Chapter 14 Red Blood Cell and Bleeding Disorders
In this chapter we will first consider diseases of red cells. Of these, by far the most important are the anemias, red cell deficiency states that most commonly have a non-neoplastic basis. We will then complete our review of blood diseases by discussing the major bleeding disorders.
Anemias
Anemia is defined as a reduction of the total circulating red cell mass below normal limits. Anemia reduces the oxygen-carrying capacity of the blood, leading to tissue hypoxia. In practice, the measurement of red cell mass is not easy, and anemia is usually diagnosed based on a reduction in the hematocrit (the ratio of packed red cells to total blood volume) and the hemoglobin concentration of the blood to levels that are below the normal range. These values correlate with the red cell mass except when there are changes in plasma volume caused by fluid retention or dehydration.
There are many classifications of anemia. We will follow one based on underlying mechanisms that is presented in Table 14-1. A second clinically useful approach classifies anemia according to alterations in red cell morphology, which often point to particular causes. Morphologic characteristics providing etiologic clues include red cell size (normocytic, microcytic, or macrocytic); degree of hemoglobinization, reflected in the color of red cells (normochromic or hypochromic); and shape. In general, microcytic hypochromic anemias are caused by disorders of hemoglobin synthesis (most often iron deficiency), while macrocytic anemias often stem from abnormalities that impair the maturation of erythroid precursors in the bone marrow. Normochromic, normocytic anemias have diverse etiologies; in some of these anemias, specific abnormalities of red cell shape (best appreciated through visual inspection of peripheral smears) provide an important clue as to the cause. The other indices can also be assessed qualitatively in smears, but precise measurement is carried out in clinical laboratories with special instrumentation. The most useful red cell indices are as follows:
TABLE 14-1 Classification of Anemia According to Underlying Mechanism
| Mechanism | Specific Examples |
|---|---|
| BLOOD LOSS | |
| Acute blood loss | Trauma |
| Chronic blood loss | Gastrointestinal tract lesions, gynecologic disturbances* |
| INCREASED RED CELL DESTRUCTION (HEMOLYSIS) | |
| Inherited genetic defects | |
| Red cell membrane disorders | Hereditary spherocytosis, hereditary elliptocytosis |
| Enzyme deficiencies | |
| Hexose monophosphate shunt enzyme deficiencies | G6PD deficiency, glutathione synthetase deficiency |
| Glycolytic enzyme deficiencies | Pyruvate kinase deficiency, hexokinase deficiency |
| Hemoglobin abnormalities | |
| Deficient globin synthesis | Thalassemia syndromes |
| Structurally abnormal globins (hemoglobinopathies) | Sickle cell disease, unstable hemoglobins |
| Acquired genetic defects | |
| Deficiency of phosphatidylinositol-linked glycoproteins | Paroxysmal nocturnal hemoglobinuria |
| Antibody-mediated destruction | Hemolytic disease of the newborn (Rh disease), transfusion reactions, drug-induced, autoimmune disorders |
| Mechanical trauma | |
| Microangiopathic hemolytic anemias | Hemolytic uremic syndrome, disseminated intravascular coagulation, thrombotic thrombocytopenia purpura |
| Cardiac traumatic hemolysis | Defective cardiac valves |
| Repetitive physical trauma | Bongo drumming, marathon running, karate chopping |
| Infections of red cells | Malaria, babesiosis |
| Toxic or chemical injury | Clostridial sepsis, snake venom, lead poisoning |
| Membrane lipid abnormalities | Abetalipoproteinemia, severe hepatocellular liver disease |
| Sequestration | Hypersplenism |
| DECREASED RED CELL PRODUCTION | |
| Inherited genetic defects | |
| Defects leading to stem cell depletion | Fanconi anemia, telomerase defects |
| Defects affecting erythroblast maturation | Thalassemia syndromes |
| Nutritional deficiencies | |
| Deficiencies affecting DNA synthesis | B12 and folate deficiencies |
| Deficiencies affecting hemoglobin synthesis | Iron deficiency anemia |
| Erythropoietin deficiency | Renal failure, anemia of chronic disease |
| Immune-mediated injury of progenitors | Aplastic anemia, pure red cell aplasia |
| Inflammation-mediated iron sequestration | Anemia of chronic disease |
| Primary hematopoietic neoplasms | Acute leukemia, myelodysplasia, myeloproliferative disorders (Chapter 13) |
| Space-occupying marrow lesions | Metastatic neoplasms, granulomatous disease |
| Infections of red cell progenitors | Parvovirus B19 infection |
| Unknown mechanisms | Endocrine disorders, hepatocellular liver disase |
G6PD, Glucose-6-phosphate dehydrogenase.
* Most often cause anemia due to iron deficiency, not bleeding per se.
Adult reference ranges for red cell indices are shown in Table 14-2.
TABLE 14-2 Adult Reference Ranges for Red Cells*
| Measurement (units) | Men | Women |
|---|---|---|
| Hemoglobin (gm/dL) | 13.6–17.2 | 12.0–15.0 |
| Hematocrit (%) | 39–49 | 33–43 |
| Red cell count (× 106/μL) | 4.3–5.9 | 3.5–5.0 |
| Reticulocyte count (%) | 0.5–1.5 | |
| Mean cell volume (fL) | 82–96 | |
| Mean cell hemoglobin (pg) | 27–33 | |
| Mean cell hemoglobin concentration (gm/dL) | 33–37 | |
| Red cell distribution width | 11.5–14.5 | |
* Reference ranges vary among laboratories. The reference ranges for the laboratory providing the result should always be used in interpreting the test result.
Whatever its cause, when sufficiently severe anemia leads to certain clinical features. Patients appear pale. Weakness, malaise, and easy fatigability are common complaints. The lowered oxygen content of the circulating blood leads to dyspnea on mild exertion. Hypoxia can cause fatty change in the liver, myocardium, and kidney. If fatty changes in the myocardium are sufficiently severe, cardiac failure can develop and compound the tissue hypoxia caused by the deficiency of O2 in the blood. On occasion, the myocardial hypoxia manifests as angina pectoris, particularly when complicated by pre-existing coronary artery disease. With acute blood loss and shock, oliguria and anuria can develop as a result of renal hypoperfusion. Central nervous system hypoxia can cause headache, dimness of vision, and faintness.
ANEMIAS OF BLOOD LOSS
Acute Blood Loss
The effects of acute blood loss are mainly due to the loss of intravascular volume, which if massive can lead to cardiovascular collapse, shock, and death. The clinical features depend on the rate of hemorrhage and whether the bleeding is external or internal. If the patient survives, the blood volume is rapidly restored by the intravascular shift of water from the interstitial fluid compartment. This fluid shift results in hemodilution and a lowering of the hematocrit. The reduction in oxygenation triggers increased secretion of erythropoietin from the kidney, which stimulates the proliferation of committed erythroid progenitors (CFU-E) in the marrow (see Fig. 13-1). It takes about 5 days for the progeny of these CFU-Es to mature and appear as newly released red cells (reticulocytes) in the peripheral blood. The iron in hemoglobin is recaptured if red cells extravasate into tissues, whereas bleeding into the gut or out of the body leads to iron loss and possible iron deficiency, which can hamper the restoration of normal red cell counts.
Significant bleeding results in predictable changes in the blood involving not only red cells, but also white cells and platelets. If the bleeding is sufficiently massive to cause a decrease in blood pressure, the compensatory release of adrenergic hormones mobilizes granulocytes from the intravascular marginal pool and results in leukocytosis (see Fig. 13-2). Initially, red cells appear normal in size and color (normocytic, normochromic). However, as marrow production increases there is a striking increase in the reticulocyte count (reticulocytosis), which reaches 10% to 15% after 7 days. Reticulocytes are larger in size than normal red cells (macrocytes) and have a blue-red polychromatophilic cytoplasm. Early recovery from blood loss is also often accompanied by thrombocytosis, which results from an increase in platelet production.
HEMOLYTIC ANEMIAS
Hemolytic anemias share the following features:
The physiologic destruction of senescent red cells takes place within mononuclear phagocytes, which are abundant in the spleen, liver, and bone marrow. This process appears to be triggered by age-dependent changes in red cell surface proteins, which lead to their recognition and phagocytosis.1 In the great majority of hemolytic anemias the premature destruction of red cells also occurs within phagocytes, an event that is referred to as extravascular hemolysis. If persistent, extravascular hemolysis leads to a hyperplasia of phagocytes manifested by varying degrees of splenomegaly.
Extravascular hemolysis is generally caused by alterations that render the red cell less deformable. Extreme changes in shape are required for red cells to navigate the splenic sinusoids successfully. Reduced deformability makes this passage difficult, leading to red cell sequestration and phagocytosis within the cords. Regardless of the cause, the principal clinical features of extravascular hemolysis are (1) anemia, (2) splenomegaly, and (3) jaundice. Some hemoglobin inevitably escapes from phagocytes, which leads to variable decreases in plasma haptoglobin, an α2-globulin that binds free hemoglobin and prevents its excretion in the urine. Because much of the pathologic destruction of red cells occurs in the spleen, individuals with extravascular hemolysis often benefit from splenectomy.
Less commonly, intravascular hemolysis predominates. Intravascular hemolysis of red cells may be caused by mechanical injury, complement fixation, intracellular parasites (e.g., falciparum malaria, Chapter 8), or exogenous toxic factors. Causes of mechanical injury include trauma caused by cardiac valves, thrombotic narrowing of the microcirculation, or repetitive physical trauma (e.g., marathon running and bongo drum beating). Complement fixation occurs in a variety of situations in which antibodies recognize and bind red cell antigens. Toxic injury is exemplified by clostridial sepsis, which results in the release of enzymes that digest the red cell membrane.
Whatever the mechanism, intravascular hemolysis is manifested by (1) anemia, (2) hemoglobinemia, (3) hemoglobinuria, (4) hemosiderinuria, and (5) jaundice. The large amounts of free hemoglobin released from lysed red cells are promptly bound by haptoglobin, producing a complex that is rapidly cleared by mononuclear phagocytes. As serum haptoglobin is depleted, free hemoglobin oxidizes to methemoglobin, which is brown in color. The renal proximal tubular cells reabsorb and catabolize much of the filtered hemoglobin and methemoglobin, but some passes out in the urine, imparting a red-brown color. Iron released from hemoglobin can accumulate within tubular cells, giving rise to renal hemosiderosis. Concomitantly, heme groups derived from hemoglobinhaptoglobin complexes are catabolized to bilirubin within mononuclear phagocytes, leading to jaundice. Unlike in extravascular hemolysis, splenomegaly is not seen.
In all types of uncomplicated hemolytic anemias, the excess serum bilirubin is unconjugated. The level of hyperbilirubinemia depends on the functional capacity of the liver and the rate of hemolysis. When the liver is normal, jaundice is rarely severe. Excessive bilirubin excreted by the liver into the gastrointestinal tract leads to increased formation and fecal excretion of urobilin (Chapter 18), and often leads to the formation of gallstones derived from heme pigments.
Morphology. Certain changes are seen in hemolytic anemias regardless of cause or type. Anemia and lowered tissue oxygen tension trigger the production of erythropoietin, which stimulates erythroid differentiation and leads to the appearance of increased numbers of erythroid precursors (normoblasts) in the marrow (Fig. 14-1). Compensatory increases in erythropoiesis result in a prominent reticulocytosis in the peripheral blood. The phagocytosis of red cells leads to hemosiderosis, which is most pronounced in the spleen, liver, and bone marrow. If the anemia is severe, extramedullary hematopoiesis can appear in the liver, spleen, and lymph nodes. With chronic hemolysis, elevated biliary excretion of bilirubin promotes the formation of pigment gallstones (cholelithiasis).
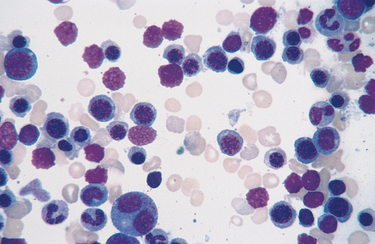
The hemolytic anemias can be classified in a variety of ways; here, we classify them according to underlying mechanisms (see Table 14-1). We begin by discussing the major inherited forms of hemolytic anemia, and then move on to the acquired forms that are most common or of particular pathophysiologic interest.
Hereditary Spherocytosis (HS)
This inherited disorder is caused by intrinsic defects in the red cell membrane skeleton that render red cells spheroid, less deformable, and vulnerable to splenic sequestration and destruction.2 The prevalence of HS is highest in northern Europe, where rates of 1 in 5000 are reported. An autosomal dominant inheritance pattern is seen in about 75% of cases. The remaining patients have a more severe form of the disease that is usually caused by the inheritance of two different defects (a state known as compound heterozygosity).
Pathogenesis.
The remarkable elasticity and durability of the normal red cell are attributable to the physicochemical properties of its specialized membrane skeleton (Fig. 14-2), which lies closely apposed to the internal surface of the plasma membrane. Its chief protein component, spectrin, consists of two polypeptide chains, α and β, which form intertwined (helical) flexible heterodimers. The “head” regions of spectrin dimers self-associate to form tetramers, while the “tails” associate with actin oligomers. Each actin oligomer can bind multiple spectrin tetramers, thus creating a two-dimensional spectrin-actin skeleton that is connected to the cell membrane by two distinct interactions. The first, involving the proteins ankyrin and band 4.2, binds spectrin to the transmembrane ion transporter, band 3. The second, involving protein 4.1, binds the “tail” of spectrin to another transmembrane protein, glycophorin A.
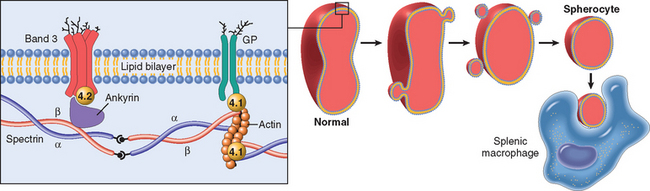
FIGURE 14-2 Role of the red cell membrane skeleton in hereditary spherocytosis. The left panel shows the normal organization of the major red cell membrane skeletal proteins. Various mutations involving α-spectrin, β-spectrin, ankyrin, band 4.2, or band 3 that weaken the interactions between these proteins cause red cells to lose membrane fragments. To accommodate the resultant change in the ratio of surface area to volume these cells adopt a spherical shape. Spherocytic cells are less deformable than normal ones and therefore become trapped in the splenic cords, where they are phagocytosed by macrophages. GP, glycophorin.
HS is caused by diverse mutations that lead to an insufficiency of membrane skeletal components. As a result of these alterations, the life span of the affected red cells is decreased on average to 10 to 20 days from the normal 120 days. The pathogenic mutations most commonly affect ankyrin, band 3, spectrin, or band 4.2, the proteins involved in the first of the two tethering interactions, presumably because this complex is particularly important in stabilizing the lipid bilayer. Most mutations cause shifts in reading frame or introduce premature stop codons, such that the mutated allele fails to produce any protein. The defective synthesis of the affected protein reduces the assembly of the skeleton as a whole and results in a decrease in the density of the membrane skeleton components. Compound heterozygosity for two defective alleles understandably results in a more severe membrane skeleton deficiency. Young HS red cells are normal in shape, but the deficiency of membrane skeleton reduces the stability of the lipid bilayer, leading to the loss of membrane fragments as red cells age in the circulation. The loss of membrane relative to cyt oplasm “forces” the cells to assume the smallest possible diameter for a given volume, namely, a sphere.
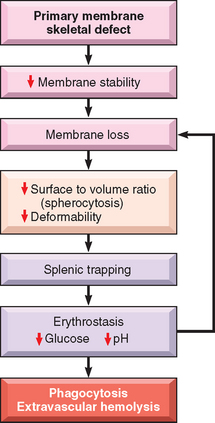
The invariably beneficial effects of splenectomy prove that the spleen has a cardinal role in the premature demise of spherocytes. The travails of spherocytic red cells are fairly well defined. In the life of the portly inflexible spherocyte, the spleen is the villain. Normal red cells must undergo extreme deformation to leave the cords of Billroth and enter the sinusoids. Because of their spheroidal shape and reduced deformability, the hapless spherocytes are trapped in the splenic cords, where they provide a happy meal for phagocytes. The splenic environment also somehow exacerbates the tendency of HS red cells to lose membrane along with K+ ions and H2O; prolonged splenic exposure (erythrostasis), depletion of red cell glucose, and diminished red cell pH have all been suggested to contribute to these abnormalities (Fig. 14-3). After splenectomy the spherocytes persist, but the anemia is corrected.
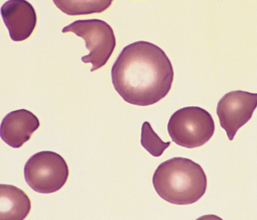
Morphology. The most specific morphologic finding is spherocytosis, apparent on smears as abnormally small, dark-staining (hyperchromic) red cells lacking the central zone of pallor (Fig. 14-4). Spherocytosis is distinctive but not pathognomonic, since other forms of membrane loss, such as in autoimmune hemolytic anemias, also cause the formation of spherocytes. Other features are common to all hemolytic anemias. These include reticulocytosis, marrow erythroid hyperplasia, hemosiderosis, and mild jaundice. Cholelithiasis (pigment stones) occurs in 40% to 50% of affected adults. Moderate splenic enlargement is characteristic (500–1000 gm); in few other hemolytic anemias is the spleen enlarged as much or as consistently. Splenomegaly results from congestion of the cords of Billroth and increased numbers of phagocytes needed to clear the spherocytes.
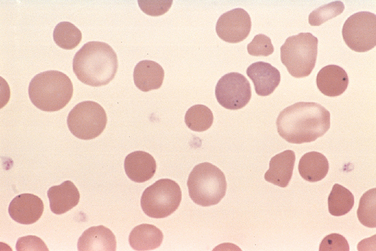
FIGURE 14-4 Hereditary spherocytosis (peripheral smear). Note the anisocytosis and several dark-appearing spherocytes with no central pallor. Howell-Jolly bodies (small dark nuclear remnants) are also present in red cells of this asplenic patient.
(Courtesy of Dr. Robert W. McKenna, Department of Pathology, University of Texas Southwestern Medical School, Dallas, TX.)
Clinical Features.
The diagnosis is based on family history, hematologic findings, and laboratory evidence. In two thirds of the patients the red cells are abnormally sensitive to osmotic lysis when incubated in hypotonic salt solutions, which causes the influx of water into spherocytes with little margin for expansion. HS red cells also have an increased mean cell hemoglobin concentration, due to dehydration caused by the loss of K+ and H2O.
The characteristic clinical features are anemia, splenomegaly, and jaundice. The severity of HS varies greatly. In a small minority (mainly compound heterozygotes) HS presents at birth with marked jaundice and requires exchange transfusions. In 20% to 30% of patients the disease is so mild as to be virtually asymptomatic; here the decreased red cell survival is readily compensated for by increased erythropoiesis. In most, however, the compensatory changes are outpaced, producing a chronic hemolytic anemia of mild to moderate severity. The generally stable clinical course is sometimes punctuated by aplastic crises, usually triggered by an acute parvovirus infection. Parvovirus infects and kills red cell progenitors, causing red cell production to cease until an effective immune response commences, generally in 1 to 2 weeks. Because of the reduced life span of HS red cells, cessation of erythropoiesis for even short time periods leads to sudden worsening of the anemia. Transfusions may be necessary to support the patient until the immune response clears the infection. Hemolytic crises are produced by intercurrent events leading to increased splenic destruction of red cells (e.g., infectious mononucleosis); these are clinically less significant than aplastic crises. Gallstones, found in many patients, can also produce symptoms. Splenectomy treats the anemia and its complications, but brings with it the risk of sepsis.
