morphine depends considerably on the circumstances. In patients who are distressed, it is pronounced, but in patients who become accustomed to chronic pain, morphine causes analgesia with little or no euphoria. Some patients report restlessness rather than euphoria under these circumstances.
Euphoria is mediated through µ receptors whereas κ-receptor activation produces dysphoria and hallucinations (see Table 41.2). Thus, different opioid drugs vary greatly in the amount of euphoria that they produce. It does not occur with codeine or with pentazocine to any marked extent.
Respiratory depression
Respiratory depression, resulting in increased arterial PCO2, occurs with a normal analgesic dose of morphine or related compounds, although in patients in severe pain the degree of respiratory depression produced may be less than anticipated. Respiratory depression is mediated by µ receptors. The depressant effect is associated with a decrease in the sensitivity of the respiratory centres to arterial Pco2 and an inhibition of respiratory rhythm generation. Changes in Pco2 are detected by chemosensitive neurons in a number of brain stem and medullary nuclei. Increased arterial CO2 (hypercapnia) thus normally results in a compensatory increase in minute ventilation rate (VE). In some of the chemosensitive regions, opioids exert a depressant effect on the hypercapnic response, making the increase in VE insufficient to counteract the increased CO2. Respiratory movements originate from activity of a rhythm generator (the pre-Bötzinger complex) within the ventral respiratory column of the medulla. µ Opioid receptors are located in this region, and local injection of opioid agonists decreases respiratory frequency.
Respiratory depression by opioids is not accompanied by depression of the medullary centres controlling cardiovascular function (in contrast to the action of anaesthetics and other general depressants). This means that respiratory depression produced by opioids is much better tolerated than a similar degree of depression caused by, say, a barbiturate. Nonetheless, respiratory depression is the most troublesome unwanted effect of these drugs and, unlike that due to general CNS depressant drugs, it occurs at therapeutic doses. It is the commonest cause of death in acute opioid poisoning.
Depression of cough reflex
Cough suppression (antitussive effect; see also Ch. 27), surprisingly, does not correlate closely with the analgesic and respiratory depressant actions of opioids, and its mechanism at the receptor level is unclear. In general, increasing substitution on the phenolic hydroxyl group of morphine increases antitussive relative to analgesic activity. Codeine and pholcodine suppress cough in subanalgesic doses but they cause constipation as an unwanted effect.
Dextromethorphan, the dextro-isomer of the opioid analgesic, levorphanol, has no affinity for opioid receptors and its cough suppression is not antagonised by naloxone. It is an uncompetitive NMDA receptor antagonist with putative actions at σ receptors and is believed to work at various sites in the brain stem and medulla to suppress cough. In addition to its antitussive action, dextromethorphan is neuroprotective (see Ch. 39) and has an analgesic action in neuropathic pain (see below).
Nausea and vomiting
Nausea and vomiting occur in up to 40% of patients to whom morphine is given, and do not seem to be separable from the analgesic effect among a range of opioid analgesics. The site of action is the area postrema (chemoreceptor trigger zone), a region of the medulla where chemical stimuli of many kinds may initiate vomiting (see Ch. 29).10 Nausea and vomiting following morphine injection are usually transient and disappear with repeated administration although, in some individuals, they persist and can limit patient compliance. Acute administration of morphine-6-glucuronide, an active metabolite of morphine, may produce less nausea and vomiting, probably because it is more polar and does not penetrate the area postrema as well as morphine.
Pupillary constriction
Pupillary constriction is caused by µ and κ receptor-mediated stimulation of the oculomotor nucleus. Pinpoint pupils are an important diagnostic feature in opioid poisoning,11 because most other causes of coma and respiratory depression produce pupillary dilatation. Tolerance does not develop to the pupillary constriction induced by opioids and therefore can be observed in opioid-dependent drug users who may have been taking opioids for a considerable time.
Effects on the gastrointestinal tract
Opioids increase tone and reduce motility in many parts of the gastrointestinal system, resulting in constipation, which may be severe and very troublesome to the patient.12 The resulting delay in gastric emptying can considerably retard the absorption of other drugs. Pressure in the biliary tract increases because of contraction of the gall bladder and constriction of the biliary sphincter. Opioids should be avoided in patients suffering from biliary colic due to gallstones, in whom pain may be increased rather than relieved. The rise in intrabiliary pressure can cause a transient increase in the concentration of amylase and lipase in the plasma.
The action of morphine on visceral smooth muscle is probably mediated mainly through the intramural nerve plexuses, because the increase in tone is reduced or abolished by atropine. It is also partly mediated by a central action of morphine, because intraventricular injection of morphine inhibits propulsive gastrointestinal movements. Methylnaltrexone bromide (see also Ch. 8) and alvimopan (not yet approved in the UK) are opioid antagonists that do not cross the blood–brain barrier. They have been developed to reduce unwanted peripheral side effects of opioids such as constipation without significantly reducing analgesia or precipitating withdrawal in dependent individuals.
Other actions of opioids
Morphine releases histamine from mast cells by an action unrelated to opioid receptors. Pethidine and fentanyl do not produce this effect. The release of histamine can cause local effects, such as urticaria and itching at the site of the injection, or systemic effects, namely bronchoconstriction and hypotension. The bronchoconstrictor effect can have serious consequences for asthmatic patients, to whom morphine should not be given. Although histamine release by morphine does not appear to be opioid-receptor mediated, itching in individuals receiving opioids given systemically has been reported to be reduced by opioid antagonists indicating another potential therapeutic use of peripherally acting antagonists.
Hypotension and bradycardia occur with large doses of most opioids, due to an action on the medulla. With morphine and similar drugs, histamine release may contribute to the hypotension.
