Chapter 12 The Heart
The human heart is a remarkably efficient, durable, and reliable pump that propels over 6000 liters of blood through the body daily and beats more than 40 million times a year, thereby providing the tissues with a steady supply of vital nutrients and facilitating the excretion of waste products. As might be anticipated, cardiac dysfunction can be associated with devastating physiologic consequences. Cardiovascular disease is the number one cause of death worldwide, with about 80% of the burden occurring in developing countries.1,2 In the United States, heart disease accounts for nearly 40% of all postnatal deaths, totaling about 750,000 individuals annually; this is nearly 1.5 times the number of deaths caused by all forms of cancer combined. It is estimated that one third of Americans have one or more types of cardiovascular disease. Moreover, 32% of heart disease deaths are “premature,” occurring in individuals younger than age 75.3 If all major forms of cardiovascular disease were eliminated, life expectancy would increase by 7 years. The yearly economic burden of ischemic heart disease, the most prevalent subgroup, is estimated to be in excess of $100 billion in the United States.
The major categories of cardiac diseases considered in this chapter include congenital heart abnormalities, ischemic heart disease, heart disease caused by systemic hypertension, heart disease caused by pulmonary diseases (cor pulmonale), diseases of the cardiac valves, and primary myocardial diseases. A few comments about pericardial diseases and cardiac neoplasms as well as cardiac transplantation are also offered. Before considering details of specific conditions, we will briefly review the anatomy of the normal heart, because many diseases cause changes in the size and appearance of one or more of its components. We will also discuss the principles of cardiac hypertrophy and failure, the common end points of many different types of heart disease; these are essential for later discussion of disease processes.
Cardiac Structure and Specializations
Heart weight varies with body height and weight; it normally averages approximately 250 to 300 gm in females and 300 to 350 gm in males, or roughly 0.4% to 0.5% of body weight. The usual thickness of the free wall of the right ventricle is 0.3 to 0.5 cm, and that of the left ventricle 1.3 to 1.5 cm. Increases in cardiac size and weight accompany many forms of heart disease. Greater heart weight or ventricular thickness indicates hypertrophy, and an enlarged chamber size implies dilation. An increase in cardiac weight or size or both (resulting from hypertrophy and/or dilation) is termed cardiomegaly.
The efficient pumping of blood by the heart to the entire body requires the normal function of each of its key components, the myocardium, valves, conduction system, and coronary arterial circulation.
MYOCARDIUM
The pumping function of the heart is accomplished by the cardiac muscle, the myocardium, composed primarily of a collection of specialized muscle cells called cardiac myocytes. Ventricular myocytes are arranged circumferentially in a spiral orientation and contract during systole and relax during diastole. The contractile unit is the sarcomere, an orderly arrangement of thick filaments composed principally of myosin, thin filaments containing actin, and regulatory proteins such as troponin and tropomyosin. Cardiac muscle cells contain strings of sarcomeres in series, which are responsible for the striated appearance of these cells. Contraction depends on a coordinated ratcheting mechanism whereby each myosin filament pulls the neighboring actin filaments toward the center of the sarcomere, leading to the shortening of the myocyte. The amount of force generated is determined by the distance each sarcomere shortens. Moderate ventricular dilation during diastole increases the extent of sarcomere shortening and the force of contraction during systole. With further dilation, however, there is a point at which effective overlap of the actin and myosin filaments is reduced and the force of contraction decreases sharply, as occurs in heart failure.
Atrial myocytes are generally smaller and arranged more haphazardly than their ventricular counterparts. Some atrial cells have distinctive electron-dense granules in the cytoplasm called specific atrial granules; these are the storage sites of atrial natriuretic peptide. Atrial natriuretic peptide can produce a variety of physiologic effects, including vasodilation, natriuresis, and diuresis, actions beneficial in pathologic states such as hypertension and congestive heart failure.4
Functional integration of cardiac myocytes is mediated by structures called intercalated discs, which link individual cells and contain specialized intercellular junctions that permit both mechanical and electrical (ionic) coupling. Within the intercalated discs, gap junctions facilitate synchronous myocyte contraction through electrical coupling by permitting relatively unrestricted passage of ions across the membranes of adjoining cells. Abnormalities in the spatial distribution of gap junctions and their respective proteins in ischemic and myocardial heart disease may contribute to electromechanical dysfunction (arrhythmia) and heart failure.5
VALVES
The four cardiac valves (tricuspid, pulmonary, mitral, and aortic) maintain unidirectional blood flow through the heart. Their function depends on the mobility, pliability, and structural integrity of their delicate flaps, called leaflets (in the tricuspid and mitral valves) or cusps (in the aortic and pulmonary valves, also known as the semilunar valves). All four valves have a similar, layered architecture: a dense collagenous core (fibrosa) close to the outflow surface and continuous with valvular supporting structures, a central core of loose connective tissue (spongiosa), a layer rich in elastin (ventricularis or atrialis depending on which chamber it faces) below the inflow surface, and an endothelial covering. The collagen is responsible for the mechanical integrity of a valve. The valve is populated throughout by interstitial cells, which produce and continuously repair the extracellular matrix (especially collagen), allowing the valve to respond and adapt to changing mechanical conditions.6,7
The function of the semilunar valves depends on the integrity and coordinated movements of the cuspal attachments. Thus, dilation of the aortic root can hinder coaptation of the aortic valve cusps during closure, yielding regurgitation. In contrast, the competence of the atrioventricular valves depends on not only the leaflets and their attachments, but also on tendinous connections to the papillary muscles of the ventricular wall. Left ventricular dilation, a ruptured tendon, or papillary muscle dysfunction can all interfere with mitral valve closure, leading to regurgitation.
Because they are thin enough to be nourished by diffusion from the heart’s blood, normal leaflets and cusps have only scant blood vessels limited to the proximal portion. Pathologic changes of valves are largely of three types: damage to collagen that weakens the leaflets, exemplified by mitral valve prolapse; nodular calcification beginning in interstitial cells, as in calcific aortic stenosis; and fibrotic thickening, the key feature in rheumatic heart disease (see later).
CONDUCTION SYSTEM
Coordinated contraction of the cardiac muscle depends on propagation of electrical impulses, which is accomplished by specialized excitatory and conducting myocytes within the cardiac conduction system that regulate heart rate and rhythm. Key components of the conduction system include (1) the sinoatrial (SA) pacemaker of the heart, the SA node, located near the junction of the right atrial appendage and the superior vena cava; (2) the AV node, located in the right atrium along the atrial septum; (3) the bundle of His, which courses from the right atrium to the summit of the ventricular septum; and its major divisions (4) the right and left bundle branches, which further arborize in the respective ventricles through the anterior-superior and posterior-inferior divisions of the left bundle and the Purkinje network. The cells of the specialized cardiac conduction system depolarize spontaneously, enabling them to function as cardiac pacemakers. Because the normal rate of spontaneous depolarization in the SA node (60 to 100 beats/minute) is faster than the other components, it normally sets the pace. The AV node serves as a kind of “gatekeeper”; by delaying the transmission of signals from the atria to the ventricles, it ensures that atria contraction precedes ventricular contraction.
The autonomic nervous system (the same part of the nervous system involved in blood pressure control) controls the rate of firing of the SA node to trigger the start of the cardiac cycle. Autonomic inputs can increase the heart rate to twice normal within only 3 to 5 seconds, and are important in cardiac responses to exercise or other states associated with increased oxygen demand.
BLOOD SUPPLY
To meet their energy needs, cardiac myocytes rely almost exclusively on oxidative phosphorylation, which is manifest by the abundant mitochondria that are found in these cells.5 Oxydative phosphorylation requires oxygen, making cardiac myocytes extremely vulnerable to ischemia. A constant supply of oxygenated blood is thus essential for cardiac function. Most of the myocardium depends on nutrients and oxygen delivered via the the coronary arteries, which arise immediately distal to the aortic valve, initially running along the external surface of the heart (epicardial coronary arteries) and then penetrating the myocardium (intramural arteries). These small penetrating arteries yield arterioles and, ultimately, provide a rich network of capillaries enveloping individual cardiac muscle cells.
The three major epicardial coronary arteries are (1) the left anterior descending (LAD) and (2) the left circumflex (LCX) arteries, both arising from branches of the left (main) coronary artery, and (3) the right coronary artery. Branches of the LAD are called “diagonal” and “septal perforators,” and those of the LCX are “obtuse marginals.” Most coronary arterial blood flow to the myocardium occurs during ventricular diastole, when the microcirculation is not compressed by cardiac contraction.
There are a number of normal variations on the anatomy of the coronary arteries, which determine the areas of myocardium that are “at risk” in coronary artery disease and are of great practical importance to the heart surgeon and the invasive cardiologist; these are discussed later.
Effects of Aging on the Heart
The number of individuals aged 65 years and older will approximately double from 2000 to 2050 (from 35 million to 79 million in the United States). Considering this, one can see that knowledge of changes that occur in the cardiovascular system with aging will become increasingly important. Changes associated with aging can affect the pericardium, cardiac chambers, valves, coronary arteries, conduction system, myocardium, and aorta (Table 12-1).
TABLE 12-1 Changes in the Aging Heart
| CHAMBERS |
| VALVES |
| EPICARDIAL CORONARY ARTERIES |
| MYOCARDIUM |
| AORTA |
With advancing age, the amount of epicardial fat increases, particularly over the anterior surface of the right ventricle and in the atrial septum. A reduction in the size of the left ventricular cavity, particularly in the base-to-apex dimension, occurs with aging and may be exacerbated by systemic hypertension and sometimes by bulging of the basal ventricular septum into the left ventricular outflow tract (termed sigmoid septum). These changes in the left ventricular cavity can produce a functional outflow obstruction similar to that seen in hypertrophic cardiomyopathy, discussed later in this chapter.
Valvular aging changes include calcification of the mitral annulus and aortic valve, the latter frequently leading to aortic stenosis. In addition, the valves can develop fibrous thickening, and the mitral leaflets tend to buckle back toward the left atrium during ventricular systole, simulating a prolapsing (myxomatous) mitral valve. Moreover, many older persons develop small filiform processes (Lambl excrescences) on the closure lines of aortic and mitral valves, probably resulting from the organization of small thrombi.
Compared with younger myocardium, “elderly” myocardium has fewer myocytes, increased collagenized connective tissue and, in some individuals, deposition of amyloid. Lipofuscin deposits (Chapter 1) and basophilic degeneration, an accumulation within cardiac myocytes of a gray-blue by-product of glycogen metabolism, may also be present. Extensive lipofuscin deposition in a small, atrophied heart is called brown atrophy; this change often accompanies cachexia, as seen in terminal cancer.
Heart Disease: Overview of Pathophysiology
Although many diseases can involve the heart and blood vessels,8,9 cardiovascular dysfunction results from one or more of six principal mechanisms, most with detectable morphologic manifestations:
Most cardiovascular disease results from a complex interplay of genetics and environmental factors that disrupt networks of genes and signaling pathways that control morphogenesis, myocyte survival and response to injury, biomechanical stress responses, contractility, or electrical conduction.10 For example, the pathogenesis of many congenital heart defects involves an underlying genetic abnormality whose expression is modified by environmental or maternal factors (see later). Moreover, genes that control the development of the heart may also regulate the response of the heart to aging or to various types of injuries and stresses. As we will discuss, certain types of adult-onset heart disease have a predominantly genetic basis, and it is suspected that genetic polymorphisms in the same genes (or other genes in the same pathways) are likely to modify the risk of many forms of heart disease. These genetic discoveries are providing new insights into the molecular causes of heart disease and are likely to increasingly become part of its diagnosis and classification.
Heart Failure
Heart failure, often called congestive heart failure (CHF), is a common, usually progressive condition with a poor prognosis. Each year in the United States, CHF affects nearly 5 million individuals (approximately 2% of the population), necessitates over 1 million hospitalizations, and is the primary or contributing cause of death of an estimated 300,000 people. It is the leading discharge diagnosis in patients over 65 years of age in the United States and has an associated annual cost of $18 billion.
CHF occurs when the heart is unable to pump blood at a rate sufficient to meet the metabolic demands of the tissues or can do so only at an elevated filling pressure. It can appear during the end stage of many forms of chronic heart disease. In this setting, it most often develops insidiously due to the cumulative effects of chronic work overload (such as in valve disease or hypertension) or ischemic heart disease (e.g., following myocardial infarction with extensive heart damage). However, acute hemodynamic stresses, such as fluid overload, acute valvular dysfunction, or a large myocardial infarction, can cause CHF to appear suddenly.
When cardiac function is impaired or the work load increases, several physiologic mechanisms maintain arterial pressure and perfusion of vital organs. The most important of these are the following:
These adaptive mechanisms may be adequate to maintain normal cardiac output in the face of heart disease, but their capacity to do so may ultimately be overwhelmed. Moreover, superimposed pathologic changes, such as myocyte apoptosis, cytoskeletal alterations, and the deposition of extracellular matrix may cause further structural and functional disturbances. Most frequently, heart failure results from progressive deterioration of myocardial contractile function (systolic dysfunction); this may be attributable to ischemic injury, pressure or volume overload due to valvular disease or hypertension, or dilated cardiomyopathy. Sometimes, however, failure results from an inability of the heart chamber to expand and fill sufficiently during diastole (diastolic dysfunction), as can occur with massive left ventricular hypertrophy, myocardial fibrosis, deposition of amyloid, or constrictive pericarditis (see below).12
CARDIAC HYPERTROPHY: PATHOPHYSIOLOGY AND PROGRESSION TO FAILURE
Increased mechanical work due to pressure or volume overload (e.g., systemic hypertension or aortic stenosis), or trophic signals (e.g., those mediated through the activation of β-adrenergic receptors) causes myocytes to increase in size (hypertrophy); cumulatively, this causes an increase in the size and weight of the heart (Fig. 12-1). Hypertrophy is dependent upon increased protein synthesis, which enables the assembly of additional sarcomeres. Hypertrophic myocytes also contain increased numbers of mitochondria and have enlarged nuclei. The latter alteration appears to be due to increases in DNA ploidy, which result from DNA replication in the absence of cell division. The pattern of hypertrophy reflects the nature of the stimulus. In response to increases in pressure (e.g., hypertension or aortic stenosis), ventricles develop pressure-overload hypertrophy, which usually causes a concentric increase in wall thickness. In pressure overload, new sarcomeres are predominantly assembled in parallel to the long axes of cells, expanding the cross-sectional area of myocytes. In contrast, volume-overload hypertrophy is characterized by ventricular dilation. This is because the new sarcomeres assembled in response to volume overload are largely positioned in series with existing sacromeres. As a result, in dilation due to volume overload the wall thickness may be increased, normal, or less than normal; thus, heart weight, rather than wall thickness, is the best measure of hypertophy in volume overloaded hearts.
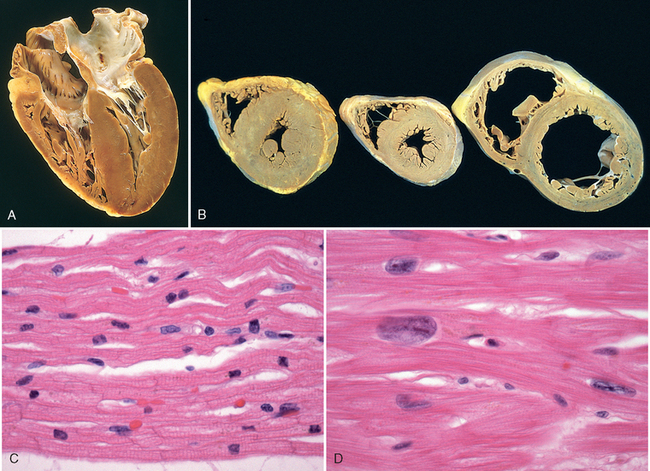
FIGURE 12-1 Left ventricular hypertrophy. A, Pressure hypertrophy due to left ventricular outflow obstruction. The left ventricle is on the lower right in this apical four-chamber view of the heart. B, Left ventricular hypertrophy with and without dilation, viewed in transverse heart sections. Compared with a normal heart (center), the pressure-hypertrophied hearts (left and in A) have increased mass and a thick left ventricular wall, while the hypertrophied, dilated heart (right) has increased mass and a normal wall thickness. C, Normal myocardium. D, Hypertrophied myocardium. Note the increases in both cell size and nuclear size in the hypertrophied myocytes.
(A,B, Reproduced by permission from Edwards WD: Cardiac anatomy and examination of cardiac specimens. In Emmanouilides GC et al. (eds): Moss and Adams Heart Disease in Infants, Children, and Adolescents: Including the Fetus and Young Adults, 5th ed. Philadelphia, Williams & Wilkins, 1995, p 86.)
Cardiac hypertrophy can be substantial in clinical heart disease. Heart weights of two to three times normal are common in patients with systemic hypertension, ischemic heart disease, aortic stenosis, mitral regurgitation, or dilated cardiomyopathy, and heart weights can be threefold to fourfold greater than normal in those with aortic regurgitation or hypertrophic cardiomyopathy.
Important changes at the tissue and cell level occur with cardiac hypertrophy. The increase in myocyte size is not accompanied by a proportional increase in capillary numbers. As a result, the supply of oxygen and nutrients to the hypertrophied heart, particularly one undergoing pressure overload hypertrophy, is more tenuous than in the normal heart. At the same time, oxygen consumption by the hypertrophied heart is elevated due to the increased workload that drives the process. Hypertrophy is also often accompanied by deposition of fibrous tissue. Molecular changes include the expression of immediate-early genes (e.g., c-fos, c-myc, c-jun, and EGR1) (Chapter 1).13 With prolonged hemodynamic overload, there may be a shift to a gene expression pattern resembling that seen during fetal cardiac development (including selective expression of embryonic/fetal forms of β-myosin heavy chain, natriuretic peptides, and collagen).
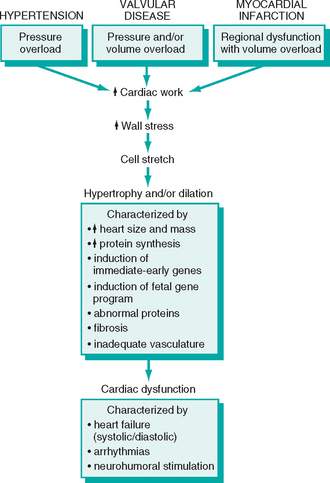
At a functional level, cardiac hypertrophy is associated with heightened metabolic demands due to increases in wall tension, heart rate, and contractility (inotropic state, or force of contraction), all of which increase cardiac oxygen consumption. As a result of these changes, the hypertrophied heart is vulnerable to decompensation, which can evolve to cardiac failure and eventually lead to death.14 The proposed sequence of initially beneficial, and later harmful, events in response to increased cardiac work is summarized in Figure 12-2. The molecular and cellular changes in hypertrophied hearts that initially mediate enhanced function may themselves contribute to the development of heart failure. This can occur through (1) abnormal myocardial metabolism,15,16 (2) alterations of intracellular handling of calcium ions, (3) apoptosis of myocytes, and (4) reprogramming of gene expression.17,18 The latter appears to occur in part through changes in expression of miRNAs, small noncoding RNAs that inhibit the expression of proteins at the level of mRNA stability or translation (Chapter 5). Cardiac hypertrophy is associated with down-regulation of miR-208 and upregulation of miR-195; of interest, enforced over-expression of miR-195 can produce cardiac hypertrophy and dilation in the mouse, whereas over-expression of miR-208 is protective even in the setting of pressure overload, suggesting a cause and effect relationship.
The degree of structural abnormality of the heart in CHF does not always reflect the level of dysfunction, and the structural, biochemical, and molecular basis for myocardial contractile failure can be obscure. Indeed, it may be impossible from morphologic examination to distinguish a damaged but functional heart from one that has failed. At autopsy, the hearts of patients with CHF are generally heavy, dilated, and thin-walled and exhibit microscopic evidence of hypertrophy, but the extent of these changes is extremely variable. In myocardial infarction, loss of pumping capacity due to myocyte death leads to work-related hypertrophy of the surrounding viable myocardium. In valvular heart disease, the increased pressure or volume overloads the myocardium globally.
Increased heart mass is correlated with excess cardiac mortality and morbidity; indeed, cardiomegaly is an independent risk factor for sudden death.19 In contrast to pathologic hypertrophy (which is often associated with contractile impairment), hypertrophy induced by regular strenuous exercise has varied effects on the heart depending on the type of exercise. Aerobic exercise (e.g., long distance running) tends to be associated with volume-load hypertrophy that may be accompanied by increases in capillary density (unlike other forms of hypertrophy) and decreases in resting heart rate and blood pressure, effects that are all beneficial. These changes are sometimes referred to as physiologic hypertrophy. Static exercise (e.g., weight lifting) is associated with pressure hypertrophy and appears more likely to be associated with deleterious changes.