Hemolytic Disease Due to Red Cell Enzyme Defects: Glucose-6-Phosphate Dehydrogenase Deficiency
The red cell is vulnerable to injury by exogenous and endogenous oxidants. Abnormalities in the hexose monophosphate shunt or glutathione metabolism resulting from deficient or impaired enzyme function reduce the ability of red cells to protect themselves against oxidative injuries and lead to hemolysis. The most important of these enzyme derangements is the hereditary deficiency of glucose-6-phosphate dehydrogenase (G6PD) activity. G6PD reduces nicotinamide adenine dinucleotide phosphate (NADP) to NADPH while oxidizing glucose-6-phosphate (Fig. 14-5). NADPH then provides reducing equivalents needed for conversion of oxidized glutathione to reduced glutathione, which protects against oxidant injury by catalyzing the breakdown of compounds such as H2O2 (Chapter 1).
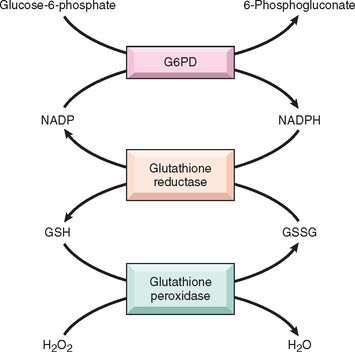
FIGURE 14-5 Role of glucose-6-phosphate dehydrogenase (G6PD) in defense against oxidant injury. The disposal of H2O2, a potential oxidant, is dependent on the adequacy of reduced glutathione (GSH), which is generated by the action of the reduced form of nicotinamide adenine dinucleotide (NADPH). The synthesis of NADPH is dependent on the activity of G6PD. GSSG, oxidized glutathione.
G6PD deficiency is a recessive X-linked trait, placing males at higher risk for symptomatic disease. Several hundred G6PD genetic variants are known, but most are harmless. Only two variants, designated G6PD− and G6PD Mediterranean, cause most of the clinically significant hemolytic anemias. G6PD− is present in about 10% of American blacks; G6PD Mediterranean, as the name implies, is prevalent in the Middle East. The high frequency of these variants in each population is believed to stem from a protective effect against Plasmodium falciparum malaria.3
G6PD variants associated with hemolysis result in misfolding of the protein, making it more susceptible to proteolytic degradation. Compared with the most common normal variant, G6PD B, the half-life of G6PD− is moderately reduced, whereas that of G6PD Mediterranean is more markedly abnormal. Because mature red cells do not synthesize new proteins, G6PD− or G6PD Mediterranean enzyme activities fall quickly to levels inadequate to protect against oxidant stress as red cells age. Thus, older red cells are much more prone to hemolysis than younger ones.
The episodic hemolysis that is characteristic of G6PD deficiency is caused by exposures that generate oxidant stress. The most common triggers are infections, in which oxygen-derived free radicals are produced by activated leukocytes. Many infections can trigger hemolysis; viral hepatitis, pneumonia, and typhoid fever are among those most likely to do so. The other important initiators are drugs and certain foods. The oxidant drugs implicated are numerous, including antimalarials (e.g., primaquine and chloroquine), sulfonamides, nitrofurantoins, and others. Some drugs cause hemolysis only in individuals with the more severe Mediterranean variant. The most frequently cited food is the fava bean, which generates oxidants when metabolized. “Favism” is endemic in the Mediterranean, Middle East, and parts of Africa where consumption is prevalent. Uncommonly, G6PD deficiency presents as neonatal jaundice or a chronic low-grade hemolytic anemia in the absence of infection or known environmental triggers.
Oxidants cause both intravascular and extravascular hemolysis in G6PD-deficient individuals. Exposure of G6PD-deficient red cells to high levels of oxidants causes the cross-linking of reactive sulfhydryl groups on globin chains, which become denatured and form membrane-bound precipitates known as Heinz bodies. These are seen as dark inclusions within red cells stained with crystal violet (Fig. 14-6). Heinz bodies can damage the membrane sufficiently to cause intravascular hemolysis. Less severe membrane damage results in decreased red cell deformability. As inclusion-bearing red cells pass through the splenic cords, macrophages pluck out the Heinz bodies. As a result of membrane damage, some of these partially devoured cells retain an abnormal shape, appearing to have a bite taken out of them (see Fig. 14-6). Other less severely damaged cells revert to a spherocytic shape due to loss of membrane surface area. Both bite cells and spherocytes are trapped in splenic cords and removed rapidly by phagocytes.
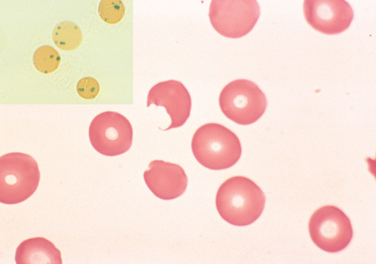
FIGURE 14-6 G6PD deficiency: effects of oxidant drug exposure (peripheral blood smear). Inset, Red cells with precipitates of denatured globin (Heinz bodies) revealed by supravital staining. As the splenic macrophages pluck out these inclusions, “bite cells” like the one in this smear are produced.
(Courtesy of Dr. Robert W. McKenna, Department of Pathology, University of Texas Southwestern Medical School, Dallas, TX.)
Acute intravascular hemolysis, marked by anemia, hemoglobinemia, and hemoglobinuria, usually begins 2 to 3 days following exposure of G6PD-deficient individuals to oxidants. The hemolysis tends to be greater in individuals with the highly unstable G6PD Mediterranean variant. Since only older red cells are at risk for lysis, the episode is self-limited, since hemolysis ceases when only younger G6PD-replete red cells remain (even if administration of an offending drug continues). The recovery phase is heralded by reticulocytosis. Since hemolytic episodes related to G6PD deficiency occur intermittently, features related to chronic hemolysis (e.g., splenomegaly, cholelithiasis) are absent.
Sickle Cell Disease
Sickle cell disease is a common hereditary hemoglobinopathy that occurs primarily in individuals of African descent. Several hundred different hemoglobinopathies caused by mutations in globin genes are known, but only those associated with sickle cell disease are prevalent enough in the United States to merit discussion. Hemoglobin, as you recall, is a tetrameric protein composed of two pairs of globin chains, each with its own heme group. Normal adult red cells contain mainly HbA (α2β2), along with small amounts of HbA2 (α2δ2) and fetal hemoglobin (HbF; α2γ2). Sickle cell disease is caused by a point mutation in the sixth codon of β-globin that leads to the replacement of a glutamate residue with a valine residue. The abnormal physiochemical properties of the resulting sickle hemoglobin (HbS) are responsible for the disease.
About 8% to 10% of African Americans, or roughly 2 million individuals, are heterozygous for HbS, a largely asymptomatic condition known as sickle cell trait. The offspring of two heterozygotes has a 1 in 4 chance of being homozygous for the sickle mutation, a state that produces symptomatic sickle cell disease. In such individuals, almost all the hemoglobin in the red cell is HbS (α2βs2). There are about 70,000 individuals with sickle cell disease in the United States. In certain populations in Africa the prevalence of heterozygosity is as high as 30%. This high frequency probably stems from protection afforded by HbS against falciparum malaria.3
Pathogenesis.
HbS molecules undergo polymerization when deoxygenated. Initially the red cell cytosol converts from a freely flowing liquid to a viscous gel as HbS aggregates form. With continued deoxygenation aggregated HbS molecules assemble into long needle-like fibers within red cells, producing a distorted sickle or holly-leaf shape.
The presence of HbS underlies the major pathologic manifestations: (1) chronic hemolysis, (2) microvascular occlusions, and (3) tissue damage. Several variables affect the rate and degree of sickling:
Sickling causes cumulative damage to red cells through several mechanisms. As HbS polymers grow, they herniate through the membrane skeleton and project from the cell ensheathed by only the lipid bilayer. This severe derangement in membrane structure causes the influx of Ca2+ions, which induce the cross-linking of membrane proteins and activate an ion channel that permits the efflux of K+ and H2O. With repeated episodes of sickling, red cells become increasingly dehydrated, dense, and rigid (Fig. 14-7). Eventually, the most severely damaged cells are converted to end-stage, nondeformable, irreversibly sickled cells, which retain a sickle shape even when fully oxygenated. The severity of the hemolysis correlates with the percentage of irreversibly sickled cells, which are rapidly sequestered and removed by mononuclear phagocytes (extravascular hemolysis). Sickled red cells are also mechanically fragile, leading to some intravascular hemolysis as well.
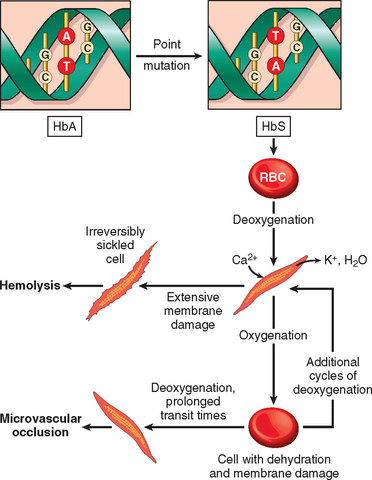
The pathogenesis of the microvascular occlusions, which are responsible for the most serious clinical features, is less certain. Microvascular occlusions are not related to the number of irreversibly sickled cells in the blood, but instead may be dependent upon more subtle red cell membrane damage and other factors, such as inflammation, that tend to slow or arrest the movement of red cells through microvascular beds (see Fig. 14-7). As mentioned above, sickle red cells express higher than normal levels of adhesion molecules and are sticky. Mediators released from granulocytes during inflammatory reactions up-regulate the expression of adhesion molecules on endothelial cells (Chapter 2) and further enhance the tendency for sickle red cells to get arrested during transit throughthe microvasculature. A possible role for inflammatory cells is suggested by observations showing that the leukocyte count correlates with the frequency of pain crises and other measures of tissue damage. The stagnation of red cells within inflamed vascular beds results in extended exposure to low oxygen tension, sickling, and vascular obstruction. Once started, it is easy to envision how a vicious cycle of sickling, obstruction, hypoxia, and more sickling ensues. Depletion of nitric oxide (NO) may also play a part in the vascular occlusions. Free hemoglobin released from lysed sickle red cells can bind and inactivate NO, which is a potent vasodilator and inhibitor of platelet aggregation. Thus, reduced NO increases vascular tone (narrowing vessels) and enhances platelet aggregation, both of which may contribute to red cell stasis, sickling, and (in some instances) thrombosis.
Morphology. In full-blown sickle cell anemia, the peripheral blood demonstrates variable numbers of irreversibly sickled cells, reticulocytosis, and target cells, which result from red cell dehydration (Fig. 14-8). Howell-Jolly bodies (small nuclear remnants) are also present in some red cells due to the asplenia (see below). The bone marrow is hyperplastic as a result of a compensatory erythroid hyperplasia. Expansion of the marrow leads to bone resorption and secondary new bone formation, resulting in prominent cheekbones and changes in the skull that resemble a crew-cut in x-rays. Extramedullary hematopoiesis can also appear. The increased breakdown of hemoglobin can cause pigment gallstones and hyperbilirubinemia.
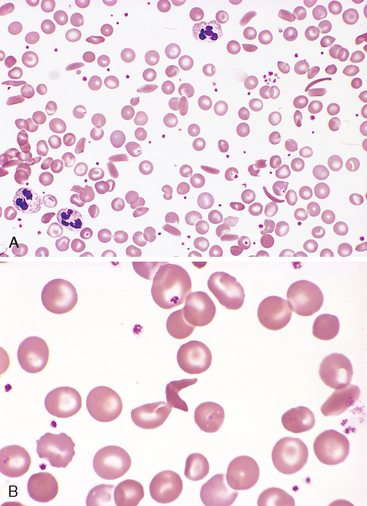
FIGURE 14-8 Sickle cell disease (peripheral blood smear). A, Low magnification shows sickle cells, anisocytosis, and poikilocytosis. B, Higher magnification shows an irreversibly sickled cell in the center.
(Courtesy of Dr. Robert W. McKenna, Department of Pathology, University of Texas Southwestern Medical School, Dallas, TX.)
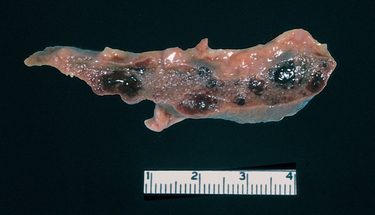
In early childhood, the spleen is enlarged up to 500 gm by red pulp congestion, which is caused by the trapping of sickled red cells in the cords and sinuses (Fig. 14-9). With time, however, the chronic erythrostasis leads to splenic infarction, fibrosis, and progressive shrinkage, so that by adolescence or early adulthood only a small nubbin of fibrous splenic tissue is left; this process is called autosplenectomy (Fig. 14-10). Infarctions caused by vascular occlusions can occur in many other tissues as well, including the bones, brain, kidney, liver, retina, and pulmonary vessels, the latter sometimes producing cor pulmonale. In adult patients, vascular stagnation in subcutaneous tissues often leads to leg ulcers; this complication is rare in children.
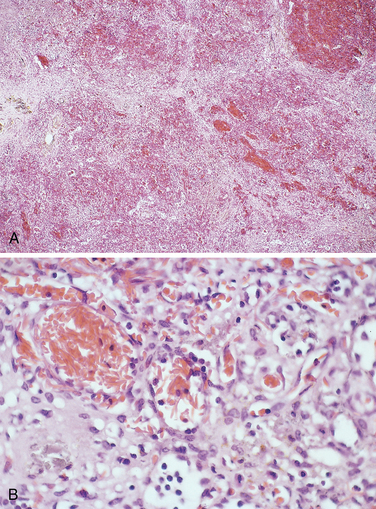
FIGURE 14-9 A, Spleen in sickle cell disease (low power). Red pulp cords and sinusoids are markedly congested; between the congested areas, pale areas of fibrosis resulting from ischemic damage are evident. B, Under high power, splenic sinusoids are dilated and filled with sickled red cells.
(Courtesy of Dr. Darren Wirthwein, Department of Pathology, University of Texas Southwestern Medical School, Dallas, TX.)
Clinical Features.
Sickle cell disease causes a moderately severe hemolytic anemia (hematocrit 18% to 30%) that is associated with reticulocytosis, hyperbilirubinemia, and the presence of irreversibly sickled cells. Its course is punctuatedby a variety of “crises.” Vaso-occlusive crises, also called pain crises, are episodes of hypoxic injury and infarction that cause severe pain in the affected region. Although infection, dehydration, and acidosis (all of which favor sickling) can act as triggers, in most instances no predisposing cause is identified. The most commonly involved sites are the bones, lungs, liver, brain, spleen, and penis. In children, painful bone crises are extremely common and often difficult to distinguish from acute osteomyelitis. These frequently manifest as the hand-foot syndrome or dactylitis of the bones of the hands or feet, or both. Acute chest syndrome is a particularly dangerous type of vaso-occlusive crisis involving the lungs, which typically presents with fever, cough, chest pain, and pulmonary infiltrates. Pulmonary inflammation (such as may be induced by a simple infection) causes blood flow to become sluggish and “spleenlike,” leading to sickling and vaso-occlusion. This compromises pulmonary function, creating a potentially fatal cycle of worsening pulmonary and systemic hypoxemia, sickling, and vaso-occlusion. Other forms of vascular obstruction, particularly stroke, can take a devastating toll. Factors proposed to contribute to stroke include the adhesion of sickle red cells to arterial vascular endothelium and vasoconstriction caused by the depletion of NO by free hemoglobin.8
Although occlusive crises are the most common cause of patient morbidity and mortality, several other acute events complicate the course. Sequestration crises occur in children with intact spleens. Massive entrapment of sickle red cells leads to rapid splenic enlargement, hypovolemia, and sometimes shock. These complications may be fatal in several cases. Survival from sequestration crises and the acute chest syndrome requires treatment with exchange transfusions. Aplastic crises stem from the infection of red cell progenitors by parvovirus B19, which causes a transient cessation of erythropoiesis and a sudden worsening of the anemia.
In addition to these dramatic crises, chronic tissue hypoxia takes a subtle but important toll. Chronic hypoxia is responsible for a generalized impairment of growth and development, as well as organ damage affecting spleen, heart, kidneys, and lungs. Sickling provoked by hypertonicity in the renal medulla causes damage that eventually leads to hyposthenuria (the inability to concentrate urine), which increases the propensity for dehydration and its attendant risks.
Increased susceptibility to infection with encapsulated organisms is another threat. This is due in large part to altered splenic function, which is severely impaired in children by congestion and poor blood flow, and completely absent in adults because of splenic infarction. Defects of uncertain etiology in the alternative complement pathway also impair the opsonization of bacteria. Pneumococcus pneumoniae and Haemophilus influenzae septicemia and meningitis, common causes of death, particularly in children, can be reduced by vaccination and prophylactic antibiotics.
It must be emphasized that there is great variation in the clinical manifestations of sickle cell disease. Some individuals are crippled by repeated vaso-occlusive crises, whereas others have only mild symptoms. The basis for this wide range in disease expression is not understood.
The diagnosis is suggested by the clinical findings and the presence of irreversibly sickled red cells and is confirmed by various tests for sickle hemoglobin. In general, these involve mixing a blood sample with an oxygen-consuming reagent, such as metabisulfite, which induces sickling of red cells if HbS is present. Hemoglobin electrophoresis is also used to demonstrate the presence of HbS and exclude other sickle syndromes, such as HbSC disease. Prenatal diagnosis is possible by analysis of fetal DNA obtained by amniocentesis or chorionic biopsy.
The outlook for patients with sickle cell disease has improved considerably over the last 10 to 20 years. About 90% of patients survive to age 20, and close to 50% survive beyond the fifth decade. A mainstay of treatment is an inhibitor of DNA synthesis, hydroxyurea, which has several beneficial effects. These include (1) an increase in red cell HbF levels, which occurs by unknown mechanisms; and (2) an anti-inflammatory effect, which stems from an inhibition of white cell production. These activities (and possibly others9) are believed to act in concert to decrease crises related to vascular occlusions in both children and adults.
Thalassemia Syndromes
The thalassemia syndromes are a heterogeneous group of disorders caused by inherited mutations that decrease the synthesis of adult hemoglobin, HbA (α2β2). The two α chains in HbA are encoded by an identical pair of α-globin genes on chromosome 16, while the two β chains are encoded by a single β-globin gene on chromosome 11. β-Thalassemia is caused by deficient synthesis of β chains, whereas α-thalassemia is caused by deficient synthesis of α chains. The hematologic consequences of diminished synthesis of one globin chain stem not only from hemoglobin deficiency but also from a relative excess of the other globin chain, particularly in β-thalassemia. Thalassemia syndromes are endemic in the Mediterranean basin, the Middle East, tropical Africa, the Indian subcontinent, and Asia, and in aggregate are among the most common inherited disorders of humans. As with sickle cell disease and other common inherited red cell disorders, their prevalence seems to be explained by the protection they afford heterozygous carriers against malaria.3 Although we will discuss the thalassemia syndromes with other inherited forms of anemia associated with hemolysis, it is important to recognize that the defects in globin synthesis that underlie these disorders also impair red cell production and contribute to the pathogenesis of these disorders.
β-Thalassemias
The β-thalassemias are caused by mutations that diminish the synthesis of β-globin chains. The clinical severity varies because of heterogeneity in the causative mutations. We will begin our discussion with the molecular lesions in β-thalassemia and then relate the clinical variants to specific underlying molecular defects.
Molecular Pathogenesis.