Effects on smooth muscle other than that of the gastrointestinal tract and bronchi are slight, although spasms of the ureters, bladder and uterus sometimes occur. The Straub tail reaction, an improbable phenomenon beloved of opioid pharmacologists, consists of a raising and stiffening of the tail of rats or mice given opioid drugs, and is due to spasm of a muscle at the base of the tail. It was through this effect that the analgesic action of pethidine was discovered.
Opioids also exert complex immunosuppressant effects, which may be important as a link between the nervous system and immune function (see Vallejo et al., 2004). The pharmacological significance of this is not yet clear, but there is evidence in humans that the immune system is depressed by long-term opioid abuse, leading to increased susceptibility to infections.
Tolerance and Dependence
Tolerance to many of the actions of opioids (i.e. an increase in the dose needed to produce a given pharmacological effect) develops within a few days during repeated administration. There is some controversy over whether significant tolerance develops to the analgesic effects of morphine, especially in palliative care patients with severe cancer pain (see McQuay, 1999; Ballantyne & Mao, 2003). Drug rotation (changing from one opioid to another) is frequently used clinically to overcome loss of effectiveness. As tolerance is likely to depend upon the level of receptor occupancy, the degree of tolerance observed may reflect the response being assessed, the intrinsic efficacy of the drug and the dose being administered.
Physical dependence refers to a state in which withdrawal of the drug causes adverse physiological effects, i.e. the abstinence syndrome.
Different adaptive cellular mechanisms underlie tolerance and dependence (see Williams et al., 2001; see also Chs. 2 and 48). These phenomena occur to some degree whenever opioids are administered for more than a few days. They must not be confused with addiction (see Ch. 48), in which physical dependence is much more pronounced and psychological dependence (or ‘craving’) is the main driving force. Addiction is rare in patients receiving opioids to control pain.
Tolerance
In animal experiments, tolerance can be detected even with a single dose of morphine. Tolerance extends to most of the pharmacological effects of morphine, including analgesia, emesis, euphoria and respiratory depression, but affects the constipating and pupil-constricting actions much less. Therefore, addicts may take 50 times the normal analgesic dose of morphine with relatively little respiratory depression but marked constipation and pupillary constriction.
The cellular mechanisms responsible for tolerance are discussed in Chapter 2. Tolerance results in part from desensitisation of the µ-opioid receptors (i.e. at the level of the drug target) as well as from long-term adaptive changes at the cellular, synaptic and network levels (see Christie, 2008). Tolerance is a general phenomenon of opioid receptor ligands, irrespective of which type of receptor they act on. Cross-tolerance occurs between drugs acting at the same receptor, but not between opioids that act on different receptors. In clinical settings, the opioid dose required for effective pain relief may increase as a result of developing tolerance, but it does not constitute a major problem.
Physical dependence
Physical dependence is characterised by a clear-cut abstinence syndrome. In experimental animals (e.g. rats), abrupt withdrawal of morphine after repeated administration for a few days, or the administration of an antagonist such as naloxone, causes an increased irritability, diarrhoea, loss of weight and a variety of abnormal behaviour patterns, such as body shakes, writhing, jumping and signs of aggression. These reactions decrease after a few days, but abnormal irritability and aggression persist for many weeks. The signs of physical dependence are much less intense if the opioid is withdrawn gradually. Humans often experience an abstinence syndrome when opioids are withdrawn after being used for pain relief over days or weeks, with symptoms of restlessness, runny nose, diarrhoea, shivering and piloerection.13 The intensity of the abstinence syndrome varies greatly, and dependence rarely progresses to addiction, in which psychological dependence (i.e. craving for the drug) is the predominant feature.
Many physiological changes have been described in relation to the abstinence syndrome. For example, spinal reflex hyperexcitability occurs in morphine-dependent animals and can be produced by chronic intrathecal as well as systemic administration of morphine. The noradrenergic pathways emanating from the LC (see Ch. 38) may also play an important role in causing the abstinence syndrome (see Ivanov & Aston-Jones, 2001), and the α2 adrenoceptor agonist clonidine (Ch. 14) can be used to alleviate it. The rate of firing of LC neurons is reduced by opioids and increased during the abstinence syndrome. In animal models, and also in humans, the abstinence syndrome is reduced by giving NMDA receptor antagonists (e.g. ketamine).
Tolerance and dependence
Pharmacokinetic Aspects
Table 41.4 summarises the pharmacokinetic properties of the main opioid analgesics. The absorption of morphine congeners by mouth is variable. Morphine itself is slowly and erratically absorbed, and is commonly given by intravenous or intramuscular injection to treat acute severe pain; oral morphine is, however, often used in treating chronic pain, and slow-release preparations are available to increase its duration of action. Oxycodone is now widely available as a slow-release oral preparation. Unfortunately, it has become popular among opioid addicts to grind up and inject such tablets as they contain large amounts of the drug. Codeine is well absorbed and normally given by mouth. Most morphine-like drugs undergo considerable first-pass metabolism, and are therefore markedly less potent when taken orally than when injected.
