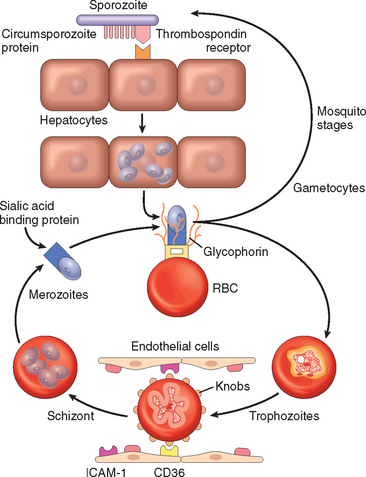
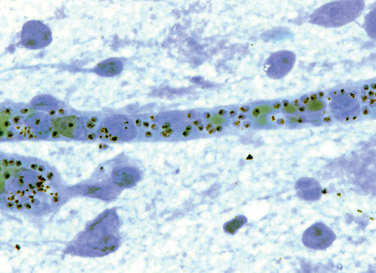
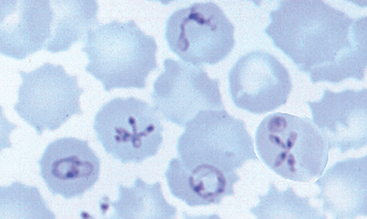
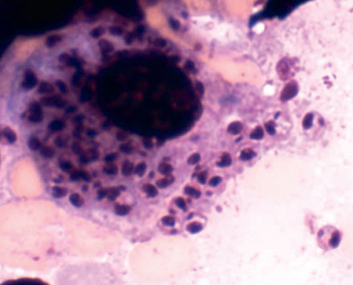
GRAM-NEGATIVE BACTERIAL INFECTIONS
Only a few gram-negative bacteria are considered in this section. A number of important gram-negative pathogens are discussed in the appropriate chapters of organ systems, including bacterial causes of gastrointestinal infections and urinary tract infections. Anaerobic gram-negative organisms are considered later in this chapter. Gram-negative bacterial infections are usually diagnosed by culture.
Neisserial Infections
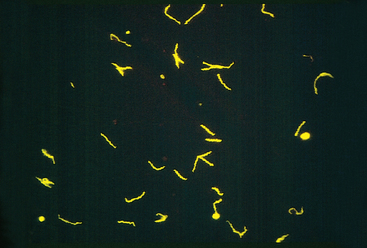
Neisseria are gram-negative diplococci that are flattened on the adjoining sides, giving the pair the shape of a coffee bean (see Fig. 8-3E). These aerobic bacteria have stringent nutritional requirements and grow best on enriched media such as lysed sheep’s blood agar (“chocolate” agar). The two clinically significant Neisseria are N. meningitidis and N. gonorrhoeae.
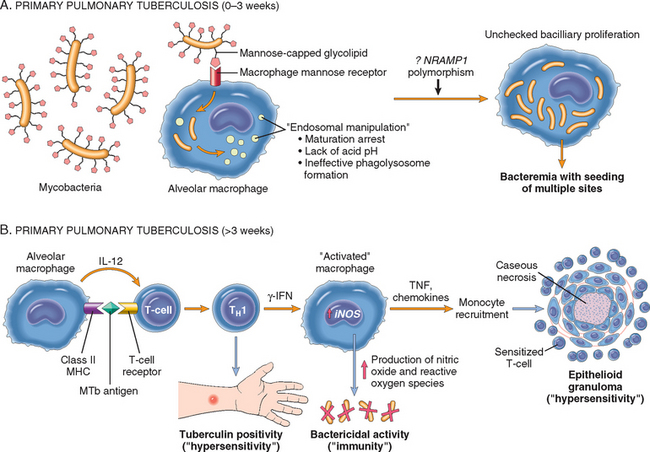
N. meningitidis is a significant cause of bacterial meningitis, particularly among children younger than 2 years of age. The organism is a common colonizer of the oropharynx and is spread by the respiratory route. Approximately 10% of the population is colonized at any one time, and each episode of colonization lasts, on average, for several months. An immune response leads to elimination of the organism in most people, and this response is protective against subsequent disease with the same serotype of bacteria. There are at least 13 serotypes of N. meningitidis. Invasive disease mainly occurs when people encounter new strains to which they are not immune, as may happen to young children or young adults living in crowded quarters such as military barracks or college dormitories. N. meningitidis disease is endemic in the United States, but epidemics occur periodically in sub-Saharan Africa and cause thousands of deaths.74
Even in the absence of pre-existing immunity, only a small fraction of people infected with N. meningitidis develop meningitis. The bacteria must invade respiratory epithelial cells and travel to the basolateral side of the cells to enter the blood.75 In the blood, the capsule of the bacteria inhibits opsonization and destruction of the bacteria by complement proteins. Despite this, the importance of complement as a first-line defense against N. meningitidis is shown by the increased rates of serious infection among people who have inherited defects in the complement proteins (C5 to C9) that form the membrane attack complex. If N. meningitidis escapes the host response, the consequences can be severe. Although antibiotic treatment of meningitis has greatly reduced mortality of N. meningitidis infection, the death rate is still about 10%. The pathology of pyogenic meningitides is discussed in Chapter 28.
