4 How drugs act
Cellular aspects—excitation, contraction and secretion
Overview
The link between a drug interacting with a molecular target and its effect at the pathophysiological level, such as a change in blood glucose concentration or the shrinkage of a tumour, involves events at the cellular level. Whatever their specialised physiological function, cells generally share much the same repertoire of signalling mechanisms. In the next three chapters, we describe the parts of this repertoire that are of particular significance in understanding drug action at the cellular level. In this chapter, we describe mechanisms that operate mainly over a short timescale (milliseconds to hours), particularly excitation, contraction and secretion, which account for many physiological responses; Chapter 5 deals with the slower processes (generally days to months), including cell division, growth, differentiation and cell death, that determine the body’s structure and constitution; Chapter 6 describes host defence mechanisms.
The short-term regulation of cell function depends mainly on the following components and mechanisms, which regulate, or are regulated by, the free concentration of Ca2+ in the cytosol, [Ca2+]i:
More detailed coverage of the topics presented in this chapter can be found in Nicholls et al. (2001), Levitan & Kaczmarek (2002) and Nestler et al. (2008).
Because [Ca2+]i plays such a key role in cell function, a wide variety of drug effects results from interference with one or more of these mechanisms. If love makes the human world go round, [Ca2+]i does the same for cells. Knowledge of the molecular and cellular details is extensive, and here we focus on the aspects that help to explain drug effects.
Regulation of Intracellular Calcium
Ever since the famous accident by Sidney Ringer’s technician, which showed that using tap water rather than distilled water to make up the bathing solution for isolated frog hearts would allow them to carry on contracting, the role of Ca2+ as a major regulator of cell function has never been in question. Many drugs and physiological mechanisms operate, directly or indirectly, by influencing [Ca2+]i. Here we consider the main ways in which it is regulated, and later we describe some of the ways in which [Ca2+]i controls cell function. Details of the molecular components and drug targets are presented in Chapter 3, and descriptions of drug effects on integrated physiological function are given in later chapters.
The study of Ca2+ regulation took a big step forward in the 1970s with the development of optical techniques based on the Ca2+-sensitive photoprotein aequorin, and fluorescent dyes such as Fura-2, which, for the first time, allowed free [Ca2+]i to be continuously monitored in living cells with a high level of temporal and spatial resolution.
Most of the Ca2+ in a resting cell is sequestered in organelles, particularly the endoplasmic or sarcoplasmic reticulum (ER or SR) and the mitochondria, and the free [Ca2+]i is kept to a low level, about 10−7 M. The Ca2+ concentration in tissue fluid, [Ca2+]o, is about 2.4 mM, so there is a large concentration gradient favouring Ca2+ entry. [Ca2+]i is kept low (a) by the operation of active transport mechanisms that eject cytosolic Ca2+ through the plasma membrane and pump it into the ER, and (b) by the normally low Ca2+ permeability of the plasma and ER membranes. Regulation of [Ca2+]i involves three main mechanisms:
These mechanisms are described in more detail below and are summarised in Figure 4.1 (see reviews by Clapham, 2007; Berridge, 2009).
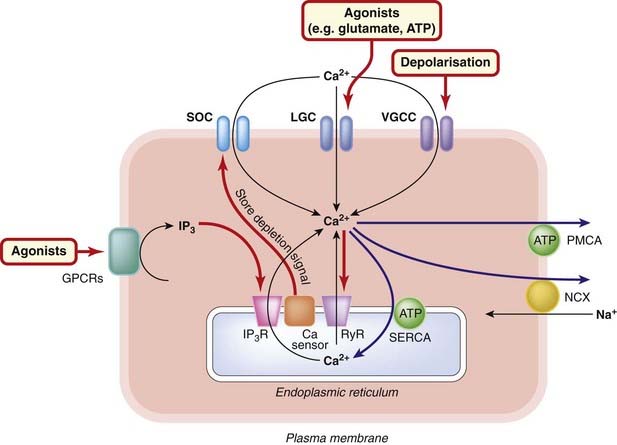
Fig. 4.1 Regulation of intracellular calcium.
The main routes of transfer of Ca2+ into, and out of, the cytosol and endoplasmic reticulum are shown for a typical cell (see text for details). Black arrows: routes into the cytosol. Blue arrows: routes out of the cytosol. Red arrows: regulatory mechanisms. The state of the ER store of Ca2+ is monitored by the sensor protein Stim1, which interacts directly with the store-operated calcium channel (SOC) to promote Ca2+ entry when the ER store is depleted. Normally, [Ca2+]i is regulated to about 10−7 mol/l in a ‘resting’ cell. Mitochondria (not shown) also function as Ca2+ storage organelles but release Ca2+ only under pathological conditions, such as ischaemia (see text). There is also evidence for an intracellular store (not shown) activated by the second messenger nicotinic acid dinucleotide phosphate. GPCR, G-protein-coupled receptor; IP3, inositol trisphosphate; IP3R, inositol trisphosphate receptor; LGC, ligand-gated cation channel; NCX, Na+–Ca2+ exchange transporter; PMCA, plasma membrane Ca2+-ATPase; RyR, ryanodine receptor; SERCA, sarcoplasmic/endoplasmic reticulum ATPase; VGCC, voltage-gated calcium channel.
Calcium Entry Mechanisms
There are four main routes by which Ca2+ enters cells across the plasma membrane:
Voltage-Gated Calcium Channels
The pioneering work of Hodgkin and Huxley on the ionic basis of the nerve action potential (see below) identified voltage-dependent Na+ and K+ conductances as the main participants. It was later found that some invertebrate nerve and muscle cells could produce action potentials that depended on Ca2+ rather than Na+, and it was then found that vertebrate cells also possess voltage-activated calcium channels capable of allowing substantial amounts of Ca2+ to enter the cell when the membrane is depolarised. These voltage-gated channels are highly selective for Ca2+ (although they also conduct Ba2+ ions, which are often used as a substitute in electrophysiological experiments), and do not conduct Na+ or K+; they are ubiquitous in excitable cells and cause Ca2+ to enter the cell whenever the membrane is depolarised, for example by a conducted action potential.
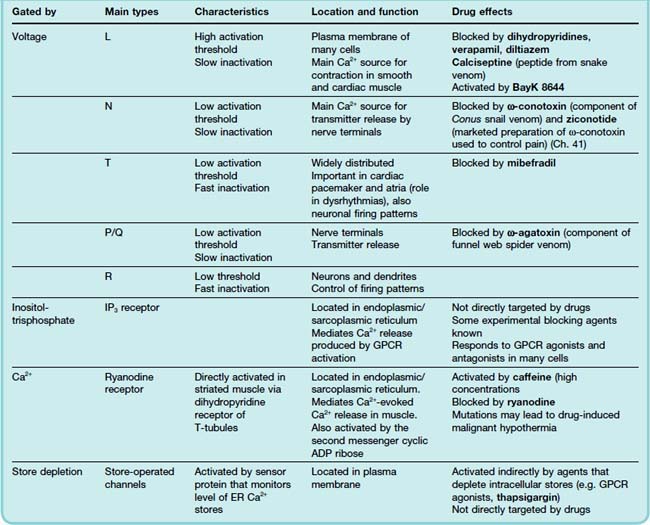
A combination of electrophysiological and pharmacological criteria have revealed five distinct subtypes of voltage-gated calcium channels: L, T, N, P/Q and R.1 The subtypes vary with respect to their activation and inactivation kinetics, their voltage threshold for activation, their conductance, and their sensitivity to blocking agents, as summarised in Table 4.1. The molecular basis for this heterogeneity has been worked out in some detail. The main pore-forming subunits (termed α1, see Fig. 3.4) occur in at least 10 molecular subtypes, and they are associated with other subunits (β, γ, δ) that also exist in different forms. Different combinations of these subunits give rise to the different physiological subtypes. In general, L channels are particularly important in regulating contraction of cardiac and smooth muscle (see below), and N channels (and also P/Q) are involved in neurotransmitter and hormone release, while T channels mediate Ca2+ entry into neurons and thereby control various Ca2+-dependent functions such as regulation of other channels, enzymes, etc. Clinically used drugs that act directly on these channels include the group of ‘Ca2+ antagonists’ consisting of dihydropyridines (e.g. nifedipine), verapamil and diltiazem (used for their cardiovascular effects; see Chs 21 and 22), and also gabapentin and pregabalin (used to treat pain and epilepsy; see Chs 41 and 44). Many drugs affect calcium channels indirectly by acting on G-protein-coupled receptors (see Ch. 3). A number of toxins act selectively on one or other type of calcium channel (Table 4.1), and these are used as experimental tools.
