32 The pituitary and the adrenal cortex
Overview
The pituitary and adrenal glands are major sites for the synthesis and release of hormones that profoundly affect the biochemistry and physiology of almost all cells, and which are crucial to the understanding of the actions of many endocrine, anti-inflammatory and other drugs. The pituitary itself is controlled by hormones released from the hypothalamus and, in turn, the hypothalamic–pituitary axis orchestrates the activity of the adrenal (and other endocrine) glands. In the first part of this chapter, we examine the control of pituitary function by hypothalamic hormones and review the physiological roles and clinical uses of both anterior and posterior pituitary hormones. The second part of the chapter concentrates on the actions of adrenal hormones and, in particular, the anti-inflammatory effect of glucocorticoids. This should be read in conjunction with the relevant sections of Chapters 3 and 26.
The Pituitary Gland
The pituitary gland comprises three different structures arising from two different embryological precursors. The anterior pituitary and the intermediate lobe are derived from the endoderm of the buccal cavity, while the posterior pituitary is derived from neural ectoderm. The main parts of the gland, the anterior and posterior lobes, receive independent neuronal input from the hypothalamus, with which they have an intimate functional relationship.
The Anterior Pituitary Gland (Adenohypophysis)
The adenohypophysis secretes a number of hormones crucial for normal physiological function. Within this tissue are specialised cells such as corticotrophs, lactotrophs (mammotrophs), somatotrophs, thyrotrophs and gonadotrophs, which secrete hormones that regulate different endocrine organs of the body (Table 32.1). Interspersed among these are other cell types, including the folliculostellate cells that exert a nurturing and regulatory influence on the hormone-secreting endocrine cells.
Table 32.1 Hormones secreted by the hypothalamus and the anterior pituitary and related drugs
| Hypothalamic factor/hormonea | Effect on anterior pituitary | Main effects of anterior pituitary hormone |
|---|---|---|
| Corticotrophin-releasing factor (CRF) | Releases adrenocorticotrophic hormone (ACTH, corticotrophin) Analogue: tetracosactide |
Stimulates secretion of adrenal cortical hormones (mainly glucocorticoids); maintains integrity of adrenal cortex |
| Thyrotrophin-releasing hormone (TRH) Analogue: protirelin |
Releases thyroid-stimulating hormone (TSH; thyrotrophin) | Stimulates synthesis and secretion of thyroid hormones, thyroxine and tri-iodothyronine; maintains integrity of thyroid gland |
| Growth hormone-releasing factor (GHRF, somatorelin) Analogue: sermorelin |
Releases growth hormone (GH; somatotrophin) Analogue: somatropin |
Regulates growth, partly directly, partly through evoking the release of somatomedins from the liver and elsewhere; increases protein synthesis, increases blood glucose, stimulates lipolysis |
| Growth hormone release-inhibiting factor (somatostatin) Analogues: octreotide, lanreotide |
Inhibits the release of GH | Prevents effects above as well as TSH release |
| Gonadotrophin (or luteinising hormone)-releasing hormone (GnRH) Analogue: gonadorelin |
Releases follicle-stimulating hormone (FSH; see Ch. 34) | Stimulates the growth of the ovum and the Graafian follicle in the female, and gametogenesis in the male; with LH, stimulates the secretion of oestrogen throughout the menstrual cycle and progesterone in the second half |
| Releases luteinising hormone (LH) or interstitial cell-stimulating hormone (see Ch. 34) | Stimulates ovulation and the development of the corpus luteum; with FSH, stimulates secretion of oestrogen and progesterone in the menstrual cycle; in male, regulates testosterone secretion | |
| Prolactin-releasing factor (PRF) | Releases prolactin | Together with other hormones, prolactin promotes development of mammary tissue during pregnancy; stimulates milk production in the postpartum period |
| Prolactin release-inhibiting factor (probably dopamine) | Inhibits the release of prolactin | Prevents effects above |
| Melanocyte-stimulating hormone (MSH)-releasing factor | Releases α-, β- and γ-MSH | Promotes formation of melanin, which causes darkening of skin; MSH is anti-inflammatory and helps to regulate feeding |
| MSH release-inhibiting factor | Inhibits the release of α-, β- and γ-MSH | Prevents effects above |
a These hormones are often spelled without the ‘h’ (e.g. corticotropin, thyrotropin, etc) in contemporary texts. We have retained the original nomenclature in this edition.
Secretion from the anterior pituitary is largely regulated by the release from the hypothalamus of ‘factors’—in effect local hormones—that reach the pituitary through the bloodstream.1 The blood supply to the hypothalamus divides to form a meshwork of capillaries, the primary plexus (Fig. 32.1), which drains into the hypophyseal portal vessels. These pass through the pituitary stalk to feed a secondary plexus of capillaries in the anterior pituitary. Peptidergic neurons in the hypothalamus secrete a variety of releasing or inhibitory hormones directly into the capillaries of the primary capillary plexus (Table 32.1 and Fig. 32.1). Most of these regulate the secretion of hormones from the anterior lobe, although the melanocyte-stimulating hormones (MSHs) are secreted mainly from the intermediate lobe.
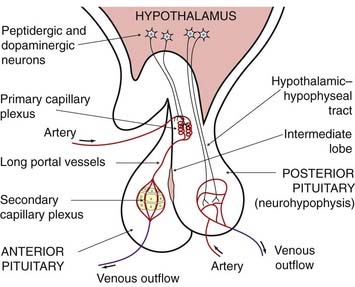
Fig. 32.1 Schematic diagram of vascular and neuronal relationships between the hypothalamus, the posterior pituitary and the anterior pituitary.
The main portal vessels to the anterior pituitary lie in the pituitary stalk and arise from the primary plexus in the hypothalamus, but some (the short portal vessels) arise from the vascular bed in the posterior pituitary (not shown).
Negative feedback pathways between the hormones of the hypothalamus, the anterior pituitary and the peripheral endocrine glands regulate the release of stimulatory hormones and integrate the functions of individual components of the endocrine system into a functional whole. In long negative feedback pathways, hormones secreted from the peripheral glands exert regulatory actions on both the hypothalamus and the anterior pituitary. The mediators of the short negative feedback pathways are anterior pituitary hormones that act directly on the hypothalamus.
The peptidergic neurons in the hypothalamus are themselves influenced by other centres within the central nervous system (CNS) mediated through pathways that release dopamine, noradrenaline, 5-hydroxytryptamine and the opioid peptides (which are particularly abundant in the hypothalamus, see Ch. 19). Hypothalamic control of the anterior pituitary is also exerted through the tuberohypophyseal dopaminergic pathway (see Ch. 38), the neurons of which lie in close apposition to the primary capillary plexus. Dopamine secreted directly into the hypophyseal portal circulation reaches the anterior pituitary in the blood.
Hypothalamic Hormones
The secretion of anterior pituitary hormones, then, is primarily regulated by the releasing factors that originate in the hypothalamus. These are listed in Table 32.1 and the most significant are described in more detail below. Somatostatin and gonadotrophin-releasing hormone are used therapeutically, the rest being used for diagnostic tests or as research tools. Many of these factors also function as neurotransmitters or neuromodulators elsewhere in the CNS (Ch. 38).
Somatostatin
Somatostatin is a peptide of 14 amino acid residues. It inhibits the release of growth hormone and thyroid-stimulating hormone (TSH, thyrotrophin) from the anterior pituitary (Fig. 32.2), and insulin and glucagon from the pancreas; it also decreases the release of most gastrointestinal hormones, and reduces gastric acid and pancreatic secretion.
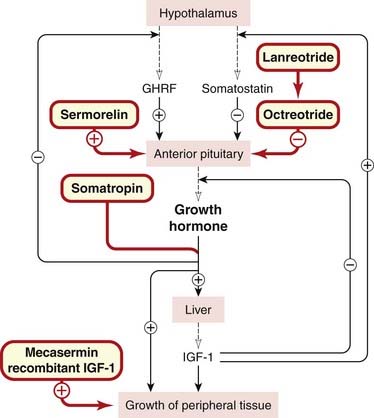
Fig. 32.2 Control of growth hormone secretion and its actions.