Whatever its basis, CHF is characterized by variable degrees of decreased cardiac output and tissue perfusion (sometimes called forward failure), as well as pooling of blood in the venous system (backward failure); the latter may cause pulmonary edema, peripheral edema, or both. Thus, many of the significant clinical features and morphologic changes noted in CHF are secondary to injuries induced by hypoxia and congestion in tissues distant from the heart.
The cardiovascular system is a closed circuit. Thus, although left-sided and right-sided failure can occur independently, failure of one side (particularly the left) often produces excessive strain on the other, terminating in global heart failure. Despite this interdependency, it is easiest to understand the pathology of heart failure by considering right- and left-sided heart failure separately.
LEFT-SIDED HEART FAILURE
Left-sided heart failure is most often caused by (1) ischemic heart disease, (2) hypertension, (3) aortic and mitral valvular diseases, and (4) myocardial diseases. The morphologic and clinical effects of left-sided CHF primarily result from congestion of the pulmonary circulation, stasis of blood in the left-sided chambers, and hypoperfusion of tissues leading to organ dysfunction.
Heart. The findings in the heart vary depending on the disease process; gross structural abnormalities such as myocardial infarcts or a deformed, stenotic, or regurgitant valve may be present. Except for failure caused by mitral valve stenosis or unusual restrictive cardiomyopathies (described later), the left ventricle is usually hypertrophied and often dilated, sometimes massively. The microscopic changes are non-specific, consisting mainly of myocyte hypertrophy and variable degrees of interstitial fibrosis. The impaired left ventricular function usually causes dilation of the left atrium and increases the risk of atrial fibrillation. This in turn results in stasis, particularly in the atrial appendage, which is a common site of thrombus formation.
Lungs. Pulmonary congestion and edema produce heavy, wet lungs, as described elsewhere (Chapters 4 and 15. Pulmonary changes include, in sequence from mildest to most severe, the following: (1) perivascular and interstitial edema, particularly in the interlobular septa, which is responsible for the characteristic Kerley B lines noted on chest roentgenogram; (2) progressive edematous widening of alveolar septa; and (3) accumulation of edema fluid in the alveolar spaces. Some red cells extravasate into the edema fluid within the alveolar spaces, where they are phagocytosed and digested by macrophages, which store the iron recovered from hemoglobin in the form of hemosiderin. These hemosiderin-laden macrophages are telltale signs of previous episodes of pulmonary edema and are often referred to as heart failure cells.
Clinically, in early left-sided heart failure symptoms may be quite subtle and are often related to pulmonary congestion and edema. Cough and dyspnea (breathlessness), initially with exertion and later at rest, are two of the earliest complaints. As failure progresses, worsening pulmonary edema may lead to orthopnea (dyspnea when lying down that is relieved by standing), requiring the patient to sleep in an upright position; or paroxysmal nocturnal dyspnea, a form of dyspnea usually occurring at night that is so severe that it induces a feeling of suffocation. Particularly in the setting of atrial fibrillation, an arrhythmia characterized by uncoordinated, chaotic contraction of the atrium, stasis greatly increases the risk of thrombosis and thomboembolic stroke.20
Decreased cardiac output causes a reduction in renal perfusion, which leads to the activation of the renin-angiotensin-aldosterone system. This in turn induces the retention of salt and water and the expansion of the interstitial and intravascular fluid volumes (Chapters 4 and 11, compensatory effects that can contribute to or exacerbate pulmonary edema. If the hypoperfusion of the kidney becomes sufficiently severe, impaired excretion of nitrogenous products may cause azotemia (called prerenal azotemia because of its vascular origin; Chapter 20). In far-advanced CHF, cerebral hypoxia can give rise to hypoxic encephalopathy (Chapter 28), with irritability, loss of attention span, and restlessness. In end-stage CHF, this can even progress to stupor and coma.
Left-sided heart failure can be divided on clinical grounds into systolic and diastolic failure. Systolic failure is defined by insufficient cardiac output (pump failure), and can thus be caused by any of the many disorders that damage or derange the contractile function of the left ventricle. In diastolic failure, cardiac output is relatively preserved at rest, but the left ventricle is abnormally stiff or otherwise restricted in its ability to relax during diastole. As a result, the heart is unable to increase its output in response to increases in the metabolic demands of peripheral tissues (e.g., during exercise). Moreover, because the left ventricle cannot expand normally, any increase in filling pressure is immediately referred back to the pulmonary circulation, producing rapid onset pulmonary edema (sometimes referred to as flash pulmonary edema), which may be severe. Diastolic failure predominantly occurs in patients over the age of 65 and for unclear reasons is more common in women. Hypertension is the most common underlying etiology. Other risk factors include diabetes mellitus, obesity, and bilateral renal artery stenosis. The reduction in the ability of the left ventricle to relax and fill may stem from myocardial fibrosis (such as occurs in cardiomyopathies and ischemic heart disease), infiltrative disorders associated with restrictive cardiomyopathies (e.g., cardiac amyloidosis), and restrictive pericarditis. Diastolic failure may also appear in elderly patients without any known predisposing factors, possibly as an exaggeration of the normal stiffening of the heart with age, as discussed previously.
RIGHT-SIDED HEART FAILURE
Most commonly, right-sided heart failure is caused by left-sided heart failure, as any increase in pressure in the pulmonary circulation incidental to left-sided failure inevitably burdens the right side of the heart. The causes of right-sided heart failure must then include all of those that induce left-sided heart failure. Pure right-sided heart failure is infrequent and usually occurs in patients with any one of a variety of disorders affecting the lungs; hence, it is often referred to as cor pulmonale. Cor pulmonale is most commonly associated with parenchymal diseases of the lung, but can also arise secondary to disorders that affect the pulmonary vasculature (e.g., primary pulmonary hypertension (Chapter 15), recurrent pulmonary thomboembolism (Chapter 4)), or that merely produce hypoxia (e.g., chronic sleep apnea, altitude sickness), with its associated pulmonary vasoconstriction. The common feature of these diverse disorders is pulmonary hypertension (discussed later), which results in hypertrophy and dilation of the right side of the heart. In extreme cases, leftward bulging of the ventricular septum can cause left ventricular dysfunction. The major morphologic and clinical effects of right-sided heart failure differ from those of left-sided heart failure in that pulmonary congestion is minimal, whereas engorgement of the systemic and portal venous systems may be pronounced.
Heart. As in left-heart failure, the morphology varies with cause. Rarely, structural defects such as valvular abnormalities or endocardial fibrosis (as in carcinoid heart disease) may be present. However, since isolated right heart failure is most often caused by lung disease, in a vast majority of cases the only findings are hypertrophy and dilation of the right atrium and ventricle.
Liver and Portal System. Congestion of the hepatic and portal vessels may produce pathologic changes in the liver, the spleen, and the gut. The liver is usually increased in size and weight (congestive hepatomegaly) due to prominent passive congestion (Chapter 4). Congestion is most prominent around central veins within hepatic lobules, which show red-brown centrilobular discoloration and paler, sometimes fatty, peripheral regions; this combination produces a characteristic appearance that is referred to as “nutmeg liver” (Chapter 4). In some instances, especially when left-sided heart failure is also present, severe central hypoxia produces centrilobular necrosis. With longstanding severe right-sided heart failure, the central areas can become fibrotic, creating so-called cardiac sclerosis and, in extreme case, cardiac cirrhosis (Chapter 18). Portal hypertension produces enlargement of the spleen (congestive splenomegaly), which often weighs from 300 to 500 gm (normal, <150 gm); it can also contribute to chronic congestion and edema of the bowel wall, which may be so severe as to interfere with the absorption of nutrients.
Pleural, Pericardial, and Peritoneal Spaces. Systemic venous congestion can lead to accumulation of fluid in the pleural, pericardial, or peritoneal spaces (effusions). Thus, pulmonary edema and pleural effusions are associated with left-sided heart failure. Large pleural effusions (over 1 liter) can cause portions of the corresponding lung to be poorly inflated (atelectasis). In addition, transudation of fluid into the peritoneal cavity may give rise to ascites.
Subcutaneous Tissues. Edema of the peripheral and dependent portions of the body, especially ankle (pedal) and pretibial edema, is a hallmark of right-sided heart filure. In chronically bedridden patients presacral edema may predominate. Generalized massive edema (anasarca) may also occur.
The clinical features of isolated right-sided heart failure are those related to systemic (and portal) venous congestion, and include hepatosplenomegaly, peripheral edema, pleural effusions, and ascites. Organs that are prominently affected in right-sided heart failure include the kidney and the brain. Congestion of the kidneys is more marked with right-sided than left-sided heart failure, leading to greater fluid retention and peripheral edema, and more pronounced azotemia. Venous congestion and hypoxia of the central nervous system can produce deficits of mental function that are essentially identical to those described in left-sided heart failure.
Although we have discussed right and left heart failure separately, it is again worth emphasizing that in many cases of chronic cardiac decompensation, the patient presents in biventricular CHF with symptoms that encompass the clinical syndromes of both right-sided and left-sided heart failure. Standard therapy for CHF relies mainly on pharmacologic approaches. Drugs that relieve fluid overload (e.g., diuretics), that block the renin-angiotensin-aldosterone axis (e.g., angiotensin converting enzyme inhibitors), and that lower adrenergic tone (e.g., β1-adrenergic blockers) are particularly useful. The efficacy of the latter two classes of drugs supports the idea that the neurohumoral changes that are seen in CHF (including elevated circulating levels of norepinephrine and renin) are maladaptive and contribute to heart failure. Newer approaches to improving cardiac function include devices that provide the heart with a mechanical assist, and resynchronization of electrical impulses to maximize cardiac efficiency. Because of the prevalence and severity of CHF, there is considerable interest in novel therapies, including cell-based approaches.21 Of note in this regard, a growing body of evidence indicates that the adult heart may have limited capacity for stem cell–mediated self-renewal. Whether and to what extent this potential can be harnessed to therapeutic advantage is not yet known.22
Congenital Heart Disease
Congenital heart disease is a general term used to describe abnormalities of the heart or great vessels that are present from birth. Most such disorders arise from faulty embryogenesis during gestational weeks 3 through 8, when major cardiovascular structures form and begin to function. The most severe anomalies are incompatible with intrauterine survival. Congenital heart defects compatible with embryologic maturation and birth generally affect individual chambers or discrete regions of the heart, with the remainder of the heart developing relatively normally. Examples are infants born with a defect in septation (“hole in the heart”), such as an atrial septal defect (ASD) or a ventricular septal defect (VSD), stenotic valvular lesions, or with abnormalities in the coronary arteries.23 Some forms of congenital heart disease produce clinically important manifestations soon after birth, which are frequently brought on by the change from fetal to postnatal circulatory patterns (with reliance on the lungs for oxygenation birth, rather than the placenta as in intrauterine life). Approximately half of congenital cardiovascular malformations are diagnosed in the first year of life, but some mild forms may not become evident until adulthood (e.g., ASD).
Incidence.
With an incidence of approximately 1% (estimates range from 4 to 50 per 1000 live births), congenital cardiovascular defects are among the most prevalent malformations and are the most common type of heart disease among children.24 The incidence is higher in premature infants and in stillborns. Twelve disorders account for about 85% of cases; their frequencies are presented in Table 12-2.
TABLE 12-2 Frequencies of Congenital Cardiac Malformations*
| Malformation | Incidence per Million Live Births | % |
|---|---|---|
| Ventricular septal defect | 4482 | 42 |
| Atrial septal defect | 1043 | 10 |
| Pulmonary stenosis | 836 | 8 |
| Patent ductus arteriosus | 781 | 7 |
| Tetralogy of Fallot | 577 | 5 |
| Coarctation of the aorta | 492 | 5 |
| Atrioventricular septal defect | 396 | 4 |
| Aortic stenosis | 388 | 4 |
| Transposition of the great arteries | 388 | 4 |
| Truncus arteriosus | 136 | 1 |
| Total anomalous pulmonary venous connection | 120 | 1 |
| Tricuspid atresia | 118 | 1 |
| TOTAL | 9757 |
* Presented as upper quartile of 44 published studies. Percentages do not add up to 100% because of rounding.
Source: Hoffman JIE, Kaplan S: The incidence of congenital heart disease. J Am Coll Cardiol 39:1890, 2002.
The number of individuals who have survived with congenital heart disease into adulthood is increasing rapidly and is presently estimated at nearly 1 million individuals in the United States.25 Many of those with congenital heart disease have benefited greatly from rapid advances in the surgical and interventional repair of various structural heart defects. Nevertheless, such repairs may not restore the heart to normal; in such instances, patients may suffer from arrhythmias or ventricular dysfunction, and require additional surgery.26 Other factors that impact the long-term outcome include risks associated with the use of prosthetic materials and devices,27 such as substitute valves or myocardial patches, and maternal risks associated with childbearing.28
Cardiac Development.
The diverse malformations seen in congenital heart disease are caused by errors that occur during cardiac development; thus, a brief review how the heart normally forms is in order before discussing the specific defects (Fig. 12-3). The fine details of this complex process are beyond our scope here. Suffice it to say that the earliest cardiac precursors originate in lateral mesoderm and move to the mid-line in two migratory waves to create a crescent of cells consisting of the first and second heart fields by about day 15 of development.29,30 Each heart field is marked by the expression of different sets of genes; for example, the first heart field expresses the transcription factors TBX5 and Hand1, whereas the second heart field expresses the transcription factor Hand2 and fibroblast growth factor-10. Both fields contain multipotent progenitor cells that can produce all of the major cell types of the heart; endocardium, myocardium, and smooth muscle cells. As an aside, there is considerable interest in the therapeutic potential of such early cardiac progenitors, which could conceivably be used to regenerate portions of the adult heart that are damaged or otherwise dysfunctional.
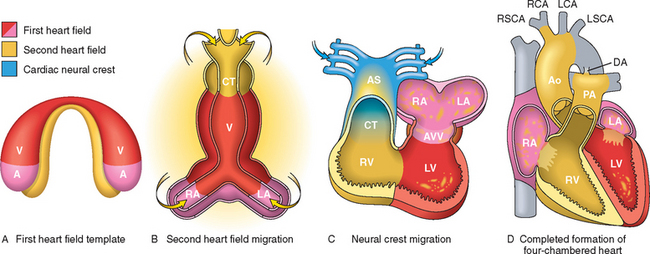
FIGURE 12-3 Human cardiac development, emphasizing the three sources of cells. A, Day 15. First heart field (FHF) cells (shown in red) form a crescent shape in the anterior embryo with second heart field (SHF) cells (shown in yellow) near the FHF. B, Day 21. SHF cells lie dorsal to the straight heart tube and begin to migrate (arrows) into the anterior and posterior ends of the tube to form the right ventricle (RV), conotruncus (CT), and part of the atria (A). C, Day 28. Following rightward looping of the heart tube, cardiac neural crest cells (shown in blue) also migrate (arrow) into the outflow tract from the neural folds to septate the outflow tract and pattern the bilaterally symmetric aortic arch arteries (III, IV, and VI). D, Day 50. Septation of the ventricles, atria, and atrioventricular valves (AVV) results in the appropriately configured four-chambered heart. Ao, aorta; AS, aortic sac; DA, ductus arteriosus; LA, left atrium; LCA, left carotid artery; LSCA, left subclavian artery; LV, left ventricle; PA, pulmonary artery; RA, right atrium; RCA, right carotid artery; RSCA, right subclavian artery; V, ventricle.
(Modified by permission from Srivastava D: Making or breaking the heart: from lineage determination to morphogenesis. Cell 126:1037, 2006.)
Even at this very early stage of development, each heart field is destined to give rise to particular portions of the heart. Cells derived from the first heart field mainly give rise to the left ventricle, whereas cells derived from the second heart field give rise to the outflow tract, right ventricle, and most of the atria. By day 20, the initial cell crescent develops into a beating tube, which loops to the right and begins to form the heart chambers by day 28. Around this time, two other critical events occur: (1) cells derived from the neural crest migrate into the outflow tract, where they participate in the septation of the outflow tract and the formation of the aortic arches; and (2) the extracellular matrix (ECM) underlying the future atrioventricular canal and outflow tract enlarges to produce swellings known as endocardial cushions. This process depends on the delamination of a subset of endocardial cells, which invade the ECM and subsequently proliferate and differentiate into the mesenchymal cells that are responsible for valve development. By day 50, further septation of the ventricles, atria, and atrioventricular valves produces the fourchambered heart.
Proper orchestration of these remarkable transformations depends on a network of transcription factors that are regulated by a number of signaling pathways, particularly the Wnt, VEGF, bone morphogenetic factor, TGF-β, fibroblast growth factor, and Notch pathways. It should also be remembered that the heart is a mechanical organ that is exposed to flowing blood from its earliest stages of development. It is likely that hemodynamic forces play an important role in cardiac development, just as they influence adaptations in the adult heart such as hypertrophy and dilation. In addition, specific micro-RNAs play critical roles in cardiac development by coordinating patterns and levels of transcription factor expression.18
Many of the genetic defects that affect heart development are autosomal dominant mutations that cause partial loss of function in one or another required factor, which are often transcription factors (discussed below). Thus, even relatively minor changes in the activity of one of the many factors necessary for normal development can lead to defects in the final product, the fully developed heart. It can be imagined (but is unproved) that transient environmental stresses during the first trimester of pregnancy that alter the activity of these same genes might give rise to defects resembling those produced by inherited mutations.
Etiology and Pathogenesis.
The main known causes of congenital heart disease consist of sporadic genetic abnormalities, which can take the form of single gene mutations, small chromosomal deletions, and additions or deletions of whole chromosomes (trisomies and monosomies). In the case of single gene mutations, the affected genes encode proteins belonging to several different functional classes, examples of which are provided in Table 12-3. Many of these mutations affect genes encoding transcription factors that are required for normal heart development. Since the affected patients are heterozygous for these mutations, it follows that a 50% reduction in the activity of these factors is probably sufficient to derange cardiac development. Some of the affected transcription factors appear to work together in large protein complexes, providing an explanation for why mutations in any one of several genes produce similar defects. For example, GATA4, TBX5, and NKX2-5, three transcription factors that are mutated in some patients with atrial and ventricular septal defects, all bind to one another and co-regulate the expression of target genes that are required for the proper development of the heart. Of further interest, GATA4 and TBX20 are also mutated in rare forms of adult-onset cardiomyopathy (discussed later), indicating that they are not only important developmentally but are also needed to maintain the function of the postnatal heart.
Other single gene mutations associated with congenital heart disease affect proteins within signaling pathways or that have structural roles. Mutations in genes encoding various components of the Notch pathway, such as JAGGED1, NOTCH1, and NOTCH2, are associated with a number of different congenital heart defects, including bicuspid aortic valve (NOTCH1, discussed later) and Tetralogy of Fallot (JAGGED1 and NOTCH2).29,30 As you will recall from Chapter 11, fibrillin mutations underlie Marfan syndrome, which is associated with valvular defects and aortic aneurysms. Although fibrillin was initially described as a structural protein, it is also an important negative regulator of TGFβ signaling, and hyperactive TGFβ signaling is at least partially responsible for the cardiovascular abnormalities in Marfan syndrome.
A notable example of a small chromosomal lesion that causes congenital heart disease is deletion of chromosome 22q11.2, which is found in up to 50% of patients with DiGeorge syndrome. In this syndrome the fourth branchial arch and the derivatives of the third and fourth pharyngeal pouches, which contribute to the formation of the thymus, parathyroids, and heart, develop abnormally. One candidate gene in the deleted region is TBX1, which encodes a transcription factor that regulates the expansion of cardiac progenitors in the second heart field. Other important genetic causes of congenital heart disease include chromosomal aneuplodies, particularly Turner syndrome (monosomy X) and trisomies 13, 18, and 21.31 Indeed, the most common genetic cause of congenital heart disease is trisomy 21 (Down syndrome),32 in which about 40% of patients have one or more heart defects, most often affecting structures derived from the endocardial cushions (e.g., the atrioventricular septae and valves). The mechanisms by which aneuploidy leads to congenital heart defects remain largely unknown, but are likely to involve the dysregulated expression of multiple genes.
Beyond these known associations, more subtle forms of genetic variation probably also contribute to congenital heart disease. This assertion is based in part on the recognition that first-degree relatives of affected patients are at increased risk for congenital heart defects compared to the general population. For example, a child of a father with a VSD has a risk of 2%; if the VSD occurred in the mother, the risk to her offspring is 6% to 10%.
Despite these genetic clues, it must be acknowledged that our understanding of the mechanisms that lead to heart defects remains rudimentary. Most affected patients have no identifiable genetic risk factor, and even in those that do, the nature and severity of the defect are highly variable. As a result, it is thought that environmental factors, alone or in combination with genetic factors, also contribute to congenital heart disease and in some cases may be the primary cause. Examples of known exposures that are associated with heart defects include congenital rubella infection, gestational diabetes, and exposure to teratogens (including some therapeutic drugs).33 There is also great interest in identifying nutritional factors that may modify risk. For instance, intake of multivitamin supplements containing folate may reduce the risk of congenital heart defects.34
Clinical Features.
