Chapter 13 Diseases of White Blood Cells, Lymph Nodes, Spleen, and Thymus
 DISORDERS OF WHITE CELLS
DISORDERS OF WHITE CELLS
The components of the hematopoietic system have been traditionally divided into the myeloid tissues, which include the bone marrow and the cells derived from it (e.g., red cells, platelets, granulocytes, and monocytes), and the lymphoid tissues, consisting of the thymus, lymph nodes, and spleen. It is important to recognize, however, that this subdivision is artificial with respect to both the normal physiology of hematopoietic cells and the diseases affecting them. For example, although bone marrow contains relatively few lymphocytes, it is the source of all lymphoid progenitors. Similarly, neoplastic disorders of myeloid progenitor cells (myeloid leukemias) originate in the bone marrow but secondarily involve the spleen and (to a lesser degree) the lymph nodes. Some red cell disorders (such as immunohemolytic anemia, discussed in Chapter 14) result from the formation of autoantibodies, signifying a primary disorder of lymphocytes. Thus, it is not possible to draw neat lines between diseases involving the myeloid and lymphoid tissues. Recognizing this difficulty, we somewhat arbitrarily divide diseases of the hematopoietic tissues into two chapters. In this chapter we discuss white cell diseases and disorders affecting the spleen and thymus. In Chapter 14 we consider diseases of red cells and those affecting hemostasis. Before delving into specific diseases, we will briefly discuss the origins of hematopoietic cells, since many disorders of white cells and red cells involve disturbances of their normal development and maturation.
Development and Maintenance of Hematopoietic Tissues
Blood cell progenitors first appear during the third week of embryonic development in the yolk sac, but definitive hematopoietic stem cells (HSCs) are believed to arise several weeks later in the mesoderm of the intraembryonic aorta/gonad/mesonephros region.1 During the third month of embryogenesis, HSCs migrate to the liver, which becomes the chief site of blood cell formation until shortly before birth. By the fourth month of development, HSCs begin to shift in location yet again, this time to the bone marrow. By birth, marrow throughout the skeleton is hematopoietically active and hepatic hematopoiesis dwindles to a trickle, persisting only in widely scattered foci that become inactive soon after birth. Until puberty, hematopoietically active marrow is found throughout the skeleton, but soon thereafter it becomes restricted to the axial skeleton. Thus, in normal adults, only about half of the marrow space is hematopoietically active.
The formed elements of blood—red cells, granulocytes, monocytes, platelets, and lymphocytes—have a common origin from HSCs, pluripotent cells that sit at the apex of a hierarchy of bone marrow progenitors (Fig. 13-1). Most evidence supporting this scheme comes from studies in mice, but human hematopoiesis is believed to proceed in a similar way. HSCs give rise to two kinds of multipotent cells, the common lymphoid and common myeloid progenitors. The common lymphoid progenitor is the source of T-cell, B-cell, and natural killer (NK) cell precursors. We will return to the origins of lymphoid cells when we discuss tumors derived from these cells. From the common myeloid progenitors arise various kinds of committed progenitors restricted to differentiation along particular lineages. These cells are referred to as colony-forming units (CFUs) (see Fig. 13-1), because they give rise to colonies composed of specific kinds of mature cells when grown in culture. From the various committed progenitors are derived the morphologically recognizable precursors, such as myeloblasts, proerythroblasts, and megakaryoblasts, which in turn give rise to mature granulocytes, red cells, and platelets.
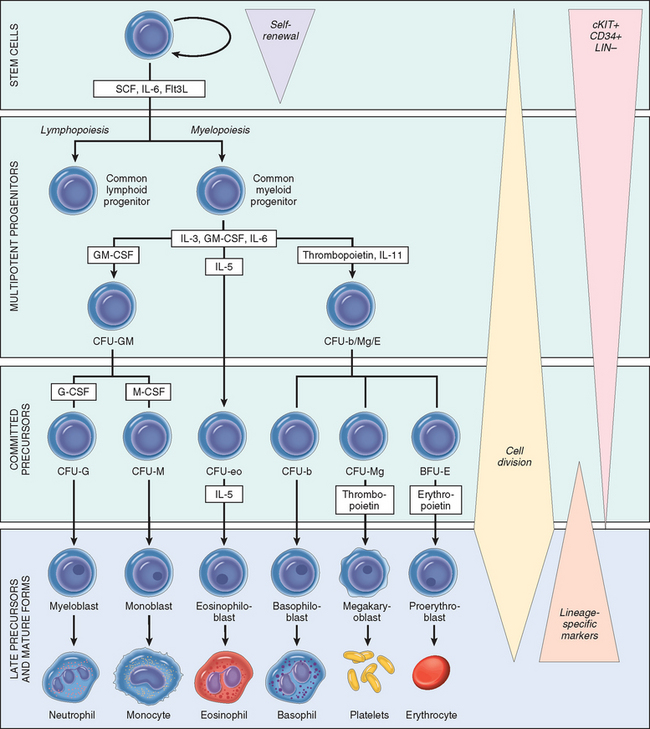
FIGURE 13-1 Differentiation of blood cells. CFU, colony forming unit; SCF, stem cell factor; Flt3L, Flt3 ligand; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; LIN–, negative for lineage-specific markers; M-CSF, macrophage colony-stimulating factor.
HSCs have two essential properties that are required for the maintenance of hematopoiesis: pluripotency and the capacity for self-renewal. Pluripotency refers to the ability of a single HSC to generate all mature hematopoietic cells. When an HSC divides at least one daughter cell must self-renew to avoid stem cell depletion. Self-renewing divisions are believed to occur within a specialized marrow niche, in which stromal cells and secreted factors nurture and somehow maintain the HSCs.2 As you may have already surmised from their ability to migrate during embryonic development, HSCs are not sessile. Particularly under conditions of marked stress, such as severe anemia, HSCs are mobilized from the bone marrow and appear in the peripheral blood. In such circumstances, additional HSC niches are sometimes induced or “unveiled” in other tissues, such as the spleen and liver, which can then become sites of extramedullary hematopoiesis.
The marrow response to short-term physiologic needs is regulated by hematopoietic growth factors through effects on the committed progenitors. Since mature blood elements are terminally differentiated cells with finite life spans, their numbers must be constantly replenished. In at least some divisions of HSCs, a single daughter cell begins to differentiate. Once past this threshold, these newly committed cells lose the capacity for self-renewal and commence an inexorable journey down a road that leads to terminal differentiation and death. However, as these progenitors differentiate they also begin to express receptors for lineage-specific growth factors, which stimulate their short-term growth and survival. Some growth factors, such as stem cell factor (also called c-KIT ligand) and FLT3-ligand, act on very early committed progenitors. Others, such as erythropoietin, granulocyte-macrophage colonystimulating factor (GM-CSF), granulocyte colony-stimulating factor (G-CSF), and thrombopoietin, act on committed progenitors with more restricted potentials. Feedback loops that are mediated through growth factors tune the marrow output, allowing the numbers of formed blood elements (red cells, white cells, and platelets) to be maintained within appropriate ranges (given in Table 13-1).
TABLE 13-1 Adult Reference Ranges for Blood Cells*
| Cell Type | |
|---|---|
| White cells (× 103/μL) | 4.8–10.8 |
| Granulocytes (%) | 40–70 |
| Neutrophils (× 103/μL) | 1.4–6.5 |
| Lymphocytes (× 103/μL) | 1.2–3.4 |
| Monocytes (× 103/μL) | 0.1–0.6 |
| Eosinophils (× 103/μL) | 0–0.5 |
| Basophils (× 103/μL) | 0–0.2 |
| Red cells (× 103/μL) | 4.3–5, men; 3.5–5.0, women |
| Platelets (× 103/μL) | 150–450 |
* Reference ranges vary among laboratories. The reference ranges for the laboratory providing the result should always be used.
Many diseases alter the production of blood cells. The marrow is the ultimate source of all cells of the innate and adaptive immune system and responds to infectious or inflammatory challenges by increasing its output of granulocytes under the direction of specific growth factors and cytokines. Conversely, other disorders are associated with defects in hematopoiesis that lead to deficiencies of one or more type of blood cell. Primary tumors of hematopoietic cells are among the most important diseases that interfere with marrow function, but specific genetic diseases, infections, toxins, and nutritional deficiencies, as well as chronic inflammation of any cause, can also decrease the production of blood cells by the marrow.
Tumors of hematopoietic origin are often associated with mutations that block progenitor cell maturation or abrogate their growth factor dependence. The net effect of such derangements is an unregulated clonal expansion of hematopoietic elements, which replace normal marrow progenitors and often spread to other hematopoietic tissues. In some instances, these tumors originate from transformed HSCs that retain the ability to differentiate along multiple lineages, whereas in other instances the origin is a more differentiated progenitor that has acquired an abnormal capacity for self-renewal. Whether this latter situation merely reflects a block in differentiation, or derives instead from the reactivation of a program of gene expression that supports the self-renewal of normal stem cells, is an area of current investigation.
Morphology. The bone marrow is a unique microenvironment that supports the orderly proliferation, differentiation, and release of blood cells. It is filled with a network of thin-walled sinusoids lined by a single layer of endothelial cells, which are underlaid by a discontinuous basement membrane and adventitial cells. Within the interstitium lie clusters of hematopoietic cells and fat cells. Differentiated blood cells enter the circulation by transcellular migration through the endothelial cells.
The normal marrow is organized in subtle, but important, ways. For example, normal megakaryocytes lie next to sinusoids and extend cytoplasmic processes that bud off into the bloodstream to produce platelets, while red cell precursors often surround macrophages (so-called nurse cells) that provide some of the iron needed for the synthesis of hemoglobin. Diseases that distort the marrow architecture, such as deposits of metastatic cancer or granulomatous disease, can cause the abnormal release of immature precursors into the peripheral blood, a finding that is referred to as leukoerythroblastosis.
Marrow aspirate smears provide the best assessment of the morphology of hematopoietic cells. The most mature marrow precursors can be identified based on their morphology alone. Immature precursors (“blast” forms) of different types are morphologically similar and must be identified definitively using lineage-specific antibodies and histochemical markers (described later under white cell neoplasms). Biopsies are a good means for estimating marrow activity. In normal adults, the ratio of fat cells to hematopoietic elements is about 1 : 1. In hypoplastic states (e.g., aplastic anemia) the proportion of fat cells is greatly increased; conversely, fat cells often disappear when the marrow is involved by hematopoietic tumors and in diseases characterized by compensatory hyperplasias (e.g., hemolytic anemias), and neoplastic proliferations such as leukemias. Other disorders (such as metastatic cancers and granulomatous diseases) induce local marrow fibrosis. Such lesions are usually inaspirable and best seen in biopsies.
DISORDERS OF WHITE CELLS
Disorders of white blood cells can be classified into two broad categories: proliferative disorders, in which there is an expansion of leukocytes, and leukopenias, which are defined as a deficiency of leukocytes. Proliferations of white cells can be reactive or neoplastic. Since the major function of leukocytes is host defense, reactive proliferation in response to an underlying primary, often microbial, disease is fairly common. Neoplastic disorders, though less frequent, are much more important clinically. In the following discussion we will first describe the leukopenic states and summarize the common reactive disorders, and then consider in some detail the malignant proliferations of white cells.
Leukopenia
The number of circulating white cells may be markedly decreased in a variety of disorders. An abnormally low white cell count (leukopenia) usually results from reduced numbers of neutrophils (neutropenia, granulocytopenia). Lymphopenia is less common; in addition to congenital immunodeficiency diseases (see Chapter 6), it is most commonly observed in advanced human immunodeficiency virus (HIV) infection, following therapy with glucocorticoids or cytotoxic drugs, autoimmune disorders, malnutrition, and certain acute viral infections. In the latter setting lymphopenia actually stems from lymphocyte activation rather than a true decrease in the number of lymphocytes in the body. You will recall that acute viral infections induce production of type I interferons, which activate T lymphocytes and change the expression of a number of surface proteins that regulate T cell migration. These changes result in the sequestration of activated T cells in lymph nodes and increased adherence to endothelial cells, both of which contribute to lymphopenia. Granulocytopenia is more common and is often associated with significantly decreased granulocyte function, and thus merits further discussion.
NEUTROPENIA, AGRANULOCYTOSIS
Neutropenia, a reduction in the number of neutrophils in the blood, occurs in a wide variety of circumstances. Agranulocytosis, a clinically significant reduction in neutrophils, has the serious consequence of making individuals susceptible to bacterial and fungal infections.
Pathogenesis.
A reduction in circulating granulocytes occurs if there is (1) inadequate or ineffective granulopoiesis, or (2) accelerated removal of neutrophils from the blood. Inadequate or ineffective granulopoiesis is observed in the setting of
Accelerated removal or destruction of neutrophils occurs with
The most common cause of agranulocytosis is drug toxicity. Certain drugs, such as alkylating agents and antimetabolites used in cancer treatment, produce agranulocytosis in a predictable, dose-related fashion. Because such drugs cause a generalized suppression of the bone marrow, production of red cells and platelets is also affected. Agranulocytosis can also occur as an idiosyncratic reaction to a large variety of agents. The roster of implicated drugs includes aminopyrine, chloramphenicol, sulfonamides, chlorpromazine, thiouracil, and phenylbutazone. The neutropenia induced by chlorpromazine and related phenothiazines results from a toxic effect on granulocytic precursors in the bone marrow. In contrast, agranulocytosis following administration of aminopyrine, thiouracil, and certain sulfonamides probably stems from antibody-mediated destruction of mature neutrophils through mechanisms similar to those involved in drug-induced immunohemolytic anemias (Chapter 14).
In some patients with acquired idiopathic neutropenia, autoantibodies directed against neutrophil-specific antigens are detected. Severe neutropenia can also occur in association with monoclonal proliferations of large granular lymphocytes (so-called LGL leukemia).3 The mechanism of this neutropenia is not clear; suppression of marrow granulocytic progenitors by products of the neoplastic cell (usually a CD8+ cytotoxic T cell) is considered most likely.
Morphology. The alterations in the bone marrow vary with cause. With excessive destruction of neutrophils in the periphery, the marrow is usually hypercellular due to a compensatory increase in granulocytic precursors. Hypercellularity is also the rule with neutropenias caused by ineffective granulopoiesis, as occurs in megaloblastic anemias and myelodysplastic syndromes. Agranulocytosis caused by agents that suppress or destroy granulocytic precursors is understandably associated with marrow hypocellularity.
Infections are a common consequence of agranulocytosis. Ulcerating necrotizing lesions of the gingiva, floor of the mouth, buccal mucosa, pharynx, or elsewhere in the oral cavity (agranulocytic angina) are quite characteristic. These are typically deep, undermined, and covered by gray to green-black necrotic membranes from which numerous bacteria or fungi can be isolated. Less frequently, similar ulcerative lesions occur in the skin, vagina, anus, or gastrointestinal tract. Severe life-threatening invasive bacterial or fungal infections may occur in the lungs, urinary tract, and kidneys. The neutropenic patient is at particularly high risk for deep fungal infections caused by Candida and Aspergillus. Sites of infection often show a massive growth of organisms with little leukocytic response. In the most dramatic instances, bacteria grow in colonies (botryomycosis) resembling those seen on agar plates.
Clinical Features.
The symptoms and signs of neutropenia are related to infection, and include malaise, chills, and fever, often followed by marked weakness and fatigability. With agranulocytosis, infections are often overwhelming and may cause death within hours to days.
Serious infections are most likely when the neutrophil count falls below 500 per mm3. Because infections are often fulminant, broad-spectrum antibiotics must be given expeditiously whenever signs or symptoms appear. In some instances, such as following myelosuppressive chemotherapy, neutropenia is treated with G-CSF, a growth factor that stimulates the production of granulocytes from marrow precursors.
Reactive (Inflammatory) Proliferations of White Cells and Lymph Nodes
LEUKOCYTOSIS
Leukocytosis refers to an increase in the number of white cells in the blood. It is a common reaction to a variety of inflammatory states.
Pathogenesis.
The peripheral blood leukocyte count is influenced by several factors, including
As was discussed in Chapter 2, leukocyte homeostasis is maintained by cytokines, growth factors, and adhesion molecules through their effects on the commitment, proliferation, differentiation, and extravasation of leukocytes and their progenitors. Table 13-2 summarizes the major mechanisms of neutrophilic leukocytosis and its causes, the most important of which is infection. In acute infection there is a rapid increase in the egress of mature granulocytes from the bone marrow pool. If the infection is prolonged, the release of interleukin-1 (IL-1), tumor necrosis factor (TNF), and other inflammatory cytokines stimulates bone marrow stromal cells and T cells to produce increased amounts of hematopoietic growth factors, which enhance the proliferation and differentiation of committed granulocytic progenitors and, over several days, cause a sustained increase in neutrophil production.
TABLE 13-2 Mechanisms and Causes of Leukocytosis
| INCREASED PRODUCTION IN THE MARROW |
| INCREASED RELEASE FROM MARROW STORES |
| DECREASED MARGINATION |
| DECREASED EXTRAVASATION INTO TISSUES |
| Glucocorticoids |
Some growth factors preferentially stimulate the production of a single type of leukocyte. For example, IL-5 mainly stimulates eosinophil production, while G-CSF induces neutrophilia. Such factors are differentially produced in response to various pathogenic stimuli and, as a result, the five principal types of leukocytosis (neutrophilia, eosinophilia, basophilia, monocytosis, and lymphocytosis) tend to be observed in different clinical settings (summarized in Table 13-3).
TABLE 13-3 Causes of Leukocytosis
| Type of Leukocytosis | Causes |
|---|---|
| Neutrophilic leukocytosis | Acute bacterial infections, especially those caused by pyogenic organisms; sterile inflammation caused by, for example, tissue necrosis (myocardial infarction, burns) |
| Eosinophilic leukocytosis (eosinophilia) | Allergic disorders such as asthma, hay fever; certain skin diseases (e.g., pemphigus, dermatitis herpetiformis); parasitic infestations; drug reactions; certain malignancies (e.g., Hodgkin and some non-Hodgkin lymphomas); collagen vascular disorders and some vasculitides; atheroembolic disease (transient) |
| Basophilic leukocytosis (basophilia) | Rare, often indicative of a myeloproliferative disease (e.g., chronic myeloid leukemia) |
| Monocytosis | Chronic infections (e.g., tuberculosis), bacterial endocarditis, rickettsiosis, and malaria; collagen vascular diseases (e.g., systemic lupus erythematosus); inflammatory bowel diseases (e.g., ulcerative colitis) |
| Lymphocytosis | Accompanies monocytosis in many disorders associated with chronic immunological stimulation (e.g., tuberculosis, brucellosis); viral infections (e.g., hepatitis A, cytomegalovirus, Epstein-Barr virus); Bordetella pertussis infection |
In sepsis or severe inflammatory disorders (such as Kawasaki disease), leukocytosis is often accompanied by morphologic changes in the neutrophils, such as toxic granulations, Döhle bodies, and cytoplasmic vacuoles (Fig. 13-2). Toxic granules, which are coarser and darker than the normal neutrophilic granules, represent abnormal azurophilic (primary) granules. Döhle bodies are patches of dilated endoplasmic reticulum that appear as sky-blue cytoplasmic “puddles.”
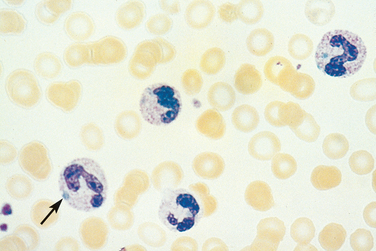
FIGURE 13-2 Reactive changes in neutrophils. Neutrophils containing coarse purple cytoplasmic granules (toxic granulations) and blue cytoplasmic patches of dilated endoplasmic reticulum (Döhle bodies, arrow) are observed in this peripheral blood smear prepared from a patient with bacterial sepsis.
In most instances it is not difficult to distinguish reactive and neoplastic leukocytoses, but uncertainties may arise in two settings. Acute viral infections, particularly in children, can cause the appearance of large numbers of activated lym phocytes that resemble neoplastic lymphoid cells. At other times, particularly in severe infections, many immature granulocytes appear in the blood, simulating a myeloid leukemia (leukemoid reaction). Special laboratory studies (discussed later) are helpful in distinguishing reactive and neoplastic leukocytoses.
LYMPHADENITIS
Following their initial development from precursors in the bone marrow (B cells) and the thymus (T cells), lymphocytes circulate through the blood and, under the influence of specific cytokines and chemokines, home to lymph nodes, spleen, tonsils, adenoids, and Peyer’s patches, which constitute the peripheral lymphoid tissues. Lymph nodes, the most widely distributed and easily accessible lymphoid tissue, are frequently examined for diagnostic purposes. They are discrete encapsulated structures that contain well-organized B-cell and T-cell zones, which are richly invested with phagocytes and antigen-presenting cells (Fig. 6-6, Chapter 6).
The activation of resident immune cells leads to morphologic changes in lymph nodes. Within several days of antigenic stimulation, the primary follicles enlarge and are transformed into pale-staining germinal centers, highly dynamic structures in which B cells acquire the capacity to make high-affinity antibodies against specific antigens. Paracortical T-cell zones may also undergo hyperplasia. The degree and pattern of the morphologic changes are dependent on the inciting stimulus and the intensity of the response. Trivial injuries and infections induce subtle changes, while more significant infections inevitably produce nodal enlargement and sometimes leave residual scarring. For this reason, lymph nodes in adults are almost never “normal” or “resting,” and it is often necessary to distinguish morphologic changes secondary to past experience from those related to present disease. Infections and inflammatory stimuli often elicit regional or systemic immune reactions within lymph nodes. Some that produce distinctive morphologic patterns are described in other chapters. Most, however, cause stereotypical patterns of lymph node reaction designated acute and chronic nonspecific lymphadenitis.