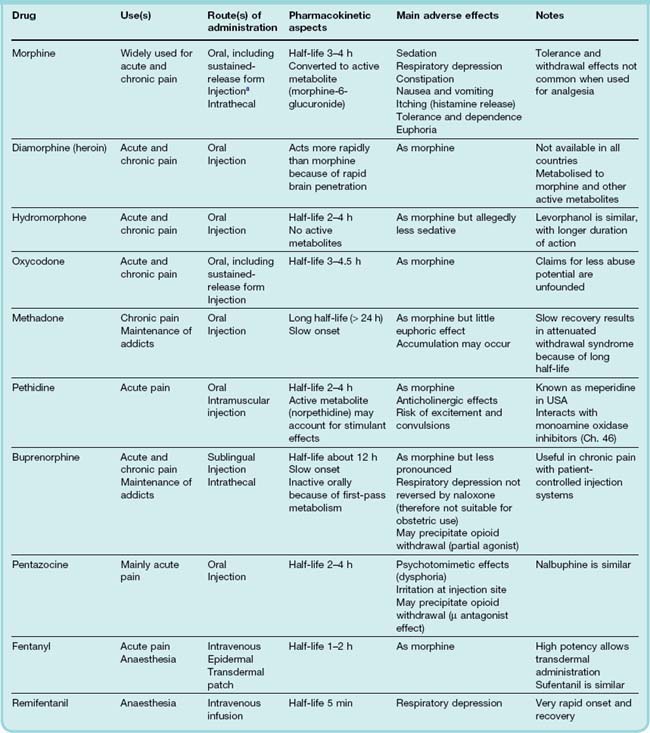
The plasma half-life of most morphine analogues is 3–6 h. Hepatic metabolism is the main mode of inactivation, usually by conjugation with glucuronide. This occurs at the 3- and 6-OH groups (see Fig 41.7), and these glucuronides constitute a considerable fraction of the drug in the bloodstream. Morphine-6-glucuronide is, surprisingly, more active as an analgesic than morphine itself, and contributes substantially to the pharmacological effect. Morphine-3-glucuronide has been claimed to antagonise the analgesic effect of morphine, but the significance of this experimental finding is uncertain as this metabolite has little or no affinity for opioid receptors. Morphine glucuronides are excreted in the urine, so the dose needs to be reduced in cases of renal failure. Glucuronides also reach the gut via biliary excretion, where they are hydrolysed, most of the morphine being reabsorbed (enterohepatic circulation). Because of low conjugating capacity in neonates, morphine-like drugs have a much longer duration of action; because even a small degree of respiratory depression can be hazardous, morphine congeners should not be used in the neonatal period, nor used as analgesics during childbirth. Pethidine (see below) is a safer alternative for this purpose.
Analogues that have no free hydroxyl group in the 3 position (i.e. diamorphine, codeine) are metabolised to morphine, which accounts for all or part of their pharmacological activity. Morphine produces very effective analgesia when administered intrathecally, and is often used in this way by anaesthetists, the advantage being that the sedative and respiratory depressant effects are reduced, although not completely avoided. Remifentanyl is rapidly hydrolysed and eliminated with a half life of 3–4 min. The advantage of this is that when given by intravenous infusion during general anaesthesia, the level of the drug can be manipulated rapidly when required (see Ch. 10 for a description of how, for intravenous infusion, both the rate of rise and the rate of decay of the plasma concentration are determined by the half-time of elimination).
For the treatment of chronic or postoperative pain, opioids are given ‘on demand’ (patient-controlled analgesia). The patients are provided with an infusion pump that they control, the maximum possible rate of administration being limited to avoid acute toxicity. Patients show little tendency to use excessively large doses and become dependent; instead, the dose is adjusted to achieve analgesia without excessive sedation, and is reduced as the pain subsides. Being in control of their own analgesia, the patients’ anxiety and distress are reduced, and analgesic consumption actually tends to decrease. In chronic pain, especially that associated with cancer, patients often experience sudden, sharp increases in the level of pain they are experiencing. This is referred to as breakthrough pain. To combat this, there is a therapeutic need to be able to increase rapidly the amount of opioid being administered. This has led to the development of touch-sensitive transdermal patches containing potent opioids such as fentanyl that rapidly release drug into the bloodstream.
The opioid antagonist, naloxone, has a shorter biological half-life than most opioid agonists. In the treatment of opioid overdose, it must be given repeatedly to avoid the respiratory depressant effect of the agonist reoccurring once the naloxone has been eliminated. Naltrexone has a longer biological half-life.
Unwanted Effects
The main unwanted effects of morphine and related drugs are listed in Table 41.4.
Acute overdosage with morphine results in coma and respiratory depression, with characteristically constricted pupils. It is treated by giving naloxone intravenously. This also serves as a diagnostic test, for failure to respond to naloxone suggests a cause other than opioid poisoning for the comatose state.14 There is a danger of precipitating a severe withdrawal syndrome with naloxone, because opioid poisoning occurs mainly in addicts.
Individual variability
Individuals vary by as much as 10-fold in their sensitivity to opioid analgesics. This can be due to altered metabolism or altered sensitivity of the receptors (for extensive review, see Rollason et al., 2008). For morphine, reduced responsiveness may result from mutations in a number of genes including that for the drug transporter, P-glycoprotein (see Chs 9 and 11), for glucuronyltransferase that metabolises morphine and for the µ receptor itself. Mutations of various cytochrome P450 (CYP) enzymes influence the metabolism of codeine, oxycodone, methadone, tramadol and dextromethorphan. Genotyping could in principle be used to identify opioid-resistant individuals, but first the contribution of genotype to clinical outcome must be confirmed in the population at large.
Other Opioid Analgesics
Diamorphine (heroin) is 3,6-diacetylmorphine; it can be considered as a prodrug as its high analgesic potency is attributable to rapid metabolism to 6-monoacetylmorphine and morphine (see Casy & Parfitt, 1986). Its effects are indistinguishable from those of morphine following oral administration. However, because of its greater lipid solubility, it crosses the blood–brain barrier more rapidly than morphine and gives a greater ‘buzz’ when injected intravenously. It is said to be less emetic than morphine, but the evidence for this is slight. It is still available in Britain for use as an analgesic, although it is banned in many countries. Its only advantage over morphine is its greater solubility, which allows smaller volumes to be given orally, subcutaneously or intrathecally. It exerts the same respiratory depressant effect as morphine and, if given intravenously, is more likely to cause dependence.