Ligand-Gated Channels
Most ligand-gated cation channels (see Ch. 3) that are activated by excitatory neurotransmitters are relatively non-selective, and conduct Ca2+ ions as well as other cations. Most important in this respect is the glutamate receptor of the NMDA type (Ch. 37), which has a particularly high permeability to Ca2+ and is a major contributor to Ca2+ uptake by postsynaptic neurons (and also glial cells) in the central nervous system. Activation of this receptor can readily cause so much Ca2+ entry that the cell dies, mainly through activation of Ca2+-dependent proteases but also by triggering apoptosis (see Ch. 5). This mechanism, termed excitotoxicity, probably plays a part in various neurodegenerative disorders (see Ch. 39).
For many years, there was dispute about the existence of ‘receptor-operated channels’ in smooth muscle, responding directly to mediators such as adrenaline (epinephrine), acetylcholine and histamine. Now it seems (see Berridge, 2009) that the P2x receptor (see Ch. 3), activated by ATP, is the only example of a true ligand-gated channel in smooth muscle, and this constitutes an important route of entry for Ca2+. As mentioned above, many mediators acting on G-protein-coupled receptors, affect Ca2+ entry indirectly, mainly by regulating voltage-gated calcium channels or potassium channels.
Store-Operated Calcium Channels (SOCs)
SOCs are very low-conductance channels that occur in the plasma membrane and open to allow entry when the ER stores are depleted, but are not sensitive to cytosolic [Ca2+]i. The linkage between the ER and the plasma membrane—for long a puzzle—was recently found to involve a Ca2+-sensor protein (Stim1) in the ER membrane, which connects directly to the channel protein (Orai1) in the plasma membrane (see Clapham, 2007).
Like the ER and SR channels, these channels can serve to amplify the rise in [Ca2+]i resulting from Ca2+ release from the stores. So far, only experimental compounds are known to block these channels, but efforts are being made to develop specific blocking agents for therapeutic use as relaxants of smooth muscle.
Calcium Extrusion Mechanisms
Active transport of Ca2+ outwards across the plasma membrane, and inwards across the membranes of the ER or SR, depends on the activity of distinct Ca2+-dependent ATPases,2 similar to the Na+/K+-dependent ATPase that pumps Na+ out of the cell in exchange for K+. Thapsigargin (derived from a Mediterranean plant, Thapsia garganica) specifically blocks the ER pump, causing loss of Ca2+ from the ER. It is a useful experimental tool but has no therapeutic significance.
Calcium is also extruded from cells in exchange for Na+, by Na+–Ca2+ exchange. The transporter that does this has been fully characterised and cloned, and (as you would expect) comes in several molecular subtypes whose functions remain to be worked out. The exchanger transfers three Na+ ions for one Ca2+, and therefore produces a net depolarising current when it is extruding Ca2+. The energy for Ca2+ extrusion comes from the electrochemical gradient for Na+, not directly from ATP hydrolysis. This means that a reduction in the Na+ concentration gradient resulting from Na+ entry will reduce Ca2+ extrusion by the exchanger, causing a secondary rise in [Ca2+]i, a mechanism that is particularly important in cardiac muscle (see Ch. 21). Digoxin, which inhibits Na+ extrusion, acts on cardiac muscle in this way (Ch. 21), causing [Ca2+]i to increase.
Calcium Release Mechanisms
There are two main types of calcium channel in the ER and SR membrane, which play an important part in controlling the release of Ca2+ from these stores.
The functions of IP3Rs and RyRs are modulated by a variety of other intracellular signals (see Berridge et al., 2003), which affect the magnitude and spatiotemporal patterning of Ca2+ signals. Fluorescence imaging techniques have revealed a remarkable level of complexity of Ca2+ signals, and much remains to be discovered about the importance of this patterning in relation to physiological and pharmacological mechanisms. The Ca2+ sensitivity of RyRs is increased by caffeine, causing Ca2+ release from the SR even at resting levels of [Ca2+]i. This is used experimentally but rarely happens in humans, because the other pharmacological effects of caffeine (see Ch. 47) occur at much lower doses. The blocking effect of dantrolene, a compound related to ryanodine, is used therapeutically to relieve muscle spasm in the rare condition of malignant hyperthermia (see Ch. 40), which is associated with inherited abnormalities in the RyR protein. There are as yet few other examples of drugs that directly affect these Ca2+ release mechanisms.
A typical [Ca2+]i signal resulting from activation of a G-protein-coupled receptor is shown in Figure 4.2. The response produced in the absence of extracellular Ca2+ represents release of intracellular Ca2+. The larger and more prolonged response when extracellular Ca2+ is present shows the contribution of SOC-mediated Ca2+ entry. The various positive and negative feedback mechanisms that regulate [Ca2+]i give rise to a variety of temporal and spatial oscillatory patterns (Fig. 4.2B) that are responsible for spontaneous rhythmic activity in smooth muscle and nerve cells (see Berridge, 2009).
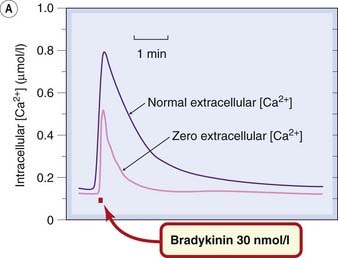
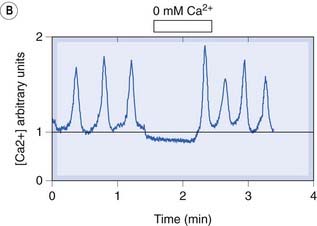
Fig. 4.2 [A] Increase in intracellular calcium concentration in response to receptor activation.
The records were obtained from a single rat sensory neuron grown in tissue culture. The cells were loaded with the fluorescent Ca2+ indicator Fura-2, and the signal from a single cell monitored with a fluorescence microscope. A brief exposure to the peptide bradykinin, which causes excitation of sensory neurons (see Ch. 41), causes a transient increase in [Ca2+]i from the resting value of about 150 nmol/l. When Ca2+ is removed from the extracellular solution, the bradykinin-induced increase in [Ca2+]i is still present but is smaller and briefer. The response in the absence of extracellular Ca2+ represents the release of stored intracellular Ca2+ resulting from the intracellular production of inositol trisphosphate. The difference between this and the larger response when Ca2+ is present extracellularly is believed to represent Ca2+ entry through store-operated ion channels in the cell membrane.
(Figure kindly provided by G M Burgess and A Forbes, Novartis Institute for Medical Research.)
[B] Spontaneous calcium oscillations in pacemaker cells from the rabbit urethra that regulate the rhythmic contractions of the smooth muscle.
The signals cease when external Ca2+ is removed, showing that activation of membrane Ca2+ channels is involved in the mechanism.
(From McHale et al. 2006 J Physiol 570:23–28.)
Other Second Messengers
 Two intracellular metabolites, cyclic ADP-ribose (cADPR) and nicotinic acid dinucleotide phosphate (NAADP; see Fliegert et al., 2007), formed from the ubiquitous coenzymes nicotinamide adenine dinucleotide (NAD) and NAD phosphate, also affect Ca2+ signalling. cADPR acts by increasing the sensitivity of RyRs to Ca2+, thus increasing the ‘gain’ of the CICR effect. NAADP releases Ca2+ from lysosomes by activating channels not yet identified but evidently distinct from the IP3R and RyR.
Two intracellular metabolites, cyclic ADP-ribose (cADPR) and nicotinic acid dinucleotide phosphate (NAADP; see Fliegert et al., 2007), formed from the ubiquitous coenzymes nicotinamide adenine dinucleotide (NAD) and NAD phosphate, also affect Ca2+ signalling. cADPR acts by increasing the sensitivity of RyRs to Ca2+, thus increasing the ‘gain’ of the CICR effect. NAADP releases Ca2+ from lysosomes by activating channels not yet identified but evidently distinct from the IP3R and RyR.
The levels of these messengers in mammalian cells may be regulated mainly in response to changes in the metabolic status of the cell, although the details are not yet clear. Abnormal Ca2+ signalling is involved in many pathophysiological conditions, such as ischaemic cell death, endocrine disorders and cardiac dysrhythmias, where the roles of cADPR and NAADP, and their interaction with other mechanisms that regulate [Ca2+]i, are the subject of much current work (see Berridge et al., 2003).