Drugs are shown in red-bordered boxes. GHRF, growth hormone-releasing factor; IGF-1, insulin-like growth factor-1.
Octreotide is a long-acting analogue of somatostatin (Ch. 19). It is used for the treatment of carcinoid and other hormone-secreting tumours (Ch. 15). It also has a place in the therapy of acromegaly (a condition in which there is oversecretion of growth hormone in an adult). It also constricts splanchnic blood vessels, and is used to treat bleeding oesophageal varices. Octreotide is generally given subcutaneously. The peak action is at 2 h, and the suppressant effect lasts for up to 8 h.
Unwanted effects include pain at the injection site and gastrointestinal disturbances. Gallstones and postprandial hyperglycaemia have also been reported, and acute hepatitis or pancreatitis has occurred in a few cases.
Lanreotide has similar effects and is also used in the treatment of thyroid tumours.
Gonadotrophin-Releasing Hormone
Gonadotrophin- (or luteinising hormone-) releasing hormone is a decapeptide that releases both follicle-stimulating hormone and luteinising hormone from gonadotrophs. It is also available as a preparation called gonadorelin, used mainly in the treatment of infertility (see Ch. 34).
Growth Hormone-Releasing Factor (Somatorelin)
Growth hormone-releasing factor (GHRF) is a peptide with 40–44 amino acid residues. The main action of GHRF is summarised in Figure 32.2. An analogue, sermorelin, may be used as a diagnostic test for growth hormone secretion. Given intravenously, subcutaneously or intranasally (generally the former), it causes secretion of growth hormone within minutes and peak concentrations in 1 h. The action is selective for the somatotrophs in the anterior pituitary, and no other pituitary hormones are affected. Unwanted effects are rare.
Thyrotrophin-Releasing Hormone (Protirelin)
Thyrotrophin-releasing hormone (TRH) from the hypothalamus releases thyroid-stimulating hormone (TSH) from the thyrotrophs. Protirelin is a synthetic TRH used for the diagnosis of thyroid disorders (see Ch. 33). Given intravenously in normal subjects, it causes an increase in plasma TSH concentration, whereas in patients with hyperthyroidism there is a blunted response because the raised blood thyroxine concentration has a negative feedback effect on the anterior pituitary. The opposite occurs with hypothyroidism, where there is an intrinsic defect in the thyroid itself.
Corticotrophin-Releasing Factor
Corticotrophin-releasing factor (CRF) is a peptide that releases adrenocorticotrophic hormone (ACTH, corticotrophin) and β-endorphin from the corticotrophs. CRF acts synergistically with antidiuretic hormone (ADH; arginine-vasopressin), and both its action and its release are inhibited by glucocorticoids (see Fig. 32.4, below). Synthetic preparations have been used to test the ability of the pituitary to secrete ACTH, and to assess whether ACTH deficiency is caused by a pituitary or a hypothalamic defect. It has also been used to evaluate hypothalamic pituitary function after therapy for Cushing’s syndrome (see Fig. 32.7, below).
Anterior Pituitary Hormones
The main hormones of the anterior pituitary are listed in Table 32.1. The gonadotrophins are dealt with in Chapter 34 and TSH in Chapter 33. The remainder are dealt with below.
Growth Hormone (Somatotrophin)
Growth hormone is secreted by the somatotroph cells and is the most abundant pituitary hormone. Secretion is high in the newborn, decreasing at 4 years to an intermediate level, which is then maintained until after puberty, after which there is a further decline. Several recombinant preparations of growth hormone, or somatropin, are available for treating growth defects and other developmental problems (see below).
Regulation of secretion
Secretion of growth hormone is regulated by the action of hypothalamic GHRF and modulated by somatostatin, as described above and outlined in Figure 32.2. One of the mediators of growth hormone action, insulin-like growth factor (IGF)-1, which is released from the liver (see below), has an inhibitory effect on growth hormone secretion by stimulating somatostatin release from the hypothalamus.
Growth hormone release, like other anterior pituitary secretions, is pulsatile, and its plasma concentration may fluctuate 10- to 100-fold. These surges occur repeatedly during the day and night, and reflect the dynamics of hypothalamic control. Deep sleep is a potent stimulus to growth hormone secretion, particularly in children.
Actions
The main effect of growth hormone (and its analogues) is to stimulate normal growth and, in doing this, it affects many tissues, acting in conjunction with other hormones secreted from the thyroid, the gonads and the adrenal cortex. It stimulates hepatic production of the IGFs—also termed somatomedins—which mediate most of its anabolic actions. Receptors for IGF-1 (the principal mediator) exist on many cell types, including liver cells and fat cells.
Growth hormone stimulates the uptake of amino acids and protein synthesis, especially in skeletal muscle. IGF-1 mediates many of these anabolic effects, acting on skeletal muscle and also on the cartilage at the epiphyses of long bones, thus influencing bone growth.
Disorders of production and clinical use
Deficiency of growth hormone results in pituitary dwarfism. In this condition, which may result from lack of GHRF or a failure of IGF generation or action, the normal proportions of the body are maintained. Growth hormone is used therapeutically in patients (often children) with growth hormone deficiency and with the short stature associated with the chromosomal disorder known as Turner’s syndrome. It may also be used to correct chronic renal insufficiency in children. Satisfactory linear growth can be achieved by giving somatropin subcutaneously, six to seven times per week, and therapy is most successful when started early. Humans are insensitive to growth hormone of other species, so human growth hormone (hGH) must be used clinically. This used to be obtained from human cadavers, but this led to the spread of Creutzfeldt–Jacob disease, a prion-mediated neurodegenerative disorder (Ch. 39). hGH is now prepared by recombinant DNA technology, which avoids this risk. Human recombinant IGF-1 is also available (mecasermin) for the treatment of growth failure in children who lack adequate amounts of this hormone. hGH is also used illicitly by athletes (see Ch. 58) to increase muscle mass. The large doses used have serious side effects, causing abnormal bone growth and cardiomegaly. It has also been tested as a means of combating the bodily changes in senescence; clinical trials have shown increases in body mass, but no functional improvement.
An excessive production of growth hormone in children results in gigantism. An excessive production in adults, which is usually the result of a benign pituitary tumour, results in acromegaly, in which there is enlargement mainly of facial structures and of the hands and feet. The dopamine agonist bromocriptine and octreotide may mitigate the condition. Another useful agent is pegvisomant, a modified version of growth hormone prepared by recombinant technology that is a highly selective antagonist of growth hormone actions.
Prolactin
Prolactin is secreted from the anterior pituitary by lactotroph (mammotroph) cells. These are abundant in the gland and increase in number during pregnancy, probably under the influence of oestrogen.
Regulation of secretion
Prolactin secretion is under tonic inhibitory control by the hypothalamus (Fig. 32.3 and Table 32.1), the inhibitory mediator being dopamine (acting on D2 receptors on the lactotrophs). The main stimulus for release is suckling; in rats, both the smell and the sounds of hungry pups are also effective triggers. Neural reflexes from the breast may stimulate the secretion from the hypothalamus of prolactin-releasing factor(s), possible candidates for which include TRH and oxytocin. Oestrogens increase both prolactin secretion and the proliferation of lactotrophs through release, from a subset of lactotrophs, of the neuropeptide galanin. Dopamine antagonists (used mainly as antipsychotic drugs; see Ch. 45) are potent stimulants of prolactin release, whereas agonists such as bromocriptine (see below and also Chs 38 and 45) suppress prolactin release. Bromocriptine is also used in parkinsonism (Ch. 39).
