The Child with Neuromuscular or Muscular Dysfunction

http://evolve.elsevier.com/wong/ncic

The Child with Cerebral Dysfunction, Ch. 37
The Child with Cognitive or Sensory Impairment, Ch. 24
Family-Centered Care of the Child with Chronic Illness or Disability, Ch. 22
Family-Centered Home Care, Ch. 25
Genetic Evaluation and Counseling, Ch. 5
The Immobilized Child, Ch. 39
Immunizations (Tetanus), Ch. 12
Myelomeningocele (Meningomyelocele), Ch. 11
Neuromuscular Dysfunction
Weakness or abnormal performance of skeletal muscle may represent a defect in the muscle itself or reflect a pathologic disorder at some point along the neural pathway from the cortex of the brain to the neuromuscular junction. The identification of the source of muscular dysfunction includes not only the testing of muscle function but also the systematic elimination of possible disorders of neural structures on which muscle function depends for its stimulus. In a few disorders muscle disease may be accompanied by a neural disorder.
Some clinical features are shared by muscle disease (myopathy), which differs in many ways from muscular dysfunction resulting from disorders of neuronal structures—brain, cranial nerve nuclei, long nerve tracts, anterior horn cells of the spinal cord, and peripheral nerves. Motor function is accomplished by means of the simple reflex arcs or by way of impulses transmitted from the cerebral cortex and other centers in the brain through the various nerve pathways of the central nervous system (CNS). The upper motor neurons consist of cells that lie in the cerebral cortex and fibers that traverse the brainstem and spinal cord to terminate at their synapses with the anterior horn cells. The lower motor neurons consist of the anterior horn cells, axons, and peripheral nerve branches. The motor unit consists of the lower motor neuron, the neuromuscular (or myoneural) junction, and the muscle fibers it supplies (Fig. 40-1). The upper motor neuronal pathways from the cerebrum to the lower motor neuron are described as (1) pyramidal—those whose fibers extend from the cortex, come together in the medulla, cross from one side to the other, then extend down the cord to synapse with anterior horn motor neurons; and (2) extrapyramidal—a complex network of motor neurons that comprise relays between motor areas of the cortex, basal ganglia, thalamus, cerebellum, and brainstem.
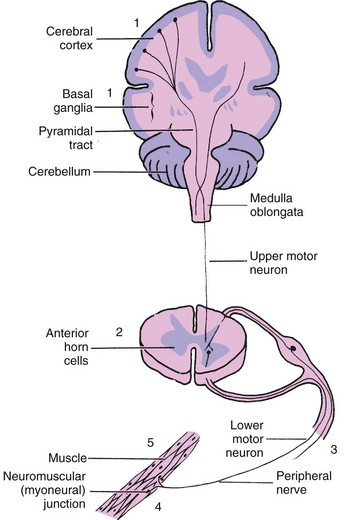
Fig. 40-1 Site of origin for neuromuscular disorders. 1, Cerebral palsy; 2, poliomyelitis, spinal muscular atrophy; 3, mononeuropathies, polyneuropathies; 4, myasthenia gravis, neurotoxic disorders; 5, muscular dystrophies.
Classification and Diagnosis
The site of pathologic disturbance determines the type of muscular dysfunction. In general, upper motor neuron lesions produce weakness associated with spasticity, increased deep tendon reflexes, and abnormal superficial reflexes. The primary disorder of upper motor neuron dysfunction is cerebral palsy (CP). Lower motor neuron lesions interrupt the reflex arc, causing weakness and atrophy of the skeletal muscles involved with associated hypotonia or flaccidity, which eventually progress to atrophy with varying degrees of contracture deformity. A disorder of the extrapyramidal pathway and the cerebellum rarely produces muscle weakness.
Lower motor neuron involvement is usually symmetric (except that of poliomyelitis and single peripheral nerve disease), whereas disorders of the pyramidal tract are more often asymmetric. Muscle wasting is characteristic of lower motor neuron lesions and more marked than in diseases of muscles. Deep tendon reflexes are briskly active in upper motor neuron disease, are diminished or absent in lower motor neuron disease, and depend on the progress of muscle degeneration in the myopathies.
These disorders can also be categorized according to onset: those in which there is acute onset of flaccid paralysis and those with more gradual onset and progressive degeneration. In most instances the sudden appearance of flaccid paralysis in a previously healthy child is due to an infectious process. Neurotoxins (e.g., botulism, tick paralysis, or heavy metal poisoning), pressure on the spinal cord from tumors or abscesses, and spinal cord injury (SCI) are less likely causes. Hereditary factors and metabolic disease are more often responsible for muscular weakness and atrophy of gradual onset.
Classification
The most useful classification of neuromuscular disorders is one that defines the site of origin of the pathologic lesion: the anterior horn cells of the spinal cord, the peripheral nerves, the neuromuscular junction, and the muscles.
Diseases of Anterior Horn Cells: Diseases and disorders that affect the anterior horn cells are the result of destruction or atrophy of the anterior horn of the spinal column along with the inability to transfer impulses from sensory neurons to motor neurons. Enteroviruses, which have a worldwide distribution, are prominent etiologic agents that selectively affect anterior horn cells. These include the polioviruses, of which there are three types: coxsackieviruses, groups A and B, and the enterocytopathogenic human orphan (ECHO) viruses. Inherited disorders, primarily the spinal muscular atrophies, cause degeneration of the anterior horn cells.
Neuropathies: Disorders affecting peripheral nerves may be mononeuropathies, which involve a single nerve and the muscles it innervates, or polyneuropathies, which involve multiple nerves and the muscles they supply. Neuropathies are caused by a number of hereditary diseases, traumatic injuries, infections, poisons, and (secondarily) some metabolic diseases. Polyneuropathy can be restricted to specific areas (as in diabetes mellitus). Some hereditary diseases involve skeletal muscles extensively. The distal limbs (feet and hands) are usually affected first, with gait disturbance and footdrop being early manifestations. The involvement gradually progresses proximally as the disorder becomes more severe.
In some polyneuropathies there is segmented or patchy loss of the myelin sheath of nerve fibers; in others the primary process appears to be progressive degeneration of nerve fibers. Examples of acute and chronic polyneuropathies are infectious polyneuritis and peroneal atrophy, respectively.
Neuromuscular Junction Disease: Disorders involving a neurohormonal deficiency interfere with transmission of nerve impulses to muscles at the neuromuscular junction. Normally nerve impulses are transmitted to skeletal muscles across the neuromuscular junction by acetylcholine. This is accomplished in three steps: (1) acetylcholine is released from vesicles in the terminal nerve endings; (2) it then diffuses across the junction and contacts receptor sites in the muscle membrane, stimulating the muscle to contract; and (3) it is removed by the action of cholinesterase. Interference at any of these three steps will block transmission of nerve impulses and prevent muscular contraction.
Several toxic substances act at the neuromuscular junction to inhibit nerve impulses to the skeletal muscles. Examples of toxins that prevent the release of acetylcholine are those that produce the paralysis of botulism and tick paralysis. Action at receptor sites is also blocked by the drug curare. Paralysis resulting from inhibition of cholinesterase release is caused by poisoning with organic phosphate insecticides.
Diseases of Muscles: Diseases of skeletal muscles can be inflammatory (such as polymyositis), the result of endocrine dysfunction (such as hypothyroidism and hyperthyroidism), or the result of congenital defects (e.g., absence of muscle, periodic paralysis, and the various muscular dystrophies [MDs] and myotonias). Inflammation occurs in a number of infectious illnesses such as trichinosis, toxoplasmosis, and those caused by the coxsackieviruses.