The varied structural anomalies in congenital heart disease fall primarily into three major categories:
A shunt is an abnormal communication between chambers or blood vessels. Abnormal channels permit the flow of blood down pressure gradients from the left (systemic) side to the right (pulmonary) side of the circulation or vice versa. When blood from the right side of the circulation flows directly into the left side (right-to-left shunt), hypoxemia and cyanosis (a dusky blueness of the skin and mucous membranes) result because of the admixture of poorly oxygenated venous blood with systemic arterial blood (called cyanotic congenital heart disease). The most important congenital causes of right-to-left shunts are tetralogy of Fallot, transposition of the great arteries, persistent truncus arteriosus, tricuspid atresia, and total anomalous pulmonary venous connection. Moreover, with right-to-left shunts, emboli arising in peripheral veins can bypass the lungs and directly enter the systemic circulation (paradoxical embolism); brain infarction and abscess are potential consequences. Severe, long-standing cyanosis also causes clubbing of the tips of the fingers and toes (called hypertrophic osteoarthropathy) and polycythemia.
In contrast, left-to-right shunts (such as ASD, VSD, and patent ductus arteriosus) increase pulmonary blood flow and are not initially associated with cyanosis. However, leftto-right shunts raise both flow volumes and pressures in the normally low-pressure, low-resistance pulmonary circulation, which can lead to right ventricular hypertrophy and atherosclerosis of the pulmonary vasculature. The muscular pulmonary arteries (<1 mm diameter) first respond to increased pressure and flow by undergoing medial hypertrophy and vasoconstriction, which maintains relatively normal distal pulmonary capillary and venous pressures, and prevents pulmonary edema. Prolonged pulmonary arterial vasoconstriction, however, stimulates the proliferation of the vascular wall cells and the consequent development of irreversible obstructive intimal lesions analogous to the arteriolar changes seen in systemic hypertension (Chapter 11). Eventually, pulmonary vascular resistance approaches systemic levels, thereby producing a new right-to-left shunt that introduces unoxygenated blood into the systemic circulation (late cyanotic congenital heart disease, or Eisenmenger syndrome).
Once irreversible pulmonary hypertension develops, the structural defects of congenital heart disease are considered irreparable. The secondary pulmonary vascular changes can eventually lead to the patient’s death. This provides the rationale for early intervention, either surgical or nonsurgical, in those with left-to-right shunts.
Some developmental anomalies of the heart (e.g., coarctation of the aorta, aortic valvular stenosis, and pulmonary valvular stenosis) produce abnormal narrowing of chambers, valves, or blood vessels and therefore are called obstructive congenital heart disease. A complete obstruction is called an atresia. In some disorders (e.g., Tetralogy of Fallot), an obstruction (pulmonary stenosis) and a shunt (right-to-left through a VSD) are both present.
The altered hemodynamics of congenital heart disease usually cause cardiac dilation or hypertrophy (or both). However, some defects induce a decrease in the volume and muscle mass of a cardiac chamber; this is called hypo-plasia if it occurs before birth and atrophy if it develops postnatally.
LEFT-TO-RIGHT SHUNTS
The most commonly encountered left-to-right shunts include ASDs, VSDs, patent ductus arteriosus, and atrioventricular septal defects, and are shown in Figure 12-4.
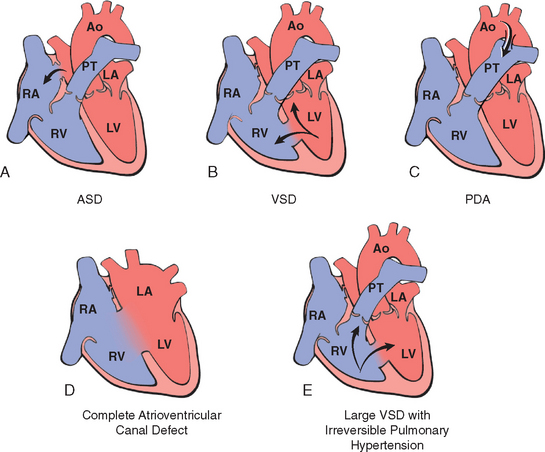
FIGURE 12-4 Schematic of congenital left-to-right shunts. Arrows indicate the direction of blood flow. A, Atrial septal defect (ASD). B, Ventricular septal defect (VSD). With VSD the shunt is left-to-right, and the pressures are the same in both ventricles. Pressure hypertrophy of the right ventricle and volume hypertrophy of the left ventricle are generally present. C, Patent ductus arteriosus (PDA). D, Atrioventricular septal defect (AVSD). E, Large VSD with irreversible pulmonary hypertension. The shunt is right-to-left (shunt reversal). Volume hypertrophy and pressure hypertrophy of the right ventricle are present. The right ventricular pressure is now sufficient to yield a right-to-left shunt. Ao, aorta; LA, left atrium; LV, left ventricle; PT, pulmonary trunk; RA, right atrium; RV, right ventricle.
Atrial Septal Defect
An atrial septal defect (ASD) is an abnormal, fixed opening in the atrial septum caused by incomplete tissue formation that allows communication of blood between the left and right atria (not to be confused with patent foramen ovale, see below). ASDs are usually asymptomatic until adulthood (Fig. 12-4A).35
Morphology. The three major types of ASDs are classified according to their location as secundum, primum, and sinus venosus. Secundum ASDs (90% of all ASDs) result from a deficient or fenestrated oval fossa near the center of the atrial septum. These are usually not associated with other anomalies, and may be of any size, be single or multiple, or be fenestrated. Primum anomalies (5% of ASDs) occur adjacent to the AV valves. Sinus venosus defects (5%) are located near the entrance of the superior vena cava and may be associated with anomalous pulmonary venous return to the right atrium.
Clinical Features.
ASDs result in a left-to-right shunt, largely because pulmonary vascular resistance is considerably less than systemic vascular resistance and because the compliance (distensibility) of the right ventricle is much greater than that of the left. Pulmonary blood flow may be two to four times normal. A murmur is often present as a result of excessive flow through the pulmonary valve. Despite the right-sided volume overload, ASDs are generally well tolerated and usually do not become symptomatic before age 30; irreversible pulmonary hypertension is unusual. Surgical or catheter-based closure of an ASD reverses the hemodynamic abnormalities and prevents complications, including heart failure, paradoxical embolization, and irreversible pulmonary vascular disease.36 Mortality is low, and long-term survival is comparable to that of a normal population.
Patent Foramen Ovale
A patent foramen ovale is a small hole created by an open flap of tissue in the atrial septum at the oval fossa.37 In the fetus, the foramen ovale is an important functional right-to-left shunt that allows oxygen-rich blood from the placenta to bypass the not yet inflated lungs by traveling directly from the right to left atrium. The hole is forced shut at birth as a result of increased blood pressure on the left side of the heart, and the tissue flap closes permanently in approximately 80% of people. In the remaining 20% of people, the unsealed flap can open when there is more pressure on the right side of the heart. Thus, sustained pulmonary hypertension or even transient increases in right-sided pressures, such as occurs during a bowel movement, coughing, or sneezing, can produce brief periods of right-to-left shunting, with the possibility of paradoxical embolism.38
Ventricular Septal Defect
Incomplete closure of the ventricular septum, allowing free communication of blood between the left to right ventricles, is the most common form of congenital cardiac anomaly (Fig. 12-4B). Most ventricular septal defects (VSDs) are associated with other congenital cardiac anomalies such as tetralogy of Fallot; only 20% to 30% are isolated.
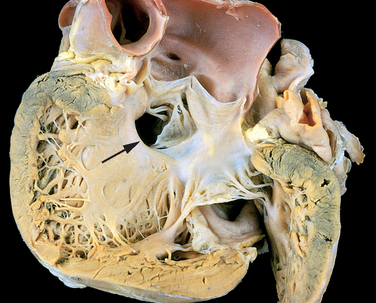
Morphology. VSDs are classified according to their size and location. Most are about the size of the aortic valve orifice. About 90% involve the region of the membranous interventricular septum (membranous VSD) (Fig. 12-5). The remainder lie below the pulmonary valve (infundibular VSD) or within the muscular septum. Although most VSDs are single, those in the muscular septum may be multiple (so-called “Swiss-cheese” septum).
Clinical Features.
The functional consequences of a VSD depend on the size of the defect and whether there are associated with right-sided malformations. Large VSDs cause difficulties virtually from birth; smaller lesions are generally well tolerated for years, and may not be recognized until much later in life. Approximately 50% of small muscular VSDs close spontaneously.39 Large defects are usually membranous or infundibular, and they generally cause significant left-to-right shunting, leading to right ventricular hypertrophy and pulmonary hypertension virtually from birth. Over time, irreversible pulmonary vascular disease develops in essentially all persons with large unclosed VSDs, ultimately resulting in shunt reversal, cyanosis, and death. Surgical or catheter-based closure of asymptomatic VSDs is generally delayed beyond infancy, in hope of spontaneous closure. Early correction, however, must be performed in babies with large defects to prevent the development of irreversible obstructive pulmonary vascular disease.
Patent Ductus Arteriosus
Patent (also called persistent) ductus arteriosus (PDA) results when the ductus arteriosus, an essential fetal structure that normally spontaneously closes, remains open after birth (see Fig. 12-4C). In the fetal circulation the ductus arteriosus shunts blood from the pulmonary artery to the aorta, which (like the patent foramen ovale) serves to bypass the lungs. About 90% of PDAs occur as an isolated anomaly. The remainder are most often associated with VSD, coarctation of the aorta, or pulmonary or aortic valve stenosis.
PDA produces a characteristic continuous harsh murmur, described as “machinery-like”. The clinical impact of a PDA depends on its diameter and the cardiovascular status of the individual.40 PDA is usually asymptomatic at birth, and a narrow PDA may have no effect on the child’s growth and development. Because the shunt is at first left-to-right, there is no cyanosis, but eventually the additional volume and pressure overload produce obstructive changes in small pulmonary arteries, leading to reversal of flow and its associated consequences.
There is general agreement that an isolated PDA should be closed as early in life as is feasible. Conversely, preservation of ductal patency (by administering prostaglandin E) assumes great importance in the survival of infants with various congenital malformations that obstruct the pulmonary or systemic outflow tracts. For example, in aortic valve atresia a PDA provides the bulk of the systemic blood flow. Depending on the context, therefore, a PDA may be either life-threatening or lifesaving.
Atrioventricular Septal Defect
Atrioventricular septal defect (AVSD, also called complete atrioventricular canal defect) results from the embryologic failure of the superior and inferior endocardial cushions of the AV canal to fuse adequately. The consequence is incomplete closure of the AV septum and malformation of the tricuspid and mitral valves (see Fig. 12-4D). The two most common forms are partial AVSD (consisting of a primum ASD and a cleft anterior mitral leaflet, causing mitral insufficiency) and complete AVSD (consisting of a large combined AV septal defect and a large common AV valve—essentially a hole in the center of the heart). In the complete form, all four cardiac chambers freely communicate, inducing volume hypertrophy of each. More than one third of all patients with a complete AVSD have Down syndrome. Surgical repair is possible.
RIGHT-TO-LEFT SHUNTS
The diseases in this group cause cyanosis early in postnatal life (cyanotic congenital heart disease). Tetralogy of Fallot, the most common in this group, and transposition of the great arteries are illustrated schematically in Figure 12-6. The others include persistent truncus arteriosus, tricuspid atresia, and total anomalous pulmonary venous connection.
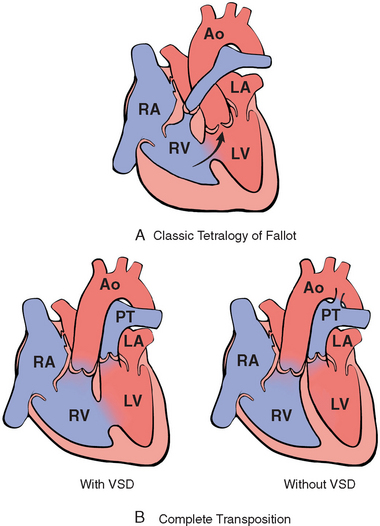
FIGURE 12-6 Schematic of the most important right-to-left shunts (cyanotic congenital heart disease). A, Classic tetralogy of Fallot. The direction of shunting across the ventricular septal defect (VSD) depends on the degree of the subpulmonary stenosis; when severe, a right-to-left shunt results (arrow). B, Transposition of the great arteries with and without VSD. Ao, aorta; LA, left atrium; LV, left ventricle; PT, pulmonary trunk; RA, right atrium; RV, right ventricle.
(Courtesy of William D. Edwards, MD, Mayo Clinic, Rochester, MN.)
Tetralogy of Fallot
The four cardinal features of the tetralogy of Fallot (TOF) are (1) VSD, (2) obstruction of the right ventricular outflow tract (subpulmonary stenosis), (3) an aorta that overrides the VSD, and (4) right ventricular hypertrophy (Fig. 12-6A). All of the features result embryologically from anterosuperior displacement of the infundibular septum.
Morphology. The heart is often enlarged and may be “boot-shaped” as a result of marked right ventricular hypertrophy, particularly of the apical region. The VSD is usually large. The aortic valve forms the superior border of the VSD, thereby overriding the defect and both ventricular chambers. The obstruction to right ventricular outflow is most often due to narrowing of the infundibulum (subpulmonic stenosis) but can be accompanied by pulmonary valvular stenosis. Sometimes there is complete atresia of the pulmonary valve and variable portions of the pulmonary arteries, such that blood flow through a patent ductus arteriosus, dilated bronchial arteries, or both, is necessary for survival. Aortic valve insufficiency or an ASD may also be present; a right aortic arch is present in about 25% of cases.
Clinical Features.
Even untreated, some patients with TOF survive into adult life (in reports of untreated patients with this condition, 10% were alive at 20 years and 3% at 40 years).49 The clinical consequences depend primarily on the severity of the subpulmonary stenosis, as this determines the direction of blood flow. If the subpulmonary stenosis is mild, the abnormality resembles an isolated VSD, and the shunt may be left-to-right, without cyanosis (so-called pink tetralogy). As the obstruction increases in severity, there is commensurately greater resistance to right ventricular outflow. As right-sided pressures approach or exceed left-sided pressures, right-to-left shunting develops, producing cyanosis (classic TOF). With increasingly severe subpulmonic stenosis, the pulmonary arteries become progressively smaller and thinner walled (hypoplastic), and the aorta grows progressively larger in diameter. As the child grows and the heart increases in size, the pulmonic orifice does not expand proportionally, making the obstruction progressively worse. Most infants with TOF are cyanotic from birth or soon thereafter. The subpulmonary stenosis, however, protects the pulmonary vasculature from pressure overload, and right ventricular failure is rare because the right ventricle is decompressed by the shunting of blood into the left ventricle and aorta. Complete surgical repair is possible for classic TOF but is more complicated for persons with pulmonary atresia and dilated bronchial arteries.
Transposition of the Great Arteries
Transposition of the great arteries (TGA) produces ventriculoarterial discordance: the aorta arises from the right ventricle, and lies anterior and to the right of the pulmonary artery, which emanates from the left ventricle (Fig. 12-7; see also Fig. 12-5B). The AV connections are normal (concordant), with the right atrium joining the right ventricle and the left atrium emptying into the left ventricle. The embryologic defect in complete TGA stems from abnormal formation of the truncal and aortopulmonary septa. The result is separation of the systemic and pulmonary circulations, a condition incompatible with postnatal life unless a shunt exists for adequate mixing of blood.
FIGURE 12-7 Transposition of the great arteries.
(Courtesy of William D. Edwards, MD, Mayo Clinic, Rochester, MN.)
The outlook for infants with TGA depends on the degree of “mixing” of the blood, the magnitude of the tissue hypoxia, and the ability of the right ventricle to maintain the systemic circulation. Patients with TGA and a VSD (∼35%) may have a stable shunt. Those with only a patent foramen ovale or ductus arteriosus (∼65%), however, have unstable shunts that tend to close and therefore require immediate intervention to create a new shunt (such as balloon atrial septostomy) within the first few days of life. Right ventricular hypertrophy becomes prominent, because this chamber functions as the systemic ventricle. Concurrently, the left ventricle becomes thin-walled (atrophic) as it supports the low-resistance pulmonary circulation. Without surgery, most patients die during the first few months of life. However, as a result of considerable improvements in surgical repair over the past several decades, many persons with TGA now survive to adulthood.41
Persistent Truncus Arteriosus
Persistent truncus arteriosus (PTA) arises from a developmental failure of separation of the embryologic truncus arteriosus into the aorta and pulmonary artery. This results in a single great artery that receives blood from both ventricles and gives rise to the systemic, pulmonary, and coronary circulations. Because there is an associated VSD and mixing of blood from the right and left ventricles, PTA produces systemic cyanosis as well as increased pulmonary blood flow, with the danger of irreversible pulmonary hypertension.
Tricuspid Atresia
Complete occlusion of the tricuspid valve orifice is known as tricuspid atresia. It results embryologically from unequal division of the AV canal; thus, the mitral valve is larger than normal, and there is almost always underdevelopment (hypoplasia) of the right ventricle. The circulation is maintained to some degree by a right-to-left shunt through an interatrial communication (ASD or patent foramen ovale) and a VSD, which affords communication between the left ventricle and the pulmonary artery that arises from the hypoplastic right ventricle. Cyanosis is present virtually from birth, and there is a high mortality in the first weeks or months of life.
Total Anomalous Pulmonary Venous Connection
Total anomalous pulmonary venous connection (TAPVC), in which the pulmonary veins fail to directly join the left atrium, results embryologically when the common pulmonary vein fails to develop or becomes atretic. Fetal development is made possible by primitive systemic venous channels that usually drain from the lung into the left innominate vein or to the coronary sinus. Either a patent foramen ovale or an ASD is always present, allowing pulmonary venous blood to enter the left atrium. Consequences of TAPVC include volume and pressure hypertrophy and dilation of the right side of the heart, and dilation of the pulmonary trunk. The left atrium is hypoplastic, but the left ventricle is usually normal in size. Cyanosis may be present as a result of mixing of well-oxygenated and poorly oxygenated blood at the site of the anomalous pulmonary venous connection and large right-to-left shunting through an ASD.
OBSTRUCTIVE CONGENITAL ANOMALIES
Congenital obstruction to blood flow may occur at the level of the heart valves or within a great vessel.42 Relatively common examples include stenosis or atresia of the aortic or pulmonary valves, and coarctation of the aorta. Obstruction can also occur within a chamber, as with subpulmonary stenosis in TOF.
Coarctation of the Aorta
Coarctation (narrowing, constriction) of the aorta ranks high in frequency among the common structural anomalies. Males are affected twice as often as females, although females with Turner syndrome frequently have a coarctation (Chapter 5). Two classic forms have been described: (1) an “infantile” form with tubular hypoplasia of the aortic arch proximal to a patent ductus arteriosus that is often symptomatic in early childhood, and (2) an “adult” form in which there is a discrete ridgelike infolding of the aorta, just opposite the closed ductus arteriosus (ligamentum arteriosum) distal to the arch vessels (Fig. 12-8). Encroachment on the aortic lumen is of variable severity, sometimes leaving only a small channel and at other times producing only minimal narrowing. Although coarctation of the aorta may occur as a solitary defect, it is accompanied by a bicuspid aortic valve in 50% of cases and may also be associated with congenital aortic stenosis, ASD, VSD, mitral regurgitation, or berry aneurysms of the circle of Willis in the brain.

FIGURE 12-8 Diagram showing coarctation of the aorta with and without patent ductus arteriosus (PDA). Ao, aorta; LA, left atrium; LV, left ventricle; PT, pulmonary trunk; RA, right atrium; RV, right ventricle; PDA, patent ductus arteriosus.
(Courtesy of William D. Edwards, MD, Mayo Clinic, Rochester, MN.)
Clinical manifestations depend on the severity of the narrowing and the patency of the ductus arteriosus. Coarctation of the aorta with a patent ductus arteriosus usually leads to manifestations early in life; indeed, it may cause signs and symptoms immediately after birth. Many infants with this anomaly do not survive the neonatal period without surgical or catheter-based intervention. In such cases, the delivery of unsaturated blood through the patent ductus arteriosus produces cyanosis localized to the lower half of the body.