The causative mutations fall into two categories: (1) β0 mutations, associated with absent β-globin synthesis, and (2) β+ mutations, characterized by reduced (but detectable) β-globin synthesis. Sequencing of β-thalassemia genes has revealed more than 100 different causative mutations, mostly consisting of point mutations. Details of these mutations and their effects are found in specialized texts. Figure 14-11 gives a few illustrative examples.
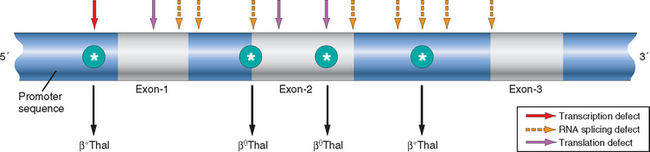
FIGURE 14-11 Distribution of β-globin gene mutations associated with β-thalassemia. Arrows denote sites where point mutations giving rise to β0 or β+ thalassemia have been identified.
Impaired β-globin synthesis results in anemia by two mechanisms (Fig. 14-12). The deficit in HbA synthesis produces “underhemoglobinized” hypochromic, microcytic red cells with subnormal oxygen transport capacity. Even more important is the diminished survival of red cells and their precursors, which results from the imbalance in α- and β-globin synthesis. Unpaired α chains precipitate within red cell precursors, forming insoluble inclusions. These inclusions cause a variety of untoward effects, but membrane damage is the proximal cause of most red cell pathology. Many red cell precursors succumb to membrane damage and undergo apoptosis. In severe β-thalassemia, it is estimated that 70% to 85% of red cell precursors suffer this fate, which leads to ineffective erythropoiesis. Those red cells that are released from the marrow also bear inclusions and membrane damage and are prone to splenic sequestration and extravascular hemolysis.
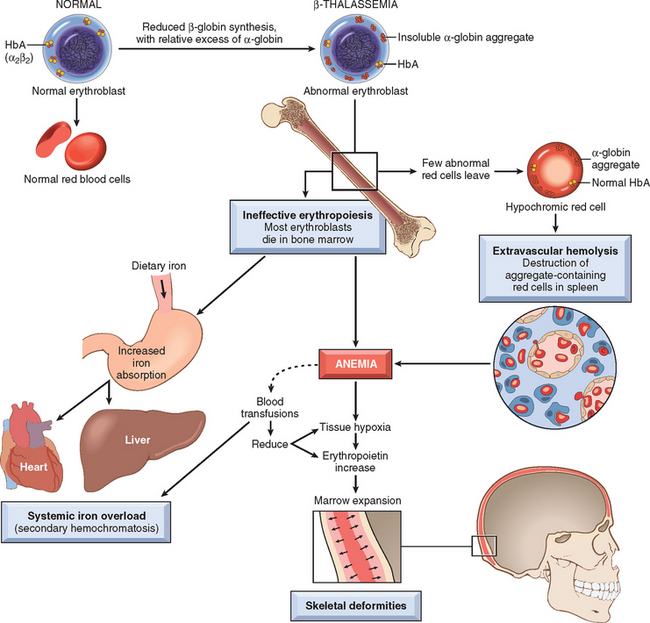
FIGURE 14-12 Pathogenesis of β-thalassemia major. Note that the aggregates of unpaired α-globin chains, a hallmark of the disease, are not visible in routinely stained blood smears. Blood transfusions are a double-edged sword, diminishing the anemia and its attendant complications, but also adding to the systemic iron overload.
In severe β-thalassemia, ineffective erythropoiesis creates several additional problems. Erythropoietic drive in the setting of severe uncompensated anemia leads to massive erythroid hyperplasia in the marrow and extensive extramedullary hematopoiesis. The expanding mass of red cell precursors erodes the bony cortex, impairs bone growth, and produces skeletal abnormalities (described later). Extramedullary hematopoiesis involves the liver, spleen, and lymph nodes, and in extreme cases produces extraosseous masses in the thorax, abdomen, and pelvis. The metabolically active erythroid progenitors steal nutrients from other tissues that are already oxygen-starved, causing severe cachexia in untreated patients.
Another serious complication of ineffective erythropoiesis is the excessive absorption of dietary iron. Ineffective erythropoiesis suppresses the circulating levels of hepcidin, a critical negative regulator of iron absorption (described later under iron deficiency anemia). Low levels of hepcidin and the iron load of repeated blood transfusions inevitably lead to severe iron overload unless preventive steps are taken. Secondary injury to parenchymal organs, particularly the iron-laden liver, often follows and sometimes induces secondary hemochromatosis (Chapter 18).
Clinical Syndromes.
The relationships of clinical phenotypes to underlying genotypes are summarized in Table 14-3. Clinical classification of β-thalassemia is based on the severity of the anemia, which in turn depends on the genetic defect (β+ or β0) and the gene dosage (homozygous or heterozygous). In general, individuals with two β-thalassemia alleles (β+/β+, β+/β0, or β0/β0) have a severe, transfusion-dependent anemia called β-thalassemia major. Heterozygotes with one βthalassemia gene and one normal gene (β+/β or β0/β) usually have a mild asymptomatic microcytic anemia. This condition is referred to as β-thalassemia minor or β-thalassemia trait. A third genetically heterogeneous variant of moderate severity is called β-thalassemia intermedia. This category includes milder variants of β+/β+ or β+/β0-thalassemia and unusual forms of heterozygous β-thalassemia. Some patients with β-thalassemia intermedia have two defective β-globin genes and an α-thalassemia gene defect, which lessens the imbalance in α- and β-chain synthesis. In other rare but informative cases, individuals have a single β-globin defect and one or two extra copies of normal α-globin genes (stemming from a gene duplication event), which worsens the chain imbalance.10 These unusual forms of the disease serve to emphasize the cardinal role of unpaired α-globin chains in the pathology. The clinical and morphologic features of β-thalassemia intermedia are not described separately but can be surmised from the following discussions of β-thalassemia major and β-thalassemia minor.
β-Thalassemia Major.
β-thalassemia major is most common in Mediterranean countries, parts of Africa, and Southeast Asia. In the United States the incidence is highest in immigrants from these areas. The anemia manifests 6 to 9 months after birth as hemoglobin synthesis switches from HbF to HbA. In untransfused patients, hemoglobin levels are 3 to 6 gm/dL. The red cells may completely lack HbA (β0/β0 genotype) or contain small amounts (β+/β+ or β0/β+ genotypes). The major red cell hemoglobin is HbF, which is markedly elevated. HbA2 levels are sometimes high but more often are normal or low.
Morphology. Blood smears show severe red cell abnormalities, including marked variation in size (anisocytosis) and shape (poikilocytosis), microcytosis, and hypochromia. Target cells (so called because hemoglobin collects in the center of the cell), basophilic stippling, and fragmented red cells are also common. Inclusions of aggregated α chains are efficiently removed by the spleen and not easily seen. The reticulocyte count is elevated, but it is lower than expected for the severity of anemia because of the ineffective erythropoiesis. Variable numbers of poorly hemoglobinized nucleated red cell precursors (normoblasts) are seen in the peripheral blood as a result of “stress” erythropoiesis and abnormal release from sites of extramedullary hematopoiesis.
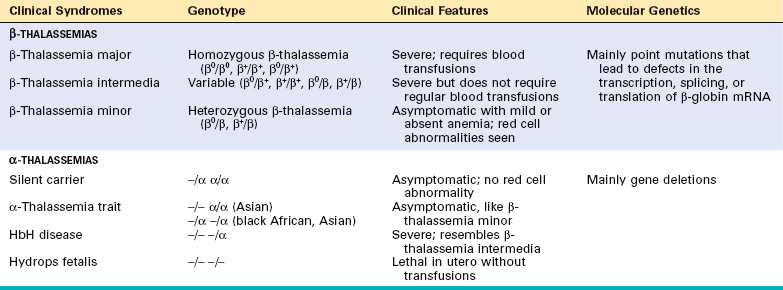
Other major alterations involve the bone marrow and spleen. In the untransfused patient there is a striking expansion of hematopoietically active marrow. In the bones of the face and skull the burgeoning marrow erodes existing cortical bone and induces new bone formation, giving rise to a “crew-cut” appearance on x-ray (Fig. 14-13). Both phagocyte hyperplasia and extramedullary hematopoiesis contribute to enlargement of the spleen, which can weigh as much as 1500 gm. The liver and the lymph nodes can also be enlarged by extramedullary hematopoiesis.
FIGURE 14-13 Thalassemia: x-ray film of the skull showing new bone formation on the outer table, producing perpendicular radiations resembling a crewcut.
(Courtesy of Dr. Jack Reynolds, Department of Radiology, University of Texas Southwestern Medical School, Dallas, TX.)
Hemosiderosis and secondary hemochromatosis, the two manifestations of iron overload (Chapter 18), occur in almost all patients. The deposited iron often damages organs, most notably the heart, liver, and pancreas.
The clinical course of β-thalassemia major is brief unless blood transfusions are given. Untreated children suffer from growth retardation and die at an early age from the effects of anemia. In those who survive long enough, the cheekbones and other bony prominences are enlarged and distorted. Hepatosplenomegaly due to extramedullary hematopoiesis is usually present. Although blood transfusions improve the anemia and suppress complications related to excessive erythropoiesis, they lead to complications of their own. Cardiac disease resulting from progressive iron overload and secondary hemochromatosis (Chapter 18) is an important cause of death, particularly in heavily transfused patients, who must be treated with iron chelators to prevent or reduce this complication. With transfusions and iron chelation, survival into the third decade is possible, but the overall outlook remains guarded. Bone marrow transplantation is the only therapy offering a cure and is being used increasingly.11 Prenatal diagnosis is possible by molecular analysis of DNA.
β-Thalassemia Minor.
β-Thalassemia minor is much more common than β-thalassemia major and understandably affects the same ethnic groups. Most patients are heterozygous carriers of a β+ or β0 allele. These patients are usually asymptomatic. Anemia, if present, is mild. The peripheral blood smear typically shows some red cell abnormalities, including hypochromia, microcytosis, basophilic stippling, and target cells. Mild erythroid hyperplasia is seen in the bone marrow. Hemoglobin electrophoresis usually reveals an increase in HbA2 (α2δ2) to 4% to 8% of the total hemoglobin (normal, 2.5% ± 0.3%), which is a reflection of an elevated ratio of δ-chain to β-chain synthesis. HbF levels are generally normal or occasionally slightly increased.
Recognition of β-thalassemia trait is important for two reasons: (1) differentiation from the hypochromic microcytic anemia of iron deficiency and (2) genetic counseling. Iron deficiency can usually be excluded through measurement of serum iron, total iron-binding capacity, and serum ferritin (as described later under iron deficiency anemia). The increase in HbA2 is diagnostically useful, particularly in individuals (such as women of childbearing age) who are at risk for both β-thalassemia trait and iron deficiency.
α-Thalassemias
The α-thalassemias are caused by inherited deletions that result in reduced or absent synthesis of α-globin chains. Normally, there are four α-globin genes, and the severity of α-thalassemia depends on how many α-globin genes are affected. As in β-thalassemias, the anemia stems both from a lack of adequate hemoglobin and the effects of excess unpaired non-α chains (β, γ, and δ), which vary in type at different ages. In newborns with α-thalassemia, excess unpaired γ-globin chains form γ4 tetramers known as hemoglobin Barts, whereas in older children and adults excess β-globin chains form β4 tetramers known as HbH. Since free β and γ chains are more soluble than free α chains and form fairly stable homotetramers, hemolysis and ineffective erythropoiesis are less severe than in β-thalassemias. A variety of molecular lesions give rise to α-thalassemia, but gene deletion is the most common cause of reduced α-chain synthesis.
Clinical Syndromes.
The clinical syndromes are determined and classified by the number of α-globin genes that are deleted. Each of the four α-globin genes normally contributes 25% of the total α-globin chains. α-Thalassemia syndromes stem from combinations of deletions that remove one to four α-globin genes. Not surprisingly, the severity of the clinical syndrome is proportional to the number of α-globin genes that are deleted. The different types of α-thalassemia and their salient clinical features are listed in Table 14-3.
Silent Carrier State.
This is associated with the deletion of a single α-globin gene, which causes a barely detectable reduction in α-globin chain synthesis. These individuals are completely asymptomatic, but they have slight microcytosis.
α-Thalassemia Trait.
This is caused by the deletion of two α-globin genes from a single chromosome (α/α α/α), or the deletion of one α-globin gene from each of the two chromosomes (α/—α α/—α) (see Table 14-3). The former genotype is more common in Asian populations, the latter in regions of Africa. Both genotypes produce similar quantitative deficiencies of α-globin and are clinically identical, but have different implications for the children of affected individuals, who are at risk of clinically significant α-thalassemia (HbH disease or hydrops fetalis) only when at least one parent has the α/—α haplotype. As a result, symptomatic α-thalassemia is relatively common in Asian populations and rare in black African populations. The clinical picture in α-thalassemia trait is identical to that described for β-thalassemia minor, that is, small red cells (microcytosis), minimal or no anemia, and no abnormal physical signs. HbA2 levels are normal or low.
Hemoglobin H Disease.
This is caused by deletion of three α-globin genes. As already discussed, HbH disease is most common in Asian populations. With only one normal α-globin gene, the synthesis of α chains is markedly reduced, and tetramers of β-globin, called HbH, form. HbH has an extremely high affinity for oxygen and therefore is not useful for oxygen delivery, leading to tissue hypoxia disproportionate to the level of hemoglobin. Additionally, HbH is prone to oxidation, which causes it to precipitate out and form intracellular inclusions that promote red cell sequestration and phagocytosis in the spleen. The result is a moderately severe anemia resembling β-thalassemia intermedia.
Hydrops Fetalis
This most severe form of α-thalassemia is caused by deletion of all four α-globin genes. In the fetus, excess γ-globin chains form tetramers (hemoglobin Barts) that have such a high affinity for oxygen that they deliver little to tissues. Survival in early development is due to the expression of ζ chains, an embryonic globin that pairs with γ chains to form a functional ζ2γ2 Hb tetramer. Signs of fetal distress usually become evident by the third trimester of pregnancy. In the past, severe tissue anoxia led to death in utero or shortly after birth; with intrauterine transfusion many such infants are now saved. The fetus shows severe pallor, generalized edema, and massive hepatosplenomegaly similar to that seen in hemolytic disease of the newborn (Chapter 10). There is a lifelong dependence on blood transfusions for survival, with the associated risk of iron overload. Bone marrow transplantation can be curative.11
Paroxysmal Nocturnal Hemoglobinuria
Paroxysmal nocturnal hemoglobinuria (PNH) is a disease that results from acquired mutations in the phosphatidylinositol glycan complementation group A gene (PIGA), an enzyme that is essential for the synthesis of certain cell surface proteins. PNH has an incidence of 2 to 5 per million in the United States. Despite its rarity, it has fascinated hematologists because it is the only hemolytic anemia caused by an acquired genetic defect. Recall that proteins are anchored into the lipid bilayer in two ways. Most have a hydrophobic region that spans the cell membrane; these are called transmembrane proteins. The others are attached to the cell membrane through a covalent linkage to a specialized phospholipid called glycosylphosphatidylinositol (GPI). In PNH, these GPI-linked proteins are deficient because of somatic mutations that inactivate PIGA. PIGA is X-linked and subject to lyonization (random inactivation of one X chromosome in cells of females; Chapter 5). As a result, a single acquired mutation in the active PIGA gene of any given cell is sufficient to produce a deficiency state. Because the causative mutations occur in a hematopoietic stem cell, all of its clonal progeny (red cells, white cells, and platelets) are deficient in GPI-linked proteins. Typically the mutant clone coexists with the progeny of normal stem cells that are not PIGA deficient.
Remarkably, most normal individuals harbor small numbers of bone marrow cells with PIGA mutations identical to those that cause PNH. It is hypothesized that these cells increase in numbers (thus producing clinically evident PNH) only in rare instances where they have a selective advantage, such as in the setting of autoimmune reactions against GPI-linked antigens.12 Such a scenario might explain the frequent association of PNH and aplastic anemia, a marrow failure syndrome (discussed later) that has an autoimmune basis in many individuals.
PNH blood cells are deficient in three GPI-linked proteins that regulate complement activity: (1) decay–accelerating factor, or CD55; (2) membrane inhibitor of reactive lysis, or CD59; and (3) C8 binding protein. Of these factors, the most important is CD59, a potent inhibitor of C3 convertase that prevents the spontaneous activation of the alternative complement pathway.
Red cells, platelets, and granulocytes deficient in these GPI-linked factors are abnormally susceptible to lysis or injury by complement. In red cells this manifests as intravascular hemolysis, which is caused by the C5b-C9 membrane attack complex. The hemolysis is paroxysmal and nocturnal in only 25% of cases; chronic hemolysis without dramatic hemoglobinuria is more typical. The tendency for red cells to lyse at night is explained by a slight decrease in blood pH during sleep, which increases the activity of complement. The anemia is variable but usually mild to moderate in severity. The loss of heme iron in the urine (hemosiderinuria) eventually leads to iron deficiency, which can exacerbate the anemia if untreated.
Thrombosis is the leading cause of disease-related death in individuals with PNH. About 40% of patients suffer from venous thrombosis, often involving the hepatic, portal, or cerebral veins. Dysfunction of platelets due to the absence of certain GPI-linked proteins contributes to the prothrombotic state, as does the absorption of NO by free hemoglobin (as discussed under sickle cell disease).13 About 5% to 10% of patients eventually develop acute myeloid leukemia or a myelodysplastic syndrome, possibly because hematopoietic stem cells have suffered some type of genetic damage.
PNH is diagnosed by flow cytometry, which provides a sensitive means for detecting red cells that are deficient in GPI-linked proteins such as CD59 (Fig. 14-14). Several therapeutic approaches are available, none of which is entirely satisfactory. Infusion of a monoclonal antibody inhibitor of C5a greatly reduces the hemolysis but exposes patients to an increased risk of serious or fatal meningococcal infections (as is true of individuals with inherited complement defects). Immunosuppressive drugs are sometimes beneficial for those with evidence of marrow aplasia. The only cure is bone marrow transplantation.
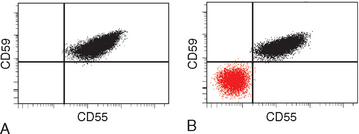
FIGURE 14-14 Paroxysmal nocturnal hemoglobinuria (PNH). A, Flow cytogram of blood from a normal individual shows that the red cells express two phosphatidylinositol glycan (PIG)–linked membrane proteins, CD55 and CD59, on their surfaces. B, Flow cytogram of blood from a patient with PNH shows a population of red cells that is deficient in both CD55 and CD59. As is typical of PNH, a second population of CD55+/CD59+ red cells that is derived from residual normal hematopoietic stem cells is also present.
(Courtesy of Dr. Scott Rodig, Department of Pathology, Brigham and Women’s Hospital, Boston, MA.)
Immunohemolytic Anemia
Hemolytic anemias in this category are caused by antibodies that bind to red cells, leading to their premature destruction. Although these disorders are commonly referred to as autoimmune hemolytic anemias, the designation immunohemolytic anemia is preferred because in some instances the immune reaction is initiated by an ingested drug. Immunohemolytic anemia can be classified based on the characteristics of the responsible antibody (Table 14-4).