Acute Nonspecific Lymphadenitis
Acute lymphadenitis in the cervical region is most often due to microbial drainage from infections of the teeth or tonsils, while in the axillary or inguinal regions it is most often caused by infections in the extremities. Acute lymphadenitis also occurs in mesenteric lymph nodes draining acute appendicitis. Unfortunately, other self-limited infections may also cause acute mesenteric adenitis and induce symptoms mimicking acute appendicitis, a differential diagnosis that plagues the surgeon. Systemic viral infections (particularly in children) and bacteremia often produce acute generalized lymphadenopathy.
Morphology. Grossly, the nodes are swollen, gray-red, and engorged. Microscopically, there is prominence of large reactive germinal centers containing numerous mitotic figures. Macrophages often contain particulate debris derived from dead bacteria or necrotic cells. When pyogenic organisms are the cause, the centers of the follicles may undergo necrosis; sometimes the entire node is converted into a bag of pus. With less severe reactions, scattered neutrophils infiltrate about the follicles and accumulate within the lymphoid sinuses. The endothelial cells lining the sinuses undergo hyperplasia.
Nodes involved by acute lymphadenitis are enlarged and painful. When abscess formation is extensive the nodes become fluctuant. The overlying skin is red. Sometimes, suppurative infections penetrate through the capsule of the node and track to the skin to produce draining sinuses. Healing of such lesions is associated with scarring.
Chronic Nonspecific Lymphadenitis
Chronic immunological stimuli produce several different patterns of lymph node reaction.
Morphology.
Follicular hyperplasia is caused by stimuli that activate humoral immune responses. It is defined by the presence of large oblong germinal centers (secondary follicles), which are surrounded by a collar of small resting naive B cells (the mantle zone) (Fig. 13-3). Germinal centers are normally polarized into two distinct regions: (1) a dark zone containing proliferating blastlike B cells (centroblasts) and (2) a light zone composed of B cells with irregular or cleaved nuclear contours (centrocytes). Interspersed between the germinal B centers is an inconspicuous network of antigen-presenting follicular dendritic cells and macrophages (often referred to as tingible-body macrophages) containing the nuclear debris of B cells, which undergo apoptosis if they fail to produce an antibody with a high affinity for antigen.
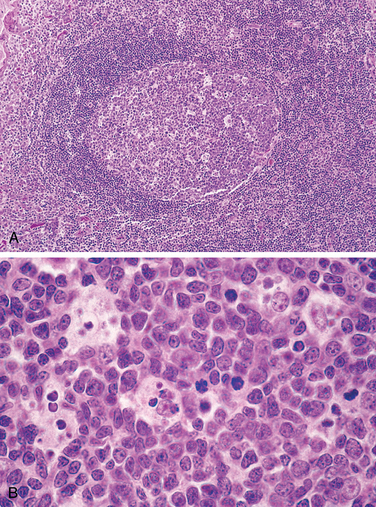
FIGURE 13-3 Follicular hyperplasia. A, Low-power view showing a reactive follicle and surrounding mantle zone. The dark-staining mantle zone is more prominent adjacent to the germinal-center light zone in the left half of the follicle. The right half of the follicle consists of the dark zone. B, High-power view of the dark zone shows several mitotic figures and numerous macrophages containing phagocytosed apoptotic cells (tingible bodies).
Causes of follicular hyperplasia include rheumatoid arthritis, toxoplasmosis, and early stages of infection with HIV. This form of hyperplasia is morphologically similar to follicular lymphoma (discussed later). Features favoring a reactive (non-neoplastic) hyperplasia include (1) preservation of the lymph node architecture, including the interfollicular T-cell zones and the sinusoids; (2) marked variation in the shape and size of the follicles; and (3) the presence of frequent mitotic figures, phagocytic macrophages, and recognizable light and dark zones, all of which tend to be absent from neoplastic follicles.
Paracortical hyperplasia is caused by stimuli that trigger T cell–mediated immune responses, such as acute viral infections (e.g., infectious mononucleosis). The T-cell regions typically contain immunoblasts, activated T cells three to four times the size of resting lymphocytes that have round nuclei, open chromatin, several prominent nucleoli, and moderate amounts of pale cytoplasm. The expanded T-cell zones encroach on and, in particularly exuberant reactions, efface the B-cell follicles. In such cases immunoblasts may be so numerous that special studies are needed to exclude a lymphoid neoplasm. In addition, there is often a hypertrophy of sinusoidal and vascular endothelial cells, sometimes accompanied by infiltrating macrophages and eosinophils.
Sinus histiocytosis (also called reticular hyperplasia) refers to an increase in the number and size of the cells that line lymphatic sinusoids. Although nonspecific, this form of hyperplasia may be particularly prominent in lymph nodes draining cancers such as carcinoma of the breast. The lining lymphatic endothelial cells are markedly hypertrophied and macrophages are greatly increased in numbers, resulting in the expansion and distension of the sinuses.
Characteristically, lymph nodes in chronic reactions are nontender, because nodal enlargement occurs slowly over time. Chronic lymphadenitis is particularly common in inguinal and axillary nodes, which drain relatively large areas of the body and are challenged frequently.
Before leaving the reactive disorders of lymphocytes, it is worth pointing out that chronic immune reactions can promote the appearance of organized collections of immune cells in nonlymphoid tissues. A classic example is seen in chronic gastritis caused by Helicobacter pylori, in which aggregates of mucosal lymphocytes are seen that simulate the appearance of Peyer’s patches. A similar phenomenon occurs in rheumatoid athritis, in which B-cell follicles often appear in the inflamed synovium. Lymphotoxin, a cytokine required for the formation of normal Peyer’s patches, is probably involved in the establishment of these “extranodal” inflammation-induced collections of lymphoid cells.4
Neoplastic Proliferations of White Cells
Malignancies are clinically the most important disorders of white cells. These diseases fall into several broad categories:
ETIOLOGIC AND PATHOGENETIC FACTORS IN WHITE CELL NEOPLASIA: OVERVIEW
As we will see in the following sections, the neoplastic disorders of white cells are extremely varied. Before we delve into this complexity, it is worth considering a few themes of general relevance to their etiology and pathogenesis.
Chromosomal Translocations and Other Acquired Mutations.
Nonrandom chromosomal abnormalities, most commonly translocations, are present in the majority of white cell neoplasms. As was discussed briefly in Chapter 7, many specific rearrangements are associated with particular neoplasms, suggesting a critical role in their genesis.
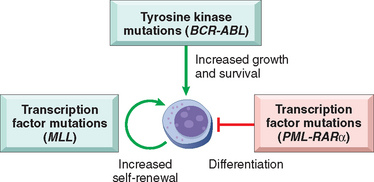
FIGURE 13-4 Molecular pathogenesis of acute leukemia. Acute leukemias arise from complementary mutations that block differentiation at early stages of white cell development, enhance self-renewal, and increase growth and survival. Important examples of each type of mutation are listed. BCR-ABL, breakpoint chromosomal region–Abelson kinase fusion gene; MLL, mixed-lineage leukemia gene; PML-RARα, promyelocytic leukemia–retinoic acid receptor α fusion gene.
Inherited Genetic Factors.
As was discussed in Chapter 7, individuals with genetic diseases that promote genomic instability, such as Bloom syndrome, Fanconi anemia, and ataxia telangiectasia, are at increased risk of acute leukemia. In addition, both Down syndrome (trisomy 21) and type I neurofibromatosis are associated with an increased incidence of childhood leukemia.
Viruses.
Three lymphotropic viruses—human T-cell leukemia virus-1 (HTLV-1), Epstein-Barr virus (EBV), and Kaposi sarcoma herpesvirus/human herpesvirus-8 (KSHV/HHV-8)—have been implicated as causative agents in particular lymphomas. The possible mechanisms of transformation by viruses were discussed in Chapter 7. HTLV-1 is associated with adult T-cell leukemia/lymphoma. EBV is found in a subset of Burkitt lymphoma, 30% to 40% of Hodgkin lymphoma (HL), many B-cell lymphomas arising in the setting of T-cell immunodeficiency, and rare NK-cell lymphomas. In addition to Kaposi sarcoma, KSHV is uniquely associated with an unusual B-cell lymphoma that presents as a malignant effusion, often in the pleural cavity.
Chronic Immune Stimulation.
Several environmental agents that cause localized chronic immune stimulation predispose to lymphoid neoplasia, which almost always arises within the inflamed tissue. Examples include the associations between H. pylori infection and gastric B-cell lymphomas (Chapter 17), and gluten-sensitive enteropathy and intestinal T-cell lymphomas. This can be contrasted with HIV infection, which is associated with an increased risk of B-cell lymphomas that may arise within virtually any organ. Early in the course, T-cell dysregulation by HIV infection causes a systemic hyperplasia of germinal center B cells that is associated with an increased incidence of germinal center B-cell lymphomas. In advanced infection (acquired immunodeficiency syndrome), severe T-cell immunodeficiency further elevates the risk for B-cell lymphomas, particularly those associated with EBV and KSHV/HHV-8.
Iatrogenic Factors.
Ironically, radiation therapy and certain forms of chemotherapy used to treat cancer increase the risk of subsequent myeloid and lymphoid neoplasms. This association stems from the mutagenic effects of ionizing radiation and chemotherapeutic drugs on hematolymphoid progenitor cells.
LYMPHOID NEOPLASMS
Definitions and Classifications
One confusing aspect of the lymphoid neoplasms concerns the use of the terms lymphocytic leukemia and lymphoma. Leukemia is used for neoplasms that present with widespread involvement of the bone marrow and (usually, but not always) the peripheral blood. Lymphoma is used for proliferations that arise as discrete tissue masses. Originally these terms were attached to what were considered distinct entities, but with time and increased understanding these divisons have blurred. Many entities called “lymphoma” occasionally have leukemic presentations, and evolution to “leukemia” is not unusual during the progression of incurable “lymphomas.” Conversely, tumors identical to “leukemias” sometimes arise as soft-tissue masses unaccompanied by bone marrow disease. Hence, when applied to particular neoplasms, the terms leukemia and lymphoma merely reflect the usual tissue distribution of each disease at presentation.
Within the large group of lymphomas, Hodgkin lymphoma is segregated from all other forms, which constitute the non-Hodgkin lymphomas (NHLs). As will be seen, Hodgkin lymphoma has distinctive pathologic features and is treated in a unique fashion. The other important group of lymphoid tumors is the plasma cell neoplasms. These most often arise in the bone marrow and only infrequently involve lymph nodes or the peripheral blood. Taken together, the diverse lymphoid neoplasms constitute a complex, clinically important group of cancers, with about 100,000 new cases being diagnosed each year in the United States.
The clinical presentation of the various lymphoid neoplasms is most often determined by the anatomic distribution of disease. Two thirds of NHLs and virtually all Hodgkin lymphomas present as enlarged nontender lymph nodes (often >2 cm). The remaining one third of NHLs present with symptoms related to the involvement of extranodal sites (e.g., skin, stomach, or brain). The lymphocytic leukemias most often come to attention because of signs and symptoms related to the suppression of normal hematopoiesis by tumor cells in the bone marrow. Finally, the most common plasma cell neoplasm, multiple myeloma, causes bony destruction of the skeleton and often presents with pain due to pathologic fractures. However, it should also be kept in mind that certain lymphoid tumors cause symptoms through the secretion of circulating factors. Specific examples include the plasma cell tumors, in which much of the pathophysiology is related to the secretion of whole antibodies or Ig fragments; and Hodgkin lymphoma, which is often associated with fever related to the release of inflammatory cytokines.
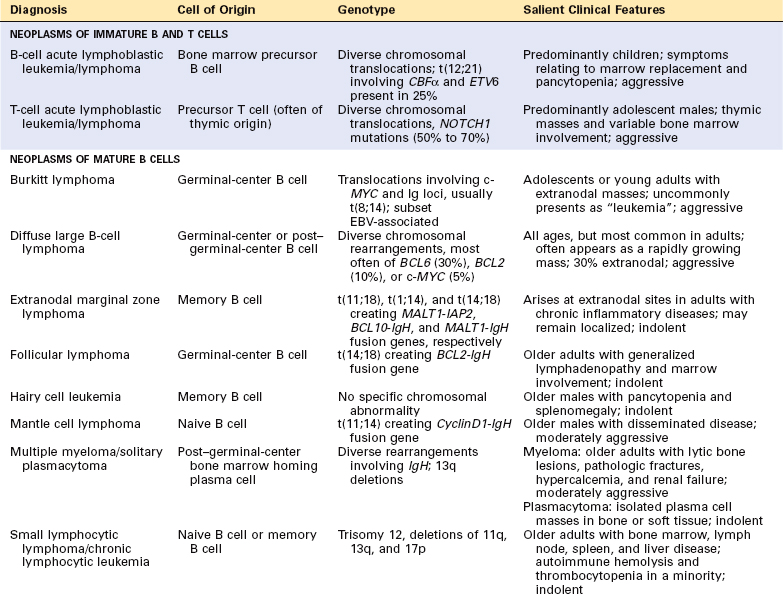
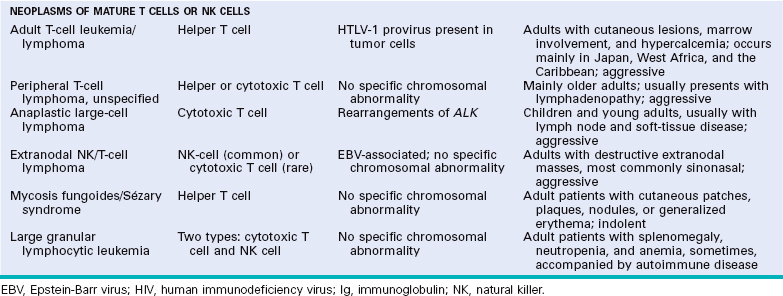
Historically, few areas of pathology evoked as much controversy as the classification of lymphoid neoplasms, but this situation has improved greatly because of advances in the use of objective molecular diagnostic tools. The current World Health Organization (WHO) classification scheme (Table 13-4) uses morphologic, immunophenotypic, genotypic, and clinical fea tures to sort the lymphoid neoplasms into five broad categories,11 which are separated according to the cell of origin:
TABLE 13-4 The WHO Classification of the Lymphoid Neoplasms
| I. PRECURSOR B-CELL NEOPLASMS |
| B-cell acute lymphoblastic leukemia/lymphoma (B-ALL) |
| II. PERIPHERAL B-CELL NEOPLASMS |
| III. PRECURSOR T-CELL NEOPLASMS |
| T-cell acute lymphoblastic leukemia/lymphoma (T-ALL) |
| IV. PERIPHERAL T-CELL AND NK-CELL NEOPLASMS |
| V. HODGKIN LYMPHOMA |
NK, natural killer.
Before we discuss the specific entities of the WHO classification, some important principles relevant to the lymphoid neoplasms should be emphasized.
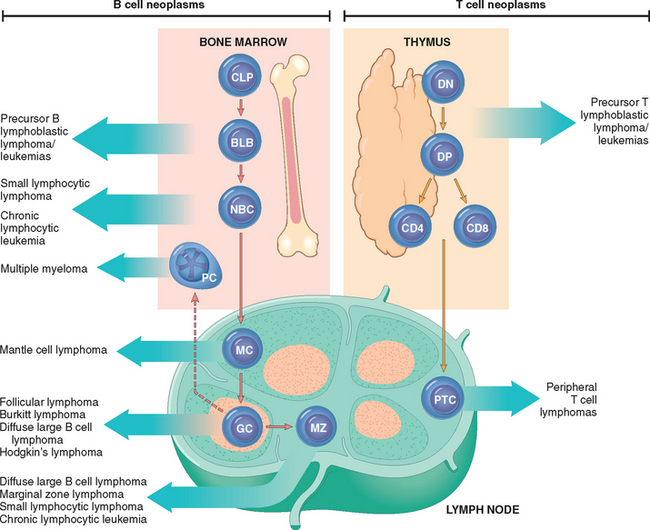
FIGURE 13-5 Origin of lymphoid neoplasms. Stages of B- and T-cell differentiation from which specific lymphoid tumors emerge are shown. CLP, common lymphoid precursor; BLB, pre-B lymphoblast; DN, CD4/CD8 double-negative pro-T cell; DP, CD4/CD8 double-positive pre-T cell; GC, germinal-center B cell; MC, mantle B cell; MZ, marginal zone B cell; NBC, naive B cell; PTC, peripheral T cell.
TABLE 13-5 Some Immune Cell Antigens Detected by Monoclonal Antibodies
| Antigen Designation | Normal Cellular Distribution |
|---|---|
| PRIMARILY T-CELL ASSOCIATED | |
| CD1 | Thymocytes and Langerhans cells |
| CD3 | Thymocytes, mature T cells |
| CD4 | Helper T cells, subset of thymocytes |
| CD5 | T cells and a small subset of B cells |
| CD8 | Cytotoxic T cells, subset of thymocytes, and some NK cells |
| PRIMARILY B-CELL ASSOCIATED | |
| CD10 | Pre-B cells and germinal-center B cells; also called CALLA |
| CD19 | Pre-B cells and mature B cells but not plasma cells |
| CD20 | Pre-B cells after CD19 and mature B cells but not plasma cells |
| CD21 | EBV receptor; mature B cells and follicular dendritic cells |
| CD23 | Activated mature B cells |
| CD79a | Marrow pre-B cells and mature B cells |
| PRIMARILY MONOCYTE- OR MACROPHAGE-ASSOCIATED | |
| CD11c | Granulocytes, monocytes, and macrophages; also expressed by hairy cell leukemias |
| CD13 | Immature and mature monocytes and granulocytes |
| CD14 | Monocytes |
| CD15 | Granulocytes; Reed-Sternberg cells and variants |
| CD33 | Myeloid progenitors and monocytes |
| CD64 | Mature myeloid cells |
| PRIMARILY NK-CELL ASSOCIATED | |
| CD16 | NK cells and granulocytes |
| CD56 | NK cells and a subset of T cells |
| PRIMARILY STEM CELL–AND PROGENITOR CELL–ASSOCIATED | |
| CD34 | Pluripotent hematopoietic stem cells and progenitor cells of many lineages |
| ACTIVATION MARKERS | |
| CD30 | Activated B cells, T cells, and monocytes; Reed-Sternberg cells and variants |
| PRESENT ON ALL LEUKOCYTES | |
| CD45 | All leukocytes; also known as leukocyte common antigen (LCA) |
CALLA, common acute lymphoblastic leukemia antigen; CD, cluster designation; EBV, Epstein-Barr virus; NK, natural killer.
We now turn to the specific entities of the WHO classification. We will begin with neoplasms of immature lymphoid cells and then move on to neoplasms of B cells, plasma cells, T cells, and NK cells. Some of the most salient molecular and clinical features of the group of neoplasms, which includes the lymphoid leukemias, non-Hodgkin lymphomas, and plasma cell tumors, are summarized in Table 13-6. We will then finish with a discussion of Hodgkin lymphoma. Throughout, the most common (and thus most important) entities will be emphasized.
Precursor B- and T-Cell Neoplasms
Acute Lymphoblastic Leukemia/Lymphoma
Acute lymphoblastic leukemia/lymphomas (ALLs) are neoplasms composed of immature B (pre-B) or T (pre-T) cells, which are referred to as lymphoblasts. About 85% are B-ALLs, which typically manifest as childhood acute “leukemias.”; The less common T-ALLs tend to present in adolescent males as thymic “lymphomas.”; There is, however, considerable overlap in the clinical behavior of B- and T-ALL; for example, B-ALL uncommonly presents as a mass in the skin or a bone, and many T-ALLs present with or evolve to a leukemic picture. Because of their morphologic and clinical similarities, the various forms of ALL will be considered here together.
ALL is the most common cancer of children. Approximately 2500 new cases are diagnosed each year in the United States, most occurring in individuals under 15 years of age. ALL is almost three times as common in whites as in blacks, and slightly more frequent in boys than in girls. Hispanics have the highest incidence of any ethnic group. B-ALL peaks in incidence at about the age of 3, perhaps because the number of normal bone marrow pre-B cells (the cell of origin) is greatest very early in life. Similarly the peak incidence of T-ALL is in adolescence, the age when the thymus reaches its maximal size. B- and T-ALL also occur less frequently in adults of all ages.
Morphology. In leukemic presentations, the marrow is hypercellular and packed with lymphoblasts, which replace the normal marrow elements. Mediastinal thymic masses occur in 50% to 70% of T-ALLs, which are also more likely to be associated with lymphadenopathy and splenomegaly. In both B- and T-ALL, the tumor cells have scant basophilic cytoplasm and nuclei somewhat larger than those of small lymphocytes (Fig. 13-6A). The nuclear chromatin is delicate and finely stippled, and nucleoli are either absent or inconspicuous. In many cases the nuclear membrane is deeply subdivided, imparting a convoluted appearance. In keeping with the aggressive clinical behavior, the mitotic rate is high. As with other rapidly growing lymphoid tumors, interspersed macrophages ingesting apoptotic tumor cells may impart a “starry sky” appearance (shown in Fig. 13-15).