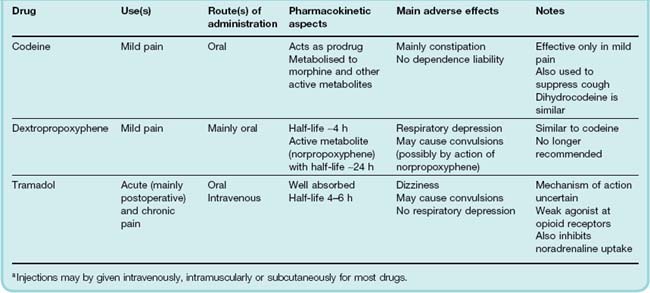
Codeine (3-methoxymorphine) is more reliably absorbed by mouth than morphine, but has only 20% or less of the analgesic potency. Furthermore, its analgesic effect does not increase appreciably at higher dose levels. It is therefore used mainly as an oral analgesic for mild types of pain (headache, backache, etc.). Unlike morphine, it causes little or no euphoria and is rarely addictive. It is often combined with paracetamol in proprietary analgesic preparations (see later section on combined use of opioids and NSAIDs). In relation to its analgesic effect, codeine produces the same degree of respiratory depression as morphine, but the limited response even at high doses means that it is seldom a problem in practice. It does, however, cause constipation. Codeine has marked antitussive activity and is often used in cough mixtures (see Ch. 27). Dihydrocodeine is pharmacologically very similar, having no substantial advantages or disadvantages over codeine. About 10% of the population is resistant to the analgesic effect of codeine, because they lack the demethylating enzyme that converts it to morphine.
Oxycodone is used in the treatment of acute and chronic pain. The suggestion that it acts on a subtype of κ opioid receptor is not generally accepted. Claims that it has less euphoric effect and less abuse potential appear unfounded. Diversion to the street market has resulted in it becoming a major drug of abuse (see Ch. 48), sometimes referred to as ‘hillbilly heroin’.
Fentanyl, alfentanyl, sufentanil and remifentanyl are highly potent phenylpiperidine derivatives, with actions similar to those of morphine but with a more rapid onset and shorter duration of action, particularly remifentanyl. They are used extensively in anaesthesia, and they may be given intrathecally. Fentanyl, alfentanyl and sufentanil are also used in patient-controlled infusion systems and in severe chronic pain, when they are administered via patches applied to the skin. The rapid onset is advantageous in breakthrough pain.
Methadone is orally active and pharmacologically similar to morphine, the main difference being that its duration of action is considerably longer (plasma half-life > 24 h). The increased duration seems to occur because the drug is bound in the extravascular compartment and slowly released. On withdrawal, the physical abstinence syndrome is less acute than with morphine, although the psychological dependence is no less pronounced. Methadone is widely used as a means of treating heroin addiction (see Ch. 48). The lower intensity of the physical abstinence syndrome makes it possible to wean addicts from heroin by giving regular oral doses of methadone—an improvement if not a cure.15 Methadone has actions at other sites in the CNS, including block of potassium channels, NMDA receptors and 5-HT receptors that may explain its CNS side effect profile. There is also interindividual variation in the response to methadone, probably due to genetic variability between individuals in its metabolism.
Pethidine (meperidine) is very similar to morphine in its pharmacological effects, except that it tends to cause restlessness rather than sedation, and it has an additional antimuscarinic action that may cause dry mouth and blurring of vision as side effects. It produces a very similar euphoric effect and is equally liable to cause dependence. Its duration of action is the same or slightly shorter than that of morphine, but the route of metabolic degradation is different. Pethidine is partly N-demethylated in the liver to norpethidine, which has hallucinogenic and convulsant effects. These become significant with large oral doses of pethidine, producing an overdose syndrome rather different from that of morphine. Pethidine is preferred to morphine for analgesia during labour, because it does not reduce the force of uterine contraction. Pethidine is only slowly eliminated in the neonate, and naloxone may be needed to reverse respiratory depression in the baby. (Morphine is even more problematic in this regard, because the conjugation reactions on which the excretion of morphine, but not of pethidine, depends are deficient in the newborn.) Severe reactions, consisting of excitement, hyperthermia and convulsions, have been reported when pethidine is given to patients receiving monoamine oxidase inhibitors. This seems to be due to inhibition of an alternative metabolic pathway, leading to increased norpethidine formation, but the details are not known.
Etorphine is a morphine analogue of remarkable potency, more than 1000 times that of morphine, but otherwise very similar in its actions. Its high potency confers no particular human clinical advantage, but it is used in veterinary practice, especially in large animals. It can be used in conjunction with sedative agents (neuroleptanalgesia) to immobilise wild animals for trapping.16
Buprenorphine is a partial agonist on µ receptors that produces strong analgesia but there is a ceiling to its respiratory depressant effect. Because of its antagonist actions, it can produce mild withdrawal symptoms in patients dependent on other opioids. It has a long duration of action and can be difficult to reverse with naloxone. It has abuse liability but, like methadone, it is also used in the treatment of heroin addiction. When heroin is injected ‘on top’ of buprenorphine, less euphoria is obtained because buprenorphine is a partial agonist that binds almost irreversibly to the receptors.
Meptazinol is an opioid of unusual chemical structure. It can be given orally or by injection and has a duration of action shorter than that of morphine. It seems to be relatively free of morphine-like side effects, causing neither euphoria nor dysphoria, nor severe respiratory depression. It does, however, produce nausea, sedation and dizziness, and has atropine-like actions. Because of its short duration of action and lack of respiratory depression, it may have advantages for obstetric analgesia.
Tramadol is widely used as an analgesic for postoperative pain. It is a weak agonist at µ opioid receptors and also a weak inhibitor of noradrenaline reuptake. It is effective as an analgesic and appears to have a better side effect profile than most opioids, although psychiatric reactions have been reported. It is given by mouth or by intramuscular or intravenous injection for moderate to severe pain.
Pentazocine is a mixed agonist–antagonist with analgesic properties similar to those of morphine. However, it causes marked dysphoria, with nightmares and hallucinations, rather than euphoria, and is now rarely used.