TABLE 14-4 Classification of Immunohemolytic Anemias
| WARM ANTIBODY TYPE (IgG ANTIBODIES ACTIVE AT 37°C) |
| COLD AGGLUTININ TYPE (IgM ANTIBODIES ACTIVE BELOW 37°C) |
| COLD HEMOLYSIN TYPE (IgG ANTIBODIES ACTIVE BELOW 37°C) |
| Rare; occurs mainly in children following viral infections |
The diagnosis of immunohemolytic anemia requires the detection of antibodies and/or complement on red cells from the patient. This is done using the direct Coombs antiglobulin test, in which the patient’s red cells are mixed with sera containingantibodies that are specific for human immunoglobulin or complement. If either immunoglobulin or complement is present on the surface of the red cells, the multivalent antibodies cause agglutination, which is easily appreciated visually as clumping. In the indirect Coombs antiglobulin test, the patient’s serum is tested for its ability to agglutinate commercially available red cells bearing particular defined antigens. This test is used to characterize the antigen target and temperature dependence of the responsible antibody. Quantitative immunological tests to measure such antibodies directly are also available.
Warm Antibody Type.
This is the most common form of immunohemolytic anemia. About 50% of cases are idiopathic (primary); the others are related to a predisposing condition (see Table 14-4) or exposure to a drug. Most causative antibodies are of the IgG class; less commonly, IgA antibodies are culpable. The red cell hemolysis is mostly extravascular. IgG-coated red cells bind to Fc receptors on phagocytes, which remove red cell membrane during “partial” phagocytosis. As in hereditary spherocytosis, the loss of membrane converts the red cells to spherocytes, which are sequestered and removed in the spleen. Moderate splenomegaly due to hyperplasia of splenic phagocytes is usually seen.
As with other autoimmune disorders, the cause of primary immunohemolytic anemia is unknown. In many cases, the antibodies are directed against the Rh blood group antigens. The mechanisms of drug-induced immunohemolytic anemia are better understood. Two different mechanisms have been described.
Cold Agglutinin Type.
This form of immunohemolytic anemia is caused by IgM antibodies that bind red cells avidly at low temperatures (0°–4°C).14 It is less common than warm antibody immunohemolytic anemia, accounting for 15% to 30% of cases. Cold agglutinin antibodies sometimes appear transiently following certain infections, such as with Mycoplasma pneumoniae, Epstein-Barr virus, cytomegalovirus, influenza virus, and human immunodeficiency virus (HIV). In these settings the disorder is self-limited and the antibodies rarely induce clinically important hemolysis. Chronic cold agglutinin immunohemolytic anemia occurs in association with certain B-cell neoplasms or as an idiopathic condition.
Clinical symptoms result from binding of IgM to red cells in vascular beds where the temperature may fall below 30°C, such as in exposed fingers, toes, and ears. IgM binding agglutinates red cells and fixes complement rapidly. As the blood recirculates and warms, IgM is released, usually before complement-mediated hemolysis can occur. However, the transient interaction with IgM is sufficient to deposit sublytic quantities of C3b, an excellent opsonin, which leads to the removal of affected red cells by phagocytes in the spleen, liver, and bone marrow. The hemolysis is of variable severity. Vascular obstruction caused by agglutinated red cells results in pallor, cyanosis, and Raynaud phenomenon (Chapter 11) in body parts exposed to cold temperature.
Cold Hemolysin Type.
Cold hemolysins are autoantibodies responsible for an unusual entity known as paroxysmal cold hemoglobinuria. This rare disorder causes substantial, sometimes fatal, intravascular hemolysis and hemoglobinuria. The autoantibodies are IgGs that bind to the P blood group antigen on the red cell surface14 in cool, peripheral regions of the body. Complement-mediated lysis occurs when the cells recirculate to warm central regions, since the complement cascade functions more efficiently at 37°C. Most cases are seen in children following viral infections; in this setting the disorder is transient, and most of those affected recover within 1 month.
Treatment of warm antibody immunohemolytic anemia centers on the removal of initiating factors (i.e., drugs); when this is not feasible, immunosuppressive drugs and splenectomy are the mainstays.15 Chronic cold agglutinin immunohemolytic anemia caused by IgM antibodies is more difficult to treat.14
Hemolytic Anemia Resulting from Trauma to Red Cells
The most significant hemolysis caused by trauma to red cells is seen in individuals with cardiac valve prostheses and microangiopathic disorders. Artificial mechanical cardiac valves are more frequently implicated than are bioprosthetic porcine valves. The hemolysis stems from shear forces produced by turbulent blood flow and pressure gradients across damaged valves. Microangiopathic hemolytic anemia is most commonly seen with disseminated intravascular coagulation, but it also occurs in thrombotic thrombocytopenic purpura (TTP), hemolytic-uremic syndrome (HUS), malignant hypertension, systemic lupus erythematosus, and disseminated cancer. The common pathogenic feature in these disorders is a microvascular lesion that results in luminal narrowing, often due to the deposition of fibrin and platelets. These vascular changes produce shear stresses that mechanically injure passing red cells. Regardless of the cause, traumatic damage leads to the appearance of red cell fragments (schistocytes), “burr cells,” “helmet cells,” and “triangle cells” in blood smears (Fig. 14-15).
ANEMIAS OF DIMINISHED ERYTHROPOIESIS
Although the anemias that stem from the inadequate production of red cells are heterogeneous, they can be classified into several major categories based on pathophysiology (see Table 14-1). The most common and important anemias associated with red cell underproduction are those caused by nutritional deficiencies, followed by those that arise secondary to renal failure and chronic inflammation. Also included are less common disorders that lead to generalized bone marrow failure, such as aplastic anemia, primary hematopoietic neoplasms (discussed in Chapter 13), and infiltrative disorders that lead to marrow replacement (such as metastatic cancer and disseminated granulomatous disease). We will first discuss the extrinsic causes of diminished erythropoiesis, which are more common and clinically important, and then move to the non-neoplastic intrinsic causes.
Megaloblastic Anemias
The common theme among the various causes of megaloblastic anemia (Table 14-5) is an impairment of DNA synthesis that leads to distinctive morphologic changes, including abnormally large erythroid precursors and red cells. The following discussion first describes the common features and then turns to the two principal types: pernicious anemia (the major form of vitamin B12 deficiency anemia) and folate deficiency anemia.
TABLE 14-5 Causes of Megaloblastic Anemia
| VITAMIN B12 DEFICIENCY |
| Decreased Intake |
| Inadequate diet, vegetarianism |
| Impaired Absorption |
| FOLIC ACID DEFICIENCY |
| Decreased Intake |
| Inadequate diet, alcoholism, infancy |
| Impaired Absorption |
| Increased Loss |
| Hemodialysis |
| Increased Requirement |
| Pregnancy, infancy, disseminated cancer, markedly increased hematopoiesis |
| Impaired Utilization |
| Folic acid antagonists |
| UNRESPONSIVE TO VITAMIN B12 OR FOLIC ACID THERAPY |
| Metabolic inhibitors of DNA synthesis and/or folate metabolism (e.g., methotrexate) |
Modified from Beck WS: Megaloblastic anemias. In Wyngaarden JB, Smith LH (eds): Cecil Textbook of Medicine, 18th ed. Philadelphia, WB Saunders, 1988, p. 900.
Some of the metabolic roles of vitamin B12 and folate are considered later. For now it suffices that vitamin B12 and folic acid are coenzymes required for the synthesis of thymidine, one of the four bases found in DNA. A deficiency of these vitamins or impairment in their metabolism results in defective nuclear maturation due to deranged or inadequate DNA synthesis, with an attendant delay or block in cell division.
Morphology. Certain peripheral blood findings are shared by all megaloblastic anemias. The presence of red cells that are macrocytic and oval (macro-ovalocytes) is highly characteristic. Because they are larger than normal and contain ample hemoglobin, most macrocytes lack the central pallor of normal red cells and even appear “hyperchromic,” but the MCHC is not elevated. There is marked variation in the size (anisocytosis) and shape (poikilocytosis) of red cells. The reticulocyte count is low. Nucleated red cell progenitors occasionally appear in the circulating blood when anemia is severe. Neutrophils are also larger than normal (macropolymorphonuclear) and hypersegmented, having five or more nuclear lobules instead of the normal three to four (Fig. 14-16).
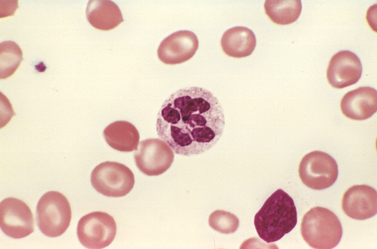
FIGURE 14-16 Megaloblastic anemia. A peripheral blood smear shows a hypersegmented neutrophil with a six-lobed nucleus.
(Courtesy of Dr. Robert W. McKenna, Department of Pathology, University of Texas Southwestern Medical School, Dallas, TX.)
The marrow is usually markedly hypercellular as a result of increased hematopoietic precursors, which often completely replace the fatty marrow. Megaloblastic changes are detected at all stages of erythroid development. The most primitive cells (promegaloblasts) are large, with a deeply basophilic cytoplasm, prominent nucleoli, and a distinctive, fine nuclear chromatin pattern (Fig. 14-17, cell A). As these cells differentiate and begin to accumulate hemoglobin, the nucleus retains its finely distributed chromatin and fails to develop the clumped pyknotic chromatin typical of normoblasts. While nuclear maturation is delayed, cytoplasmic maturation and hemoglobin accumulation proceed at a normal pace, leading to nuclear-to-cytoplasmic asynchrony. Because DNA synthesis is impaired in all proliferating cells, granulocytic precursors also display dysmaturation in the form of giant metamyelocytes and band forms. Megakaryocytes, too, can be abnormally large and have bizarre, multilobate nuclei.
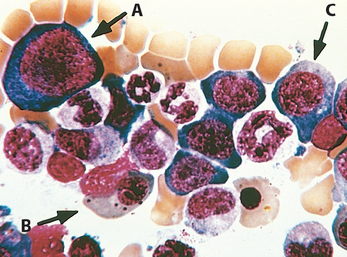
FIGURE 14-17 Megaloblastic anemia (bone marrow aspirate). A to C, Megaloblasts in various stages of differentiation. Note that the orthochromatic megaloblast (B) is hemoglobinized (as revealed by cytoplasmic color), but in contrast to normal orthochromatic normoblasts, the nucleus is not pyknotic. The early erythroid precursors (A,C) and the granulocytic precursors are also large and have abnormally immature chromatin.
(Courtesy of Dr. Jose Hernandez, Department of Pathology, University of Texas Southwestern Medical School, Dallas, TX.)
The marrow hyperplasia is a response to increased levels of growth factors, such as erythropoietin. However, the derangement in DNA synthesis causes most precursors to undergo apoptosis in the marrow (an example of ineffective hematopoiesis) and leads to pancytopenia. The anemia is further exacerbated by a mild degree of red cell hemolysis of uncertain etiology.
Anemias of Vitamin B12 Deficiency: Pernicious Anemia
Pernicious anemia is a specific form of megaloblastic anemia caused by autoimmune gastritis and an attendant failure of intrinsic factor production, which leads to vitamin B12 deficiency. We first review vitamin B12 metabolism, since this helps to place pernicious anemia in perspective relative to the other causes of vitamin B12 deficiency anemia.
Normal Vitamin B12Metabolism.
Vitamin B12 is a complex organometallic compound known as cobalamin. Under normal circumstances humans are totally dependent on dietary vitamin B12. Microorganisms are the ultimate origin of cobalamin in the food chain. Plants and vegetables contain little cobalamin, save that contributed by microbial contamination, and strictly vegetarian or macrobiotic diets do not provide adequate amounts of this essential nutrient. The daily requirement is 2 to 3 μg. A diet that includes animal products contains significantly larger amounts and normally results in the accumulation of intrahepatic stores of vitamin B12 that are sufficient to last for several years.
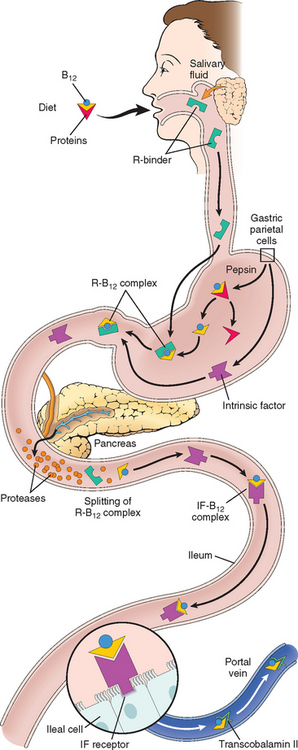
Absorption of vitamin B12 requires intrinsic factor, which is secreted by the parietal cells of the fundic mucosa (Fig. 14-18). Vitamin B12 is freed from binding proteins in food through the action of pepsin in the stomach and binds to salivary proteins called cobalophilins, or R-binders. In the duodenum, bound vitamin B12 is released by the action of pancreatic proteases. It then associates with intrinsic factor. This complex is transported to the ileum, where it is endocytosed by ileal enterocytes that express intrinsic factor receptors on their surfaces. Within ileal cells, vitamin B12 associates with a major carrier protein, transcobalamin II, and is secreted into the plasma. Transcobalamin II delivers vitamin B12 to the liver and other cells of the body, including rapidly proliferating cells in the bone marrow and the gastrointestinal tract. In addition to this major pathway, there is also a poorly understood alternative uptake mechanism that is not dependent on intrinsic factor or an intact terminal ileum. Up to 1% of a large oral dose can be absorbed by this pathway, making it feasible to treat pernicious anemia with high doses of oral vitamin B12.
Biochemical Functions of Vitamin B2.
Only two reactions in humans are known to require vitamin B12. In one, methylcobalamin serves as an essential cofactor in the conversion of homocysteine to methionine by methionine synthase (Fig. 14-19). In the process, methylcobalamin yields a methyl group that is recovered from N5-methyltetrahydrofolic acid (N5-methyl FH4), the principal form of folic acid in plasma. In the same reaction, N5-methyl FH4 is converted to tetrahydrofolic acid (FH4). FH4 is crucial, since it is required (through its derivative N5,10-methylene FH4) for the conversion of deoxyuridine monophosphate (dUMP) to deoxythymidine monophosphate (dTMP), an immediate precursor of DNA. It is postulated that the fundamental cause of the impaired DNA synthesis in vitamin B12 deficiency is the reduced availability of FH4, most of which is “trapped” as N5-methyl FH4. The FH4 deficit may be further exacerbated by an “internal” folate deficiency caused by a failure to synthesize metabolically active polyglutamylated forms. This stems from the requirement for vitamin B12 in the synthesis of methionine, which contributes a carbon group needed in the metabolic reactions that create folate polyglutamates (Fig. 14-20). Whatever the mechanism, lack of folate is the proximate cause of anemia in vitamin B12 deficiency, since the anemia improves with administration of folic acid.
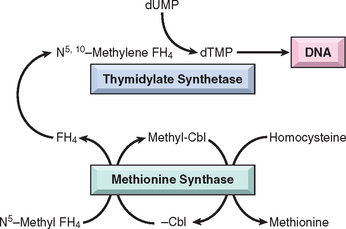
FIGURE 14-19 Relationship of N5-methyl FH4, methionine synthase, and thymidylate synthetase. In cobalamin (Cbl) deficiency, folate is sequestered as N5-methyl FH4. This ultimately deprives thymidylate synthetase of its folate coenzyme (N5,10-methylene FH4), thereby impairing DNA synthesis. FH4, tetrahydrofolic acid.
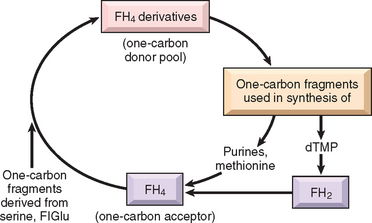
FIGURE 14-20 Role of folate derivatives in the transfer of one-carbon fragments for synthesis of biologic macromolecules. FH4, tetrahydrofolic acid; FH2, dihydrofolic acid; FIGlu, formiminoglutamate; dTMP, deoxythymidine monophosphate.
The neurologic complications associated with vitamin B12 deficiency are more enigmatic, since they are not improved by folate administration. The other known reaction that depends on vitamin B12 is the isomerization of methylmalonyl coenzyme A to succinyl coenzyme A, which requires adenosylcobalamin as a prosthetic group on the enzyme methylmalonyl–coenzyme A mutase. A deficiency of vitamin B12 thus leads to increased plasma and urine levels of methylmalonic acid. Interruption of this reaction and the consequent buildup of methylmalonate and propionate (a precursor) could lead to the formation and incorporation of abnormal fatty acids into neuronal lipids. It has been suggested that this biochemical abnormality predisposes to myelin breakdown and thereby produces the neurologic complications of vitamin B12 deficiency (Chapter 28). However, rare individuals with hereditary deficiencies of methylmalonyl–coenzyme A mutase, while having complications related to methylmalonyl acidemia, do not suffer from the neurologic abnormalities seen in vitamin B12 deficiency, casting doubt on this explanation.
Having completed our overview of vitamin B12 metabolism, we can now turn to pernicious anemia.
Incidence.
Although somewhat more prevalent in Scandinavian and other Caucasian populations, pernicious anemia occurs in all racial groups, including blacks and Hispanics. It is a disease of older adults; the median age at diagnosis is 60 years, and it is rare in people younger than 30. A genetic predisposition is strongly suspected, but no definable genetic pattern of transmission has been discerned. As described below, many affected individuals have a tendency to form antibodies against multiple self-antigens.
Pathogenesis.
Pernicious anemia is believed to result from an autoimmune attack on the gastric mucosa. Histologically, there is a chronic atrophic gastritis marked by a loss of parietal cells, a prominent infiltrate of lymphocytes and plasma cells, and megaloblastic changes in mucosal cells similar to those found in erythroid precursors. Three types of autoantibodies are present in many, but not all, patients. About 75% of patients have a type I antibody that blocks the binding of vitamin B12 to intrinsic factor. Type I antibodies are found in both plasma and gastric juice. Type II antibodies prevent binding of the intrinsic factor–vitamin B12 complex to its ileal receptor. These antibodies are also found in a large proportion of patients with pernicious anemia. Type III antibodies, present in 85% to 90% of patients, recognize the α and β subunits of the gastric proton pump, which is normally localized to the microvilli of the canalicular system of the gastric parietal cell. These antibodies are not specific for pernicious anemia or other autoimmune diseases, since they are found in as many as 50% of elderly persons with idiopathic chronic gastritis not associated with pernicious anemia.
Autoantibodies are of diagnostic utility, but they are not thought to be the primary cause of the gastric pathology. Rather, it seems that an autoreactive T-cell response initiates gastric mucosal injury and triggers the formation of autoantibodies, which may exacerbate the epithelial injury. When the mass of intrinsic factor–secreting cells falls below a threshold (and reserves of stored vitamin B12 are depleted), anemia develops. In an animal model of autoimmune gastritis mediated by CD4+ T cells, a pattern of autoantibodies resembling that seen in pernicious anemia develops, thus supporting the primacy of T-cell autoimmunity. The common association of pernicious anemia with other autoimmune disorders, particularly autoimmune thyroiditis and adrenalitis, is also consistent with an underlying immune basis. The tendency to develop multiple autoimmune disorders, including pernicious anemia, is linked to specific sequence variants of NALP1,16 an innate immune receptor that maps to chromosome 17p13.
Vitamin B12 deficiency is associated with disorders other than pernicious anemia. Most of these impair absorption of the vitamin at one of the steps outlined earlier (see Table 14-5). With achlorhydria and loss of pepsin secretion (which occurs in some elderly individuals), vitamin B12 is not readily released from proteins in food. With gastrectomy, intrinsic factor is not available for uptake in the ileum. With loss of exocrine pancreatic function, vitamin B12 cannot be released from R-binder–vitamin B12 complexes. Ileal resection or diffuse ileal disease can remove or damage the site of intrinsic factor–vitamin B12 complex absorption. Tapeworms compete with the host for B12 and can induce a deficiency state. In some settings, such as pregnancy, hyperthyroidism, disseminated cancer, and chronic infection, an increased demand for vitamin B12 can produce a relative deficiency, even with normal absorption.