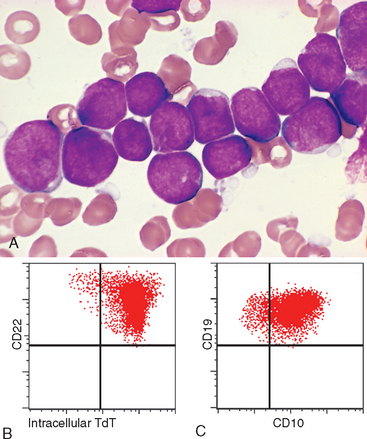
FIGURE 13-6 A, Acute lymphoblastic leukemia/lymphoma. Lymphoblasts with condensed nuclear chromatin, small nucleoli, and scant agranular cytoplasm. B and C represent the phenotype of the ALL shown in A, analyzed by flow cytometry. B, Note that the lymphoblasts represented by the red dots express terminal deoxynucleotidyl-transferase (TdT) and the B-cell marker CD22. C, The same cells are positive for two other markers, CD10 and CD19, commonly expressed on pre-B lymphoblasts. Thus, this is a B-ALL.
(A, Courtesy of Dr. Robert W. McKenna; Department of Pathology, University of Texas Southwestern Medical School, Dallas, TX; B and C, courtesy of Dr. Louis Picker, Oregon Health Science Center, Portland, OR.)
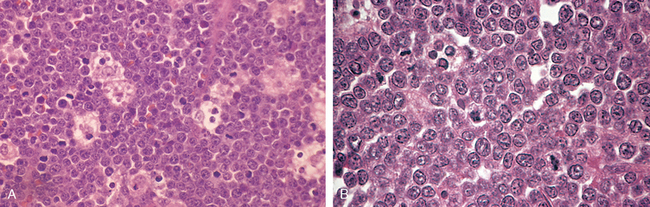
FIGURE 13-15 Burkitt lymphoma. A, At low power, numerous pale tingible body macrophages are evident, producing a “starry sky” appearance. B, At high power, tumor cells have multiple small nucleoli and high mitotic index. The lack of significant variation in nuclear shape and size lends a monotonous appearance.
(B, Courtesy of Dr. José Hernandez, Department of Pathology, University of Texas Southwestern Medical School, Dallas, TX.)
Because of differing responses to chemotherapy, ALL must be distinguished from acute myeloid leukemia (AML), a neoplasm of immature myeloid cells that can cause identical signs and symptoms. Compared with myeloblasts, lymphoblasts have more condensed chromatin, less conspicuous nucleoli, and smaller amounts of cytoplasm that usually lacks granules. However, these morphologic distinctions are not absolute and definitive diagnosis relies on stains performed with antibodies specific for B- and T-cell antigens (Fig. 13-6B and C). Histochemical stains are also helpful, in that (in contrast to myeloblasts) lymphoblasts are myeloperoxidase-negative and often contain periodic acid–Schiff-positive cytoplasmic material.
Immunophenotype.
Immunostaining for terminal deoxynucleotidyl-transferase (TdT), a specialized DNA polymerase that is expressed only in pre-B and pre-T lymphoblasts, is positive in more than 95% of cases (Fig. 13-6B). B- and T-ALLs are distinguished with stains for B- and T-cell–specific markers (summarized below).
B-ALLs are arrested at various stages of pre-B cell development. The lymphoblasts usually express the pan B-cell marker CD19 and the transcription factor PAX5, as well as CD10. In very immature B-ALLs, CD10 is negative. Alternatively, more mature “late pre-B” ALLs express CD10, CD19, CD20, and cytoplasmic IgM heavy chain (μ chain).
Similarly, T-ALLs are arrested at various stages of pre-T cell development. In most cases the cells are positive for CD1, CD2, CD5, and CD7. The more immature tumors are usually negative for surface CD3, CD4, and CD8, whereas “late” pre-T cell tumors are positive for these markers.
Molecular Pathogenesis.
Approximately 90% of ALLs have numerical or structural chromosomal changes. Most common is hyperploidy (>50 chromosomes), but hypoploidy and a variety of balanced chromosomal translocations are also seen. These alterations frequently correlate with immunophenotype and sometimes prognosis. For example, hyperdiploidy and hypodiploidy are seen only in B-ALL. In addition, B- and T-ALL are associated with completely different sets of translocations, indicating that they are pathogenetically distinct. RNA profiling using “gene chips” has also shown that certain chromosomal translocations correlate with unique patterns of gene expression.
Many of the chromosomal aberrations seen in ALL dysregulate the expression and function of transcription factors that are required for normal B- and T-cell development. Up to 70% of T-ALLs have gain-of-function mutations in NOTCH1, a gene that is essential for T-cell development.14 On the other hand, a high fraction of B-ALLs have loss-of-function mutations in genes that are required for B-cell development, such as PAX5, E2A, and EBF,15 or a balanced t(12;21) involving the genes TEL and AML1, two genes that are needed in very early hematopoietic precursors. All of these varied mutations seem to disturb the differentiation of lymphoid precursors and promote maturation arrest. As we will see, similar themes are relevant in the genesis of AML.
In keeping with the multistep origin of cancer (Chapter 7), single mutations are not sufficient to produce ALL. This conclusion stems in part from studies of identical twins with concordant B-ALL.16 In these rare cases, the ALLs in both twins share a common chromosomal aberration and are derived from a single clone transmitted from one twin to the other by transfusion in utero. Despite the presence of the leukemogenic aberration at birth, ALL most often makes its clinical appearance in such patients between 4 and 12 years of age. This lengthy prodrome is most consistent with the existence of a “pre-leukemic” clone that must acquire additional mutations before ALL can develop. The identity of these complementary mutations is incomplete, but aberrations that increase growth and survival, such as activating mutations in tyrosine kinases, are commonly present.
Clinical Features.
It should be emphasized that although ALL and AML are genetically and immunophenotypically distinct, they are clinically very similar. In both, the accumulation of neoplastic “blasts” in the bone marrow suppresses normal hematopoiesis by physical crowding, competition for growth factors, and other poorly understood mechanisms. The common features and those more characteristic of ALL are the following:
Prognosis.
Pediatric ALL is one of the great success stories of oncology. With aggressive chemotherapy about 95% of children with ALL obtain a complete remission, and 75% to 85% are cured. Despite these achievements, however, ALL remains the leading cause of cancer deaths in children, and only 35% to 40% of adults are cured. Several factors have been consistently associated with a worse prognosis: (1) age under 2, largely because of the strong association of infantile ALL with translocations involving the MLL gene; (2) presentation in adolescence or adulthood; (3) peripheral blood blast counts greater than 100,000, which probably reflects a high tumor burden; and (4) the presence of particular cytogenetic aberrations such as the t(9;22) (the Philadelphia chromosome).17 The t(9;22) is present in only 3% of childhood ALL, but up to 25% of adult cases, which partially explains the poor outcome in adults. Favorable prognostic markers include (1) an age of 2 to 10 years, (2) a low white cell count, (3) hyperploidy, (4) trisomy of chromosomes 4, 7, and 10, and (5) the presence of a t(12;21).17 Notably, the molecular detection of residual disease after therapy is predictive of a worse outcome in both B- and T-ALL and is being used to guide new clinical trials.12
Although most chromosomal aberrations in ALL alter the function of transcription factors, the t(9;22) instead creates a fusion gene that encodes a constitutively active BCR-ABL tyrosine kinase (described in more detail under chronic myeloid leukemia). In B-ALL, the BCR-ABL protein is usually 190 kDa in size and has stronger tyrosine kinase activity than the form of BCR-ABL that is found in chronic myeloid leukemia, in which a BCR-ABL protein of 210 kDa in size is usually seen. Treatment of t(9;22)-positive ALLs with BCR-ABL kinase inhibitors leads to clinical responses, but patients relapse quickly because of acquired mutations in BCR-ABL that render the tumor cells drug-resistant.18 BCR-ABL-positive B-ALL generates mutations at a high rate, a phenomenon referred to as genomic instability that contributes to the clinical progression and therapeutic resistance of many aggressive malignant tumors.
Peripheral B-Cell Neoplasms
Chronic Lymphocytic Leukemia (CLL)/Small Lymphocytic Lymphoma (SLL)
These two disorders differ only in the degree of peripheral blood lymphocytosis. Most affected patients have sufficient lymphocytosis to fulfill the diagnostic requirement for CLL (absolute lymphocyte count >4000 per mm3). CLL is the most common leukemia of adults in the Western world. There are about 15,000 new cases of CLL each year in the United States. The median age at diagnosis is 60 years, and there is a 2 : 1 male predominance. In contrast, SLL constitutes only 4% of NHLs. CLL/SLL is much less common in Japan and other Asian countries than in the West.
Morphology. Lymph nodes are diffusely effaced by an infiltrate of predominantly small lymphocytes 6 to 12 μm in diameter with round to slightly irregular nuclei, condensed chromatin, and scant cytoplasm (Fig. 13-7). Admixed are variable numbers of larger activated lymphocytes that often gather in loose aggregates referred to as proliferation centers, which contain mitotically active cells. When present, proliferation centers are pathognomonic for CLL/SLL. The blood contains large numbers of small round lymphocytes with scant cytoplasm (Fig. 13-8). Some of these cells are usually disrupted in the process of making smears, producing so-called smudge cells. The bone marrow is almost always involved by interstitial infiltrates or aggregates of tumor cells. Infiltrates are also virtually always seen in the splenic white and red pulp and the hepatic portal tracts (Fig. 13-9).
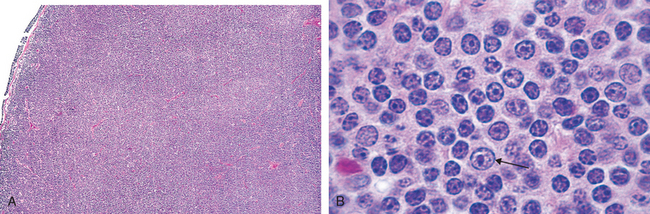
FIGURE 13-7 Small lymphocytic lymphoma/chronic lymphocytic leukemia (lymph node). A, Low-power view shows diffuse efface-ment of nodal architecture. B, At high power the majority of the tumor cells are small round lymphocytes. A “prolymphocyte,” a larger cell with a centrally placed nucleolus, is also present in this field (arrow).
(A, Courtesy of Dr. José Hernandez, Department of Pathology, University of Texas Southwestern Medical School, Dallas, TX.)
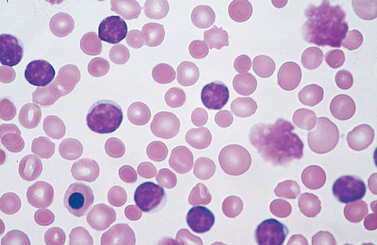
FIGURE 13-8 Chronic lymphocytic leukemia. This peripheral blood smear is flooded with small lymphocytes with condensed chromatin and scant cytoplasm. A characteristic finding is the presence of disrupted tumor cells (smudge cells). A coexistent autoimmune hemolytic anemia (Chapter 14) explains the presence of spherocytes (hyperchromatic, round erythrocytes). A nucleated erythroid cell is present in the lower left-hand corner of the field. In this setting, circulating nucleated red cells could stem from premature release of progenitors in the face of severe anemia, marrow infiltration by tumor (leukoerythroblastosis), or both.
Immunophenotype.
CLL/SLL has a distinctive immunophenotype. The tumor cells express the pan-B cell markers CD19 and CD20, as well as CD23 and CD5, the latter a marker that is found on a small subset of normal B cells. Low-level expression of surface Ig (usually IgM or IgM and IgD) is also typical.
Molecular Pathogenesis.
Unlike most other lymphoid malignancies, chromosomal translocations are rare in CLL/SLL. The most common findings are deletions of 13q14.3, 11q, and 17p, and trisomy 12q. Molecular characterization of the region deleted on chromosome 13 has implicated two microRNAs, miR-15a and miR-16-1, as possible tumor suppressor genes.19 DNA sequencing has revealed that the Ig genes of some CLL/SLL are somatically hypermutated, whereas others are not, suggesting that the cell of origin may be either a postgerminal center memory B cell or a naive B cell. For unclear reasons, tumors with unmutated Ig segments (those putatively of naive B-cell origin) pursue a more aggressive course.20
The growth of CLL/SLL cells is largely confined to proliferation centers, where tumor cells must receive critical cues from the microenvironment. Stromal cells in proliferation centers seem to express a variety of factors that stimulate the activity of the transcription factor NF-κB,21 which promotes cell growth and survival.
Clinical Features.
Patients are often asymptomatic at diagnosis. When symptoms appear, they are nonspecific and include easy fatigability, weight loss, and anorexia. Generalized lymphadenopathy and hepatosplenomegaly are present in 50% to 60% of symptomatic patients. The leukocyte count is highly variable; leukopenia can be seen in individuals with SLL and marrow involvement, while counts in excess of 200,000 per mm3 are sometimes seen in CLL patients with heavy tumor burdens. At the other end of the spectrum are asymptomatic patients that have in their peripheral blood monoclonal CD5+ B cells in numbers that are too few to merit the diagnosis of CLL. These abnormal B cells often have some of the same chromosomal aberrations that are seen in CLL, such as 13q deletions and trisomy 12, yet only about 1% of such patients progress to symptomatic CLL per year, presumably due to acquisition of additional genetic lesions that have yet to be identified. A small monoclonal Ig “spike” is present in the blood of some patients.
CLL/SLL disrupts normal immune function through uncertain mechanisms. Hypogammaglobulinemia is common and contributes to an increased susceptibility to infections, particularly those caused by bacteria. Conversely, 10% to 15% of patients develop hemolytic anemia or thrombocytopenia due to autoantibodies made by non-neoplastic B cells.
The course and prognosis are extremely variable and depend primarily on the clinical stage. Overall median survival is 4 to 6 years, but over 10 years in individuals with minimal tumor burdens at diagnosis. Other variables that correlate with a worse outcome include (1) the presence of deletions of 11q and 17p, (2) a lack of somatic hypermutation, and (3) the expression of ZAP-70, a protein that augments signals produced by the Ig receptor.19 Patients are generally treated with “gentle” chemotherapy to control symptoms. Immunotherapy with antibodies against proteins found on the surface of CLL/SLL cells, such as CD20 and CD52, is finding increasing use.22 Bone marrow transplantation is being offered to the relatively young.
An additional factor in patient survival is the tendency of CLL/SLL to transform to more aggressive tumors. Most commonly this takes the form of a prolymphocytic transform-ation (15% to 30% of patients) or a transformation to diffuse large B-cell lymphoma, so-called Richter syndrome (∼5% to 10% of patients). Prolymphocytic transformation is marked by worsening cytopenias, increasing splenomegaly, and the appearance of increased numbers of “prolymphocytes” (large cells with a single prominent, centrally placed nucleolus) in the peripheral blood. Transformation to diffuse large B-cell lymphoma is often heralded by the development of a rapidly enlarging mass within a lymph node or the spleen. These transformations probably stem from the acquisition of additional, still unknown mutations that increase growth. Both prolymphocytic and large-cell transformation are ominous events, with most patients surviving less than 1 year.23
Follicular Lymphoma
Follicular lymphoma is the most common form of indolent NHL in the United States, affecting 15,000 to 20,000 individuals per year. It usually presents in middle age and afflicts males and females equally. It is less common in Europe and rare in Asian populations. The tumor likely arises from germinal center B cells and is strongly associated with chromosomal translocations involving BCL2.
Morphology. In most cases, at low magnification, a predominantly nodular or nodular and diffuse growth pattern is observed in involved lymph nodes (Fig. 13-10A). Two principal cell types are present in varying proportions: (1) small cells with irregular or cleaved nuclear contours and scant cytoplasm, referred to as centrocytes (small cleaved cells); and (2) larger cells with open nuclear chromatin, several nucleoli, and modest amounts of cytoplasm, referred to as centroblasts (Fig. 13-10B). In most follicular lymphomas, small cleaved cells are in the majority. Peripheral blood involvement sufficient to produce lymphocytosis (usually under 20,000 cells per mm3) is seen in about 10% of cases. Bone marrow involvement occurs in 85% of cases and characteristically takes the form of paratrabecular lymphoid aggre gates. The splenic white pulp (Fig. 13-11) and hepatic portal triads are also frequently involved.
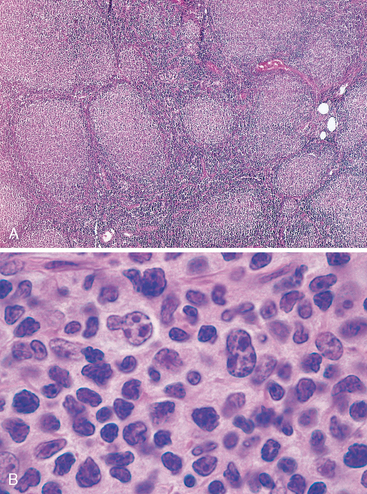
FIGURE 13-10 Follicular lymphoma (lymph node). A, Nodular aggregates of lymphoma cells are present throughout lymph node. B, At high magnification, small lymphoid cells with condensed chromatin and irregular or cleaved nuclear outlines (centrocytes) are mixed with a population of larger cells with nucleoli (centroblasts).
(A, Courtesy of Dr. Robert W. McKenna, Department of Pathology, University of Texas Southwestern Medical School, Dallas, TX.)
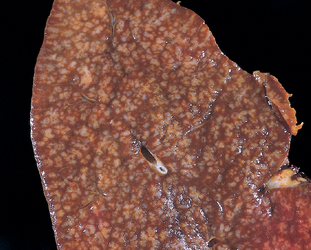
FIGURE 13-11 Follicular lymphoma (spleen). Prominent nodules represent white pulp follicles expanded by follicular lymphoma cells. Other indolent B-cell lymphomas (small lymphocytic lymphoma, mantle cell lymphoma, marginal zone lymphoma) can produce an identical pattern of involvement.
(Courtesy of Dr. Jeffrey Jorgenson, Department of Hematopathology, M.D. Anderson Cancer Center, Houston, TX.)
Immunophenotype.
The neoplastic cells closely resemble normal germinal center B cells, expressing CD19, CD20, CD10, surface Ig, and BCL6. Unlike CLL/SLL and mantle cell lymphoma, CD5 is not expressed. BCL2 is expressed in more than 90% of cases, in distinction to normal follicular center B cells, which are BCL2-negative (Fig. 13-12).
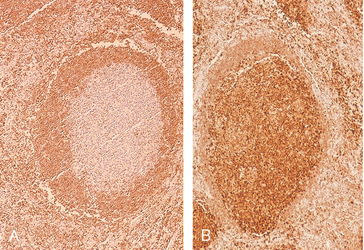
FIGURE 13-12 BCL2 expression in reactive and neoplastic follicles. BCL2 protein was detected by using an immunohistochemical technique that produces a brown stain. In reactive follicles (A), BCL2 is present in mantle zone cells but not follicular-center B cells, whereas follicular lymphoma cells (B) show strong BCL2 staining.
(Courtesy of Dr. Jeffrey Jorgenson, Department of Hematopathology, M.D. Anderson Cancer Center, Houston, TX.)
Molecular Pathogenesis.
The hallmark of follicular lymphoma is a (14;18) translocation that juxtaposes the IgH locus on chromosome 14 and the BCL2 locus on chromosome 18. The t(14;18) is seen in up to 90% of follicular lymphomas, and leads to overexpression of BCL2 (see Fig. 13-12). BCL2 antagonizes apoptosis (Chapter 7) and promotes the survival of follicular lymphoma cells. Notably, while normal germinal centers contain numerous B cells undergoing apoptosis, follicular lymphoma is characteristically devoid of apoptotic cells.
Particularly early in the disease, follicular lymphoma cells growing in lymph nodes are found within a network of reactive follicular dendritic cells admixed with macrophages and T cells. Expression profiling studies have shown that differences in the genes expressed by these reactive cells are predictive of outcome, implying that the response of follicular lymphoma cells to therapy is somehow influenced by the surrounding microenvironment.24,25
Clinical Features.
Follicular lymphoma tends to present with painless, generalized lymphadenopathy. Involvement of extranodal sites, such as the gastrointestinal tract, central nervous system, or testis, is relatively uncommon. Although incurable, it usually follows an indolent waxing and waning course. Survival (median, 7–9 years) is not improved by aggressive therapy; hence, the usual approach is to palliate patients with low-dose chemotherapy or immunotherapy (such as anti-CD20 antibody) when they become symptomatic.
Histologic transformation occurs in 30% to 50% of follicular lymphomas, most commonly to diffuse large B-cell lymphoma. Less commonly, tumors resembling Burkitt lymphoma emerge that are associated with chromosomal translocations involving c-MYC. Like normal germinal center B cells, follicular lymphomas have ongoing somatic hypermutation, which may promote transformation by causing point mutations or chromosomal aberrations. The median survival is less than 1 year after transformation.
Diffuse Large B-Cell Lymphoma
Diffuse large B-cell lymphoma (DLBCL) is the most common form of NHL. Each year in the United States there are about 25,000 new cases. There is a slight male predominance. The median patient age is about 60 years, but DLBCL also occurs in young adults and children.
