Infectious Enterocolitis
Enterocolitis can present with a broad range of symptoms including diarrhea, abdominal pain, urgency, perianal discomfort, incontinence, and hemorrhage (Table 17-7). This global problem is responsible for more than 12,000 deaths per day among children in developing countries and half of all deaths before age 5 worldwide. Bacterial infections, such as enterotoxigenic Escherichia coli, are frequently responsible, but the most common pathogens vary with age, nutrition, and host immune status as well as environmental influences (Table 17-7). For example, epidemics of cholera are common in areas with poor sanitation, as a result of inadequate public health measures, or as a consequence of natural disasters or war. Pediatric infectious diarrhea, which may result in severe dehydration and metabolic acidosis, is commonly caused by enteric viruses.
CHOLERA
Vibrio cholerae are comma-shaped, Gram-negative bacteria that cause cholera, a disease that has been endemic in the Ganges Valley of India and Bangladesh for all of recorded history. Since 1817, seven great pandemics have spread along trade routes to large parts of Europe, Australia, and the Americas,56 but, for unknown reasons these pandemics resolved and cholera retreated back to the Ganges Valley. Cholera also persists within the Gulf of Mexico.
V. cholerae is primarily transmitted by contaminated drinking water. However, it can also be present in food and causes sporadic cases of seafood-associated disease in North America. There is a marked seasonal variation in most climates due to rapid growth of Vibrio bacteria at warm temperatures; the only animal reservoirs are shellfish and plankton. Relatively few V. cholerae serotypes are pathogenic, but other species of Vibrio can also cause disease. For example, V. parahaemolyticus is the most common cause of seafood-associated gastroenteritis in North America.57
Pathogenesis.
Despite the severe diarrhea, Vibrio organisms are non-invasive and remain within the intestinal lumen. A preformed enterotoxin, cholera toxin, encoded by a virulence phage and released by the Vibrio organism, causes disease, but flagellar proteins, which are involved in motility and attachment, are necessary for efficient bacterial colonization. Hemagglutinin, a metalloproteinase, is important for bacterial detachment and shedding in the stool. The mechanism by which cholera toxin induces diarrhea is well understood (Fig. 17-27). Cholera toxin is composed of five B subunits and a single A subunit. The B subunit binds GM1 ganglioside on the surface of intestinal epithelial cells, and is carried by endocytosis to the endoplasmic reticulum, a process called retrograde transport.58 Here, the A subunit is reduced by protein disulfide isomerase, and a fragment of the A subunit is unfolded and released. This peptide fragment is then transported into the cytosol using host cell machinery that moves misfolded proteins from the endoplasmic reticulum to the cytosol. Such unfolded proteins are normally disposed of via the proteasome, but the A subunit refolds to avoid degradation. The refolded A subunit peptide then interacts with cytosolic ADP ribosylation factors (ARFs) to ribosylate and activate the stimulatory G protein Gsα. This stimulates adenylate cyclase and the resulting increases in intracellular cAMP open the cystic fibrosis transmembrane conductance regulator, CFTR, which releases chloride ions into the lumen. This causes secretion of bicarbonate, sodium, and water, leading to massive diarrhea. Chloride and sodium absorption are also inhibited by cAMP. Remarkably, mucosal biopsies show only minimal alterations.
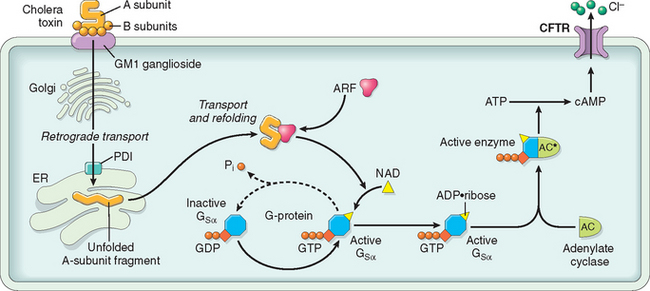
FIGURE 17-27 Mechanisms of cholera toxin transport and signaling. After retrograde toxin transport to the endoplasmic reticulum (ER), the A subunit is released by the action of protein disulfide isomerase (PDI) and is then able to access the epithelial cell cytoplasm. In concert with an ADP-ribosylation factor (ARF), the A subunit then ADP-ribosylates Gsα, which locks it in the active, GTP-bound state. This leads to adenylate cyclase (AC) activation, and the cAMP produced opens CFTR to drive chloride secretion and diarrhea.