Loperamide is an opioid that does not enter the brain and therefore lacks analgesic activity. It inhibits peristalsis, and is used to control diarrhoea (see Ch. 29).
Opioid Antagonists
Naloxone was the first pure opioid antagonist, with affinity for all three classical opioid receptors (µ > κ ≥ δ). It blocks the actions of endogenous opioid peptides as well as those of morphine-like drugs, and has been extensively used as an experimental tool to determine the physiological role of these peptides, particularly in pain transmission.
Given on its own, naloxone produces very little effect in normal subjects but produces a rapid reversal of the effects of morphine and other opioids. It has little effect on pain threshold under normal conditions but causes hyperalgesia under conditions of stress or inflammation, when endogenous opioids are produced. This occurs, for example, in patients undergoing dental surgery, or in animals subjected to physical stress. Naloxone also inhibits acupuncture analgesia, which is known to be associated with the release of endogenous opioid peptides. Analgesia produced by PAG stimulation is also prevented.
The main clinical uses of naloxone are to treat respiratory depression caused by opioid overdosage, and occasionally to reverse the effect of opioid analgesics, used during labour, on the respiration of the newborn baby. It is usually given intravenously, and its effects are produced immediately. It is rapidly metabolised by the liver, and its effect lasts only 2–4 h, which is considerably shorter than that of most morphine-like drugs and therefore it may have to be given repeatedly.
Naloxone has no important unwanted effects of its own but precipitates withdrawal symptoms in addicts. It can be used to detect opioid addiction.
Naltrexone is very similar to naloxone but with the advantage of a much longer duration of action (half-life about 10 h). It may be of value in addicts who have been ‘detoxified’, because it nullifies the effect of a dose of opioid should the patient’s resolve fail. For this purpose, it is available in a slow-release subcutaneous implant formulation. It is also effective in reducing alcohol consumption in heavy drinkers, the rationale being that part of the high from alcohol comes from the release of endogenous opioid peptides. It may also have beneficial effects in septic shock. It is effective in treating chronic itching (pruritus) as occurs in chronic liver disease. Again, this may indicate the involvement of endogenous opioid peptides in the pathophysiology of such itch conditions.
Methylnaltrexone bromide and alvimopan are µ opioid-receptor antagonists that do not cross the blood–brain barrier. They can be used in combination with opioid agonists to block unwanted effects, most notably reduced gastrointestinal motility, nausea and vomiting.
Specific antagonists at µ, δ and κ receptors are available for experimental use (Table 41.3) but they are not used clinically.
Opioid antagonists
Paracetamol
Non-steroidal anti-inflammatory drugs (NSAIDs, covered in detail in Ch. 26) are widely used to treat painful inflammatory conditions and to reduce fever. Paracetamol (known as acetaminophen in the USA) deserves special mention. It was first synthesised more than a century ago, and since the 1950s has (alongside aspirin) been the most widely used over-the-counter remedy for minor aches and pains. Paracetamol differs from other NSAIDs in producing analgesic and antipyretic effects while lacking anti-inflammatory effects. It also lacks the tendency of other NSAIDs to cause gastric ulceration and bleeding. The reason for the difference between paracetamol and other NSAIDs is unclear. Biochemical tests showed it to be only a weak cyclo-oxygenase (COX) inhibitor, with some selectivity for brain COX. It remains contentious whether paracetamol relieves pain centrally by inhibiting COX-3 (not a separate gene product but a splice variant of COX-1) or by inhibiting COX-2 at low rates of enzyme activity (see Davies et al., 2004; Graham & Scott, 2005).
Paracetamol is well absorbed by mouth, and its plasma half-life is about 3 h. It is metabolised by hydroxylation, conjugated mainly as glucuronide, and excreted in the urine. In therapeutic doses, it has few adverse effects. However, in overdose, paracetamol causes severe liver damage, which is commonly fatal (see Chs 26 and 57), and the drug is often used in attempted suicide.
Use of Opioids and Nsaids in Combination
The rationale behind co-administration of two drugs that produce analgesia by different mechanisms is that, if the effects are additive, less of each drug can therefore be given but the same degree of analgesia produced. This has the effect of reducing the intensity of the unwanted side effects produced by each drug. In the case of opioids (e.g. codeine) in combination with paracetamol or aspirin, the combination appears to produce synergy rather than simple additivity. The combination of dextropropoxyphene and paracetamol has been withdrawn in the UK due to concerns about overdosing.
Treatment of Neuropathic Pain
Neuropathic pain is the severe, debilitating pain that occurs in conditions such as trigeminal neuralgia, diabetic neuropathy, postherpetic neuralgia and phantom limb pain affecting millions of people worldwide. It is often stated that neuropathic pain is opioid resistant. However, recent clinical studies have shown opioids such as morphine, oxycodone, levorphanol and tramadol to be effective in the treatment of neuropathic pain, provided an adequate dose can be reached that provides analgesia without excessive side effects.
Several non-opioid drugs that are also used clinically for effects other than analgesia have been found to be effective in neuropathic pain (see Dworkin et al., 2007), largely as a result of serendipitous observations rather than a rational programme of drug discovery.
Tricyclic antidepressants, particularly amitriptyline, nortriptyline and desipramine (Ch. 46) are widely used. These drugs act centrally by inhibiting noradrenaline reuptake and are highly effective in relieving neuropathic pain in some, but not all, cases. Their action is independent of their antidepressant effects. Drugs such as venlafaxine, which inhibit 5-HT and noradrenaline uptake, are also effective and have a different side effect profile, but selective serotonin reuptake inhibitors show little or no benefit.