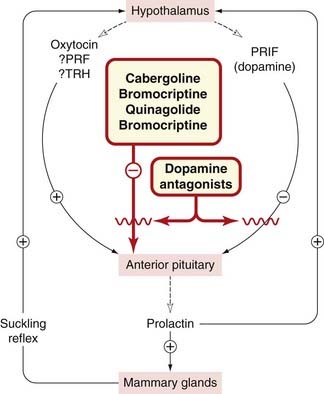
Actions
There are at least three specific receptor subtypes that bind prolactin, and these are not only found in the mammary gland but are widely distributed throughout the body, including the brain, ovary, heart and lungs. The main function of prolactin in women is the control of milk production. At parturition, when the blood level of oestrogen falls, the prolactin concentration rises and lactation is initiated. Maintenance of lactation depends on suckling (see above), which causes a 10- to 100-fold increase within 30 min.
Together with other hormones, prolactin is responsible for the proliferation and differentiation of mammary tissue during pregnancy. It inhibits gonadotrophin release and/or the response of the ovaries to these trophic hormones. This is one of the reasons why ovulation does not usually occur during breastfeeding, and it is believed to constitute a natural contraceptive mechanism.
 According to one rather appealing hypothesis, the high postpartum concentration of prolactin reflects its biological function as a ‘parental’ hormone. Certainly, broodiness and nest-building activity can be induced in birds, mice and rabbits by prolactin injections. Prolactin also exerts other, apparently unrelated, actions, including stimulating mitogenesis in lymphocytes. There is some evidence that it may play a part in regulating immune responses.
According to one rather appealing hypothesis, the high postpartum concentration of prolactin reflects its biological function as a ‘parental’ hormone. Certainly, broodiness and nest-building activity can be induced in birds, mice and rabbits by prolactin injections. Prolactin also exerts other, apparently unrelated, actions, including stimulating mitogenesis in lymphocytes. There is some evidence that it may play a part in regulating immune responses.
Modification of prolactin secretion
Prolactin itself is not used clinically. Bromocriptine, a dopamine receptor agonist, is used to decrease excessive prolactin secretion (hyperprolactinaemia). It is well absorbed orally, and peak concentrations occur after 2 h. Unwanted reactions include nausea and vomiting. Dizziness, constipation and postural hypotension may also occur. Cabergoline and quinagolide are similar.
Adrenocorticotrophic Hormone
Adrenocorticotrophic hormone (ACTH, corticotrophin) is the anterior pituitary secretion that controls the synthesis and release of the glucocorticoids of the adrenal cortex (see Table 32.1). It is a 39-residue peptide derived from the precursor pro-opiomelanocortin (POMC; Ch. 19) by sequential proteolytic processing. Failure of ACTH action because of defects in its receptor or intracellular signalling pathways can lead to severe glucocorticoid deficiency (Chan et al., 2008). Details of the regulation of ACTH secretion are shown in Figure 32.4.
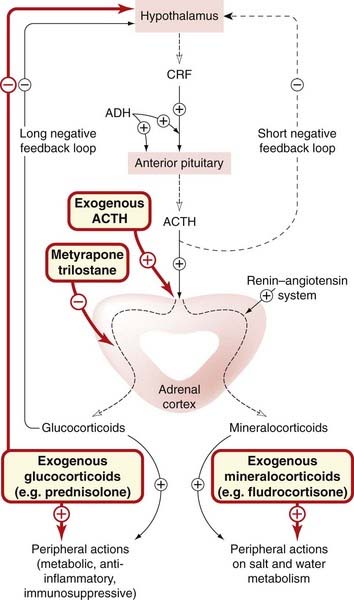
Fig. 32.4 Regulation of synthesis and secretion of adrenal corticosteroids.
The long negative feedback loop is more important than the short loop (dashed lines). Adrenocorticotrophic hormone (ACTH, corticotrophin) has only a minimal effect on mineralocorticoid production. Drugs are shown in red-bordered boxes. ADH, antidiuretic hormone (vasopressin); CRF, corticotrophin-releasing factor.
This hormone occupies (together with cortisone) an important place in the history of inflammation therapy because of the work of Hench and his colleagues in the 1940s, who first observed that both substances had anti-inflammatory effects in patients with rheumatoid disease. The effect of ACTH was thought to be secondary to stimulation of the adrenal cortex but, interestingly, the hormone also has anti-inflammatory actions in its own right, through activation of macrophage (melanocortin) MC3 receptors (Getting et al., 2002).
Adrenocorticotrophic hormone itself is not often used in therapy today, because its action is less predictable than that of the corticosteroids and it may provoke antibody formation. Tetracosactide, a synthetic polypeptide that consists of the first 24 N-terminal residues of human ACTH, has the same drawbacks but is now widely used in its stead for assessing the competency of the adrenal cortex (see below).
The concentration of ACTH in the blood is reduced by glucocorticoids, forming the basis of the dexamethasone suppression test.
Actions
Tetracosactide and ACTH have two actions on the adrenal cortex:
The main use of tetracosactide is in the diagnosis of adrenal cortical insufficiency. The drug is given intramuscularly or intravenously, and the concentration of hydrocortisone in the plasma is measured by radioimmunoassay.
Melanocyte-Stimulating Hormone (MSH)
α-, β- and γ-MSH are peptide hormones with structural similarity to ACTH and are derived from the same precursor. Together, these peptides are referred to as melanocortins, because their first recognised action was to stimulate the production of melanin by specialised skin cells called melanocytes. As such, they play an important part in determining hair coloration, skin colour and reaction to ultraviolet light.
Melanocyte-stimulating hormone acts on melanocortin receptors, of which five (MC1–5) have been cloned. These are G-protein-coupled receptors that activate cAMP synthesis. Melanin formation is under the control of the MC1 receptor, and excessive α-MSH production can provoke abnormal proliferation of melanocytes and may predispose to melanoma.
Melanocortins exhibit numerous other biological effects. For example, α-MSH inhibits the release of interleukin IL-1β and tumour necrosis factor (TNF)-α, reduces neutrophil infiltration, and exhibits anti-inflammatory and antipyretic activity. Levels of α-MSH are increased in synovial fluid of patients with rheumatoid arthritis. MC1 and MC3 receptors mediate the immunomodulatory effect of MSH. Agonists at these receptors with potential anti-inflammatory activity are being sought. Central injection of α-MSH also causes changes in animal behaviour, such as increased grooming and sexual activity as well as reduced feeding.
γ-MSH increases blood pressure, heart rate and cerebral blood flow following intracerebroventricular or intravenous injection. These effects are likely mediated by the MC4 receptor.
Two naturally occurring ligands for melanocortin receptors (agouti-signalling protein and agouti-related peptide, together called the agouti) have been discovered in human tissues. These are proteins that competitively antagonise the effect of MSH at melanocortin receptors.
The anterior pituitary gland and hypothalamus 
Adrenocorticotrophic hormone (corticotrophin) and the adrenal steroids
Posterior Pituitary Gland (Neurohypophysis)
The neurohypophysis consists largely of the terminals of nerve cells that lie in the supraoptic and paraventricular nuclei of the hypothalamus. Their axons form the hypothalamic–hypophyseal tract, and the fibres terminate in dilated nerve endings in close association with capillaries in the posterior pituitary gland (Fig. 32.1). Peptides, synthesised in the hypothalamic nuclei, pass down these axons into the posterior pituitary, where they are stored and eventually secreted into the bloodstream.
The two main hormones of the posterior pituitary are oxytocin (which contracts the smooth muscle of the uterus; for details see Ch. 34) and ADH (also called vasopressin; see Chs 22 and 28). Several similar peptides have been synthesised that vary in their antidiuretic, vasopressor and oxytocic (uterine stimulant) properties.
Antidiuretic Hormone
Regulation of secretion and physiological role
Antidiuretic hormone released from the posterior pituitary has a crucial role in the control of the water content of the body through its action on the cells of the distal part of the nephron and the collecting tubules in the kidney (see Ch. 28). The hypothalamic nuclei that control fluid balance lie close to the nuclei that synthesise and secrete ADH.
One of the main stimuli to ADH release is an increase in plasma osmolarity (which produces a sensation of thirst). A decrease in circulating blood volume (hypovolaemia) is another, and here the stimuli arise from stretch receptors in the cardiovascular system or from angiotensin release. Diabetes insipidus is a condition in which large volumes of dilute urine are produced because ADH secretion is reduced or absent, or because of a reduced sensitivity of the kidney to the hormone.