N. gonorrhoeae is an important cause of sexually transmitted disease (STD), infecting about 700,000 people each year in the United States. It is second only to C. trachomatis as a bacterial causative agent of STDs. Infection in men causes urethritis. In women, N. gonorrhoeae infection is often asymptomatic and so may go unnoticed. Untreated infection can lead to pelvic inflammatory disease, which can cause infertility or ectopic pregnancy (Chapter 22). Infection is diagnosed by PCR tests, in addition to culture.
Although N. gonorrhoeae infection usually manifests locally in the genital or cervical mucosa, pharynx, or anorectum, disseminated infections may occur. Like N. meningitidis, N. gonorrhoeae is much more likely to become disseminated in people who lack the complement proteins that form the membrane attack complex. Disseminated infection of adults and adolescents usually causes septic arthritis accompanied by a rash of hemorrhagic papules and pustules. Neonatal N. gonorrhoeae infection causes blindness and, rarely, sepsis. The eye infection, which is preventable by instillation of silver nitrate or antibiotics in the newborn’s eyes, remains an important cause of blindness in some developing nations.
Pathogenesis.
Neisseria use antigenic variation as a strategy to escape the immune response. The existence of multiple serotypes of N. meningitidis results in meningitis in some people on exposure to a new strain, as discussed above. In addition, Neisseria species also generate antigenic variation by special genetic mechanisms, which permit a single bacterial clone to change its expressed antigens (see below) and escape immune defenses.19 Neisseria organisms adhere to and invade nonciliated epithelial cells at the site of entry (nasopharynx, urethra, or cervix). Two surface proteins of Neisseria, both of which bind the bacteria to host cells, undergo antigenic variation through different mechanisms. Although both N. meningitidis and N. gonorrhoeae use these mechanisms, they seem to be more important in N. gonorrhoeae.
Whooping Cough
Whooping cough, caused by the gram-negative coccobacillus Bordetella pertussis, is an acute, highly communicable illness characterized by paroxysms of violent coughing followed by a loud inspiratory “whoop.” B. pertussis vaccination, whether with killed bacteria or the newer acellular vaccine, has been effective in preventing whooping cough. Since the 1980s, however, rates of pertussis have been increasing in the United States particularly in adolescents and adults, despite continued high rates of vaccination.76 The cause of this increase is not known, but antigenic divergence of clinical strains from vaccine strains and waning immunity in young adults may play a role. In parts of the developing world, where vaccination is not widely practiced, pertussis kills hundreds of thousands of children each year. The diagnosis is best made by PCR, because culture is less sensitive.
Pathogenesis.
Bordetella pertussis colonizes the brush border of the bronchial epithelium and also invades macrophages. Coordinated expression of virulence factors is regulated by the Bordetella virulence gene locus (bvg).77 BVGS is a transmembrane protein that “senses” signals that induce expression of virulence factors. On activation, BVGS phosphorylates the protein BVGA, which regulates transcription of mRNA for adhesins and toxins. The filamentous hemagglutinin adhesin binds to carbohydrates on the surface of respiratory epithelial cells, as well as to CR3 (Mac-1) integrins on macrophages. Pertussis toxin is an exotoxin composed of five distinct proteins, including a catalytic peptide S1 that shows homology with the catalytic peptides of cholera toxin and E. coli heat-labile toxin.78 Like cholera toxin, pertussis toxin ADP-ribosylates and inactivates guanine nucleotide–binding proteins, so these G proteins no longer transduce signals from host plasma membrane receptors. The toxin produced by B. pertussis paralyzes the cilia, thus impairing an important pulmonary defense.