The outlook is different with coarctation of the aorta without a patent ductus arteriosus, unless it is very severe. Most children are asymptomatic, and the disease may go unrecognized until well into adult life. Typically there is hypertension in the upper extremities; in contrast, there are weak pulses and hypotension in the lower extremities, associated with manifestations of arterial insufficiency (i.e., claudication and coldness). Particularly characteristic in adults is the development of collateral circulation between the precoarctation arterial branches and the postcoarctation arteries through enlarged intercostal and internal mammary arteries, which produce radiographically visible erosions (“notching”) of the undersurfaces of the ribs.
With significant coarctations, murmurs are present throughout systole; sometimes a thrill may be present. There is cardiomegaly due to left ventricular pressure-overload hypertrophy. With uncomplicated coarctation of the aorta, surgical resection and end-to-end anastomosis or replacement of the affected aortic segment by a prosthetic graft yields excellent results.
Pulmonary Stenosis and Atresia
This relatively frequent malformation constitutes an obstruction at the pulmonary valve, which may be mild to severe; the lesion can be isolated or part of a more complex anomaly—either tetralogy of Fallot or transposition of the great arteries. Right ventricular hypertrophy often develops, and there is sometimes poststenotic dilation of the pulmonary artery due to injury of the wall by “jetting” blood. With coexistent subpulmonary stenosis (as in tetralogy of Fallot), the high ventricular pressure is not transmitted to the valve, and the pulmonary trunk is not dilated and may in fact be hypoplastic. When the valve is entirely atretic, there is no communication between the right ventricle and lungs. In such cases the anomaly is associated with a hypoplastic right ventricle and an ASD; blood reaches the lungs through a patent ductus arteriosus. Mild stenosis may be asymptomatic and compatible with long life, whereas symptomatic cases require surgical correction.
Aortic Stenosis and Atresia
Congenital narrowing and obstruction of the aortic valve can occur at three locations: valvular, subvalvular, and supravalvular. With valvular aortic stenosis the cusps may be hypoplastic (small), dysplastic (thickened, nodular), or abnormal in number (usually acommissural or unicommissural). In severe congenital aortic stenosis or atresia, obstruction of the left ventricular outflow tract leads to underdevelopment (hypoplasia) of the left ventricle and ascending aorta, sometimes accompanied by dense, porcelain-like left ventricular endocardial fibroelastosis. The ductus must be open to allow blood flow to the aorta and coronary arteries. This constellation of findings, called the hypoplastic left heart syndrome, is nearly always fatal in the first week of life, when the ductus closes, unless a palliative procedure is done. Less severe degrees of congenital aortic stenosis may be compatible with long survival. Congenital aortic stenosis is an isolated lesion in 80% of cases.
Subaortic stenosis can be caused by a thickened ring (discrete type) or collar (tunnel type) of dense endocardial fibrous tissue below the level of the cusps. Supravalvular aortic stenosis is an inherited form of aortic dysplasia in which the ascending aortic wall is greatly thickened, causing luminal constriction. In some cases it is a component of a multiorgan developmental disorder resulting from deletions on chromosome 7 that include the gene for elastin. Other features of the syndrome include hypercalcemia, cognitive abnormalities, and hallmark facial anomalies (Williams-Beuren syndrome).43 Mutations in the elastin gene probably cause supravalvular aortic stenosis by disrupting elastin–smooth muscle cell interactions during arterial morphogenesis.
Subaortic stenosis is usually associated with a prominent systolic murmur and sometimes a thrill. Pressure hypertrophy of the left ventricle develops as a consequence of the obstruction to blood flow, but congenital stenoses are well tolerated unless very severe. Mild stenoses can be managed conservatively with antibiotic prophylaxis (to prevent endocarditis) and avoidance of strenuous activity, but owing to left ventricular hypertrophy the threat of sudden death with exertion always looms.
Ischemic Heart Disease
Ischemic heart disease (IHD) is the leading cause of death worldwide for both men and women (7 million total per year). IHD is the generic designation for a group of pathophysiologically related syndromes resulting from myocardial ischemia—an imbalance between the supply (perfusion) and demand of the heart for oxygenated blood. Ischemia brings not only an insufficiency of oxygen, but also reduces the availability of nutrients and the removal of metabolites (Chapter 1). For this reason, ischemia is generally less well tolerated by the heart than pure hypoxia, such as may be seen with severe anemia, cyanotic heart disease, or advanced lung disease.
In more than 90% of cases, the cause of myocardial ischemia is reduced blood flow due to obstructive atherosclerotic lesions in the coronary arteries. Thus, IHD is often termed coronary artery disease (CAD) or coronary heart disease. In most cases there is a long period (up to decades) of silent, slow progression of coronary lesions before symptoms appear. Thus, the syndromes of IHD are only the late manifestations of coronary atherosclerosis that may have started during childhood or adolescence (Chapter 11).
IHD usually presents as one or more of the following clinical syndromes:
In addition to coronary atherosclerosis, myocardial ischemia may be caused by coronary emboli, blockage of small myocardial blood vessels, and lowered systemic blood pressure (e.g., shock). Moreover, in the setting of coronary arterial obstruction, ischemia can be aggravated by an increase in cardiac energy demand (e.g., as occurs with myocardial hypertrophy or increased heart rate [tachycardia]), by diminished availability of blood or oxygen due to shock, or by hypoxemia. Some conditions have several deleterious effects; for example, tachycardia increases oxygen demand (because of more contractions per unit time) and decreases supply (by decreasing the relative time spent in diastole, when cardiac perfusion occurs).
Epidemiology.
IHD in its various forms is the leading cause of death for both males and females in the United States and other industrialized nations. Each year nearly 500,000 Americans die of IHD. As troublesome as these numbers may be, they represent an improvement over those that prevailed 2 to 3 decades ago. Since its peak in 1963, the overall death rate from IHD has fallen in the United States by approximately 50%. This remarkable improvement has resulted primarily from (1) prevention, achieved by modification of important risk factors, such as smoking, elevated blood cholesterol, and hypertension, and (2) diagnostic and therapeutic advances, allowing earlier, more effective, and safer treatments. The latter include new medications, coronary care units, thrombolysis for MI, percutaneous transluminal coronary angioplasty, endovascular stents, coronary artery bypass graft (CABG) surgery, and improved control of heart failure and arrhythmias. Additional risk reduction may potentially be achieved by maintenance of normal blood glucose levels in diabetic patients, control of obesity, and prophylactic anticoagulation of middle-aged men with aspirin. Nevertheless, continuing this encouraging trend in the 21st century will be challenging, in view of the predicted doubling of the number of individuals over age 65 by 2050 and the increased longevity of “baby boomers,” the “obesity epidemic,” and other factors. Interestingly, the genetic determinants of coronary atherosclerosis and IHD may not be identical, since MI occurs in only a small fraction of individuals with coronary disease. For example, the risk of MI but not coronary atherosclerosis is associated with genetic variants that modify leukotriene B4 metabolism.44
Pathogenesis.
The dominant cause of the IHD syndromes is insufficient coronary perfusion relative to myocardial demand, due to chronic, progressive atherosclerotic narrowing of the epicardial coronary arteries, and variable degrees of superimposed acute plaque change, thrombosis, and vasospasm. The individual elements and their interactions are discussed below.
Chronic Atherosclerosis.
More than 90% of patients with IHD have atherosclerosis of one or more of the epicardial coronary arteries. The clinical manifestations of coronary atherosclerosis are generally due to progressive narrowing of the lumen leading to stenosis (“fixed” obstructions) or to acute plaque disruption with thrombosis, both of which compromise blood flow. A fixed lesion obstructing 75% or greater of the lumen is generally required to cause symptomatic ischemia precipitated by exercise (most often manifested as chest pain, known as angina); with this degree of obstruction, compensatory coronary arterial vasodilation is no longer sufficient to meet even moderate increases in myocardial demand. Obstruction of 90% of the lumen can lead to inadequate coronary blood flow even at rest. The progressive myocardial ischemia induced by slowly developing occlusions may stimulate the formation of collateral vessels over time, which can protect against myocardial ischemia and infarction and mitigate the effects of high-grade stenoses.45
Although only a single major coronary epicardial trunk may be affected, two or all three—the left anterior descending (LAD), the left circumflex (LCX), and the right coronary artery (RCA)—are often involved by atherosclerosis. Clinically significant stenosing plaques may be located anywhere within these vessels but tend to predominate within the first several centimeters of the LAD and LCX and along the entire length of the RCA. Sometimes the major secondary epicardial branches are also involved (i.e., diagonal branches of the LAD, obtuse marginal branches of the LCX, or posterior descending branch of the RCA), but atherosclerosis of the intramural (penetrating) branches is rare.
Acute Plaque Change.
The risk of an individual developing clinically important IHD depends in part on the number, distribution, structure, and degree of obstruction of atheromatous plaques. However, the varied clinical manifestations of IHD cannot be explained by the anatomic disease burden alone. This is particularly true for the so-called acute coronary syndromes, unstable angina, acute MI, and sudden death. The acute coronary syndromes are typically initiated by an unpredictable and abrupt conversion of a stable atherosclerotic plaque to an unstable and potentially life-threatening atherothrombotic lesion through rupture, superficial erosion, ulceration, fissuring, or deep hemorrhage (Chapter 11). In most instances, the plaque change causes the formation of a superimposed thrombus that partially or completely occludes the affected artery.46, 47 These acute events are often associated with intralesional inflammation, which you will remember mediates the initiation, progression, and acute complications of atherosclerosis (discussed in Chapter 11). For purposes of simplicity, the spectrum of acute alterations in atherosclerotic lesions will be termed either plaque disruption or plaque change.
Consequences of Myocardial Ischemia.
In each syndrome the critical consequence is downstream myocardial ischemia. Stable angina results from increases in myocardial oxygen demand that outstrip the ability of stenosed coronary arteries to increase oxygen delivery; it is usually not associated with plaque disruption. Unstable angina is caused by plaque rupture complicated by partially occlusive thrombosis and vasoconstriction, which lead to severe but transient reductions in coronary blood flow. In some cases, microinfarcts can occur distal to disrupted plaques due to thromboemboli. In MI, acute plaque change induces total thrombotic occlusion and the subsequent death of heart muscle. Finally, sudden cardiac death frequently involves an atherosclerotic lesion in which a disrupted plaque causes regional myocardial ischemia that induces a fatal ventricular arrhythmia. Each of these important syndromes is discussed in detail below, followed by an examination of the important myocardial consequences.
ANGINA PECTORIS
Angina pectoris (literally, chest pain) is characterized by paroxysmal and usually recurrent attacks of substernal or precordial chest discomfort (variously described as constricting, squeezing, choking, or knifelike) caused by transient (15 seconds to 15 minutes) myocardial ischemia that falls short of inducing
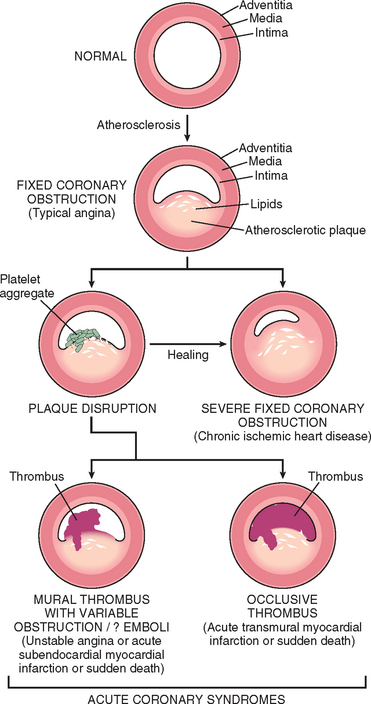
FIGURE 12-9 Schematic of sequential progression of coronary artery lesions and their association with various acute coronary syndromes.
(Modified and redrawn from Schoen FJ: Interventional and Surgical Cardiovascular Pathology: Clinical Correlations and Basic Principles. Philadelphia, WB Saunders, 1989, p 63.)
myocyte necrosis. The three overlapping patterns of angina pectoris—(1) stable or typical angina, (2) Prinzmetal variant angina, and (3) unstable or crescendo angina—are caused by varying combinations of increased myocardial demand, decreased myocardial perfusion, and coronary arterial pathology. Moreover, not all ischemic events are perceived by patients (silent ischemia).48
Stable angina, the most common form, is also called typical angina pectoris. It is caused by an imbalance in coronary perfusion (due to chronic stenosing coronary atherosclerosis) relative to myocardial demand, such as that produced by physical activity, emotional excitement, or any other cause of increased cardiac workload. Typical angina pectoris is usually relieved by rest (which decreases demand) or administering nitroglycerin, a strong vasodilator (which increases perfusion).
Prinzmetal variant angina is an uncommon from of episodic myocardial ischemia that is caused by coronary artery spasm. Although individuals with Prinzmetal variant angina may well have significant coronary atherosclerosis, the anginal attacks are unrelated to physical activity, heart rate, or blood pressure. Prinzmetal angina generally responds promptly to vasodilators, such as nitroglycerin and calcium channel blockers.
Unstable or crescendo angina refers to a pattern of increasingly frequent pain, often of prolonged duration, that is precipitated by progressively lower levels of physical activity or that even occurs at rest. In most patients, unstable angina is caused by the disruption of an atherosclerotic plaque with superimposed partial (mural) thrombosis and possibly embolization or vasospasm (or both). Unstable angina thus serves as a warning that an acute MI may be imminent; indeed, this syndrome is sometimes referred to as preinfarction angina.
MYOCARDIAL INFARCTION (MI)
MI, also known as “heart attack,” is the death of cardiac muscle due to prolonged severe ischemia. It is by far the most important form of IHD. About 1.5 million individuals in the United States suffer an MI annually.
Incidence and Risk Factors.
MI can occur at virtually any age, but its frequency rises progressively with increasing age and when predispositions to atherosclerosis are present. Nearly 10% of myocardial infarcts occur in people under age 40, and 45% occur in people under age 65. Blacks and whites are equally affected. Throughout life, men are at significantly greater risk than women.49 Indeed, except for those having some predisposing atherogenic condition, women are protected against MI and other heart diseases during the reproductive years. However, the decrease of estrogen following menopause is associated with rapid development of CAD, and IHD is the most common cause of death in elderly women. Postmenopausal hormonal replacement therapy is not currently felt to protect against atherosclerosis and IHD (Chapter 11).50
Pathogenesis.
We now consider the basis for and consequences of myocardial ischemia.
Coronary Arterial Occlusion.
In the typical case of MI, the following sequence of events is considered most likely (see Chapter 11 for more detail):
Compelling evidence for this sequence has been obtained from (1) autopsy studies of patients dying of acute MI, (2) angiographic studies demonstrating a high frequency of thrombotic occlusion early after MI, (3) the high success rate of coronary revascularization (i.e., thrombolysis, angioplasty, stent placement, and surgery) following MI, and (4) the demonstration of residual disrupted atherosclerotic lesions by angiography after thrombolysis. Coronary angiography performed within 4 hours of the onset of an MI shows a thrombosed coronary artery in almost 90% of cases. However, when angiography is delayed until 12 to 24 hours after onset, occlusion is seen only about 60% of the time, suggesting that some occlusions resolve due to fibrinolysis, relaxation of spasm, or both.
In approximately 10% of cases, transmural MI occurs in the absence of the typical coronary vascular pathology. In such situations, other mechanisms may be responsible for the reduced coronary blood flow, including:
Myocardial Response.
Coronary arterial obstruction compromises the blood supply to a region of myocardium (Fig. 12-10), causing ischemia, myocardial dysfunction, and potentially myocyte death. The anatomic region supplied by that artery is referred to as the area at risk. The outcome depends predominantly on the severity and duration of flow deprivation (Fig. 12-11).
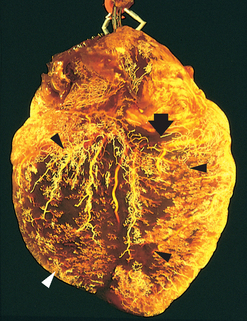
FIGURE 12-10 Postmortem angiogram showing the posterior aspect of the heart of a patient who died during the evolution of acute myocardial infarction, demonstrating total occlusion of the distal right coronary artery by an acute thrombus (arrow) and a large zone of myocardial hypoperfusion involving the posterior left and right ventricles, as indicated by arrowheads, and having almost absent filling of capillaries. The heart has been fixed by coronary arterial perfusion with glutaraldehyde and cleared with methyl salicylate, followed by intracoronary injection of silicone polymer (yellow). Photograph courtesy of Lewis L. Lainey.
(Reproduced by permission from Schoen FJ: Interventional and Surgical Cardiovascular Pathology: Clinical Correlations and Basic Principles. Philadelphia, WB Saunders, 1989, p. 60.)
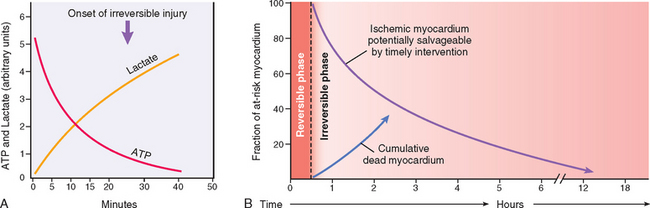
FIGURE 12-11 Temporal sequence of early biochemical findings and progression of necrosis after onset of severe myocardial ischemia. A, Early changes include loss of adenosine triphosphate (ATP) and accumulation of lactate. B, For approximately 30 minutes after the onset of even the most severe ischemia, myocardial injury is potentially reversible. Thereafter, progressive loss of viability occurs that is complete by 6 to 12 hours. The benefits of reperfusion are greatest when it is achieved early, and are progressively lost when reperfusion is delayed.
(Modified with permission from Antman E: Acute myocardial infarction. In Braunwald E et al. (eds): Heart Disease: A Textbook of Cardiovascular Medicine, 6th ed. Philadelphia, WB Saunders, 2001, pp 1114–1231.)
The early biochemical consequence of myocardial ischemia is the cessation of aerobic metabolism within seconds, leading to inadequate production of high-energy phosphates (e.g., creatine phosphate and adenosine triphosphate) and accumulation of potentially noxious metabolites (such as lactic acid) (Fig. 12-11A). Because of the exquisite dependence of myocardial function on oxygen, severe ischemia induces loss of contractility within 60 seconds. This cessation of function can precipitate acute heart failure long before myocardial cell death. As detailed in Chapter 1, ultrastructural changes (including myofibrillar relaxation, glycogen depletion, cell and mitochondrial swelling) also develop within a few minutes of the onset of ischemia. Nevertheless, these early changes are potentially reversible. As demonstrated both experimentally and in clinical studies, only severe ischemia lasting 20 to 30 minutes or longer leads to irreversible damage (necrosis) of cardiac myocytes. Ultrastructural evidence of irreversible myocyte injury (primary structural defects in the sarcolemmal membrane) develops only after prolonged, severe myocardial ischemia (such as occurs when blood flow is 10% or less of normal).
A key feature that marks the early phases of myocyte necrosis is the disruption of the integrity of the sarcolemmal membrane, which allows intracellular macromolecules to leak out of cells into the cardiac interstitium and ultimately into the microvasculature and lymphatics in the region of the infarct. Tests that measure the levels of myocardial proteins in the blood are important in the diagnosis and management of MI (see later). With prolonged severe ischemia, injury to the microvasculature then follows. The temporal progression of these events is summarized in Table 12-4.
TABLE 12-4 Approximate Time of Onset of Key Events in Ischemic Cardiac Myocytes
| Feature | Time |
|---|---|
| Onset of ATP depletion | Seconds |
| Loss of contractility | <2 min |
| ATP reduced | |
| to 50% of normal | 10 min |
| to 10% of normal | 40 min |
| Irreversible cell injury | 20–40 min |
| Microvascular injury | >1 hr |
ATP, adenosine triphosphate.
In most cases of acute MI, permanent damage to the heart occurs when the perfusion of the myocardium is severely reduced for an extended interval (usually at least 2 to 4 hours), (Fig. 12-11B). This delay in the onset of permanent myocardial injury provides the rationale for rapid diagnosis in acute MI—to permit early coronary intervention, the purpose of which is to establish reperfusion and salvage as much “at risk” myocardium as possible.