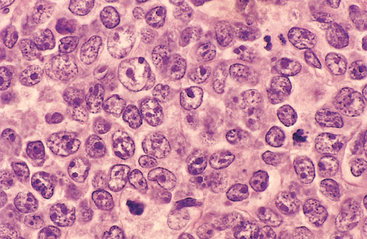
Morphology. The common features are a relatively large cell size (usually four to five times the diameter of a small lymphocyte) and a diffuse pattern of growth (Fig. 13-13). In other respects, substantial morphologic variation is seen. Most commonly, the tumor cells have a round or oval nucleus that appears vesicular due to margination of chromatin to the nuclear membrane, but large multilobated or cleaved nuclei are prominent in some cases. Nucleoli may be two to three in number and located adjacent to the nuclear membrane, or single and centrally placed. The cytoplasm is usually moderately abundant and may be pale or basophilic. More anaplastic tumors may even contain multinucleated cells with large inclusion-like nucleoli that resemble Reed-Sternberg cells (the malignant cell of Hodgkin lymphoma).
Immunophenotype.
These mature B-cell tumors express CD19 and CD20 and show variable expression of germinal center B-cell markers such as CD10 and BCL6. Most have surface Ig.
Molecular Pathogenesis.
Cytogenetic, gene expression profiling, and immunohistochemical studies indicate that DLBCL is heterogeneous.26,27 One frequent pathogenic event is dysregulation of BCL6, a DNA-binding zinc-finger transcriptional repressor that is required for the formation of normal germinal centers. About 30% of DLBCLs contain various translocations that have in common a breakpoint in BCL6 at chromosome 3q27. Acquired mutations in BCL6 promoter sequences that abrogate BCL6 autoregulation (an important negative-regulatory mechanism) are seen even more frequently. It is hypothesized that both types of lesions are inadvertent byproducts of somatic hypermutation that result in overexpression of BCL6, which has several important consequences. BCL6 represses the expression of factors that promote germinal center B-cell differentiation and growth arrest, and thereby holds cells in a relatively undifferentiated, proliferative state.28,29 BCL6 can also silence the expression of p53, the “guardian of the genome” (Chapter 7).30 This “anti-p53” activity may serve to prevent the activation of DNA repair mechanisms in germinal center B cells undergoing somatic hypermutation and class switch recombination. Each of these activities is believed to contribute to the development of DLBCL. Mutations similar to those found in BCL6 are also seen in multiple other oncogenes, including c-MYC,10 suggesting that somatic hypermutation in DLBCL cells is “mistargeted” to a wide variety of loci.
Another 10% to 20% of tumors are associated with the t(14;18), which (as discussed under follicular lymphoma) leads to the overexpression of the anti-apoptotic protein BCL2. Tumors with BCL2 rearrangements almost always lack BCL6 rearrangements, suggesting that these rearrangements define two distinct molecular classes of DLBCL. Some tumors with BCL2 rearrangements may arise from unrecognized underlying follicular lymphomas, which (as discussed already) frequently transform to DLBCL.
Special Subtypes Associated with Oncogenic Herpesviruses.
Several other subtypes of DLBCL are sufficiently distinctive to merit brief discussion.
Clinical Features.
DLBCL typically presents as a rapidly enlarging mass at a nodal or extranodal site. It can arise virtually anywhere in the body. Waldeyer ring, the oropharyngeal lymphoid tissue that includes the tonsils and adenoids, is involved commonly. Primary or secondary involvement of the liver and spleen may take the form of large destructive masses (Fig. 13-14). Extranodal sites include the gastrointestinal tract, skin, bone, brain, and other tissues. Bone marrow involvement is relatively uncommon and usually occurs late in the course. Rarely, a leukemic picture emerges.
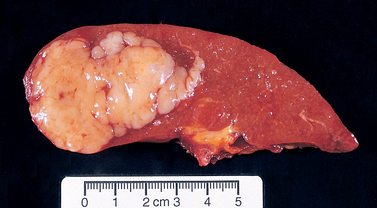
FIGURE 13-14 Diffuse large B-cell lymphoma involving the spleen. The isolated large mass is typical. In contrast, indolent B-cell lymphomas usually produce multifocal expansion of white pulp (see Fig. 13-11).
(Courtesy of Dr. Mark Fleming, Department of Pathology, Children’s Hospital, Boston, MA.)
DLBCLs are aggressive tumors that are rapidly fatal without treatment. With intensive combination chemotherapy, 60% to 80% of patients achieve a complete remission, and 40% to 50% are cured. Immunotherapy with anti-CD20 antibody seems to improve both the initial response and the overall outcome, particularly in the elderly. Individuals with limited disease fare better than those with widespread disease or bulky tumor masses. Expression profiling has identified distinct molecular subtypes with differing clinical outcomes and led to new targeted therapeutic approaches directed at components of the NF-κB and B cell receptor signaling pathways.26,27
Burkitt Lymphoma
Within this category fall (1) African (endemic) Burkitt lymphoma, (2) sporadic (nonendemic) Burkitt lymphoma, and (3) a subset of aggressive lymphomas occurring in individuals infected with HIV. Burkitt lymphomas occurring in each of these settings are histologically identical but differ in some clinical, genotypic, and virologic characteristics.
Morphology. Involved tissues are effaced by a diffuse infiltrate of intermediate-sized lymphoid cells 10 to 25 μm in diameter with round or oval nuclei, coarse chromatin, several nucleoli, and a moderate amount of cytoplasm (Fig. 13-15). The tumor exhibits a high mitotic index and contains numerous apoptotic cells, the nuclear remnants of which are phagocytosed by interspersed benign macrophages. These phagocytes have abundant clear cytoplasm, creating a characteristic “starry sky” pattern. When the bone marrow is involved, aspirates reveal tumor cells with slightly clumped nuclear chromatin, two to five distinct nucleoli, and royal blue cytoplasm containing clear cytoplasmic vacuoles.
Immunophenotype.
These are tumors of mature B cells that express surface IgM, CD19, CD20, CD10, and BCL6, a phenotype consistent with a germinal center B-cell origin. Unlike other tumors of germinal center origin, Burkitt lymphoma almost always fails to express the anti-apoptotic protein BCL2.
Molecular Pathogenesis.
All forms of Burkitt lymphoma are associated with translocations of the c-MYC gene on chromosome 8. The translocation partner is usually the IgH locus [t(8;14)] but may also be the Ig κ [t(2;8)] or γ [t(8;22)] light-chain loci. The breakpoints in the IgH locus in sporadic Burkitt lymphoma are usually found in the class switch regions, whereas the breakpoints in endemic Burkitt lymphoma tend to lie within more 5′ V(D)J sequences. The basis for this subtle molecular distinction is not known, but both types of translocations can be induced in germinal center B cells by AID,8,9 which you will recall is a specialized DNA-modifying enzyme that is required for both Ig class switching and somatic hypermutation. The net effect of these translocations is similar; the c-MYC coding sequence is repositioned adjacent to strong Ig promoter and enhancer elements, which drive increased c-MYC expression. In addition, the translocated c-MYC allele often harbors point mutations that further increase its activity.31 Burkitt lymphomas also commonly have mutations that inactivate p53, an event that increases the frequency of c-MYC translocations in germinal center B cells.9 Hence, it is possible that pre-existent defects in p53 set the stage for the acquisition of c-MYC translocations.
Essentially all endemic tumors are latently infected with EBV, which is also present in about 25% of HIV-associated tumors and 15% to 20% of sporadic cases. The configuration of the EBV DNA is identical in all tumor cells within individual cases, indicating that infection precedes transformation. Although this places EBV at the “scene of the crime,” its precise role in the genesis of Burkitt lymphoma remains poorly understood.
About 5% of DLBCLs have c-MYC translocations, and in such instances DLBCL may be difficult to distinguish from Burkitt lymphoma by conventional diagnostic tests. This distinction can be important, since DLBCL and Burkitt lymphoma are often treated with different chemotherapeutic regimens. Gene expression profiling may provide a more accurate assay for differentiating between these two tumors in difficult cases.32
Clinical Features.
Both endemic and sporadic Burkitt lymphomas are found mainly in children or young adults; overall, it accounts for about 30% of childhood NHLs in the United States. Most tumors manifest at extranodal sites. Endemic Burkitt lymphoma often presents as a mass involving the mandible and shows an unusual predilection for involvement of abdominal viscera, particularly the kidneys, ovaries, and adrenal glands. In contrast, sporadic Burkitt lymphoma most often appears as a mass involving the ileocecum and peritoneum. Involvement of the bone marrow and peripheral blood is uncommon, especially in endemic cases. Burkitt lymphoma is very aggressive but responds well to intensive chemotherapy. Most children and young adults can be cured. The outcome is more guarded in older adults.
Plasma Cell Neoplasms and Related Disorders
These B-cell proliferations contain neoplastic plasma cells that virtually always secrete a monoclonal Ig or Ig fragment. Collectively, the plasma cell neoplasms (often referred to as dyscrasias) account for about 15% of the deaths caused by lymphoid neoplasms. The most common and deadly of these neoplasms is multiple myeloma, of which there are about 15,000 new cases per year in the United States.
A monoclonal Ig identified in the blood is referred to as an M component, in reference to myeloma. Since complete M components have molecular weights of 160,000 or higher, they are restricted to the plasma and extracellular fluid and excluded from the urine in the absence of glomerular damage. However, unlike normal plasma cells, in which the production and coupling of heavy and light chains are tightly balanced, neoplastic plasma cells often synthesize excess light or heavy chains along with complete Igs. Occasionally only light chains or heavy chains are produced. The free light chains are small enough to be excreted in the urine, where they are called Bence-Jones proteins. Free light chains can be detected and measured in the urine or the blood, the latter with new, highly sensitive tests that are in the process of being evaluated.
Terms used to describe the abnormal Igs include monoclonal gammopathy, dysproteinemia, and paraproteinemia. The following clinicopathologic entities are associated with monoclonal gammopathies.
With this background, we now turn to some of the specific clinicopathologic entities. Primary amyloidosis was discussed along with other disorders of the immune system in Chapter 6.
Multiple Myeloma.
Multiple myeloma is a plasma cell neoplasm characterized by multifocal involvement of the skeleton. Although bony disease dominates, it can spread late in its course to lymph nodes and extranodal sites such as the skin. Multiple myeloma causes 1% of all cancer deaths in Western countries. Its incidence is higher in men and people of African descent. It is chiefly a disease of the elderly, with a peak age of incidence of 65 to 70 years.
Molecular Pathogenesis.
The Ig genes in myeloma cells always show evidence of somatic hypermutation. On this basis, the cell of origin is considered to be a post-germinal center B cell that homes to the bone marrow and has differentiated into a plasma cell. Of interest, some studies suggest that the tumor originates in and is maintained by stem-like cells resembling small B lymphocytes that rely on signals generated by the “hedgehog” pathway for self-renewal.33,34
The proliferation and survival of myeloma cells are dependent on several cytokines, most notably IL-6. IL-6 is an important growth factor for plasma cells that is produced by the tumor cells themselves and resident marrow stromal cells. High serum levels of IL-6 are seen in patients with active disease and are associated with a poor prognosis. Myeloma cell growth and survival are also augmented by direct physical interactions with bone marrow stromal cells, which is a focus of new therapeutic approaches.35
Factors produced by neoplastic plasma cells mediate bone destruction, the major pathologic feature of multiple myeloma. Of particular importance, myeloma-derived MIP1α upregulates the expression of the receptor activator of NF-κB ligand (RANKL) by bone marrow stromal cells, which in turn activates osteoclasts.36 Other factors released from tumor cells, such as modulators of the Wnt pathway, are potent inhibitors of osteoblast function. The net effect is a marked increase in bone resorption, which leads to hypercalcemia and pathologic fractures.37
Many myelomas have rearrangements involving the Ig heavy-chain gene on chromosome 14q32.38,39 Common translocation partners include FGFR3 (fibroblast growth factor receptor 3) on chromosome 4p16, a gene encoding a tyrosine kinase receptor implicated in the control of cellular proliferation; the cell cycle–regulatory genes cyclin D1 on chromosome 11q13 and cyclin D3 on chromosome 6p21; the gene for the transcription factor c-MAF on chromosome 16q23; and the gene encoding the transcription factor MUM1/IRF4 on chromosome 6p25. As may be gathered from the involvement of two different D cyclin genes, dysreglation of D cyclins is a common feature.38 The other most frequent karyotypic abnormalities are deletions of 13q. Consistent with the diversity of chromosomal aberrations, gene expression profiling studies suggest that myeloma is molecularly quite heterogeneous.40
Morphology. Multiple myeloma usually presents as destructive plasma cell tumors (plasmacytomas) involving the axial skeleton. The bones most commonly affected (in descending order of frequency) are the vertebral column, ribs, skull, pelvis, femur, clavicle, and scapula. Lesions begin in the medullary cavity, erode cancellous bone, and progressively destroy the bony cortex, often leading to pathologic fractures; these are most common in the vertebral column, but may occur in any affected bone. The bone lesions appear radiographically as punched-out defects, usually 1 to 4 cm in diameter (Fig. 13-16), and grossly consist of soft, gelatinous, red tumor masses. Less commonly, widespread myelomatous bone disease produces diffuse demineralization (osteopenia) rather than focal defects.
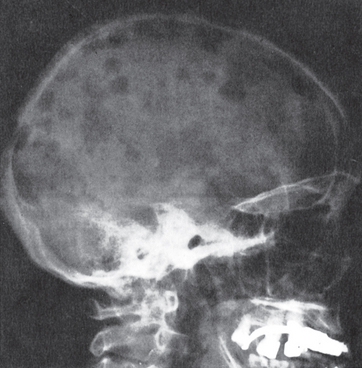
FIGURE 13-16 Multiple myeloma of the skull (radiograph, lateral view). The sharply punched-out bone lesions are most obvious in the calvarium.
Even away from overt tumor masses, the marrow contains an increased number of plasma cells, which usually constitute more than 30% of the cellularity. The plasma cells may infiltrate the interstitium or be present in sheets that completely replace normal elements. Like their benign counterparts, malignant plasma cells have a perinuclear clearing due to a prominent Golgi apparatus and an eccentrically placed nucleus (Fig. 13-17). Relatively normal-appearing plasma cells, plasmablasts with vesicular nuclear chromatin and a prominent single nucleolus, or bizarre, multinucleated cells may predominate. Other cytologic variants stem from the dysregulated synthesis and secretion of Ig, which often leads to intracellular accumulation of intact or partially degraded protein. Such variants include flame cells with fiery red cytoplasm, Mott cells with multiple grapelike cytoplasmic droplets, and cells containing a variety of other inclusions, including fibrils, crystalline rods, and globules. The globular inclusions are referred to as Russell bodies (if cytoplasmic) or Dutcher bodies (if nuclear). In advanced disease, plasma cell infiltrates may be present in the spleen, liver, kidneys, lungs, lymph nodes, and other soft tissues.
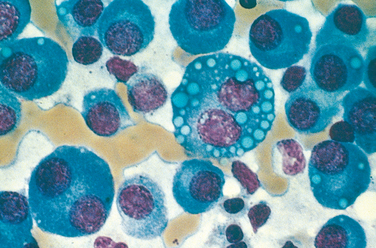
FIGURE 13-17 Multiple myeloma (bone marrow aspirate). Normal marrow cells are largely replaced by plasma cells, including forms with multiple nuclei, prominent nucleoli, and cytoplasmic droplets containing Ig.
Commonly, the high level of M proteins causes red cells in peripheral blood smears to stick to one another in linear arrays, a finding referred to as rouleaux formation. Rouleaux formation is characteristic but not specific, in that it may be seen in other conditions in which Ig levels are elevated, such as lupus erythematosus and early HIV infection. Rarely, tumor cells flood the peripheral blood, giving rise to plasma cell leukemia.
Bence Jones proteins are excreted in the kidney and contribute to a form of renal disease called myeloma kidney. This important complication is discussed in detail in Chapter 20.
Clinical Features.
The clinical features of multiple myeloma stem from (1) the effects of plasma cell growth in tissues, particularly the bones; (2) the production of excessive Igs, which often have abnormal physicochemical properties; and (3) the suppression of normal humoral immunity.
Bone resorption often leads to pathologic fractures and chronic pain. The attendant hypercalcemia can give rise to neurologic manifestations, such as confusion, weakness, lethargy, constipation, and polyuria, and contributes to renal dysfunction. Decreased production of normal Igs sets the stage for recurrent bacterial infections. Cellular immunity is relatively unaffected. Of great significance is renal insufficiency, which trails only infections as a cause of death. The pathogenesis of renal failure (discussed in Chapter 20), which occurs in up to 50% of patients, is multifactorial. However, the single most important factor seems to be Bence Jones proteinuria, since the excreted light chains are toxic to renal tubular epithelial cells. Certain light chains (particularly those of the γ6 and γ3 families) are prone to cause amyloidosis of the AL type (Chapter 6), which can exacerbate renal dysfunction and deposit in other tissue as well.
In 99% of patients, laboratory analyses reveal increased levels of Igs in the blood and/or light chains (Bence Jones proteins) in the urine. The monoclonal Igs are usually first detected as abnormal protein “spikes” in serum or urine electrophoresis and then further characterized by immunofixation (Fig. 13-18). Most myelomas are associated with more than 3 gm/dL of serum Ig and/or more than 6 gm/dL of urine Bence Jones protein. The most common monoclonal Ig (“M protein”) is IgG (∼55% of patients), followed by IgA (∼25% of cases). Myelomas expressing IgM, IgD, or IgE occur but are rare. Excessive production and aggregation of M proteins, usually of the IgA and or IgG3 subtype, leads to symptoms related to hyperviscosity (described under lymphoplasmacytic lymphoma) in about 7% of patients. Both free light chains and a serum M protein are observed together in 60% to 70% of patients. However, in about 20% of patients only free light chains are present. Around 1% of myelomas are nonsecretory; hence, the absence of detectable M proteins does not completely exclude the diagnosis.
FIGURE 13-18 M protein detection in multiple myeloma. Serum protein electrophoresis (SP) is used to screen for a monoclonal immunoglobulin (M protein). Polyclonal IgG in normal serum (denoted by the arrow) appears as a broad band; in contrast, serum from a patient with multiple myeloma contains a single sharp protein band (denoted by the arrowhead) in this region of the electropherogram. The suspected monoclonal Ig is confirmed and characterized by immunofixation. In this procedure, proteins separated by electrophoresis within a gel are reacted with specific antisera. After extensive washing, proteins that are cross-linked by antisera are retained and detected with a protein stain. Note the sharp band in the patient serum is cross-linked by antisera specific for IgG heavy chain (G) and kappa light chain (κ), indicating the presence of an IgGκ M protein. Levels of polyclonal IgG, IgA (A), and lambda light chain (γ) are also decreased in the patient serum relative to normal, a finding typical of multiple myeloma.
(Courtesy of Dr. David Sacks, Department of Pathology, Brigham and Women’s Hospital, Boston, MA.)
The clinicopathologic diagnosis of multiple myeloma rests on radiographic and laboratory findings. It can be strongly suspected when the distinctive radiographic changes are present, but definitive diagnosis requires a bone marrow examination. Marrow involvement often gives rise to a normocytic normochromic anemia, sometimes accompanied by moderate leukopenia and thrombocytopenia.
The prognosis is variable but generally poor. The median survival is 4 to 6 years, and cures have yet to be achieved. Patients with multiple bony lesions, if untreated, rarely survive for more than 6 to 12 months, whereas patients with “smoldering myeloma” may be asymptomatic for many years. Translocations involving cyclin D1 are associated with a good outcome, whereas deletions of 13q, deletions of 17p, and the t(4;14) all portend a more aggressive course.41
Cytotoxic agents induce remission in 50% to 70% of patients, and new therapeutic approaches are bringing hope. Myeloma cells are sensitive to inhibitors of the proteasome,42 a cellular organelle that degrades unwanted and misfolded proteins. You will recall from Chapter 1 that misfolded proteins activate apoptotic pathways. Myeloma cells are prone to the accumulation of misfolded, unpaired Ig chains. Proteasome inhibitors may induce cell death by exacerbating this inherent tendency, and also seem to retard bone resorption through effects on stromal cells.43 Thalidomide and related compounds also have activity against myeloma, apparently by altering interactions between myeloma cells and bone marrow stromal cells and by inhibiting angiogenesis.35 Biphosphonates, drugs that inhibit bone resorption, reduce pathologic fractures and limit the hypercalcemia. Bone marrow transplantation prolongs life but has not yet proven to be curative.
Solitary Myeloma (Plasmacytoma).
About 3% to 5% of plasma cell neoplasms present as a solitary lesion of bone or soft tissue. The bone lesions tend to occur in the same locations as in multiple myeloma. Extra-osseous lesions are often located in the lungs, oronasopharynx, or nasal sinuses. Modest elevations of M proteins in the blood or urine may be found in some patients. Solitary osseous plasmacytoma almost inevitably progresses to multiple myeloma, but this can take 10 to 20 years or longer. In contrast, extra-osseous plasmacytomas, particularly those involving the upper respiratory tract, are frequently cured by local resection.
Smoldering Myeloma.
This entity defines a middle ground between multiple myeloma and monoclonal gammopathy of uncertain significance. Plasma cells make up 10% to 30% of the marrow cellularity, and the serum M protein level is greater than 3 gm/dL, but patients are asymptomatic. About 75% of patients progress to multiple myeloma over a 15-year period.44
Monoclonal Gammopathy of Uncertain Significance (MGUS).
MGUS is the most common plasma cell dyscrasia,45 occurring in about 3% of persons older than 50 years of age and in about 5% of individuals older than 70 years of age. By definition, patients are asymptomatic and the serum M protein level is less than 3 gm/dL. Approximately 1% of patients with MGUS develop a symptomatic plasma cell neoplasm, usually multiple myeloma, per year,46 a rate of conversion that remains roughly constant over time. Of pathogenic interest, the clonal plasma cells in MGUS often contain the same chromosomal translocations and deletions that are found in full-blown multiple myeloma,47 indicating that MGUS is an early stage of myeloma development. As in patients with smoldering myeloma, progression to multiple myeloma is unpredictable; hence, periodic assessment of serum M component levels and Bence Jones proteinuria is warranted.