The Role of Mitochondria
Under normal conditions, mitochondria accumulate Ca2+ passively as a result of the intramitochondrial potential, which is strongly negative with respect to the cytosol. This negativity is maintained by active extrusion of protons, and is lost—thus releasing Ca2+ into the cytosol—if the cell runs short of ATP, for example under conditions of hypoxia. This only happens in extremis, and the resulting Ca2+ release contributes to the cytotoxicity associated with severe metabolic disturbance. Cell death resulting from brain ischaemia or coronary ischaemia (see Chs 21 and 39) involves this mechanism, along with others that contribute to an excessive rise in [Ca2+]i.
Calcium regulation 
Calmodulin
Calcium exerts its control over cell functions by virtue of its ability to regulate the activity of many different proteins, including enzymes (particularly kinases and phosphatases), channels, transporters, transcription factors, synaptic vesicle proteins and many others. In most cases, a Ca2+-binding protein serves as an intermediate between Ca2+ and the regulated functional protein, the best known such binding protein being the ubiquitous calmodulin (see Clapham, 2007). This regulates at least 40 different functional proteins—indeed a powerful fixer. Calmodulin is a dimer, with four Ca2+ binding sites. When all are occupied, it undergoes a conformational change, exposing a ‘sticky’ hydrophobic domain that lures many proteins into association, thereby affecting their functional properties.
Excitation
Excitability describes the ability of a cell to show a regenerative all-or-nothing electrical response to depolarisation of its membrane, this membrane response being known as an action potential. It is a characteristic of most neurons and muscle cells (including striated, cardiac and smooth muscle) and of many endocrine gland cells. In neurons and muscle cells, the ability of the action potential, once initiated, to propagate to all parts of the cell membrane, and often to spread to neighbouring cells, explains the importance of membrane excitation in intra- and intercellular signalling. In the nervous system, and in striated muscle, action potential propagation is the mechanism responsible for communication over long distances at high speed, indispensable for large, fast-moving creatures. In cardiac and smooth muscle, as well as in some central neurons, spontaneous rhythmic activity occurs. In gland cells, the action potential, where it occurs, serves to amplify the signal that causes the cell to secrete. In each type of tissue, the properties of the excitation process reflect the special characteristics of the ion channels that underlie the process. The molecular nature of ion channels, and their importance as drug targets, is considered in Chapter 3; here we discuss the cellular processes that depend primarily on ion channel function. For more detail, see Hille (2001).
The ‘Resting’ Cell
The resting cell is not resting at all but very busy controlling the state of its interior, and it requires a continuous supply of energy to do so. In relation to the topics discussed in this chapter, the following characteristics are especially important:
Under resting conditions, all cells maintain a negative internal potential between about −30 mV and −80 mV, depending on the cell type. This arises because (a) the membrane is relatively impermeable to Na+, and (b) Na+ ions are actively extruded from the cell in exchange for K+ ions by an energy-dependent transporter, the Na+ pump (or Na+–K+-ATPase). The result is that the intracellular K+ concentration, [K+]i, is higher, and [Na+]i is lower, than the respective extracellular concentrations. In many cells, other ions, particularly Cl−, are also actively transported and unequally distributed across the membrane. In many cases (e.g. in neurons), the membrane permeability to K+ is relatively high, and the membrane potential settles at a value of −60 to −80 mV, close to the equilibrium potential for K+ (Fig. 4.3). In other cells (e.g. smooth muscle), anions play a larger part, and the membrane potential is generally lower (−30 to −50 mV) and less dependent on K+.
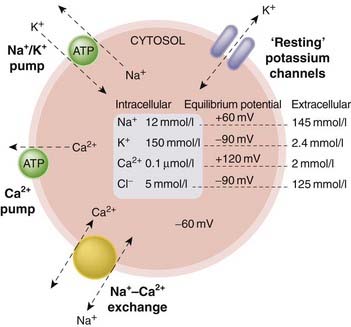
Fig. 4.3 Simplified diagram showing the ionic balance of a typical ‘resting’ cell.
The main transport mechanisms that maintain the ionic gradients across the plasma membrane are the ATP-driven Na+–K+ and Ca2+ pumps and the Na+–Ca2+ exchange transporter. The membrane is relatively permeable to K+, because potassium channels are open at rest, but impermeable to other cations. The unequal ion concentrations on either side of the membrane give rise to the ‘equilibrium potentials’ shown. The resting membrane potential, typically about −60 mV but differing between different cell types, is determined by the equilibrium potentials and the permeabilities of the various ions involved, and by the ‘electrogenic’ effect of the transporters. For simplicity, anions and other ions, such as protons, are not shown, although these play an important role in many cell types.
Electrical and Ionic Events Underlying the Action Potential
Our present understanding of electrical excitability rests firmly on the work of Hodgkin, Huxley and Katz on squid axons, published in 1949–1952. Their experiments (see Katz, 1966) revealed the existence of voltage-gated ion channels (see above) and showed that the action potential is generated by the interplay of two processes:
Because of the inequality of Na+ and K+ concentrations on the two sides of the membrane, an increase in Na+ permeability causes an inward (depolarising) current of Na+ ions, whereas an increase in K+ permeability causes an outward current. The separability of these two currents can be most clearly demonstrated by the use of drugs blocking sodium and potassium channels, as shown in Figure 4.4. During the physiological initiation or propagation of a nerve impulse, the first event is a small depolarisation of the membrane, produced either by transmitter action or by the approach of an action potential passing along the axon. This opens sodium channels, allowing an inward current of Na+ ions to flow, which depolarises the membrane still further. The process is thus a regenerative one, and the increase in Na+ permeability is enough to bring the membrane potential close to ENa. The increased Na+ conductance is transient, because the channels inactivate rapidly and the membrane returns to its resting state.
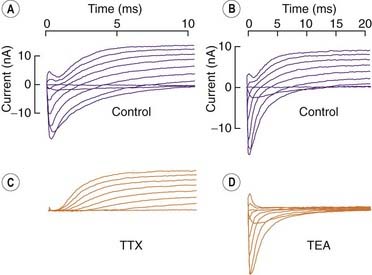
Fig. 4.4 Separation of sodium and potassium currents in the nerve membrane.
Voltage clamp records from the node of Ranvier of a single frog nerve fibre. At time 0, the membrane potential was stepped to a depolarised level, ranging from −60 mV (lower trace in each series) to +60 mV (upper trace in each series) in 15-mV steps. [A] [B] Control records from two fibres. [C] Effect of tetrodotoxin (TTX), which abolishes Na+ currents. [D] Effect of tetraethylammonium (TEA), which abolishes K+ currents.
(From Hille B 1970. Ionic channels in nerve membranes. Prog Biophys Mol Biol 21: 1–32.)
In many types of cell, including most nerve cells, repolarisation is assisted by the opening of voltage-dependent potassium channels. These function in much the same way as sodium channels, but their activation kinetics are about 10 times slower and they do not inactivate appreciably. This means that the potassium channels open later than the sodium channels, and contribute to the rapid termination of the action potential. The behaviour of the sodium and potassium channels during an action potential is shown in Figure 4.5.
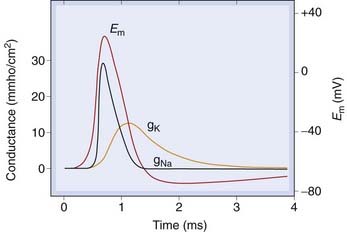
Fig. 4.5 Behaviour of sodium and potassium channels during a conducted action potential.
Rapid opening of sodium channels occurs during the action potential upstroke. Delayed opening of potassium channels, and inactivation of sodium channels, causes repolarisation. Em, membrane potential; gNa, gK, membrane conductance to Na+, K+.
The foregoing account, based on Hodgkin and Huxley’s work 60 years ago, involves only Na+ and K+ channels. Subsequently (see Hille, 2001), voltage-gated calcium channels (see Fig. 4.1) were discovered. These function in basically the same way as sodium channels; they contribute to action potential generation in many cells, particularly cardiac and smooth muscle cells, but also in neurons and secretory cells. Ca2+ entry through voltage-gated calcium channels plays a key role in intracellular signalling, as described above.