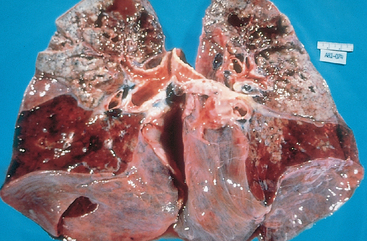
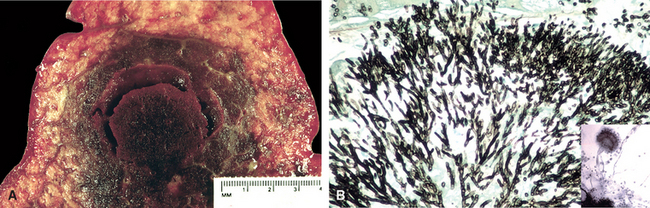
Morphology. Bordetella bacteria cause a laryngotracheobronchitis that in severe cases features bronchial mucosal erosion, hyperemia, and copious mucopurulent exudate (Fig. 8-25). Unless superinfected, the lung alveoli remain open and intact. In parallel with a striking peripheral lymphocytosis (up to 90%), there is hypercellularity and enlargement of the mucosal lymph follicles and peribronchial lymph nodes.
Pseudomonas Infection
Pseudomonas aeruginosa is an opportunistic aerobic gram-negative bacillus that is a frequent, deadly pathogen of people with cystic fibrosis, severe burns, or neutropenia.93 Many people with cystic fibrosis die of pulmonary failure secondary to chronic infection with P. aeruginosa. P. aeruginosa can be very resistant to antibiotics, making these infections difficult to treat. P. aeruginosa often infects extensive skin burns, which can be a source of sepsis. P. aeruginosa is a common cause of hospital-acquired infections; it has been cultured from washbasins, respirator tubing, nursery cribs, and even antiseptic-containing bottles. P. aeruginosa also causes corneal keratitis in wearers of contact lenses, endocarditis and osteomyelitis in intravenous drug abusers, external otitis (swimmer’s ear) in healthy individuals, and severe external otitis in diabetics.
Pathogenesis.
P. aeruginosa has pili and adherence proteins that bind to epithelial cells and lung mucin, and expresses an endotoxin that causes the symptoms and signs of gram-negative sepsis. Pseudomonas also has a number of distinctive virulence factors. In the lungs of people with cystic fibrosis, these bacteria secrete a mucoid exopolysaccharide called alginate, forming a slimy biofilm that protects bacteria from antibodies, complement, phagocytes, and antibiotics. The organisms also secrete an exotoxin and several other virulence factors. Exotoxin A, like diphtheria toxin, inhibits protein synthesis by ADP-ribosylating the ribosomal protein EF-2.80 P. aeruginosa also releases exoenzyme S, which ADPribosylates RAS and other G proteins that regulate cell growth and metabolism. The organisms also secrete a phospholipase C that lyses red cells and degrades pulmonary surfactant, and an elastase that degrades IgGs and extracellular matrix proteins. These enzymes may be important in tissue invasion and destruction of the cornea in keratitis. Finally, P. aeruginosa produces iron-containing compounds that are extremely toxic to endothelial cells and so may cause the vascular lesions that are characteristic of this infection.81
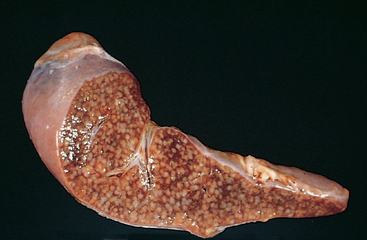
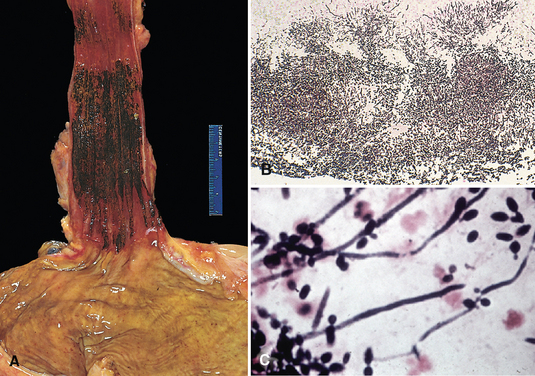
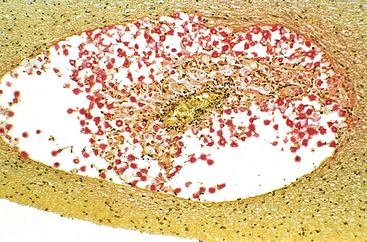
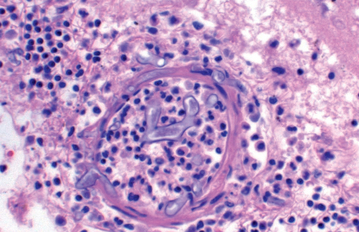
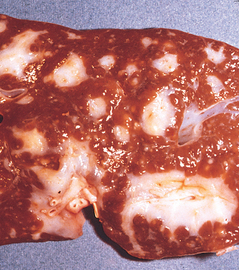
Morphology. Pseudomonas causes a necrotizing pneumonia that is distributed through the terminal airways in a fleur-de-lis pattern, with striking pale necrotic centers and red, hemorrhagic peripheral areas. On microscopic examination, masses of organisms cloud the tissue with a bluish haze, concentrating in the walls of blood vessels, where host cells undergo coagulative necrosis (Fig. 8-26). This picture of gram-negative vasculitis accompanied by thrombosis and hemorrhage, although not pathognomonic, is highly suggestive of P. aeruginosa infection.