Gabapentin and its congener, pregabalin, are antiepileptic drugs (Ch. 44) that are also effective in the treatment of neuropathic pain. They bind to α2δ1 and α2δ2 subunits of voltage-activated calcium channels (see Ch. 4) and reduce neurotransmitter release. There has been considerable debate about how exactly these drugs inhibit calcium channel function: it may be by inhibiting channel opening or by interfering with the trafficking of the calcium channels to the plasma membrane. The α2δ subunits are upregulated in damaged sensory neurons, thus explaining why these agents are more effective across a range of pain states associated with nerve damage than in other forms of pain.
Carbamazepine, another type of antiepileptic drug, is effective in trigeminal neuralgia but evidence for effectiveness against other neuropathic pains is lacking. Carbamazepine blocks voltage-gated sodium channels (see Ch. 4) being slightly more potent in blocking Nav1.8 than Nav1.7 and Nav1.3 channels; all of these channel subtypes are thought to be upregulated by nerve damage and contribute to the sensation of pain. At higher concentrations, it inhibits voltage-activated calcium channels.
Other antiepileptic agents such as valproic acid, lamotrogine, oxcarbazepine and topiramate may have efficacy in some neuropathic pain states.
Lidocaine (lignocaine), a local anaesthetic drug (Ch. 42) with a short plasma half-life given either topically in a patch or intravenously, can give long-lasting relief in neuropathic pain states. It probably acts by blocking spontaneous discharges from damaged sensory nerve terminals, but the reason for its persistent analgesic effect is not clear. Some antidysrhythmic drugs (e.g. mexiletine, tocainide, flecainide; see Ch. 21) are effective orally (see Challapalli et al., 2005).
Other analgesic drugs
Drugs used to treat neuropathic pain
Treatment of Fibromyalgia
Fibromyalgia is a chronic disorder characterised by widespread musculoskeletal pain, fatigue and insomnia. Its cause is unknown, with no obvious characteristic pathology being apparent. It is associated with allodynia (painful sensation in response to stimuli that normally would be innocuous). As with neuropathic pain, classical analgesics (i.e NSAIDs and opioids), while bringing some relief, are not very effective in treating this disorder. Various antidepressant drugs (e.g amitriptyline, citalopram, milnacipram, duloxetine, venlafaxine; see Ch. 46), antiepileptic agents (e.g. gabapentin, pregabalin; see Ch. 44), benzodiazepines (e.g. clonazepam, zopiclone; see Ch. 43) and dopamine agonists (e.g. ropinirole; see Ch. 39) are currently used for this disorder—this long list reflecting their uncertain efficacy.
Other Pain-Relieving Drugs
Ketamine, a dissociative anaesthetic (Ch. 40), memantine and dextromethorphan work by blocking NMDA receptor channels, and probably reduce the wind-up phenomenon in the dorsal horn (Fig. 41.3). Given intrathecally, ketamine’s effects on memory and cognitive function are largely avoided.
Ziconotide, a synthetic analogue of the N-type calcium channel blocking peptide ω-conotoxin MVIIA, is effective when administered by the intrathecal route. It is used in patients whose pain does not respond to other analgesic agents. Blockers of low-voltage-activated T-type calcium channels may also be effective analgesics in some pain states.
Cannabinoids acting at CB1 receptors are effective pain-relieving agents in animal pain models, including models of acute, antinociceptive, inflammatory and neuropathic pain. Although in clinical trials on neuropathic pain these drugs are able to reduce pain perception, the effect is generally weak and clinical relevance remains under evaluation (see Hosking & Zajicek, 2008). The strongest evidence of their benefit is for central neuropathic pain in multiple sclerosis. Sativex is an extract of the cannabis plant containing Δ9-tetrahydrocannabinol (THC) and cannabidiol that has been suggested to have improved therapeutic efficacy. CB2 receptor agonists may also be potential analgesic agents.
Botulinum toxin injections are effective in relieving back pain and the pain associated with spasticity. This effect is due mainly to a relief of muscle spasm (Ch. 13).
New Approaches
As in other fields of neuropharmacology, increasing knowledge of the various chemical mediators and signalling pathways responsible for pain sensation suggests many new approaches to the control of pain. Pain treatment is currently far from perfect, and many new approaches are being explored. For reviews of current areas of drug development, see Hill (2006) and Dray (2008).
Clinical uses of analgesic drugs (1) 
Clinical uses of analgesic drugs (2)
References and Further Reading
Fields H.L., Basbaum A.I., Heinricher M.M. Central nervous system mechanisms of pain modulation. In: McMahon S.B., Koltzenburg M., editors. Wall & Melzack’s Textbook of Pain. fifth ed. Edinburgh: Elsevier; 2006:125-142. (Detailed account of central pathways that inhibit or enhance transmission in the dorsal horn)
Julius D., McCleskey E.W. Cellular and molecular properties of primary afferent neurons. In: McMahon S.B., Koltzenburg M., editors. Wall & Melzack’s textbook of pain. fifth ed. Edinburgh: Elsevier; 2006:35-48. (Describes receptors, ion channels and signalling mechanisms of nociceptive neurons)
McMahon S.B., Koltzenburg M., editors. Wall & Melzack’s textbook of pain, fifth ed, Edinburgh: Elsevier, 2006. (Large multiauthor reference book)
Millan M.J. Descending control of pain. Prog. Neurobiol.. 2002;66:355-474. (Comprehensive review article covering inhibitory and facilitatory mechanisms in great detail)
Tracey I. Imaging pain. Br. J. Anaesth.. 2008;101:32-39. (Description of brain imaging studies on which parts of the brain process pain information)
Yaksh T.L. Spinal systems and pain processing: development of novel analgesic drugs with mechanistically defined models. Trends Pharmacol. Sci.. 1999;20:329-337. (Good general review article on spinal cord mechanisms—more general than its title suggests)
Chahine M., Ziane R., Vijayaragavan K., et al. Regulation of Nav channels in sensory neurons. Trends Pharmacol. Sci.. 2005;26:496-502. (Discusses role of regulation of expression and gating characteristics of voltage-gated sodium channels in pathogenesis of pain)
Cox J.J., Reimann F., Nicholas A.K., et al An SCN9A channelopathy causes congenital inability to experience pain Nature. 444:2006:894-898 (Describes families in which there are mutations of Nav1.7 which renders the person unable to experience pain)
Flockerzi V., Nilius B. Transient receptor potential (TRP) channels. (Eds.). Handb. Exp. Pharmacol. 179:2007 (An entire volume given over to this topic with individual chapters written by experts in the field)
Jarvis M.F. Contributions of P2X3 homomeric and heteromeric channels to acute and chronic pain. Expert. Opin. Ther. Targets. 2003;7:513-522.