Channel Function
The discharge patterns of excitable cells vary greatly. Skeletal muscle fibres are quiescent unless stimulated by the arrival of a nerve impulse at the neuromuscular junction. Cardiac muscle fibres discharge spontaneously at a regular rate (see Ch. 21). Neurons may be normally silent, or they may discharge spontaneously, either regularly or in bursts; smooth muscle cells show a similar variety of firing patterns. The frequency at which different cells normally discharge action potentials also varies greatly, from 100 Hz or more for fast-conducting neurons, down to about 1 Hz for cardiac muscle cells. These very pronounced functional variations reflect the different characteristics of the ion channels expressed in different cell types. Rhythmic fluctuations of [Ca2+]i underlie the distinct firing patterns that occur in different types of cell (see Berridge, 2009).
Drugs that alter channel characteristics, either by interacting directly with the channel itself or indirectly through second messengers, affect the function of many organ systems, including the nervous, cardiovascular, endocrine, respiratory and reproductive systems, and are a frequent theme in this book. Here we describe some of the key mechanisms involved in the regulation of excitable cells.
In general, action potentials are initiated by membrane currents that cause depolarisation of the cell. These currents may be produced by synaptic activity, by an action potential approaching from another part of the cell, by a sensory stimulus or by spontaneous pacemaker activity. The tendency of such currents to initiate an action potential is governed by the excitability of the cell, which depends mainly on the state of (a) the voltage-gated sodium and/or calcium channels, and (b) the potassium channels of the resting membrane. Anything that increases the number of available sodium or calcium channels, or reduces their activation threshold, will tend to increase excitability, whereas increasing the resting K+ conductance reduces it. Agents that do the reverse, by blocking channels or interfering with their opening, will have the opposite effect. Some examples are shown in Figures 4.6 and 4.7 and in Table 4.1. Inherited mutations of channel proteins are responsible for a wide variety of (mostly rare) neurological and other genetic disorders (see Ashcroft, 2000, 2006).
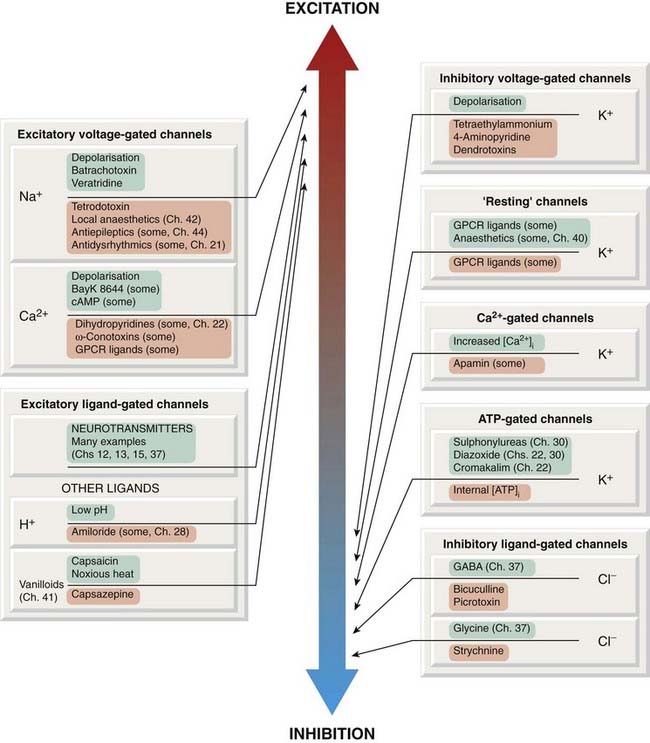
Fig. 4.6 Ion channels associated with excitatory and inhibitory membrane effects, and some of the drugs and other ligands that affect them.
Channel openers are shown in green boxes, blocking agents and inhibitors in pink boxes. GPCR, G-protein-coupled receptor.
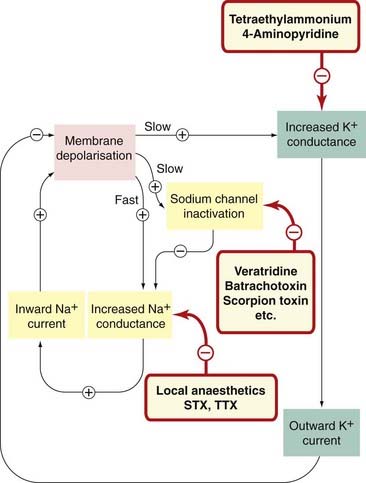
Fig. 4.7 Sites of action of drugs and toxins that affect channels involved in action potential generation.
Many other mediators affect these channels indirectly via membrane receptors, through phosphorylation or altered expression. STX, saxitoxin; TTX, tetrodotoxin.
Use Dependence and Voltage Dependence
Voltage-gated channels can exist in three functional states (Fig. 4.8): resting (the closed state that prevails at the normal resting potential), activated (the open state favoured by brief depolarisation) and inactivated (the blocked state resulting from a trap door-like occlusion of the open channel by a floppy intracellular appendage of the channel protein). After the action potential has passed, many sodium channels are in the inactivated state; after the membrane potential returns to its resting value, the inactivated channels take time to revert to the resting state and thus become available for activation once more. In the meantime, the membrane is temporarily refractory. Each action potential causes the channels to cycle through these states. The duration of the refractory period determines the maximum frequency at which action potentials can occur. Drugs that block sodium channels, such as local anaesthetics (Ch. 42), antidysrhythmic drugs (Ch. 21) and antiepileptic drugs (Ch. 44), commonly show a selective affinity for one or other of these functional states of the channel, and in their presence the proportion of channels in the high-affinity state is increased. Of particular importance are drugs that bind most strongly to the inactivated state of the channel and thus favour the adoption of this state, thus prolonging the refractory period and reducing the maximum frequency at which action potentials can be generated. This type of block is called use dependent, because the binding of such drugs increases as a function of the rate of action potential discharge, which governs the rate at which inactivated—and therefore drug-sensitive—channels are generated. This is important for some antidysrhythmic drugs (see Ch. 21) and for antiepileptic drugs (Ch. 44), because high-frequency discharges can be inhibited without affecting excitability at normal frequencies. Drugs that readily block sodium channels in their resting state (e.g. local anaesthetics, Ch. 42) prevent excitation at low as well as high frequencies.
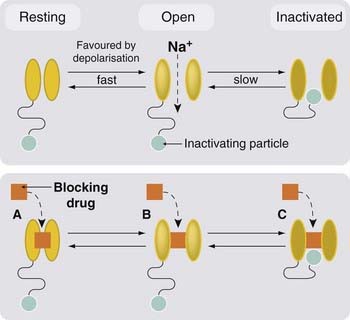
Fig. 4.8 Resting, activated and inactivated states of voltage-gated channels, exemplified by the sodium channel.
Membrane depolarisation causes a rapid transition from the resting (closed) state to the open state. The inactivating particle (part of the intracellular domain of the channel protein) is then able to block the channel. Blocking drugs (e.g. local anaesthetics and antiepileptic drugs) often show preference for one of the three channel states, and thus affect the kinetic behaviour of the channels, with implications for their clinical application.
Most sodium channel-blocking drugs are cationic at physiological pH and are therefore affected by the voltage gradient across the cell membrane. They block the channel from the inside, so that their blocking action is favoured by depolarisation. This phenomenon, known as voltage dependence, is also of relevance to the action of antidysrhythmic and antiepileptic drugs, because the cells that are the seat of dysrhythmias or seizure activity are generally somewhat depolarised and therefore more strongly blocked than ‘healthy’ cells. Similar considerations apply also to drugs that block potassium or calcium channels, but we know less about the importance of use and voltage dependence for these than we do for sodium channels.
Sodium Channels
In most excitable cells, the regenerative inward current that initiates the action potential results from activation of voltage-gated sodium channels. The early voltage clamp studies by Hodgkin and Huxley on the squid giant axon, described above, revealed the essential functional properties of these channels. Later, advantage was taken of the potent and highly selective blocking action of tetrodotoxin (TTX, see Ch. 42) to label and purify the channel protein, and subsequently to clone it, revealing the complex structure shown in Figure 3.18, with four similar domains each comprising six membrane-spanning helices (reviewed by Catterall, 2000). One of these helices, S4, contains several basic amino acids and forms the voltage sensor, and moves outwards, thus opening the channel, when the membrane is depolarised. One of the intracellular loops is designed to swing across and block the channel when S4 is displaced, thus inactivating the channel.
It was known from physiological studies that the sodium channels of heart and skeletal muscle differ in various ways from those of neurons. In particular, cardiac sodium channels (and also those of some sensory neurons) are relatively insensitive to TTX, and slower in their kinetics, compared with most neuronal sodium channels. Nine distinct molecular subtypes have so far been identified, more than enough to explain the functional diversity.