The progression of ischemic necrosis in the myocardium is summarized in Figure 12-12. Ischemia is most pronounced in the subendocardium; thus, irreversible injury of ischemic myocytes occurs first in the subendocardial zone. With more extended ischemia, a wavefront of cell death moves through the myocardium to involve progressively more of the transmural thickness and breadth of the ischemic zone. The precise location, size, and specific morphologic features of an acute MI depend on:
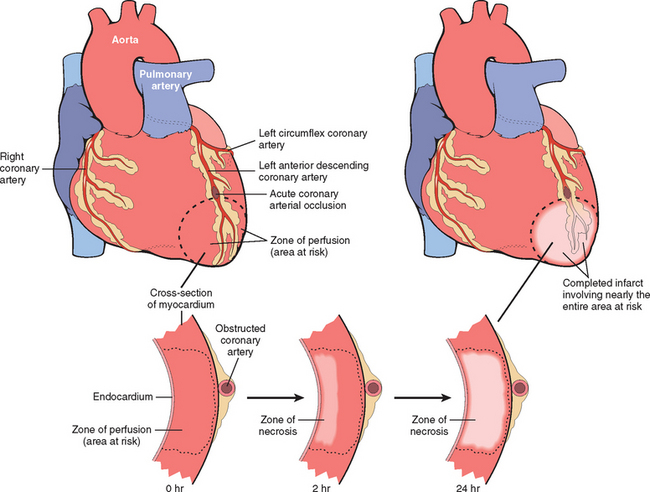
FIGURE 12-12 Progression of myocardial necrosis after coronary artery occlusion. Necrosis begins in a small zone of the myocardium beneath the endocardial surface in the center of the ischemic zone. The area that depends on the occluded vessel for perfusion is the “at risk” myocardium (shaded). Note that a very narrow zone of myocardium immediately beneath the endocardium is spared from necrosis because it can be oxygenated by diffusion from the ventricle.
Necrosis is usually complete within 6 hours of the onset of severe myocardial ischemia. However, in instances where the coronary arterial collateral system, stimulated by chronic is-chemia, is better developed and thereby more effective, the progression of necrosis may follow a more protracted course (possibly over 12 hours or longer).
Knowledge of the areas of myocardium perfused by the three major coronary arteries helps correlate sites of vascular obstruction with regions of myocardial infarction. Typically, the left anterior descending branch of the left coronary artery (LAD) supplies most of the apex of the heart (distal end of the ventricles), the anterior wall of the left ventricle, and the anterior two thirds of the ventricular septum. By convention, the coronary artery (either the right coronary artery [RCA] or the left circumflex artery [LCX]) that perfuses the posterior third of the septum is called “dominant” (even though the LAD and LCX collectively perfuse the majority of the left ventricular myocardium). In a right dominant circulation, present in approximately four fifths of individuals, the LCX generally perfuses only the lateral wall of the left ventricle, and the RCA supplies the entire right ventricular free wall, the posterobasal wall of the left ventricle, and the posterior third of the ventricular septum. Thus, occlusions of the RCA (as well as the left coronary artery) can cause left ventricular damage. The right and left coronary arteries function as end arteries, although anatomically most hearts have numerous intercoronary anastomoses (connections called the collateral circulation). Little blood courses through the collateral circulation in the normal heart. However, when one artery is severely narrowed, blood flows via collaterals from the high- to the low-pressure system, and causes the channels to enlarge. Thus, progressive dilation and growth of collaterals, stimulated by ischemia, may play a role in providing blood flow to areas of the myocardium otherwise deprived of adequate perfusion.
Transmural Versus Subendocardial Infarction.
The distribution of myocardial necrosis correlates with the location and cause of the decreased perfusion (Fig. 12-16). Most myocardial infarcts are transmural, in which the ischemic necrosis involves the full or nearly full thickness of the ventricular wall in the distribution of a single coronary artery. This pattern of infarction is usually associated with a combination of chronic coronary atherosclerosis, acute plaque change, and superimposed thrombosis (as discussed previously). In contrast, a subendocardial (nontransmural) infarct constitutes an area of ischemic necrosis limited to the inner one third to one half of the ventricular wall. As the subendocardial zone is normally the least perfused region of myocardium, this area is most vulnerable to any reduction in coronary flow. A subendocardial infarct can occur as a result of a plaque disruption followed by a coronary thrombus that becomes lysed before myocardial necrosis extends across the full thickness of the wall; in this case the infarct will be limited to the distribution of the coronary artery that suffered plaque change. However, subendocardial infarcts can also result from prolonged, severe reduction in systemic blood pressure, as in shock superimposed on chronic, otherwise noncritical, coronary stenoses. In the subendocardial infarcts that occur as a result of global hypotension, myocardial damage is usually circumferential, rather than being limited to the distribution of a single major coronary artery. Owing to the characteristic electrocardiographic changes resulting from myocardial ischemia/necrosis in various distributions, transmural infarcts are often referred to as “ST elevation infarcts” and subendocardial infarcts are known as “non-ST elevation infarcts.”
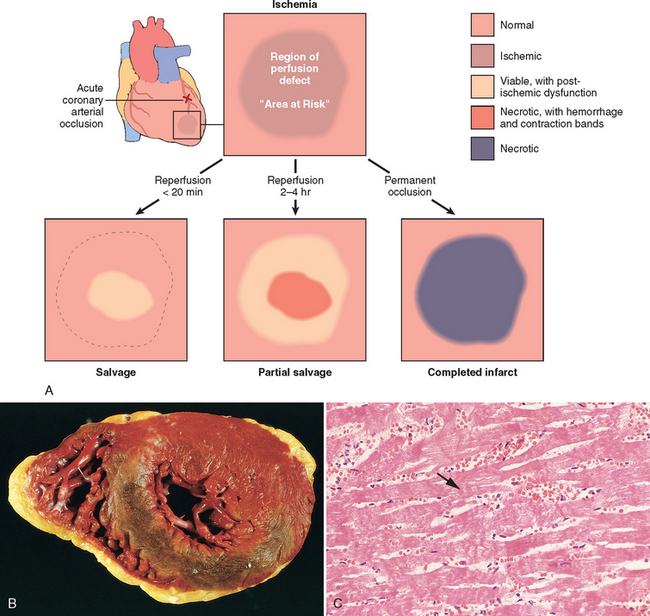
FIGURE 12-16 Consequences of myocardial ischemia followed by reperfusion. A, Schematic illustration of the progression of myocardial ischemic injury and its modification by restoration of flow (reperfusion). Hearts suffering brief periods of ischemia of longer than 20 minutes followed by reperfusion do not develop necrosis (reversible injury). Brief ischemia followed by reperfusion results in stunning. If coronary occlusion is extended beyond 20 minutes’ duration, a wavefront of necrosis progresses from subendocardium to subepicardium over time. Reperfusion before 3 to 6 hours of ischemia salvages ischemic but viable tissue. This salvaged tissue may also demonstrate stunning. Reperfusion beyond 6 hours does not appreciably reduce myocardial infarct size. B, Gross and C, microscopic appearance of myocardium modified by reperfusion. B, Large, densely hemorrhagic, anterior wall acute myocardial infarction in a patient with left anterior descending artery thrombus treated with streptokinase, a fibrinolytic agent (triphenyl tetrazolium chloride–stained heart slice). Specimen oriented with posterior wall at top. C, Myocardial necrosis with hemorrhage and contraction bands, visible as dark bands spanning some myofibers (arrow). This is the characteristic appearance of markedly ischemic myocardium that has been reperfused.
Morphology. The temporal evolution of the morphologic changes in acute MI and subsequent healing are summarized in Table 12-5.
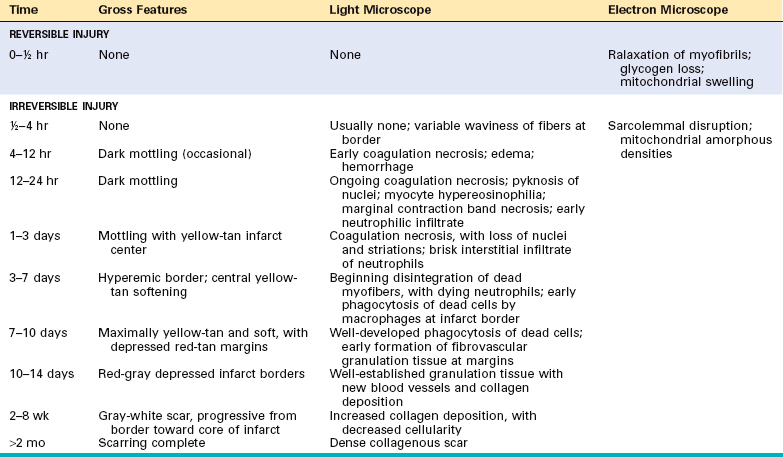
Nearly all transmural infarcts involve at least a portion of the left ventricle (comprising the free wall and ventricular septum) and encompass nearly the entire perfusion zone of the occluded coronary artery save for a narrow rim (∼0.1 mm) of preserved subendocardial myocardium that is sustained by the diffusion of oxygen and nutrients from the ventricular lumen.
Of MIs caused by a right coronary obstruction, 15% to 30% extend from the posterior free wall of the septal portion of the left ventricle into the adjacent right ventricular wall. Isolated infarction of the right ventricle is unusual (1% to 3% of cases), as is infarction of the atria.
The frequencies of involvement of each of the three main arterial trunks and the corresponding sites of myocardial lesions resulting in infarction (in the typical right dominant heart) are as follows (Fig. 12-13A):
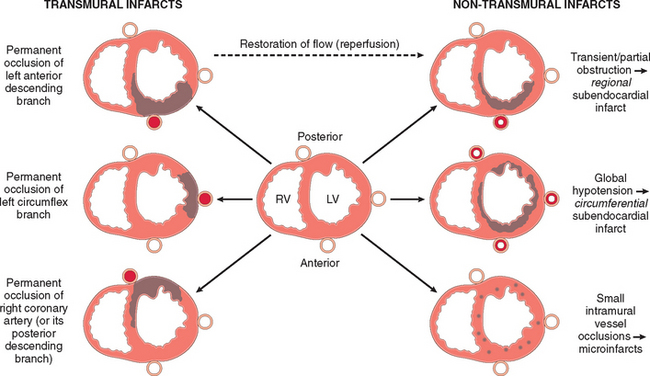
FIGURE 12-13 Distribution of myocardial ischemic necrosis correlated with the location and nature of decreased perfusion. Left, The positions of transmural acute infarcts resulting from occlusions of the major coronary arteries; top to bottom, left anterior descending, left circumflex, and right coronary arteries. Right, The types of infarcts that result from a partial or transient occlusion, global hypotension, or intramural small vessel occlusions.
Other locations of critical coronary arterial lesions causing infarcts are sometimes encountered, such as the left main coronary artery, the secondary branches of the left anterior descending coronary artery, or the marginal branches of the left circumflex coronary artery.
The gross and microscopic appearance of an infarct depends on the duration of survival of the patient following the MI. Areas of damage undergo a progressive sequence of morphologic changes that consist of typical ischemic coagulative necrosis (the predominant mechanism of cell death in MI, although apoptosis may also occur), followed by inflammation and repair that closely parallels tissue responses to injury at other sites.
Early recognition of acute MI can be difficult, particularly when death has occurred within a few hours after the onset of symptoms. MIs less than 12 hours old are usually not apparent on gross examination. If the patient died at least 2 to 3 hours after the infarct, however, it is possible to highlight the area of necrosis by immersion of tissue slices in a solution of triphenyltetrazolium chloride. This histochemical stain imparts a brick-red color to intact, noninfarcted myocardium where dehydrogenase (e.g., lactate dehydrogenase) activity is preserved. Because dehydrogenases leak out through the damaged membranes of dead cell, an infarct appears as an unstained pale zone (Fig. 12-14). By 12 to 24 hours an infarct can be identified grossly in transverse slices as a reddish-blue area of discoloration caused by stagnated, trapped blood. Thereafter, the infarct becomes progressively more sharply defined, yellow-tan, and soft. By 10 days to 2 weeks, it is rimmed by a hyperemic zone of highly vascularized granulation tissue. Over the succeeding weeks, the injured region evolves to a fibrous scar.
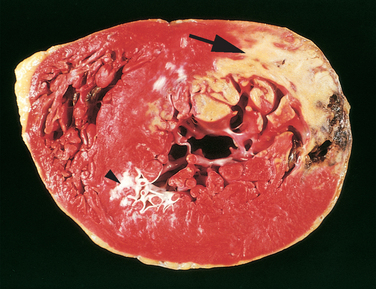
FIGURE 12-14 Acute myocardial infarct, predominantly of the posterolateral left ventricle, demonstrated histochemically by a lack of staining by triphenyltetrazolium chloride in areas of necrosis (arrow). The staining defect is due to the enzyme leakage that follows cell death. Note the myocardial hemorrhage at one edge of the infarct that was associated with cardiac rupture, and the anterior scar (arrowhead), indicative of old infarct. Specimen is oriented with the posterior wall at the top.
The histopathologic changes also proceed in a fairly predictable sequence (summarized in Fig. 12-15). The typical changes of coagulative necrosis become detectable in the first 6 to 12 hours. “Wavy fibers” may be present at the periphery of the infarct; these changes probably result from the forceful systolic tugs of the viable fibers on immediately adjacent, noncontractile dead fibers, which stretches and folds them. An additional sublethal ischemic change may be seen in the margins of infarcts: so-called vacuolar degeneration or myocytolysis, which takes the form of large vacuolar spaces within cells that probably contain water. The necrotic muscle elicits acute inflammation (most prominent between 1 and 3 days). Thereafter macrophages remove the necrotic myocytes (most pronounced at 3 to 7 days), and the damaged zone is progressively replaced by the ingrowth of highly vascularized granulation tissue (most prominent at 1 to 2 weeks); as healing progresses, this is replaced by fibrous tissue. In most instances, scarring is well advanced by the end of the sixth week, but the efficiency of repair depends on the size of the original lesion.
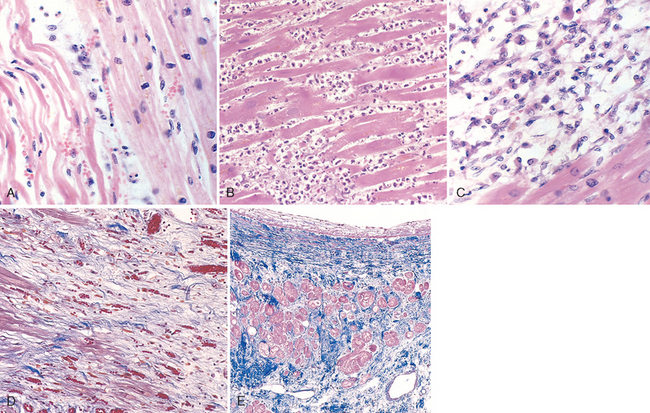
FIGURE 12-15 Microscopic features of myocardial infarction and its repair. A, One-day-old infarct showing coagulative necrosis and wavy fibers (elongated and narrow, as compared with adjacent normal fibers at right). Widened spaces between the dead fibers contain edema fluid and scattered neutrophils. B, Dense polymorphonuclear leukocytic infiltrate in area of acute myocardial infarction of 3 to 4 days’ duration. C, Nearly complete removal of necrotic myocytes by phagocytosis (approximately 7 to 10 days). D, Granulation tissue characterized by loose collagen and abundant capillaries. E, Well-healed myocardial infarct with replacement of the necrotic fibers by dense collagenous scar. A few residual cardiac muscle cells are present.
Since healing requires the participation of inflammatory cells that migrate to the region of damage through intact blood vessels, which often survive only at the infarct margins, the infarct heals from its margins toward its center. Thus, a large infarct may not heal as quickly or as completely as a small one. A healing infarct may appear nonuniform, with the most advanced healing at the periphery. Once a lesion is completely healed, it is impossible to determine its age (i.e., the dense fibrous scar of 8-week-old and 10-year-old infarcts may look identical).
Infarcts may expand beyond their original borders over a period of days to weeks via a process of repetitive necrosis of adjacent regions (extension). In such cases, there is a central zone in which healing is more advanced than the periphery of the infarct. This contrasts with the appearance of a simple infarct described above, in which the most advanced repair is peripheral. Infarct extension may occur because of retrograde propagation of a thrombus, proximal vasospasm, progressively impaired cardiac contractility that renders flow through moderate stenoses insufficient, the deposition of platelet-fibrin microemboli, or an arrhythmia that impairs cardiac function.
We now consider interventions that seek to limit infarct size by salvaging myocardium that is not yet necrotic.
Infarct Modification by Reperfusion.
The most effective way to “rescue” ischemic myocardium threatened by infarction is to restore myocardial blood flow as rapidly as possible, a process referred to as reperfusion.51 Although this can often be accomplished, reperfusion may also trigger deleterious complications, including arrhythmias, myocardial hemorrhage with contraction bands, irreversible cell damage superimposed on the original ischemic injury (reperfusion injury), microvascular injury, and prolonged ischemic dysfunction (myocardial stunning); these are discussed below and summarized in Figures 12-16 and 12-17. Coronary intervention (i.e., thrombolysis, angioplasty, stent placement, or coronary artery bypass graft [CABG] surgery) is often used in an attempt to dissolve, mechanically alter, or bypass the lesion that initiated the acute MI. The purpose of these treatments is to restore blood flow to the area at risk for infarction and rescue the ischemic (but not yet necrotic) heart muscle. Because loss of myocardial viability in infarction is progressive, occurring over a period of at least several hours (see Fig. 12-11B and Fig. 12-17A), early reperfusion can salvage myocardium and thereby limit infarct size, with consequent improvement in both short- and long-term function and survival. The potential benefit of reperfusion is related to (1) the rapidity with which the coronary obstruction is alleviated (the first 3 to 4 hours following onset are critical) and (2) the extent of correction of the vascular occlusion and the underlying causal lesion. For example, thrombolysis can remove a thrombus occluding a coronary artery, but does not alter the underlying atherosclerotic plaque that initiated it. In contrast, percutaneous transluminal coronary angioplasty (PTCA) with stent placement not only eliminates a thrombotic occlusion but also can relieve some of the original obstruction and instability caused by the underlying disrupted plaque. CABG provides flow around a blocked vessel.
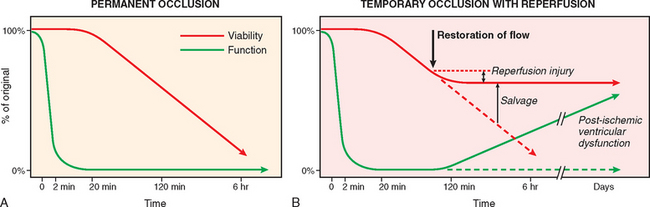
FIGURE 12-17 Effects of reperfusion on myocardial viability and function. Following coronary occlusion, contractile function is lost within 2 minutes and viability begins to diminish after approximately 20 minutes. If perfusion is not restored (A), then nearly all myocardium in the affected region will die. B, If flow is restored, then some necrosis is prevented, myocardium is salvaged, and at least some function will return. The earlier reperfusion occurs, the greater the degree of salvage. However, the process of reperfusion itself may induce some damage (reperfusion injury), and return of function of salvaged myocardium may be delayed for hours to days (post-ischemic ventricular dysfunction).
Recall that (1) severe ischemia does not cause immediate cell death even in the most severely affected regions of myocardium, and (2) not all regions of myocardium are equally ischemic. Therefore, the outcome following the restoration of blood flow may vary from region to region. As indicated in Figure 12-16A, reperfusion of myocardium within 20 minutes of the onset of ischemia may completely prevent necrosis. Reperfusion after a longer interval may not prevent all necrosis but can salvage at least some myocytes that would have otherwise died.
The typical appearance of reperfused myocardium is illustrated in Figure 12-16B and C. A reperfused infarct is usually hemorrhagic because the vasculature is injured during the period of ischemia and leaks when flow is restored. Microscopic examination reveals that myocytes that were irreversibly injured at the time of reperfusion often contain contraction bands, intensely eosinophilic intracellular “stripes” composed of closely packed sarcomeres. These result from the exaggerated contraction of myofibrils when perfusion is reestablished, at which time the interior of dead cells with damaged plasma membranes are exposed to a high concentration of calcium ions from the plasma. Thus, reperfusion not only salvages reversibly injured cells but also alters the morphology of lethally injured cells.
In addition to its benefits, reperfusion may also have some deleterious effects on the vulnerable ischemic myocardium (reperfusion injury; see Fig. 12-17B).52 The clinical significance of myocardial reperfusion injury is uncertain. As discussed in Chapter 1, reperfusion injury may be mediated by oxidative stress, calcium overload, and potentially inflammation initiated during reperfusion. Reperfusion-induced microvascular injury causes not only hemorrhage but also endothelial swelling that occludes capillaries and may limit the reperfusion of critically injured myocardium (called no-reflow).
Biochemical abnormalities may also persist for a period of days to several weeks in myocytes that are rescued from ischemia by reperfusion. These are thought to underlie a phenomenon referred to as stunned myocardium, a state of reversible cardiac failure that usually recovers after several days.53 Reperfusion also frequently induces arrhythmias. Myocardium that is subjected to chronic, sublethal ischemia may also enter into a state of lowered metabolism and function that is referred to as hibernation.54 The function of hibernating myocardium may be restored by revascularization (e.g., by CABG surgery, angioplasty, or stenting). Paradoxically, repetitive short-lived transient severe ischemia may protect the myocardium against infarction (a phenomenon known as preconditioning) by mechanisms that are not understood.55
Clinical Features.