Antidiuretic hormone receptors
There are three classes of receptor for ADH: V1, V2 and V3. V2 receptors stimulate adenylyl cyclase, which mediates its main physiological actions in the kidney, whereas the V1 and V3 receptors are coupled to the phospholipase C/inositol trisphosphate system.
Actions
Renal actions
Antidiuretic hormone binds to V2 receptors in the basolateral membrane of the cells of the distal tubule and collecting ducts of the nephron. Its main effect in the collecting duct is to increase the rate of insertion of water channels (aquaporins) into the lumenal membrane, thus increasing the permeability of the membrane to water (see Ch. 28). It also activates urea transporters and transiently increases Na+ absorption, particularly in the distal tubule.
Several drugs affect the action of ADH. Non-steroidal anti-inflammatory drugs and carbamazepine increase, and lithium, colchicine and vinca alkaloids decrease, ADH effects. The effects of the last two agents are secondary to their action on the microtubules required for translocation of water channels. The antagonist demeclocycline counteracts the action of ADH on renal tubules and can be used to treat patients with water retention combined with urinary salt loss (and thus hyponatraemia) caused by excessive secretion of the hormone. This syndrome of inappropriate ADH secretion (‘SIADH’) is seen in some patients with lung or other malignancies or following head injury. More specific antagonists of V2 receptors are also used for SIADH and in some patients with heart failure (Ch. 22).
Other non-renal actions
Antidiuretic hormone causes contraction of smooth muscle, particularly in the cardiovascular system, by acting on V1 receptors (see Ch. 22). The affinity of these receptors for ADH is lower than that of the V2 receptors, and smooth muscle effects are seen only with doses larger than those affecting the kidney. ADH also stimulates blood platelet aggregation and mobilisation of coagulation factors. In the CNS, ADH acts as a neuromodulator and neurotransmitter. When released into the pituitary portal circulation, it promotes the release of ACTH from the anterior pituitary by an action on V3 receptors (Fig. 32.4).
Pharmacokinetic aspects
ADH, as well as various peptide analogues, is used clinically either for the treatment of diabetes insipidus or as a vasoconstrictor. The analogues have been developed to (a) increase the duration of action and (b) shift the potency between V1 and V2 receptors.
The main substances used are vasopressin (ADH itself; short duration of action, weak selectivity for V2 receptors, given by subcutaneous or intramuscular injection, or by intravenous infusion), desmopressin (increased duration of action, V2-selective and therefore fewer pressor effects, can be given by several routes including nasal spray) and terlipressin (increased duration of action, low but protracted vasopressor action and minimal antidiuretic properties). Felypressin is a short-acting vasoconstrictor that is injected with local anaesthetics such as prilocaine to prolong their action (see Ch. 42).
Vasopressin itself is rapidly eliminated, with a plasma half-life of 10 min and a short duration of action. Metabolism is by tissue peptidases, and 33% is removed by the kidney. Desmopressin is less subject to degradation by peptidases, and its plasma half-life is 75 min.
Unwanted effects
There are few unwanted effects and they are mainly cardiovascular in nature: intravenous vasopressin may cause spasm of the coronary arteries with resultant angina, but this risk can be minimised if the antidiuretic peptides are administered intranasally.
Clinical uses of antidiuretic hormone (vasopressin) and analogues 
The Adrenal Cortex
The adrenal glands consist of two parts: the inner medulla, which secretes catecholamines (see Ch. 14), and the outer cortex, which secretes adrenal steroids. The cortex, which concerns us in this section, comprises three concentric zones: the zona glomerulosa (the outermost layer) that elaborates mineralocorticoids, the zona fasciculata that elaborates glucocorticoids, and the innermost zona reticularis. While the principal adrenal steroids are those with glucocorticoid and mineralocorticoid2 activity, some sex steroids (mainly androgens) are also secreted by the gland but are not considered further in this chapter.
The mineralocorticoids regulate water and electrolyte balance, and the main endogenous hormone is aldosterone. The glucocorticoids have widespread actions on intermediate metabolism, affecting carbohydrate and protein metabolism, as well as potent regulatory effects on host defence mechanisms (Ch. 6). The adrenal secretes a mixture of glucocorticoids; the main hormone in humans is hydrocortisone (also, confusingly, called cortisol), and in rodents, corticosterone. The mineralocorticoid and glucocorticoid actions are not completely separated in naturally occurring steroids, some glucocorticoids having quite substantial effects on water and electrolyte balance. In fact, hydrocortisone and aldosterone are equiactive on mineralocorticoid receptors, but, in mineralocorticoid-sensitive tissues such as the kidney, the action of 11β-hydroxysteroid dehydrogenase converts hydrocortisone to the inactive metabolite cortisone,3 thereby protecting the receptor from inappropriate activation.
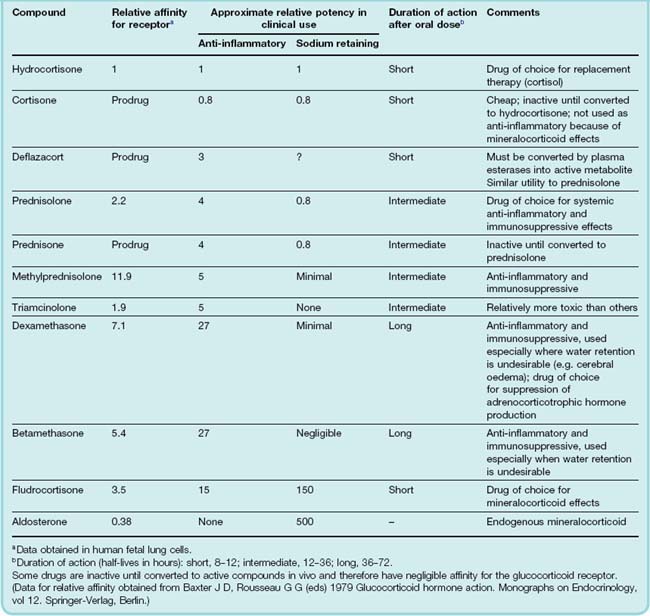
With the exception of replacement therapy (see below), glucocorticoids are most commonly employed for their anti-inflammatory and immunosuppressive properties (see Ch. 26). Under these circumstances, all their metabolic and other actions are seen as unwanted side effects. Synthetic steroids have been developed in which it has been possible to separate, to some degree, the glucocorticoid from the mineralocorticoid actions (see Table 32.2), but it has not been possible to separate the anti-inflammatory from the other actions of the glucocorticoids completely.
Table 32.2 Comparison of the main corticosteroid agents used for systemic therapy (using hydrocortisone as a standard)
The adrenal gland is essential to life, and animals deprived of these glands are able to survive only under rigorously controlled conditions. In humans, a deficiency in corticosteroid production, termed Addison’s disease, is characterised by muscular weakness, low blood pressure, depression, anorexia, loss of weight and hypoglycaemia. Addison’s disease may have an autoimmune aetiology, or it may result from destruction of the gland by chronic inflammatory conditions such as tuberculosis.
When corticosteroids are produced in excess, the clinical picture depends on which species predominates. Excessive glucocorticoid activity results in Cushing’s syndrome, the manifestations of which are outlined in Figure 32.7. This can be caused by hypersecretion from the adrenal glands or by prolonged therapeutic use of glucocorticoids. An excessive production of mineralocorticoids results in disturbances of Na+ and K+ balance. This may occur with hyperactivity or tumours of the adrenals (primary hyperaldosteronism, or Conn’s syndrome, an uncommon but important cause of hypertension; see Ch. 22), or with excessive activation of the renin–angiotensin system such as occurs in some forms of kidney disease, cirrhosis of the liver or congestive cardiac failure (secondary hyperaldosteronism).