Clinical Features.
Most exposed individuals are asymptomatic or develop only mild diarrhea. In those with severe disease there is an abrupt onset of watery diarrhea and vomiting following an incubation period of 1 to 5 days. The voluminous stools resemble rice water and are sometimes described as having a fishy odor. The rate of diarrhea may reach 1 liter per hour, leading to dehydration, hypotension, muscular cramping, anuria, shock, loss of consciousness, and death. Most deaths occur within the first 24 hours after presentation. Although the mortality for severe cholera is about 50% without treatment, timely fluid replacement can save more than 99% of patients. Oral rehydration is often sufficient.59 Because of an improved understanding of the host and Vibrio proteins involved, new therapies are being developed including CFTR inhibitors that block chloride secretion and prevent diarrhea.60 Prophylactic vaccination is a long-term goal.61
CAMPYLOBACTER ENTEROCOLITIS
Campylobacter jejuni is the most common bacterial enteric pathogen in developed countries57 and is an important cause of traveler’s diarrhea. Most infections are associated with ingestion of improperly cooked chicken, but outbreaks can also be caused by unpasteurized milk or contaminated water.
Pathogenesis.
The pathogenesis of Campylobacter infection remains poorly defined, but four major virulence properties contribute: motility, adherence, toxin production, and invasion. Flagella allow Campylobacter to be motile. This facilitates adherance and colonization, which are necessary for mucosal invasion. Cytotoxins that cause epithelial damage and a cholera toxin–like enterotoxin are also released by some C. jejuni isolates. Dysentery is generally associated with invasion and only occurs with a small minority of Campylobacter strains. Enteric fever occurs when bacteria proliferate within the lamina propria and mesenteric lymph nodes.
Campylobacter infection can result in reactive arthritis, primarily in patients with HLA-B27. Other extra-intestinal complications, including erythema nodosum and Guillain-Barré syndrome, a flaccid paralysis caused by autoimmune-induced inflammation of peripheral nerves, are not HLA-linked.62 Molecular mimicry has been implicated in the pathogenesis of Guillain-Barré syndrome, as serum antibodies to C. jejuni lipopolysaccharide cross-react with peripheral and central nervous system gangliosides. Moreover, 15% to 50% of individuals with Guillain-Barré syndrome have positive stool cultures or circulating antibodies to Campylobacter.63 Fortunately, Guillain-Barré syndrome develops in 0.1% or less of those infected with Campylobacter.
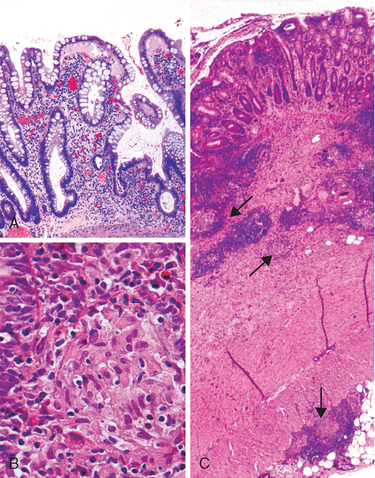
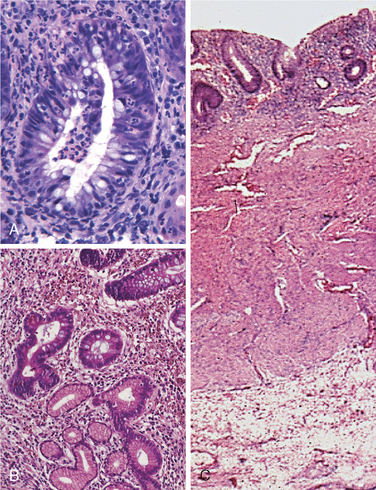
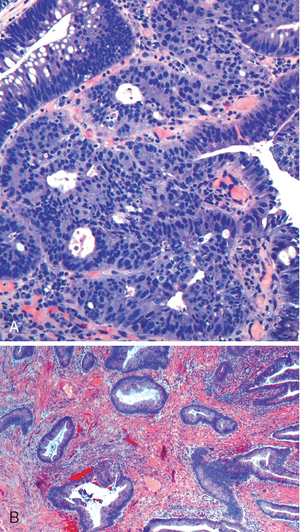
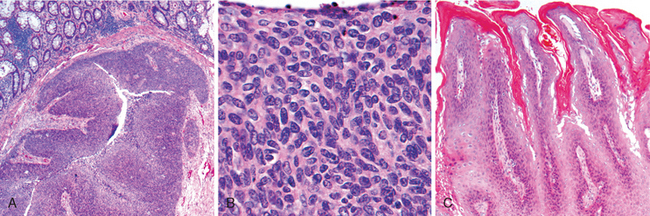
Morphology. Campylobacter are comma-shaped, flagellated, Gram-negative organisms. Diagnosis is primarily by stool culture, since biopsy findings are nonspecific, and reveal acute self-limited colitis with features common to many forms of infectious colitis.64 Mucosal and intraepithelial neutrophil infiltrates are prominent, particularly within the superficial mucosa (Fig. 17-28A); cryptitis (neutrophil infiltration of the crypts) and crypt abscesses (crypts with accumulations of luminal neutrophils) may also be present. Importantly, crypt architecture is preserved (Fig. 17-28D), although this can be difficult to assess in cases with severe mucosal damage.
