44 Antiepileptic drugs
Overview
In this chapter, we describe the nature of epilepsy, the neurobiological mechanisms underlying it and the animal models that can be used to study it. We then proceed to describe the various classes of drugs that are used to treat it, the mechanisms by which they work and their pharmacological characteristics. More information on the topics covered can be obtained from specialist textbooks (e.g. Engel & Pedley, 2007; Browne & Holmes, 2008; Hart & Sander, 2008).
Centrally acting muscle relaxants are discussed briefly at the end of the chapter.
Introduction
Epilepsy is a very common disorder, characterised by seizures, which take various forms and result from episodic neuronal discharges, the form of the seizure depending on the part of the brain affected. Epilepsy affects 0.5–1% of the population. Often, there is no recognisable cause, although it may develop after brain damage, such as trauma, stroke, infection or tumour growth, or other kinds of neurological disease, including various inherited neurological syndromes. Epilepsy is treated mainly with drugs, although brain surgery may be used for suitable severe cases. Current antiepileptic drugs are effective in controlling seizures in about 70% of cases, but their use is often limited by side effects. In addition to their use in patients with epilepsy, antiepileptic drugs are used to treat or prevent convulsions caused by other brain diseases, for example trauma (including following neurosurgery), infection (as an adjunct to antibiotics), brain tumours and stroke. For this reason, they are sometimes termed anticonvulsants rather than antiepileptics. Increasingly, some antiepileptic drugs have been found to have beneficial effects in non-convulsive disorders such as neuropathic pain (Ch. 41) and bipolar depression (Ch. 46). Many new antiepileptic drugs have been developed over the past 20 or so years in attempts to improve their efficacy and side-effect profile. Improvements have been steady rather than spectacular, and epilepsy remains a difficult problem, despite the fact that controlling reverberative neuronal discharges would seem, on the face of it, to be a much simpler problem than controlling those aspects of brain function that determine emotions, mood and cognitive function.
The Nature of Epilepsy
The term ‘epilepsy’ is used to define a group of neurological disorders all of which exhibit periodic seizures. For information on the underlying causes of epilepsy and factors which precipitate periodic seizures see Browne & Holmes (2008) and Hart & Sander (2008). As explained later, not all seizures involve convulsions. Seizures are associated with episodic high-frequency discharge of impulses by a group of neurons (sometimes referred to as the focus) in the brain. What starts as a local abnormal discharge may then spread to other areas of the brain. The site of the primary discharge and the extent of its spread determine the symptoms that are produced, which range from a brief lapse of attention to a full convulsive fit lasting for several minutes, as well as odd sensations or behaviours. The particular symptoms produced depend on the function of the region of the brain that is affected. Thus, involvement of the motor cortex causes convulsions, involvement of the hypothalamus causes peripheral autonomic discharge, and involvement of the reticular formation in the upper brain stem leads to loss of consciousness.
Abnormal electrical activity during and following a seizure can be detected by electroencephalography (EEG) recording from electrodes distributed over the surface of the scalp. Various types of seizure can be recognised on the basis of the nature and distribution of the abnormal discharge (Fig. 44.1). Modern brain imaging techniques, such as magnetic resonance imaging and positron emission tomography, are now routinely used in the diagnosis of epilepsy (see Fig. 44.2) to identify structural abnormalities (e.g. lesions, tumours) that cause certain epilepsies (see Deblaere & Achten, 2008).
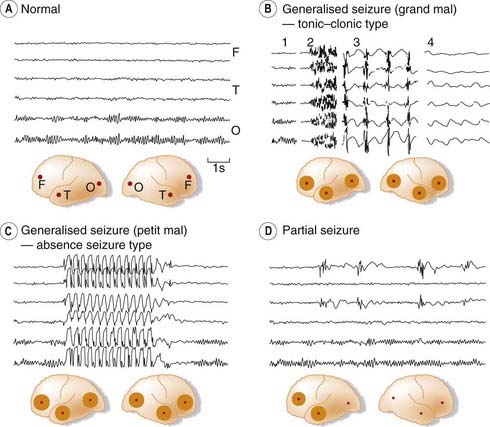
Fig. 44.1 Electroencephalography (EEG) records in epilepsy.
[A] Normal EEG recorded from frontal (F), temporal (T) and occipital (O) sites on both sides, as shown in the inset diagram. The α rhythm (10/s) can be seen in the occipital region. [B] Sections of EEG recorded during a generalised tonic–clonic (grand mal) seizure: 1, normal record; 2, onset of tonic phase; 3, clonic phase; 4, postconvulsive coma. [C] Generalised absence seizure (petit mal) showing sudden brief episode of 3/s ‘spike and wave’ discharge. [D] Partial seizure with synchronous abnormal discharges in left frontal and temporal regions.
(From Eliasson S G et al. 1978 Neurological pathophysiology, 2nd edn. Oxford University Press, New York.)
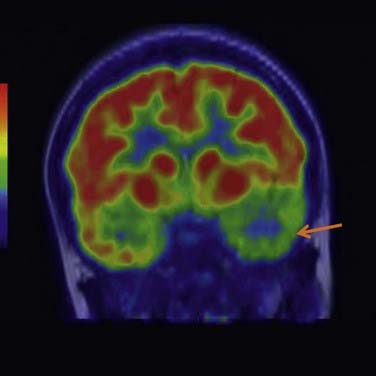
Fig. 44.2 Positron emission tomography (PET) image using [18F]-fluoro-2-deoxyglucose (FDG) of the brain of a female patient suffering from temporal lobe epilepsy.
The interictal area of hypometabolism in the left temporal lobe (indicated by the arrow) is suggestive of the site of the epileptic focus.
(Image kindly provided by Prof. John Duncan and Prof. Peter Ell, UCL Institute of Neurology, London.)
Types of Epilepsy
The clinical classification of epilepsy is done on the basis of the characteristics of the seizure rather than on the cause or underlying pathology. There are two major categories, namely partial and generalised seizures, although there is some overlap and many varieties of each. Either form is classified as simple (if consciousness is not lost) or complex (if consciousness is lost).
Partial Seizures
Partial seizures are those in which the discharge begins locally and often remains localised. The symptoms depend on the brain region or regions involved, and include involuntary muscle contractions, abnormal sensory experiences or autonomic discharge, or effects on mood and behaviour, often termed psychomotor epilepsy. The EEG discharge in this type of epilepsy is normally confined to one hemisphere (Fig. 44.1D). Partial seizures can often be attributed to local cerebral lesions, and their incidence increases with age. In complex partial seizures, loss of consciousness may occur at the outset of the attack, or somewhat later, when the discharge has spread from its site of origin to regions of the brain stem reticular formation. In some individuals, a partial seizure can, during the seizure, develop into a generalised seizure (see below)—referred to as partial seizures with secondary generalisation—when the abnormal neuronal activity spreads across the whole brain.