Glucocorticoids
Synthesis and release
Glucocorticoids are not stored in the adrenal. They are synthesised under the influence of circulating ACTH secreted from the anterior pituitary gland (Fig. 32.4) and released in a pulsatile fashion into the blood. While they are always present, there is a well-defined circadian rhythm in the secretion in healthy humans, with the net blood concentration being highest early in the morning, gradually diminishing throughout the day and reaching a low point in the evening or night. ACTH secretion itself (also pulsatile in nature) is regulated by CRF released from the hypothalamus and vasopressin from the posterior gland. The release of both ACTH and CRF, in turn, is reflexly inhibited by the ensuing rising concentrations of glucocorticoids in the blood. This functional hypothalamic–pituitary–adrenal unit is referred to as the HPA axis.
Opioid peptides also exercise a tonic inhibitory control on the secretion of CRF, and psychological factors can affect the release of both vasopressin and CRF, as can stimuli such as excessive heat or cold, injury or infections. This is the principal mechanism whereby the HPA axis is activated in response to a threatening environment.
The precursor of glucocorticoids is cholesterol (Fig. 32.5). The initial conversion of cholesterol to pregnenolone is the rate-limiting step and is itself regulated by ACTH. Some of the reactions in the biosynthetic pathway can be inhibited by drugs. Metyrapone prevents the β-hydroxylation at C11, and thus the formation of hydrocortisone and corticosterone. Synthesis is blocked at the 11-deoxycorticosteriod stage, and as these substances have no effects on the hypothalamus and pituitary, there is a marked increase in ACTH in the blood. Metyrapone can therefore be used to test ACTH production, and may also be used to treat patients with Cushing’s syndrome. Trilostane (also of use in Cushing’s syndrome and primary hyperaldosteronism) blocks an earlier enzyme in the pathway—the 3β-dehydrogenase.
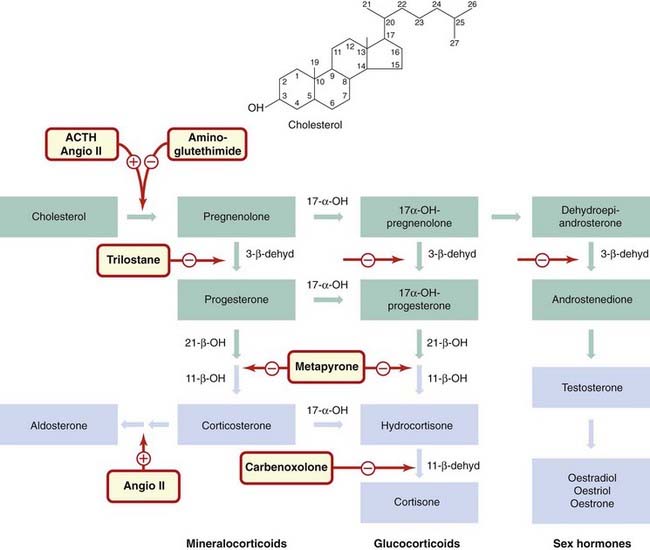
Fig. 32.5 Biosynthesis of corticosteroids, mineralocorticoids and sex hormones.
All steroid hormones are synthesised from cholesterol. Successive steps of hydroxylation and dehydrogenation are important in the biosynthetic pathway and are targets for drugs. Intermediates are shown in green boxes; interconversions occur between the pathways. Blue boxes indicate circulating hormones. Drugs are shown in red-bordered boxes adjacent to their sites of action. Glucocorticoids are produced by cells of the zona fasciculata, and their synthesis is stimulated by adrenocorticotrophic hormone (ACTH); aldosterone is produced by cells of the zona glomerulosa, and its synthesis is stimulated by angiotensin II (angio II). Metyrapone inhibits glucocorticoid synthesis, aminoglutethimide and trilostane block synthesis of all three types of adrenal steroid (see text for details). Carbenoxolone inhibits the interconversion of hydrocortisone and cortisone in the kidney. Enzymes: 17-α-OH, 17-α-hydroxylase; 3-β-dehyd, 3-β-dehydrogenase; 21-β-OH, 21-β-hydroxylase; 11-β-OH, 11-β-hydroxylase; 11-β-dehyd, 11-β-hydroxysteroid dehydrogenase.
Aminoglutethimide inhibits the initial step in the biosynthetic pathway and has the same overall effect as metyrapone. Ketoconazole, an antifungal agent (Ch. 52), used in higher doses also inhibits steroidogenesis and may be of value in the specialised treatment of Cushing’s syndrome.
Glucocorticoids
Common drugs used systemically include hydrocortisone, prednisolone and dexamethasone.
Metabolic actions
Regulatory actions
Mechanism of action
The glucocorticoid effects relevant to this discussion are initiated by interaction of the drugs with specific intracellular glucocorticoid receptors belonging to the nuclear receptor superfamily (although there may be other binding proteins or sites; see Norman et al., 2004). This superfamily (see Ch. 3) also includes the receptors for mineralocorticoids, the sex steroids, thyroid hormones, vitamin D3 and retinoic acid. The actual mechanism of transcriptional control is complex with at least four mechanisms operating within the nucleus. These are summarised diagrammatically in Figure 32.6.
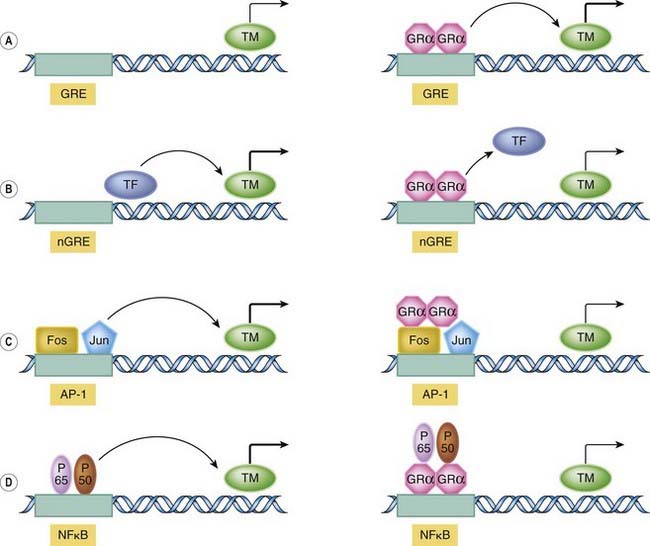
Fig. 32.6 Molecular mechanism of action of glucocorticoids.
The schematic figure shows three possible ways by which the liganded glucocorticoid receptor can control gene expression following translocation to the nucleus. [A] Basic transactivation mechanism. Here, the transcriptional machinery (TM) is presumed to be operating at a low level. The liganded glucocorticoid receptor (GR) dimer binds to one or more ‘positive’ glucocorticoid response elements (GREs) within the promoter sequence (shaded zone) and upregulates transcription. [B] Basic transrepression mechanism. The transcriptional machinery is constitutively driven by transcription factors (TF). In binding to the ‘negative’ GRE (nGRE), the receptor complex displaces these factors and expression falls. [C] Fos/Jun mechanism. Transcription is driven at a high level by Fos/Jun transcription factors binding to their AP-1 regulatory site. This effect is reduced in the presence of the GR. [D] Nuclear factor (NF)κβ mechanism. The transcription factors P65 and P50 bind to the NFκβ site, promoting gene expression. This is prevented by the presence of the GR, which binds the transcription factors, preventing their action (this may occur in the cytoplasm also). (For further details of the structure of the glucocorticoid receptor, see Ch. 3.)
(Modified from Oakley R H, Cidlowski J A in Goulding N J, Flower R J (eds) 2001 Glucocorticoids. Birkhauser Verlag.)
In addition to controlling gene expression, the liganded receptor itself, in either a monomeric or a dimeric form, may trigger important signal transduction events while still in the cytosolic compartment (there may even be a subpopulation of receptors that reside there permanently). One of these cytosolic effects, germane to the anti-inflammatory action of these drugs, is the release, following phosphorylation, of the protein annexin-1, which has potent inhibitory effects on leukocyte trafficking and other biological actions. The significance of such ‘receptor-mediated, non-genomic’ actions is that they can happen very rapidly (within seconds), as they do not entail changes in protein synthesis that require a longer time frame.