Lymphoplasmacytic Lymphoma.
Lymphoplasmacytic lymphoma is a B-cell neoplasm of older adults that usually presents in the sixth or seventh decade of life. Although bearing a superficial resemblance to CLL/SLL, it differs in that a substantial fraction of the tumor cells undergo terminal differentiation to plasma cells. Most commonly, the plasma cell component secretes monoclonal IgM, often in amounts sufficient to cause a hyperviscosity syndrome known as Waldenström macroglobulinemia. Unlike multiple myeloma, heavy- and light-chain synthesis is usually balanced and complications stemming from the secretion of free light chains (e.g., renal failure and amyloidosis) are rare. A further important distinction is that bone destruction is not observed in this disease.
Morphology. Typically, the marrow contains a diffuse sparse-to-heavy infiltrate of lymphocytes, plasma cells, and plasmacytoid lymphocytes in varying proportions, often accompanied by mast cell hyperplasia (Fig. 13-19). Some tumors also contain a population of larger lymphoid cells with more vesicular nuclear chromatin and prominent nucleoli. Periodic acid–Schiff-positive inclusions containing Ig are frequently seen in the cytoplasm (Russell bodies) or the nucleus (Dutcher bodies) of some of the plasma cells. At diagnosis the tumor has usually disseminated to the lymph nodes, spleen, and liver. Infiltration of the nerve roots, meninges, and more rarely the brain can also occur with disease progression.
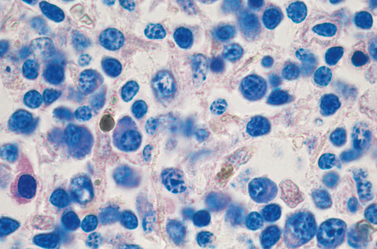
Immunophenotype and Molecular Pathogenesis.
The lymphoid component expresses B-cell markers such as CD20 and surface Ig, whereas the plasma cell component secretes the same Ig that is expressed on the surface of the lymphoid cells. This is usually IgM but can also be IgG or IgA. These tumors usually lack chromosomal translocations; the most common cytogenetic abnormality is a deletion involving chromosome 6q.
Clinical Features.
The dominant presenting complaints are nonspecific and include weakness, fatigue, and weight loss. Approximately half the patients have lymphadenopathy, hepatomegaly, and splenomegaly. Anemia caused by marrow infiltration is common. About 10% of patients have autoimmune hemolysis caused by cold agglutinins, IgM antibodies that bind to red cells at temperatures of less than 37°C (described in Chapter 14).
Patients with IgM-secreting tumors have additional complaints stemming from the physicochemical properties of IgM. Because of its large size, at high concentrations IgM greatly increases the viscosity of the blood, giving rise to a hyperviscosity syndrome characterized by the following:
Lymphoplasmacytic lymphoma is an incurable progressive disease. Since most IgM is intravascular, symptoms caused by the high IgM levels (such as hyperviscosity and hemolysis) can be alleviated by plasmapheresis. Tumor growth can be controlled for a time with low doses of chemotherapeutic drugs and immunotherapy with anti-CD20 antibody. Transformation to large-cell lymphoma occurs but is uncommon. Median survival is about 4 years.
Mantle Cell Lymphoma
Mantle cell lymphoma is an uncommon lymphoid neoplasm that makes up about 2.5% of NHL in the United States and 7% to 9% of NHL in Europe. It usually presents in the fifth to sixth decades of life and shows a male predominance. As the name implies, the tumor cells closely resemble the normal mantle zone B cells that surround germinal centers.
Morphology. Nodal tumor cells may surround reactive germinal centers to produce a nodular appearance at low power, or diffusely efface the node. Typically, the proliferation consists of a homogeneous population of small lymphocytes with irregular to occasionally deeply clefted (cleaved) nuclear contours (Fig. 13-20). Large cells resembling centroblasts and proliferation centers are absent, distinguishing mantle cell lymphoma from follicular lymphoma and CLL/SLL, respectively. In most cases the nuclear chromatin is condensed, nucleoli are inconspicuous, and the cytoplasm is scant. Occasionally, tumors composed of intermediate-sized cells with more open chromatin and a brisk mitotic rate are observed; immunophenotyping is necessary to distinguish these “blastoid” variants of mantle cell lymphoma from ALL.
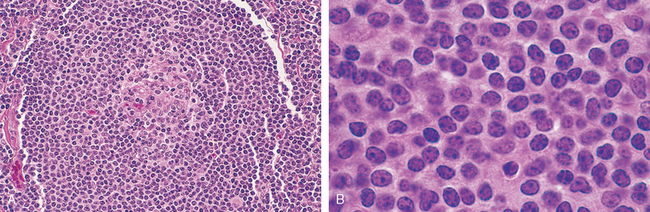
FIGURE 13-20 Mantle cell lymphoma. A, At low power, neoplastic lymphoid cells surround a small, atrophic germinal center, producing a mantle zone pattern of growth. B, High-power view shows a homogeneous population of small lymphoid cells with somewhat irregular nuclear outlines, condensed chromatin, and scant cytoplasm. Large cells resembling prolymphocytes (seen in chronic lymphocytic leukemia) and centroblasts (seen in follicular lymphoma) are absent.
At diagnosis the majority of patients have generalized lymphadenopathy, and 20% to 40% have peripheral blood involvement. Frequent sites of extranodal involvement include the bone marrow, spleen, liver, and gut. Occasionally, mucosal involvement of the small bowel or colon produces polyp-like lesions (lymphomatoid polyposis); of all forms of NHL, mantle cell lymphoma is most likely to spread in this fashion.
Immunophenotype.
Mantle cell lymphomas express high levels of cyclin D1. Most tumors are also express CD19, CD20, and moderately high levels of surface Ig (usually IgM and IgD with κ or γ light chain). It is usually CD5+ and CD23−, which helps to distinguish it from CLL/SLL. The IgH genes lack somatic hypermutation, supporting an origin from a naive B cell.
Molecular Pathogenesis.
Cyclin D1 overexpression is caused by an (11;14) translocation involving the IgH locus on chromosome 14 and the cyclin D1 locus on chromosome 11. This translocation is detected in about 70% of cases by standard karyotyping and in virtually all tumors by fluorescence in situ hybridization. The resulting up-regulation of cyclin D1 promotes G1- to S-phase progression during the cell cycle, as was described in Chapter 7.
Clinical Features.
The most common presentation is painless lymphadenopathy. Symptoms related to involvement of the spleen (present in ∼50% of cases) and the gut are also common. The prognosis is poor; the median survival is only 3 to 4 years. This lymphoma is not curable with conventional chemotherapy, and most patients eventually succumb to organ dysfunction caused by tumor infiltration. The blastoid variant and a “proliferative” expression profiling signature are associated with even shorter survivals.26 Bone marrow transplantation and proteasome inhibitors are new therapeutic approaches showing some promise.
Marginal Zone Lymphomas
The category of marginal zone lymphoma encompasses a heterogeneous group of B-cell tumors that arise within lymph nodes, spleen, or extranodal tissues. The extranodal tumors were initially recognized at mucosal sites and are often referred to as mucosa-associated lymphoid tumors (or “maltomas”). In most cases, the tumor cells show evidence of somatic hypermutation and are considered to be of memory B-cell origin.
Although all marginal zone lymphomas share certain features, those occurring at extranodal sites deserve special attention because of their unusual pathogenesis and three exceptional characteristics.
These characteristics suggest that extranodal marginal zone lymphomas arising in chronically inflamed tissues lie on a continuum between reactive lymphoid hyperplasia and full-blown lymphoma. The disease begins as a polyclonal immune reaction. With the acquisition of still-unknown initiating mutations, a B-cell clone emerges that still depends on antigen-stimulated T-helper cells for signals that drive growth and survival. At this stage, withdrawal of the responsible antigen causes tumor involution. A clinically relevant example is found in gastric “maltoma,” in which antibiotic therapy directed against H. pylori often leads to tumor regression (Chapter 17). With time, however, tumors may acquire additional mutations that render their growth and survival antigen-independent, such as the (11;18), (14;18), or (1;14) chromosomal translocations, which are relatively specific for extranodal marginal zone lymphomas. All of these translocations up-regulate the expression and function of BCL10 or MALT1, protein components of a signaling complex that activates NF-κB and promotes the growth and survival of B cells.5 With further clonal evolution, spread to distant sites and transformation to diffuse large B-cell lymphoma may occur. This theme of polyclonal to monoclonal transition during lymphomagenesis is also applicable to the pathogenesis of EBV-induced lymphoma and is discussed more fully in Chapter 7.
Hairy Cell Leukemia
This rare but distinctive B-cell neoplasm constitutes about 2% of all leukemias. It is predominantly a disease of middle-aged white males, with a median age of 55 and a male-to-female ratio of 5 : 1.
Morphology. Hairy cell leukemia derives its picturesque name from the appearance of the leukemic cells, which have fine hairlike projections that are best recognized under the phase-contrast microscope (Fig. 13-21). On routine peripheral blood smears, hairy cells have round, oblong, or reniform nuclei and moderate amounts of pale blue cytoplasm with threadlike or bleblike extensions. The number of circulating cells is highly variable. The marrow is involved by a diffuse interstitial infiltrate of cells with oblong or reniform nuclei, condensed chromatin, and pale cytoplasm. Because these cells are enmeshed in an extracellular matrix composed of reticulin fibrils, they usually cannot be aspirated (a clinical difficulty referred to as a “dry tap”) and are only seen in marrow biopsies. The splenic red pulp is usually heavily infiltrated, leading to obliteration of white pulp and a beefy red gross appearance. Hepatic portal triads are also involved frequently.
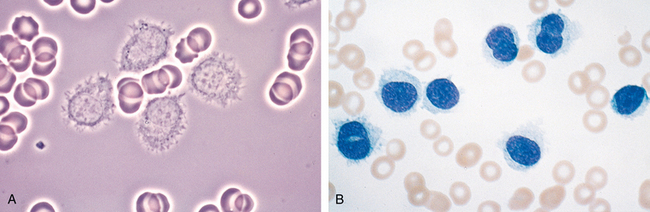
Immunophenotype and Molecular Pathogenesis.
Hairy cell leukemias typically express the pan-B-cell markers CD19 and CD20, surface Ig (usually IgG), and certain relatively distinctive markers, such as CD11c, CD25, and CD103. Analysis of Ig gene sequences has revealed a high incidence of somatic hypermutation, suggesting a post-germinal center memory B-cell origin.
Clinical Features.
Clinical manifestations result largely from infiltration of the bone marrow, liver, and spleen. Splenomegaly, often massive, is the most common and sometimes the only abnormal physical finding. Hepatomegaly is less common and not as marked; lymphadenopathy is rare. Pancytopenia resulting from marrow involvement and splenic sequestration is seen in more than half the cases. About one third of those affected present with infections. There is an increased incidence of atypical mycobacterial infections, possibly related to frequent unexplained monocytopenia.
Hairy cell leukemia follows an indolent course. For unclear reasons, this tumor is exceptionally sensitive to “gentle” chemotherapeutic regimens, which produce long-lasting remissions. Tumors often relapse after 5 or more years, yet generally respond well when retreated with chemotherapy. The overall prognosis is excellent.
Peripheral T-Cell and NK-Cell Neoplasms
These categories include a heterogeneous group of neoplasms having phenotypes resembling mature T cells or NK cells. Peripheral T-cell tumors make up about 5% to 10% of NHLs in the United States and Europe, but are more common in Asia. NK-cell tumors are rare in the West, but also more common in the Far East. Only the most common diagnoses and those of particular pathogenetic interest will be discussed.
Peripheral T-Cell Lymphoma, Unspecified
Although the WHO classification includes a number of distinct peripheral T-cell neoplasms, many of these lymphomas are not easily categorized and are lumped into a “wastebasket” diagnosis, peripheral T-cell lymphoma, unspecified. As might be expected, no morphologic feature is pathognomonic, but certain findings are characteristic. These tumors efface lymph nodes diffusely and are typically composed of a pleomorphic mixture of variably sized malignant T cells (Fig. 13-22). There is often a prominent infiltrate of reactive cells, such as eosinophils and macrophages, probably attracted by tumor-derived cytokines. Brisk neoangiogenesis may also be seen.
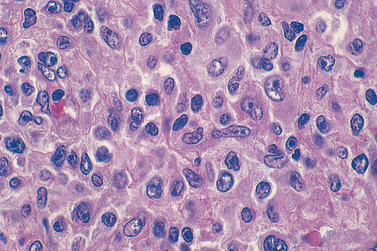
FIGURE 13-22 Peripheral T-cell lymphoma, unspecified (lymph node). A spectrum of small, intermediate, and large lymphoid cells, many with irregular nuclear contours, is visible.
The diagnosis requires immunophenotyping. By definition, all peripheral T-cell lymphomas have a mature T-cell phenotype. They usually express CD2, CD3, CD5, and either αβ or γδ T-cell receptors. Some also express CD4 or CD8; such tumors are taken to be of helper or cytotoxic T-cell origin, respectively. However, many tumors have phenotypes that do not resemble any known normal T cell. In difficult cases where the differential diagnosis lies between lymphoma and a florid reactive process, DNA analysis can be used to confirm the presence of clonal T-cell receptor rearrangements.
Most patients present with generalized lymphadenopathy, sometimes accompanied by eosinophilia, pruritus, fever, and weight loss. Although cures of peripheral T-cell lymphoma have been reported, these tumors have a significantly worse prognosis than comparably aggressive mature B-cell neoplasms (e.g., diffuse large B-cell lymphoma).
Anaplastic Large-Cell Lymphoma (ALK Positive)
This uncommon entity is defined by the presence of rearrangements in the ALK gene on chromosome 2p23. These rearrangements break the ALK locus and lead to the formation of chimeric genes encoding ALK fusion proteins, constitutively active tyrosine kinases that trigger a number of signaling pathways, including the JAK/STAT pathway.48
As the name implies, this tumor is typically composed of large anaplastic cells, some containing horseshoe-shaped nuclei and voluminous cytoplasm (so-called hallmark cells) (Fig. 13-23A). The tumor cells often cluster about venules and infiltrate lymphoid sinuses, mimicking the appearance of metastatic carcinoma. ALK is not expressed in normal lymphocytes or other lymphomas; thus, the detection of ALK protein in tumor cells (Fig. 13-23B) is a reliable indicator of an ALK gene rearrangement.
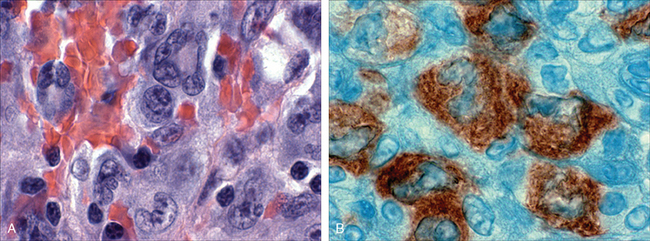
FIGURE 13-23 Anaplastic large-cell lymphoma. A, Several “hallmark” cells with horseshoe-like or “embryoid” nuclei and abundant cytoplasm lie near the center of the field. B, Immunohistochemical stain demonstrating the presence of ALK fusion protein.
(Courtesy of Dr. Jeffrey Kutok, Department of Pathology, Brigham and Women’s Hospital, Boston, MA.)
T-cell lymphomas with ALK rearrangements tend to occur in children or young adults, frequently involve soft tissues, and carry a very good prognosis (unlike other aggressive peripheral T-cell neoplasms). The cure rate with chemotherapy is 75% to 80%. Inhibitors of ALK are under development and offer an excellent opportunity for the development of a selective, targeted therapy. Morphologically similar tumors lacking ALK rearrangements occur in older adults and have a poor prognosis, similar to that of peripheral T-cell lymphoma, unspecified.
Adult T-Cell Leukemia/Lymphoma
This neoplasm of CD4+ T cells is only observed in adults infected by human T-cell leukemia retrovirus type 1 (HTLV-1), which was discussed in Chapter 7. It occurs mainly in regions where HTLV-1 is endemic, namely southern Japan, West Africa, and the Caribbean basin. Common findings include skin lesions, generalized lymphadenopathy, hepatosplenomegaly, peripheral blood lymphocytosis, and hypercalcemia. The appearance of the tumor cells varies, but cells with multilobated nuclei (“cloverleaf” or “flower” cells) are frequently observed. The tumor cells contain clonal HTLV-1 provirus, which is believed to play a critical pathogenic role. Notably, HTLV-1 encodes a protein called Tax that is a potent activator of NF-κB,49 which (as we have discussed) enhances lymphocyte growth and survival.
Most patients present with rapidly progressive disease that is fatal within months to 1 year despite aggressive chemotherapy. Less commonly, the tumor involves only the skin and follows a much more indolent course, like that of mycosis fungoides (described below). It should be noted that in addition to adult T-cell leukemia/lymphoma, HTLV-1 infection sometimes gives rise to a progressive demyelinating disease of the central nervous system and spinal cord (see Chapter 28).
Mycosis Fungoides/Sézary Syndrome
Mycosis fungoides and Sézary syndrome are different manifestations of a tumor of CD4+ helper T cells that home to the skin. Clinically, the cutaneous lesions of mycosis fungoides typically progress through three somewhat distinct stages, an inflammatory premycotic phase, a plaque phase, and a tumor phase (all discussed in more detail in Chapter 25). Histologically, the epidermis and upper dermis are infiltrated by neoplastic T cells, which often have a cerebriform appearance due to marked infolding of the nuclear membrane. Late disease progression is characterized by extracutaneous spread, most commonly to lymph nodes and bone marrow.
Sézary syndrome is a variant in which skin involvement is manifested as a generalized exfoliative erythroderma. In contrast to mycosis fungoides, the skin lesions rarely proceed to tumefaction, and there is an associated leukemia of “Sézary” cells with characteristic cerebriform nuclei.
The tumor cells characteristically express the adhesion molecule CLA and the chemokine receptors CCR4 and CCR10, all of which contribute to the homing of normal CD4+ T cells to the skin. Although cutaneous disease dominates the clinical picture, sensitive molecular analyses have shown that the tumor cells circulate through the blood, marrow, and lymph nodes even early in the course. Nevertheless, these are indolent tumors, with a median survival of 8 to 9 years. Transformation to aggressive T-cell lymphoma occurs occasionally as a terminal event.
Large Granular Lymphocytic Leukemia
T-cell and NK-cell variants of this rare neoplasm are recognized, both of which occur mainly in adults. Individuals with T-cell disease usually present with mild to moderate lymphocytosis and splenomegaly. Lymphadenopathy and hepatomegaly are usually absent. NK-cell disease often presents in an even more subtle fashion, with little or no lymphocytosis or splenomegaly.
The tumor cells are large lymphocytes with abundant blue cytoplasm and a few coarse azurophilic granules, best seen in peripheral blood smears. The marrow usually contains sparse interstitial lymphocytic infiltrates, which can be difficult to appreciate without immunohistochemical stains. Infiltrates are also usually present in the spleen and liver. As might be expected, T-cell variants are CD3+, whereas NK-cell large granular lymphocytic leukemias are CD3−, CD56+.
Despite the relative paucity of marrow infiltration, neutropenia and anemia dominate the clinical picture. Neutropenia is often accompanied by a striking decrease in late myeloid forms in the marrow. Rarely, pure red cell aplasia is seen. There is also an increased incidence of rheumatologic disorders. Some patients with Felty syndrome, a triad of rheumatoid arthritis, splenomegaly, and neutropenia, have this disorder as an underlying cause. The basis for these varied clinical abnormalities is unknown, but autoimmunity, provoked in some way by the tumor, seems likely.
The course is variable, being largely dependent on the severity of the cytopenias and their responsiveness to low-dose chemotherapy or steroids. In general, tumors of T-cell origin pursue an indolent course, whereas NK-cell tumors behave more aggressively.
Extranodal NK/T-Cell Lymphoma
This neoplasm is rare in the United States and Europe, but constitutes as many as 3% of NHLs in Asia. It presents most commonly as a destructive nasopharygeal mass; less common sites of presentation include the testis and the skin. The tumor cell infiltrate typically surrounds and invades small vessels, leading to extensive ischemic necrosis. The tumor cell size is variable but usually includes a large-cell component. In touch preparations, large azurophilic granules are seen in the cytoplasm of the tumor cells that resemble those found in normal NK cells.
This form of lymphoma is highly associated with EBV. Within individual patients, all of the tumor cells contain identical EBV episomes, indicating that the tumor originates from a single EBV-infected cell. How EBV gains entry is uncertain, since the tumor cells fail to express CD21, a surface protein that serves as the B-cell EBV receptor. Most tumors are CD3− and lack T-cell receptor rearrangements and express NK-cell markers, including a restricted set of killer-cell Ig-like receptors, supporting an NK-cell origin. No consistent chromosome aberration has been described, and relatively little is known about the molecular pathogenesis beyond the involvement of EBV.
Most extranodal NK/T-cell lymphomas are highly aggressive neoplasms that respond well to radiation therapy but are resistant to chemotherapy. Thus, the prognosis is poor in patients with advanced disease.
This ends the discussion of the lymphocytic leukemias and the NHLs. We will now turn to the second major category of lymphoid neoplasms, Hodgkin lymphoma.