Morphology. The findings in the bone marrow and blood in pernicious anemia are similar to those described earlier for all megaloblastic anemias. The stomach typically shows diffuse chronic gastritis (Chapter 17). The most characteristic alteration is atrophy of the fundic glands, affecting both chief cells and parietal cells, the latter being virtually absent. The glandular lining epithelium is replaced by mucus-secreting goblet cells that resemble those lining the large intestine, a form of metaplasia referred to as intestinalization. Some of the cells as well as their nuclei may increase to double the normal size, a form of “megaloblastic” change exactly analogous to that seen in the marrow. With time, the tongue may become shiny, glazed, and “beefy” (atrophic glossitis). The gastric atrophy and metaplastic changes are due to autoimmunity and not vitamin B12 deficiency; hence, parenteral administration of vitamin B12 corrects the megaloblastic changes in the marrow and the epithelial cells of the alimentary tract, but gastric atrophy and achlorhydria persist.
Central nervous system lesions are found in about three fourths of all cases of florid pernicious anemia but can also be seen in the absence of overt hematologic findings. The principal alterations involve the spinal cord, where there is demyelination of the dorsal and lateral tracts, sometimes followed by loss of axons. These changes give rise to spastic paraparesis, sensory ataxia, and severe paresthesias in the lower limbs. Less frequently, degenerative changes occur in the ganglia of the posterior roots and in peripheral nerves (Chapter 28).
Clinical Features.
Pernicious anemia is insidious in onset, so the anemia is often quite severe by the time the affected person seeks medical attention. The course is progressive unless halted by therapy.
The diagnosis is based on (1) a moderate to severe megaloblastic anemia, (2) leukopenia with hypersegmented granulocytes, (3) low serum vitamin B12, and (4) elevated levels of homocysteine and methylmalonic acid in the serum. The diagnosis is confirmed by a striking increase in reticulocytes and an improvement in hematocrit levels beginning about 5 days after parenteral administration of vitamin B12. Serum antibodies to intrinsic factor are highly specific for pernicious anemia. Their presence attests to the cause rather than the presence or absence of vitamin B12 deficiency.
Persons with atrophic and metaplastic changes in the gastric mucosa associated with pernicious anemia are at increased risk of developing gastric carcinoma (Chapter 17). As mentioned, serum homocysteine levels are raised in individuals with vitamin B12 deficiency. Elevated homocysteine levels are a risk factor for atherosclerosis and thrombosis, and it is suspected that vitamin B12 deficiency may increase the incidence of vascular disease. With parenteral or high-dose oral vitamin B12, the anemia can be cured and the peripheral neurologic changes reversed or at least halted in their progression, but the changes in the gastric mucosa and the risk of carcinoma are unaffected.
Anemia of Folate Deficiency
A deficiency of folic acid (more properly, pteroylmonoglutamic acid) results in a megaloblastic anemia having the same characteristics as that caused by vitamin B12 deficiency. FH4 derivatives act as intermediates in the transfer of one-carbon units such as formyl and methyl groups to various compounds (see Fig. 14-20). FH4 serves as an acceptor of one-carbon fragments from compounds such as serine and formiminoglutamic acid. The FH4 derivatives so generated in turn donate the acquired one-carbon fragments in reactions synthesizing various metabolites. FH4, then, can be viewed as the biologic “middleman” in a series of swaps involving one-carbon moieties. The most important metabolic processes depending on such transfers are (1) purine synthesis; (2) the conversion of homocysteine to methionine, a reaction also requiring vitamin B12; and (3) deoxythymidylate monophosphate synthesis. In the first two reactions, FH4 is regenerated from its one-carbon carrier derivatives and is available to accept another one-carbon moiety and reenter the donor pool. In the synthesis of dTMP, a dihydrofolate is produced that must be reduced by dihydrofolate reductase for reentry into the FH4 pool. The reductase step is significant, since this enzyme is susceptible to inhibition by various drugs. Among the molecules whose synthesis is dependent on folates, dTMP is perhaps the most important biologically, since it is required for DNA synthesis. It should be apparent from this discussion that suppressed synthesis of DNA, the common denominator of folic acid and vitamin B12 deficiency, is the immediate cause of megaloblastosis.
Etiology.
The three major causes of folic acid deficiency are (1) decreased intake, (2) increased requirements, and (3) impaired utilization (see Table 14-5). Humans are entirely dependent on dietary sources for their folic acid requirement, which is 50 to 200 μg daily. Most normal diets contain ample amounts. The richest sources are green vegetables such as lettuce, spinach, asparagus, and broccoli. Certain fruits (e.g., lemons, bananas, melons) and animal sources (e.g., liver) contain lesser amounts. The folic acid in these foods is largely in the form of folylpolyglutamates. Despite their abundance in raw foods, polyglutamates are sensitive to heat; boiling, steaming, or frying of foods for 5 to 10 minutes destroys up to 95% of the folate content. Intestinal conjugases split the polyglutamates into monoglutamates that are readily absorbed in the proximal jejunum. During intestinal absorption they are modified to 5-methyltetrahydrofolate, the normal transport form of folate. The body’s reserves of folate are relatively modest, and a deficiency can arise within weeks to months if intake is inadequate.
Decreased intake can result from either a nutritionally inadequate diet or impairment of intestinal absorption. A normal diet contains folate in excess of the minimal daily adult requirement. Inadequate dietary intakes are almost invariably associated with grossly deficient diets. Such dietary inadequacies are most frequently encountered in chronic alcoholics, the indigent, and the very elderly. In alcoholics with cirrhosis, other mechanisms of folate deficiency such as trapping of folate within the liver, excessive urinary loss, and disordered folate metabolism have also been implicated. Under these circumstances, the megaloblastic anemia is often accompanied by general malnutrition and manifestations of other avitaminoses, including cheilosis, glossitis, and dermatitis. Malabsorption syndromes, such as sprue, can lead to inadequate absorption of this nutrient, as can diffuse infiltrative diseases of the small intestine (e.g., lymphoma). In addition, certain drugs, particularly the anticonvulsant phenytoin and oral contraceptives, interfere with absorption.
Despite normal intake of folic acid, a relative deficiency can be encountered when requirements are increased. Conditions in which this is seen include pregnancy, infancy, hematologic derangements associated with hyperactive hematopoiesis (hemolytic anemias), and disseminated cancer. In all these circumstances the demands of increased DNA synthesis render normal intake inadequate.
Folic acid antagonists, such as methotrexate, inhibit dihydrofolate reductase and lead to a deficiency of FH4. With inhibition of folate metabolism, all rapidly growing cells are affected, but particularly the cells of the bone marrow and the gastrointestinal tract. Many chemotherapeutic drugs used in the treatment of cancer damage DNA or inhibit DNA synthesis through other mechanisms; these can also cause megaloblastic changes in rapidly dividing cells.
As mentioned at the outset, the megaloblastic anemia that results from a deficiency of folic acid is identical to that encountered in vitamin B12 deficiency. Thus, the diagnosis of folate deficiency can be made only by demonstration of decreased folate levels in the serum or red cells. As in vitamin B12 deficiency, serum homocysteine levels are increased, but methylmalonate concentrations are normal. However, neurologic changes do not occur.
Although prompt hematologic response heralded by reticulocytosis follows the administration of folic acid, it should be remembered that the hematologic symptoms of vitamin B12 deficiency anemia also respond to folate therapy. However, folate does not prevent (and may even exacerbate) the neurologic deficits typical of the vitamin B12 deficiency states. It is thus essential to exclude vitamin B12 deficiency in megaloblastic anemia before initiating therapy with folate.
Iron Deficiency Anemia
Deficiency of iron is the most common nutritional disorder in the world. Although the prevalence of iron deficiency anemia is higher in developing countries, this form of anemia is also common in the United States, particularly in toddlers, adolescent girls, and women of childbearing age. The factors underlying the iron deficiency differ somewhat in various population groups and can be best considered in the context of normal iron metabolism.17
Iron Metabolism.
The normal daily Western diet contains about 10 to 20 mg of iron, most in the form of heme contained in animal products, with the remainder being inorganic iron in vegetables. About 20% of heme iron (in contrast to 1% to 2% of nonheme iron) is absorbable, so the average Western diet contains sufficient iron to balance fixed daily losses. The total body iron content is normally about 2 gm in women and as high as 6 gm in men, and can be divided into functional and storage compartments (Table 14-6). About 80% of the functional iron is found in hemoglobin; myoglobin and iron-containing enzymes such as catalase and the cytochromes contain the rest. The storage pool represented by hemosiderin and ferritin contains about 15% to 20% of total body iron. Healthy young females have smaller stores of iron than do males, primarily because of blood loss during menstruation, and often develop iron deficiency due to excessive losses or increased demands associated with menstruation and pregnancy, respectively.
TABLE 14-6 Iron Distribution in Healthy Young Adults (mg)
| Pool | Men | Women |
|---|---|---|
| Total | 3450 | 2450 |
| Functional | ||
| Hemoglobin | 2100 | 1750 |
| Myoglobin | 300 | 250 |
| Enzymes | 50 | 50 |
| Storage | ||
| Ferritin, hemosiderin | 1000 | 400 |
Iron in the body is recycled extensively between the functional and storage pools (Fig. 14-21). It is transported in plasma by an iron-binding glycoprotein called transferrin, which is synthesized in the liver. In normal individuals, transferrin is about one third saturated with iron, yielding serum iron levels that average 120 μg/dL in men and 100 μg/dL in women. The major function of plasma transferrin is to deliver iron to cells, including erythroid precursors, which require iron to synthesize hemoglobin. Erythroid precursors possess high-affinity receptors for transferrin, which mediate iron import through receptor-mediated endocytosis.
FIGURE 14-21 Iron metabolism. Iron absorbed from the gut is bound to plasma transferrin and transported to the marrow, where it is delivered to developing red cells and incorporated into hemoglobin. Mature red cells are released into the circulation and, after 120 days, are ingested by macrophages, primarily in the spleen, liver, and bone marrow. Here iron is extracted from hemoglobin and recycled to plasma transferrin. At equilibrium, iron absorbed from the gut is balanced by losses in shed keratinocytes, enterocytes, and (in women) endometrium.
Free iron is highly toxic (as described in Chapter 18), and it is therefore important that storage iron be sequestered. This is achieved by binding iron in the storage pool tightly to either ferritin or hemosiderin. Ferritin is a ubiquitous protein-iron complex that is found at highest levels in the liver, spleen, bone marrow, and skeletal muscles. In the liver, most ferritin is stored within the parenchymal cells; in other tissues, such as the spleen and the bone marrow, it is found mainly in macrophages. Hepatocyte iron is derived from plasma transferrin, whereas storage iron in macrophages is derived from the breakdown of red cells. Intracellular ferritin is located in the cytosol and in lysosomes, in which partially degraded protein shells of ferritin aggregate into hemosiderin granules. Iron in hemosiderin is chemically reactive and turns blue-black when exposed to potassium ferrocyanide, which is the basis for the Prussian blue stain. With normal iron stores, only trace amounts of hemosiderin are found in the body, principally in macrophages in the bone marrow, spleen, and liver. In iron-overloaded cells, most iron is stored in hemosiderin.
Since plasma ferritin is derived largely from the storage pool of body iron, its levels correlate well with body iron stores. In iron deficiency, serum ferritin is always below 12 μg/L, whereas in iron overload values approaching 5000 μg/L can be seen. Of physiologic importance, the storage iron pool can be readily mobilized if iron requirements increase, as may occur after loss of blood.
Iron balance is maintained largely by regulating the absorption of dietary iron in the proximal duodenum. Iron is both essential for cellular metabolism and highly toxic in excess, and total body iron stores must therefore be regulated meticulously. There is no regulated pathway for iron excretion, which is limited to the 1 to 2 mg lost each day through the shedding of mucosal and skin epithelial cells. In contrast, as body iron stores rise, absorption falls, and vice versa. The pathways responsible for the absorption of iron are now understood in reasonable detail (Fig. 14-22), and differ partially for nonheme and heme iron.17 Luminal nonheme iron is mostly in the Fe3+ (ferric) state and must first be reduced to Fe2+ (ferrous) iron by ferrireductases, such as b cytochromes and STEAP3. Fe2+ iron is then transported across the apical membrane by divalent metal transporter 1 (DMT1). The absorption of nonheme iron is variable and often inefficient, being inhibited by substances in the diet that bind and stabilize Fe3+ iron and enhanced by substances that stabilize Fe2+ iron (described below). Frequently, less than 5% of dietary nonheme iron is absorbed. In contrast, about 25% of the heme iron derived from hemoglobin, myoglobin, and other animal proteins is absorbed. Heme iron is moved across the apical membrane into the cytoplasm through transporters that are incompletely characterized. Here, it is metabolized to release Fe2+ iron, which enters a common pool with nonheme Fe2+ iron. Iron that enters the duodenal cells can follow one of two pathways: transport to the blood or storage as mucosal iron. This distribution is influenced by body iron stores, as we shall discuss below. Fe2+ iron destined for the circulation, is transported from the cytoplasm across the basolateral enterocyte membrane by ferriportin. This process is coupled to the oxidation of Fe2+ iron to Fe3+ iron, which is carried out by the iron oxidases hephaestin and ceruloplasmin. Newly absorbed Fe3+ iron binds rapidly to the plasma protein transferrin, which delivers iron to red cell progenitors in the marrow (see Fig. 14-21). Both DMT1 and ferriportin are widely distributed in the body and are involved in iron transport in other tissues as well. For example, DMT1 also mediates the uptake of “functional” iron (derived from endocytosed transferrin) across lysosomal membranes into the cytosol of red cell precursors in the bone marrow, and ferriportin plays an important role in the release of storage iron from macrophages.
FIGURE 14-22 Regulation of iron absorption. Duodenal epithelial cell uptake of heme and nonheme iron is depicted. When the storage sites of the body are replete with iron and erythropoietic activity is normal, plasma hepcidin levels are high. This leads to downregulation of ferriportin and trapping of most of the absorbed iron, which is lost when duodenal epithelial cells are shed into the gut. Conversely, when body iron stores decrease or when erythropoiesis is stimulated, hepcidin levels fall and ferriportin activity increases, allowing a greater fraction of the absorbed iron to be transferred to plasma transferrin. DMT1, divalent metal transporter 1.
Iron absorption is regulated by hepcidin, a small circulating peptide that is synthesized and released from the liver in response to increases in intrahepatic iron levels.17 Hepcidin inhibits iron transfer from the enterocyte to plasma by binding to ferriportin and causing it to be endocytosed and degraded. As a result, as hepcidin levels rise, iron becomes trapped within duodenal cells in the form of mucosal ferritin and is lost as these cells are sloughed. Thus, when the body is replete with iron, high hepcidin levels inhibit its absorption into the blood. Conversely, with low body stores of iron, hepcidin synthesis falls and this in turn facilitates iron absorption. By inhibiting ferriportin, hepcidin not only reduces iron uptake from enetrocytes but also suppresses iron release from macrophages, which are an important source of the iron that is used by erythroid precursors to make hemoglobin. This, as we shall see, is important in the pathogenesis of anemia of chronic diseases.
Alterations in hepcidin have a central role in diseases involving disturbances of iron metabolism. As will be described subsequently, the anemia of chronic disease is caused in part by inflammatory mediators that increase hepatic hepcidin production.18 A rare form of microcytic anemia is caused by mutations that disable TMPRSS6, a hepatic transmembrane serine protease that normally suppresses hepcidin production when iron stores are low. Affected patients have high hepcidin levels, resulting in reduced iron absorption and failure to respond to iron therapy. Conversely, hepcidin activity is inappropriately low in both primary and secondary hemochromatosis, a syndrome caused by systemic iron overload. Secondary hemochromatosis can occur in diseases associated with ineffective erythropoiesis, such as β-thalassemia major and myelodysplastic syndromes (Chapter 13). Through incompletely understood mechanisms, ineffective erythropoiesis suppresses hepatic hepcidin production, even when iron stores are high. As discussed in Chapter 18, the various inherited forms of primary hematochromatosis are associated with mutations in hepcidin or the genes that regulate hepcidin expression.
Etiology.
Iron deficiency can result from (1) dietary lack, (2) impaired absorption, (3) increased requirement, or (most importantly) (4) chronic blood loss. To maintain a normal iron balance, about 1 mg of iron must be absorbed from the diet every day. Because only 10% to 15% of ingested iron is absorbed, the daily iron requirement is 7 to 10 mg for adult men and 7 to 20 mg for adult women. Since the average daily dietary intake of iron in the Western world is about 15 to 20 mg, most men ingest more than adequate iron, whereas many women consume marginally adequate amounts of iron. The bioavailability of dietary iron is as important as the overall content. Heme iron is much more absorbable than inorganic iron, the absorption of which is influenced by other dietary contents. Absorption of inorganic iron is enhanced by ascorbic acid, citric acid, amino acids, and sugars in the diet, and inhibited by tannates (found in tea), carbonates, oxalates, and phosphates.
Dietary lack is rare in developed countries, where on average about two thirds of the dietary iron is in the readily absorbed heme form provided by meat. The situation is different in developing countries, where food is less abundant and most iron in the diet is found in plants in the poorly absorbable inorganic form. Dietary iron inadequacy occurs in even privileged societies in the following groups:
Impaired absorption is found in sprue, other causes of fat malabsorption (steatorrhea), and chronic diarrhea. Gastrectomy impairs iron absorption by decreasing hydrochloric acid and transit time through the duodenum. Specific items in the diet, as is evident from the preceding discussion, can also affect absorption.
Increased requirement is an important cause of iron deficiency in growing infants, children, and adolescents, as well as premenopausal women, particularly during pregnancy. Economically deprived women having multiple, closely spaced pregnancies are at exceptionally high risk.
Chronic blood loss is the most common cause of iron deficiency in the Western world. External hemorrhage or bleeding into the gastrointestinal, urinary, or genital tracts depletes iron reserves. Iron deficiency in adult men and postmenopausal women in the Western world must be attributed to gastrointestinal blood loss until proven otherwise. To prematurely ascribe iron deficiency in such individuals to any other cause is to run the risk of missing an occult gastrointestinal cancer or other bleeding lesion. An alert clinician investigating unexplained iron deficiency anemia occasionally discovers an occult bleed or cancer and thereby saves a life.
Pathogenesis.
Whatever its basis, iron deficiency produces a hypochromic microcytic anemia. At the outset of chronic blood loss or other states of negative iron balance, reserves in the form of ferritin and hemosiderin may be adequate to maintain normal hemoglobin and hematocrit levels as well as normal serum iron and transferrin saturation. Progressive depletion of these reserves first lowers serum iron and transferrin saturation levels without producing anemia. In this early stage there is increased erythroid activity in the bone marrow. Anemia appears only when iron stores are completely depleted and is accompanied by low serum iron, ferritin, and transferrin saturation levels.
Morphology. The bone marrow reveals a mild to moderate increase in erythroid progenitors. A diagnostically significant finding is the disappearance of stainable iron from macrophages in the bone marrow, which is best assessed by performing Prussian blue stains on smears of aspirated marrow. In peripheral blood smears, the red cells are small (microcytic) and pale (hypochromic). Normal red cells with sufficient hemoglobin have a zone of central pallor measuring about one third of the cell diameter. In established iron deficiency the zone of pallor is enlarged; hemoglobin may be seen only in a narrow peripheral rim (Fig. 14-23). Poikilocytosis in the form of small, elongated red cells (pencil cells) is also characteristically seen.