Diagnostic Tools
Several general diagnostic tools aid in differentiating diseases with similar manifestations. In addition, a number of more definitive tests are used to establish a specific diagnosis. The neurologic examination is a basic test that helps assess the extent of motor and sensory function.
The electromyogram (EMG) measures the electric potentials generated in individual muscles. A small metal disk is placed on the skin overlying the muscle to be tested, or a sterile needle electrode is inserted directly into the muscle. The electric activity generated in the skeletal muscles is measured at rest, with slight voluntary contraction, and with maximum contraction. The electric activity is amplified and displayed on a cathode ray oscilloscope. Needle electrodes are sensitive enough to pick up the activity of a single muscle fiber; thus this is usually the method of choice. However, the procedure is traumatic for children. It is often not useful because it requires cooperation. A topical anesthetic such as EMLA (eutectic mixture of local anesthetics) or LMX4 (4% lidocaine) applied to the EMG sites can decrease the amount of pain.
Nerve conduction velocity, or the velocity of electric impulse conduction along motor or sensory nerves, is often measured in conjunction with the EMG. Certain diseases affect the peripheral nerves, prolonging the conduction time from the point of stimulation of the nerve to the muscle and increasing the duration of the evoked potential of the muscle.
Muscle biopsy is the most useful laboratory examination to confirm and classify muscle disorders. The vastus lateralis is the most commonly sampled muscle. Procedural sedation may be accomplished with a number of medications used singly or in combination: midazolam and morphine; ketamine and midazolam; pentobarbital; etomidate; inhaled nitrous oxide; or midazolam and fentanyl. (See Surgical Procedures, Chapter 27.) Serum enzyme measurements are helpful in diagnosing and monitoring the course of muscular disease, but these are used as adjuncts in the diagnosis of most neuromuscular diseases. The intracellular enzyme creatine (phosphokinase) kinase (CK) is present in muscle tissues, including cardiac muscle, and the brain. It is released in large amounts in some muscular diseases, such as MD. CK is not elevated in neurogenic disease.
Cerebral Palsy
 A new definition describes CP as a “group of permanent disorders of the development of movement and posture, causing activity limitation, that are attributed to nonprogressive disturbances that occurred in the developing fetal or infant brain” (Rosenbaum, Paneth, Leviton, et al, 2007). In addition to motor disorders, the condition often involves disturbances of sensation, perception, communication, cognition, and behavior; secondary musculoskeletal problems; and epilepsy (Rosenbaum, Paneth, Leviton, et al, 2007). The cause, clinical features, and course are variable and are characterized by abnormal muscle tone and coordination as the primary disturbances. It is the most common permanent physical disability of childhood, and the incidence is reported to range from 2.4 to 3.6 in 1000 live births in various studies in the United States (Hirtz, Thurman, Gwinn-Hardy, et al, 2007; Yeargin-Allsopp, Van Naarden Braun, Doernberg, et al, 2008). In the 1960s the prevalence of CP rose approximately 20%, which most likely reflected the improved survival of extremely low–birth-weight (ELBW) and very low–birth-weight (VLBW) infants. However, in the past two decades there has been a decrease in the incidence of CP among ELBW and VLBW infants (Hack and Costello, 2008).
A new definition describes CP as a “group of permanent disorders of the development of movement and posture, causing activity limitation, that are attributed to nonprogressive disturbances that occurred in the developing fetal or infant brain” (Rosenbaum, Paneth, Leviton, et al, 2007). In addition to motor disorders, the condition often involves disturbances of sensation, perception, communication, cognition, and behavior; secondary musculoskeletal problems; and epilepsy (Rosenbaum, Paneth, Leviton, et al, 2007). The cause, clinical features, and course are variable and are characterized by abnormal muscle tone and coordination as the primary disturbances. It is the most common permanent physical disability of childhood, and the incidence is reported to range from 2.4 to 3.6 in 1000 live births in various studies in the United States (Hirtz, Thurman, Gwinn-Hardy, et al, 2007; Yeargin-Allsopp, Van Naarden Braun, Doernberg, et al, 2008). In the 1960s the prevalence of CP rose approximately 20%, which most likely reflected the improved survival of extremely low–birth-weight (ELBW) and very low–birth-weight (VLBW) infants. However, in the past two decades there has been a decrease in the incidence of CP among ELBW and VLBW infants (Hack and Costello, 2008).
 Critical Thinking Case Study—Cerebral Palsy
Critical Thinking Case Study—Cerebral Palsy
Etiology
A variety of prenatal, perinatal, and postnatal factors contribute to the development of CP, singly or multifactorially (Box 40-1). The human brain undergoes development during the prenatal period and up to 2 years of age. A brain insult or injury occurring during this period may result in CP.
Although the prevalent traditional hypothesis has been that CP results from perinatal problems, especially birth asphyxia, it is now believed that CP results more often from existing prenatal brain abnormalities. However, the exact cause of these abnormalities remains unknown. It has been estimated that as many as 70% to 80% of the cases of CP are caused by unknown prenatal factors (Krigger, 2006). Intrauterine exposure to maternal chorioamnionitis is associated with an increased risk of CP in infants of normal birth weight and preterm infants (Hermansen and Hermansen, 2006); however, not all term infants exposed to chorioamnionitis develop CP (Grether, Nelson, Walsh, et al, 2003; Wu, Escobar, Grether, et al, 2003).
In general, infants exposed to maternal and perinatal infections are at increased risk for the development of CP as a result of the effects on the developing brain. Although CP occurs in term births, preterm birth of ELBW and VLBW infants continues to be the single most important risk factor for CP. Still, in some cases no identifiable cause is determined. Periventricular leukomalacia and intracerebral hemorrhage in low-birth-weight infants are significant risk factors in the development of CP. Perinatal ischemic stroke is also associated with a later diagnosis of CP (Golomb, Saha, Garg, et al, 2007). White matter abnormalities such as focal lesions are present in a large number of preterm infants subsequently diagnosed with CP (Johnston, 2007). Damage occurring as a result of shaken baby syndrome may also result in CP in survivors. Additional factors that may contribute to the development of CP postnatally include bacterial meningitis, viral encephalitis, motor vehicle accidents, and child abuse (Krigger, 2006). In summary, as many as 80% of the total cases of CP may be linked to a perinatal or neonatal brain lesion or brain maldevelopment, regardless of the cause (Krageloh-Mann and Cans, 2009).
A number of biochemical disorders may cause motor abnormalities often seen in CP and may be initially misdiagnosed as CP (Nehring, 2010).
Pathophysiology
It is difficult to establish a precise location of neurologic lesions on the basis of etiology or clinical signs because there is no characteristic pathologic picture. In some cases there are gross malformations of the brain. In others there may be evidence of vascular occlusion, atrophy, loss of neurons, and laminar degeneration that produce narrower gyri, wider sulci, and low brain weight. Anoxia appears to play the most significant role in the pathologic state of brain damage, which is often secondary to other causative mechanisms.