Hodgkin Lymphoma
Hodgkin lymphoma (HL) encompasses a group of lymphoid neoplasms that differ from NHL in several respects (Table 13-7). While NHLs frequently occur at extranodal sites and spread in an unpredictable fashion, HL arises in a single node or chain of nodes and spreads first to anatomically contiguous lymphoid tissues. For this reason, the staging of HL is much more important in guiding therapy than it is in NHL. HL also has distinctive morphologic features. It is characterized by the presence of neoplastic giant cells called Reed-Sternberg cells. These cells release factors that induce the accumulation of reactive lymphocytes, macrophages, and granulocytes, which typically make up greater than 90% of the tumor cellularity. In the vast majority of HLs, the neoplastic Reed-Sternberg cells are derived from germinal center or post-germinal center B cells.
TABLE 13-7 Differences between Hodgkin and Non-Hodgkin Lymphomas
| Hodgkin Lymphoma | Non-Hodgkin Lymphoma |
|---|---|
| More often localized to a single axial group of nodes (cervical, mediastinal, para-aortic) | More frequent involvement of multiple peripheral nodes |
| Orderly spread by contiguity | Noncontiguous spread |
| Mesenteric nodes and Waldeyer ring rarely involved | Waldeyer ring and mesenteric nodes commonly involved |
| Extra-nodal presentation rare | Extra-nodal presentation common |
Hodgkin lymphoma accounts for 0.7% of all new cancers in the United States; there are about 8000 new cases each year. The average age at diagnosis is 32 years. It is one of the most common cancers of young adults and adolescents, but also occurs in the aged. It was the first human cancer to be successfully treated with radiation therapy and chemotherapy, and is curable in most cases.
Classification.
The WHO classification recognizes five subtypes of HL:
In the first four subtypes—nodular sclerosis, mixed cellularity, lymphocyte-rich, and lymphocyte depletion—the Reed-Sternberg cells have a similar immunophenotype. These subtypes are often lumped together as classical forms of HL. In the remaining subtype, lymphocyte predominance, the Reed-Sternberg cells have a distinctive B-cell immunophenotype that differs from that of the “classical” types.
Morphology. Identification of Reed-Sternberg cells and their variants is essential for the diagnosis. Diagnostic Reed-Sternberg cells are large cells (≥45 μm in diameter) with multiple nuclei or a single nucleus with multiple nuclear lobes, each with a large inclusion-like nucleolus about the size of a small lymphocyte (5–7 μm in diameter) (Fig. 13-24A). The cytoplasm is abundant. Several Reed-Sternberg cell variants are also recognized. Mononuclear variants contain a single nucleus with a large inclusion-like nucleolus (Fig. 13-24B). Lacunar cells (seen in the nodular sclerosis subtype) have more delicate, folded, or multilobate nuclei and abundant pale cytoplasm that is often disrupted during the cutting of sections, leaving the nucleus sitting in an empty hole (a lacuna) (Fig. 13-24C). In classical forms of HL, Reed-Sternberg cells undergo a peculiar form of cell death in which the cells shrink and become pyknotic, a process described as “mummification.” Lymphohistocytic variants (L&H cells) with polypoid nuclei, inconspicuous nucleoli, and moderately abundant cytoplasm are characteristic of the lymphocyte predominance subtype (Fig. 13-24D).
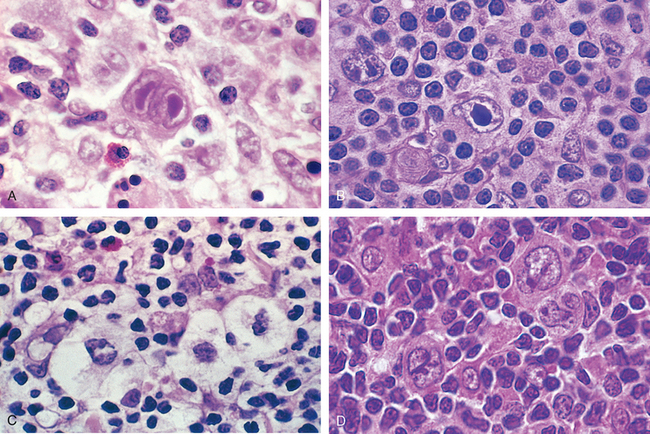
FIGURE 13-24 Reed-Sternberg cells and variants. A, Diagnostic Reed-Sternberg cell, with two nuclear lobes, large inclusion-like nucleoli, and abundant cytoplasm, surrounded by lymphocytes, macrophages, and an eosinophil. B, Reed-Sternberg cell, mononuclear variant. C, Reed-Sternberg cell, lacunar variant. This variant has a folded or multilobated nucleus and lies within a open space, which is an artifact created by disruption of the cytoplasm during tissue sectioning. D, Reed-Sternberg cell, lymphohistiocytic variant. Several such variants with multiply infolded nuclear membranes, small nucleoli, fine chromatin, and abundant pale cytoplasm are present.
(A, Courtesy of Dr. Robert W. McKenna, Department of Pathology, University of Texas Southwestern Medical School, Dallas, TX.)
HL must be distinguished from other conditions in which cells resembling Reed-Sternberg cells can be seen, such as infectious mononucleosis, solid-tissue cancers, and large-cell NHLs. The diagnosis of HL depends on the identification of Reed-Sternberg cells in a typical prominent background of non-neoplastic inflammatory cells. The Reed-Sternberg cells of HL also have a characteristic immunohistochemical profile.
With this as background, we turn to the subclasses of HL, pointing out some of the salient morphologic and immunophenotypic features of each (summarized in Table 13-8). The clinical manifestations common to all will be presented later.
TABLE 13-8 Subtypes of Hodgkin Lymphoma
| Subtype | Morphology and Immunophenotype | Typical Clinical Features |
|---|---|---|
| Nodular sclerosis | Frequent lacunar cells and occasional diagnostic RS cells; background infiltrate composed of T lymphocytes, eosinophils, macrophages, and plasma cells; fibrous bands dividing cellular areas into nodules. RS cells CD15+, CD30+; usually EBV− | Most common subtype; usually stage I or II disease; frequent mediastinal involvement; equal occurrence in males and females (F = M), most patients young adults |
| Mixed cellularity | Frequent mononuclear and diagnostic RS cells; background infiltrate rich in T lymphocytes, eosinophils, macrophages, plasma cells; RS cells CD15+, CD30+; 70% EBV+ | More than 50% present as stage III or IV disease; M greater than F; biphasic incidence, peaking in young adults and again in adults older than 55 |
| Lymphocyte rich | Frequent mononuclear and diagnostic RS cells; background infiltrate rich in T lymphocytes; RS cells CD15+, CD30+; 40% EBV+ | Uncommon; M greater than F; tends to be seen in older adults |
| Lymphocyte depletion | Reticular variant: Frequent diagnostic RS cells and variants and a paucity of background reactive cells; RS cells CD15+, CD30+; most EBV+ | Uncommon; more common in older males, HIV-infected individuals, and in developing countries; often presents with advanced disease |
| Lymphocyte predominance | Frequent L&H (popcorn cell) variants in a background of follicular dendritic cells and reactive B cells; RS cells CD20+, CD15−, C30−; EBV− | Uncommon; young males with cervical or axillary lymphadenopathy; mediastinal |
L&H, lymphohistiocytic; RS cell, Reed-Sternberg cell.
Nodular Sclerosis Type. This is the most common form of HL, constituting 65% to 70% of cases. It is characterized by the presence of lacunar variant Reed-Sternberg cells and the deposition of collagen in bands that divide involved lymph nodes into circumscribed nodules (Fig. 13-25). The fibrosis may be scant or abundant. The Reed-Sternberg cells are found in a polymorphous background of T cells, eosinophils, plasma cells and macrophages. Diagnostic Reed-Sternberg cells are often uncommon. The Reed-Sternberg cells in this and other “classical” HL subtypes have a characteristic immunophenotype; they are positive for PAX5 (a B-cell transcription factor), CD15, and CD30, and negative for other B-cell markers, T-cell markers, and CD45 (leukocyte common antigen). As in other forms of HL, involvement of the spleen, liver, bone marrow, and other organs and tissues can appear in due course in the form of irregular tumor nodules resembling those seen in lymph nodes. This subtype is uncommonly associated with EBV.
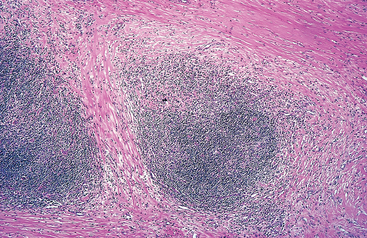
FIGURE 13-25 Hodgkin lymphoma, nodular sclerosis type. A low-power view shows well-defined bands of pink, acellular collagen that subdivide the tumor into nodules.
(Courtesy of Dr. Robert W. McKenna, Department of Pathology, University of Texas Southwestern Medical School, Dallas, TX.)
The nodular sclerosis type occurs with equal frequency in males and females. It has a propensity to involve the lower cervical, supraclavicular, and mediastinal lymph nodes of adolescents or young adults. The prognosis is excellent.
Mixed-Cellularity Type. This form of HL constitutes about 20% to 25% of cases. Involved lymph nodes are diffusely effaced by a heterogeneous cellular infiltrate, which includes T cells, eosinophils, plasma cells, and benign macrophages admixed with Reed-Sternberg cells (Fig. 13-26). Diagnostic ReedSternberg cells and mononuclear variants are usually plentiful. The Reed-Sternberg cells are infected with EBV in about 70% of cases. The immunophenotype is identical to that observed in the nodular sclerosis type.
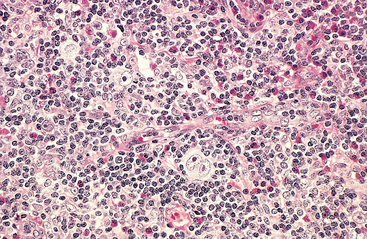
FIGURE 13-26 Hodgkin lymphoma, mixed-cellularity type. A diagnostic, binucleate Reed-Sternberg cell is surrounded by reactive cells, including eosinophils (bright red cytoplasm), lymphocytes, and histiocytes.
(Courtesy of Dr. Robert W. McKenna, Department of Pathology, University of Texas Southwestern Medical School, Dallas, TX.)
Mixed-cellularity HL is more common in males. Compared with the lymphocyte predominance and nodular sclerosis subtypes, it is more likely to be associated with older age, systemic symptoms such as night sweats and weight loss, and advanced tumor stage. Nonetheless, the overall prognosis is very good.
Lymphocyte-Rich Type. This is an uncommon form of classical HL in which reactive lymphocytes make up the vast majority of the cellular infiltrate. In most cases, involved lymph nodes are diffusely effaced, but vague nodularity due to the presence of residual B-cell follicles is sometimes seen. This entity is distinguished from the lymphocyte predominance type by the presence of frequent mononuclear variants and diagnostic Reed-Sternberg cells with a “classical” immunophenotypic profile. It is associated with EBV in about 40% of cases and has a very good to excellent prognosis.
Lymphocyte Depletion Type. This is the least common form of HL, amounting to less than 5% of cases. It is characterized by a paucity of lymphocytes and a relative abundance of Reed-Sternberg cells or their pleomorphic variants. The immunophenotype of the Reed-Sternberg cells is identical to that seen in other classical types of HL. Immunophenotyping is essential, since most tumors suspected of being lymphocyte depletion HL actually prove to be large-cell NHLs. The Reed-Sternberg cells are infected with EBV in over 90% of cases.
Lymphocyte depletion HL occurs predominantly in the elderly, in HIV+ individuals of any age, and in nonindustrialized countries. Advanced stage and systemic symptoms are frequent, and the overall outcome is somewhat less favorable than in the other subtypes.
Lymphocyte Predominance Type. This uncommon “nonclassical” variant of HL accounts for about 5% of cases. Involved nodes are effaced by a nodular infiltrate of small lymphocytes admixed with variable numbers of macrophages (Fig. 13-27). “Classical” Reed-Sternberg cells are usually difficult to find. Instead, this tumor contains so-called L&H (lymphocytic and histiocytic) variants, which have a multilobed nucleus resembling a popcorn kernel (“popcorn cell”). Eosinophils and plasma cells are usually scant or absent.
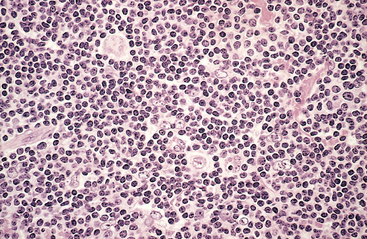
FIGURE 13-27 Hodgkin lymphoma, lymphocyte predominance type. Numerous mature-looking lymphocytes surround scattered, large, pale-staining lymphohistiocytic variants (“popcorn” cells).
(Courtesy of Dr. Robert W. McKenna, Department of Pathology, University of Texas Southwestern Medical School, Dallas, TX.)
In contrast to the Reed-Sternberg cells found in classical forms of HL, L&H variants express B-cell markers typical of germinal-center B cells, such as CD20 and BCL6, and are usually negative for CD15 and CD30. The typical nodular pattern of growth is due to the presence of expanded B-cell follicles, which are populated with L&H variants, numerous reactive B cells, and follicular dendritic cells. The IgH genes of the L&H variants show evidence of ongoing somatic hypermutation, a modification that occurs only in germinal-center B cells. In 3% to 5% of cases, this type transforms into a tumor resembling diffuse large B-cell lymphoma. EBV is not associated with this subtype.
A majority of patients are males, usually younger than 35 years of age, who typically present with cervical or axillary lymphadenopathy. Mediastinal and bone marrow involvement is rare. In some series, this form of HL is more likely to recur than the classical subtypes, but the prognosis is excellent.
Molecular Pathogenesis.
The origin of the neoplastic Reed-Sternberg cells of classical HL has been explained through elegant studies relying on molecular analysis of single isolated Reed-Sternberg cells and variants. In the vast majority of cases, the Ig genes of Reed-Sternberg cells have undergone both V(D)J recombination and somatic hypermutation, establishing an origin from a germinal center or post-germinal-center B cell.50 Despite having the genetic signature of a B cell, the Reed-Sternberg cells of classical HL fail to express most B cell–specific genes, including the Ig genes. The cause of this wholesale reprogramming of gene expression has yet to be fully explained.51
Activation of the transcription factor NF-κB is a common event in classical HL. NF-κB is activated either by EBV infection or by some other mechanism and turns on genes that promote lymphocyte survival and proliferation. EBV+ tumor cells express latent membrane protein-1 (LMP-1), a protein encoded by the EBV genome that transmits signals that up-regulate NF-κB. Activation of NF-κB also occurs in EBV− tumors, in some instances as a result of acquired mutations in IκB,52 a negative regulator of NF-κB. It is hypothesized that activation of NF-κB by EBV or other mechanisms rescues “crippled” germinal-center B cells that cannot express Igs from apoptosis, setting the stage for the acquisition of other unknown mutations that collaborate to produce ReedSternberg cells. Little is known about the basis for the morphology of Reed-Sternberg cells and variants, but it is intriguing that EBV-infected B cells resembling ReedSternberg cells are found in the lymph nodes of individuals with infectious mononucleosis, strongly suggesting that EBV-encoded proteins play a part in the remarkable metamorphosis of B cells into Reed-Sternberg cells.
The florid accumulation of reactive cells in tissues involved by classical HL occurs in response to a wide variety of cytokines (such as IL-5, IL-10, IL-13, and TGF-β) and chemokines (such as TARC, MDC, IP-10, and CCL28) that are secreted by Reed-Sternberg cells.53 Once attracted, the reactive cells produce factors that support the growth and survival of the tumor cells and further modify the reactive cell response. For example, eosinophils and T cells express ligands that activate the CD30 and CD40 receptors found on Reed-Sternberg cells, producing signals that up-regulate NF-κB. Other examples of “cross-talk” between Reed-Sternberg cells and surrounding reactive cells are provided in Figure 13-28.
FIGURE 13-28 Proposed signals mediating “cross-talk” between Reed-Sternberg cells and surrounding normal cells in classical forms of Hodgkin lymphoma. CD30L, CD30 ligand; bFGF, basic fibroblast growth factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; HGF, hepatocyte growth factor (binds to the c-MET receptor); TGFβ, transforming growth factor β; TNFβ, tumor necrosis factor β (lymphotoxin); Tc, CD8+ cytotoxic T cell; TH1 and TH2, CD4+ T helper cell subsets.
Reed-Sternberg cells are aneuploid and possess diverse clonal chromosomal aberrations. Copy number gains in the c-REL proto-oncogene on chromosome 2p are particularly common and may contribute to increases in NF-κB activity.54
Clinical Features.
HL most commonly present as painless lymphadenopathy. Patients with the nodular sclerosis or lymphocyte predominance types tend to present with stage I–II disease and are usually free of systemic manifestations. Patients with disseminated disease (stages III–IV) or the mixed-cellularity or lymphocyte depletion subtypes are more likely to have constitutional symptoms, such as fever, night sweats, and weight loss. Cutaneous anergy resulting from depressed cell-mediated immunity is seen in most cases. The mix of factors released from Reed-Sternberg cells (see Fig. 13-28) suppress TH1 immune responses and may contribute to immune dysregulation.
The spread of HL is remarkably stereotyped: nodal disease first, then splenic disease, hepatic disease, and finally involvement of the marrow and other tissues. Because of this behavior, radiation therapy can be curative for persons with early-stage disease. Thus, the staging of HL (Table 13-9) not only determines the prognosis, but also guides therapy. Staging involves physical examination, radiologic imaging of the abdomen, pelvis, and chest, and biopsy of the bone marrow. Systemic treatment is preferred whenever the staging is equivocal.
TABLE 13-9 Clinical Staging of Hodgkin and Non-Hodgkin Lymphomas (Ann Arbor Classification)
| Stage | Distribution of Disease |
|---|---|
| I | Involvement of a single lymph node region (I) or a single extra-lymphatic organ or site (IE). |
| II | Involvement of two or more lymph node regions on the same side of the diaphragm alone (II) or localized involvement of an extra-lymphatic organ or site (IIE). |
| III | Involvement of lymph node regions on both sides of the diaphragm without (III) or with (IIIE) localized involvement of an extra-lymphatic organ or site. |
| IV | Diffuse involvement of one or more extra-lymphatic organs or sites with or without lymphatic involvement. |
| All stages are further divided on the basis of the absence (A) or presence (B) of the following symptoms: unexplained fever, drenching night sweats, and/or unexplained weight loss of greater than 10% of normal body weight. | |
Data from Carbone PT et al.: Symposium (Ann Arbor): Staging in Hodgkin’s disease. Cancer Res 31:1707, 1971.
With current treatment protocols, tumor stage rather than histologic type is the most important prognostic variable. The cure rate of patients with stages I and IIA is close to 90%. Even with advanced disease (stages IVA and IVB), disease-free survival at 5 years is 60% to 70%.
Progress in the treatment of HL has created a new set of problems. Long-term survivors of chemotherapy and radiation therapy have an increased risk of developing second cancers. Myelodysplastic syndromes, AML, and lung cancer head the list, but also included are NHL, breast cancer, gastric cancer, sarcoma, and melanoma. Most of the risk of solid tumors is attributable to radiation therapy, which has also been linked to pulmonary fibrosis and accelerated atherosclerosis. The risk of breast cancer is particularly high in females treated with radiation to the chest during adolescence. Alkylating chemotherapeutic drugs seem to be responsible for the increased risk of AML and myelodysplasia. Fortunately, newer combinations of chemotherapeutic drugs and more judicious use of radiation therapy seem to largely avoid these complications and are equally curative.
MYELOID NEOPLASMS
The common feature of this heterogeneous group of neoplasms is an origin from hematopoietic progenitor cells. These diseases primarily involve the marrow and to a lesser degree the secondary hematopoietic organs (the spleen, liver, and lymph nodes), and usually present with symptoms related to altered hematopoiesis. Three broad categories of myeloid neoplasia exist:
The pathogenesis of myeloid neoplasms is best understood in the context of normal hematopoiesis, which (you will remember from Fig. 13-1) involves a hierarchy of hematopoietic stem cells, committed progenitors, and more differentiated elements. Normal hematopoiesis is finely tuned by homeostatic feedback mechanisms involving cytokines and growth factors that modulate the production of red cells, white cells, and platelets in the marrow. These mechanisms are deranged in marrows involved by myeloid neoplasms, which “escape” from normal homeostatic controls on growth and survival and suppress the function of residual normal stem cells. The specific manifestations of the different myeloid neoplasms are influenced by
We will return to these themes as each type of myeloid neoplasm is discussed.
Given that all myeloid neoplasms originate from transformed hematopoietic progenitors, it is not surprising that divisions between these neoplasms are sometimes blurred. Myeloid neoplasms, like other malignancies, tend to evolve over time to more aggressive forms of disease. In particular, both myelodysplastic syndromes and myeloproliferative disorders often “transform” to AML. In one of the most important myeloproliferative disorders, chronic myeloid leukemia, transformation to acute lymphoblastic leukemia is also seen, indicating that it originates from a transformed pluripotent hematopoietic stem cell.
Acute Myeloid Leukemia
Acute myeloid leukemia (AML) is a tumor of hematopoietic progenitors caused by acquired oncogenic mutations that impede differentiation, leading to the accumulation of immature myeloid blasts in the marrow. The arrest in myeloid development leads to marrow failure and complications related to anemia, thrombocytopenia, and neutropenia. AML occurs at all ages, but the incidence rises throughout life, peaking after 60 years of age. There are about 13,000 new cases each year in the United States.