Julius D., Basbaum A.I. Molecular mechanisms of nociception. Nature.. 2001;413:203-210. (Review article focusing mainly on receptors and channels involved in activation of sensory nerves by noxious stimuli)
Lai J., Porreca F., Hunter J.C., et al. Voltage-gated sodium channels and hyperalgesia. Annu. Rev. Pharmacol.. 2004;44:371-397. (Review summarising the role of particular sodium channel subtypes in the pathogenesis of pain, and the use of local anaesthetic and antidysrhythmic drugs in controlling neuropathic pain)
Patapoutian A., Tate S., Woolf C.J. Transient receptor potential channels: targeting pain at the source. Nat. Rev. Drug. Discov.. 2009;8:55-68. (Reviews the function of TRPs in pain and discusses their potential as drug targets for the treatment of pain)
Chemical mediators and receptors
Calixto J.B., Medeiros R., Fernandes E.S. Kinin B1 receptors: key G-protein-coupled receptors and their role in inflammatory and painful processes Br. J. Pharmacol. 143:2004:803-818 (Review emphasising the role of inducible B1 receptors in various conditions, including inflammatory pain)
Fleetwood-Walker S.M., Proudfoot C.W.J., Garry E.M., et al. Cold comfort pharm. Trends Pharmacol. Sci.. 2007;28:621-628.
Hall A., Billinton A., Giblin G.M. EP1 antagonists for the treatment of inflammatory pain. Curr. Opin. Drug. Discov. Devel.. 2007;10:597-612.
Hill R.G., Oliver K.R. Neuropeptide and kinin antagonists. Handb. Exp. Pharmacol.. 2007;177:181-216.
Ji R.-R., Kohno T., Moore K.A., et al. Central sensitisation and LTP: do pain and memory share similar mechanisms? Trends Neurosci.. 2003;25:696-705. (Review that emphasises the mechanistic parallels between pain and memory)
Liu X.J., Salter M.W. Purines and pain mechanisms: recent developments. Curr. Opin. Investig. Drugs. 2005;6:65-75.
McMahon S.B., Bennett D.H.L., Bevan S.J. Inflammatory mediators and modulators of pain. In: McMahon S.B., Koltzenburg M., editors. Wall & Melzack’s textbook of pain. fifth ed. Edinburgh: Elsevier; 2006:49-72. (Review of the actions of many peripheral mediators on nociceptive nerve terminals)
Marceau F., Regoli D. Bradykinin receptor ligands: therapeutic perspectives. Nat. Rev. Drug. Discov.. 2004;3:845-852.
Pezet P., McMahon S.B. Neurotrophins: mediators and modulators of pain. Annu. Rev. Neurosci.. 2006;29:507-538. (Comprehensive review of roles and mechanisms of action of NGF and BDNF in pain)
Samad T.A., Sapirstein A., Woolf C.J. Prostanoids and pain: unravelling mechanisms and revealing therapeutic targets. Trends. Mol. Med.. 2002;8:390-396. (Useful review article)
Sawynok J. Adenosine and ATP receptors. Handb. Exp. Pharmacol.. 2007;177:309-328.
Ballantyne J.C., Mao J. Opioid therapy for chronic pain. N. Engl. J. Med.. 2003;349:1943-1953. (Considers whether or not tolerance is a problem when opioids are used to treat chronic pain)
Casy A.F., Parfitt R.T. Opioid analgesics. New York: Plenum Press; 1986. (This book focuses on the chemistry of opioid drugs)
Christie M.J. Cellular neuroadaptations to chronic opioids: tolerance, withdrawal and addiction. Br. J. Pharmacol.. 2008;154:384-396.
Chu L.F., Angst M.S., Clark D. Opioid-induced hyperalgesia in humans: molecular mechanisms and clinical considerations. Clin. J. Pain. 2008;24:479-496.
Corbett A.D., Henderson G., McKnight A.T., et al. 75 years of opioid research: the exciting but vain search for the holy grail. Br. J. Pharmacol.. 2006;147:S153-S162. (Comprehensive historical review of opioid research)
Fields H. State-dependent opioid control of pain. Nat. Rev. Neurosci.. 2004;5:565-575.
Hashimoto K., Ishiwata K. Sigma receptor ligands: possible application as therapeutic drugs and as radiopharmaceuticals. Curr. Pharm. Des.. 2006;12:3857-3876.
Ivanov A., Aston-Jones G. Local opiate withdrawal in locus coeruleus neurons in vitro. J. Neurophysiol.. 2001;85:2388-2397.
Kelly E., Bailey C.P., Henderson G. Agonist-selective mechanisms of GPCR desensitization. Br. J. Pharmacol.. 2008;153(Suppl. 1):S379-388.
Kieffer B.L. Opioids: first lessons from knockout mice. Trends Pharmacol. Sci.. 1999;20:19-26.
McQuay H. Opioids in pain management. Lancet. 1999;353:2229-2232. (Discusses whether or not tolerance occurs to opioids in clinical situations)
Milligan G. G-protein-coupled receptor dimerization: function and ligand pharmacology. Mol. Pharmacol.. 2004;66:1-7.