There are a few exceptions. In some cases the manifestations or etiology are related to anatomic areas. For example, CP associated with preterm birth is usually spastic diplegia caused by hypoxic infarction or hemorrhage with periventricular leukomalacia in the area adjacent to the lateral ventricles. The athetoid (extrapyramidal) type of CP is most likely to be associated with birth asphyxia but can also be caused by kernicterus and metabolic genetic disorders such as mitochondrial disorders and glutaricaciduria (Johnston, 2007). Hemiplegic (hemiparetic) CP is often associated with a focal cerebral infarction (stroke) secondary to an intrauterine or perinatal thromboembolism, usually a result of maternal thrombosis or hereditary clotting disorder (Johnston, 2007). Cerebral hypoplasia and, sometimes, severe neonatal hypoglycemia are related to ataxic CP. Generalized cortical and cerebral atrophy often cause severe quadriparesis with cognitive impairment and microcephaly.
Clinical Classification
A revision of the Winter classification was proposed in 2005 to reflect the child’s actual clinical problems and their severity, an assessment of the child’s physical and quality-of-life status across time, and long-term support needs (Bax, Goldstein, Rosenbaum, et al, 2005; Nehring, 2010). The proposed new definition has four major dimensions of classification (Bax, Goldstein, Rosenbaum, et al, 2005):
Motor abnormalities—Nature and typology of the motor disorder; functional motor abilities
Associated impairments—Seizures; hearing or vision impairment; attentional, behavioral, communicative, and/or cognitive deficits; oral motor and speech function
Anatomic and radiologic findings—Anatomic distribution or parts of the body affected by motor impairments or limitations; radiologic findings sometimes including white matter lesions or brain anomaly noted on computed tomography (CT) or magnetic resonance imaging (MRI)
Causation and timing—Identification of a clearly identified cause such as a postnatal event (e.g., meningitis, traumatic brain injury)
CP has four primary types of movement disorders: spastic, dyskinetic, ataxic, and mixed (Box 40-2) (Nehring, 2010). The most common clinical type, spastic CP, represents an upper motor neuron muscular weakness. See Box 40-3 for types of spastic CP. The reflex arc is intact, and the characteristic physical signs are increased stretch reflexes, increased muscle tone, and (often) weakness. Early neurologic manifestations are usually generalized hypotonia or decreased tone that lasts for a few weeks or may extend for months or even as long as a year.
Clinical Manifestations
The alert observer may suspect CP when a child demonstrates some of the following groups of manifestations (Box 40-3).
Delayed Gross Motor Development: Delayed gross motor development is a universal manifestation of CP. The child shows a delay in all motor accomplishments, and the discrepancy between motor ability and expected achievement tends to increase with successive developmental milestones as growth advances. It is especially significant if other developmental behaviors, such as language and personal-social achievement, are normal. Delayed development of the ability to balance may also slow the progression of milestones.
Abnormal Motor Performance: Neuromotor dysfunction is particularly evident in motor performance. An early sign is preferential unilateral hand use that may be apparent at approximately 6 months of age. Hand dominance does not normally develop until the preschool years. Abnormal crawling with propulsion by hand movements only and with lower extremities and hips hiked along, much like a “bunny hop,” occurs in diplegia. Children with hemiplegia have an asymmetric crawl, using the unaffected arm and leg to propel themselves on either the buttocks or the abdomen. Spasticity may cause the child to stand or walk on the toes. Uncoordinated or involuntary movements are characteristic of dyskinetic CP, and facial grimacing and writhing movements of the tongue, fingers, and toes are signs of athetosis. Other significant signs of motor dysfunction are poor sucking and feeding difficulties, with persistent tongue thrust. Head staggering, tremor on reaching, and truncal ataxia are also common. Hand preference in the first 2 years of life is reported to be a sign of hemiplegic CP (Berker and Yalçin, 2008).
Alterations of Muscle Tone: Increased or decreased resistance to passive movements is a sign of abnormal muscle tone. The child may exhibit opisthotonic postures (exaggerated arching of the back) and may feel stiff on handling or dressing. Also, there is difficulty in diapering because of spasticity of the hip adductor muscles and lower extremities. When pulled to a sitting position, the child may extend the entire body and be rigid and unbending at the hip and knee joints. This is an early sign of spasticity.
Abnormal Posture: Children with spastic CP assume abnormal postures at rest or when their position is changed. From an early age, a child lying in a prone position will maintain the hips higher than the trunk with the legs and arms flexed or drawn under the body. In the supine position spasticity is evident by scissoring (legs in crossed position; knees, hips, and ankles stiff) and extension of the legs, with the feet plantar flexed. This posture is exaggerated when the child is suspended vertically or when others try to make the child bear weight. Depending on the degree of impairment, spasticity may be mild or severe. A persistent infantile resting and sleeping posture (i.e., arms abducted at shoulders, elbows flexed, and hands fisted) is a sign of spasticity when it remains constant after 4 to 5 months of age. The hemiparetic child may rest with the affected arm adducted and held against the torso, with the elbow pronated and slightly flexed and the hand closed.
Reflex Abnormalities: Persistence of primitive reflexes is one of the earliest clues to CP (e.g., obligatory tonic neck reflex at any age or nonobligatory persistence beyond 6 months of age, and the persistence or even hyperactivity of the Moro, plantar, and palmar grasp reflexes). Hyperreflexia, ankle clonus, and stretch reflexes can be elicited from many muscle groups on fast passive movements (e.g., resistance to passive abduction when the hips are suddenly separated [adductor catch]).
Associated Disabilities and Problems: Some of the disabilities associated with CP are visual impairment, hearing impairment, behavioral problems, communication and speech difficulties, seizures, and intellectual impairment. Additional sensory deficits such as hypersensitivity, hyposensitivity, and balance difficulties may occur in children with CP (Nehring, 2010).
Intellectual impairment is a concern, although children with CP have a wide range of intelligence and 50% to 60% are within normal limits. Speech difficulties are often interpreted as a sign of cognitive impairment. Assessing the intelligence of a child with CP is often difficult because of the motor and sensory deficits. Tests carried out periodically over time should determine the degree of intelligence. Many persons with CP who have severely limiting physical involvement actually have the least intellectual impairment. As a group, children with athetosis and ataxia are intellectually superior to those with other types of CP. The incidence of severe or profound impairment is highest in rigid, atonic, and quadriparetic CP.
The manifestations of attention deficit hyperactivity disorder may occur in children with CP. The primary presenting symptoms are poor attention span, marked distractibility, hyperactive behavior, and defects of integration. (See Chapter 18.) Seizures are more likely to accompany postnatally acquired hemiplegia. They are an unusual finding in ataxia and diplegia. The most common types of seizures are generalized tonic-clonic seizures and minor motor types (Nehring, 2010).
Poor control of oral musculature may contribute to a number of problems. Abnormal posture and motor performance and alterations in muscle tone affect chewing, swallowing, and talking. Occupational and speech-language therapy interventions may be necessary to assist some children with feeding and speech. Coughing and choking, especially while eating, may predispose the child with CP to aspiration, which may not be readily apparent. Respiratory problems may result from and coexist with feeding difficulties in children with CP; respiratory symptoms observed during feedings include apnea, dyspnea, tachypnea, coughing and choking, and hypoxemia (Nehring, 2010). Many children with CP may also have gastroesophageal reflux.
Motor impairment associated with CP contributes to other problems. Children with CP who are nonambulatory have an increased risk of developing orthopedic complications such as unilateral or bilateral hip dislocations, scoliosis, and joint contractures resulting from unbalanced muscle tone. A variety of factors, including decreased mobility, decreased fluid intake, a fear of toileting, poor positioning on the toilet, and lack of fiber intake may be responsible for constipation (Nehring, 2010). Stool softeners, laxatives, and a bowel management program may be required to prevent chronic constipation.