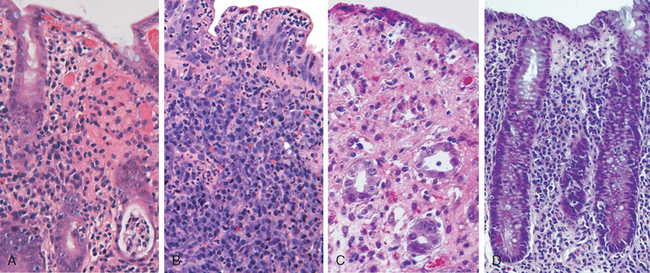
FIGURE 17-28 Bacterial enterocolitis. A, Campylobacter jejuni infection produces acute, self-limited colitis. Neutrophils can be seen within surface and crypt epithelium and a crypt abscess is present at the lower right. B, In Yersinia infection the surface epithelium can be eroded by neutrophils and the lamina propria is densely infiltrated by sheets of plasma cells admixed with lymphocytes and neutrophils. C, Enterohemorrhagic E. coli O157:H7 results in an ischemia-like morphology with surface atrophy and erosion. D, Enteroinvasive E. coli infection is a similar to other acute, self-limited colitides. Note the maintenance of normal crypt architecture and spacing, despite abundant intraepithelial neutrophils.
Clinical Features.
Ingestion of as few as 500 C. jejuni organisms can cause disease after an incubation period of up to 8 days. Watery diarrhea, either acute or following an influenza-like prodrome, is the primary symptom, and dysentery develops in 15% of patients. Patients may shed bacteria for 1 month or more after clinical resolution. Antibiotic therapy is generally not required.
SHIGELLOSIS
Shigella are Gram-negative bacilli that were initially isolated during the Japanese red diarrhea epidemic of 1897. Four major strains are now recognized. Shigella are unencapsulated, nonmotile, facultative anaerobes that belong to the Enterobacteriaceae and are closely related to enteroinvasive E. coli. Although humans are the only known reservoir, Shigella spp. remain one of the most common causes of bloody diarrhea. It is estimated that 165 million cases occur worldwide each year.65 Given the infective dose of fewer than several hundred organisms and the presence of as many as 109 organisms in each gram of stool during acute disease, Shigella are highly transmissible by the fecal-oral route or via contaminated water and food.
In the United States and Europe, children in daycare centers, migrant workers, travelers to developing countries, and those in nursing homes are most commonly affected.66,67 Most Shigella infections and deaths occur in children under 5 years of age, and in countries where Shigella is endemic it is responsible for approximately 10% of all pediatric diarrheal disease and as many as 75% of diarrheal deaths.65,68
Pathogenesis.