An epileptic focus in the motor cortex results in attacks, sometimes called jacksonian epilepsy,1 consisting of repetitive jerking of a particular muscle group, beginning on one side of the body, often in the thumb, big toe or angle of the mouth, which spreads and may involve much of the body within about 2 min before dying out. The patient loses voluntary control of the affected parts of the body but does not necessarily lose consciousness. In psychomotor epilepsy, which is often associated with a focus in the temporal lobe, the attack may consist of stereotyped purposive movements such as rubbing or patting movements, or much more complex behaviour such as dressing, walking or hair combing. The seizure usually lasts for a few minutes, after which the patient recovers with no recollection of the event. The behaviour during the seizure can be bizarre and accompanied by a strong emotional response.
Generalised Seizures
Generalised seizures involve the whole brain, including the reticular system, thus producing abnormal electrical activity throughout both hemispheres. Immediate loss of consciousness is characteristic of generalised seizures. There are a number of types of generalised seizure—two important categories are tonic–clonic seizures (formerly referred to as grand mal, Fig. 44.1B) and absence seizures (petit mal, Fig. 44.1C); others include myoclonic, tonic, atonic and clonic seizures.
A tonic–clonic seizure consists of an initial strong contraction of the whole musculature, causing a rigid extensor spasm and an involuntary cry. Respiration stops, and defaecation, micturition and salivation often occur. This tonic phase lasts for about 1 min, during which the face is suffused and becomes blue (an important clinical distinction from syncope, the main disorder from which fits must be distinguished, where the face is ashen pale), and is followed by a series of violent, synchronous jerks that gradually die out in 2–4 min. The patient stays unconscious for a few more minutes and then gradually recovers, feeling ill and confused. Injury may occur during the convulsive episode. The EEG shows generalised continuous high-frequency activity in the tonic phase and an intermittent discharge in the clonic phase (Fig. 44.1B).
Absence seizures occur in children; they are much less dramatic but may occur more frequently (many seizures each day) than tonic–clonic seizures. The patient abruptly ceases whatever he or she was doing, sometimes stopping speaking in mid-sentence, and stares vacantly for a few seconds, with little or no motor disturbance. Patients are unaware of their surroundings and recover abruptly with no after effects. The EEG pattern shows a characteristic rhythmic discharge during the period of the seizure (Fig. 44.1C). The rhythmicity appears to be due to oscillatory feedback between the cortex and the thalamus, the special properties of the thalamic neurons being dependent on the T-type calcium channels that they express (see Shin, 2006). The pattern differs from that of partial seizures, where a high-frequency asynchronous discharge spreads out from a local focus. Accordingly (see below), the drugs used specifically to treat absence seizures act mainly by blocking T-type calcium channels, whereas drugs effective against other types of epilepsy act mainly by blocking sodium channels or enhancing GABA-mediated inhibition.
A particularly severe kind of epilepsy, Lennox–Gastaut syndrome, occurs in children and is associated with progressive mental retardation, possibly a reflection of excitotoxic neurodegeneration (see Ch. 39).
About one-third of cases of epilepsy are familial and involve genetic mutations. While some are due to a single mutation, most result from polygenetic mutations (see Weber & Lerche, 2008). Most genes associated with familial epilepsies encode neuronal ion channels closely involved in controlling action potential generation (see Ch. 4), such as voltage-gated sodium and potassium channels, GABAA receptors and nicotinic acetylcholine receptors. Some other genes encode proteins that interact with ion channels.
Status epilepticus refers to continuous uninterrupted seizures, requiring emergency medical treatment.
Neural Mechanisms and Animal Models of Epilepsy
 The underlying neuronal abnormality in epilepsy is poorly understood. In general, excitation will naturally tend to spread throughout a network of interconnected neurons but is normally prevented from doing so by inhibitory mechanisms. Thus epileptogenesis can arise if excitatory transmission is facilitated or inhibitory transmission is reduced (exemplified by GABAA receptor antagonists causing convulsions; see Ch. 37). In certain respects, epileptogenesis resembles long-term potentiation (Ch. 37), and similar types of use-dependent synaptic plasticity may be involved (see Kulmann et al., 2000). Because detailed studies are difficult to carry out on epileptic patients, many different animal models of epilepsy have been investigated (see Sarkisian, 2001). These include a variety of genetic strains that show epilepsy-like characteristics (e.g. mice that convulse briefly in response to certain sounds, baboons that show photically induced seizures and beagles with an inherited abnormality that closely resembles human epilepsy). Recently, several transgenic mouse strains have been reported that show spontaneous seizures. They include knockout mutations of various ion channels, receptors and other synaptic proteins. Local cortical damage (e.g. by applying aluminium oxide paste or crystals of a cobalt salt) results in focal epilepsy. Local application of penicillin crystals has a similar effect, probably by interfering with inhibitory synaptic transmission. Convulsant drugs such as pentylenetetrazol (PTZ) are often used, particularly in the testing of antiepileptic agents, and seizures caused by electrical stimulation of the whole brain are used for the same purpose. It has been found empirically that drugs that inhibit PTZ-induced convulsions and raise the threshold for production of electrically induced seizures are generally effective against absence seizures, whereas those that reduce the duration and spread of electrically induced convulsions are effective in focal types of epilepsy such as tonic–clonic seizures.
The underlying neuronal abnormality in epilepsy is poorly understood. In general, excitation will naturally tend to spread throughout a network of interconnected neurons but is normally prevented from doing so by inhibitory mechanisms. Thus epileptogenesis can arise if excitatory transmission is facilitated or inhibitory transmission is reduced (exemplified by GABAA receptor antagonists causing convulsions; see Ch. 37). In certain respects, epileptogenesis resembles long-term potentiation (Ch. 37), and similar types of use-dependent synaptic plasticity may be involved (see Kulmann et al., 2000). Because detailed studies are difficult to carry out on epileptic patients, many different animal models of epilepsy have been investigated (see Sarkisian, 2001). These include a variety of genetic strains that show epilepsy-like characteristics (e.g. mice that convulse briefly in response to certain sounds, baboons that show photically induced seizures and beagles with an inherited abnormality that closely resembles human epilepsy). Recently, several transgenic mouse strains have been reported that show spontaneous seizures. They include knockout mutations of various ion channels, receptors and other synaptic proteins. Local cortical damage (e.g. by applying aluminium oxide paste or crystals of a cobalt salt) results in focal epilepsy. Local application of penicillin crystals has a similar effect, probably by interfering with inhibitory synaptic transmission. Convulsant drugs such as pentylenetetrazol (PTZ) are often used, particularly in the testing of antiepileptic agents, and seizures caused by electrical stimulation of the whole brain are used for the same purpose. It has been found empirically that drugs that inhibit PTZ-induced convulsions and raise the threshold for production of electrically induced seizures are generally effective against absence seizures, whereas those that reduce the duration and spread of electrically induced convulsions are effective in focal types of epilepsy such as tonic–clonic seizures.