MI is diagnosed by clinical symptoms, laboratory tests for the presence of myocardial proteins in the plasma, and characteristic electrocardiographic changes. Patients with MI often present with a rapid, weak pulse and profuse sweating (diaphoresis). Dyspnea due to impaired contractility of the ischemic myocardium and the resultant pulmonary congestion and edema is common. However, in about 10% to 15% of patients the onset is entirely asymptomatic and the disease is discovered only by electrocardiographic changes or laboratory tests that show evidence of myocardial damage (see below). Such “silent” MIs are particularly common in elderly patients and in the setting of diabetes mellitus.
The laboratory evaluation of MI is based on measuring the blood levels of proteins that leak out of fatally injured myocytes; these molecules include myoglobin, cardiac troponins T and I, the MB fraction of creatine kinase (CK-MB), lactate dehydrogenase, and many others (Fig. 12-18).56 The diagnosis of myocardial injury is established when blood levels of these cardiac biomarkers are increased in the clinical setting of acute ischemia. The rate of appearance of these markers in the peripheral circulation depends on several factors, including their intracellular location and molecular weight, the blood flow and lymphatic drainage in the area of the infarct, and the rate of elimination of the marker from the blood.
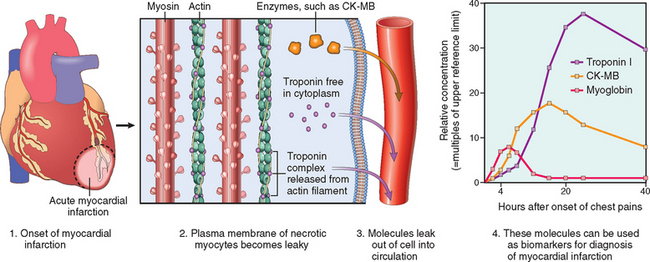
FIGURE 12-18 Release of myocyte proteins in myocardial infarction. Some of these proteins (e.g., troponin I, C, or T and creatine phosphokinase, MB fraction [CK-MB]) are used as diagnostic biomarkers.
The most sensitive and specific biomarkers of myocardial damage are cardiac-specific proteins, particularly Troponins I and T (proteins that regulate calcium-mediated contraction of cardiac and skeletal muscle). Troponins I and T are not normally detectable in the circulation. Following an MI, levels of both begin to rise at 2 to 4 hours and peak at 48 hours. Formerly the “gold standard,” cardiac creatine kinase remains useful. Creatine kinase, an enzyme that is present in brain, myocardium, and skeletal muscle, is a dimer composed of two isoforms designated “M” and “B.” MM homodimers are found predominantly in cardiac and skeletal muscle; BB homodimers in brain, lung, and many other tissues; and MB heterodimers principally in cardiac muscle, with lesser amounts also being found in skeletal muscle. As a result, the MB form of creatine kinase (CK-MB) is sensitive but not specific, since it is also elevated when skeletal muscle is injured. CK-MB begins to rise within 2 to 4 hours of the onset of MI, peaks at about 24 hours, and returns to normal within approximately 72 hours. Although the diagnostic sensitivities of cardiac troponin and CK-MB measurements are similar in the early stages of MI, elevated troponin levels persist for approximately 7 to 10 days after acute MI, well after CK-MB levels have returned to normal. Troponin and CK-MB levels peak earlier in patients whose hearts are successfully reperfused, because proteins are washed out of the necrotic tissue more rapidly. Unchanged levels of CK-MB and troponin over a period of 2 days essentially excludes the diagnosis of MI.
Consequences and Complications of MI.
Extraordinary progress has been made in the treatment of patients with acute MI. Concurrent with the decrease in the overall mortality of IHD since the 1960s, the in-hospital death rate has declined from around 30% to approximately 7% in patients receiving timely therapy. Half of the deaths associated with acute MI occur within 1 hour of onset; most of these individuals never reach the hospital. Therapies given routinely in the setting of acute MI include aspirin and heparin (to prevent further thrombosis); oxygen (to minimize ischemia); nitrates (to induce vasodilation and reverse vasospasm); beta-adrenergic inhibitors (beta-blockers, to diminish cardiac oxygen demand and decrease the risk of arrythmias); angiotensinogen converting enzyme (ACE) inhibitors (to limit venticular dilation); and maneuvers that aim to open up blocked vessels, including the administration of fibrinolytic agents, coronary angioplasty with or without stenting, and emergent CABG surgery. The choice of therapy depends on the clinical picture and the expertise of the treating institution. Angioplasty is highly effective in skilled hands, while fibinolytic therapy can be given with almost equivalent efficacy by simple infusion. In general, factors associated with a poor prognosis include advanced age, female gender, diabetes mellitus, and, as a result of the cumulative loss of functional myocardium, previous MI.
Despite these interventions, many patients have one or more complications following acute MI, including the following (some of which are illustrated in Fig. 12-19):
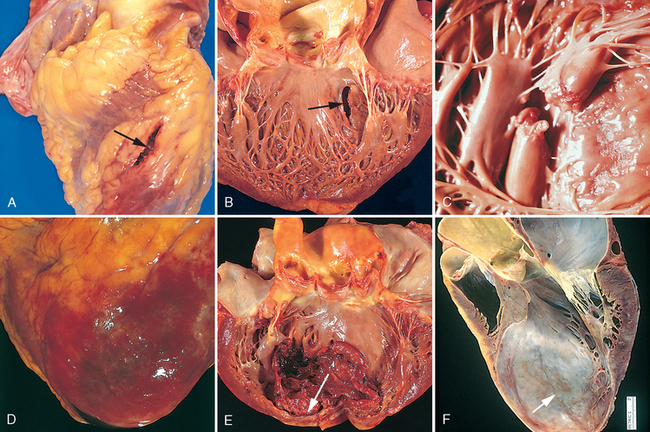
FIGURE 12-19 Complications of myocardial infarction. Cardiac rupture syndromes (A–C). A, Anterior myocardial rupture in an acute infarct (arrow). B, Rupture of the ventricular septum (arrow). C, Complete rupture of a necrotic papillary muscle. D, Fibrinous pericarditis, showing a dark, roughened epicardial surface overlying an acute infarct. E, Early expansion of anteroapical infarct with wall thinning (arrow) and mural thrombus. F, Large apical left ventricular aneurysm. The left ventricle is on the right in this apical four-chamber view of the heart.
(A–E, Reproduced by permission from Schoen FJ: Interventional and Surgical Cardiovascular Pathology: Clinical Correlations and Basic Principles. Philadelphia, WB Saunders, 1989; F, Courtesy of William D. Edwards, MD, Mayo Clinic, Rochester, MN.)
The risk of specific postinfarct complications and the prognosis depend primarily on the infarct size, location, and thickness (subendocardial or transmural). Large transmural infarcts yield a higher probability of cardiogenic shock, arrhythmias, and late CHF. Patients with anterior transmural infarcts are at greatest risk for free-wall rupture, expansion, mural thrombi, and aneurysm. In contrast, posterior transmural infarcts are more likely to be complicated by conduction blocks, right ventricular involvement, or both; when acute VSDs occur in this area they are more difficult to manage. Overall, however, patients with anterior infarcts have a worse clinical course than those with inferior (posterior) infarcts. With subendocardial infarcts, only rarely do pericarditis, rupture, and aneurysms occur.
In addition to the sequence of repair in the infarcted tissues described above, the noninfarcted segments of the ventricle undergo hypertrophy and dilation; collectively, these changes are termed ventricular remodeling. The compensatory hypertrophy of noninfarcted myocardium is initially hemodynamically beneficial. However, this adaptive effect may be overwhelmed by ventricular dilation (with or without ventricular aneurysm) and increased oxygen demand, which can exacerbate ischemia and depress cardiac function. There may also be changes in ventricular shape and stiffening of the ventricle due to scar formation and hypertrophy that further diminish cardiac output. Some of these deleterious effects appear to be reduced by ACE inhibitors, which lessen the ventricular dilation that occurs after MI.
Long-term prognosis after MI depends on many factors, the most important of which are the quality of residual left ventricular function and the extent of vascular obstructions in vessels that perfuse the viable myocardium. The overall total mortality within the first year is about 30%. Thereafter there is a 3% to 4% mortality among survivors with each passing year. Infarct prevention through control of risk factors in individuals who have never experienced MI (primary prevention) and prevention of reinfarction in those who have recovered from an acute MI (secondary prevention) are important strategies that have received much attention and achieved considerable success.
The relationship of the causes, pathophysiology, and consequences of MI are summarized in Figure 12-20, including the possible outcomes of chronic IHD and sudden death, to be discussed next.
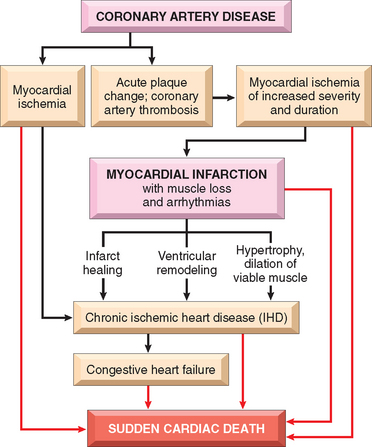
CHRONIC IHD
The designation chronic IHD is used here to describe progressive heart failure as a consequence of ischemic myocardial damage. The term ischemic cardiomyopathy is often used by clinicians to describe chronic IHD. In most instances there has been prior MI and sometimes previous coronary arterial interventions and/or bypass surgery. Chronic IHD usually appears postinfarction due to the functional decompensation of hypertrophied noninfarcted myocardium (see earlier discussion of cardiac hypertrophy). However, in other cases severe obstructive coronary artery disease may present as chronic IHD in the absence of prior infarction.
Morphology. Hearts from patients with chronic IHD are usually enlarged and heavy, due to left ventricular hypertrophy and dilation. Invariably there is some degree of obstructive coronary atherosclerosis. Discrete scars representing healed infarcts are usually present. The mural endocardium may have patchy, fibrous thickenings, and mural thrombi may be present. Microscopic findings include myocardial hypertrophy, diffuse subendocardial vacuolization, and fibrosis.
Clinically, progressive CHF may occur in patients who have had past episodes of MI or anginal attacks. In some individuals, however, progressive myocardial damage is silent, and heart failure is the first indication of IHD. The diagnosis rests largely on the exclusion of other cardiac diseases. Patients with chronic IHD account for nearly half of cardiac transplant recipients.
SUDDEN CARDIAC DEATH
Sudden cardiac death (SCD) strikes down about 300,000 to 400,000 individuals annually in the United States. It is defined as unexpected death from cardiac causes in individuals without symptomatic heart disease or early after symptom onset (usually within 1 hour). SCD is usually the consequence of a lethal arrhythmia (e.g., asystole, ventricular fibrillation). It most frequently occurs in the setting of IHD; in some cases, SCD is the first clinical manifestation of IHD.
Acute myocardial ischemia is the most common trigger for fatal arrhythmias.57 Although ischemic injury can affect the conduction system and create electromechanical cardiac instability, fatal arrhythmias usually result from acute ischemia-induced electrical instability of myocardium that is distant from the conduction system. Arrythmogenic foci are often located adjacent to scars left by old MIs.
Nonatherosclerotic conditions associated with SCD include
Morphology. Marked coronary atherosclerosis with a critical (>75%) stenosis involving one or more of the three major vessels is present in 80% to 90% of SCD victims; only 10% to 20% of cases are of nonatherosclerotic origin. Usually there are high-grade stenoses (>90%); in approximately one half, acute plaque disruption is observed, and in approximately 25% diagnostic changes of acute MI are seen.58 This suggests that many patients who die suddenly are suffering an MI, but the short interval from onset to death precludes the development of diagnostic myocardial changes. However, in one study of those who had been successfully resuscitated from a sudden cardiac arrest, a new MI occurred in only 39% of the patients.59 Thus, most SCD is not associated with acute MI; most of these deaths are thought to result from myocardial ischemia–induced irritability that initiates malignant ventricular arrhythmias. Scars of previous infarcts and subendocardial myocyte vacuolization indicative of severe chronic ischemia are common in such patients.
Heritable conditions associated with SCD are of importance, since they may provide a basis for intervention in surviving family members.60 Some of these disorders are associated with recognizable anatomic abnormalities (e.g., congenital anomalies, hypertrophic cardiomyopathy, mitral valve prolapse). However, other heritable arrhythmias can precipitate sudden death in the absence of structural cardiac pathology (so-called primary electrical disorders). These syndromes can only be diagnosed definitively by genetic testing, which is performed in those with a positive family history or an unexplained nonlethal arrhythmia.
The primary electrical abnormalities of the heart that predispose to SCD include long QT syndrome, Brugada syndrome, short QT syndrome, catecholaminergic polymorphic ventricular tachycardia, Wolff-Parkinson-White syndrome, congenital sick sinus syndrome, and isolated cardiac conduction disease.61 The most important of these disorders are the so-called channelopathies, which are caused by mutations in genes that are required for normal ion channel function.62 These disorders (mostly with autosomal-dominant inheritance) either involve genes that encode the ion channels (including Na+, K+, and Ca+), or accessory proteins that are essential for the normal function of the same channels, which are responsible for conducting the electrical currents that mediate contraction of the heart. The prototype is the long QT syndrome, characterized by prolongation of the QT segment in electrocardiograms and susceptibility to malignant ventricular arrhythmias. Mutations in seven different genes account for the majority of cases of long QT syndrome. The most frequent mutations are in the gene encoding KCNQ1 and result in decreased potassium currents. Ion channels are needed for the normal function of many tissues, and certain channelopathies are also associated with skeletal muscle disorders and diabetes; however, the most common cardiac channelopathies are isolated disorders of the heart.
The prognosis of many patients vulnerable to SCD, including those with chronic IHD, is markedly improved by implantation of a pacemaker or an automatic cardioverter defibrillator, which senses and electrically counteracts an episode of ventricular fibrillation.63
Hypertensive Heart Disease
Hypertensive heart disease (HHD) stems from the increased demands placed on the heart by hypertension, which causes pressure overload and ventricular hypertrophy. Although most commonly seen in the left heart as the result of systemic hypertension, pulmonary hypertension can cause right-sided HHD, or cor pulmonale.
SYSTEMIC (LEFT-SIDED) HYPERTENSIVE HEART DISEASE
In hypertension, hypertrophy of the heart is an adaptive response to pressure overload that can lead to myocardial dysfunction, cardiac dilation, CHF, and in some cases sudden death. The minimal criteria for the diagnosis of systemic HHD are the following: (1) left ventricular hypertrophy (usually concentric) in the absence of other cardiovascular pathology and (2) a history or pathologic evidence of hypertension. The Framingham Study established unequivocally that even mild hypertension (levels only slightly above 140/90 mm Hg), if sufficiently prolonged, induces left ventricular hypertrophy. Approximately 25% of the population of the United States suffers from hypertension of at least this degree. The pathogenesis of hypertension is discussed in Chapter 11.
Morphology. Hypertension induces left ventricular pressure overload hypertrophy, initially without ventricular dilation. As a result, the left ventricular wall thickening increases the weight of the heart disproportionately to the increase in overall cardiac size (Fig. 12-21A). The thickness of the left ventricular wall may exceed 2.0 cm, and the heart weight may exceed 500 gm. In time the increased thickness of the left ventricular wall imparts a stiffness that impairs diastolic filling, often inducing left atrial enlargement.
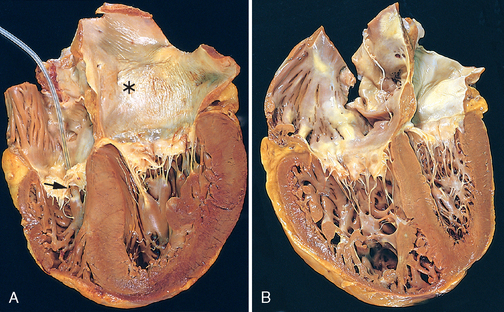
FIGURE 12-21 Hypertensive heart disease, systemic and pulmonary. A, Systemic (left-sided) hypertensive heart disease. There is marked concentric thickening of the left ventricular wall causing reduction in lumen size. The left ventricle and left atrium (asterisk) is on the right in this apical four-chamber view of the heart. A pacemaker is present in the right ventricle (arrow). B, Pulmonary (right-sided) hypertensive heart disease (cor pulmonale). The right ventricle is markedly dilated and has a thickened free wall and hypertrophied trabeculae (apical four-chamber view of heart, right ventricle on left). The shape of the left ventricle (to the right) has been distorted by the enlarged right ventricle.
Microscopically, the earliest change of systemic HHD is an increase in the transverse diameter of myocytes, which may be difficult to appreciate on routine microscopy. At a more advanced stage variable degrees of cellular and nuclear enlargement become apparent, often accompanied by interstitial fibrosis. The biochemical, molecular, and morphologic changes that occur in hypertensive hypertrophy are similar to those noted in other conditions associated with myocardial pressure overload.
Compensated systemic HHD may be asymptomatic, producing only electrocardiographic or echocardiographic evidence of left ventricular enlargement. In many patients, systemic HHD comes to attention due to new atrial fibrillation induced by left atrial enlargement or CHF. Depending on the severity, duration, and underlying basis of the hypertension, and on the adequacy of therapeutic control, the patient may (1) enjoy normal longevity and die of unrelated causes, (2) develop IHD due to the potentiating effects of hypertension on coronary atherosclerosis, (3) suffer renal damage or cerebrovascular stroke as direct effects of hypertension, or (4) experience progressive heart failure or SCD. Effective control of hypertension can prevent or lead to regression of cardiac hypertrophy and its associated risks.
PULMONARY (RIGHT-SIDED) HYPERTENSIVE HEART DISEASE (COR PULMONALE)
Normally, because the pulmonary vasculature is the low pressure side of the circulation, the right ventricle has a thinner and more compliant wall than the left ventricle.64 Cor pulmonale, as isolated pulmonary HHD is frequently called, stems from pressure overload of the right ventricle,65 and is characterized by right ventricular hypertrophy, dilation, and potentially failure secondary to pulmonary hypertension. The most frequent causes are disorders of the lungs, especially chronic respiratory diseases such as emphysema, or primary pulmonary hypertension (Table 12-6). It should be remembered, however, that pulmonary venous hypertension most commonly occurs as a complication of left-sided heart diseases of various etiologies.
TABLE 12-6 Disorders Predisposing to Cor Pulmonale
| DISEASES OF THE PULMONARY PARENCHYMA |
| DISEASES OF THE PULMONARY VESSELS |
| DISORDERS AFFECTING CHEST MOVEMENT |
| DISORDERS INDUCING PULMONARY ARTERIAL CONSTRICTION |
Cor pulmonale may be acute or chronic. Acute cor pulmonale can follow massive pulmonary embolism. Chronic cor pulmonale results from right ventricular hypertrophy (and dilation) secondary to prolonged pressure overload, such as can occur with chronic lung diseases and a variety of other conditions, many of which are discussed in more detail in Chapter 15.
Morphology. In acute cor pulmonale there is marked dilation of the right ventricle without hypertrophy. On cross-section the normal crescent shape of the right ventricle is transformed to a dilated ovoid. In chronic cor pulmonale the right ventricular wall thickens, sometimes up to 1.0 cm or more (Fig. 12-21B). More subtle right ventricular hypertrophy may take the form of thickening of the muscle bundles in the outflow tract, immediately below the pulmonary valve, or thickening of the moderator band, the muscle bundle that connects the ventricular septum to the anterior right ventricular papillary muscle. Sometimes, the hypertrophied right ventricle compresses the left ventricular chamber, or leads to regurgitation and fibrous thickening of the tricuspid valve. Normally, the myocytes of the right ventricle are haphazardly arranged and the wall contains transmural fat; in right ventricular hypertrophy, fat in the wall disappears and the myocytes align themselves circumferentially.
Valvular Heart Disease
Valvular disease can come to clinical attention due to stenosis, insufficiency (regurgitation or incompetence), or both. Stenosis is the failure of a valve to open completely, which impedes forward flow. Insufficiency, in contrast, results from failure of a valve to close completely, thereby allowing reversed flow. These abnormalities can be present alone or coexist, and may involve only a single valve (isolated disease) or more than one valve (combined disease). Functional regurgitation is used to describe the incompetence of a valve stemming from an abnormality in one of its support structures. For example, dilation of the right or left ventricle can pull the ventricular papillary muscles down and outward, thereby preventing proper closure of otherwise normal mitral or tricuspid leaflets. Similarly, dilation of the aortic or pulmonary artery may pull the valve commissures apart and prevent full closure of the aortic or pulmonary valve cusps. Functional mitral valve regurgitation is particularly common in IHD (ischemic mitral regurgitation).66
The clinical consequences of valve dysfunction vary depending on the valve involved, the degree of impairment, how fast it develops, and the rate and quality of compensatory mechanisms. For example, sudden destruction of an aortic valve cusp by infection (infective endocarditis; see later) can cause acute, massive regurgitation that can be rapidly fatal. In contrast, rheumatic mitral stenosis usually develops indolently over years, and its clinical effects are often remarkably well tolerated. Certain conditions can complicate valvular heart disease by increasing the demands on the heart; for example, pregnancy can exacerbate valve disease and lead to an unfavorable maternal or fetal outcome.67 Valvular stenosis or insufficiency often produces secondary changes, both proximal and distal to the affected valve. Generally, valvular stenosis leads to pressure overload of the heart, whereas valvular insufficiency leads to volume overload of the heart. In addition, the ejection of blood through narrowed stenotic valves can produce high speed “jets” of blood that injure the endocardium where they impact.