FIGURE 14-23 Hypochromic microcytic anemia of iron deficiency (peripheral blood smear). Note the small red cells containing a narrow rim of peripheral hemoglobin. Scattered fully hemoglobinized cells, present due to recent blood transfusion, stand in contrast.
(Courtesy of Dr. Robert W. McKenna, Department of Pathology, University of Texas Southwestern Medical School, Dallas, TX.)
Clinical Features.
The clinical manifestations of the anemia are nonspecific and were detailed earlier. The dominating signs and symptoms frequently relate to the underlying cause of the anemia, for example, gastrointestinal or gynecologic disease, malnutrition, pregnancy, and malabsorption. In severe and long-standing iron deficiency, depletion of iron-containing enzymes in cells throughout the body also causes other changes, including koilonychia, alopecia, atrophic changes in the tongue and gastric mucosa, and intestinal malabsorption. Depletion of iron from the central nervous system may lead to the appearance of pica, in which affected individuals consume non-foodstuffs such as clay or food ingredients such as flour, and periodically move their limbs during sleep. Esophageal webs appear together with microcytic hypochromic anemia and atrophic glossitis to complete the triad of major findings in the rare Plummer-Vinson syndrome (Chapter 17).
The diagnosis of iron deficiency anemia ultimately rests on laboratory studies. Both the hemoglobin and hematocrit are depressed, usually to a moderate degree, in association with hypochromia, microcytosis, and modest poikilocytosis. The serum iron and ferritin are low, and the total plasma iron-binding capacity (reflecting elevated transferrin levels) is high. Low serum iron with increased iron-binding capacity results in a reduction of transferrin saturation to below 15%. Reduced iron stores inhibit hepcidin synthesis, and its serum levels fall. In uncomplicated iron deficiency, oral iron supplementation produces an increase in reticulocytes in about 5 to 7 days that is followed by a steady increase in blood counts and the normalization of red cell indices.
Anemia of Chronic Disease
Impaired red cell production associated with chronic diseases is perhaps the most common cause of anemia among hospitalized patients in the United States. It is associated with a reduction in the proliferation of erythroid progenitors and impaired iron utilization. The chronic illnesses associated with this form of anemia can be grouped into three categories:
The anemia of chronic disease occurs in the setting of persistent systemic inflammation and is associated with low serum iron, reduced total iron-binding capacity, and abundant stored iron in tissue macrophages. Several effects of inflammation contribute to the observed abnormalities. Most notably, certain inflammatory mediators, particularly interleukin-6 (IL-6), stimulate an increase in the hepatic production of hepcidin.17,18 As was discussed under the anemia of iron deficiency, hepcidin inhibits ferriportin function in macrophages and reduces the transfer of iron from the storage pool to developing erythroid precursors in the bone marrow. As a result, the erythroid precursors are starved for iron in the midst of plenty. In addition, these progenitors do not proliferate adequately because erythropoietin levels are inappropriately low for the degree of anemia. The precise mechanism underlying this alteration is uncertain, but transgenic mice expressing high levels of hepcidin develop a microcytic anemia associated with low erythropoietin levels,19 suggesting that hepcidin directly or indirectly suppresses erythropoietin production.
What might be the reason for iron sequestration in the setting of inflammation? The best guess is that it serves to enhance the body’s ability to fend off certain types of infection, particularly those caused by bacteria (such as H. influenzae) that require iron for pathogenicity. In this regard it is interesting to consider that hepcidin is structurally related to defensins, a family of peptides that have intrinsic antibacterial activity. This connection further highlights the uncertain but intriguing interrelationship between inflammation, innate immunity, and iron metabolism.
The anemia is usually mild, and the dominant symptoms are those of the underlying disease. The red cells can be normocytic and normochromic, or hypochromic and microcytic, as in anemia of iron deficiency. The presence of increased storage iron in marrow macrophages, a high serum ferritin level, and a reduced total iron-binding capacity readily rule out iron deficiency as the cause of anemia. Only successful treatment of the underlying condition reliably corrects the anemia. However, some patients, particularly those with cancer, benefit from administration of erythropoietin.
Aplastic Anemia
Aplastic anemia refers to a syndrome of chronic primary hematopoietic failure and attendant pancytopenia (anemia, neutropenia, and thrombocytopenia). In the majority of patients autoimmune mechanisms are suspected,20 but inherited or acquired abnormalities of hematopoietic stem cells also seem to contribute in at least a subset of patients.
Etiology.
The most common circumstances associated with aplastic anemia are listed in Table 14-7. Most cases of “known” etiology follow exposure to chemicals and drugs. Certain drugs and agents (including many cancer chemotherapy drugs and the organic solvent benzene) cause marrow suppression that is dose related and reversible. In other instances, aplastic anemia arises in an unpredictable, idiosyncratic fashion following exposure to drugs that normally cause little or no marrow suppression. The implicated drugs include chloramphenicol and gold salts.
TABLE 14-7 Major Causes of Aplastic Anemia
| ACQUIRED |
| Idiopathic |
| Chemical Agents |
| Idiosyncratic |
| Physical Agents |
| Whole-body irradiation |
| Viral Infections |
| INHERITED |
Persistent marrow aplasia can also appear after a variety of viral infections, most commonly viral hepatitis of the non-A, non-B, non-C, non-G type, which is associated with 5% to 10% of cases. Why aplastic anemia develops in certain individuals is not understood.
Whole-body irradiation can destroy hematopoietic stem cells in a dose-dependent fashion. Persons who receive therapeutic irradiation or are exposed to radiation in nuclear accidents (e.g., Chernobyl) are at risk for marrow aplasia.
Inherited defects underlie some forms of aplastic aplasia. Fanconi anemia is a rare autosomal recessive disorder caused by defects in a multiprotein complex that is required for DNA repair (Chapter 7).21 Marrow hypofunction becomes evident early in life and is often accompanied by multiple congenital anomalies, such as hypoplasia of the kidney and spleen and bone anomalies, which most commonly involve the thumbs or radii. Inherited defects in telomerase are found in 5% to 10% of adult-onset aplastic anemia.22 You will recall from Chapters 1 and 7 that telomerase is required for cellular immortality and limitless replication. It might be anticipated, therefore, that partial deficits in telomerase activity could result in premature hematopoietic stem cell exhaustion and marrow aplasia. Even more common than telomerase mutations are abnormally short telomeres, which are found in the marrow cells of as many as half of those affected with aplastic anemia. It is unknown whether this shortening is due to other unappreciated telomerase defects or is a consequence of excessive stem cell replication.
In most instances, however, no initiating factor can be identified; about 65% of cases fall into this idiopathic category.
Pathogenesis.
The pathogenesis of aplastic anemia is not fully understood. Indeed, it is unlikely that a single mechanism underlies all cases. However, two major etiologies have been invoked: an extrinsic, immune-mediated suppression of marrow progenitors; and an intrinsic abnormality of stem cells (Fig. 14-24).
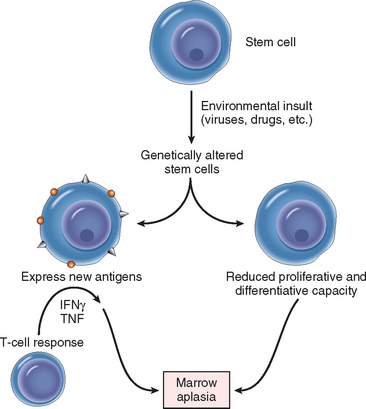
FIGURE 14-24 Pathophysiology of aplastic anemia. Damaged stem cells can produce progeny expressing neoantigens that evoke an autoimmune reaction, or give rise to a clonal population with reduced proliferative capacity. Either pathway could lead to marrow aplasia. See text for abbreviations.
Experimental studies have increasingly focused on a model in which activated T cells suppress hematopoietic stem cells. Stem cells may first be antigenically altered by exposure to drugs, infectious agents, or other unidentified environmental insults. This provokes a cellular immune response, during which activated TH1 cells produce cytokines such as interferon-γ (IFNγ) and TNF that suppress and kill hematopoietic progenitors. This scenario is supported by several observations. Expression analysis of the few remaining marrow stem cells from aplastic anemia marrows has revealed that genes involved in apoptosis and death pathways are up-regulated; of note, the same genes are up-regulated in normal stem cells exposed to interferon-γ. Even more compelling (and clinically relevant) evidence comes from experience with immunosuppressive therapy. Antithymocyte globulin and other immunosuppressive drugs such as cyclosporine produce responses in 60% to 70% of patients. It is proposed that these therapies work by suppressing or killing autoreactive T-cell clones. The antigens recognized by the autoreactive T cells are not well defined. In some instances GPI-linked proteins may be the targets, possibly explaining the previously noted association of aplastic anemia and PNH.
Alternatively, the notion that aplastic anemia results from a fundamental stem cell abnormality is supported by the presence of karyotypic aberrations in many cases; the occasional transformation of aplasias into myeloid neoplasms, typically myelodysplasia or acute myeloid leukemia; and the association with abnormally short telomeres. Some marrow insult (or a predisposition to DNA damage) presumably results in sufficient injury to limit the proliferative and differentiative capacity of stem cells. If the damage is extensive enough, aplastic anemia results. These two mechanisms are not mutually exclusive, since genetically altered stem cells might also express “neoantigens” that could serve as targets for a T-cell attack.
Morphology. The markedly hypocellular bone marrow is largely devoid of hematopoietic cells; often only fat cells, fibrous stroma, and scattered lymphocytes and plasma cells remain. Marrow aspirates often yield little material (a “dry tap”); hence, aplasia is best appreciated in marrow biopsies (Fig. 14-25). Other nonspecific pathologic changes are related to granulocytopenia and thrombocytopenia, such as mucocutaneous bacterial infections and abnormal bleeding, respectively. If the anemia necessitates multiple transfusions, systemic hemosiderosis can appear.
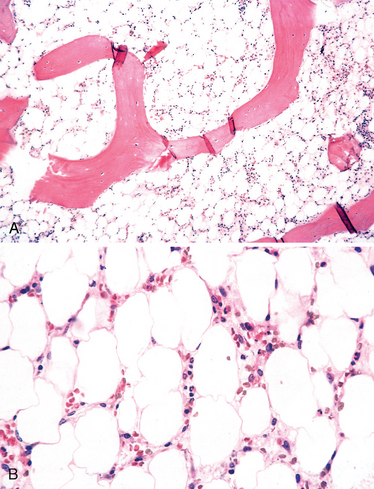
Clinical Features.
Aplastic anemia can occur at any age and in either sex. The onset is usually insidious. Initial manifestations vary somewhat, depending on which cell line is predominantly affected, but pancytopenia ultimately appears, with the expected consequences. Anemia can cause progressive weakness, pallor, and dyspnea; thrombocytopenia is heralded by petechiae and ecchymoses; and neutropenia manifests as frequent and persistent minor infections or the sudden onset of chills, fever, and prostration. Splenomegaly is characteristically absent; if it is present, the diagnosis of aplastic anemia should be seriously questioned. The red cells are usually slightly macrocytic and normochromic. Reticulocytopenia is the rule.
The diagnosis rests on examination of a bone marrow biopsy. It is important to distinguish aplastic anemia from other causes of pancytopenia, such as “aleukemic” leukemia and myelodysplastic syndromes (Chapter 13), which can have identical clinical manifestations. In aplastic anemia, the marrow is hypocellular (and usually markedly so), whereas myeloid neoplasms are associated with hypercellular marrows filled with neoplastic progenitors.
The prognosis is variable.20 Bone marrow transplantation is the treatment of choice in those with a suitable donor and provides a 5-year survival of over 75%. Older patients or those without suitable donors often respond well to immunosuppressive therapy.
Pure Red Cell Aplasia
As the name implies, pure red cell aplasia is a primary marrow disorder in which only erythroid progenitors are suppressed. In severe cases, red cell progenitors are completely absent from the marrow. It may occur in association with neoplasms, particularly thymoma and large granular lymphocytic leukemia (Chapter 13), drug exposures, autoimmune disorders, and parvovirus infection (a circumstance that is discussed below). With the exception of those with parvovirus infection, it is likely that most cases have an autoimmune basis. When a thymoma is present, resection leads to hematologic improvement in about half of the patients, possibly because the tumor is a source of marrow suppressive cells. In patients without thymoma, immunosuppressive therapy is often beneficial. Plasmapheresis may also be helpful in unusual patients with pathogenic autoantibodies, such as neutralizing antibodies to erythropoietin that appear de novo or following the administration of recombinant erythropoietin.
A special form of red cell aplasia occurs in individuals infected with parvovirus B19, which preferentially infects and destroys red cell progenitors. Normal individuals clear parvovirus infections within 1 to 2 weeks; as a result, the aplasia is transient and clinically unimportant. However, in persons with moderate to severe hemolytic anemias, even a brief cessation of erythropoiesis results in rapid worsening of the anemia, producing an aplastic crisis. In those who are severely immunosuppressed (such as persons with advanced HIV infection), an ineffective immune response sometimes permits the infection to persist, leading to chronic red cell aplasia and a moderate to severe anemia.
Other Forms of Marrow Failure
Myelophthisic anemia describes a form of marrow failure in which space-occupying lesions replace normal marrow elements. The commonest cause is metastatic cancer, most often carcinomas arising in the breast, lung, and prostate. However, any infiltrative process (e.g., granulomatous disease) involving the marrow can produce identical findings. It should be remembered that myelophthisic anemia is also a feature of the spent phase of myeloproliferative disorders (Chapter 13). All of the responsible diseases cause marrow distortion and fibrosis, which act to displace normal marrow elements and disturb mechanisms that regulate the egress of red cells and granulocytes from the marrow. The latter effect causes the abnormal release of nucleated erythroid precursors and immature granulocytic forms (leukoerythroblastosis) into peripheral smears, and the appearance of teardrop-shaped red cells, which are believed to be deformed during their tortuous escape from the fibrotic marrow.
Chronic renal failure, whatever its cause, is almost invariably associated with an anemia that tends to be roughly proportional to the severity of the uremia. The basis of anemia in renal failure is multifactorial, but the dominant cause is the diminished synthesis of erythropoietin by the damaged kidneys, which leads to inadequate red cell production. Other contributors are an extracorpuscular defect that reduces red cell life span, and iron deficiency due to platelet dysfunction and increased bleeding, which is often encountered in uremia. Administration of recombinant erythropoietin results in a significant improvement of the anemia, although an optimal response may require concomitant iron replacement therapy. Hepatocellular liver disease, whether toxic, infectious, or cirrhotic, is associated with anemia attributed to decreased marrow function. Folate and iron deficiencies caused by poor nutrition and excessive bleeding often exacerbate anemia in this setting. Erythroid progenitors are preferentially affected; depression of the white cell count and platelets is less common but also occurs. The anemia is often slightly macrocytic due to lipid abnormalities associated with liver failure, which cause red cell membranes to acquire phospholipid and cholesterol as they circulate in the peripheral blood. Endocrine disorders, particularly hypothyroidism, may also be associated with a mild normochromic, normocytic anemia.
Polycythemia
Polycythemia denotes an abnormally high red cell count, usually with a corresponding increase in the hemoglobin level. The increase in red cell count is relative when there is hemoconcentration due to decreased plasma volume, or absolute when there is an increase in the total red cell mass. Relative polycythemia results from dehydration, such as occurs with deprivation of water, prolonged vomiting or diarrhea, or excessive use of diuretics. It is also associated with an obscure condition of unknown etiology called stress polycythemia, or Gaisböck syndrome. Affected individuals are usually hypertensive, obese, and anxious (“stressed”). Absolute polycythemia is primary when it results from an intrinsic abnormality of hematopoietic precursors and secondary when the red cell progenitors are responding to increased levels of erythropoietin. A pathophysiologic classification of polycythemia divided along these lines is given in Table 14-8.
TABLE 14-8 Pathophysiologic Classification of Polycythemia
| RELATIVE |
| Reduced plasma volume (hemoconcentration) |
| ABSOLUTE |
| Primary (Low Erythropoietin) |
| Secondary (High Erythropoietin) |
HIF-1α, hypoxia-induced factor 1α.
The most common cause of primary polycythemia is polycythemia vera, a myeloproliferative disorder associated with mutations that lead to erythropoietin-independent growth of red cell progenitors (considered in Chapter 13). Another much less common form of primary polycythemia results from familial mutations in the erythropoietin receptor that induce erythropoietin-independent receptor activation. One such individual has won Olympic gold medals in cross-country skiing, having benefited from this natural form of blood doping! Secondary polycythemias are caused by compensatory or pathologic increases in erythropoietin secretion. Causes of the latter include erythropoietin-secreting tumors and rare, but illustrative, inherited defects that lead to the stabilization of HIF-1α, a hypoxia-induced factor that stimulates the transcription of the erythropoietin gene.23
Bleeding Disorders: Hemorrhagic Diatheses
Excessive bleeding can result from (1) increased fragility of vessels, (2) platelet deficiency or dysfunction, and (3) derangement of coagulation, alone or in combination. Before discussing specific bleeding disorders, it is helpful to review the common laboratory tests used in the evaluation of a bleeding diathesis. It should be recalled from the discussion in Chapter 4 that the normal hemostatic response involves the blood vessel wall, the platelets, and the clotting cascade. Tests used to evaluate different aspects of hemostasis are the following:
More specialized tests are available to measure the levels of specific clotting factors, fibrinogen, fibrin split products, and the presence of circulating anticoagulants.
BLEEDING DISORDERS CAUSED BY VESSEL WALL ABNORMALITIES
Disorders within this category, sometimes called nonthrombocytopenic purpuras, are relatively common but do not usually cause serious bleeding problems. Most often, they induce small hemorrhages (petechiae and purpura) in the skin or mucous membranes, particularly the gingivae. On occasion, however, more significant hemorrhages can occur into joints, muscles, and subperiosteal locations, or take the form of menorrhagia, nosebleeds, gastrointestinal bleeding, or hematuria. The platelet count, the bleeding time, and tests of coagulation (PT, PTT) usually yield normal results.
The varied clinical conditions in which abnormalities in the vessel wall cause bleeding include the following:
Among these conditions, serious bleeding is most often associated with hereditary telangiectasia. The bleeding in each is nonspecific, and the diagnosis of these entities is based on the recognition of other more specific associated findings.
BLEEDING RELATED TO REDUCED PLATELET NUMBER: THROMBOCYTOPENIA
Reduction in platelet number constitutes an important cause of generalized bleeding. A count below 100,000 platelets/μL is generally considered to constitute thrombocytopenia. However, spontaneous bleeding does not become evident until platelet counts fall below 20,000 platelets/μL. Platelet counts in the range of 20,000 to 50,000 platelets/μL can aggravate post-traumatic bleeding. Bleeding resulting from thrombocytopenia is associated with a normal PT and PTT.
It hardly needs reiteration that platelets are critical for hemostasis, since they form temporary plugs that stop bleeding and promote key reactions in the coagulation cascade (as discussed in Chapter 4). Spontaneous bleeding associated with thrombocytopenia most often involves small vessels. Common sites for such hemorrhages are the skin and the mucous membranes of the gastrointestinal and genitourinary tracts. Most feared, however, is intracranial bleeding, which is a threat to any patient with a markedly depressed platelet count.
The many causes of thrombocytopenia can be classified into four major categories (Table 14-9).
TABLE 14-9 Causes of Thrombocytopenia
| DECREASED PRODUCTION OF PLATELETS |
| DECREASED PLATELET SURVIVAL |
| SEQUESTRATION |
| Hypersplenism |
| DILUTION |
| Transfusions |
Chronic Immune Thrombocytopenic Purpura (ITP)
Chronic ITP is caused by autoantibodies to platelets. It can occur in the setting of a variety of predisposing conditions and exposures (secondary) or in the absence of any known risk factors (primary or idiopathic). The contexts in which chronic ITP occurs secondarily are numerous and include individuals with systemic lupus erythematosus (Chapter 6), HIV infection, and B-cell neoplasms such as chronic lymphocytic leukemia (Chapter 13). The diagnosis of primary chronic ITP is made only after secondary causes are excluded.