The kindling model may approximate the human condition more closely than directly evoked seizure models. Low-intensity electrical stimulation of certain regions of the limbic system, such as the amygdala, with implanted electrodes normally produces no seizure response. If a brief period of stimulation is repeated daily for several days, however, the response gradually increases until very low levels of stimulation will evoke a full seizure, and eventually seizures begin to occur spontaneously. Once produced, the kindled state persists indefinitely. This change is prevented by NMDA receptor antagonists, and may involve processes similar to those that cause long-term potentiation of synaptic transmission in the hippocampus (see Ch. 37). In human focal epilepsies, surgical removal of a damaged region of cortex may fail to cure the condition, as though the abnormal discharge from the region of primary damage had somehow produced a secondary hyperexcitability elsewhere in the brain. Furthermore, prophylactic treatment with antiepileptic drugs for 2 years following severe head injury reduces the subsequent incidence of post-traumatic epilepsy, which suggests that a phenomenon similar to kindling may underlie this form of epilepsy.
The kainate model entails a single injection of the glutamate receptor agonist kainic acid into the amygdaloid nucleus of a rat. After transient intense stimulation, spontaneous seizures begin to occur 2–4 weeks later, and then continue indefinitely. It is believed that excitotoxic damage to inhibitory neurons is responsible, associated with structural remodelling of excitatory synaptic connections, changes that may also be a factor in human epilepsies.
Neurons from which the epileptic discharge originates display an unusual type of electrical behaviour termed the paroxysmal depolarising shift (PDS), during which the membrane potential suddenly decreases by about 30 mV and remains depolarised for up to a few seconds before returning to normal. A burst of action potentials often accompanies this depolarisation (Fig. 44.3). This event probably results from the abnormally exaggerated and prolonged action of an excitatory transmitter. Activation of NMDA receptors (see Ch. 37) produces ‘plateau-shaped’ depolarising responses very similar to the PDS, as well as initiating seizure activity. This membrane response occurs because of the voltage-dependent blocking action of Mg2+ on channels operated by NMDA receptors (see Ch. 37). Glutamate must undoubtedly participate in the epileptic discharge, but efforts to develop glutamate antagonists as antiepileptic drugs have met with little success. It is known that repeated seizure activity can lead to neuronal degeneration, possibly due to excitotoxicity (Ch. 39).
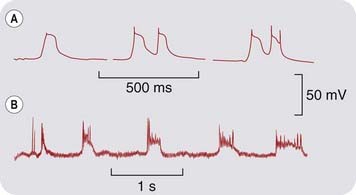
Fig. 44.3 ‘Paroxysmal depolarising shift’ (PDS) compared with experimental activation of glutamate receptors of the NMDA type.
[A] PDS recorded with an intracellular microelectrode from cortical neurons of anaesthetised cats. Seizure activity was induced by topical application of penicillin. [B] Intracellular recording from the caudate nucleus of an anaesthetised cat. The glutamate analogue NMDA was applied by ionophoresis from a nearby micropipette. Note the periodic waves of depolarisation, associated with a burst of action potentials, which closely resemble the PDS.
(From: [A] Matsumoto H, Marsan C A 1964 Exp Neurol 9: 286; [B] Herrling P L et al. 1983 J Physiol 339: 207.)
Studies on experimental epilepsy in the kindling or kainate models have revealed a deficit in various biochemical markers of GABA-mediated inhibitory transmission, and an increase of markers associated with glutamate-mediated excitation (see Jarrott, 1999). Human studies have shown less consistent changes, although studies on brain samples removed at operation suggest that the epileptic focus contains more glutamate than normal; the GABA content is not affected. Potassium-stimulated glutamate release is also increased in the epileptic focus compared with in normal tissue.
Neurotrophins, particularly brain-derived neurotrophic factor (BDNF), may play a role in epileptogenesis. BDNF, which acts on a membrane receptor tyrosine kinase (TrkB; Ch. 3), enhances membrane excitability and also stimulates synapse formation. Deletion of the neurotrophin receptor, TrkB, in mice prevents seizures from developing in the kindling model. Production and release of BDNF is increased in the kindling models, and there is also evidence for its involvement in human epilepsy. Specific blocking agents represent a possible future strategy for treating epilepsy but remain to be identified.
Nature of epilepsy 
Antiepileptic Drugs
Antiepileptic (sometimes known as anticonvulsant) drugs are used to treat epilepsy as well as non-epileptic convulsive disorders.
With optimal drug therapy, epilepsy is controlled completely in about 75% of patients, but about 10% (50 000 in Britain) continue to have seizures at intervals of 1 month or less, which severely disrupts their life and work. There is therefore a need to improve the efficacy of therapy.
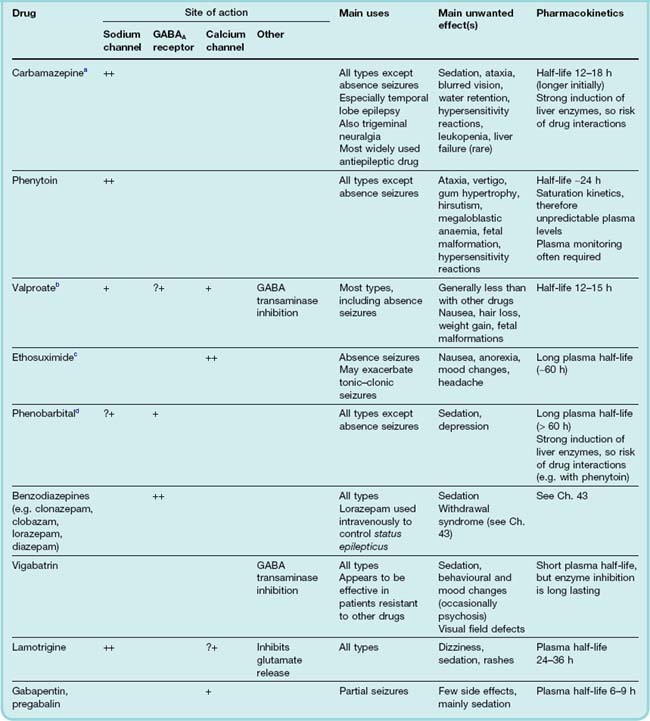
Patients with epilepsy usually need to take drugs continuously for many years, so avoidance of side effects is particularly important. Nevertheless, some drugs that have considerable adverse effects are still quite widely used even though they are not drugs of choice for newly diagnosed patients.2 There is clearly a need for more specific and effective drugs, and a number of new drugs have recently been introduced for clinical use or are in late stages of clinical trials. Long-established antiepileptic drugs (see Table 44.1) include phenytoin, carbamazepine, valproate, ethosuximide and phenobarbital, together with various benzodiazepines, such as diazepam, clonazepam and clobazam. Newer drugs in current use include vigabatrin, gabapentin, pregabalin, lamotrigine, felbamate, tiagabine, topiramate, levetiracetam, oxcarbazepine, zonisamide and rufinamide. Those new drugs in late stages of development that have novel mechanisms of action are described briefly towards the end of this section. The number of new antiepileptic drugs reflects the efforts being made to improve on the far from ideal properties of the earlier drugs. In general, the newer drugs are less likely to interact pharmacokinetically with other drugs (see Ch. 56) and have fewer adverse effects. The appropriate use of drugs from this large available menu depends on many clinical factors (for recent clinical use updates, see Macleod & Appleton, 2007; Azar & Abou-Khalil, 2008).