Increased incidence of dental caries results from (1) improper dental hygiene, (2) congenital enamel defects (hypoplasia of primary teeth), (3) high carbohydrate intake and retention, (4) dietary imbalance with poor nutritional intake, (5) inadequate fluoride, and (6) difficulty in mouth closure and drooling. Spastic or clonic movements can cause gagging or biting down on the toothbrush, thus interfering with cleaning techniques. Oral hypersensitivity is also common, which causes the child to resist dental hygiene. Malocclusion can occur in as many as 90% of these children. Gingivitis is secondary to inadequate dental hygiene and may be further complicated by the use of antiepileptic drugs (AEDs) such as phenytoin (Nehring, 2010).
Skin breakdown may occur with prolonged positioning, especially with underweight children with bony prominences and those who are unable to reposition themselves or who may have insensate areas of skin.
Nystagmus and amblyopia are common and may require surgery, corrective lenses, or both. Hearing impairment is also common in children with CP. Some loss is caused by sensorineural involvement. Affected infants may spend increased amounts of time lying flat. This predisposes them to otitis media, which may result in conductive hearing loss. (See Chapter 24.)
Diagnostic Evaluation
Infants at risk according to known etiologic factors associated with CP warrant careful assessment during early infancy to identify the signs of muscular dysfunction as early as possible. The neurologic examination and history are the primary modalities for diagnosis. Neuroimaging of the child with suspected brain abnormality and CP is now recommended for diagnostic assessment, with MRI being the preferred method to identify the lesions or abnormalities associated with CP (Ashwal, Russman, Blasco, et al, 2004). Metabolic and genetic testing is recommended if no structural abnormality is identified by neuroimaging; laboratory tests are no longer recommended in the diagnostic process for CP (Ashwal, Russman, Blasco, et al, 2004).
Early recognition is made more difficult by the lack of reliable neonatal neurologic signs. However, the nurse should monitor infants with known etiologic risk factors and evaluate them closely in the first 2 years of life. Box 40-4 lists some warning signs, but these are not diagnostic. Because cortical control of movement does not occur until later in infancy, motor impairment associated with voluntary control is usually not apparent until after 2 to 4 months of age at the earliest. More often the diagnosis cannot be confirmed until the age of 2 years because motor tone abnormalities may be indicative of another neuromuscular illness. In addition, some children who show signs consistent with CP before 2 years do not demonstrate such signs after 2 years (Nehring, 2010). However, there is no consensus regarding an age cut-off for the onset of symptoms (Ashwal, Russman, Blasco, et al, 2004).
Establishing a diagnosis may be easier with the persistence of primitive reflexes: (1) either the asymmetric tonic neck reflex or persistent Moro reflex (beyond 4 months of age), and (2) the crossed extensor reflex. The tonic neck reflex normally disappears between 4 and 6 months of age. An obligatory response is considered abnormal. This is elicited by turning the infant’s head to one side and holding it there for 20 seconds. When a crying infant is unable to move from the asymmetric posturing of the tonic neck reflex, it is considered obligatory and an abnormal response. The crossed extensor reflex, which normally disappears by 4 months, is elicited by applying a noxious stimulus to the sole of one foot with the knee extended. Normally the contralateral foot responds with extensor, abduction, and then adduction movements. The possibility of CP is suggested if these reflexes occur after 4 months.
A number of assessment instruments are now available to evaluate muscle spasticity (Modified Ashworth Scale); functional independence in self-care, mobility, and cognition (Functional Independence Measure and WeeFIM [specific to children]); self-initiated movements over time (Gross Motor Function Measure); and capability and performance of functional activities in self-care, mobility, and social function (Pediatric Evaluation of Disability Inventory) (Krigger, 2006).
A thorough knowledge of normal variations of motor development is required for detecting abnormal progress, and a careful history is necessary to detect possible etiologic factors. Observe the child’s spontaneous movements and behavior, including posture; attitude; and muscle size, function, and tone. Because children with CP often have sensory deficits, it is appropriate to evaluate the child for hearing and vision deficits.
Therapeutic Management: General Concepts
The goals of therapy for children with CP are early recognition and promotion of an optimum developmental course to enable affected children to attain their potential within the limits of their dysfunction. The disorder is permanent, and therapy is chiefly symptomatic and preventive.
The beneficial influences of a habilitation program on both child and family are based on recognizing the disability as early as possible and implementing treatment. Parents are essential to a treatment program. Consider their goals and desires, their cooperation, and their confidence in all aspects of management. With early diagnosis parents can begin to provide the sensorimotor experiences essential to cognitive development, since CNS structures depend on stimulation and use to attain and maintain their functional integrity.
The broad aims of therapy are to (1) establish locomotion, communication, and self-help; (2) gain optimum appearance and integration of motor functions; (3) correct associated defects as early and effectively as possible; (4) provide educational opportunities adapted to the individual child’s needs and capabilities; and (5) promote socialization experiences with other affected and unaffected children. Each child is evaluated and managed on an individual basis. The plan of therapy may involve a variety of settings, facilities, and specially trained persons. The scope of the child’s needs requires multidisciplinary planning and care coordination among professionals and the child’s family (Box 40-5). The outcome for the child and family with CP is normalization and promotion of self-care activities that empower the child and family to achieve maximum potential.
Mobilizing Devices: Many children with CP wear ankle-foot orthoses (AFOs) (braces) and a variety of orthotics. Orthotics are molded to fit the feet and are worn inside the shoes. Devices are often used to help prevent or reduce deformity, increase the energy efficiency of gait, and control alignment. Some of the more commonly used mobility devices include wheeled scooter boards that allow children to propel themselves while the abdomen or total body is supported and the legs are positioned with wedges to prevent scissoring. Wheeled go-carts provide good sitting balance and serve as an early “wheelchair” experience for young children. Strollers can be equipped with custom seats for dependent mobilization. Special devices for independent mobilization that may or may not allow the upper extremities to remain free are particularly valuable for children with lower extremity involvement (Fig. 40-2). A number of wheelchairs can be customized to meet the needs and preferences of older children. (See Mobilization Devices, Chapter 39.)
Surgery: Surgical intervention is usually reserved for the child who does not respond to the more conservative measures such as orthotics, but it is also indicated for the child whose spasticity causes progressive deformities. Orthopedic surgery may be required to correct contracture or spastic deformities, to provide stability for an uncontrollable joint, to address bone malalignment (e.g., lever arm dysfunction), and to provide balanced muscle power. This includes tendon-lengthening procedures (especially heel-cord lengthening), release of spastic wrist flexor muscles, and correction of hip and adductor muscle spasticity or contracture to improve locomotion. Orthopedic surgery is generally not performed until after the child is 6 years of age (Nehring, 2010). Surgery is used primarily to improve function rather than for cosmetic purposes and is followed by physical therapy. Surgery may also be performed to improve feedings, correct gastroesophageal reflux disease, and correct associated dental problems (Nehring, 2010).
Neurosurgical procedures are used only in selected cases. Selective dorsal rhizotomy has provided marked improvement in some children with CP (Nordmark, Josenby, Lagergren, et al, 2008). However, achieving the benefits from the surgery requires intensive physical therapy and family commitment. Because the procedure results in flaccid muscles, the child must relearn to sit, stand, and walk.