Rollason V., Samer C., Piquet V., et al. Pharmacogenetics of analgesics: towards the personalization of prescription. Pharmacogenomics. 2008;9:905-933.
Sawynok J. Topical and peripherally acting analgesics. Pharmacol. Rev.. 2003;55:1-20. (Review of the numerous mechanisms by which drugs interfere with nociceptive mechanisms in the periphery)
Vallejo R., de Leon-Casasola O., Benyamin R. Opioid therapy and immunosuppression: a review. Am. J. Ther.. 2004;11:354-365.
Williams J.T., Christie M.J., Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiol. Rev.. 2001;81:299-343. (Very comprehensive review of the molecular and cellular mechanisms underlying opioid tolerance and dependence)
Davies N.M., Good R.L., Roupe K.A., et al. Cyclooxygenase-3: axiom, dogma, anomaly or splice error—not as easy as 1, 2, 3. J. Pharm. Pharm. Sci.. 2004;7:217-226. (Update on the confusing role of COX-3 as a target for paracetamol)
Graham G.G., Scott K.F. Mechanisms of action of paracetamol. Am. J. Ther.. 2005;12:46-55.
Neuropathic pain and new drug targets
Challapalli, V., Tremont-Lukats, I.W., McNicol, E.D., et al., 2005. Systemic administration of local anesthetic agents to relieve neuropathic pain. Cochrane Database Syst. Rev. Issue 4. Art. No.: CD003345. DOI: 10.1002/14651858.CD003345.pub2.
Dray A. Neuropathic pain: emerging treatments. Br. J. Anaesth.. 2008;101:48-58. (Extensive review of potential drug therapies for neuropathic pain)
Dworkin R.H., O’Connor A.B., Backonja M., et al. Pharmacologic management of neuropathic pain: evidence-based recommendations. Pain.. 2007;132:237-251. (An evaluation of the clinical effectiveness of current drugs used to treat neuropathic pain)
Hefti F.F., Rosenthal A., Walicke P.A., et al. Novel class of pain drugs based on antagonism of NGF. Trends. Pharmacol. Sci.. 2006;27:85-91.
Hill R.G. Analgesic drugs in development. In: McMahon S.B., Koltzenburg M., editors. Wall & Melzack’s textbook of pain. fifth ed. Edinburgh: Elsevier; 2006:541-552. (Balanced account of current approaches to develop novel analgesic drugs)
Hosking R.D., Zajicek J.P. Therapeutic potential of cannabis in pain medicine. Br. J. Anaesth.. 2008;101:59-68.
1Defined as pain that outlasts the precipitating tissue injury. Many clinical pain states fall into this category. The dissociation of pain from noxious input is most evident in ‘phantom limb’ pain, which occurs after amputations and may be very severe. At the other extreme, noxious input with no pain, there are many well-documented reports of mystics and showmen who subject themselves to horrifying ordeals with knives, burning embers, nails and hooks (undoubtedly causing massive afferent input) without apparently suffering pain.
2Anyone who has rubbed their eyes after cutting up chilli peppers will know this.
3The receptor was originally known as the vanilloid receptor because many capsaicin-like compounds are based on the structure of vanillic acid.
4P2X3 knockout mice are, in contrast, fairly normal in this respect, presumably because other mechanisms take over.
5And, sceptics may argue, face similar obstacles in relation to specificity and unwanted effects.
6While ‘diamorphine’ is the recommended International Nonproprietary Name (rINN), this drug is better known as heroin.
7The σ ‘receptor’ is no longer considered to be an opioid receptor. It was postulated in order to account for the dysphoric effects (anxiety, hallucinations, bad dreams, etc.) produced by some opioids. It is now accepted that these effects result from drug-induced block of the NMDA receptor channel pore, an effect that is also produced by agents such as ketamine (see Ch. 40). Subsequently, the term σ receptor has also been used to describe other, non-NMDA receptor sites and a subdivision into σ1 and σ2 subtypes proposed (Hashimoto & Ishiwata, 2006). These proteins may be novel drug targets for psychiatric disorders.
8It may seem obvious today that if there is a receptor then there is likely also to be an endogenous ligand for that receptor but it was the search for, and subsequent discovery of, the enkephalins that gave credence to this idea. There are, however, exceptions to this rule. For example, although several endogenous ligands for the benzodiazepine ‘receptor’ or binding site on the GABAA receptor have been suggested, none so far has achieved universal acceptance (see Ch. 43).
9The opioid receptors are unusual among G-protein-coupled receptors. First, in that there are many (20 or more) opioid peptides but only four receptors. In contrast, 5-hydroxytryptamine, for example, is a single mediator interacting with many (about 14) receptors, which is the more common pattern. Second, all four receptors couple to the same types of G-protein (Gi/Go) and therefore activate the same spectrum of cellular effector mechanisms. In contrast, other receptor families (e.g. muscarinic receptors) couple to different types of G-proteins and therefore give rise to different cellular responses (see Ch. 13).
10The chemically related compound apomorphine is more strongly emetic than morphine, through its action as a dopamine agonist; despite its name, it is inactive on opioid receptors.
11The exception is pethidine, which causes pupillary dilatation because it blocks muscarinic receptors.
12In treating pain, constipation is considered as an undesirable side effect. However, opioids such as codeine and morphine can be used to treat diarrhoea.
13Causing goose pimples. This is the origin of the phrase ‘cold turkey’ used to describe the effect of morphine withdrawal.
14Naloxone is less effective in reversing the effects of buprenorphine as this agonist binds very tightly to the receptors.
15The benefits come mainly from removing the risks of self-injection and the need to finance the drug habit through crime.
16The required dose of etorphine, even for an elephant, is small enough to be incorporated into a dart or pellet.