Actions
General metabolic and systemic effects
The main metabolic effects are on carbohydrate and protein metabolism. The glucocorticoids cause both a decrease in the uptake and utilisation of glucose and an increase in gluconeogenesis, resulting in a tendency to hyperglycaemia (see Ch. 30). There is a concomitant increase in glycogen storage, which may be a result of insulin secretion in response to the increase in blood sugar. Overall, there is decreased protein synthesis and increased protein breakdown, particularly in muscle, and this can lead to wasting. Glucocorticoids also have a ‘permissive’ effect on the cAMP-dependent lipolytic response to catecholamines and other hormones. Such hormones cause lipase activation through a cAMP-dependent kinase, the synthesis of which requires the presence of glucocorticoids (see below). Large doses of glucocorticoids given over a long period result in the redistribution of body fat characteristic of Cushing’s syndrome (Fig. 32.7).
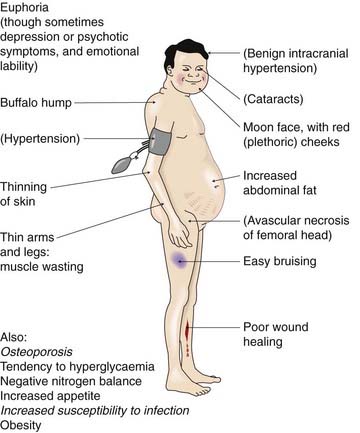
This is caused by excessive exposure to glucocorticoids, and may be caused by disease (e.g. an adrenocorticotrophic hormone-secreting tumour) or by prolonged administration of glucocorticoid drugs (iatrogenic Cushing’s). Italicised effects are particularly common. Less frequent effects, related to dose and duration of therapy, are shown in parentheses.
(Adapted from Baxter JD, Rousseau G G (eds) 1979 Glucocorticoid hormone action. Monographs on Endocrinology, vol 12. Springer-Verlag, Berlin.)
Glucocorticoids tend to produce a negative calcium balance by decreasing Ca2+ absorption in the gastrointestinal tract and increasing its excretion by the kidney. This may contribute to osteoporosis (see below). In higher, non-physiological concentrations, the glucocorticoids have some mineralocorticoid actions (see below), causing Na+ retention and K+ loss—possibly by swamping the protective 11β-hydroxysteriod dehydrogenase and acting at mineralocorticoid receptors.
Negative feedback effects on the anterior pituitary and hypothalamus
Both endogenous and exogenous glucocorticoids have a negative feedback effect on the secretion of CRF and ACTH (see Fig. 32.4). Administration of exogenous glucocorticoids depresses the secretion of CRF and ACTH, thus inhibiting the secretion of endogenous glucocorticoids and potentially causing atrophy of the adrenal cortex. If therapy is prolonged, it may take many months to return to normal function when the drugs are stopped.
Anti-inflammatory and immunosuppressive effects
That endogenous glucocorticoids maintain a low-level anti-inflammatory tonus can be readily demonstrated by observing the heightened response seen in adrenalectomised animals to even mild inflammatory stimuli. A failure of appropriate secretion in response to injury or infection may underlie certain chronic inflammatory human pathologies.
Exogenous glucocorticoids are the anti-inflammatory drugs par excellence, and when given therapeutically inhibit both the early and the late manifestations of inflammation, i.e. not only the initial redness, heat, pain and swelling, but also the later stages of wound healing and repair, and the proliferative reactions seen in chronic inflammation. They reverse virtually all types of inflammatory reaction, whether caused by invading pathogens, by chemical or physical stimuli, or by inappropriately deployed immune responses such as are seen in hypersensitivity or autoimmune disease. When used prophylactically to suppress graft rejection, glucocorticoids suppress the initiation and generation of an immune response mounted against this new ‘invader’ more efficiently than an established response in which clonal proliferation has already occurred.
Given that the glucocorticoids are able to modify the expression of so many genes, and that the extent and direction of regulation varies between tissues and even at different times during disease, you will not be surprised to learn that their anti-inflammatory effects are fearsomely complex. Some prominent actions may be highlighted, but these should not be considered a complete list.
Actions on inflammatory cells include:
Action on the mediators of inflammatory and immune responses (Ch. 17) include:
Inflammation is an important protective response designed to ensure the survival of an infected or injured host. It therefore strikes many as odd that we should not only have potent anti-inflammatory hormones circulating constantly in the blood, but that these should be dramatically increased during such threatening episodes. A useful explanatory paradigm is that of Munck et al. (1984): according to this idea, the anti-inflammatory and immunosuppressive actions may play a crucial counter-regulatory role, in that they prevent excessive activation of inflammation and other powerful defence reactions that might, if unchecked, themselves threaten homeostasis. Certainly, this view is borne out by experimental work. While these drugs are of great value in treating conditions characterised by hypersensitivity and unwanted inflammation, they carry the hazard that they are able to suppress the same defence reactions that provide protection to infection and promote healing.
Mechanism of action of the glucocorticoids
Unwanted effects
Unwanted effects occur with large doses or prolonged administration of glucocorticoids rather than replacement therapy, and are a serious problem. The major effects are as follows:
Sudden withdrawal of the drugs after prolonged therapy may result in acute adrenal insufficiency through suppression of the patient’s capacity to synthesise corticosteroids.5 Careful procedures for phased withdrawal should be followed. Recovery of full adrenal function usually takes about 2 months, although it can take 18 months or more.
Pharmacokinetic aspects
There are many glucocorticoid drugs in therapeutic use. Although cortisol, the endogenous hormone, is often used, synthetic derivatives are even more common. These have different physicocochemical properties as well as varying potency and have been optimised for administration by different routes. They may be administered orally, systemically or intra-articularly; given by aerosol into the respiratory tract, administered as drops into the eye or the nose, or applied in creams or ointments to the skin. Topical administration diminishes the likelihood of systemic toxic effects unless large quantities are used. When prolonged use of systemic glucocorticoids is necessary, therapy on alternate days may decrease suppression of the HPA axis and other unwanted effects.
As small lipophilic molecules, glucocorticoids probably enter their target cells by simple diffusion. Hydrocortisone has a plasma half-life of 90 min, although its main biological effects have a 2–8 h latency. Biological inactivation, which occurs in liver cells and elsewhere, is initiated by reduction of the C4–C5 double bond. Cortisone and prednisone are inactive until converted in vivo to hydrocortisone and prednisolone, respectively.
Endogenous glucocorticoids are transported in the plasma bound to corticosteroid-binding globulin (CBG) and to albumin. CBG accounts for about 77% of bound hydrocortisone, but many synthetic glucocorticoids are not bound at all. Albumin has a lower affinity for hydrocortisone but binds both natural and synthetic steroids. Both CBG-bound and albumin-bound steroids are biologically inactive.
The clinical use of systemic glucocorticoids is given in the clinical box. Dexamethasone has a special use: it can be used to test HPA axis function in the dexamethasone suppression test. A relatively low dose, usually given at night, should suppress the hypothalamus and pituitary, and result in reduced ACTH secretion and hydrocortisone output, as measured in the plasma about 9 hours later. Failure of suppression implies hypersecretion of ACTH or of glucocorticoids (Cushing’s syndrome).
Clinical uses of glucocorticoids
Pharmacokinetics and unwanted actions of the glucocorticoids
Mineralocorticoids
The main endogenous mineralocorticoid is aldosterone. Its chief action is to increase Na+ reabsorption by the distal tubules in the kidney, with concomitant increased excretion of K+ and H+ (see Ch. 28). An excessive secretion of mineralocorticoids, as in Conn’s syndrome, causes marked Na+ and water retention, with increased extracellular fluid volume, and sometimes hypokalaemia, alkalosis and hypertension. Decreased secretion, as in Addison’s disease, causes Na+ loss (desalinisation) and a marked decrease in extracellular fluid volume. There is a concomitant decrease in the excretion of K+, resulting in hyperkalaemia.