Valvular abnormalities may be congenital (discussed earlier) or acquired. Acquired stenoses of the aortic and mitral valves account for approximately two thirds of all cases of valve disease. Valvular stenosis is almost always due to a chronic abnormality of the valve cusp that becomes clinically evident after many years. Relatively few disorders produce valvular stenosis. In contrast, valvular insufficiency can result from intrinsic disease of the valve cusps or damage to or distortion of the supporting structures (e.g., the aorta, mitral annulus, tendinous cords, papillary muscles, ventricular free wall). Valvular insufficiency has many causes and may appear acutely, as with rupture of the cords, or chronically in disorders associated with leaflet scarring and retraction.
The causes of acquired heart valve diseases are summarized in Table 12-7 and discussed in the following sections. The most frequent causes of the major functional valvular lesions are:
TABLE 12-7 Major Etilogies of Acquired Heart Valve Disease
| Mitral Valve Disease | Aortic Valve Disease |
|---|---|
| MITRAL STENOSIS | AORTIC STENOSIS |
| Postinflammatory scarring (rheumatic heart disease) | Postinflammatory scarring (rheumatic heart disease) |
| Senile calcific aortic stenosis | |
| Calcification of congenitally deformed valve | |
| MITRAL REGURGITATION | AORTIC REGURGITATION |
| Abnormalities of Leaflets and Commissures | |
| Postinflammatory scarring | Postinflammatory scarring (rheumatic heart disease) |
| Infective endocarditis | Infective endocarditis |
| Mitral valve prolapse | Marfan syndrome |
| Drugs (e.g., fen-phen) | |
| Abnormalities of Tensor Apparatus | Aortic Disease |
| Rupture of papillary muscle | Degenerative aortic dilation |
| Papillary muscle dysfunction (fibrosis) | Syphilitic aortitis |
| Rupture of chordae tendineae | Ankylosing spondylitis |
| Rheumatoid arthritis | |
| Marfan syndrome | |
| Abnormalities of Left Ventricular Cavity and/or Annulus | |
| LV enlargement (myocarditis, dilated cardiomyopathy) | |
| Calcification of mitral ring | |
LV, Left ventricular.
Modified from Schoen FJ: Surgical pathology of removed natural and prosthetic valves. Hum Pathol 18:558, 1987.
VALVULAR DEGENERATION ASSOCIATED WITH CALCIFICATION
Heart valves are subjected to high levels of repetitive mechanical stress, particularly at the hinge points of the cusps and leaflets, as a result of (1) 40 million or more cardiac contractions per year, (2) substantial tissue deformations during each contraction, and (3) transvalvular pressure gradients in the closed phase of each contraction of approximately 120 mm Hg for the mitral and 80 mm Hg for the aortic valve. It is therefore not surprising that these delicate structures can suffer cumulative damage and dystrophic calcification (deposits of calcium phosphate salts) that lead to clinically important dysfunction.68
Calcific Aortic Stenosis
The most common of all valvular abnormalities, acquired aortic stenosis, is usually the consequence of age-associated “wear and tear” of either anatomically normal valves or congenitally bicuspid valves (∼1% of the population).69 The prevalence of aortic stenosis, estimated at 2%, is increasing with the rising average age of the population. Aortic stenosis of previously normal valves (termed senile calcific aortic stenosis) usually comes to clinical attention in the seventh to ninth decades of life, whereas stenotic bicuspid valves tend to present in patients 50 to 70 years of age.
Prior work attributed aortic valve calcification to wear and tear degeneration and dystrophic and passive accumulation of hydroxyapatite, the same calcium salt that is found in bone.70 More recent studies suggest that chronic injury due to hyperlipidemia, hypertension, inflammation, and other factors implicated in atherosclerosis may have a role and perhaps even precede the calcification. It is clear, however, that the valve injury of calcific aortic stenosis differs in some respects from atherosclerosis. Most notably, instead of accumulating smooth muscle cells, the abnormal valves contain cells resembling osteoblasts that synthesize bone matrix proteins and promote the deposition of calcium salts. Bicuspid valves incur greater mechanical stress than normal tricuspid valves, which may explain why they become stenotic more rapidly.
Morphology. The morphologic hallmark of nonrheumatic, calcific aortic stenosis (with either tricuspid or bicuspid valves) is heaped-up calcified masses within the aortic cusps that ultimately protrude through the outflow surfaces into the sinuses of Valsalva, preventing the opening of the cusps. The free edges of the cusps are usually not involved (Fig. 12-22A). The calcific process begins in the valvular fibrosa, at the points of maximal cusp flexion (near the margins of attachment). Microscopically, the layered architecture of the valve is largely preserved. An earlier, hemodynamically inconsequential stage of the calcification process is called aortic valve sclerosis. In aortic stenosis the functional valve area is decreased sufficiently by large nodular calcific deposits to cause measurable obstruction to outflow; this subjects the left ventricular myocardium to progressively increasing pressure overload.
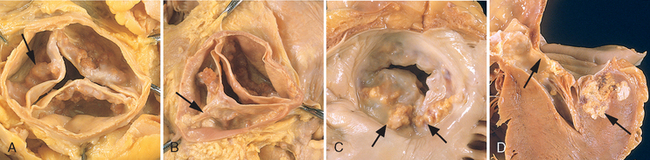
FIGURE 12-22 Calcific valvular degeneration. A, Calcific aortic stenosis of a previously normal valve (viewed from aortic aspect). Nodular masses of calcium are heaped up within the sinuses of Valsalva (arrow). Note that the commissures are not fused, as in postrheumatic aortic valve stenosis (see Fig. 12-27E). B, Calcific aortic stenosis of a congenitally bicuspid valve. One cusp has a partial fusion at its center, called a raphe (arrow). C and D, Mitral annular calcification, with calcific nodules at the base (attachment margin) of the anterior mitral leaflet (arrows). C, Left atrial view. D, Cut section of myocardium.
In contrast to rheumatic (and congenital) aortic stenosis (see Fig. 12-24E), commissural fusion is not usually seen. The mitral valve is generally normal, although some patients may have direct extension of aortic valve calcific deposits onto the anterior mitral leaflet. In contrast, virtually all patients with rheumatic aortic stenosis also have concomitant and characteristic structural abnormalities of the mitral valve (see later).
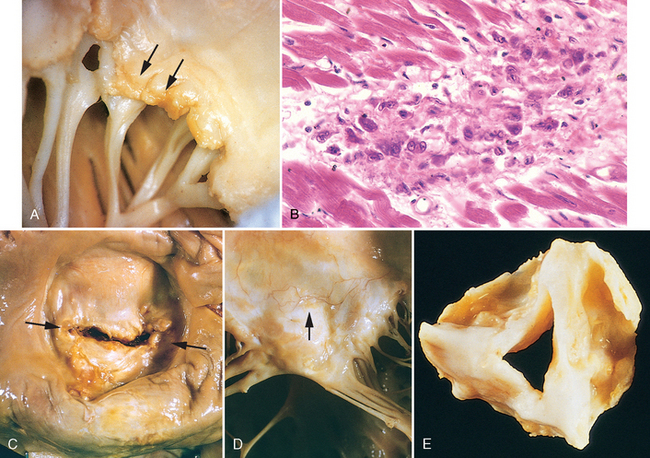
FIGURE 12-24 Acute and chronic rheumatic heart disease. A, Acute rheumatic mitral valvulitis superimposed on chronic rheumatic heart disease. Small vegetations (verrucae) are visible along the line of closure of the mitral valve leaflet (arrows). Previous episodes of rheumatic valvulitis have caused fibrous thickening and fusion of the chordae tendineae. B, Microscopic appearance of Aschoff body in a patient with acute rheumatic carditis. The myocardial interstitium has a circumscribed collection of mononuclear inflammatory cells, including some large macrophages with prominent nucleoli and a binuclear macrophage, associated with necrosis. C and D, Mitral stenosis with diffuse fibrous thickening and distortion of the valve leaflets and commissural fusion (arrows, C), and thickening of the chordae tendineae (D). Note neovascularization of anterior mitral leaflet (arrow, D). E, Rheumatic aortic stenosis, demonstrating thickening and distortion of the cusps with commissural fusion.
(E, Reproduced from Schoen FJ, St. John-Sutton M: Contemporary issues in the pathology of valvular heart disease. Hum Pathol 18:568, 1967.)
Clinical Features.
In calcific aortic stenosis (superimposed on a previously normal or bicuspid aortic valve), the obstruction to left ventricular outflow leads to gradual narrowing of the valve orifice (valve area approximately 0.5 to 1 cm2 in severe aortic stenosis; normal, ∼4 cm2) and an increasing pressure gradient across the calcified valve, reaching 75 to 100 mm Hg in severe cases. Left ventricular pressures rise to 200 mm Hg or more in such instances, producing concentric left ventricular (pressure overload) hypertrophy. The hypertrophied myocardium tends to be ischemic (as a result of diminished microcirculatory perfusion, often complicated by coronary atherosclerosis), and angina pectoris may appear. Both systolic and diastolic myocardial function may be impaired; eventually, cardiac decompensation and CHF may ensue. The onset of symptoms (angina, CHF, or syncope, for which the pathophysiologic basis is poorly understood) in aortic stenosis heralds cardiac decompensation and carries a poor prognosis; ∼50% with angina will die within 5 years, and 50% with CHF will die within 2 years, if the obstruction is not alleviated by surgical valve replacement. Medical therapy is ineffective in severe symptomatic aortic stenosis. In contrast, asymptomatic patients with aortic stenosis generally have an excellent prognosis.
Calcific Stenosis of Congenitally Bicuspid Aortic Valve
With a prevalence of approximately 1%, bicuspid aortic valve (BAV) is the most frequent congenital cardiovascular malformation in humans.71 Although BAV is usually uncomplicated early in life, late complications of BAV include aortic stenosis or regurgitation, infective endocarditis, and aortic dilation and/or dissection. BAVs are predisposed to progressive degenerative calcification, similar to that occurring in aortic valves with initially normal anatomy (see Fig. 12-22B). Bicuspid aortic valves are responsible for approximately 50% of cases of aortic stenosis in adults.72 Structural abnormalities of the aortic wall commonly accompany BAV, even when the valve is hemodynamically normal, and this may potentiate aortic dilation or aortic dissection (see later). Recent studies have confirmed previous reports of familial clustering of BAV and left ventricular outflow tract obstruction malformations, and their association with other cardiovascular malformations.73
In a congenitally bicuspid aortic valve, there are only two functional cusps, usually of unequal size, with the larger cusp having a midline raphe, resulting from incomplete commissural separation during development; less frequently the cusps are of equal size and the raphe is absent. The raphe is frequently a major site of calcific deposits. Once stenosis is present, the clinical course is similar to that described above for calcific aortic stenosis. Valves that become bicuspid because of an acquired deformity (e.g., rheumatic valve disease) have a fused commissure that produces a conjoined cusp that is generally twice the size of the nonconjoined cusp. BAVs may also become incompetent as a result of aortic dilation, cusp prolapse, or infective endocarditis. The mitral valve is generally normal in patients with a congenitally bicuspid aortic valve.
Mitral Annular Calcification
Degenerative calcific deposits can develop in the peripheral fibrous ring (annulus) of the mitral valve. Grossly, these appear as irregular, stony hard, occasionally ulcerated nodules (2–5 mm in thickness) that lie behind the leaflets (see Fig. 12-22C and D). The process generally does not affect valvular function or otherwise become clinically important. In unusual cases, however, mitral annular calcification may lead to (1) regurgitation by interfering with physiologic contraction of the valve ring, (2) stenosis by impairing opening of the mitral leaflets, or (3) arrhythmias and occasionally sudden death by penetration of calcium deposits to a depth sufficient to impinge on the atrioventricular conduction system. Because calcific nodules may also provide a site for thrombi that can embolize, patients with mitral annular calcification have an increased risk of stroke, and the calcific nodules can also be the nidus for infective endocarditis. Heavy calcific deposits are sometimes visualized on echocardiography or seen as a distinctive, ringlike opacity on chest radiographs. Mitral annular calcification is most common in women over age 60 and individuals with mitral valve prolapse (see below) or elevated left ventricular pressure (as in systemic hypertension, aortic stenosis, or hypertrophic cardiomyopathy).
MITRAL VALVE PROLAPSE (MYXOMATOUS DEGENERATION OF THE MITRAL VALVE)
In mitral valve prolapse (MVP), one or both mitral valve leaflets are “floppy” and prolapse, or balloon back, into the left atrium during systole.74 The key histologic change in the tissue is called myxomatous degeneration. MVP affects approximately 3% of adults in the United States; it is most often an incidental finding on physical examination (particularly in young women), but in a small minority of affected individuals may lead to serious complications.
Morphology. The characteristic anatomic change in MVP is interchordal ballooning (hooding) of the mitral leaflets or portions thereof (Fig. 12-23A–C). The affected leaflets are often enlarged, redundant, thick, and rubbery. The associated tendinous cords may be elongated, thinned, or even ruptured, and the annulus may be dilated. The tricuspid, aortic, or pulmonary valves may also be affected. Histologically, there is attenuation of the collagenous fibrosa layer of the valve, on which the structural integrity of the leaflet depends, accompanied by marked thickening of the spongiosa layer with deposition of mucoid (myxomatous) material (Fig. 12-23E). Secondary changes reflect the stresses and injury incident to the billowing leaflets: (1) fibrous thickening of the valve leaflets, particularly where they rub against each other; (2) linear fibrous thickening of the left ventricular endocardial surface where the abnormally long cords snap or rub against it; (3) thickening of the mural endocardium of the left ventricle or atrium as a consequence of friction-induced injury induced by the prolapsing, hyper-mobile leaflets; (4) thrombi on the atrial surfaces of the leaflets or the atrial walls; and (5) focal calcifications at the base of the posterior mitral leaflet. Mild myxomatous degeneration can also occur in mitral valves secondary to regurgitation of other etiologies (e.g., ischemic dysfunction).
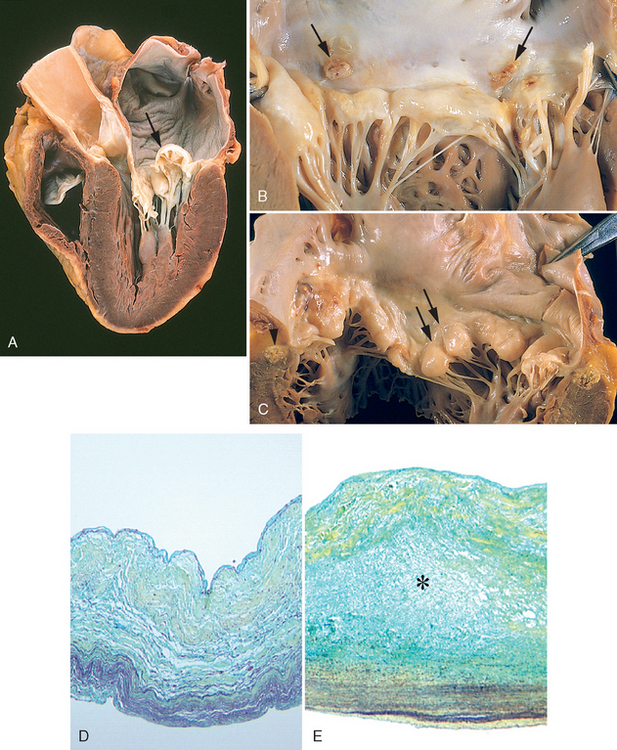
FIGURE 12-23 Myxomatous degeneration of the mitral valve. A, Long axis of left ventricle demonstrating hooding with prolapse of the posterior mitral leaflet into the left atrium (arrow). The left ventricle is on right in this apical four-chamber view. B, Opened valve, showing pronounced hooding of the posterior mitral leaflet with thrombotic plaques at sites of leaflet–left atrium contact (arrows). C, Opened valve with pronounced hooding from patient who died suddenly (double arrows). Note also mitral annular calcification (arrowhead). Normal heart valve (D) and myxomatous mitral valve (E) (Movat pentachrome stain, in which collagen is yellow, elastin is black, and proteoglycans are blue). In myxomatous valves, collagen in the fibrosa is loose and disorganized, proteolgycans (asterisk) are deposited in the spongiosa, and elastin in the atrialis is disorganized.
(A, Courtesy of William D. Edwards, MD, Mayo Clinic, Rochester, MN; D,E, From Rabkin E, et al: Activated interstitial myofibroblasts express catabolic enzymes and mediate matrix remodeling in myxomatous heart valves. Circulation 104:2525–2532, 2001.)
Pathogenesis.
The basis for the changes that weaken the valve leaflets and associated structures is unknown in most cases. Uncommonly, MVP is associated with heritable disorders of connective tissue including Marfan syndrome, which is usually caused by mutations in fibrillin-1 (FBN-1) (Chapter 5). As you will recall, defects in FBN-1 alter cell-matrix interactions and also dysregulate TGF-β signaling.75 Of interest, mice engineered to express mutated FBN-1 develop a form of mitral valve prolapse that can be prevented by inhibitors of TGF-β,76 indicating that excess TGF-β can cause the characteristic structural laxity and myxomatous change. Whether similar mechanisms contribute to sporadic MVP is unknown. Studies utilizing genetic linkage analysis have also mapped autosomal-dominant forms of MVP to several other genetic loci that may be involved in remodeling of the valvular extracellular matrix.77
Clinical Features.
Most individuals diagnosed with MVP are asymptomatic, and the condition is discovered incidentally by detection of a midsystolic click on physical examination. The diagnosis can be confirmed by echocardiography. Those cases with mitral regurgitation are also associated with a systolic murmur. A minority of patients have chest pain mimicking angina, dyspnea, and fatigue. Although the great majority of persons with MVP have no untoward effects, approximately 3% develop one of four serious complications: (1) infective endocarditis; (2) mitral insufficiency, sometimes with chordal rupture; (3) stroke or other systemic infarct, resulting from embolism of leaflet thrombi; or (4) arrhythmias, both ventricular and atrial.
The risk of complications is very low when MVP is discovered incidentally in young asymptomatic patients, and higher in men, older patients, and those with arrhythmias or mitral regurgitation. For patients with symptoms or at high risk for serious complications, valve surgery is often done; indeed, MVP is presently the most common cause for surgical repair or replacement of the mitral valve.
RHEUMATIC FEVER AND RHEUMATIC HEART DISEASE
Rheumatic fever (RF) is an acute, immunologically mediated, multisystem inflammatory disease that occurs a few weeks after an episode of group A streptococcal pharyngitis.78 Acute rheumatic carditis is a frequent manifestation during the active phase of RF and may progress over time to chronic rheumatic heart disease (RHD), of which valvular abnormalities are key manifestations.
RHD is characterized principally by deforming fibrotic valvular disease, particularly mitral stenosis, of which it is virtually the only cause. The incidence and mortality rate of RF and RHD have declined remarkably in many parts of the world over the past century, as a result of improved socioeconomic conditions and rapid diagnosis and treatment of streptococcal pharyngitis. Nevertheless, in developing countries, and in many crowded, economically depressed urban areas in the Western world, RHD remains an important public health problem, affecting an estimated 15 million people. Rheumatic fever only rarely follows infections by streptococci at other sites, such as the skin.
Morphology. Key pathologic features of acute RF and chronic RHD are shown in Figure 12-24. During acute RF, focal inflammatory lesions are found in various tissues. Distinctive lesions occur in the heart, called Aschoff bodies, which consist of foci of lymphocytes (primarily T cells), occasional plasma cells, and plump activated macrophages called Anitschkow cells (pathognomonic for RF). These macrophages have abundant cytoplasm and central round-toovoid nuclei in which the chromatin is disposed in a central, slender, wavy ribbon (hence the designation “caterpillar cells”), and may become multinucleated.
During acute RF, diffuse inflammation and Aschoff bodies may be found in any of the three layers of the heart, causing pericarditis, myocarditis, or endocarditis (pancarditis).