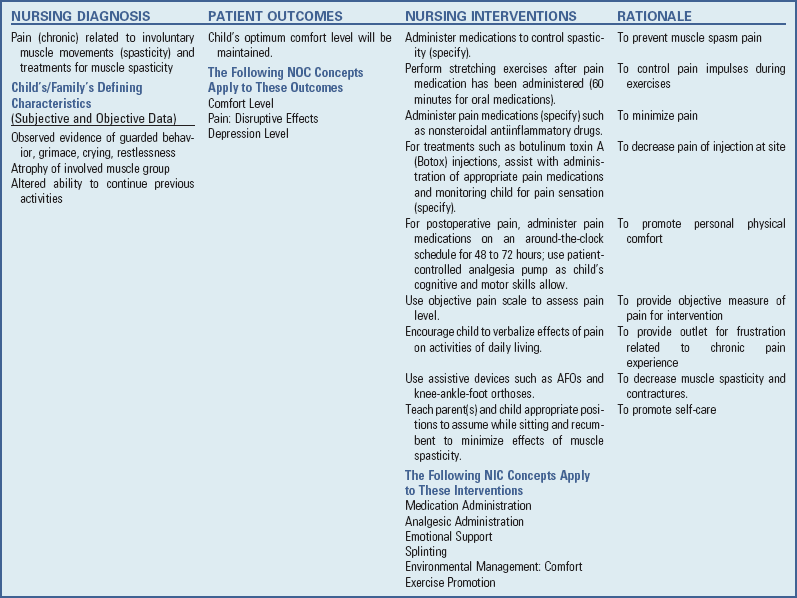
Medication: Intense pain may occur with muscle spasms in patients with CP. Children with CP may also experience pain as a result of painful procedures such as injection with botulinum toxin type A (Botox), surgical procedures intended to reduce contracture deformities, abdominal pain related to position and gastroesophageal reflux, and pain associated with physical therapy (McKearnan, Kieckhefer, Engel, et al, 2004). Therefore pain management is an important aspect of the care of the child with CP.
Inhaled nitrous oxide or oral midazolam may be used for sedation during botulinum toxin A injections. In one study children who received inhaled nitrous oxide had fewer side effects than those receiving midazolam (Zier, Rivard, Krach, et al, 2008).
Pharmacologic agents given orally (dantrolene sodium [Dantrium], baclofen [Lioresal], and diazepam [Valium]) have had little effectiveness in improving muscle coordination in children with CP. However, they are effective in decreasing overall spasticity. The most common side effects of these agents include hepatotoxicity (dantrolene), drowsiness, fatigue, and muscle weakness. Less commonly, diaphoresis and constipation may occur with oral baclofen; other possible complications include hallucinations, mood changes, seizures, nausea, and urinary incontinence. Diazepam is used frequently but should be restricted to older children and adolescents.
Botulinum toxin A is also used to reduce spasticity in targeted muscles of the upper and lower extremities (Lukban, Rosales, and Dressler, 2009). Botulinum toxin A is injected into a selected muscle (commonly the quadriceps, gastrocnemius, or medial hamstrings), where it acts to inhibit the release of acetylcholine into a specific muscle group, thereby preventing muscle movement. When it is administered early in the course of the illness, this may prevent affected muscle contractures, particularly in lower extremities, thus avoiding surgical procedures with possible adverse effects. The goal is to allow stretching of the muscle as it relaxes and permits ambulation with an AFO. The major reported adverse effects of botulinum toxin A injection include pain at the injection site and a temporary weakness (Lukban, Rosales, and Dressler, 2009; Roscigno, 2002). Prime candidates for botulinum toxin A injections are children with spasticity confined to the lower extremities. The onset of action occurs within 24 to 72 hours, with a peak effect observed at 2 weeks and a duration of action of 3 to 6 months (Green, Greenberg, and Hurwitz, 2003).
The neurosurgical and pharmacologic approach to managing the spasticity associated with CP involves the implantation of a pump to infuse baclofen directly into the intrathecal space surrounding the spinal cord to provide relief of spasticity. High doses of oral baclofen are associated with significant side effects, including drowsiness and confusion, yet are often unable to provide adequate relief of spasticity. Direct infusion of baclofen into the intrathecal space provides relief without as many side effects (Krach, 2001).
Patients may be screened before pump placement by the infusion of a “test dose” of intrathecal baclofen delivered via a lumbar puncture. Close monitoring for side effects (hypotonia, somnolence, seizures, nausea, vomiting, headache, and catheter- or pump-related problems) (Albright, Gilmartin, Swift, et al, 2003) and relief of spasticity occurs for several hours after the infusion. If a positive effect occurs, the patient is considered a candidate for pump placement.
The pump is placed in the subcutaneous space of the midabdomen. An intrathecal catheter is tunneled from the lumbar area to the abdomen and connected to the pump. The pump is filled with baclofen and programmed to provide a set dose using a telemetry wand and a computer. The patient remains hospitalized for 3 to 7 days to adjust the dosage and ensure proper healing. Outpatient visits to refill the pump and make dosage adjustments occur about every 4 to 6 weeks, depending on the patient’s response to the treatment. Benefits of intrathecal baclofen include fewer systemic side effects, dosage titration for maximizing effects, and reversibility of therapy with removal of the pump if so desired. Abrupt withdrawal of intrathecal baclofen, especially at high doses, may result in adverse effects such as rebound spasticity, pruritus, hyperthermia, rhabdomyolysis, disseminated intravascular coagulation, multiorgan failure, and death; in some cases intrathecal baclofen withdrawal may mimic sepsis (Zuckerbraun, Ferson, Albright, et al, 2004). Treatment of withdrawal centers on reestablishing the medication dosage, with improvements observed within 1 to 2 hours. Hospitalization and surgery may be required for withdrawal as a result of pump or catheter failure.
AEDs such as carbamazepine (Tegretol), divalproex (valproate sodium and valproic acid; Depakote), oxcarbazepine (Trileptal), and lamotrigine (Lamictal) are prescribed routinely for children who have seizures. Gabapentin (Neurontin) has been used in adults with SCI to decrease spasticity with success. No studies are available on the effectiveness of the drug in children (Krach, 2001). The α2-adrenergic agonists clonidine (Catapres) and tizanidine (Zanaflex) have been used to decrease spasticity in adults with SCI and multiple sclerosis; however, their use in children does not appear to have gained widespread acceptance in the United States. Oral tizanidine given in conjunction with botulinum type A has been reported to be more effective than oral baclofen and botulinum type A in one study of children with CP (Dai, Wasay, and Awan, 2008). Monitor all medications for maintenance of therapeutic levels and avoidance of subtherapeutic or toxic levels.
Children with CP have been treated with a number of complementary and alternative medicine strategies, including Chinese herbs, acupuncture, growth hormone therapy, aquatic exercise, equine-assisted therapy, and hyperbaric oxygen (Nehring, 2010). Gasalberti (2006) reported some alternative therapies being used in children with disabilities that the practitioner may overlook during a health history but that may be beneficial to such children. These include pet therapy, massage, hippotherapy (horse riding), music, and color-light therapy. Other alternative therapies that may be used by families with children with disabilities include vitamins, prayer, meditation, hypnosis, and guided imagery.
Technical Aids: A wide variety of technical aids are available to improve the functioning of children with CP. These include electromechanical toys that employ the concept of biofeedback and operate from a head unit. The toy is manipulated only when the head and trunk are in correct alignment. Computerized toys and games can also enhance eye-hand coordination.
Microcomputers combined with voice synthesizers help children with speech difficulties to “speak.” These and other devices print messages onto screen monitors and paper. These devices have made it apparent that some children have been erroneously considered to be cognitively impaired. Microcomputers have also increased the possibilities for increased mobility via wheelchairs and specially designed mobilization devices.