Regulation of aldosterone synthesis and release
The regulation of the synthesis and release of aldosterone is complex. Control depends mainly on the electrolyte composition of the plasma and on the angiotensin II system (Fig. 32.4; Chs 22 and 28). Low plasma Na+ or high plasma K+ concentrations affect the zona glomerulosa cells of the adrenal directly, stimulating aldosterone release. Depletion of body Na+ also activates the renin–angiotensin system (see Fig. 22.4). One of the effects of angiotensin II is to increase the synthesis and release of aldosterone (see Fig. 28.5).
Mechanism of action
Like other steroid hormones, aldosterone acts through specific intracellular receptors of the nuclear receptor family. Unlike the glucocorticoid receptor, which occurs in most tissues, the mineralocorticoid receptor is restricted to a few tissues, such as the kidney and the transporting epithelia of the colon and bladder. Cells containing mineralocorticoid receptors also contain the 11β-hydroxysteroid dehydrogenase type 2 enzyme (see above), which converts hydrocortisone (cortisol) into inactive cortisone. This has a low affinity for the mineralocorticoid receptors, thus ensuring that the cells are appropriately affected only by the mineralocorticoid hormone itself. Interestingly, this enzyme is inhibited by carbenoxolone (previously used to treat gastric ulcers; see Ch. 29), a compound derived from liquorice. If this inhibition is marked, it allows corticosterone to act on the mineralocorticoid receptor, producing a syndrome similar to Conn’s syndrome (primary hyperaldosteronism) except that the circulating aldosterone concentration is not raised.
As with the glucocorticoids, the interaction of aldosterone with its receptor initiates transcription and translation of specific proteins, resulting in an increase in the number of sodium channels in the apical membrane of the cell, and subsequently an increase in the number of Na+-K+-ATPase molecules in the basolateral membrane (see Fig. 28.5), causing increased K+ excretion (see Ch. 28). In addition to the genomic effects, there is evidence for a rapid non-genomic effect of aldosterone on Na+ influx, through an action on the Na+-H+ exchanger in the apical membrane.
Clinical use of mineralocorticoids and antagonists
The main clinical use of mineralocorticoids is in replacement therapy. The most commonly used drug is fludrocortisone (Table 32.2 and Fig. 32.4), which can be taken orally. Spironolactone is a competitive antagonist of aldosterone, and it also prevents the mineralocorticoid effects of other adrenal steroids on the renal tubule (Ch. 28). Side effects include gynaecomastia and impotence, because spironolactone also has some blocking action on androgen and progesterone receptors. It is used to treat primary or secondary hyperaldosteronism and, in conjunction with other drugs, in the treatment of resistant hypertension and of heart failure (Ch. 22) and resistant oedema (Ch. 28). Eplerenone has a similar indication and mechanism of action, although fewer side effects.
New Directions in Glucocorticoid Therapy
Glucocorticoids are highly effective in controlling inflammation, but severely limited by their unwanted effects. The ideal solution would be a glucocorticoid possessing the anti-inflammatory but not the unwanted metabolic or other effects.
For many years, the pharmaceutical industry pursued this goal using simple strategies based on the development of structural analogues of hydrocortisone. While this yielded many new active and interesting compounds (several of which are in clinical use today), they never achieved ‘separation’ of the glucocorticoid actions.
Recently, investigators have taken another tack. It has been noted that glucocorticoids suppress inflammation largely by downregulating genes (e.g. cytokine genes) that promote the inflammatory response, whereas many of the side effects are caused by overexpression of metabolic and other genes (causing, for example, diabetes). Because these effects are brought about through different molecular pathways, researchers have sought steroids that may exhibit one set of actions without the other. At the time of writing, modest successes have been achieved with these ‘dissociated’ steroids (see Schacke et al., 2002, 2005), but it is too early to tell whether they will really make a difference in the clinic.
A related idea has been to manipulate the histone deacetylase enzymes that are responsible for facilitating the transcriptional regulation of genes following nuclear receptor binding to response elements (Hayashi et al., 2004). One current notion is that there may be a specific isoform of this enzyme that deals with gene upregulation, and that if this could be inhibited, it would lessen the possibility of those unwanted effects.
Another approach has been to focus on the actual mechanism of receptor activation. It is clear that not all glucocorticoids bind to the receptor in the same way, and so the dynamics of the resulting liganded complex may vary (Adcock, 2003). This could be exploited to alter the ability of the steroid–receptor complex to initiate transcriptional and other changes in a way that could be beneficial to the profile of the drug.
These (and other) ideas have been reviewed by Song et al., 2005, but despite their ingenuity it is depressing to report that none has yet made a major difference to the tolerability of these most useful drugs.
References and Further Reading
The hypothalamus and pituitary
Birnbaumer M. Vasopressin receptors. Trends Endocrinol. Metab.. 2000;11:406-410.
Chini B., Manning M., Guillon G. Affinity and efficacy of selective agonists and antagonists for vasopressin and oxytocin receptors: an ‘easy guide’ to receptor pharmacology. Prog. Brain Res.. 2008;170:513-517. (The title is self-explanatory! Also deals with the prospects for new drugs in this area)
Clark R.G., Robinson C.A.F. Up and down the growth hormone cascade. Cytokine Growth Factor Rev.. 1996;1:65-80. (A review covering the cascade that controls the primary regulators of growth and metabolism, namely growth hormone and the insulin-like growth factors)
Drolet G., Rivest S. Corticotropin-releasing hormone and its receptors; an evaluation at the transcription level in vivo. Peptides. 2001;22:761-767.
Freeman M.E., Kanyicska B., Lerant A., Nagy G. Prolactin: structure, function and regulation of secretion. Physiol. Res.. 2000;80:1524-1585. (Comprehensive review of prolactin and its receptors)
Guillemin R. Hypothalamic hormones a.k.a. hypothalamic releasing factors. J. Endocrinol.. 2005;184:11-28. (This little review focuses on the history of research in this area and covers the discovery and characterisation of the principal releasing factors. Something to read if you are drawn to this area)
Lamberts S.W.J., van der Lely A.-J., de Herder W.W., Hofland L.J. Octreotide. N. Engl. J. Med.. 1996;334:246-254. (A review covering somatostatin receptors, somatostatin analogues and treatment of tumours expressing somatostatin receptors with octreotide)
Maybauer M.O., Maybauer D.M., Enkhbaatar P., Traber D.L. Physiology of the vasopressin receptors. Best Pract. Res. Clin. Anaesthesiol.. 2008;22:253-263. (A small review written mainly from a clinical perspective. Discusses future therapeutic uses for receptor agonists)
Okada S., Kopchick J.J. Biological effects of growth hormone and its antagonist. Trends Mol. Med.. 2001;7:126-132.
Prakash A., Goa K.L. Sermorelin: a review of its use in the diagnosis and treatment of children with idiopathic growth hormone deficiency. BioDrugs. 1999;12:139-157. (Mainly a clinical appraisal of sermorelin utility in treating growth deficiency in contrast to growth hormone itself)
Schneider F., Tomek W., Grundker C. Gonadotropin-releasing hormone (GnRH) and its natural analogues: a review. Theriogenology. 2006;66:691-709. (Focuses mainly on the use of such agents in veterinary medicine)
Thibonnier M., Coles P., Thibonnier A., et al. The basic and clinical pharmacology of nonpeptide vasopressin receptor antagonists. Annu. Rev. Pharmacol.. 2001;41:175-202. (Authoritative account of ADH receptors and the search for new antagonists)
Vance M.L. Hypopituitarism. N. Engl. J. Med.. 1994;330:1651-1662. (Review of causes, clinical features and hormone replacement therapy of hypopituitarism)
Wikberg J.E.S., Muceniece R., Mandrika I., et al. New aspects on the melanocortins and their receptors. Pharmacol. Res.. 2000;42:393-420. (Detailed review of the varied biological roles of melanocortins and their receptors)
Chan L.F., Clark A.J., Metherell L.A. Familial glucocorticoid deficiency: advances in the molecular understanding of ACTH action. Horm. Res.. 2008;69:75-82. (This paper and the one below discuss research into the role of the ACTH signalling system in familial glucocorticoid deficiency. An expert piece of scientific detective work. The second paper is more accessible)
Clark A.J., Metherell L.A., Cheetham M.E., Huebner A. Inherited ACTH insensitivity illuminates the mechanisms of ACTH action. Trends Endocrinol. Metab.. 2005;16:451-457.