In addition to channel blocking compounds such as tetrodotoxin, other compounds affect sodium channel gating. For example, the plant alkaloid veratridine and the frog skin poison batrachotoxin cause persistent activation, while various scorpion toxins prevent inactivation, mechanisms resulting in enhanced neuronal excitability.
Therapeutic agents that act by blocking sodium channels include local anaesthetic drugs (Ch. 42), antiepileptic drugs (Ch. 44) and antidysrhythmic drugs (Ch. 21). The sodium channel-blocking actions of these drugs were in most cases discovered long after their clinical applications were recognised; many of them lack specificity and produce a variety of unwanted side effects. The use of induced mutations in cloned sodium channels expressed in cell lines is now revealing which regions of the very large channel molecule are involved in the binding of particular agents, knowledge that should allow more specific drugs to be designed in the future.
Potassium Channels
In a typical resting cell (see above), the membrane is selectively permeable to K+, and the membrane potential (about −60 mV) is somewhat positive to the K+ equilibrium (about −90 mV). This resting permeability comes about because potassium channels are open. If more potassium channels open, the membrane hyperpolarises and the cell is inhibited, whereas the opposite happens if potassium channels close. As well as affecting excitability in this way, potassium channels also play an important role in regulating the duration of the action potential and the temporal patterning of action potential discharges; altogether, these channels play a central role in regulating cell function. As mentioned in Chapter 3, the number and variety of potassium channel subtypes is extraordinary, implying that evolution has been driven by the scope for biological advantage to be gained from subtle variations in the functional properties of these channels. A recent résumé lists over 60 different pore-forming subunits, plus another 20 or so auxiliary subunits. An impressive evolutionary display, maybe, but hard going for most of us. Here we outline the main types that are known to be important pharmacologically. For more details, and information on potassium channels and the various drugs and toxins that affect them, see Shieh et al. (2000) and Jenkinson (2006).
Potassium channels fall into three main classes (Table 4.2),3 of which the structures are shown in Figure 3.18.
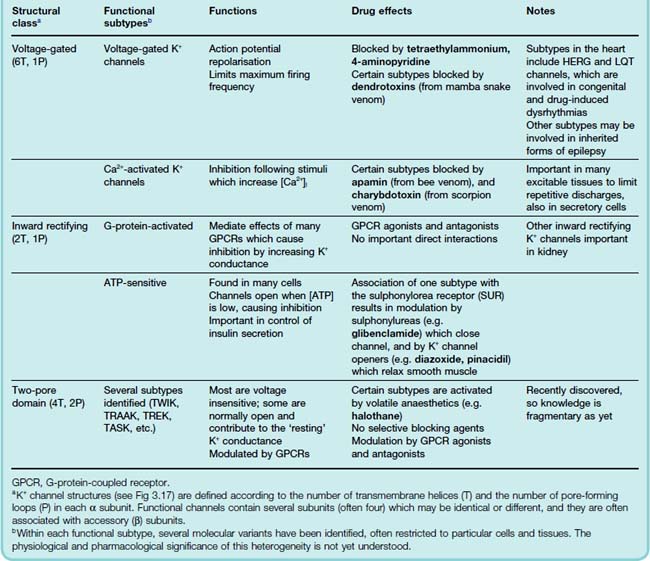
Inherited abnormalities of potassium channels (channelopathies) contribute to a rapidly growing number of cardiac, neurological and other diseases. These include the long QT syndrome associated with mutations in cardiac voltage-gated potassium channels, causing episodes of ventricular arrest that can result in sudden death. Certain familial types of deafness and epilepsy are associated with mutations in voltage-gated potassium channels. (Ashcroft, 2000, 2006).
Ion channels and electrical excitability
Muscle Contraction
Effects of drugs on the contractile machinery of smooth muscle are the basis of many therapeutic applications, for smooth muscle is an important component of most physiological systems, including blood vessels and the gastrointestinal, respiratory and urinary tracts. For many decades, smooth muscle pharmacology with its trademark technology—the isolated organ bath—held the centre of the pharmacological stage, and neither the subject nor the technology shows any sign of flagging, even though the stage has become much more crowded. Cardiac muscle contractility is also the target of important drug effects, whereas striated muscle contractility is only rarely affected by drugs.
Although in each case the basic molecular basis of contraction is similar, namely an interaction between actin and myosin, fuelled by ATP and initiated by an increase in [Ca2+]i, there are differences between these three kinds of muscle that account for their different responsiveness to drugs and chemical mediators.
These differences (Fig. 4.9) involve (a) the linkage between membrane events and increase in [Ca2+]i, and (b) the mechanism by which [Ca2+]i regulates contraction.
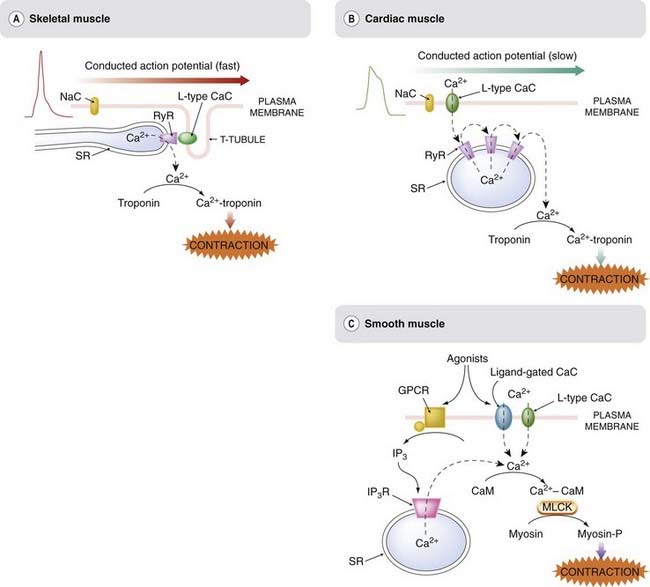
Fig. 4.9 Comparison of excitation–contraction coupling in [A] striated muscle, [B] cardiac muscle and [C] smooth muscle.
Striated and cardiac muscle differ mainly in the mechanism by which membrane depolarisation is coupled to Ca2+ release. In striated muscle, the T-tubule membrane is coupled closely to the sarcoplasmic reticulum (SR) via the L-type CaC and the ryanodine receptor (RyR). In cardiac muscle, Ca2+ entry via voltage-gated calcium channels initiates a regenerative release through activation of the Ca2+-sensitive RyRs. In smooth muscle, contraction can be produced either by Ca2+ entry through voltage- or ligand-gated calcium channels, or by inositol trisphosphate (IP3)-mediated Ca2+ release from the SR. The mechanism by which Ca2+ activates contraction is different, and operates more slowly, in smooth muscle compared with in striated or cardiac muscle. CaC, calcium channel; CaM, calmodulin; SR, sarcoplasmic reticulum; GPCR, G-protein-coupled receptor; MLCK, myosin light-chain kinase; NaC, voltage-gated sodium channel; RyR, ryanodine receptor.
Skeletal Muscle
Skeletal muscle possesses an array of transverse T-tubules extending into the cell from the plasma membrane. The action potential of the plasma membrane depends on voltage-gated sodium channels, as in most nerve cells, and propagates rapidly from its site of origin, the motor endplate (see Ch. 13), to the rest of the fibre. The T-tubule membrane contains L-type calcium channels, which respond to membrane depolarisation conducted passively along the T-tubule when the plasma membrane is invaded by an action potential. These calcium channels are located extremely close to ryanodine receptors (RyRs; see Ch. 3) in the adjacent SR membrane, and activation of these RyRs causes release of Ca2+ from the SR. There is evidence of direct coupling between the calcium channels of the T-tubule and the RyRs of the SR (as shown in Fig. 4.9); however, Ca2+ entry through the T-tubule channels into the restricted zone between these channels and associated RyRs may also contribute. Through this link, depolarisation rapidly activates the RyRs, releasing a short puff of Ca2+ from the SR into the sarcoplasm. The Ca2+ binds to troponin, a protein that normally blocks the interaction between actin and myosin. When Ca2+ binds, troponin moves out of the way and allows the contractile machinery to operate. Ca2+ release is rapid and brief, and the muscle responds with a short-lasting ‘twitch’ response. This is a relatively fast and direct mechanism compared with the arrangement in cardiac and smooth muscle (see below), and consequently less susceptible to pharmacological modulation. The few examples of drugs that directly affect skeletal muscle contraction are shown in Table 4.1.