Classification.
AML is quite heterogeneous, reflecting the complexities of myeloid cell differentiation. A new proposed classification from the WHO subdivides AML into four categories (Table 13-10).11 The first includes forms of AML that are associated with particular genetic aberrations, which are important because they correlate with prognosis and guide therapy. Also included are categories of AML arising after a myelodysplastic disorder (MDS) or with MDS-like features, and therapy-related AML. AMLs in these two categories have distinct genetic features and respond poorly to therapy. A fourth “wastebasket” category includes AMLs lacking any of these features. These are classified according to the earlier French-American-British (FAB) classification, which divide AMLs into subtypes based on the degree of differentiation and the lineage of the leukemic blasts. Although it has limited utility, the FAB classification is still commonly referred to in practice. In recognition of this, Table 13-10 correlates (to the extent possible) the FAB and WHO classifications. Given the increasing role of cytogenetic and molecular features in directing therapy, a further shift toward genetic classification of AML is both inevitable and desirable.
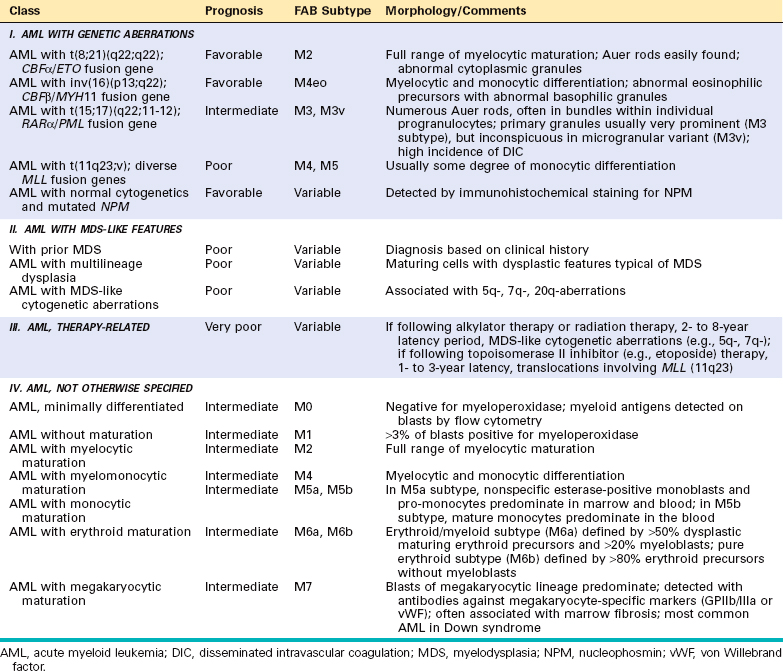
Morphology. The diagnosis of AML is based on the presence of at least 20% myeloid blasts in the bone marrow. Several types of myeloid blasts are recognized, and individual tumors may have more than one type of blast or blasts with hybrid features. Myeloblasts have delicate nuclear chromatin, two to four nucleoli, and more voluminous cytoplasm than lymphoblasts (Fig. 13-29A). The cytoplasm often contains fine, peroxidase-positive azurophilic granules. Auer rods, distinctive needle-like azurophilic granules, are present in many cases; they are particularly numerous in AML with the t(15;17) (acute promyelocytic leukemia) (Fig. 13-30A). Monoblasts (Fig. 13-30B) have folded or lobulated nuclei, lack Auer rods, and are nonspecific esterase-positive. In some AMLs, blasts show megakaryocytic differentiation, which is often accompanied by marrow fibrosis caused by the release of fibrogenic cytokines. Rarely, the blasts of AML show erythroid differentiation.

FIGURE 13-29 A, Acute myeloid leukemia without maturation (FAB M1 subtype). Myeloblasts have delicate nuclear chromatin, prominent nucleoli, and fine azurophilic granules in the cytoplasm. B, In the flow cytometric analysis shown, the myeloid blasts, represented by the red dots, express CD34, a marker of multipotent stem cells, but do not express CD64, a marker of mature myeloid cells. C, The same myeloid blasts express CD33, a marker of immature myeloid cells, and a subset express CD15, a marker of more mature myeloid cells. Thus, these blasts are myeloid cells showing limited maturation.
(A, Courtesy of Dr. Robert W. McKenna Department of Pathology, University of Texas Southwestern Medical School, Dallas, TX; B and C, courtesy of Dr. Louis Picker, Oregon Health Science Center, Portland, OR.)
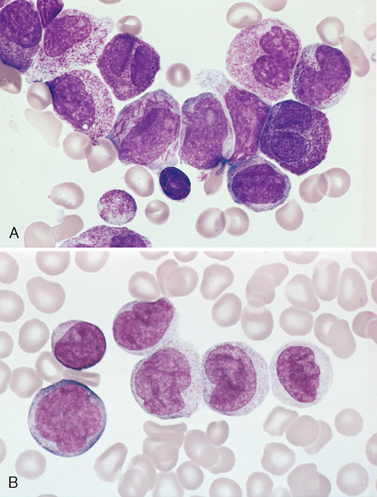
FIGURE 13-30 Acute myeloid leukemia subtypes. A, Acute promyelocytic leukemia with the t(15;17) (FAB M3 subtype). Bone marrow aspirate shows neoplastic promyelocytes with abnormally coarse and numerous azurophilic granules. Other characteristic findings include the presence of several cells with bilobed nuclei and a cell in the center of the field that contains multiple needle-like Auer rods. B, Acute myeloid leukemia with monocytic differentiation (FAB M5b subtype). Peripheral smear shows one monoblast and five promonocytes with folded nuclear membranes.
(Courtesy of Dr. Robert W. McKenna, Department of Pathology, University of Texas Southwestern Medical School, Dallas, TX.)
The number of leukemic cells in the blood is highly variable. Blasts may be more than 100,000 per mm3, but are under 10,000 per mm3 in about 50% of patients. Occasionally, blasts are entirely absent from the blood (aleukemic leukemia). For this reason, a bone marrow examination is essential to exclude acute leukemia in pancytopenic patients.
Immunophenotype.
Because it can be difficult to distinguish myeloblasts and lymphoblasts morphologically, the diagnosis of AML is confirmed by performing stains for myeloidspecific antigens (see Fig. 13-29B, C).
Cytogenetics.
Cytogenetic analysis has a central role in the classification of AML. Karyotypic aberrations are detected in 50% to 70% of cases with standard techniques and in approximately 90% of cases using special high-resolution banding. Particular chromosomal abnormalities correlate with certain clinical features. AMLs arising de novo in younger adults are commonly associated with balanced chromosomal translocations, particularly t(8;21), inv(16), and t(15;17). In contrast, AMLs following MDS or exposure to DNA-damaging agents (such as chemotherapy or radiation therapy) often have deletions or monosomies involving chromosomes 5 and 7 and usually lack chromosomal translocations. The exception to this rule is AML occurring after treatment with topoisomerase II inhibitors, which is strongly associated with translocations involving the MLL gene on chromosome 11q23. AML in the elderly is also more likely to be associated with “bad” aberrations, such as deletions of chromosomes 5q and 7q.
Molecular Pathogenesis.
Many recurrent genetic aberrations seen in AML disrupt genes encoding transcription factors that are required for normal myeloid differentiation. For example, the two most common chromosomal rearrangements, t(8;21) and inv(16), disrupt the CBF1α and CBF1β genes, respectively. These two genes encode polypeptides that bind one another to form a CBF1α/CBF1β transcription factor that is required for normal hematopoiesis.55 The t(8;21) and the inv(16) create chimeric genes encoding fusion proteins that interfere with the function of CBF1α/CBF1β and block the maturation of myeloid cells. It should be noted, however, that “knockout” mice lacking either CBF1α or CBF1β and “knock-in” mice expressing the CBF1α or CBF1β fusion proteins succumb to hematopoietic failure, not leukemia. Thus, genetic lesions that merely block the maturation of myeloid progenitors are not by themselves sufficient to cause AML.
In line with this idea, there is increasing evidence that mutated tyrosine kinases collaborate with transcription factor aberrations to produce AML. One example is found in AML with the t(15;17), acute promyelocytic leukemia. The t(15;17) creates yet another fusion gene that encodes a part of the retinoic acid receptor-α (RARα) fused to a portion of a protein called PML (after the tumor). In the presence of physiologic amounts of retinoic acid, normal RARα interacts with other transcription factors to activate genes that are needed for granulocytic differentiation. However, the PML-RARα fusion protein interacts instead with transcriptional repressors, which results in an inhibition of granulocytic maturation.56 AMLs with the t(15;17) also have frequent activating mutations in FLT3, a receptor tyrosine kinase that transmits signals that increase cellular proliferation and survival. The combination of PML-RARα and activated FLT3 is a potent inducer of AML in mice,57 whereas neither gene alone is sufficient. Identical FLT3 mutations are also found in other forms of AML, particularly those associated with NPM (nucleophosmin) mutations,58 and activating mutations in another tyrosine kinase receptor, c-KIT, are found in about 25% of AMLs associated with the inv(16) or the t(8;21).59 Thus, aberrant tyrosine kinase activation is a common (and possibly universal) feature of AML.
The t(15;17) not only has pathogenic significance, but also guides therapy, since tumors with this translocation respond to pharmacologic doses of all-trans retinoic acid (ATRA). ATRA binds to the PML-RARα fusion protein and antagonizes its inhibitory effect on the transcription of target genes. Remarkably, the resulting activation of transcription overcomes the block in differentiation, and within 1 to 2 days the neoplastic promyelocytes begin to differentiate into neutrophils, which rapidly die. The response to ATRA proves that the major effect of PML-RARα is to block differentiation, and stands as one of the most successful uses of a targeted therapy in a human cancer.
Clinical Features.
Most patients present within weeks or a few months of the onset of symptoms with complaints related to anemia, neutropenia, and thrombocytopenia, most notably fatigue, fever, and spontaneous mucosal and cutaneous bleeding. You will remember that these findings are very similar to those produced by ALL. Thrombocytopenia results in a bleeding diathesis, which is often prominent. Cutaneous petechiae and ecchymoses, serosal hemorrhages into the linings of the body cavities and viscera, and mucosal hemorrhages into the gingivae and urinary tract are common. Procoagulants and fibrinolytic factors released by leukemic cells, especially in AML with the t(15;17), exacerbate the bleeding tendency. Infections are frequent, particularly in the oral cavity, skin, lungs, kidneys, urinary bladder, and colon, and are often caused by opportunists such as fungi, Pseudomonas, and commensals.
Signs and symptoms related to involvement of tissues other than the marrow are usually less striking in AML than in ALL, but tumors with monocytic differentiation often infiltrate the skin (leukemia cutis) and the gingiva; this probably reflects the normal tendency of monocytes to extravasate into tissues. Central nervous system spread is less common than in ALL. AML occasionally presents as a localized soft-tissue mass known variously as a myeloblastoma, granulocytic sarcoma, or chloroma. Without systemic treatment, such tumors inevitably progress to full-blown AML over time.
Prognosis.
AML is a difficult disease to treat. About 60% of patients achieve complete remission with chemotherapy, but only 15% to 30% remain free of disease for 5 years. AMLs with t(8;21) or inv(16) have a relatively good prognosis with conventional chemotherapy, particularly in the absence of c-KIT mutations.59 In contrast, the prognosis is dismal for AMLs that follow MDS or genotoxic therapy, or that occur in the elderly, possibly because in these contexts the disease arises out of a background of hematopoietic stem cell damage or depletion. These “high-risk” forms of AML (as well as relapsed AML of all types) are treated with bone marrow transplantation when possible.
It is hoped that new approaches based on a better understanding of molecular pathogenesis will improve this situation. The best current example is AML with the t(15;17), which (as we have discussed) is treated with pharmacologic doses of ATRA combined with conventional chemotherapy, or, more recently, with arsenic salts, which appear to cause PML-RARα to be degraded. New therapies that target other molecular lesions in AML (e.g., the activated FLT3 and c-KIT tyrosine kinases) are being evaluated.
Myelodysplastic Syndromes
The term “myelodysplastic syndrome” (MDS) refers to a group of clonal stem cell disorders characterized by maturation defects that are associated with ineffective hematopoiesis and a high risk of transformation to AML. In MDS the bone marrow is partly or wholly replaced by the clonal progeny of a neoplastic multipotent stem cell that retains the capacity to differentiate but does so in an ineffective and disordered fashion. These abnormal cells stay within the bone marrow and hence the patients have peripheral blood cytopenias.
MDS may be either primary (idiopathic) or secondary to previous genotoxic drug or radiation therapy (t-MDS). t-MDS usually appears from 2 to 8 years after the genotoxic exposure. All forms of MDS can transform to AML, but transformation occurs with highest frequency and most rapidly in t-MDS. Although characteristic morphologic changes are typically seen in the marrow and the peripheral blood, the diagnosis frequently requires correlation with other laboratory tests. Cytogenetic analysis is particularly helpful, since certain chromosomal aberrations (discussed below) are often observed.
Molecular Pathogenesis.
The pathogenesis is poorly understood.60 In MDS, bone marrow progenitors undergo apoptotic cell death at an increased rate, the hallmark of ineffective hematopoiesis. Given this, it is difficult to understand how MDS progenitors gain a selective advantage over any remaining normal marrow progenitors, suggesting that the tumor may arise out of a background of stem cell damage or depletion. Both primary MDS and t-MDS are associated with similar clonal chromosomal abnormalities, including monosomies 5 and 7, deletions of 5q, 7q, and 20q, and trisomy 8.
Morphology. Although the marrow is usually hypercellular at diagnosis, it is sometimes normocellular or, less commonly, hypocellular. The most characteristic finding is disordered (dysplastic) differentiation affecting the erythroid, granulocytic, monocytic, and megakaryocytic lineages to varying degrees (Fig. 13-31). Within the erythroid series, common abnormalities include ringed sideroblasts, erythroblasts with iron-laden mitochondria visible as perinuclear granules in Prussian blue–stained aspirates or biopsies; megaloblastoid maturation, resembling that seen in vitamin B12 and folate deficiency (Chapter 14); and nuclear budding abnormalities, recognized as nuclei with misshapen, often polyploid, outlines. Neutrophils frequently contain decreased numbers of secondary granules, toxic granulations, and/or Döhle bodies. Pseudo-Pelger-Hüet cells, neutrophils with only two nuclear lobes, are commonly observed, and neutrophils are seen occasionally that completely lack nuclear segmentation. Megakaryocytes with single nuclear lobes or multiple separate nuclei (pawn ball megakaryocytes) are also characteristic. Myeloid blasts may be increased but make up less than 20% of the overall marrow cellularity. The blood often contains pseudo-Pelger-Hüet cells, giant platelets, macrocytes, and poikilocytes, accompanied by a relative or absolute monocytosis. Myeloid blasts usually make up less than 10% of the leukocytes in the blood.
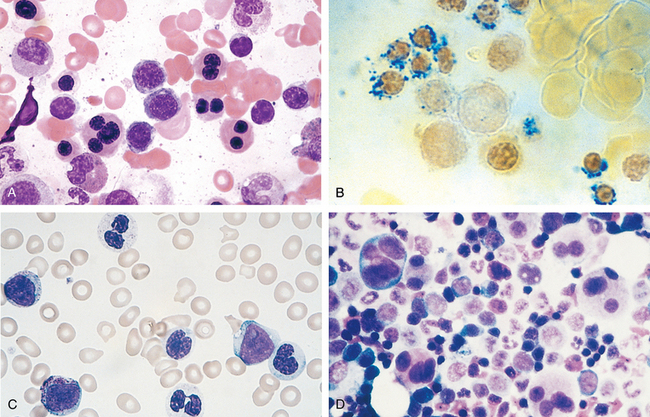
FIGURE 13-31 Myelodysplasia. Characteristic forms of dysplasia are shown. A, Nucleated red cell progenitors with multilobated or multiple nuclei. B, Ringed sideroblasts, erythroid progenitors with iron-laden mitochondria seen as blue perinuclear granules (Prussian blue stain). C, Pseudo-Pelger-Hüet cells, neutrophils with only two nuclear lobes instead of the normal three to four, are observed at the top and bottom of this field. D, Megakaryocytes with multiple nuclei instead of the normal single multilobated nucleus. (A, B, D, Marrow aspirates; C, peripheral blood smear.)
Clinical Course.
Primary MDS is predominantly a disease of the elderly; the mean age of onset is 70 years. In up to half of the cases, it is discovered incidentally on routine blood testing. When symptomatic, it presents with weakness, infections, and hemorrhages, all due to pancytopenia.
Primary MDS is divided into five morphologic categories in the WHO classification,11 details of which are beyond our scope. Subtypes defined by having a higher proportion of blasts are associated with more severe cytopenias, an increased risk of progression to AML, and a worse prognosis. The presence of multiple clonal chromosomal abnormalities and the severity of peripheral blood cytopenias are independent risk factors also portending a worse outcome.
The median survival in primary MDS varies from 9 to 29 months, but some individuals in good prognostic groups may live for 5 years or more. Overall, progression to AML occurs in 10% to 40% of individuals and is usually accompanied by the appearance of additional cytogenetic abnormalities. Patients often succumb to the complications of thrombocytopenia (bleeding) and neutropenia (infection). The outlook is even grimmer in t-MDS, which has a median survival of only 4 to 8 months. In t-MDS, cytopenias tend to be more severe and progression to AML is often rapid.
Treatment options are fairly limited. In younger patients, allogeneic bone marrow transplantation offers hope for reconstitution of normal hematopoiesis and long-term survival. Older patients with MDS are treated supportively with antibiotics and blood product transfusions. Thalidomide-like drugs (which appear to alter the interaction of MDS progenitors with bone marrow stromal cells) and DNA methylase inhibitors improve the effectiveness of hematopoiesis and the peripheral blood counts in a subset of patients.60
Myeloproliferative Disorders
The common pathogenic feature of the myeloproliferative disorders is the presence of mutated, constitutively activated tyrosine kinases.61,62 Hematopoietic growth factors act on normal progenitors by binding to surface receptors and activating tyrosine kinases, which turn on pathways that promote growth and survival. The mutated tyrosine kinases found in the myeloproliferative disorders circumvent normal controls and lead to the growth factor–independent proliferation and survival of marrow progenitors. Because the tyrosine kinase mutations underlying the various myeloproliferative disorders do not impair differentiation, the most common consequence is an increase in the production of one or more mature blood elements. Most myeloproliferative disorders originate in multipotent myeloid progenitors, whereas others arise in pluripotent stem cells that give rise to both lymphoid and myeloid cells.
There is a considerable degree of clinical and morphologic overlap among the myeloproliferative disorders. The common features include
Certain myeloproliferative disorders are strongly associated with activating mutations in specific tyrosine kinases. This insight and the availability of kinase inhibitors have increased the importance of molecular tests for tyrosine kinase mutations, both for purposes of diagnosis and the selection of therapy. We will confine our discussion to the more common myeloproliferative disorders, which are classified based on clinical, laboratory, and molecular criteria. Systemic mastocytosis, a distinctive myeloproliferative disorder that is associated with mutations in the c-KIT tyrosine kinase, is discussed under disorders of the skin (Chapter 25). The association of various myeloproliferative disorders with specific tyrosine kinase mutations (including several too rare to merit discussion) is summarized in Table 13-11.

Chronic Myeloid Leukemia
Chronic myeloid leukemia (CML) is distinguished from other myeloproliferative disorders by the presence of a chimeric BCR-ABL gene derived from portions of the BCR gene on chromosome 22 and the ABL gene on chromosome 9. BCR-ABL directs the synthesis of a constitutively active BCR-ABL tyrosine kinase (Fig. 13-32),63 which in CML is usually 210 kDa in size. In more than 90% of cases, BCR-ABL is created by a reciprocal (9;22)(q34;q11) translocation (the so-called Philadelphia chromosome [Ph]). In the remaining cases the BCR-ABL fusion gene is formed by cytogenetically complex or cryptic rearrangments and must be detected by other methods, such as fluorescence in situ hybridization or PCR-based tests. The cell of origin is a pluripotent hematopoietic stem cell.
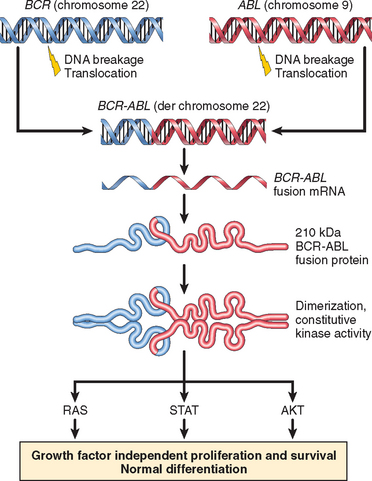
FIGURE 13-32 Molecular pathogenesis of chronic myeloid leukemia. Breakage and joining of BCR and ABL creates a chimeric BCR-ABL fusion gene that encodes a constitutively active BCR-ABL tyrosine kinase. BCR-ABL activates multiple downstream pathways, which drive growth factor–independent proliferation and survival of bone marrow progenitors. Because BCR-ABL does not interfere with differentiation, the net result is an increase in mature elements in the peripheral blood, particularly granulocytes and platelets.
Molecular Pathogenesis.