Bronchial obstruction caused by mucus plugging and subsequent P. aeruginosa infection are frequent complications of cystic fibrosis. Despite antibiotic treatment and the host immune response, chronic P. aeruginosa infection may result in bronchiectasis and pulmonary fibrosis (Chapter 15).
In skin burns, P. aeruginosa proliferates widely, penetrating deeply into the veins and spreading hematogenously. Well-demarcated necrotic and hemorrhagic oval skin lesions, called ecthyma gangrenosum, often appear. Disseminated intravascular coagulation (DIC) is a frequent complication of bacteremia.
Plague
Yersinia pestis is a gram-negative facultative intracellular bacterium that is transmitted from rodents to humans by fleabites or, less often, from one human to another by aerosols. It causes an invasive, frequently fatal infection called plague. Plague, also named Black Death, caused three great pandemics that killed an estimated 100 million people in Egypt and Byzantium in the sixth century; one quarter of Europe’s population in the fourteenth and fifteenth centuries; and tens of millions in India, Myanmar, and China at the beginning of the twentieth century. Currently, 1000 to 3000 cases of plague occur each year worldwide. Wild rodents in the rural western United States are infected with Y. pestis, and 10 to 15 human cases occur per year. Y. enterocolitica and Y. pseudotuberculosis are genetically similar to Y. pestis; these bacteria cause fecal-orally transmitted ileitis and mesenteric lymphadenitis.
Pathogenic Yersinia proliferate within lymphoid tissue. These organisms have a complex of genes, called the Yop virulon, which enable the bacteria to kill host phagocytes.82 The Yop virulon encodes proteins that assemble into a type III secretion system, which is a hollow syringe-like structure that projects from the bacterial surface, binds to host cells, and injects bacterial toxins, called Yops (Yersinia outercoat proteins), into the cell. YopE, YopH, and YopT block phagocytosis by inactivating molecules that regulate actin polymerization. YopJ inhibits the signaling pathways that are activated by LPS, blocking the production of inflammatory cytokines. Y. pestis ensures its own spread by forming a biofilm that obstructs the gut of the infected flea. The flea must regurgitate before it feeds, and thus infects the rodent or human that it is biting.
Morphology. Yersinia pestis causes lymph node enlargement (buboes), pneumonia, or sepsis with a striking neutrophilia. The distinctive histologic features include (1) massive proliferation of the organisms, (2) early appearance of protein-rich and polysaccharide-rich effusions with few inflammatory cells but with marked tissue swelling, (3) necrosis of tissues and blood vessels with hemorrhage and thrombosis, and (4) neutrophilic infiltrates that accumulate adjacent to necrotic areas as healing begins.