Bastl C., Hayslett J.P. The cellular action of aldosterone in target epithelia Kidney Int. 42:1992:250-264 (A detailed review covering the aldosterone receptor and regulation of gene expression, aldosterone action on electrogenic and electroneutral Na+ transport, and on K+ and H+ secretion)
Adcock I.M. Glucocorticoids: new mechanisms and future agents. Curr. Allergy Asthma. Rep.. 2003;3:249-257. (Excellent review of advances in glucocorticoid pharmacology)
Borski R.J. Nongenomic membrane actions of glucocorticoids in vertebrates. Trends Endocrinol. Metab.. 2000;11:427-436. (A thought-provoking account of the non-genomic effects of glucocorticoids)
Buckingham J.C. Stress and the hypothalamo–pituitary–immune axis. Int. J. Tissue React.. 1998;20:23-34. (Clear review of the complexities of the effect of stress on HPA axis function)
D’Acquisto F., Perretti M., Flower R.J. Annexin-A1: a pivotal regulator of the innate and adaptive immune systems. Br. J. Pharmacol.. 2008;155:152-169. (Reviews the role of the glucocorticoid-regulated protein annexin-A1 in mediating the anti-inflammatory action of glucocorticoid drugs)
de Kloet E.R. Stress in the brain. Eur. J. Pharmacol.. 2000;405:187-198.
Eberhardt W., Kilz T., Akool el-S., Müller R., Pfeilschifter J. Dissociated glucocorticoids equipotently inhibit cytokine- and cAMP-induced matrix degrading proteases in rat mesangial cells. Biochem. Pharmacol.. 2005;70:433-445. (A research paper on the action of dissociated glucocorticoids on the expression of proteolytic enzymes during inflammation as an example of their action)
Falkenstein E., Tillmann H.C., Christ M., et al. Multiple actions of steroid hormones—a focus on rapid, nongenomic effects. Pharmacol. Rev.. 2000;52:513-556.
Funder J.W. Glucocorticoid and mineralocorticoid receptors: biology and clinical relevance. Annu. Rev. Med.. 1997;48:231-240. (Succinct review of glucocorticoid and mineralocorticoid receptors, differences in glucocorticoid receptor—and mineralocorticoid receptor-mediated transcription and responses, and steroid resistance)
Getting S.J., Christian H.C., Flower R.J., Perretti M. Activation of melanocortin type 3 receptor as a molecular mechanism for adrenocorticotropic hormone efficacy in gouty arthritis. Arthritis Rheum.. 2002;46:2765-2775. (Original paper that demonstrates that ACTH has intrinsic anti-inflammatory actions that are independent of the adrenals)
Hayashi R., Wada H., Ito K., Adcock I.M. Effects of glucocorticoids on gene transcription. Eur. J. Pharmacol.. 2004;500:51-62. (Good basic review of glucocorticoid action; easy to read)
Kirwan J., Power L. Glucocorticoids: action and new therapeutic insights in rheumatoid arthritis. Curr. Opin. Rheumatol.. 2007;19:233-237. (Written mainly from the viewpoint of a practising rheumatologist, this review offers interesting insights into the use of these drugs to modify severe chronic arthritic disease)
Lamberts S.W.J., Bruining H.A., de Jong F.S. Corticosteroid therapy in severe illness. N. Engl. J. Med.. 1997;337:1285-1292. (Review with succinct coverage of normal response of adrenal to illness, followed by more detail on clinical therapy)
Munck A., Guyre P.M., Holbrook N.J. Physiological functions of glucocorticoids in stress and their relation to pharmacological actions. Endocr. Rev.. 1984;5:25-44. (Seminal review suggesting that the anti-inflammatory/immunosuppressive actions of the glucocorticoids have a physiological function; required reading if you want to understand glucocorticoid physiology and pharmacology)
Norman A.W., Mizwicki M.T., Norman D.P. Steroid-hormone rapid actions, membrane receptors and a conformational ensemble model. Nat. Rev. Drug Discov.. 2004;3:27-41. (Fairly advanced reading but contains many useful tables and excellent diagrams; well worth the effort if this subject interests you)
Rhodes D., Klug A. Zinc fingers. Sci. Am.. 1993;268:32-39. (Clear discussion of the role of zinc fingers, such as those utilised by nuclear receptors, in regulating gene transcription; excellent diagrams, of course)
Schacke H., Docke W.D., Asadullah K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol. Ther.. 2002;96:23-43. (Useful review dealing with side effects and ‘dissociated steroids’)
Schacke H., Rehwinkel H., Asadullah K. Dissociated glucocorticoid receptor ligands: compounds with an improved therapeutic index. Curr. Opin. Investig. Drugs. 2005;6:503-507. (More details about the dissociated steroid concept, with several examples)
Schafer-Korting M., Kleuser B., Ahmed M., et al. Glucocorticoids for human skin: new aspects of the mechanism of action. Skin Pharmacol. Physiol.. 2005;18:103-114. (The title is self-explanatory: good discussions of different mechanisms of glucocorticoid action)
Song I.H., Gold R., Straub R.H., et al. New glucocorticoids on the horizon: repress, don’t activate!. J. Rheumatol.. 2005;32:1199-1207. (Good summary of the various approaches taken to circumvent glucocorticoid side effects)
Tak P.P., Firestein G.S. NF-kappaB: a key role in inflammatory diseases. J. Clin. Invest.. 2001;107:7-11. (Succinct and very readable account of the role of nuclear factor (NF)κβ in inflammation)
Tsai M.-J., O’Malley B.W. Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu. Rev. Biochem.. 1994;63:451-486. (Detailed review, by one of the pioneers of the field, of the molecular biology of these receptors, including gene activation and gene silencing)
Whittington P.F., Barnes H.V., Bayless T.M. Medical management of Crohn’s disease in adolescence. Gastroenterology. 1977;72:1338-1344.
Wilckens T. Glucocorticoids and immune dysfunction: physiological relevance and pathogenic potential of hormonal dysfunction. Trends Pharmacol. Sci.. 1995;16:193-197. (Covers glucocorticoid interaction with their receptors, heat shock protein 90, AP-1 and nuclear factor NFκβ transcription factors; clear diagrams)
Buckingham J.C., Gillies G.E., Cowell A.M., editors. Stress, stress hormones and the immune system. Chichester: John Wiley, 1997. (Another excellent source book for information that covers the concepts of stress, the release of cortisol and its subsequent physiological actions)
Goulding N.J., Flower R.J., editors. Milestones in drug therapy: glucocorticoids. Basel: Birkhäuser Verlag, 2001. (A useful source of information on all aspects of glucocorticoid biology and pharmacology, containing chapters by some of the leaders in the field)
1The word ‘factor’ was originally coined at a time when their structure and function were not known. These are blood-borne messengers, and as such are clearly hormones. Nevertheless, the term ‘factor’, however irrational, lingers on.
2So named because early experimenters noticed that two crude fractions of adrenal gland extracts caused changes in blood glucose or salt and water retention.
3Oddly, it was cortisone that was originally demonstrated to have potent anti-inflammatory activity in the classic studies by Hench and his colleagues in 1949. The reason for this apparent anomaly is that an isoform of 11β-hydroxysteroid dehydrogenase present in some tissues can transform this steroid back into cortisol (i.e. hydrocortisone), thus restoring biological activity.
4However, some of the diseases for which glucocorticoids are indicated themselves retard growth. In a classical trial, glucocorticoid treatment increased growth in adolescents with inflammatory bowel disease as the disease resolved (Whittington et al., 1977).
5Patients on long-term glucocorticoid therapy are advised to carry a card stating, ‘I am a patient on STEROID TREATMENT which must not be stopped abruptly’.