Mechanism of Action
Antiepileptic drugs aim to inhibit the abnormal neuronal discharge rather than to correct the underlying cause. Three main mechanisms of action appear to be important (see Rogawski & Löscher, 2004a):
Antiepileptic drugs may exert more than one beneficial action, prime examples being valproate and topiramate (see Table 44.1). The relative importance and contribution of each of these actions to the therapeutic effect is somewhat uncertain.
As with drugs used to treat cardiac dysrhythmias (Ch. 21), the aim is to prevent the paroxysmal discharge without affecting normal transmission. It is clear that properties such as use-dependence and voltage-dependence of channel-blocking drugs (see Ch. 4) are important in achieving this selectivity, but our understanding remains fragmentary.
Enhancement of GABA action
Several antiepileptic drugs (e.g. phenobarbital and benzodiazepines) enhance the activation of GABAA receptors, thus facilitating the GABA-mediated opening of chloride channels (see Chs 3 and 43).3 Vigabatrin acts by irreversibly inhibiting the enzyme GABA transaminase located within astrocytes, which is responsible for inactivating GABA (see Ch. 37), and tiagabine inhibits GABA uptake into neurons and glial cells, producing an increase in the extracellular concentration of GABA, and enhancing its action as an inhibitory transmitter. Gabapentin was designed as a brain penetrating agonist at GABAA receptors, but ironically was found to be an effective antiepileptic drug, not by affecting GABA receptors or the transporter, but by acting on calcium channels (see below).
Inhibition of sodium channel function
A large number of antiepileptic drugs (see Table 44.1) affect membrane excitability by an action on voltage-dependent sodium channels (see Chs 4 and 42), which carry the inward membrane current necessary for the generation of an action potential. Their blocking action shows the property of use-dependence; in other words, they block preferentially the excitation of cells that are firing repetitively, and the higher the frequency of firing, the greater the block produced. This characteristic, which is relevant to the ability of drugs to block the high-frequency discharge that occurs in an epileptic fit without unduly interfering with the low-frequency firing of neurons in the normal state, arises from the ability of blocking drugs to discriminate between sodium channels in their resting, open and inactivated states (see Chs 4 and 42). Depolarisation of a neuron (such as occurs in the PDS described above) increases the proportion of the sodium channels in the inactivated state. Antiepileptic drugs bind preferentially to channels in this state, preventing them from returning to the resting state, and thus reducing the number of functional channels available to generate action potentials.
Inhibition of calcium channels
Drugs that are effective against absence seizures (ethosuximide, valproate, clonazepam) all appear to share the ability to block T-type low-voltage-activated calcium channels. T-type channel activity is important in determining the rhythmic discharge of thalamic neurons associated with absence seizures (Khosravani et al., 2004).
Gabapentin, though designed as a simple analogue of GABA that would be sufficiently lipid soluble to penetrate the blood–brain barrier, owes its antiepileptic effect mainly to an action on P/Q-type calcium channels. By binding to a particular channel subunit (α2δ1), it reduces the trafficking to the plasma membrane of calcium channels containing this subunit, thereby reducing calcium entry into the nerve terminals and reducing the release of various neurotransmitters and modulators.
Other mechanisms
Many of the newer antiepileptic drugs were developed empirically on the basis of activity in animal models. Their mechanism of action at the cellular level is not fully understood.4
Levetiracetam appears to act in a manner different from all other antiepileptic drugs, its target being a synaptic vesicle protein involved in neurotransmitter release (see below).
While a drug may appear to work by one of the major mechanisms described above, close scrutiny often reveals other actions that may also be therapeutically relevant. For example, phenytoin not only causes use-dependent block of sodium channels (see above) but also affects other aspects of membrane function, including calcium channels and post-tetanic potentiation, as well as intracellular protein phosphorylation by calmodulin-activated kinases, which could also interfere with membrane excitability and synaptic function.
One theme, which has become familiar in earlier chapters in the central nervous system section of this book, is that antagonists at ionotropic excitatory amino acid receptors have not, despite showing efficacy in animal models, proved useful in the clinic, because the margin between the desired anticonvulsant effect and unacceptable side effects, such as loss of motor coordination, is too narrow.
Mechanism of action of antiepileptic drugs
Carbamazepine
Carbamazepine, one of the most widely used antiepileptic drugs, is chemically derived from the tricyclic antidepressant drugs (see Ch. 46) and was found in a routine screening test to inhibit electrically evoked seizures in mice. Pharmacologically and clinically, its actions resemble those of phenytoin, although it appears to be particularly effective in treating certain partial seizures (e.g. psychomotor epilepsy). It is also used to treat other conditions, such as neuropathic pain (Ch. 41) and manic-depressive illness (Ch. 46).
Pharmacokinetic aspects
Carbamazepine is slowly but well absorbed after oral administration. Its plasma half-life is about 30 h when it is given as a single dose, but it is a strong inducer of hepatic enzymes, and the plasma half-life shortens to about 15 h when it is given repeatedly. Some of its metabolites have antiepileptic properties. A slow-release preparation is used for patients who experience transient side effects coinciding with plasma concentration peaks following oral dosing (see below).
Unwanted effects
Carbamazepine produces a variety of unwanted effects ranging from drowsiness, dizziness and ataxia to more severe mental and motor disturbances. It can also cause water retention (and hence hyponatraemia; Ch. 28) and a variety of gastrointestinal and cardiovascular side effects. The incidence and severity of these effects is relatively low, however, compared with other drugs. Treatment is usually started with a low dose, which is built up gradually to avoid dose-related toxicity. Severe bone marrow depression, causing neutropenia, and other severe forms of hypersensitivity reaction can occur, especially in people of Asian origin (see Ch. 11).