Cardiac Muscle
Cardiac muscle (see review by Bers, 2002) differs from skeletal muscle in several important respects. The nature of the cardiac action potential, the ionic mechanisms underlying its inherent rhythmicity, and the effects of drugs on the rate and rhythm of the heart are described in Chapter 21. Cardiac muscle cells lack T-tubules, and there is no direct coupling between the plasma membrane and the SR. The cardiac action potential varies in its configuration in different parts of the heart, but commonly shows a ‘plateau’ lasting several hundred milliseconds following the initial rapid depolarisation. The plasma membrane contains many L-type calcium channels, which open during this plateau and allow Ca2+ to enter the cell, although not in sufficient quantities to activate the contractile machinery directly. Instead, this initial Ca2+ entry acts on RyRs (a different molecular type from those of skeletal muscle) to release Ca2+ from the SR, producing a secondary and much larger wave of Ca2+. Because the RyRs of cardiac muscle are themselves activated by Ca2+, the [Ca2+]i wave is a regenerative, all-or-nothing event. The initial Ca2+ entry that triggers this event is highly dependent on the action potential duration, and on the functioning of the membrane L-type channels. Some of the drugs that affect it are shown in Table 4.1. With minor differences, the mechanism by which Ca2+ activates the contractile machinery is the same as in skeletal muscle. Mutations of ryanodine receptors are implicated in various disorders of skeletal and cardiac muscle function (see Priori & Napolitano, 2005), but so far, no therapeutically useful drugs have emerged from this line of inquiry.
Smooth Muscle
The properties of smooth muscle vary considerably in different organs, and the mechanisms linking membrane events and contraction are correspondingly variable and more complex than in other kinds of muscle. Spontaneous rhythmic activity occurs in many organs, by mechanisms producing oscillations of [Ca2+]i (see Berridge, 2009). The action potential of smooth muscle is generally a rather lazy and vague affair compared with the more military behaviour of skeletal and cardiac muscle, and it propagates through the tissue much more slowly and uncertainly. The action potential is, in most cases, generated by L-type calcium channels rather than by voltage-gated sodium channels, and this is one important route of Ca2+ entry. In addition, many smooth muscle cells possess P2x receptors, ligand-gated cation channels, which allow Ca2+ entry when activated by ATP released from autonomic nerves (see Ch. 12). Smooth muscle cells also store Ca2+ in the ER, from which it can be released when the IP3R is activated (see Ch. 3). IP3 is generated by activation of many types of G-protein-coupled receptor. Thus, in contrast to skeletal and cardiac muscle, Ca2+ release and contraction can occur in smooth muscle when such receptors are activated without necessarily involving depolarisation and Ca2+ entry through the plasma membrane.
The contractile machinery of smooth muscle is activated when the myosin light chain undergoes phosphorylation, causing it to become detached from the actin filaments. This phosphorylation is catalysed by a kinase, myosin light-chain kinase (MLCK), which is activated when it binds to Ca2+–calmodulin (see p. 52). A second enzyme, myosin phosphatase, reverses the phosphorylation and causes relaxation. The activity of MLCK and myosin phosphatase thus exerts a balanced effect, promoting contraction and relaxation, respectively. Both enzymes are regulated by cyclic nucleotides (cAMP and cGMP; see Ch. 3), and many drugs that cause smooth muscle contraction or relaxation mediated through G-protein-coupled receptors or through guanylyl cyclase-linked receptors act in this way. Figure 4.10 summarises the main mechanisms by which drugs control smooth muscle contraction. The complexity of these control mechanisms and interactions explains why pharmacologists have been entranced for so long by smooth muscle. Many therapeutic drugs work by contracting or relaxing smooth muscle, particularly those affecting the cardiovascular, respiratory and gastrointestinal systems, as discussed in later chapters, where details of specific drugs and their physiological effects are given.
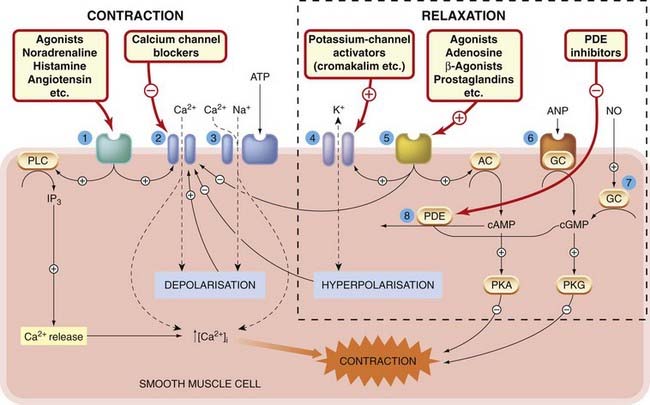
Fig. 4.10 Mechanisms controlling smooth muscle contraction and relaxation.
1. G-protein-coupled receptors for excitatory agonists, mainly regulating inositol trisphosphate formation and calcium channel function. 2. Voltage-gated calcium channels. 3. P2x receptor for ATP (ligand-gated cation channel). 4. Potassium channels. 5. G-protein-coupled receptors for inhibitory agonists, mainly regulating cAMP formation and potassium and calcium channel function. 6. Receptor for atrial natriuretic peptide (ANP), coupled directly to guanylyl cyclase (GC). 7. Soluble guanylyl cyclase, activated by nitric oxide (NO). 8. Phosphodiesterase (PDE), the main route of inactivation of cAMP and cGMP. AC, adenylate cyclase; PKA, protein kinase A; PKG, protein kinase G; PLC, phospholipase C.
Muscle contraction
Release of Chemical Mediators
Much of pharmacology is based on interference with the body’s own chemical mediators, particularly neurotransmitters, hormones and inflammatory mediators. Here we discuss some of the common mechanisms involved in the release of such mediators, and it will come as no surprise that Ca2+ plays a central role. Drugs and other agents that affect the various control mechanisms that regulate [Ca2+]i will therefore also affect mediator release, and this accounts for many of the physiological effects that they produce.
Chemical mediators that are released from cells fall into two main groups (Fig. 4.11):
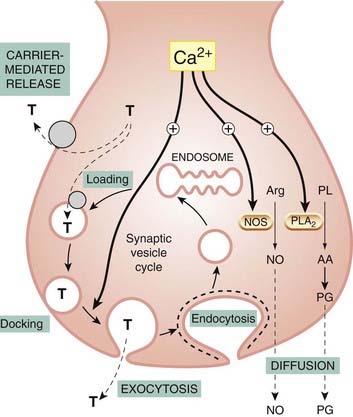
Fig. 4.11 Role of exocytosis, carrier-mediated transport and diffusion in mediator release.
The main mechanism of release of monoamine and peptide mediators is Ca2+-mediated exocytosis, but carrier-mediated release from the cytosol also occurs. T represents a typical amine transmitter, such as noradrenaline (norepinephrine) or 5-hydroxytryptamine. Nitric oxide (NO) and prostaglandins (PGs) are released by diffusion as soon as they are formed, from arginine (Arg) and arachidonic acid (AA), respectively, through the action of Ca2+-activated enzymes, nitric oxide synthase (NOS) and phospholipase A2 (PLA2) (see Chs 17 and 20 for more details).
Calcium ions play a key role in both cases, because a rise in [Ca2+]i initiates exocytosis and is also the main activator of the enzymes responsible for the synthesis of diffusible mediators.
In addition to mediators that are released from cells, some are formed from precursors in the plasma, two important examples being kinins (Ch. 17) and angiotensin (Ch. 22), which are peptides produced by protease-mediated cleavage of circulating proteins.
Exocytosis
Exocytosis, occurring in response to an increase of [Ca2+]i, is the principal mechanism of transmitter release (see Fig. 4.11) in the peripheral and central nervous systems, as well as in endocrine cells and mast cells. The secretion of enzymes and other proteins by gastrointestinal and exocrine glands and by vascular endothelial cells is also basically similar. Exocytosis (see Burgoyne & Morgan, 2002) involves fusion between the membrane of synaptic vesicles and the inner surface of the plasma membrane. The vesicles are preloaded with stored transmitter, and release occurs in discrete packets, or quanta, each representing the contents of a single vesicle. The first evidence for this (see Nicholls et al., 2000) came from the work of Katz and his colleagues in the 1950s, who recorded spontaneous ‘miniature endplate potentials’ at the frog neuromuscular junction, and showed that each resulted from the spontaneous release of a packet of the transmitter, acetylcholine. They also showed that release evoked by nerve stimulation occurred by the synchronous release of several hundred such quanta, and was highly dependent on the presence of Ca2+ in the bathing solution. Unequivocal evidence that the quanta represented vesicles releasing their contents by exocytosis came from electron microscopic studies, in which the tissue was rapidly frozen in mid-release, revealing vesicles in the process of extrusion, and from elegant electrophysiological measurements showing that membrane capacitance (reflecting the area of the presynaptic membrane) increased in a stepwise way as each vesicle fused, and then gradually returned as the vesicle membrane was recovered from the surface. There is also biochemical evidence showing that, in addition to the transmitter, other constituents of the vesicles are released at the same time.