Many other electronic devices allow independent functioning. Sensors can be activated and deactivated using a head-stick, a voluntary muscle such as the tongue, or any other voluntary muscle movement over which the child has control. (See Figs. 24-4 and 24-5.) The application of this technology makes it possible for older persons with CP to eventually function in their own apartments and can be extended into the workplace.
Associated Problems: Children with CP often have sensory deficits, which require the attention of appropriate specialists. Speech-language therapy involves the services of a speech-language pathologist (SLP) who may also assist with feeding problems. (See Chapter 24.) Dental care is especially important for children with CP and often is overlooked. Regular visits to the dentist and dental prophylaxis, including brushing, fluoride, and flossing (after several teeth are present), should begin as soon as the teeth erupt. This is especially important for children given phenytoin, who often develop gum hyperplasia. Additional problems common among children with CP include constipation caused by neurologic deficits and lack of exercise; poor bladder control and urinary retention; chronic respiratory tract infections and aspiration pneumonia, which occur as a result of gastroesophageal reflux, abnormal muscle tone, immobility, and altered positioning; and skin problems as a result of altered positioning, poor nutrition, and immobility. Hip dislocation occurs often in children with CP. Latex allergy has also been reported in children with CP (Nehring, 2010).
Therapeutic Management: Therapies, Education, Recreation
Physical Therapy: Physical therapy is one of the most commonly used treatment modalities in children with CP. In general, physical therapy is directed toward good skeletal alignment for the child with spasticity; training in purposeful acts, even in the face of involuntary motion, for the child with athetosis; and gait training and maximum development of proprioceptive sense for the child with ataxia.
An active therapy program involves the family, the physical therapist (PT), the occupational therapist (OT), and other members of the health care team. Developing a treatment program that can be carried out at home is of utmost importance. The major approach uses traditional types of therapeutic exercises that consist of stretching, passive, active, and resistive movements applied to specific muscle groups or joints to maintain or increase range of motion, strength, or endurance.
No therapeutic approach is able to achieve spectacular changes in the ultimate outcome of motor disability. Early efforts focus on alleviating abnormal postures by positioning and range-of-motion exercises. Passive range-of-motion exercises, stretching, and elongation exercises are valuable at any age, even when the child is too young to cooperate. Some active extension can be performed when the child is old enough to cooperate, with passive motion applied to complete joint extension. Prevention of contracture deformity is a prime function of physical therapy. Seating and mobility are other key goals.
Functional and Adaptive Training (Occupational Therapy): Training in manual skills and activities of daily living (ADLs) proceeds along developmental lines and according to the child’s functional level. Sitting, balancing, crawling, and walking are encouraged at appropriate ages and are accompanied by stimulation of protective extension and equilibrium reactions. Hand activities are begun early to improve motor function and provide the child with sensory experiences and information about the environment. As the child progresses from simple feeding and self-care activities, training is extended to include other tasks (e.g., cooking or use of keyboard or computer mouse) that are within the child’s developmental and functional capabilities.
Incorporating play into the therapeutic program often requires great ingenuity and inventiveness from those involved in the child’s care. Objects and toys are chosen to provide needed sensory input using a variety of shapes, forms, and textures. Nurses can help parents integrate therapy into play activities in natural ways.
Children with CP may need considerable help (and patience) in learning to feed and dress themselves and care for personal hygiene needs. A feeding program may be developed by an OT in conjunction with an SLP. Children should be fed in the normal eating position. When they have difficulty sucking and swallowing, it is tempting to hold them in a semireclining posture to make use of gravity flow. However, this method does not promote active swallowing, and the neck hyperextension may even interfere with swallowing. A more flexed sitting position, with the arms brought forward to decrease the tendency toward back and neck extension, is more natural during bottle- or spoon-feeding and encourages active swallowing.
Because jaw control is compromised, more normal control can be achieved if the feeder stabilizes the oral mechanism from the side or front of the face. When directed from the front, the middle finger of the nonfeeding hand is placed posterior to the bony portion of the chin, the thumb is placed below the bottom lip, and the index finger is placed parallel to the child’s mandible (Fig. 40-3). Manual jaw control from the side assists with head control, correction of neck and trunk hyperextension, and jaw stabilization. The middle finger of the nonfeeding hand is placed posterior to the bony portion of the chin, the index finger is placed on the chin below the lower lip, and the thumb is placed obliquely across the cheek to provide lateral jaw stability (Fig. 40-4).
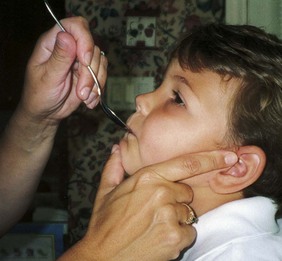
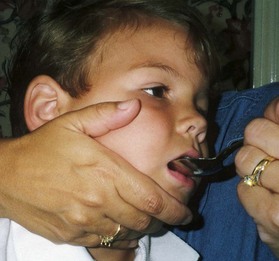
In all ADLs it is important to capitalize on the child’s assets and compensate for liabilities. The level of expected independence is related to both gross and fine motor manipulation. Even when complete independence in a specific activity is not realistic, the child should learn any part of the task that he or she can master. However, motor function is not the sole purpose of learning to be as independent as possible. Any accomplishment promotes self-reliance and self-esteem for healthier personality development.
Speech Therapy: Speech training under the supervision of an SLP begins early, before the child learns poor habits of communication. Parents and others can help by following the directions of the speech therapist and by talking to the child slowly while using pictures or handling objects about which the adult is speaking. Feeding techniques such as forcing the child to use the lips and tongue in eating facilitate speech. An example of this technique is placing food at the side of the tongue, first one side, then the other; making the child use the lips to take food from a spoon rather than placing it directly on the tongue. If severe dysarthria prevents articulate speech and the child has reasonable intelligence, the child learns nonverbal communication (e.g., sign language). (See Chapter 24.)
Education: As in all aspects of care, educational requirements are determined by the child’s needs and potential. This includes the severity of the child’s disease and the presence and degree of associated conditions that affect learning and participation, such as learning impairment, abnormal actions or behavior, impaired vision or hearing, and seizures. Children with mild to moderate involvement are generally able to participate, for varying amounts of time, in regular classes. Resource rooms are available in most schools to provide more individualized attention to a child’s particular needs. Integration of these children into regular classrooms should be the initial goal. Teachers’ assistants often work one-on-one with children in both settings. A training program may be appropriate for those children who are unable to benefit from formal education. Prevocational and vocational counseling and guidance are arranged at adolescence. Education is geared toward the child’s assets at any phase or in any setting. Nurses should be aware of early intervention programs and provisions for special education and related services for children (see Box 24-3) and should support parents in their efforts to obtain appropriate educational services for the child.
Recreation: Recreational activities are also a necessary part of growing up. Recreational outlets and after-school activities should be an option for the child who is unable to participate in regular athletic and other peer activities. Some can compete in athletic and artistic endeavors, and many games and pastimes are suited to their capabilities. Sports, physical fitness, and recreation programs are encouraged for children with CP, and young children should be exposed to all physical activities available to children without disabilities. Individual sports such as the martial arts (e.g., TaeKwonDo) in which groups are small and the emphasis is on discipline and balance are also enjoyable to many children with CP. Many states mandate adaptive physical education classes.