Shigella are resistant to the harsh acidic environment of the stomach, which translates into an extremely low infective dose. Once in the intestine, organisms are taken up by M, or microfold, epithelial cells, which are specialized for sampling and presentation of luminal antigens. Shigella proliferate intracellularly, escape into the lamina propria, and are phagocytosed by macrophages, in which they induce apoptosis. The ensuing inflammatory process damages surface epithelia and allows Shigella within the intestinal lumen to gain access to the colonocyte basolateral membrane, which is the only surface through which infection can occur in epithelial cells (other than M cells). All Shigella spp. carry virulence plasmids, some of which encode a type III secretion system capable of directly injecting bacterial proteins into the host cytoplasm. S. dysenteriae serotype 1 also release the Shiga toxin Stx, which inhibits eukaryotic protein synthesis resulting in host cell damage and death.69
Morphology. Shigella infections are most prominent in the left colon, but the ileum may also be involved, perhaps reflecting the abundance of M cells in the dome epithelium over the Peyer’s patches. The mucosa is hemorrhagic and ulcerated, and pseudomembranes may be present. The histology of early cases is similar to other acute self-limited colitides, such as Campylobacter colitis, but because of the tropism for M cells, aphthous-appearing ulcers similar to those seen in Crohn disease may occur. The potential for confusion with chronic inflammatory bowel disease is significant, particularly if there is distortion of crypt architecture.
Clinical Features.
After an incubation period of as long as 4 days, Shigella causes self-limited disease characterized by about 6 days of diarrhea, fever, and abdominal pain. The initially watery diarrhea progresses to a dysenteric phase in approximately 50% of patients, and constitutional symptoms can persist for as long as 1 month. The subacute presentation that develops in a minority of adults is characterized by several weeks of waxing and waning diarrhea that can mimic new-onset ulcerative colitis.68 While duration is typically shorter in children, severity is often much greater. Confirmation of Shigella infection requires stool culture.
Complications of Shigella infection are uncommon and include Reiter syndrome, a triad of sterile arthritis, urethritis, and conjunctivitis that preferentially affects HLA-B27-positive men between 20 and 40 years of age. Hemolytic-uremic syndrome, which is typically associated with enterohemorrhagic E. coli (EHEC), may also occur after infection with S. dysenteriae serotype 1 that secrete Shiga toxin69-71; only Shigella organisms that secrete the toxin are associated with hemolytic-uremic syndrome (Chapter 20). Antibiotic treatment shortens the clinical course and reduces the duration over which organisms are shed in the stool, but antidiarrheal medications are contraindicated because they can prolong symptoms and delay Shigella clearance.
SALMONELLOSIS
Salmonella, which are classified within the Enterobacteriaceae family of Gram-negative bacilli, are divided into Salmonella typhi, the causative agent of typhoid fever (discussed in the next section) and nontyphoid Salmonella. Nontyphoid Salmonella infection is usually due to S. enteritidis; more than 1 million cases occur each year in the United States, and the prevalence is even greater in other countries. Infection is most common in young children and the elderly, with peak incidence in summer and fall. Transmission is usually through contaminated food, particularly raw or undercooked meat, poultry, eggs, and milk.
Pathogenesis.
Very few viable Salmonella are necessary to cause infection, and the absence of gastric acid, as in individuals with atrophic gastritis or those on acid-suppressive therapy, further reduces the required inoculum. Salmonella possess virulence genes that encode a type III secretion system capable of transferring bacterial proteins into M cells and enterocytes. The transferred proteins activate host cell Rho GTPases, thereby triggering actin rearrangement and bacterial uptake that allow bacterial growth within phagosomes. In addition, flagellin, the core protein of bacterial flagellae, activates TLR5 on host cells and increases the local inflammatory response.72 Similarly, bacterial lipopolysaccharide activates TLR4, although some Salmonella strains express a virulence factor that prevents TLR4 activation from occurring. Salmonella also secrete a molecule that induces epithelial release of the eicosanoid hepoxilin A3, thereby drawing neutrophils into the intestinal lumen and potentiating mucosal damage.73 Mucosal TH17 immune responses limit infection to the colon.
Clinical Features.
Salmonella infections are clinically indistinguishable from those caused by other enteric pathogens, and symptoms range from loose stools to cholera-like profuse diarrhea to dysentery. Fever often resolves within 2 days, but diarrhea can persist for a week and organisms can be shed in the stool for several weeks after resolution. Antibiotic therapy is not recommended in most cases, because it can prolong the carrier state or even cause relapse and does not typically shorten the duration of diarrhea.74 Most Salmonella infections are self-limited, but deaths do occur. The risk of severe illness and complications is increased in patients with malignancies, immunosuppression, alcoholism, cardiovascular dysfunction, sickle cell disease, and hemolytic anemia.