Pathogenesis.
The autoantibodies, most often directed against platelet membrane glycoproteins IIb-IIIa or Ib-IX, can be demonstrated in the plasma and bound to the platelet surface in about 80% of patients. In the overwhelming majority of cases, the antiplatelet antibodies are of the IgG class.
As in autoimmune hemolytic anemias, antiplatelet antibodies act as opsonins that are recognized by IgG Fc receptors expressed on phagocytes (Chapter 6), leading to increased platelet destruction. The thrombocytopenia is usually markedly improved by splenectomy, indicating that the spleen is the major site of removal of opsonized platelets. The splenic red pulp is also rich in plasma cells, and part of the benefit of splenectomy (a common treatment for chronic ITP) may stem from the removal of a source of autoantibodies. In some instances the autoantibodies may also bind to and damage megakaryocytes, leading to decreases in platelet production that further exacerbate the thrombocytopenia.
Morphology. The principal changes of thrombocytopenic purpura are found in the spleen, bone marrow, and blood, but they are not specific. Secondary changes related to the bleeding diathesis may be found in any tissue or structure in the body.
The spleen is of normal size. Typically, there is congestion of the sinusoids and enlargement of the splenic follicles, often associated with prominent reactive germinal centers. In many instances scattered megakaryocytes are found within the sinuses. This may represent a very mild form of extramedullary hematopoiesis that is driven by elevated levels of thrombopoietin. The marrow reveals a modestly increased number of megakaryocytes. Some are apparently immature, with large, nonlobulated, single nuclei. These findings are not specific for ITP but merely reflect accelerated thrombopoiesis, being found in most forms of thrombocytopenia resulting from increased platelet destruction. The importance of bone marrow examination is to rule out thrombocytopenias resulting from bone marrow failure or other primary bone marrow disorders. The secondary changes relate to the hemorrhages that are dispersed throughout the body. The peripheral blood often reveals abnormally large platelets (megathrombocytes), which are a sign of accelerated thrombopoiesis.
Clinical Features.
Chronic ITP occurs most commonly in adult women typically under 40 years of age. The female-to-male ratio is 3 : 1. It is often insidious in onset and is characterized by bleeding into the skin and mucosal surfaces. Cutaneous bleeding is seen in the form of pinpoint hemorrhages (petechiae), which are especially prominent in the dependent areas where the capillary pressure is higher. Petechiae can become confluent, giving rise to ecchymoses. Often there is a history of easy bruising, nosebleeds, bleeding from the gums, and hemorrhages into soft tissues from relatively minor trauma. The disease may manifest first with melena, hematuria, or excessive menstrual flow. Subarachnoid hemorrhage and intracerebral hemorrhage are serious and sometimes fatal complications, but fortunately they are rare in treated patients. Splenomegaly and lymphadenopathy are uncommon in primary disease, and their presence should lead one to consider other diagnoses, such as ITP secondary to a B-cell neoplasm.
The clinical signs and symptoms are not specific but rather reflective of the thrombocytopenia. The findings of a low platelet count, normal or increased megakaryocytes in the bone marrow, and large platelets in the peripheral blood are taken as presumptive evidence of accelerated platelet destruction. The PT and PTT are normal. Tests for platelet autoantibodies are not widely available. Therefore, the diagnosis is one of exclusion and can be made only after other causes of thrombocytopenia, such as those listed inTable 14-9, have been ruled out.
Almost all patients respond to glucocorticoids (which inhibit phagocyte function), but many eventually relapse. In such individuals, splenectomy normalizes the platelet count in about two thirds of patients, but with the attendant in-creased risk of bacterial sepsis. Immunomodulatory agents such as intravenous immunoglobulin or anti-CD20 antibody (rituximab) are often effective in patients who relapse after splenectomy or for whom splenectomy is contraindicated.
Acute Immune Thrombocytopenic Purpura
Like chronic ITP, this condition is caused by autoantibodies to platelets, but its clinical features and course are distinct. Acute ITP is mainly a disease of childhood occurring with equal frequency in both sexes. Symptoms appear abruptly and usually follow a viral illness, which typically occurs about 2 weeks before the onset of the thrombocytopenia. Unlike chronic ITP, acute ITP is self-limited, usually resolving spontaneously within 6 months. Glucocorticoids are given only if the thrombocytopenia is severe. In about 20% of children, usually those without a viral prodrome, thrombocytopenia persists; these less fortunate children have a childhood form of chronic ITP that follows a course similar to the adult disease.
Drug-Induced Thrombocytopenia
Drugs can induce thrombocytopenia through direct effects on platelets and secondary to immunologically mediated platelet destruction. The drugs most commonly implicated are quinine, quinidine, and vancomycin, all of which bind platelet glycoproteins and in one way or another create antigenic determinants that are recognized by antibodies.24 Much more rarely, drugs such as gold salts induce true autoantibodies through unknown mechanisms. Thrombocytopenia, which may be severe, is also a common consequence of platelet inhibitory drugs that bind glycoprotein IIb/IIIa; it is hypothesized that these drugs induce conformational changes in glycoprotein IIb/IIIa and create an immunogenic epitope.
Heparin-induced thrombocytopenia (HIT) has a distinctive pathogenesis and is of particular importance because of its potential for severe clinical consequences.25 Thrombocytopenia occurs in about 5% of persons receiving heparin. Most develop so-called type I thrombocytopenia, which occurs rapidly after the onset of therapy and is of little clinical importance, sometimes resolving despite the continuation of therapy. It most likely results from a direct platelet-aggregating effect of heparin. Type II thrombocytopenia is less common but of much greater clinical significance. It occurs 5 to 14 days after therapy begins (or sooner if the person has been sensitized to heparin) and, paradoxically, often leads to life-threatening venous and arterial thrombosis. This severe form of HIT is caused by antibodies that recognize complexes of heparin and platelet factor 4, which is a normal component of platelet granules. Binding of antibody to these complexes activates platelets and promotes thrombosis, even in the setting of thrombocytopenia. Unless therapy is immediately discontinued and an alternative nonheparin anticoagulant instituted, clots within large arteries may lead to vascular insufficiency and limb loss, and emboli from deep venous thrombosis can cause fatal pulmonary thromboembolism. The risk of severe HIT is lowered, but not completely eliminated, by the use of low-molecular-weight heparin preparations. Unfortunately, once severe HIT develops even low-molecular-weight heparins exacerbate the thrombotic tendency and must be avoided.
HIV-Associated Thrombocytopenia
Thrombocytopenia is one of the most common hematologic manifestation of HIV infection. Both impaired platelet production and increased destruction contribute. CD4 and CXCR4, the receptor and coreceptor, respectively, for HIV, are found on megakaryocytes, allowing these cells to be infected. HIV-infected megakaryocytes are prone to apoptosis and their ability to produce platelets is impaired. HIV infection also causes B-cell hyperplasia and dysregulation, which predisposes to the development of autoantibodies. In some instances the antibodies are directed against platelet membrane glycoprotein IIb-III complexes. As in other immune cytopenias, the autoantibodies opsoninize platelets, promoting their destruction by mononuclear phagocytes in the spleen and elsewhere. The deposition of immune complexes on platelets may also contribute to the accelerated loss of platelets in some patients who are HIV infected.
Thrombotic Microangiopathies: Thrombotic Thrombocytopenic Purpura (TTP) and Hemolytic-Uremic Syndrome (HUS)
The term thrombotic microangiopathy encompasses a spectrum of clinical syndromes that includes TTP and HUS. According to its original description, TTP was defined as the pentad of fever, thrombocytopenia, microangiopathic hemolytic anemia, transient neurologic deficits, and renal failure. HUS is also associated with microangiopathic hemolytic anemia and thrombocytopenia but is distinguished by the absence of neurologic symptoms, the prominence of acute renal failure, and its frequent occurrence in children. With time, experience, and increased mechanistic insight, however, these distinctions have blurred. Many adult patients with “TTP” lack one or more of the five criteria, and some patients with “HUS” have fever and neurologic dysfunction. It is now appreciated that HUS and TTP are both caused by insults that lead to the excessive activation of platelets, which deposit as thrombi in microcirculatory beds. These intravascular thrombi cause a microangiopathic hemolytic anemia and widespread organ dysfunction, and the attendant consumption of platelets leads to thrombocytopenia. It is believed that the varied clinical manifestations of TTP and HUS are related to differing proclivities for thrombus formation in tissues.
Although certain features of the various thrombotic microangiopathies overlap, the triggers for the pathogenic platelet activation are distinctive and provide a more satisfying and clinically relevant way of thinking about these disorders; these are summarized in Table 14-10. TTP is usually associated with a deficiency in a plasma enzyme called ADAMTS13, also designated “vWF metalloprotease.” ADAMTS13 normally degrades very high-molecular-weight multimers of von Willebrand factor (vWF). In its absence, these multimers accumulate in plasma and tend to promote platelet activation and aggregation. Superimposition of endothelial cell injury (caused by some other condition) may further promote the formation of platelet microaggregates, thus initiating or exacerbating clinically evident TTP.
TABLE 14-10 Thrombotic Microangiopathies: Causes and Associations
| THROMBOTIC THROMBOCYTOPENIC PURPURA |
| Deficiency of ADAMTS13 |
| HEMOLYTIC UREMIC SYNDROME |
| Epidemic: Escherichia coli strain O157 : H7 infection |
| Nonepidemic: alternative complement pathway inhibitor deficiencies (complement factor H, membrane cofactor protein (CD46), or factor I) |
| Miscellaneous associations |
HIV, human immunodeficiency virus.
The deficiency of ADAMTS13 can be inherited or acquired. In the acquired form, an autoantibody that inhibits the metalloprotease activity of ADAMTS13 is present.26 Less commonly, patients inherit an inactivating mutation in ADAMTS13.27 In those with hereditary ADAMTS13 deficiency, the onset is often delayed until adolescence and the symptoms are episodic. Thus, factors other than ADAMTS13 (e.g., some superimposed vascular injury or prothrombotic state) must be involved in triggering full-blown TTP.
TTP is an important diagnosis to consider in any patient presenting with thrombocytopenia and microangiopathic hemolytic anemia, since delays in diagnosis can be fatal. With plasma exchange, which removes autoantibodies and provides functional ADAMTS13, TTP (which once was uniformly fatal) can be treated successfully in more than 80% of patients.
In contrast, HUS is associated with normal levels of ADAMTS13 and is initiated by several other distinct defects.28 Epidemic, “typical” HUS is strongly associated with infectious gastroenteritis caused by Escherichia coli strain O157:H7, which elaborates a Shiga-like toxin. This toxin is absorbed from the inflamed gastrointestinal mucosa into the circulation, where it alters endothelial cell function in some manner that results in platelet activation and aggregation. Children and the elderly are at highest risk. Those affected present with bloody diarrhea, and a few days later HUS makes its appearance. With appropriate supportive care complete recovery is possible, but irreversible renal damage and death can occur in more severe cases.
Nonepidemic, “atypical” HUS is often associated with defects in complement factor H, membrane cofactor protein (CD46), or factor I, three proteins that normally act to prevent excessive activation of the alternative complement pathway. Deficiencies of these proteins can be caused by inherited defects or acquired inhibitory autoantibodies and are associated with a remitting, relapsing course. Unlike TTP, the basis for the platelet activation in HUS is unclear; presumably, both Shiga-like toxin produced by pathogenic E. coli and defects in complement-regulatory proteins alter endothelial cell function in some way that promotes platelet activation.
Thrombotic microangiopathies resembling HUS can also be seen following exposures to other agents that damage endothelial cells (e.g., certain drugs and radiation therapy). The prognosis in these settings is guarded, because the HUS is often complicated by chronic, life-threatening conditions.
While DIC (discussed later) and thrombotic microangiopathies share features such as microvascular occlusion and microangiopathic hemolytic anemia, they are pathogenically distinct. In TTP and HUS (unlike in DIC), activation of the coagulation cascade is not of primary importance, and hence laboratory tests of coagulation, such as the PT and PTT, are usually normal.
BLEEDING DISORDERS RELATED TO DEFECTIVE PLATELET FUNCTIONS
Qualitative defects of platelet function can be inherited or acquired. Several inherited disorders characterized by abnormal platelet function and normal platelet count have been described. A brief discussion of these rare diseases is warranted because they provide excellent models for investigating the molecular mechanisms of platelet function.
Inherited disorders of platelet function can be classified into three pathogenically distinct groups: (1) defects of adhesion, (2) defects of aggregation, and (3) disorders of platelet secretion (release reaction).
Among the acquired defects of platelet function, two are clinically significant. The first is caused by ingestion of aspirin and other nonsteroidal anti-inflammatory drugs. Aspirin is a potent, irreversible inhibitor of the enzyme cyclooxygenase, which is required for the synthesis of thromboxane A2 and prostaglandins (Chapter 2). These mediators play important roles in platelet aggregation and subsequent release reactions (Chapter 4). The antiplatelet effects of aspirin form the basis for its use in the prophylaxis of coronary thrombosis (Chapter 12). Uremia (Chapter 20) is the second condition exemplifying an acquired defect in platelet function. The pathogenesis of platelet dysfunction in uremia is complex and involves defects in adhesion, granule secretion, and aggregation.29
HEMORRHAGIC DIATHESES RELATED TO ABNORMALITIES IN CLOTTING FACTORS
Inherited or acquired deficiencies of virtually every coagulation factor have been reported as causes of bleeding diatheses. Unlike the petechial bleeding seen with thrombocytopenia, bleeding due to isolated coagulation factor deficiencies most commonly manifests as large post-traumatic ecchymoses or hematomas, or prolonged bleeding after a laceration or any form of surgical procedure. Bleeding into the gastrointestinal and urinary tracts, and particularly into weight-bearing joints (hemarthrosis), is common. Typical stories include the patient who oozes blood for days after a tooth extraction or who develops a hemarthrosis after minor stress on a knee joint.
Hereditary deficiencies typically affect a single clotting factor. The most common and important inherited deficiencies of coagulation factors affect factor VIII (hemophilia A), and factor IX (hemophilia B). Deficiencies of vWF (von Willebrand disease) are also discussed here, as this factor influences both coagulation and platelet function. Rare inherited deficiencies of each of the other coagulation factors have also been described. All cause bleeding except for factor XII deficiency; presumably, in vivo the extrinsic pathway and thrombin-mediated activation of factors XI and IX compensate for the absence of factor XII.
Acquired deficiencies usually involve multiple coagulation factors simultaneously and can be based on decreased protein synthesis or a shortened half-life. Vitamin K deficiency (Chapter 9) results in the impaired synthesis of factors II, VII, IX, and X and protein C. Many of these factors are made in the liver and are therefore deficient in severe parenchymal liver disease. Alternatively, in DIC, multiple coagulation factors are consumed and are therefore deficient. Acquired deficiencies of single factors occur, but they are rare. These are usually caused by inhibitory autoantibodies.
The Factor VIII-vWF Complex
The two most common inherited disorders of bleeding, hemophilia A and von Willebrand disease, are caused by qualitative or quantitative defects involving factor VIII and vWF, respectively. Before we discuss these disorders it will be helpful to review the structure and function of these two proteins, which exist together in the plasma as part of a single large complex.
Factor VIII and vWF are encoded by separate genes and are synthesized in different cells. Factor VIII is an essential cofactor of factor IX, which converts factor X to factor Xa (Fig. 14-26; also see Chapter 4). It is made in several tissues; sinusoidal endothelial cells and Kupffer cells in the liver, and tubular epithelial cells in the kidney, seem to be particularly important sources. Once factor VIII reaches the circulation, it binds to vWF, which is produced by endothelial cells and, to a lesser degree, by megakaryocytes, which are the source of the vWF that is found in platelet α-granules. vWF stabilizes factor VIII, which has a half-life of about 2.4 hours when free and 12 hours when bound to vWF in the circulation.
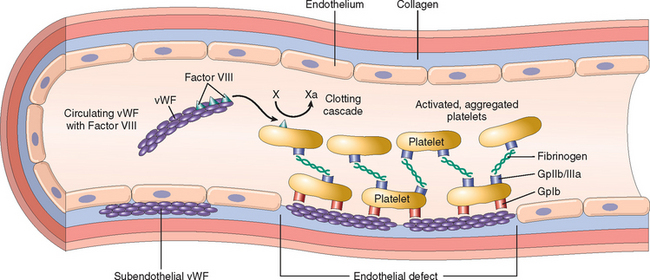
FIGURE 14-26 Structure and function of factor VIII–von Willebrand factor (vWF) complex. Factor VIII is synthesized in the liver and kidney, and vWF is made in endothelial cells and megakaryocytes. The two associate to form a complex in the circulation. vWF is also present in the subendothelial matrix of normal blood vessels and the α-granules of platelets. Following endothelial injury, exposure of subendothelial vWF causes adhesion of platelets, primarily via the glycoprotein lb (GpIb) platelet receptor. Circulating vWF and vWF released from the α-granules of activated platelets can bind exposed subendothelial matrix, further contributing to platelet adhesion and activation. Activated platelets form hemostatic aggregates; fibrinogen (and possibly vWF) participates in aggregation through bridging interactions with the glycoprotein IIb/IIIa (GpIIb/IIIa) platelet receptor. Factor VIII takes part in the coagulation cascade as a cofactor in the activation of factor X on the surface of activated platelets.
Circulating vWF exists as multimers containing as many as 100 subunits that can exceed 20 × 106 daltons in molecular mass. In addition to factor VIII, these multimers interact with several other proteins involved in hemostasis, including collagen, heparin, and possibly platelet membrane glycoproteins. The most important function of vWF is to promote the adhesion of platelets to the subendothelial matrix. This occurs through bridging interactions between platelet glycoprotein Ib-IX, vWF, and matrix components such as collagen. Some vWF is secreted from endothelial cells directly into the subendothelial matrix, where it lies ready to promote platelet adhesion if the endothelial lining is disrupted (see Fig. 14-26). Endothelial cells and platelets also release vWF into the circulation. Upon vascular injury, this second pool of vWF binds collagen in the subendothelial matrix to further augment platelet adhesion. vWF multimers may also promote platelet aggregation by binding to activated GpIIb/IIIa integrins; this activity may be of particular importance under conditions of high shear stress (such as occurs in small vessels).
Factor VIII and vWF protein levels are measured by immunological techniques. Factor VIII function is assessed by conducting coagulation assays with mixtures of patient plasma and factor VIII-deficient plasma. vWF function is assessed using the ristocetin agglutination test. This assay is performed by mixing the patient’s plasma with formalin-fixed platelets and ristocetin, a small molecule that binds and “activates” vWF. Ristocetin induces multivalent vWF multimers to bind platelet glycoprotein Ib-IX and form interplatelet “bridges.” The resulting clumping (agglutination) of platelets is measured in a device called an aggregometer. Thus, the degree to which patient plasma promotes ristocetin-dependent platelet agglutination is a measure of vWF activity.
Von Willebrand Disease
Von Willebrand disease is the most common inherited bleeding disorder of humans, affecting about 1% of adults in the United States. In most of those affected, the bleeding tendency is mild and often goes unnoticed until some hemostatic stress, such as surgery or a dental procedure, reveals its presence. The most common symptoms are spontaneous bleeding from mucous membranes (e.g., epistaxis); excessive bleeding from wounds; menorrhagia; and a prolonged bleeding time in the presence of a normal platelet count. It is usually transmitted as an autosomal dominant disorder, but rare autosomal recessive variants have been described.