In nerve terminals specialised for fast synaptic transmission, Ca2+ enters through voltage-gated calcium channels, mainly of the N and P type (see above), and the synaptic vesicles are ‘docked’ at active zones—specialised regions of the presynaptic membrane from which exocytosis occurs, situated close to the relevant calcium channels and opposite receptor-rich zones of the postsynaptic membrane (see Stanley, 1997). Elsewhere, where speed is less critical, Ca2+ may come from intracellular stores as described above, and the spatial organisation of active zones is less clear. It is common for secretory cells, including neurons, to release more than one mediator (for example, a ‘fast’ transmitter such as glutamate and a ‘slow’ transmitter such as a neuropeptide) from different vesicle pools (see Ch. 12). The fast transmitter vesicles are located close to active zones, while the slow transmitter vesicles are further away. Release of the fast transmitter, because of the tight spatial organisation, occurs as soon as the neighbouring calcium channels open, before the Ca2+ has a chance to diffuse throughout the terminal, whereas release of the slow transmitter requires the Ca2+ to diffuse more widely. As a result, release of fast transmitters occurs impulse by impulse, even at low stimulation frequencies, whereas release of slow transmitters builds up only at higher stimulation frequencies. The release rates of the two therefore depend critically on the frequency and patterning of firing of the presynaptic neuron (Fig. 4.12). In non-excitable cells (e.g. most exocrine and endocrine glands), the slow mechanism predominates and is activated mainly by Ca2+ release from intracellular stores.
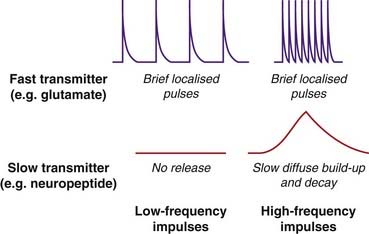
Fig. 4.12 Time course and frequency dependence of the release of ‘fast’ and ‘slow’ transmitters.
Fast transmitters (e.g. glutamate) are stored in synaptic vesicles that are ‘docked’ close to voltage-gated calcium channels in the membrane of the nerve terminal, and are released in a short burst when the membrane is depolarised (e.g. by an action potential). Slow transmitters (e.g. neuropeptides) are stored in separate vesicles further from the membrane. Release is slower, because they must first migrate to the membrane, and occurs only when [Ca2+]i builds up sufficiently.
Calcium causes exocytosis by binding to the vesicle-bound protein synaptotagmin, and this favours association between a second vesicle-bound protein, synaptobrevin, and a related protein, synaptotaxin, on the inner surface of the plasma membrane. This association brings the vesicle membrane into close apposition with the plasma membrane, causing membrane fusion. This group of proteins, known collectively as SNAREs, plays a key role in exocytosis.
Having undergone exocytosis, the empty vesicle5 is recaptured by endocytosis and returns to the interior of the terminal, where it fuses with the larger endosomal membrane. The endosome buds off new vesicles, which take up transmitter from the cytosol by means of specific transport proteins and are again docked on the presynaptic membrane. This sequence, which typically takes several minutes, is controlled by various trafficking proteins associated with the plasma membrane and the vesicles, as well as cytosolic proteins. Further details about exocytosis and vesicle recycling are given by Nestler et al. (2008) and Südhof (2004). So far, there are few examples of drugs that affect transmitter release by interacting with synaptic proteins, although the botulinum neurotoxins (see Ch. 13) produce their effects by proteolytic cleavage of SNARE proteins.
Non-Vesicular Release Mechanisms
If this neat and tidy picture of transmitter packets ready and waiting to pop obediently out of the cell in response to a puff of Ca2+ seems a little too good to be true, rest assured that the picture is not quite so simple. Acetylcholine, noradrenaline (norepinephrine) and other mediators can leak out of nerve endings from the cytosolic compartment, independently of vesicle fusion, by utilising carriers in the plasma membrane (Fig. 4.11). Drugs such as amphetamines, which release amines from central and peripheral nerve terminals (see Chs 14 and 38), do so by displacing the endogenous amine from storage vesicles into the cytosol, whence it escapes via the monoamine transporter in the plasma membrane, a mechanism that does not depend on Ca2+.
Nitric oxide (see Ch. 20) and arachidonic acid metabolites (e.g. prostaglandins; Ch. 17) are two important examples of mediators that are released by diffusion across the membrane or by carrier-mediated extrusion, rather than by exocytosis. The mediators are not stored but escape from the cell as soon as they are synthesised. In both cases, the synthetic enzyme is activated by Ca2+, and the moment-to-moment control of the rate of synthesis depends on [Ca2+]i. This kind of release is necessarily slower than the classic exocytotic mechanism, but in the case of nitric oxide is fast enough for it to function as a true transmitter (see Ch. 20).
Mediator release
Epithelial Ion Transport
Fluid-secreting epithelia include the renal tubule, salivary glands, gastrointestinal tract and airways epithelia. In each case, epithelial cells are arranged in sheets separating the interior (blood-perfused) compartment from the exterior lumen compartment, into which, or from which, secretion takes place. Fluid secretion involves two main mechanisms, which often coexist in the same cell and indeed interact with each other. Greger (2000) and Ashcroft (2000) give more detailed accounts. The two mechanisms (Fig. 4.13) are concerned, respectively, with Na+ transport and Cl− transport.
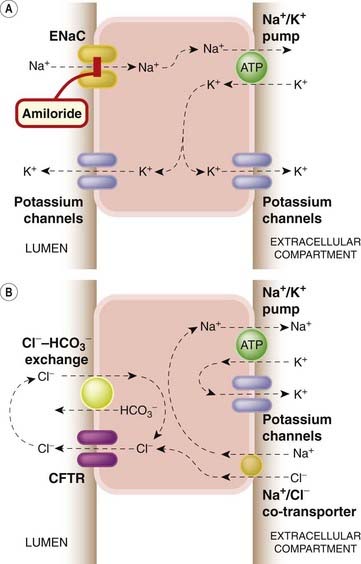
Fig. 4.13 Mechanisms of epithelial ion transport.
Such mechanisms are important in renal tubules (see Ch. 28 for more details) and also in many other situations, such as the gastrointestinal and respiratory tracts. [A] Sodium transport. A special type of epithelial sodium channel (ENaC) controls entry of Na+ into the cell from the lumenal surface, the Na+ being actively pumped out at the apical surface by the Na+–K+ exchange pump. K+ moves passively via potassium channels. [B] Chloride transport. Cl− leaves the cell via a special membrane channel, the cystic fibrosis transmembrane conductance regulator (CFTR), after entering the cell either from the apical surface via the Na+/Cl− co-transporter, or at the lumenal surface via the Cl−/HCO3− co-transporter.
In the case of Na+ transport, secretion occurs because Na+ enters the cell passively at one end and is pumped out actively at the other, with water following passively. Critical to this mechanism is a class of highly regulated epithelial sodium channels (ENaCs) that allow Na+ entry.
Epithelial sodium channels (see De la Rosa et al., 2000) are widely expressed, not only in epithelial cells but also in neurons and other excitable cells, where their function is largely unknown. They are regulated mainly by aldosterone, a hormone produced by the adrenal cortex that enhances Na+ reabsorption by the kidney (Ch. 28). Aldosterone, like other steroid hormones, exerts its effects by regulating gene expression (see Ch. 3), and causes an increase in ENaC expression, thereby increasing the rate of Na+ and fluid transport. ENaCs are selectively blocked by certain diuretic drugs, notably amiloride (see Ch. 28), a compound that is widely used to study the functioning of ENaCs in other situations.