Tyrosine kinases are normally regulated by ligand-mediated dimerization and autophosphorylation, which creates an activated kinase capable of phosphorylating other protein substrates (discussed in Chapters 3 and 7. The BCR moiety of BCR-ABL contains a dimerization domain that self-associates, leading to the activation of the ABL tyrosine kinase moiety. The ABL kinase in turn phosphorylates proteins that induce signaling through the same pro-growth and pro-survival pathways that are turned on by hematopoietic growth factors, including the RAS, JAK/STAT, and AKT pathways. For unknown reasons, BCR-ABL preferentially drives the proliferation of granulocytic and megakaryocytic progenitors, and also causes the abnormal release of immature granulocytic forms from the marrow into the blood.
Morphology. The marrow is markedly hypercellular because of massively increased numbers of maturing granulocytic precursors, which usually include an elevated proportion of eosinophils and basophils. Megakaryocytes are also increased and usually include small, dysplastic forms. Erythroid progenitors are present in normal or mildly decreased numbers. A characteristic finding is the presence of scattered macrophages with abundant wrinkled, green-blue cytoplasm so-called sea-blue histiocytes. Increased deposition of reticulin is typical, but overt marrow fibrosis is rare early in the course. The blood reveals a leukocytosis, often exceeding 100,000 cells/mm3 (Fig. 13-33), which consists predominantly of neutrophils, band forms, metamyelocytes, myelocytes, eosinophils, and basophils. Blasts usually make up less than 10% of the circulating cells. Platelets are also usually increased, sometimes markedly. The spleen is often greatly enlarged as a result of extensive extramedullary hematopoiesis (Fig. 13-34) and often contains infarcts of varying age. Extramedullary hematopoiesis can also produce mild hepatomegaly and lymphadenopathy.
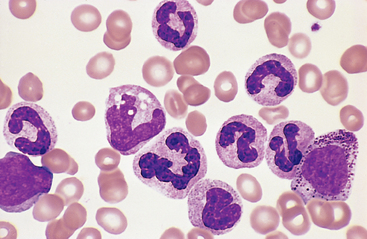

Clinical Features.
CML is primarily a disease of adults but also occurs in children and adolescents. The peak incidence is in the fifth to sixth decades of life. There are about 4500 new cases per year in the United States.
The onset is insidious. Mild-to-moderate anemia and hypermetabolism due to increased cell turnover lead to fatigability, weakness, weight loss, and anorexia. Sometimes the first symptom is a dragging sensation in the abdomen caused by splenomegaly, or the acute onset of left upper quadrant pain due to splenic infarction. CML is best differentiated from other myeloproliferative disorders by detection of the BCR-ABL fusion gene through either chromosomal analysis or PCR-based tests.
The natural history is one of slow progression; even without treatment, the median survival is about 3 years. After a variable period averaging 3 years, about 50% of patients enter an “accelerated phase” marked by increasing anemia and thrombocytopenia, sometimes accompanied by a rise in the number of basophils in the blood. Additional clonal cytogenetic abnormalities, such as trisomy 8, isochromosome 17q, or duplication of the Ph chromosome, often appear. Within 6 to 12 months, the accelerated phase terminates in a picture resembling acute leukemia (blast crisis). In the other 50% of patients, blast crises occur abruptly without an accelerated phase. In 70% of crises, the blasts are of myeloid origin (myeloid blast crisis), whereas in most of the remainder the blasts are of pre–B cell origin (lymphoid blast crisis). This is taken as evidence that CML orginates from a pluripotent stem cell with both myeloid and lymphoid potential. Recently, it has been observed that in greater than 85% of cases CML is associated with the appearance of mutations that interfere with the activity of Ikaros, a transcription factor that regulates the differentiation of hematopoietic progenitors.64 The same types of Ikaros mutations are also seen in BCR-ABL-positive ALL, suggesting that these two varieties of aggressive leukemia have a similar pathogenic basis.
Understanding of the molecular pathogenesis of CML has led to the use of drugs that target BCR-ABL. Treatment with one BCR-ABL inhibitor, imatinib, results in sustained hematologic remissions in greater than 90% of patients, with few side effects.65 Imatinib markedly decreases the number of BCR-ABL-positive cells in the marrow and elsewhere, but does not extinguish the CML “stem cell,” a primitive cell that resembles a normal hematopoietic stem cell. As a result, it is not clear if imatinib is ever truly curative. However, imatinib therapy controls blood counts and substantially decreases the risk of transformation to the accelerated phase and blast crisis, which is the greatest threat to the patient. It is thought that by lowering the proliferative drive of the BCR-ABL-positive progenitors, imatinib decreases the rate at which these cells acquire mutations that lead to disease progression.66 For relatively young patients, allogeneic bone marrow transplantation performed in the stable phase is curative in about 75% of cases and remains the treatment of choice. The outlook is much more dire once the accelerated phase or blast crisis supervenes. Transplantation is not as effective, and (as in BCR-ABLpositive ALL) the disease rapidly becomes resistant to BCR-ABL inhibitors.
Polycythemia Vera
Polycythemia vera (PCV) is characterized by increased marrow production of red cells, granulocytes, and platelets (panmyelosis), but it is the increase in red cells (polycythemia) that is responsible for most of the clinical symptoms. PCV must be differentiated from relative polycythemia resulting from hemoconcentration and other causes of absolute polycythemia (discussed in Chapter 14). PCV is strongly associated with activating point mutations in the tyrosine kinase JAK2.61,62 JAK2 participates in the JAK/STAT pathway, which lies downstream of multiple hematopoietic growth factor receptors, including the erythropoietin receptor.
Molecular Pathogenesis.
In PCV the progenitor cells have markedly decreased requirements for erythropoietin and other hematopoietic growth factors due to constitutive JAK2 signaling. Accordingly, serum erythropoietin levels in PCV are very low, whereas secondary forms of absolute polycythemia have high erythropoietin levels. The elevated hematocrit leads to increased blood viscosity and sludging. These hemodynamic factors, together with thrombocytosis and abnormal platelet function, make patients with PCV prone to both thrombosis and bleeding.
More than 97% of cases are associated with a mutation in JAK2 that results in a valine-to-phenylalanine substitution at residue 617; other JAK2 mutations are found in most (and perhaps all) of the remaining cases. The mutated forms of JAK2 found in PCV render hematopoietic cell lines growth factor–independent, and when expressed in murine bone marrow progenitors cause a PCV-like syndrome that is associated with marrow fibrosis.61,62 In 25% to 30% of cases the tumor cells contain two mutated copies of JAK2, a homozygous genotype that is associated with higher white cell counts, more significant splenomegaly, symptomatic pruritus, and a greater rate of progression to the spent phase.67
The proliferative drive in PCV (and other myeloproliferative disorders associated with JAK2 mutations) is less than in CML, which is associated with more pronounced marrow hypercellularity, leukocytosis, and splenomegaly. Presumably, JAK2 signals are quantitatively weaker or qualitatively different from those produced by BCR-ABL (see Fig. 13-32).
Morphology. The marrow is hypercellular, but some residual fat is usually present. The increase in red cell progenitors is subtle and usually accompanied by an increase in granulocytic precursors and megakaryocytes as well. At diagnosis, a moderate to marked increase in reticulin fibers is seen in about 10% of marrows. Mild organomegaly is common, being caused early in the course largely by congestion; at this stage extramedullary hematopoiesis is minimal. The peripheral blood often contains increased numbers of basophils and abnormally large platelets.
Late in the course, PCV often progresses to a spent phase characterized by extensive marrow fibrosis that displaces hematopoietic cells. This is accompanied by increased extramedullary hematopoiesis in the spleen and liver, often leading to prominent organomegaly (Fig. 13-35). Transformation to AML, with its typical features, occurs in about 1% of patients.
Clinical Features.
PCV is uncommon, having an incidence of 1 to 3 per 100,000 per year. It appears insidiously, usually in adults of late middle age. Most symptoms are related to the increased red cell mass and hematocrit. Usually, there is also an increased total blood volume. Together, these factors cause abnormal blood flow, particularly on the low-pressure venous side of the circulation, which becomes greatly distended. Patients are plethoric and cyanotic due to stagnation and deoxygenation of blood in peripheral vessels. Headache, dizziness, hypertension, and gastrointestinal symptoms are common. Intense pruritus and peptic ulceration may occur, both possibly resulting from the release of histamine from basophils. High cell turnover gives rise to hyperuricemia; symptomatic gout is seen in 5% to 10% of cases.
More ominously, the abnormal blood flow and platelet function lead to an increased risk of both major bleeding and thrombotic episodes. About 25% of patients first come to attention due to deep venous thrombosis, myocardial infarction, or stroke. Thromboses sometimes also occur in the hepatic veins (producing Budd-Chiari syndrome) and the portal and mesenteric veins (leading to bowel infarction). It should be remembered that thrombotic complications sometimes precede the appearance of the typical hematologic findings.68 Minor hemorrhages (epistaxis, bleeding gums) are common, and life-threatening hemorrhages occur in 5% to 10% of cases.
The hemoglobin concentration ranges from 14 to 28 gm/dL, and the hematocrit is usually 60% or more. Sometimes chronic bleeding leads to iron deficiency, which can suppress erythropoiesis sufficiently to lower the hematocrit into the normal range, an example of two defects counteracting one another to “correct” a laboratory abnormality. The white cell count ranges from 12,000 to 50,000 cells/mm3, and the platelet count is often greater than 500,000 platelets/mm3. The platelets usually exhibit morphologic abnormalities such as giant forms and are often defective in functional aggregation studies.
Without treatment, death from bleeding or thrombosis occurs within months of diagnosis. However, simply maintaining the red cell mass at nearly normal levels by phlebotomy extends the median survival to about 10 years. JAK2 inhibitors are in preclinical development and represent a promising form of targeted therapy.
Extended survival with treatment has revealed that PCV tends to evolve to a “spent phase,” during which clinical and anatomic features of primary myelofibrosis develop. The disease undergoes this transition in about 15% to 20% of patients after an average period of 10 years. It is marked by the appearance of obliterative fibrosis in the bone marrow (myelofibrosis) and extensive extramedullary hematopoiesis, principally in the spleen, which enlarges greatly. The mechanisms underlying the progression to the spent phase are not known.
In about 2% of patients, PCV transforms to AML. Surprisingly, the AML clone often lacks JAK2 mutations,69 suggesting that JAK2 mutations are late secondary events, rather than early initiators of PCV. Unlike CML, transformation to ALL is rarely observed, consistent with the cell of origin being a progenitor committed to myeloid differentiation.
Essential Thrombocytosis
Essential thrombocytosis (ET) is often associated with activating point mutations in JAK2 (50% of cases) or MPL (5–10% of cases), a receptor tyrosine kinase that is normally activated by thrombopoietin.61 It manifests clinically with elevated platelet counts and is separated from PCV and primary myelofibrosis based on the absence of polycythemia and marrow fibrosis, respectively. In those cases without tyrosine kinase mutations, causes of reactive thrombocytosis, such as inflammatory disorders and iron deficiency, must be excluded before the diagnosis can be established.
Constitutive JAK2 or MPL signaling renders the progenitors thrombopoietin-independent and leads to hyperprolifer-ation. The JAK2 mutation is the same as that found in almost all cases of PCV. Why some patients with JAK2 mutations present with PCV and others with ET is not understood. Some cases of “ET” may in fact be PCV disguised by iron deficiency (which is more common in individuals diagnosed with ET), but this is probably true of only a small fraction of patients. Mutations in other tyrosine kinases are being sought in those cases in which JAK2 and MPL are apparently normal.
Bone marrow cellularity is usually only mildly increased, but megakaryocytes are often markedly increased in number and include abnormally large forms. Delicate reticulin fibrils are often seen, but the overt fibrosis of primary myelofibrosis (see below) is absent. Peripheral smears usually reveal abnormally large platelets (Fig. 13-36), often accompanied by mild leukocytosis. Modest degrees of extramedullary hematopoiesis may occur, producing mild organomegaly in about 50% of patients. Uncommonly, a spent phase of marrow fibrosis or transformation to AML supervenes.
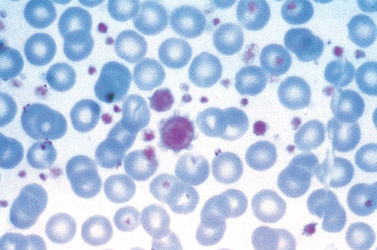
FIGURE 13-36 Essential thrombocytosis. Peripheral blood smear shows marked thrombocytosis, including giant platelets approximating the size of surrounding red cells.
The incidence of ET is 1 to 3 per 100,000 per year. It usually occurs past the age of 60 but may also be seen in young adults. Dysfunctions of platelets derived from the neoplastic clone can lead to thrombosis and hemorrhage, the major clinical manifestations. Platelets are not only increased in numbers but also frequently demonstrate qualitative abnormalities in functional tests. The types of thrombotic events resemble those observed in PCV; they include deep venous thrombosis, portal and hepatic vein thrombosis, and myocardial infarction. One characteristic symptom is erythromelalgia, a throbbing and burning of hands and feet caused by occlusion of small arterioles by platelet aggregates, which may also be seen in PCV.
ET is an indolent disorder with long asymptomatic periods punctuated by occasional thrombotic or hemorrhagic crises. Median survival times are 12 to 15 years. Thrombotic complications are most likely in patients with very high platelet counts and homozygous JAK2 mutations.67 Therapy consists of “gentle” chemotherapeutic agents that suppress thrombopoiesis.
Primary Myelofibrosis
The hallmark of primary myelofibrosis is the development of obliterative marrow fibrosis. The replacement of the marrow by fibrosis suppresses bone marrow hematopoiesis, leading to cytopenias and extensive neoplastic extramedullary hematopoiesis. Histologically, the appearance is identical to the spent phase that occurs occasionally late in the course of other myeloproliferative disorders. This similarity also extends to the underlying molecular pathogenesis.
Molecular Pathogenesis.
Activating JAK2 mutations are present in 50% to 60% of cases and activating MPL mutations in an additional 1% to 5% of cases.61 The chief pathologic feature is the extensive deposition of collagen in the marrow by non-neoplastic fibroblasts. The fibrosis inexorably displaces hematopoietic elements, including stem cells, from the marrow and eventually leads to marrow failure. It is probably caused by the inappropriate release of fibrogenic factors from neoplastic megakaryocytes. Two factors synthesized by megakaryocytes have been implicated: platelet-derived growth factor and TGF-β. As you recall, platelet-derived growth factor and TGF-β are fibroblast mitogens. In addition, TGF-β promotes collagen deposition and causes angiogenesis, both of which are observed in myelofibrosis. As marrow fibrosis progresses, circulating hematopoietic stem cells take up residence in niches in secondary hematopoietic organs, such as the spleen, the liver, and the lymph nodes, leading to the appearance of extramedullary hematopoiesis. For incompletely understood reasons, blood cell production at extramedullary sites is disordered. This factor and the concomitant suppression of marrow function result in moderate to severe cytopenias. It is not clear whether primary myelofibrosis (particularly when associated with JAK2 or MPL mutations) is truly distinct from PCV and ET, or merely reflects unusually rapid progression of these MPDs to the spent phase.61
Morphology. Early in the course, the marrow is often hypercellular due to increases in maturing cells of all lineages, a feature reminiscent of PCV. Morphologically, the erythroid and granulocytic precursors appear normal, but megakaryocytes are large, dysplastic, and abnormally clustered. At this stage fibrosis is minimal, and the blood may show leukocytosis and thrombocytosis. With progression, the marrow becomes more hypocellular and diffusely fibrotic. Clusters of atypical megakaryocytes are seen, and hematopoietic elements are often found within dilated sinusoids, which is a manifestation of severe architectural distortion cause by the fibrosis. Very late in the course, the fibrotic marrow space may be converted into bone, a change called “osteosclerosis.” These features are identical to those seen in the spent phase of other myeloproliferative disorders.
Fibrotic obliteration of the marrow space leads to extensive extramedullary hematopoiesis, principally in the spleen, which is usually markedly enlarged, sometimes up to 4000 gm. Grossly, such spleens are firm and diffusely red to gray. As in CML, subcapsular infarcts are common (see Fig. 13-40). Initially, extramedullary hematopoiesis is confined to the sinusoids, but later it expands into the cords. The liver may be enlarged moderately by sinusoidal foci of extramedullary hematopoiesis. Hematopoiesis can also appear within lymph nodes, but significant lymphadenopathy is uncommon.
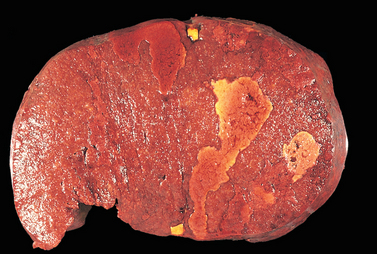
FIGURE 13-40 Splenic infarcts. Multiple well-circumscribed infarcts are present in this spleen, which is massively enlarged (2820 gm; normal: 150–200 gm) by extramedullary hematopoiesis secondary to a myeloproliferative disorder (myelofibrosis). Recent infarcts are hemorrhagic, whereas older, more fibrotic infarcts are a pale yellow-gray color.
The marrow fibrosis is reflected in several characteristic blood findings (Fig. 13-37). Marrow distortion leads to the premature release of nucleated erythroid and early granulocyte progenitors (leukoerythroblastosis), and immature cells also enter the circulation from sites of extramedullary hematopoiesis. Teardrop-shaped red cells (dacryocytes), cells that were probably damaged during the birthing process in the fibrotic marrow, are also often seen. Although characteristic of primary myelofibrosis, leukoerythroblastosis and teardrop red cells are seen in many infiltrative disorders of the marrow, including granulomatous diseases and metastatic tumors. Other common, albeit nonspecific, blood findings include abnormally large platelets and basophilia.
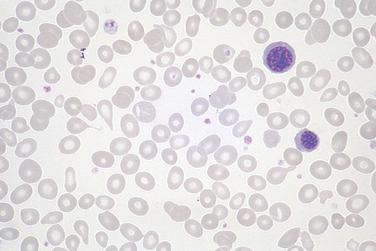
FIGURE 13-37 Primary myelofibrosis (peripheral blood smear). Two nucleated erythroid precursors and several teardrop-shaped red cells (dacryocytes) are evident. Immature myeloid cells were present in other fields. An identical picture can be seen in other diseases producing marrow distortion and fibrosis.
Clinical Features.
Primary myelofibrosis is less common than PCV and ET and usually occurs in individuals older than 60 years of age. Except when preceded by another myeloproliferative disorder, it comes to attention because of progressive anemia and splenomegaly, which produces a sensation of fullness in the left upper quadrant. Nonspecific symptoms such as fatigue, weight loss, and night sweats result from an increase in metabolism associated with the expanding mass of hematopoietic cells. Hyperuricemia and secondary gout due to a high rate of cell turnover can complicate the picture.
Laboratory studies typically show a moderate to severe normochromic normocytic anemia accompanied by leukoerythroblastosis. The white cell count is usually normal or reduced, but can be markedly elevated (80,000–100,000 cells/mm3) early in the course. The platelet count is usually normal or elevated at the time of diagnosis, but thrombocytopenia supervenes as the disease progresses. These blood findings are not specific; bone marrow biopsy is essential for diagnosis.
Primary myelofibrosis is a much more difficult disease to treat than PCV or ET. The course is variable, but the median survival is in the range of 3 to 5 years. Threats to life include intercurrent infections, thrombotic episodes, bleeding related to platelet abnormalities, and transformation to AML, which occurs in 5% to 20% of cases. When myelofibrosis is extensive, AML sometimes arises at extramedullary sites, including lymph nodes and soft tissues. Bone marrow transplantation is being used in some younger patients, and kinase inhibitors represent a future hope for targeted therapy.
LANGERHANS CELL HISTIOCYTOSIS
The term histiocytosis is an “umbrella” designation for a variety of proliferative disorders of dendritic cells or macrophages. Some, such as rare “histiocytic” lymphomas, are clearly malignant, whereas others, such as reactive proliferations of macrophages in lymph nodes, are clearly benign. Lying between these two extremes are the Langerhans cell histiocytoses, a spectrum of proliferations of a special type of immature dendritic cell called the Langerhans cell (see Chapter 6). In most instances, these proliferations are monoclonal and therefore likely to be neoplastic in origin.
Regardless of the clinical picture, the proliferating Langerhans cells have abundant, often vacuolated cytoplasm and vesicular nuclei containing linear grooves or folds (Fig. 13-38A). The presence of Birbeck granules in the cytoplasm is characteristic. Birbeck granules are pentalaminar tubules, often with a dilated terminal end producing a tennis racket–like appearance (Fig. 13-38B), which contain the protein langerin. In addition, the tumor cells also typically express HLA-DR, S-100, and CD1a.
FIGURE 13-38 Langerhans cell histiocytosis. A, Langerhans cells with folded or grooved nuclei and moderately abundant pale cytoplasm are mixed with a few eosinophils. B, An electron micrograph shows rodlike Birbeck granules with characteristic periodicity and dilated terminal end.
(B, Courtesy of Dr. George Murphy, Department of Pathology, Brigham and Women’s Hospital, Boston, MA.)
Langerhans cell histiocytosis presents as several clinicopathologic entities:
One factor that contributes to the homing of neoplastic Langerhans cells is the aberrant expression of chemokine receptors.70,71 For example, while normal epidermal Langerhans cells express CCR6, their neoplastic counterparts express both CCR6 and CCR7. This allows the neoplastic cells to migrate into tissues that express the relevant chemokines—CCL20 (a ligand for CCR6) in skin and bone, and CCL19 and 21 (ligands for CCR7) in lymphoid organs.