Carbamazepine is a powerful inducer of hepatic microsomal enzymes, and thus accelerates the metabolism of many other drugs, such as phenytoin, oral contraceptives, warfarin and corticosteroids. In general, it is inadvisable to combine it with other antiepileptic drugs. Oxcarbazepine is a prodrug that is metabolised to a compound closely resembling carbamazepine, with similar actions but less tendency to induce drug-metabolising enzymes. Another related drug, eslicarbazepine, is in development and may also have less effect on metabolising enzymes.
Phenytoin
Phenytoin is the most important member of the hydantoin group of compounds, which are structurally related to the barbiturates. It is highly effective in reducing the intensity and duration of electrically induced convulsions in mice, although ineffective against PTZ-induced convulsions. Despite its many side effects and unpredictable pharmacokinetic behaviour, phenytoin is widely used, being effective against various forms of partial and generalised seizures, although not against absence seizures, which may even get worse.
Pharmacokinetic aspects
Phenytoin has certain pharmacokinetic peculiarities that need to be taken into account when it is used clinically. It is well absorbed when given orally, and about 80–90% of the plasma content is bound to albumin. Other drugs, such as salicylates, phenylbutazone and valproate, inhibit this binding competitively (see Ch. 56). This increases the free phenytoin concentration but also increases hepatic clearance of phenytoin, so may enhance or reduce the effect of the phenytoin in an unpredictable way. Phenytoin is metabolised by the hepatic mixed function oxidase system and excreted mainly as glucuronide. It causes enzyme induction, and thus increases the rate of metabolism of other drugs (e.g. oral anticoagulants). The metabolism of phenytoin itself can be either enhanced or competitively inhibited by various other drugs that share the same hepatic enzymes. Phenobarbital produces both effects, and because competitive inhibition is immediate whereas induction takes time, it initially enhances and later reduces the pharmacological activity of phenytoin. Ethanol has a similar dual effect.
The metabolism of phenytoin shows the characteristic of saturation (see Ch. 10), which means that over the therapeutic plasma concentration range the rate of inactivation does not increase in proportion to the plasma concentration. The consequences of this are that:
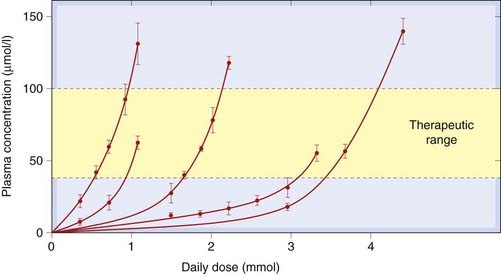
Fig. 44.4 Non-linear relationship between daily dose of phenytoin and steady-state plasma concentration in five individual human subjects.
The daily dose required to achieve the therapeutic range of plasma concentrations (40–100 µmol/l) varies greatly between individuals, and for any one individual the dose has to be adjusted rather precisely to keep within the acceptable plasma concentration range.
(Redrawn from Richens A, Dunlop A, 1975 Lancet 2: 247.)
The range of plasma concentration over which phenytoin is effective without causing excessive unwanted effects is quite narrow (approximately 40–100 µmol/l). The very steep relationship between dose and plasma concentration, and the many interacting factors, mean that there is considerable individual variation in the plasma concentration achieved with a given dose. A radioimmunoassay for phenytoin in plasma is available, and regular monitoring of plasma concentration has helped considerably in achieving an optimal therapeutic effect. The past tendency was to add further drugs in cases where a single drug failed to give adequate control. It is now recognised that much of the unpredictability can be ascribed to pharmacokinetic variability, and regular monitoring of plasma concentration has reduced the use of polypharmacy.
Unwanted effects
Side effects of phenytoin begin to appear at plasma concentrations exceeding 100 µmol/l and may be severe above about 150 µmol/l. The milder side effects include vertigo, ataxia, headache and nystagmus, but not sedation. At higher plasma concentrations, marked confusion with intellectual deterioration occurs; a paradoxical increase in seizure frequency is a particular trap for the unwary prescriber. These effects occur acutely and are quickly reversible. Hyperplasia of the gums often develops gradually, as does hirsutism and coarsening of the features, which probably result from increased androgen secretion. Megaloblastic anaemia, associated with a disorder of folate metabolism, sometimes occurs, and can be corrected by giving folic acid (Ch. 25). Hypersensitivity reactions, mainly rashes, are quite common. Phenytoin has also been implicated as a cause of the increased incidence of fetal malformations in children born to epileptic mothers, particularly the occurrence of cleft palate, associated with the formation of an epoxide metabolite. Severe idiosyncratic reactions, including hepatitis, skin reactions and neoplastic lymphocyte disorders, occur in a small proportion of patients.
Valproate
Valproate is a simple monocarboxylic acid, chemically unrelated to any other class of antiepileptic drug, and in 1963 it was discovered quite accidentally to have anticonvulsant properties in mice. It inhibits most kinds of experimentally induced convulsions and is effective in many kinds of epilepsy, being particularly useful in certain types of infantile epilepsy, where its low toxicity and lack of sedative action are important, and in adolescents who exhibit both tonic–clonic or myoclonic seizures as well as absence seizures, because valproate (unlike most antiepileptic drugs) is effective against each. Like carbamazepine, valproate is also used in psychiatric conditions such as bipolar depressive illness (Ch. 46).
Valproate works by several mechanisms, the relative importance of which remains to be clarified. It causes a significant increase in the GABA content of the brain and is a weak inhibitor of two enzyme systems that inactivate GABA, namely GABA transaminase and succinic semialdehyde dehydrogenase, but in vitro studies suggest that these effects would be very slight at clinical dosage. Other more potent inhibitors of these enzymes (e.g. vigabatrin; see below) also increase GABA content and have an anticonvulsant effect in experimental animals. There is some evidence that it enhances the action of GABA by a postsynaptic action, but no clear evidence that it affects inhibitory synaptic responses. It inhibits sodium channels, but less so than phenytoin, and inhibits T-type calcium channels which might explain why it is effective against absence seizures.
Valproate is well absorbed orally and excreted, mainly as the glucuronide, in the urine, the plasma half-life being about 15 h.
Unwanted effects
Valproate causes thinning and curling of the hair in about 10% of patients. The most serious side effect is hepatotoxicity. An increase in plasma glutamic oxaloacetic transaminase, which signals liver damage of some degree, commonly occurs, but proven cases of valproate-induced hepatitis are rare. The few cases of fatal hepatitis in valproate-treated patients may well have been caused by other factors. Valproate is teratogenic, causing spina bifida and other neural tube defects.