Chloride transport is particularly important in the airways and gastrointestinal tract. In the airways, it is essential for fluid secretion, whereas in the colon it mediates fluid reabsorption, the difference being due to the different arrangement of various transporters and channels with respect to the polarity of the cells. The simplified diagram in Figure 4.13B represents the situation in the pancreas, where secretion depends on Cl− transport. The key molecule in Cl− transport is the cystic fibrosis transmembrane conductance regulator (CFTR; see Hwang & Sheppard, 1999), so named because early studies on the inherited disorder cystic fibrosis showed it to be associated with impaired Cl− conductance in the membrane of secretory epithelial cells, and the CFTR gene, identified through painstaking genetic linkage studies and isolated in 1989, was found to encode a Cl−-conducting ion channel. Severe physiological consequences follow from the impairment of secretion, particularly in the airways but also in many other systems, such as sweat glands and pancreas. Studies on the disease-associated mutations of the CFTR gene have revealed much about the molecular mechanisms involved in Cl− transport, but as yet no significant therapeutic advance. So far, no drugs are known that interact specifically with CFTRs.
Both Na+ and Cl− transport are regulated by intracellular messengers, notably by Ca2+ and cAMP, the latter exerting its effects by activating protein kinases and thereby causing phosphorylation of channels and transporters. CFTR itself is activated by cAMP. In the gastrointestinal tract, increased cAMP formation causes a large increase in the rate of fluid secretion, an effect that leads to the copious diarrhoea produced by cholera infection (see Ch. 3) and also by inflammatory conditions in which prostaglandin formation is increased (see Ch. 17). Activation of G-protein-coupled receptors, which cause release of Ca2+, also stimulates secretion, possibly also by activating CFTR. Many examples of therapeutic drugs that affect epithelial secretion by activating or blocking G-protein-coupled receptors appear in later chapters.
Epithelial ion transport
References and Further Reading
Katz B. Nerve, muscle and synapse. New York: McGraw Hill; 1966. (A classic account of the ground-breaking electrophysiological experiments that established the basis of nerve and muscle function)
Levitan I.B., Kaczmarek L.K. The neuron: cell and molecular biology, third ed. New York: Oxford University Press; 2002. (Useful textbook covering ion channels and synaptic mechanisms as well as other aspects of neuronal function)
Nestler E.J., Hyman S.E., Malenka R.C. Molecular neuropharmacology, second ed. New York: McGraw-Hill; 2008. (Excellent modern textbook)
Nicholls J.G., Fuchs P.A., Martin A.R., Wallace B.G. From neuron to brain. Sunderland: Sinauer Associates; 2001. (Excellent, well-written textbook of neuroscience)
Second messengers and calcium regulation
Berridge M.J. Elementary and global aspects of calcium signalling J. Physiol.499:1997:291-306 (Review of Ca2+ signalling, emphasising the various intracellular mechanisms that produce spatial and temporal patterning—‘sparks’, ‘waves’, etc.)
Berridge M.J. Inositol trisphosphate and calcium signalling mechanisms. Biochim. Biophys. Acta. Mol. Cell Res.. 2009;1793:933-940. (Clear and readable up-to-date account of the mechanisms and versatility of calcium signalling)
Berridge M.J., Bootman M.D., Roderick H.L. Calcium signalling: dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol.. 2003;4:517-529.
Clapham D.E. Calcium signalling. Cell. 2007;131:1047-1056. (Excellent, readable and well-illustrated short review article—recommended)
Fliegert R., Gasser A., Guse A.H. Regulation of calcium signalling by adenine-based second messengers. Biochem. Soc. Trans.. 2007;35:109-114. (Summary of second messenger role of cADPR and NAADP)
Mikoshiba K. IP3 receptor/Ca2+ channel: from discovery to new signaling concepts J. Neurochem. 102:2007:1426-1446 (Interesting account of the discovery of the IP3 receptor and its functional role)
Ashcroft F.M. Ion channels and disease. San Diego: Academic Press; 2000. (A very useful textbook that describes the physiology of different kinds of ion channels, and relates it to their molecular structure; the book emphasises the importance of ‘channelopathies’, genetic channel defects associated with disease states)
Ashcroft F.M. From molecule to malady. Nature. 2006;440:440-447. (Brief summary and update of the above account of channelopathies)
Catterall W.A. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26:13-25. (Useful, authoritative review article)
De la Rosa D.A., Canessa C.M., Fyfe G.K., Zhang P. Structure and regulation of amiloride-sensitive sodium channels. Annu. Rev. Physiol.. 2000;62:573-594. (General review on the nature and function of ‘epithelial’ sodium channels)
Goldstein S.A.N., Bockenhauer D., Zilberberg N. Potassium leak channels and the KCNK family of two-P-domain subunits. Nat. Rev. Neurosci.. 2001;2:175-184. (Review on this important class of potassium channels)
Hille B. Ionic channels of excitable membranes. Sunderland: Sinauer Associates; 2001. (A clear and detailed account of the basic principles of ion channels, with emphasis on their biophysical properties)
Jenkinson D.H. Potassium channels—multiplicity and challenges Br. J. Pharmacol. 147 Suppl. 2006:63-71 (Useful short article on the many types of K+ channel)
Reimann F., Ashcroft F.M. Inwardly rectifying potassium channels. Curr. Opin. Cell. Biol.. 1999;11:503-508. (Review describing the various mechanisms by which the inwardly rectifying potassium channels are modulated)
Shieh C.-C., Coghlan M., Sullivan J.P., Gopalakrishnan M. Potassium channels: molecular defects, diseases and therapeutic opportunities. Pharmacol. Rev.. 2000;52:557-593. (Comprehensive review of potassium channel pathophysiology and pharmacology)
Berridge M.J. Smooth muscle cell calcium activation mechanisms. J. Physiol.. 2008;586:5047-5061. (Excellent review article describing the various mechanisms by which calcium signals control activity in different types of smooth muscle—complicated but clear)
Bers D.M. Cardiac excitation–contraction coupling. Nature. 2002;415:198-205. (Short, well-illustrated review article)
Priori S.G., Napolitano C. Cardiac and skeletal muscle disorders caused by mutations in the intracellular Ca2+ release channels. J. Clin. Invest.. 2005;115:2033-2038. (Focuses on RyR mutations in various inherited diseases)
Burgoyne R.D., Morgan A. Secretory granule exocytosis. Physiol. Rev.. 2002;83:581-632. (Comprehensive review of the molecular machinery responsible for secretory exocytosis)
Greger R. The role of CFTR in the colon. Annu. Rev. Physiol.. 2000;62:467-491. (A useful résumé of information about CFTR and epithelial secretion, more general than its title suggests)
Hwang T.-C., Sheppard D.N. Molecular pharmacology of the CFTR channel. Trends Pharmacol. Sci.. 1999;20:448-453. (Description of approaches aimed at finding therapeutic drugs aimed at altering the function of the CFTR channel)
Stanley E.E. The calcium channel and the organization of the presynaptic transmitter release face. Trends Neurosci.. 1997;20:404-409. (Discusses the microphysiology of vesicular release)
Südhof T.C. The synaptic vesicle cycle. Annu. Rev. Neurosci.. 2004;27:509-547. (Summarises the mechanism of vesicular release at the molecular level)
1P and Q are so similar that they usually get lumped together. The terminology is less than poetic: L stands for long-lasting; T stands for transient; N stands for neither long-lasting nor transient; and P, Q and R carry on alphabetically from N, with O (of course) omitted.
2Clapham (2007) likens these pumps to Sisyphus, condemned endlessly to push a stone up a hill (also consuming ATP, no doubt), only for it to roll down again.
3Potassium channel terminology is confusing, to put it mildly. Electrophysiologists have named K+ currents prosaically on the basis of their functional properties (IKV, IKCa, IKATP, IKIR, etc.); geneticists have named genes somewhat fancifully according to the phenotypes associated with mutations (shaker, ether-a-go-go, etc.), while molecular biologists have introduced a rational but unmemorable nomenclature on the basis of sequence data (KCNK, KCNQ, etc., with numerical suffixes). The rest of us have to make what we can of the unlovely jargon of labels such as HERG (which—don’t blink—stands for Human Ether-a-go-go Related Gene), TWIK, TREK and TASK.
4Carrier-mediated release can also occur with neurotransmitters that are stored in vesicles but is quantitatively less significant than exocytosis (see Ch. 13).
5The vesicle contents may not always discharge completely. Instead, vesicles may fuse transiently with the cell membrane and release only part of their contents (see Burgoyne & Morgan, 2002) before becoming disconnected (termed kiss-and-run exocytosis).