Numerous developmental centers have facilities for indoor and outdoor activities designed to appeal to children of all ages. If these are not available, they should be developed. However, such programs require adequate supervision to avoid any harmful effects. Recreational activities serve to stimulate children’s interest and curiosity, help them adjust to their disability, improve their functional abilities, and build self-esteem. Competitive sports are also becoming increasingly available to children with disabilities and offer an added dimension to physical activities. For more information access the United Cerebral Palsy website (www.ucp.org) and go to the Sports and Leisure link.
Prognosis: The prognosis for the child with CP depends largely on the type and severity of the condition. Children with mild to moderate involvement (85%) have the capability of achieving ambulation between the ages of 2 and 7 years (Berker and Yalçin, 2008). If the child does not achieve independent ambulation by this time, chances are poor for ambulation and independence. Approximately 30% to 50% of individuals with CP have significant cognitive impairments, and an even higher percentage have mild cognitive and learning deficits (Green, Greenberg, and Hurwitz, 2003; Liptak and Accardo, 2004). However, many children with severe spastic tetraplegic CP have normal intelligence. Individuals with CP and severe cognitive impairment had lower survival rates in an Australian study (Blair, Watson, Badawi, et al, 2001). Growth is affected in children with spastic tetraplegia, and many children remain below the 5th percentile for age and sex.
As children with CP become adults, about 30% remain in the home and are cared for by a parent or caregiver; 50% of individuals with spastic tetraplegia live in independent settings and function at appropriate social levels considering their disability (Green, Greenberg, and Hurwitz, 2003). Vocational rehabilitation and higher education are possible for adults with CP. One study found that 53% of all persons with CP were able to work outside the home in regular jobs; one third of the severely disabled adults with CP worked outside the home (Murphy, Molnar, and Lankasky, 2000). Children with severe CP mobility impairment and feeding problems often succumb to respiratory tract infection in childhood. The few survival rate studies on children or adults with CP show that survival is influenced by existing comorbidities (Nehring, 2010).
Nursing Care Management
Assessment: Nursing assessment includes risk identification of infants with etiologic factors that are associated with CP. Ongoing assessment of infants for abnormal muscle tone, inability to achieve developmental milestones, and persistence of neonatal reflexes alert the nurse to investigate further.
Reinforce Therapeutic Plan and Assist in Normalization: Because children are being treated at an earlier age, parents are participating at an earlier stage in treatment programs for their disabled child. They learn the proper handling and home care of young children with CP and need carefully programmed steps so that their expanded parental role can be melded into the established relationship. Close work with other multidisciplinary team members is essential. Nurses reinforce the therapeutic plan and assist the family in devising and modifying equipment and activities to continue the therapy program in the home. (See also Chapter 24.)
Some children have difficulty keeping their heads upright. Because of this, they can neither explore much of their environment nor process the information. Parents need to be complimented on their efforts to provide a stimulating environment for these children. These infants are at risk for delayed development in holding up their heads, righting their shoulders and trunks for stable posture, sitting, pulling, standing, and crawling. Most parents of children with impaired movements benefit from support and practical suggestions for feeding, moving, holding, and encouraging the infant to explore hands and feet and to play. Helping parents incorporate therapeutic suggestions into typical daily activities is an important normalizing strategy. (See Chapter 22 for a discussion of normalization.)
Although practical advice is important, the nurse, OT, or PT should offer suggestions at a pace that can be absorbed by the parents. Encourage the parents to define their concerns, acknowledge the concerns as genuine, and ask the parents what approach(es) they have tried and for how long. In this way the nurse is able to find out what works, what does not work, and what the parents would like to try next. Give the parents positive feedback for their observations of the infant, the progress they note, and how they differentiate the child’s needs.
Address Health Maintenance Needs: Because children with CP expend so much energy in their efforts to accomplish ADLs, more frequent rest periods should be arranged to avoid the fatigue that may aggravate their limited capabilities. Meeting the child’s nutritional needs may be a challenge because of gastroesophageal reflux, feeding and swallowing difficulties, chronic constipation and subsequent anorexia, and absence or diminished ability to independently feed himself or herself (Jones, Morgan, and Shelton, 2007). As a result of being ELBW or VLBW in combination with these feeding problems, children with CP are at risk for growth failure (failure to thrive), and the nurse must ensure an adequate caloric intake. Children with spasticity expend more energy and often require more energy intake than same age counterparts to maintain adequate growth. Nutritional supplements such as high-calorie milk products (e.g., Pediasure, Ensure, Boost) may be necessary to provide adequate caloric intake. Additional nutritional concerns include providing adequate intake of fruits and fiber to enhance gastrointestinal motility, routinely monitoring child’s growth on a standardized growth chart, and avoiding overfeeding and obesity (Jones, Morgan, and Shelton, 2007).
Routine assessment of skin status is imperative in children with CP who are limited in movement or who must remain in assistive devices such as a wheelchair for a prolonged period. The overall nutritional status may also be a risk factor for skin breakdown. Care must be taken to ensure that adequate objective skin assessments are routinely performed. If skin breakdown does occur, consult a skin and wound specialist for treatment and further prevention.
Gastrostomy feedings may be necessary to supplement regular feedings and ensure adequate weight gain, particularly in children at risk for growth failure and chronic malnutrition, those with severe CP and subsequent oral feeding difficulties, and children whose well-being is affected by illness and decreased fluid or medication intake (Rogers, 2004). Oral feedings may be continued to maintain oral motor skills. Weight gain is perceived as an important measure of adequate oral feeding efficiency (Rogers, 2004).
Parents may need assistance and advice with medication administration through a gastrostomy tube to prevent clotting. Pills may be crushed and mixed with small amounts of water but not other liquids, such as formula or elixir medications, since these may act together to form a sludge that can interfere with gastrostomy tube function. When crushed pills or tablets are administered, flush the feeding tube with more water after instilling the dissolved pill in water. The pharmacist can provide information regarding crushed pills and tablets and elixirs, which should not be mixed together when administered via gastrostomy or nasogastric tube. A skin-level gastrostomy is particularly suited for the child with CP. (See also Gastrostomy Feeding, Chapter 27.)
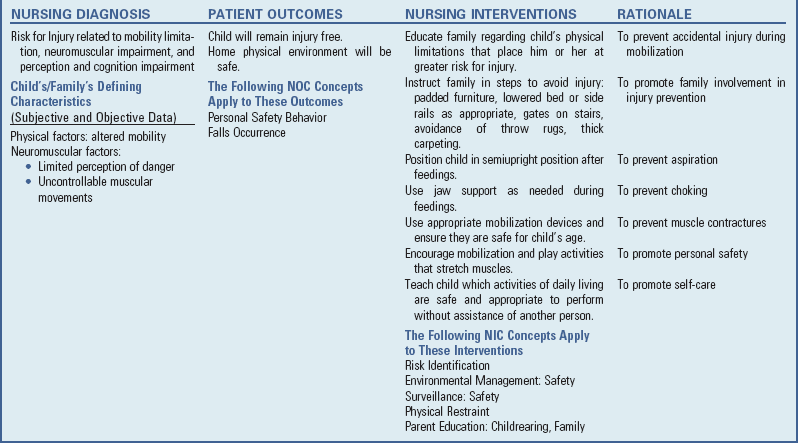
Safety precautions are implemented, such as having children wear protective helmets if they are subject to falls or capable of injuring their heads on hard objects. Because the child with CP is at risk for altered proprioception and subsequent falls, parents should adapt the home and play environment to the child’s particular needs to prevent bodily harm. Transportation of the child with motor problems and restricted mobility may be especially challenging for the family and child. Attention must be given to the child’s safety when riding in a motor vehicle; a federally approved safety restraint should be used at all times. Lovette (2008) recommends that children with CP ride in a rear-facing position as long as possible because of their poor head, neck, and trunk control. This author also provides a list of options for special car restraint systems for children with CP, including several restraints that are suitable for children with a hip spica cast.