Ethosuximide
Ethosuximide is another drug developed empirically by modifying the barbituric acid ring structure. Pharmacologically and clinically, however, it is different from the drugs so far discussed, in that it is active against PTZ-induced convulsions in animals and against absence seizures in humans, with little or no effect on other types of epilepsy. It supplanted trimethadione, the first drug found to be effective in absence seizures, which had major side effects. Ethosuximide is used clinically for its selective effect on absence seizures.
The mechanism of action of ethosuximide and trimethadione appears to differ from that of other antiepileptic drugs. The main effect is inhibition of T-type calcium channels, which may play a role in generating the 3/second firing rhythm in thalamic relay neurons that is characteristic of absence seizures.
Ethosuximide is well absorbed, and metabolised and excreted much like phenobarbital, with a plasma half-life of about 60 h. Its main side effects are nausea and anorexia, sometimes lethargy and dizziness, and it is said to precipitate tonic–clonic seizures in susceptible patients. Very rarely, it can cause severe hypersensitivity reactions.
Phenobarbital
Phenobarbital was one of the first barbiturates to be developed, and its antiepileptic properties were recognised in 1912. In its action against experimentally induced convulsions and clinical forms of epilepsy, it closely resembles phenytoin; it affects the duration and intensity of artificially induced seizures, rather than the seizure threshold, and is (like phenytoin) ineffective in treating absence seizures. Primidone, now rarely used, acts by being metabolised to phenobarbital. It often causes hypersensitivity reactions. The clinical uses of phenobarbital are virtually the same as those of phenytoin, although phenytoin is preferred because of the absence of sedation. It is now seldom used clinically because of sedation. For some years, it was widely used in children, including as prophylaxis following febrile convulsions in infancy, but it can cause behavioural disturbances and hyperkinesias. It is, however, widely used in veterinary practice.
Pharmacokinetic aspects
Phenobarbital is well absorbed, and about 50% of the drug in the blood is bound to plasma albumin. It is eliminated slowly from the plasma (half-life 50–140 h). About 25% is excreted unchanged in the urine. Because phenobarbital is a weak acid, its ionisation and hence renal elimination are increased if the urine is made alkaline (see Ch. 9). The remaining 75% is metabolised, mainly by oxidation and conjugation, by hepatic microsomal enzymes. Phenobarbital is a powerful inducer of liver CYP enzymes, and it lowers the plasma concentration of several other drugs (e.g. steroids, oral contraceptives, warfarin, tricyclic antidepressants) to an extent that is clinically important.
Unwanted effects
The main unwanted effect of phenobarbital is sedation, which often occurs at plasma concentrations within the therapeutic range for seizure control. This is a serious drawback, because the drug may have to be used for years on end. Some degree of tolerance to the sedative effect seems to occur, but objective tests of cognition and motor performance show impairment even after long-term treatment. Other unwanted effects that may occur with clinical dosage include megaloblastic anaemia (similar to that caused by phenytoin), mild hypersensitivity reactions and osteomalacia. Like other barbiturates, it must not be given to patients with porphyria (see Ch. 56). In overdose, phenobarbital produces coma and respiratory and circulatory failure, as do all barbiturates.
Benzodiazepines
Benzodiazepines can be used to treat both acute seizures, especially in children—diazepam often being administered rectally and status epilepticus (a life-threatening condition in which epileptic seizures occur almost without a break) for which agents such as diazepam, lorazepam or clonazepam are administered intravenously. The advantage in status epilepticus is that they act very rapidly compared with other antiepileptic drugs. With most benzodiazepines (see Ch. 43), the sedative effect is too pronounced for them to be used for maintenance therapy and tolerance develops over 1–6 months. Clonazepam is unique among the benzodiazepines in that in addition to acting at the GABAA receptor, it also inhibits T-type calcium channels. Both it and the related compound clobazam are claimed to be relatively selective as antiepileptic drugs. Sedation is the main side effect of these compounds, and an added problem may be the withdrawal syndrome, which results in an exacerbation of seizures if the drug is stopped abruptly.
Newer Antiepileptic Drugs
Vigabatrin
Vigabatrin, the first ‘designer drug’ in the epilepsy field, is a vinyl-substituted analogue of GABA that was designed as an inhibitor of the GABA-metabolising enzyme GABA transaminase. Vigabatrin is extremely specific for this enzyme and works by forming an irreversible covalent bond. In animal studies, vigabatrin increases the GABA content of the brain and also increases the stimulation-evoked release of GABA, implying that GABA transaminase inhibition can increase the releasable pool of GABA and effectively enhance inhibitory transmission. In humans, vigabatrin increases the content of GABA in the cerebrospinal fluid. Although its plasma half-life is short, it produces a long-lasting effect because the enzyme is blocked irreversibly, and the drug can be given by mouth once daily.
Vigabatrin has been reported to be effective in a substantial proportion of patients resistant to the established drugs. However, a drawback of vigabatrin is the development of peripheral visual field defect in a proportion of patients on long-term therapy. Therefore the benefit of using this drug in refractory epilepsy must be weighed against the potential risk of developing visual problems. Vigabatrin may cause depression, and occasionally psychotic disturbances and hallucinations, in a minority of patients.
Lamotrigine
Lamotrigine, although chemically unrelated, resembles phenytoin and carbamazepine in its pharmacological effects, acting on sodium channels as well as possibly calcium channels and inhibiting the release of excitatory amino acids. It appears that, despite its similar mechanism of action, lamotrigine has a broader therapeutic profile than the earlier drugs, with significant efficacy against absence seizures (it is also used to treat unrelated psychiatric disorders). Its main side effects are nausea, dizziness and ataxia, and hypersensitivity reactions (mainly mild rashes, but occasionally more severe). Its plasma half-life is about 24 h, with no particular pharmacokinetic anomalies, and it is taken orally.
Felbamate
Felbamate is an analogue of an obsolete anxiolytic drug, meprobamate. It is active in many animal seizure models and has a broader clinical spectrum than earlier antiepileptic drugs, but its mechanism of action at the cellular level is uncertain. It has only a weak effect on sodium channels and some effect on GABA, but causes some block of the NMDA receptor channel (Ch. 37). Its acute side effects are mild, mainly nausea, irritability and insomnia, but it occasionally causes severe reactions resulting in aplastic anaemia or hepatitis. For this reason, its recommended use is limited to intractable epilepsy (e.g. in children with Lennox–Gastaut syndrome) that is unresponsive to other drugs. Its plasma half-life is about 24 h, and it can enhance the plasma concentration of other antiepileptic drugs given concomitantly. Carisbamate, a new drug currently in clinical trials, was designed with the intention of producing a drug similar to felbamate that does not cause aplastic anaemia.