In bubonic plague the infected fleabite is usually on the legs and is marked by a small pustule or ulcer. The draining lymph nodes enlarge dramatically within a few days and become soft, pulpy, and plum colored, and may infarct or rupture through the skin. In pneumonic plague there is a severe, confluent, hemorrhagic and necrotizing bronchopneumonia, often with fibrinous pleuritis. In septicemic plague lymph nodes throughout the body as well as organs rich in mononuclear phagocytes develop foci of necrosis. Fulminant bacteremias also induce DIC with widespread hemorrhages and thrombi.
Chancroid (Soft Chancre)
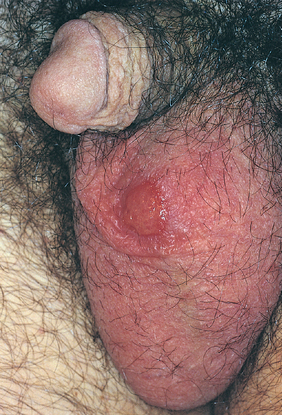
Chancroid is an acute, sexually transmitted, ulcerative infection caused by Hemophilus ducreyi.83 The disease is most common in tropical and subtropical areas among lower socioeconomic groups and men who have regular contact with prostitutes. Chancroid is one of the most common causes of genital ulcers in Africa and Southeast Asia, where it probably serves as an important cofactor in the transmission of HIV infection. Chancroid is uncommon in the United States, with 20 to 50 cases per year reported to the CDC in the past several years. The organism must be cultured in special conditions and PCR-based tests are not widely available, so chancroid may be underdiagnosed.
Morphology. Four to seven days after inoculation the person develops a tender, erythematous papule involving the external genitalia. In males the primary lesion is usually on the penis; in females most lesions occur in the vagina or the periurethral area. Over the course of several days the surface of the primary lesion erodes to produce an irregular ulcer, which is more apt to be painful in males than in females. In contrast to the primary chancre of syphilis, the ulcer of chancroid is not indurated, and multiple lesions may be present. The base of the ulcer is covered by shaggy, yellow-gray exudate. The regional lymph nodes, particularly in the inguinal region, become enlarged and tender in about 50% of cases within 1 to 2 weeks of the primary inoculation. In untreated cases the inflamed and enlarged nodes (buboes) may erode the overlying skin to produce chronic, draining ulcers.
Microscopically, the ulcer of chancroid contains a superficial zone of neutrophilic debris and fibrin, with an underlying zone of granulation tissue containing areas of necrosis and thrombosed vessels. A dense, lymphoplasmacytic inflammatory infiltrate is present beneath the layer of granulation tissue. Coccobacilli are sometimes demonstrable in Gram or silver stains, but they are often obscured by other bacteria that colonize the ulcer base.
Granuloma Inguinale
Granuloma inguinale, or donovanosis, is a chronic inflammatory disease caused by Klebsiella granulomatis (formerly called Calymmatobacterium donovani), a minute, encapsulated, coccobacillus. The organism is sexually transmitted. Granuloma inguinale is uncommon in the United States and western Europe but is endemic in rural areas in certain tropical and subtropical regions. Untreated cases are characterized by the development of extensive scarring, often associated with lymphatic obstruction and lymphedema (elephantiasis) of the external genitalia. Culture of the organism is difficult, and PCR assays are still in development, so the diagnosis is made by microscopic examination of smears or biopsy samples of the ulcer.
Morphology. Granuloma inguinale begins as a raised, papular lesion on the moist, stratified squamous epithelium of the genitalia or, rarely, the oral mucosa or pharynx. The lesion eventually ulcerates and develops abundant granulation tissue, which is manifested grossly as a protuberant, soft, painless mass. As the lesion enlarges, its borders become raised and indurated. Disfiguring scars may develop in untreated cases and are sometimes associated with urethral, vulvar, or anal strictures. Regional lymph nodes typically are spared or show only nonspecific reactive changes, in contrast to chancroid.
Microscopic examination of active lesions reveals marked epithelial hyperplasia at the borders of the ulcer, sometimes mimicking carcinoma (pseudoepitheliomatous hyperplasia). A mixture of neutrophils and mononuclear inflammatory cells is present at the base of the ulcer and beneath the surrounding epithelium. The organisms are demonstrable in Giemsa-stained smears of the exudate as minute, encapsulated coccobacilli (Donovan bodies) in macrophages. Silver stains (e.g., the Warthin-Starry stain) may also be used to demonstrate the organism.