TYPHOID FEVER
Typhoid fever, also referred to as enteric fever, is caused by Salmonella typhi and Salmonella paratyphi. It affects up to 30 million individuals worldwide each year. The majority of cases in endemic countries are due to S. typhi, while infection by S. paratyphi is more common among travelers,75 perhaps because travelers tend to be vaccinated against S. typhi (there are no effective S. paratyphi vaccines). In endemic areas children and adolescents are affected most often, but there is no age preference in developed countries. Infection is strongly associated with travel to India, Mexico, the Philippines, Pakistan, El Salvador, and Haiti.76 Like Shigella, humans are the sole reservoir for S. typhi and S. paratyphi and transmission occurs from person to person or via food or contaminated water. Gallbladder colonization with S. typhi or S. paratyphi may be associated with gallstones and the chronic carrier state.
Pathogenesis.
S. typhi are able to survive in gastric acid and, once in the small intestine, are taken up by and invade M cells. Organisms are then engulfed by mononuclear cells in the underlying lymphoid tissue. Unlike S. enteritidis, S. typhi can then disseminate via lymphatic and blood vessels. This causes reactive hyperplasia of phagocytes and lymphoid tissues throughout the body.
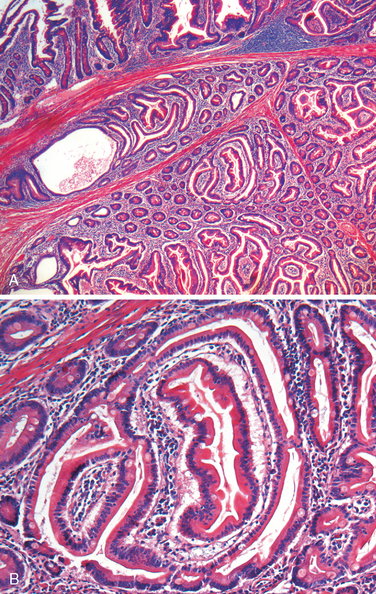
Morphology. Infection causes Peyer’s patches in the terminal ileum to enlarge into sharply delineated, plateau-like elevations up to 8 cm in diameter. Draining mesenteric lymph nodes are also enlarged. Neutrophils accumulate within the superficial lamina propria, and macrophages containing bacteria, red blood cells, and nuclear debris mix with lymphocytes and plasma cells in the lamina propria. Mucosal shedding creates oval ulcers, oriented along the axis of the ileum, that may perforate. The draining lymph nodes also harbor organisms and are enlarged due to phagocyte accumulation.
The spleen is enlarged and soft, with uniformly pale red pulp, obliterated follicular markings, and prominent phagocyte hyperplasia. The liver shows small, randomly scattered foci of parenchymal necrosis in which hepatocytes are replaced by macrophage aggregates, called typhoid nodules, that may also develop in the bone marrow and lymph nodes.
Clinical Features.
Patients experience anorexia, abdominal pain, bloating, nausea, vomiting, and bloody diarrhea followed by a short asymptomatic phase that gives way to bacteremia and fever with flu-like symptoms.77 Blood cultures are positive in more than 90% of affected individuals during the febrile phase, which may prompt antibiotic treatment and prevent further disease progression.76 In patients who do not receive treatment the febrile phase is followed by up to 2 weeks of sustained high fevers and abdominal tenderness that may mimic appendicitis. Rose spots, small erythematous maculopapular lesions, are seen on the chest and abdomen.77 Symptoms abate after several weeks in those who survive, although relapse can occur.77 Systemic dissemination may cause extra-intestinal complications including encephalopathy, meningitis, seizures, endocarditis, myocarditis, pneumonia, and cholecystitis. Patients with sickle cell disease are particularly susceptible to Salmonella osteomyelitis.
YERSINIA
Three Yersinia species are human pathogens. Y. enterocolitica and Y. pseudotuberculosis cause GI disease and are discussed here; Y. pestis, the agent of pulmonic and bubonic plague, is discussed in Chapter 8. GI Yersinia infections are more common in Europe than North America and are most frequently linked to ingestion of pork, raw milk, and contaminated water. Y. enterocolitica is far more common than Y. pseudotuberculosis, and infections tend to cluster in the winter, possibly related to inadequately cooked foods served at holiday gatherings.