Gabapentin and Pregabalin
Gabapentin is effective against partial seizures. Its side effects (mainly sedation and ataxia) are less severe than with many antiepileptic drugs. The absorption of gabapentin from the intestine depends on the L-amino acid carrier system and shows the property of saturability, which means that increasing the dose does not proportionately increase the amount absorbed. This makes gabapentin relatively safe and free of side effects associated with overdosing. Its plasma half-life is about 6 h, requiring dosing two to three times daily. It is free of interactions with other drugs. It is also used as an analgesic to treat neuropathic pain (Ch. 41). Pregabalin, an analogue of gabapentin, is more potent but otherwise very similar. As these drugs are excreted unchanged in the urine they must be used with care in patients whose renal function is impaired.
Tiagabine
Tiagabine is an analogue of GABA that is able to penetrate the blood–brain barrier. It is an equipotent inhibitor of both neuronal and glial GABA transporter GAT1, thus inhibiting the removal of GABA from the synapse. It enhances the extracellular GABA concentration, as measured in microdialysis experiments, and also potentiates and prolongs GABA-mediated synaptic responses in the brain. It has a short plasma half-life, and its main side effects are drowsiness and confusion. Tiagabine is mainly used as an add-on therapy for partial seizures.
Topiramate
Topiramate is a recently introduced drug that, mechanistically, appears to do a little of everything, blocking sodium and calcium channels, enhancing the action of GABA, blocking AMPA receptors and, for good measure, weakly inhibiting carbonic anhydrase. Its spectrum of action resembles that of phenytoin, and it is claimed to produce less severe side effects, as well as being devoid of the pharmacokinetic properties that cause trouble with phenytoin. Its main drawback is that (like many antiepileptic drugs) it is teratogenic in animals, so it should not be used in women of child-bearing age (see below). Currently, it is mainly used as add-on therapy in refractory cases of epilepsy.
Levetiracetam
Levetiracetam was developed as an analogue of piracetam, a drug used to improve cognitive function, and discovered by accident to have antiepileptic activity in animal models. Unusually, it lacks activity in conventional models such as electroshock and PTZ tests, but is effective in the audiogenic and kindling models. It is believed to interfere with neurotransmitter release by binding to synaptic vesicle protein 2A (SV2A), a protein thought to be involved in synaptic vesicle docking and fusion. Brivaracetam, a related antiepileptic agent, also binds to SV2A with ten-fold higher affinity. Levetiracetam is excreted unchanged in the urine.
Zonisamide
Zonisamide is a sulfonamide compound originally intended as an antibacterial drug and found accidentally to have antiepileptic properties. It is believed to act by blocking sodium channels and T-type calcium channels but may well have other effects such as enhancing GABA function. It is free of major unwanted effects, although it causes drowsiness, and of serious interaction with other drugs. It tends to suppress appetite and cause weight loss, and is sometimes used for this purpose. Zonisamide has a long plasma half-life of 60–80 h, and is partly excreted unchanged and partly converted to a glucuronide metabolite. It is licensed for use as an adjunct treatment of partial and generalised seizures but may be effective as a monotherapy.
Rufinamide
Rufinamide is a triazole derivative structurally unrelated to other antiepileptic drugs. It appears to act by enhancing sodium channel inactivation and may also inhibit GABA reuptake. It is licensed for treating Lennox–Gastaut syndrome and may also be effective in partial seizures. It has low plasma protein binding and is not metabolised by CYP enzymes.
Stiripentol
Stiripentol has some efficacy as an adjunctive therapy in children. It enhances GABA release and prolongs GABA-mediated synaptic events in a manner similar to phenobarbital.
The major antiepileptic drugs
The main drugs in current use are carbamazepine, phenytoin, valproate, ethosuximide and benzodiazepines.
Development of New Drugs
There are a number of new antiepileptic agents currently being evaluated in clinical trials (see Bialer et al., 2009). Several of these appear to act by novel mechanisms. Retigabine is an activator of neuronal KCNQ (Kv7) potassium channels that underlie the M current which controls membrane excitability. It also appears to be effective in treating some pain states. Lacosamide may enhance sodium channel inactivation, but unlike other antiepileptic drugs it appears to affect slow rather than rapid inactivation processes. Ganaxolone, structurally resembling endogenous neurosteroids (see Ch. 37), is a positive allosteric modulator of GABAA receptors containing δ subunits (see Ch. 37). Tonabersat is a neuronal gap junction inhibitor.
Novel targets for new antiepileptic agents are discussed by Meldrum & Rogawski (2007). The identification of epileptogenic mutations of genes encoding specific ion channels and other functional proteins (see Weber & Lerche, 2008) is expected to lead to new drugs aimed at these potential targets—a field to watch.
Other Uses of Antiepileptic Drugs
Antiepileptic drugs have proved to have much wider clinical applications than was originally envisaged, and clinical trials have shown many of them to be effective in the following conditions:
This surprising multiplicity of clinical indications may reflect the fact that similar neurobiological mechanisms, involving synaptic plasticity and increased excitability of interconnected populations of neurons, underlie each of these disorders (see Rogawski & Löscher, 2004b).
Clinical uses of antiepileptic drugs 
Antiepileptic Drugs and Pregnancy
There are several important implications for women taking antiepileptic drugs. By inducing hepatic CYP3A4 enzymes, some antiepileptic drugs may increase oral contraceptive metabolism, thus reducing their effectiveness. Taken during pregnancy, drugs such as phenytoin, carbamazepine, lamotrogine, topiramate and valproate are thought to produce teratogenic effects. It remains to be clarified if newer agents also have this problem. Induction of CYP enzymes may result in vitamin K deficiency in the newborn (Ch. 24). Phenytoin, valproate and topiramate may also induce fetal abnormalities if taken during pregnancy.
Muscle Spasm and Muscle Relaxants
Many diseases of the brain and spinal cord produce an increase in muscle tone, which can be painful and disabling. Spasticity resulting from birth injury or cerebral vascular disease, and the paralysis produced by spinal cord lesions, are examples. Multiple sclerosis is a neurodegenerative disease that is triggered by inflammatory attack of the CNS. When the disease has progressed for some years it can cause muscle stiffness and spasms as well as other symptoms such as pain, fatigue, difficulty passing urine and tremors. Local injury or inflammation, as in arthritis, can also cause muscle spasm, and chronic back pain is also often associated with local muscle spasm.