Inflammation of the endocardium and the left-sided valves typically results in fibrinoid necrosis within the cusps or along the tendinous cords. Overlying these necrotic foci are small (1- to 2-mm) vegetations, called verrucae, along the lines of closure. These vegetations place RHD within a small group of disorders that are associated with vegetative valve disease, each with its own characteristic morphologic features (Fig. 12-25). Subendocardial lesions, perhaps exacerbated by regurgitant jets, may induce irregular thickenings called MacCallum plaques, usually in the left atrium.
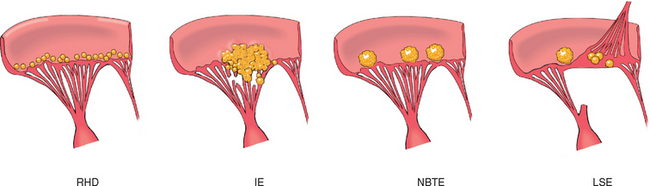
FIGURE 12-25 Comparison of the four major forms of vegetative endocarditis. The rheumatic fever phase of rheumatic heart disease (RHD) is marked by small, warty vegetations along the lines of closure of the valve leaflets. Infective endocarditis (IE) is characterized by large, irregular masses on the valve cusps that can extend onto the chordae (see Fig. 12-25). Nonbacterial thrombotic endocarditis (NBTE) typically exhibits small, bland vegetations, usually attached at the line of closure. One or many may be present (see Fig. 12-27). Libman-Sacks endocarditis (LSE) has small or medium-sized vegetations on either or both sides of the valve leaflets.
The cardinal anatomic changes of the mitral valve in chronic RHD are leaflet thickening, commissural fusion and shortening, and thickening and fusion of the tendinous cords (Fig. 12-24D). In chronic disease the mitral valve is virtually always involved. The mitral valve is affected alone in 65% to 70% of cases, and along with the aortic valve in another 25% of cases. Tricuspid valve involvement is infrequent, and the pulmonary valve is only rarely affected. Because of the increase in calcific aortic stenosis (see earlier) and the reduced frequency of RHD, rheumatic aortic stenosis now accounts for less than 10% of cases of acquired aortic stenosis. Fibrous bridging across the valvular commissures and calcification create “fish mouth” or “buttonhole” stenoses. With tight mitral stenosis, the left atrium progressively dilates and may harbor mural thrombi in the appendage or along the wall, either of which can embolize. Long-standing congestive changes in the lungs may induce pulmonary vascular and parenchymal changes and in time lead to right ventricular hypertrophy. The left ventricle is largely unaffected by isolated pure mitral stenosis. Microscopically, in the mitral leaflets there is organization of the acute inflammation and subsequent diffuse fibrosis and neovascularization that obliterate the originally layered and avascular leaflet architecture. Aschoff bodies are rarely seen in surgical specimens or autopsy tissue from patients with chronic RHD, as a result of the long times between the initial insult and the development of the chronic deformity.
Pathogenesis.
Acute rheumatic fever results from immune responses to group A streptococci, which happen to cross-react with host tissues. Antibodies directed against the M proteins of streptococci have been shown to cross-react with self antigens in the heart. In addition, CD4+ T cells specific for streptococcal peptides also react with self proteins in the heart, and produce cytokines that activate macrophages (such as those found in Aschoff bodies). Damage to heart tissue may thus be caused by a combination of antibody- and T cell–mediated reactions (Chapter 6).
Clinical Features.
RF is characterized by a constellation of findings that includes as major manifestations: (1) migratory polyarthritis of the large joints, (2) pancarditis, (3) subcutaneous nodules, (4) erythema marginatum of the skin, and (5) Sydenham chorea, a neurologic disorder with involuntary rapid, purposeless movements. The diagnosis is established by the so-called Jones criteria: evidence of a preceding group A streptococcal infection, with the presence of two of the major manifestations listed above or one major and two minor manifestations (nonspecific signs and symptoms that include fever, arthralgia, or elevated blood levels of acute-phase reactants).80
Acute RF typically appears 10 days to 6 weeks after an episode of pharyngitis caused by group A streptococci in about 3% of infected patients. It occurs most often in children between ages 5 and 15, but first attacks can occur in middle to later life. Although pharyngeal cultures for streptococci are negative by the time the illness begins, antibodies to one or more streptococcal enzymes, such as streptolysin O and DNase B, can be detected in the sera of most patients with RF. The predominant clinical manifestations are carditis and arthritis, the latter more common in adults than in children. Clinical features related to acute carditis include pericardial friction rubs, weak heart sounds, tachycardia, and arrhythmias. Myocarditis may cause cardiac dilation that can evolve to functional mitral valve insufficiency or even heart failure. Approximately 1% of patients die from fulminant RF. Arthritis typically begins with migratory polyarthritis (accompanied by fever) in which one large joint after another becomes painful and swollen for a period of days and then subsides spontaneously, leaving no residual disability.
After an initial attack there is increased vulnerability to reactivation of the disease with subsequent pharyngeal infections, and the same manifestations are likely to appear with each recurrent attack. Damage to the valves is cumulative. Turbulence induced by ongoing valvular deformities begets additional fibrosis. Clinical manifestations appear years or even decades after the initial episode of RF and depend on which cardiac valves are involved. In addition to various cardiac murmurs, cardiac hypertrophy and dilation, and heart failure, individuals with chronic RHD may suffer from arrhythmias (particularly atrial fibrillation in the setting of mitral stenosis), thromboembolic complications, and infective endocarditis (see below). The long-term prognosis is highly variable. Surgical repair or prosthetic replacement of diseased valves has greatly improved the outlook for persons with RHD.
INFECTIVE ENDOCARDITIS
Infective endocarditis (IE) is a serious infection characterized by colonization or invasion of the heart valves or the mural endocardium by a microbe.81 This leads to the formation of vegetations composed of thrombotic debris and organisms, often associated with destruction of the underlying cardiac tissues. The aorta, aneurysmal sacs, other blood vessels, and prosthetic devices can also become infected. Although fungi and other classes of microorganisms can be responsible, most cases are caused by bacterial infections (bacterial endocarditis). Prompt diagnosis and effective treatment of IE is important.
Traditionally, IE has been classified on clinical grounds into acute and subacute forms. This subdivision reflects the range of the disease severity and tempo, which are determined in large part by the virulence of the infecting microorganism and whether underlying cardiac disease is present. Acute infective endocarditis is typically caused by infection of a previously normal heart valve by a highly virulent organism that produces necrotizing, ulcerative, destructive lesions. These infections are difficult to cure with antibiotics and usually require surgery. Death within days to weeks ensues in many patients with acute IE, despite treatment. In contrast, in subacute IE, the organisms are of lower virulence. These organisms cause insidious infections of deformed valves that are less destructive. In such cases the disease may pursue a protracted course of weeks to months, and cures are often produced with antibiotics.
Etiology and Pathogenesis.
As mentioned above, IE can develop on previously normal valves, especially with highly virulent organisms, but a variety of cardiac and vascular abnormalities predispose to this form of infection. In years past, rheumatic heart disease was the major antecedent disorder, but more common now are mitral valve prolapse, degenerative calcific valvular stenosis, bicuspid aortic valve (whether calcified or not), artificial (prosthetic) valves, and unrepaired and repaired congenital defects.82 The causative organisms differ somewhat in the major high-risk groups. Endocarditis of native but previously damaged or otherwise abnormal valves is caused most commonly (50% to 60% of cases) by Streptococcus viridans, which is part of the normal flora of the oral cavity. In contrast, more virulent S. aureus organisms commonly found on the skin can infect either healthy or deformed valves and are responsible for 10% to 20% of cases overall; S. aureus is the major offender in intravenous drug abusers with IE. The roster of the remaining bacteria includes enterococci and the so-called HACEK group (Haemophilus, Actinobacillus, Cardiobacterium, Eikenella, and Kingella), all commensals in the oral cavity. Prosthetic valve endocarditis is caused most commonly by coagulase-negative staphylococci (e.g., S. epidermidis). Other agents causing endocarditis include gram-negative bacilli and fungi. In about 10% to 15% of all cases of endocarditis, no organism can be isolated from the blood (“culture-negative” endocarditis).
Foremost among the factors predisposing to the development of endocarditis are those that lead to bacteremia. The source of the organism may be an obvious infection elsewhere, a dental or surgical procedure, a contaminated needle shared by intravenous drug users, or seemingly trivial breaks in the epithelial barriers of the gut, oral cavity, or skin. The risk can be lowered in those with predisposing factors (e.g., valve abnormalities, conditions causing bacteremia) by prophylaxis with antibiotics.
Morphology. The hallmark of IE is the presence of friable, bulky, potentially destructive vegetations containing fibrin, inflammatory cells, and bacteria or other organisms on the heart valves (Figs. 12-25B and 12-26). The aortic and mitral valves are the most common sites of infection, although the valves of the right heart may also be involved, particularly in intravenous drug abusers. The vegetations may be single or multiple and may involve more than one valve. Vegetations sometimes erode into the underlying myocardium and produce an abscess (ring abscess). Emboli may be shed from the vegetations at any time; because the embolic fragments may contain large numbers of virulent organisms, abscesses often develop at the sites where the emboli lodge, leading to sequelae such as septic infarcts or mycotic aneurysms.
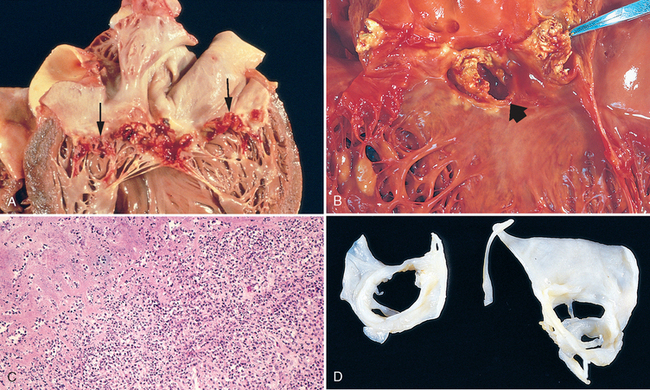
FIGURE 12-26 Infective (bacterial) endocarditis. A, Endocarditis of mitral valve (subacute, caused by Streptococcus viridans). The large, friable vegetations are denoted by arrows. B, Acute endocarditis of congenitally bicuspid aortic valve (caused by Staphylococcus aureus) with extensive cuspal destruction and ring abscess (arrow). C, Histologic appearance of vegetation of endocarditis with extensive acute inflammatory cells and fibrin. Bacterial organisms were demonstrated by tissue Gram stain. D, Healed endocarditis, demonstrating mitral valvular destruction but no active vegetations.
(C, Reproduced from Schoen FJ: Surgical pathology of removed natural and prosthetic heart valves. Hum Pathol 18:558, 1987.)
The vegetations of subacute endocarditis are associated with less valvular destruction than those of acute endocarditis, although the distinction between the two forms may blur. Microscopically, the vegetations of typical subacute IE often have granulation tissue indicative of healing at their bases. With time, fibrosis, calcification, and a chronic inflammatory infiltrate can develop.
Clinical Features.
Fever is the most consistent sign of IE. Acute endocarditis has a stormy onset with rapidly developing fever, chills, weakness, and lassitude. However, fever may be slight or absent, particularly in the elderly, and the only manifestations may be nonspecific fatigue, loss of weight, and a flu-like syndrome. Complications of IE generally begin within the first few weeks of onset. They may be immunologically mediated, as exemplified by glomerulonephritis caused by the deposition of antigen-antibody complexes (Chapter 20). Murmurs are present in 90% of patients with left-sided IE and may stem from a new valvular defect or represent a preexisting abnormality. The so-called Duke criteria (Table 12-8) provide a standardized assessment of individuals with suspected IE that takes into account predisposing factors, physical findings, blood culture results, echocardiographic findings, and laboratory information.83 Earlier diagnosis and effective treatment has nearly eliminated some previously common clinical manifestations of long-standing IE—for example, micro-thromboemboli (manifest as splinter or subungual hemorrhages), erythematous or hemorrhagic nontender lesions on the palms or soles (Janeway lesions), subcutaneous nodules in the pulp of the digits (Osler nodes), and retinal hemorrhages in the eyes (Roth spots).
TABLE 12-8 Diagnostic Criteria for Infective Endocarditis*
| PATHOLOGIC CRITERIA |
| CLINICAL CRITERIA |
| Major |
|
Blood culture(s) positive for a characteristic organism or persistently positive for an unusual organism
|
| Minor |
|
Vascular lesions, including arterial petechiae, subungual/splinter hemorrhages, emboli, septic infarcts, mycotic aneurysm, intracranial hemorrhage, Janeway lesions†
|
* Diagnosis by these guidelines, often called the Duke Criteria, requires either pathologic or clinical criteria; if clinical criteria are used, 2 major, 1 major + 3 minor, or 5 minor criteria are required for diagnosis.
† Janeway lesions are small erythematous or hemorrhagic, macular, nontender lesions on the palms and soles and are the consequence of septic embolic events.
‡ Osler nodes are small, tender subcutaneous nodules that develop in the pulp of the digits or occasionally more proximally in the fingers and persist for hours to several days.
§ Roth spots are oval retinal hemorrhages with pale centers.
Modified from Durack DT et al: Am J Med, 96:200, 1994 and Karchmer AW: In Braunwald E, Zipes DP, Libby P (eds): Heart Disease. A Textbook of Cardiovascular Medicine, 6th ed. Philadelphia, WB Saunders, 2001, p 1723.
NONINFECTED VEGETATIONS
Noninfected (sterile) vegetations are caused by nonbacterial thrombotic endocarditis and the endocarditis of systemic lupus erythematosus (SLE), called Libman-Sacks endocarditis (see below).
Nonbacterial Thrombotic Endocarditis (NBTE)
NBTE is characterized by the deposition of small sterile thrombi on the leaflets of the cardiac valves (Figs. 12-25C and 12-27). The lesions are 1 mm to 5 mm in size, and occur singly or multiply along the line of closure of the leaflets or cusps. Histologically they are composed of bland thrombi that are loosely attached to the underlying valve. The vegetations are not invasive and do not elicit any inflammatory reaction. Thus, the local effect of the vegetations is usually unimportant, but they may be the source of systemic emboli that produce infarcts in the brain, heart, or elsewhere.
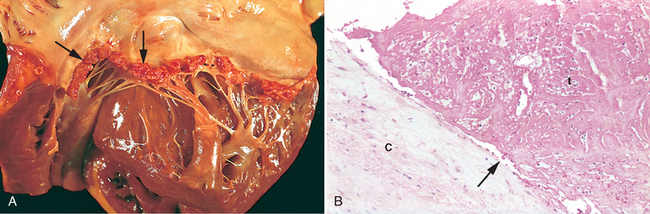
FIGURE 12-27 Nonbacterial thrombotic endocarditis (NBTE). A, Nearly complete row of thrombotic vegetations along the line of closure of the mitral valve leaflets (arrows). B, Photomicrograph of NBTE, showing bland thrombus, with virtually no inflammation in the valve cusp (c) or the thrombotic deposit (t). The thrombus is only loosely attached to the cusp (arrow).
NBTE is often encountered in debilitated patients, such as those with cancer or sepsis—hence the previously used term marantic endocarditis. It frequently occurs concomitantly with deep venous thromboses, pulmonary emboli, or other findings consistent with an underlying systemic hypercoagulable state (Chapter 4). Indeed, there is a striking association with mucinous adenocarcinomas, which may relate to the procoagulant effects of tumor-derived mucin or tissue factor, and NBTE can be a part of the Trousseau syndrome of migratory thrombophlebitis (Chapter 7). Endocardial trauma, as from an indwelling catheter, is another well-recognized predisposing condition, and right-sided valvular and endocardial thrombotic lesions frequently track along the course of Swan-Ganz pulmonary artery catheters.
Endocarditis of Systemic Lupus Erythematosus (Libman-Sacks Disease)
Mitral and tricuspid valvulitis with small, sterile vegetations, called Libman-Sacks endocarditis, is occasionally encountered in SLE. The lesions are small (1–4 mm in diameter) single or multiple, sterile, pink vegetations that often have a warty (verrucous) appearance. They may be located on the undersurfaces of the atrioventricular valves, on the valvular endocardium, on the chords, or on the mural endocardium of atria or ventricles. Histologically the vegetations consist of a finely granular, fibrinous eosinophilic material that may contain hematoxylin bodies, homogeneous remnants of nuclei damaged by anti-nuclear antigen bodies (see Chapter 6). An intense valvulitis may be present, characterized by fibrinoid necrosis of the valve substance that is often contiguous with the vegetation. Active leaflet vegetations can be difficult to distinguish from those of infective endocarditis (see Fig. 12-25); fibrosis and serious deformities can result that resemble chronic rheumatoid heart disease and require surgery.
Thrombotic heart valve lesions with sterile vegetations or rarely fibrous thickening commonly occur with the antiphospholipid syndrome (discussed in Chapter 4), which you will recall can also lead to a hypercoagulable state.84 The mitral valve is more frequently involved than the aortic valve; regurgitation is the usual functional abnormality.
CARCINOID HEART DISEASE
Carcinoid heart disease is the cardiac manifestation of the systemic syndrome caused by carcinoid tumors. It generally involves the endocardium and valves of the right heart. Cardiac lesions are present in one half of patients with the carcinoid syndrome, which is characterized by episodic flushing of the skin, cramps, nausea, vomiting, and diarrhea (see Chapter 17).
Morphology. The cardiovascular lesions associated with the carcinoid syndrome are distinctive, consisting of firm plaquelike endocardial fibrous thickenings on the inside surfaces of the cardiac chambers and the tricuspid and pulmonary valves; occasionally they involve the major blood vessels of the right side, the inferior vena cava and the pulmonary artery (Fig. 12-28). The plaquelike thickenings are composed predominantly of smooth muscle cells and sparse collagen fibers embedded in an acid mucopolysaccharide-rich matrix material. Elastic fibers are not present in the plaques. Structures underlying the plaques are intact.
FIGURE 12-28 Carcinoid heart disease. A, Characteristic endocardial fibrotic lesion involving the right ventricle and tricuspid valve. B, Microscopic appearance of carcinoid heart disease with intimal thickening. Movat stain shows myocardial elastic tissue (black) underlying the acid mucopolysaccharide-rich lesion (blue-green).
Although the mechanisms of the fibrosis are not understood, it appears that the clinical and pathologic findings relate to the elaboration by carcinoid tumors of a variety of bioactive products, such as serotonin (5-hydroxytryptamine), kallikrein, bradykinin, histamine, prostaglandins, and tachykinins. Plasma levels of serotonin and urinary excretion of the serotonin metabolite 5-hydroxyindoleacetic acid correlate with the severity of the right heart lesions.
The key bioactive mediators released into the portal circulation by gut carcinoid tumors are readily metabolized by the liver and do not reach the heart in high concentration. Thus, gastrointestinal carcinoids (with venous drainage via the portal system) do not usually induce carcinoid heart disease, unless there are extensive hepatic metastases that release the relevant mediators directly into the interior vena cava. Restriction of the cardiac changes to the right side of the heart is explained by inactivation of both serotonin and bradykinin during passage through the lungs by monoamine oxidase present in the pulmonary vascular endothelium. In contrast, primary carcinoid tumors in organs outside of the portal system of venous drainage that empty directly into the inferior vena cava (e.g., ovary and lung) can induce the syndrome in the absence of hepatic metastases.
The most common cardiac manifestation is tricuspid insufficiency, followed by pulmonary valve insufficiency, which usually occurs in combination with tricuspid disease. Stenoses of the right-sided valves may also develop, whereas left-sided valvular disease is only seen under unusual circumstances, such as when there is patent foramen ovale with right to left shunting or primary or metastatic carcinoid tumor involving the lung. Left-sided valvular abnormalities with pathologic features similar to those seen in the carcinoid syndrome have been reported to complicate the use of drugs that have serotonergic activity. These include fenfluramine (part of the “fen-phen” combination of appetite suppressants), some antiparkinsonian drugs, and methysergide or ergotamine, used to treat migraine headaches.85
COMPLICATIONS OF ARTIFICIAL VALVES
Replacement of damaged cardiac valves with prostheses is a common and often lifesaving mode of therapy.86 Artificial valves are primarily of two types: (1) mechanical prostheses, consisting of different kinds of rigid mechanical valves, such as caged balls, tilting disks, or hinged semicircular flaps that are composed of nonphysiologic material; and (2) tissue valves, usually bioprostheses, consisting of chemically treated animal tissue, especially porcine aortic valve tissue, which has been preserved in a dilute glutaraldehyde solution and subsequently mounted on a prosthetic frame. Tissue valves are flexible and function similarly to natural semilunar valves.
Approximately 60% of substitute valve recipients develop a serious prosthesis-related problem within 10 years postoperatively. The nature of these complications differs among types (Table 12-9 and Fig. 12-29).87
TABLE 12-9 Complications of Cardiac Valve Prostheses