Von Willebrand disease is molecularly heterogeneous.30 More than 20 variants have been described, which can be grouped into two major categories:
Type 1 and type 3 von Willebrand disease are associated with a reduced quantity of circulating vWF. Type 1, an autosomal dominant disorder characterized by a mild to moderate quantitative vWF deficiency, accounts for about 70% of all cases. Incomplete penetrance and variable expressivity are commonly observed, but it generally is associated with mild disease. Type 3 (an autosomal recessive disorder) is associated with extremely low levels of functional vWF and correspondingly severe clinical manifestations. Because a severe deficiency of vWF has a marked effect on the stability of factor VIII, some of the bleeding characteristics resemble those seen in hemophilia. The nature of the mutations in the majority of individuals with type 1 disease is poorly defined. In some cases missense mutations have been found. Type 3 disease is usually caused by deletions or frameshift mutations involving both alleles.
Type 2 von Willebrand disease is characterized by qualitative defects in vWF; there are several subtypes, of which type 2A is the most common. It is inherited as an autosomal dominant disorder. vWF is expressed in normal amounts, but missense mutations are present that lead to defective multimer assembly. Large and intermediate multimers, representing the most active forms of vWF, are missing from plasma. Type 2 von Willebrand disease accounts for 25% of all cases and is associated with mild to moderate bleeding.
Patients with von Willebrand disease have defects in platelet function despite a normal platelet count. The plasma level of active vWF, measured as the ristocetin cofactor activity, is reduced. Because vWF stabilizes factor VIII, a deficiency of vWF gives rise to a secondary decrease in factor VIII levels. This may be reflected by a prolongation of the PTT in von Willebrand disease types 1 and 3. However, except in rare type 3 patients, adverse complications typical of severe factor VIII deficiency, such as bleeding into the joints, are not seen.
To summarize, in von Willebrand disease inherited defects in vWF lead to secondary abnormalities in platelet adhesion and clot formation. Even within families in which a single defective vWF allele is segregating, a wide variability in clinical expression is common. This is due in part to additional genetic factors that influence circulating levels of vWF, which vary greatly in normal populations. Persons facing hemostatic challenges (dental work, surgery) can be treated with desmopressin, which stimulates vWF release, or infusions of plasma concentrates containing factor VIII and vWF.
Hemophilia A (Factor VIII Deficiency)
Hemophilia A is the most common hereditary disease associated with life-threatening bleeding.31 It is caused by mutations in factor VIII, which is an essential cofactor for factor IX in the coagulation cascade (Chapter 4). Hemophilia A is inherited as an X-linked recessive trait and thus affects mainly males and homozygous females. Rarely, excessive bleeding occurs in heterozygous females, presumably as a result of inactivation of the X chromosome bearing the normal factor VIII allele by chance in most cells (unfavorable lyonization). About 30% of patients have no family history; their disease is caused by new mutations.
Hemophilia A exhibits a wide range of clinical severity that correlates well with the level of factor VIII activity. Those with less than 1% of normal levels have severe disease; those with 2% to 5% of normal levels have moderately severe disease; and those with 6% to 50% of normal levels have mild disease. The varying degrees of factor VIII deficiency are largely explained by heterogeneity in the causative mutations. As with β-thalassemia, the genetic lesions include deletions, nonsense mutations that create stop codons, and mutations that cause errors in mRNA splicing. The most severe deficiencies result from an inversion involving the X chromosome that completely abolishes the synthesis of factor VIII. Less commonly, severe hemophilia A is associated with point mutations in factor VIII that impair the function of the protein. In such cases factor VIII levels seem normal by immunoassay. Mutations permitting some active factor VIII to be synthesized are associated with mild to moderate disease. The disease in such patients may be modified by other genetic factors that influence factor VIII expression levels, which vary widely in normal individuals.
In all symptomatic cases there is a tendency toward easy bruising and massive hemorrhage after trauma or operative procedures. In addition, “spontaneous” hemorrhages frequently occur in regions of the body normally subject to trauma, particularly the joints, where they are known as hemarthroses. Recurrent bleeding into the joints leads to progressive deformities that can be crippling. Petechiae are characteristically absent.
Patients with hemophilia A typically have a prolonged PTT and a normal PT. These tests point to an abnormality of the intrinsic coagulation pathway. Factor VIII–specific assays are required for diagnosis.
Given that one arm of the coagulation cascade, the extrinsic pathway, is intact in hemophilia A, it is reasonable to ask: Why do patients bleed? Obviously, test tube assays of coagulation are imperfect surrogates for what occurs in vivo, and it must be that in the face of factor VIII deficiency, fibrin deposition is inadequate to achieve hemostasis. It is beyond our scope to discuss this issue in detail; it seems that the chief role of the extrinsic pathway in hemostasis is to initiate a limited burst of thrombin activation upon tissue injury. This initial procoagulant stimulus is reinforced and amplified by a critical feedback loop in which thrombin activates factors XI and IX of the intrinsic pathway (Chapter 4). In the absence of factor VIII, this feedback loop is inactive and insufficient thrombin (and fibrin) is generated to create a stable clot. In addition, high levels of thrombin are required to activate TAFI (thrombin-activated fibrinolysis inhibitor), a factor that inhibits fibrinolysis. Thus, both inadequate coagulation (fibrinogenesis) and inappropriate clot removal (fibrinolysis) contribute to the bleeding diathesis in hemophilia. The precise explanation for the tendency of hemophiliacs to bleed at particular sites (joints, muscles, and the central nervous system) remains uncertain.
Hemophilia A is treated with infusions of recombinant factor VIII. About 15% of patients with severe hemophilia A develop antibodies that bind and inhibit factor VIII, probably because the protein is perceived as foreign, having never been “seen” by the immune system. These antibody inhibitors can be a very difficult therapeutic challenge. Before the development of recombinant factor VIII therapy, thousands of hemophiliacs received plasma-derived factor VIII concentrates containing HIV, and many developed AIDS (Chapter 6). The risk of HIV transmission has been eliminated but tragically too late for an entire generation of hemophiliacs. Efforts to develop somatic gene therapy for hemophilia are continuing.
Hemophilia B (Christmas Disease, Factor IX Deficiency)
Severe factor IX deficiency produces a disorder clinically indistinguishable from factor VIII deficiency (hemophilia A). This should not be surprising, given that factors VIII and IX function together to activate factor X. A wide spectrum of mutations involving the gene that encodes factor IX is found in hemophilia B. Like hemophilia A it is inherited as an X-linked recessive trait and shows variable clinical severity. In about 15% of these patients, factor IX is present but nonfunctional. As with hemophilia A, the PTT is prolonged and the PT is normal. Diagnosis of Christmas disease (named after the first patient identified with this condition, and not the holiday) is possible only by assay of the factor levels. The disease is treated with infusions of recombinant factor IX.
DISSEMINATED INTRAVASCULAR COAGULATION (DIC)
DIC is an acute, subacute, or chronic thrombohemorrhagic disorder characterized by the excessive activation of coagulation, which leads to the formation of thrombi in the microvasculature of the body. It occurs as a secondary complication of many different disorders. Sometimes the coagulopathy is localized to a specific organ or tissue. As a consequence of the thrombotic diathesis there is consumption of platelets, fibrin, and coagulation factors and, secondarily, activation of fibrinolysis. DIC can present with signs and symptoms relating to the tissue hypoxia and infarction caused by the myriad microthrombi; with hemorrhage caused by the depletion of factors required for hemostasis and the activation of fibrinolytic mechanisms; or both.
Etiology and Pathogenesis.
At the outset, it must be emphasized that DIC is not a primary disease. It is a coagulopathy that occurs in the course of a variety of clinical conditions. In discussing the general mechanisms underlying DIC, it is useful to briefly review the normal process of blood coagulation and clot removal (see Chapter 4 for more details).
Clotting can be initiated by either of two pathways: (1) the extrinsic pathway, which is triggered by the release of tissue factor (“tissue thromboplastin”); and (2) the intrinsic pathway, which involves the activation of factor XII by surface contact with collagen or other negatively charged substances. Both pathways, through a series of intermediate steps, result in the generation of thrombin, which in turn converts fibrinogen to fibrin. At the site of injury, thrombin further augments local fibrin deposition by directly activating the intrinsic pathway and factors that inhibit fibrinolysis.
Once clotting is initiated, it is critically important that it be limited to the site of injury. Remarkably, as thrombin is swept away in the bloodstream and encounters normal vessels, it is converted to an anticoagulant through binding to thrombomodulin, a protein found on the surface of endothelial cells. The thrombin-thrombomodulin complex activates protein C, which is an important inhibitor of two procoagulants, factor V and factor VIII. Other activated coagulation factors are removed from the circulation by the liver, and as you will recall, the blood also contains several potent fibrinolytic factors, such as plasmin. These and additional checks and balances normally ensure that just enough clotting occurs at the right place and time.
From this brief review it should be clear that DIC could result from pathologic activation of the extrinsic and/or intrinsic pathways of coagulation or the impairment of clot-inhibiting mechanisms. Since the latter rarely constitute primary mechanisms of DIC, we will focus on the abnormal initiation of clotting.
Two major mechanisms trigger DIC: (1) release of tissue factor or thromboplastic substances into the circulation, and (2) widespread injury to the endothelial cells. Thromboplastic substances can be derived from a variety of sources, such as the placenta in obstetric complications and the cytoplasmic granules of acute promyelocytic leukemia cells (Chapter 13). Mucus released from certain adenocarcinomas can directly activate factor X, independent of factor VII.
Endothelial injury can initiate DIC in several ways. Injuries that cause endothelial cell necrosis expose the subendothelial matrix, leading to the activation of platelets and both arms of the coagulation pathway. However, even subtle endothelial injuries can unleash procoagulant activity. One mediator of such effects is TNF, which is implicated in DIC occurring with sepsis. TNF induces endothelial cells to express tissue factor on their cell surfaces and to decrease the expression of thrombomodulin, shifting the checks and balances that govern hemostasis towards coagulation. In addition, TNF upregulates the expression of adhesion molecules on endothelial cells, thereby promoting the adhesion of leukocytes, which can damage endothelial cells by releasing reactive oxygen species and preformed proteases. Widespread endothelial injury may also be produced by deposition of antigen-antibody complexes (e.g., systemic lupus erythematosus), temperature extremes (e.g., heat stroke, burns), or microorganisms (e.g., meningococci, rickettsiae). Even subtle endothelial injury can unleash procoagulant activity by enhancing membrane expression of tissue factor.
DIC is most likely to be associated with obstetric complications, malignant neoplasms, sepsis, and major trauma. The triggers in these conditions are often multiple and interrelated. For example, in bacterial infections endotoxins can injure endothelial cells and inhibit the expression of thrombomodulin directly or through production of TNF; stimulate the release of thromboplastins from inflammatory cells; and activate factor XII. Antigen-antibody complexes formed in response to the infection can activate the classical complement pathway, giving rise to complement fragments that secondarily activate both platelets and granulocytes. In massive trauma, extensive surgery, and severe burns, the major trigger is the release of tissue thromboplastins. In obstetric conditions, thromboplastins derived from the placenta, dead retained fetus, or amniotic fluid may enter the circulation. Hypoxia, acidosis, and shock, which often coexist in very ill patients, can also cause widespread endothelial injury, and supervening infections can complicate the problems further. Among cancers, acute promyelocytic leukemia and adenocarcinomas of the lung, pancreas, colon, and stomach are most frequently associated with DIC.
The possible consequences of DIC are twofold (Fig. 14-27). Firstly, there is widespread deposition of fibrin within the microcirculation. This leads to ischemia of the more severely affected or more vulnerable organs and a microangiopathic hemolytic anemia, which results from the fragmentation of red cells as they squeeze through the narrowed microvasculature. Secondly, the consumption of platelets and clotting factors and the activation of plasminogen leads to a hemorrhagic diathesis. Plasmin not only cleaves fibrin, but it also digests factors V and VIII, thereby reducing their concentration further. In addition, fibrin degradation products resulting from fibrinolysis inhibit platelet aggregation, fibrin polymerization, and thrombin. All of these derangements contribute to the hemostatic failure seen in DIC.
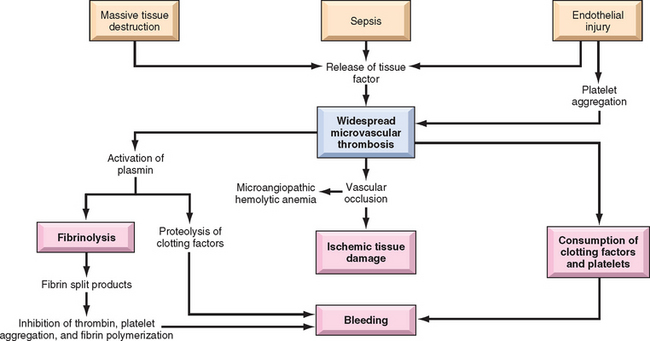
Morphology. Thrombi are most often found in the brain, heart, lungs, kidneys, adrenals, spleen, and liver, in decreasing order of frequency, but any tissue can be affected. Affected kidneys may have small thrombi in the glomeruli that evoke only reactive swelling of endothelial cells or, in severe cases, microinfarcts or even bilateral renal cortical necrosis. Numerous fibrin thrombi may be found in alveolar capillaries, sometimes associated with pulmonary edema and fibrin exudation, creating “hyaline membranes” reminiscent of acute respiratory distress syndrome (Chapter 15). In the central nervous system, fibrin thrombi can cause microinfarcts, occasionally complicated by simultaneous hemorrhage, which can sometimes lead to variable neurologic signs and symptoms. The manifestations in the endocrine glands are of considerable interest. In meningococcemia, fibrin thrombi within the microcirculation of the adrenal cortex are the probable basis for the massive adrenal hemorrhages seen in Waterhouse-Friderichsen syndrome (Chapter 24). Similarly, Sheehan postpartum pituitary necrosis (Chapter 24) is a form of DIC complicating labor and delivery. In toxemia of pregnancy (Chapter 22) the placenta exhibits widespread microthrombi, providing a plausible explanation for the premature atrophy of the cytotrophoblast and syncytiotrophoblast that is encountered in this condition. An unusual form of DIC occurs in association with giant hemangiomas, in which thrombi form within the neoplasm because of stasis and recurrent trauma to fragile blood vessels.
Clinical Features.
The onset can be fulminant, as in endotoxic shock or amniotic fluid embolism, or insidious and chronic, as in cases of carcinomatosis or retention of a dead fetus. Overall, about 50% of the affected are obstetric patients having complications of pregnancy. In this setting the disorder tends to be reversible with delivery of the fetus. About 33% of the affected patients have carcinomatosis. The remaining cases are associated with the various entities previously listed.
It is almost impossible to detail all the potential clinical presentations, but a few common patterns are worthy of description. These include microangiopathic hemolytic anemia; dyspnea, cyanosis, and respiratory failure; convulsions and coma; oliguria and acute renal failure; and sudden or progressive circulatory failure and shock. In general, acute DIC, associated with obstetric complications or major trauma, for example, is dominated by a bleeding diathesis, whereas chronic DIC, such as occurs in cancer patients, tends to present with thrombotic complications. The diagnosis is based on clinical observation and laboratory studies, including measurement of fibrinogen levels, platelets, the PT and PTT, and fibrin degradation products.
The prognosis is highly variable and largely depends on the underlying disorder. The only definitive treatment is to remove or treat the inciting cause. The management requires meticulous maneuvering between the Scylla of thrombosis and the Charybdis of bleeding diathesis. Administration of anticoagulants or procoagulants has been advocated in specific settings, but not without controversy.
1 Kay M. Immunoregulation of cellular life span. Ann N Y Acad Sci. 2005;1057:85.
2 Eber S, Lux SE. Hereditary spherocytosis—defects in proteins that connect the membrane skeleton to the lipid bilayer. Semin Hematol. 2004;41:118.
3 Kwiatkowski DP. How malaria has affected the human genome and what human genetics can teach us about malaria. Am J Hum Genet. 2005;77:171.
4 Browne PV, Hebbel RP. CD36-positive stress reticulocytosis in sickle cell anemia. J Lab Clin Med. 1996;127:340.
5 Zennadi R, et al. Epinephrine-induced activation of LW-mediated sickle cell adhesion and vaso-occlusion in vivo. Blood. 2007;110:2708.
6 El Nemer W, et al. Endothelial Lu/BCAM glycoproteins are novel ligands for red blood cell alpha4beta1 integrin: role in adhesion of sickle red blood cells to endothelial cells. Blood. 2007;109:3544.
7 Belcher JD, et al. Transgenic sickle mice have vascular inflammation. Blood. 2003;101:3953.
8 Kato GJ, et al. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev. 2007;21:37.
9 Platt OS. Hydroxyurea for the treatment of sickle cell anemia. N Engl J Med. 2008;358:1362.
10 Premawardhena A, et al. A novel molecular basis for beta thalassemia intermedia poses new questions about its pathophysiology. Blood. 2005;106:3251.
11 Schrier SL, Angelucci E. New strategies in the treatment of the thalassemias. Annu Rev Med. 2005;56:157.
12 Luzzatto L. Paroxysmal nocturnal hemoglobinuria: an acquired X-linked genetic disease with somatic-cell mosaicism. Curr Opin Genet Dev. 2006;16:317.
13 Hill A, et al. Recent developments in the understanding and management of paroxysmal nocturnal haemoglobinuria. Br J Haematol. 2007;137:181.
14 Gertz MA. Management of cold haemolytic syndrome. Br J Haematol. 2007;138:422.
15 King KE, Ness PM. Treatment of autoimmune hemolytic anemia. Semin Hematol. 2005;42:131.
16 Jin Y, et al. NALP1 in vitiligo-associated multiple autoimmune disease. N Engl J Med. 2007;356:1216.
17 Andrews NC, Schmidt PJ. Iron homeostasis. Annu Rev Physiol. 2007;69:69.
18 Roy CN, Andrews NC. Anemia of inflammation: the hepcidin link. Curr Opin Hematol. 2005;12:107.
19 Roy CN, et al. Hepcidin antimicrobial peptide transgenic mice exhibit features of the anemia of inflammation. Blood. 2007;109:4038.
20 Young NS, et al. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood. 2006;108:2509.
21 Taniguchi T, D’Andrea AD. Molecular pathogenesis of Fanconi anemia: recent progress. Blood. 2006;107:4223.
22 Yamaguchi H, et al. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N Engl J Med. 2005;352:1413.
23 Lee FS, et al. Oxygen sensing: recent insights from idiopathic erythrocytosis. Cell Cycle. 2006;5:941.
24 Aster RH, Bougie DW. Drug-induced immune thrombocytopenia. N Engl J Med. 2007;357:580.
25 Levy JH, Husting JM. Heparin-induced thrombocytopenia, a prothrombotic disease. Hematol Oncol Clin North Am. 2007;21:65.
26 Zheng XL, Sadler JE. Pathogenesis of thrombotic microangiopathies. Annu Rev Pathol. 2008;3:249.
27 Kokame K, Miyata T. Genetic defects leading to hereditary thrombotic thrombocytopenic purpura. Semin Hematol. 2004;41:34.
28 Tsai HM. The molecular biology of thrombotic microangiopathy. Kidney Int. 2006;70:16.
29 Sohal AS, et al. Uremic bleeding: pathophysiology and clinical risk factors. Thromb Res. 2006;118:417.
30 Sadler JE, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. J Thromb Haemost. 2006;4:2103.
31 Castaldo G, et al. Haemophilia A: molecular insights. Clin Chem Lab Med. 2007;45:450.