Certain centrally acting drugs are available that have the effect of reducing the background tone of the muscle without seriously affecting its ability to contract transiently under voluntary control. The distinction between voluntary movements and ‘background tone’ is not clear-cut, and the selectivity of those drugs is not complete. Postural control, for example, is usually jeopardised by centrally acting muscle relaxants. Furthermore, drugs that affect motor control generally produce rather widespread effects on the central nervous system, and drowsiness and confusion turn out to be very common side effects of these agents. The main groups of drugs that have been used to control muscle tone are:
Baclofen (see Ch. 37) is a chlorophenyl derivative of GABA originally prepared as a lipophilic GABA-like agent in order to assist penetration of the blood–brain barrier, which is impermeable to GABA itself. Baclofen is a selective agonist at GABAB receptors (see Ch. 37). The antispastic action of baclofen is exerted mainly on the spinal cord, where it inhibits both monosynaptic and polysynaptic activation of motor neurons. It is effective when given by mouth, and is used in the treatment of spasticity associated with multiple sclerosis or spinal injury. However, it is ineffective in cerebral spasticity caused by birth injury.
Baclofen produces various unwanted effects, particularly drowsiness, motor incoordination and nausea, and it may also have behavioural effects. It is not useful in epilepsy.
Tizanidine is an α2 adrenoceptor agonist that relieves spasticity associated with multiple sclerosis and spinal cord injury.
Anecdotal evidence suggests that smoking cannabis (Ch. 18) relieves the painful muscle spasms associated with multiple sclerosis. A full-scale controlled trial of Δ9-tetrahydrocannabinol (also known as THC or dronabinol; see Ch. 18), however, showed no significant effect on muscle spasm, tremor, bladder control or disability, although the patients reported subjective improvements (Zajicek et al., 2003). More recently a number of different cannabinoids have been tested, including a 1 : 1 mixture of THC and cannabidiol (sativex), and nabilone. Such studies suggest that cannabinoids may be of limited use in some individuals suffering from multiple sclerosis.
References and Further Reading
Browne T.R., Holmes G.L. Handbook of epilepsy. Philadelphia: Lippincott, Williams & Wilkins; 2008. (A compact textbook covering most areas of epilepsy and its treatment)
Engel J.E., Pedley T.A., editors. Epilepsy: a comprehensive textbook, second ed, Vol. I–III. Philadelphia: Wolters Kluwer/Lippincott, Williams and Wilkins, 2007. (At just over 3000 pages, this three-volume set provides a comprehensive coverage of all aspects of epilepsy)
Hart Y.M., Sander J.W. Epilepsy questions and answers. Weybridge: Merit Publishing International; 2008. (The question-and-answer format of this book makes obtaining specific information relatively easy)
Pathogenesis and types of epilepsy
Deblaere K., Achten E. Structural magnetic resonance imaging in epilepsy. Eur. Radiol.. 2008;18:119-129. (Describes the use of brain imaging in the diagnosis of epilepsy)
Jarrott B. Epileptogenesis: biochemical aspects. Handb. Exp. Pharmacol.. 1999;138:87-121. (Review article describing possible neurochemical mechanisms underlying epilepsy—mostly speculative)
Khosravani H., Altier C., Simms B., et al. Gating effects of mutations in the Cav3.2 T-type calcium channel associated with childhood absence epilepsy. J. Biol. Chem.. 2004;279:9681-9684. (Study showing that calcium channel mutations seen in childhood absence seizures cause abnormal neuronal discharges in transgenic mice)
Kulmann D.M., Asztely F., Walker M.C. The role of mammalian ionotropic receptors in synaptic plasticity: LTP, LTD and epilepsy. Cell. Mol. Life. Sci.. 2000;57:1551-1561. (Draws parallels between epileptogenesis and other well-studied forms of synaptic plasticity)
Manning J.-P.A., Richards D.A., Bowery N.G. Pharmacology of absence epilepsy. Trends Pharmacol. Sci.. 2003;24:542-549. (Short review emphasising the difference between absence seizures and other types of epilepsy)
Sarkisian M.R. Overview of the current animal models for human seizure and epileptic disorders. Epilepsy Behav.. 2001;2:201-216.
Shin H.-S. T-type Ca2+ channels and absence epilepsy. Cell Calcium. 2006;40:191-196.
Weber Y.G., Lerche H. Genetic mechanisms in idiopathic epilepsies. Dev. Med. Child Neurol.. 2008;50:648-654. (Reviews how mutations in voltage- and ligand-gated ion channels are associated with idiopathic epilepsy syndromes)
Azar N.J., Abou-Khalil B.W. Considerations in the choice of an antiepileptic drug in the treatment of epilepsy. Semin. Neurol.. 2008;28:305-316. (Describes the current Food and Drug Administration approval for antiepileptic drug use in the USA)
Bialer M., Johannessen S.I., Levy R.H., et al. Progress report on new antiepileptic drugs: a summary of the Ninth Eilat Conference (EILAT IX). Epilepsy Res.. 2009;83:1-43. (Provides an update on new drugs currently in late stages of development)
Macleod S., Appleton R.E. The new antiepileptic drugs. Arch. Dis. Child. Educ. Pract. Ed.. 2007;92:182-188. (Focuses on the clinical usefulness of newer antiepileptic drugs)
Meldrum B.S., Rogawski M.A. Molecular targets for antiepileptic drug development. Neurotherapeutics. 2007;4:18-61. (Review of novel targets for antiepileptic drugs)
Rogawski M.A., Löscher W. The neurobiology of antiepileptic drugs. Nat. Rev. Neurosci.. 2004;5:553-564. (Good general review article covering basic pharmacology and mechanisms of action of the major antiepileptic drugs)
Rogawski M.A., Löscher W. The neurobiology of antiepileptic drugs for the treatment of nonepileptic conditions. Nat. Med.. 2004;10:685-692.
Zajicek J., Fox P., Sanders H., et al. Cannabinoids for treatment of spasticity and other symptoms related to multiple sclerosis: multi-centre randomised placebo-controlled trial. Lancet. 2003;362:1517-1526. (Full-scale trial showing very limited efficacy of cannabinoids in multiple sclerosis)
1After Hughlings Jackson, a distinguished 19th-century Yorkshire neurologist who published his outstanding work in the Annals of the West Riding Lunatic Asylum.
2Bromide was the first antiepileptic agent. Its propensity to induce sedation and other unwanted side effects has resulted in it being largely withdrawn from human medicine, although it is still approved for human use in some countries (e.g. Germany) and may have uses in childhood epilepsies. It is still widely used in veterinary practice to treat epilepsy in dogs and cats.
3Absence seizures, paradoxically, are often exacerbated by drugs that enhance GABA activity (see Manning et al., 2003) and better treated by drugs acting by different mechanisms such as T-type calcium channel inhibition.
4The highly complex actions of current antiepileptic drugs are apt to make discouraging reading for those engaged in trying to develop new drugs on simple rational principles. Serendipity, not science, appears to be the path to therapeutic success.