Appropriate immunizations should be administered to prevent childhood illnesses and protect against respiratory tract infections such as influenza. Depending on the level of involvement, dental problems may be more common in children with CP, which creates a need for meticulous attention to all aspects of dental care.
Support the Family: The nursing interventions that are probably most valuable to the family are support and help in coping with the emotional aspects of the chronic disorder, many of which are discussed in Chapter 22. Initially the parents need information and support in understanding the implications of the diagnosis and all the feelings it engenders. Later they need clarification regarding what they can expect from the child and from health care professionals. Educating families in the principles of family-centered care and parent-professional collaboration is essential. The family also may require help modifying the home environment for care of the child. (See Chapter 25.) Transportation to the practitioner’s office and other health care agencies often requires special considerations.
Care management for the child and family with CP is an important nursing role. In many cases the family assumes complete care of the child and becomes quite adept at caring for her or his individual needs. The home health nurse or case manager has an important role in the support and encouragement of families who assume the primary care of a child with CP. Having a child with CP implies numerous problems of daily management and changes in family life, and the nurse can stress principles of normalization. (See Normalization, Chapter 22.)
The nurse can support the parents by acknowledging and addressing their concerns and frustrations and by noting and appreciating their problem-solving skills and their approaches to helping the child. Siblings of a child with a disability are also affected and may respond with overt or less evident behavioral problems. The family needs a relationship with nurses who can provide continued contact, support, and encouragement through the long process of habilitation.
Parents can also find help and support from parent groups, where they can share experiences, accomplishments, problems, and concerns while deriving comfort and practical information. For example, parents can understand from others what it is like to have a child with CP. United Cerebral Palsy* has branches in most communities and provides a variety of services for children and families.
Care of the Hospitalized Child: CP is not a disorder that requires hospitalization; therefore when children with CP are hospitalized, they are usually admitted for an associated illness or for corrective surgery.
To facilitate the care and management of hospitalized children with CP, the therapy program should be continued (insofar as their condition allows) while they are hospitalized. This should be incorporated into the multidisciplinary care plan, with every effort expended to make certain the ground that has been so laboriously gained is not lost. Encouraging the parent to room-in and actively participate in the child’s care facilitates a continuation of the home therapy program and helps the child adjust to an unfamiliar environment. However, it is equally important to remember that a hospitalization may be the first time a parent can defer care to a nurse and not be the primary caregiver. This respite may be crucial to the parent’s well-being. Respect the parent’s preference in this regard (see Nursing Care Plan).
 NURSING CARE PLAN
NURSING CARE PLAN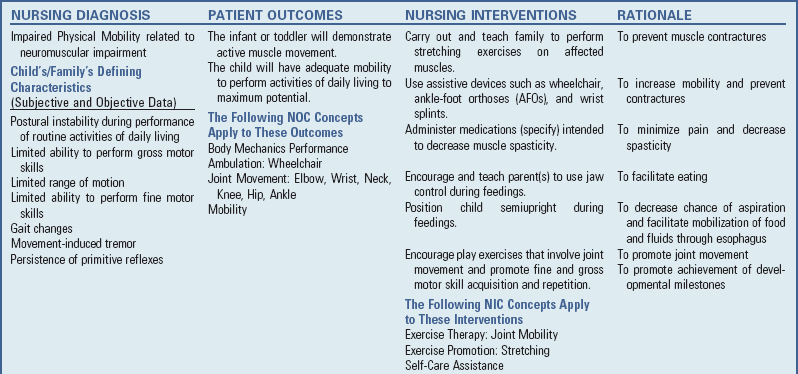
Hypotonia
Decreased muscle tone may be observed in the neonatal period and is one of the most common presenting symptoms in neuromuscular disorders. Hypotonia in neonates born before 37 weeks may be due to neuromuscular immaturity or perinatal maternal medications. (See also Chapter 10.) Monitor such infants over time for neuromuscular tone and make further evaluation if physiologic immaturity is not determined to be a contributing factor. Hypotonia may also indicate a variety of systemic conditions. Common causes are cerebral trauma or perinatal hypoxia, but most neuromuscular disorders with hypotonia as the presenting symptom, especially Down syndrome and infantile spinal muscular atrophy (SMA), are genetically determined.
Clinical Manifestations
Hypotonia is marked by diminished muscle tone and weakness in both spontaneous and passive motion and reflex testing. The affected infant, when placed in a supine position, assumes a characteristic “frog-leg posture” or lies in some other unusual position at rest. Normally the neonate or infant who is held in horizontal suspension (i.e., with the examiner’s hand supporting the infant under the chest) responds by slightly raising the head with the back relatively straight, the arms flexed and slightly abducted, and the knees partly flexed. The hypotonic infant droops over the supporting hand with head and extremities hanging loosely, resembling an inverted U. The muscles feel atrophied when palpated, and there is marked head lag when the infant is pulled to a sitting position. Poor sucking may be noted. Floppy infant syndrome was the term traditionally used to describe infants with hypotonia.
Diagnostic Evaluation
The infant with hypotonia presents a diagnostic challenge. The child’s and family’s history and the physical examination offer important clues to the general category of causes, such as central or motor neuron disorders. Laboratory diagnosis may include a CK level specific for skeletal muscle. Molecular deoxyribonucleic acid (DNA) analysis may eliminate the need for additional invasive tests when a definitive diagnosis is made for a hereditary myopathy or neuropathy (Sarnat, 2007). Nerve conduction velocity, EMG, and muscle biopsy may be used in diagnostic testing. Accurate diagnosis is essential for appropriate treatment, genetic implications, and family counseling.
Therapeutic and Nursing Care Management
The management of an infant with hypotonia is determined by the cause of the hypotonia. It is a nursing responsibility to record and report findings that suggest hypotonia in an infant so that further evaluation can be carried out and therapeutic measures implemented if indicated.
Infantile Spinal Muscular Atrophy (Werdnig-Hoffmann Disease)
Progressive infantile SMA (Werdnig-Hoffmann disease), or SMA type 1, is a disorder characterized by progressive weakness and wasting of skeletal muscles caused by degeneration of anterior horn cells. It is inherited as an autosomal recessive trait and is the most common paralytic form of the floppy infant syndrome. The sites of the pathologic condition are the anterior horn cells of the spinal cord and the motor nuclei of the brainstem, but the primary effect is atrophy of skeletal muscles.
Clinical Manifestations
The age of onset is variable, but the earlier the onset, the more disseminated and severe the motor weakness. The disorder may be manifested early—often at birth or in utero—and almost always before 6 months of age; death may occur as a result of respiratory failure by age 2 years (Iannaccone and Burghes, 2002; Lunn and Wang, 2008). The manifestations (Box 40-6) and prognosis are categorized according to the age of onset, severity of weakness, and clinical course; some children may fluctuate between exhibiting symptoms of types 1 and 2 or between types 2 and 3 in terms of clinical function (Sarnat, 2007). Some experts also categorize SMA according to the highest level of motor function (Lunn and Wang, 2008); type 1 would be “nonsitters,” type 2 “sitters,” and type 3 “walkers” (Iannaccone, 2007). A severe rare fetal form of SMA, classified as type 0, is reported to be lethal in the perinatal period (Sarnat, 2007).