Pathogenesis.
Yersinia invade M cells and use bacterial adhesion proteins, adhesins, to bind to host cell β1 integrins. A pathogenicity island encodes an iron uptake system that mediates iron capture and transport; similar iron transport systems are also present in E. coli, Klebsiella, Salmonella, and enterobacteria. In Yersinia, iron enhances virulence and stimulates systemic dissemination, explaining why individuals with hemolytic anemia or hemochromatosis are more likely to develop sepsis and are at greater risk of death.78
Morphology. Yersinia infections preferentially involve the ileum, appendix, and right colon (Fig. 17-28B). The organisms multiply extracellularly in lymphoid tissue, resulting in regional lymph node and Peyer’s patch hyperplasia and bowel wall thickening.79 The mucosa overlying lymphoid tissue may become hemorrhagic, and aphthous-appearing ulcers may develop, along with neutrophil infiltrates (see Fig. 17-28B) and granulomas, increasing the potential for diagnostic confusion with Crohn disease.
Clinical Features.
People infected with Yersinia generally present with abdominal pain, but fever and diarrhea may also occur. Nausea, vomiting, and abdominal tenderness are common, and Peyer’s patch invasion with subsequent involvement of regional lymphatics can mimic acute appendicitis in teenagers and young adults. Enteritis and colitis predominate in younger children. Extra-intestinal symptoms of pharyngitis, arthralgia, and erythema nodosum occur frequently. Yersinia can be detected in stool cultures on Yersinia-selective agar or, in cases with extra-intestinal disease, cultures of lymph nodes or blood.80 Postinfectious complications, including sterile arthritis, Reiter syndrome, myocarditis, glomerulonephritis, and thyroiditis, have been reported.
ESCHERICHIA COLI
Escherichia coli are Gram-negative bacilli that colonize the healthy GI tract; most are nonpathogeneic, but a subset cause human disease. The latter are classified according to morphology, mechanism of pathogenesis, and in vitro behavior. Subgroups with major clinical relevance include enterotoxigenic E. coli (ETEC), enterohemorrhagic E. coli (EHEC), enteroinvasive E. coli (EIEC), and enteroaggregative E. coli (EAEC).
Enterotoxigenic E. Coli.
ETEC organisms are the principal cause of traveler’s diarrhea and spread via contaminated food or water. Infection is common in underdeveloped regions, and children younger than 2 years of age are particularly susceptible. ETEC produce heat-labile toxin (LT) and heat-stable toxin (ST), and both induce chloride and water secretion while inhibiting intestinal fluid absorption. The LT toxin is similar to cholera toxin and activates adenylate cyclase, resulting in increased intracellular cAMP. This stimulates chloride secretion and, simultaneously, inhibits absorption. ST toxins, which have homology to the mammalian regulatory protein guanylin, bind to guanylate cyclase and increase intracellular cGMP with resulting effects on transport that are similar to those produced by LT. Like cholera, the histopathology induced by ETEC infection is limited. Clinical symptoms include secretory, noninflammatory diarrhea, dehydration, and, in severe cases, shock.
Enterohemorrhagic E. Coli.
EHEC are categorized as E. coli O157:H7 and non-O157:H7 serotypes. Large outbreaks of E. coli O157:H7 in developed countries have been associated with the consumption of inadequately cooked ground beef, sometimes from fast-food establishments. Contaminated milk and vegetables are also vehicles for infection. Both O157:H7 and non-O157:H7 serotypes produce Shiga-like toxins, and the morphology (see Fig. 17-28C) and clinical symptoms are thus similar to S. dysenteriae. O157:H7 strains of EHEC are more likely than non-O157:H7 serotypes to cause large outbreaks, bloody diarrhea, and hemolytic-uremic syndrome.
Enteroinvasive E. Coli.
EIEC organisms are bacteriologically similar to Shigella and are transmitted via food, water, or by person-to-person contact. While EIEC do not produce toxins, they invade epithelial cells and cause nonspecific features of acute self-limited colitis (see Fig. 17-28D). EIEC infections are most common among young children in developing countries and are occasionally associated with outbreaks in developed countries.
Enteroaggregative E. Coli.
EAEC organisms were identified on the basis of their unique pattern of adherence to epithelial cells. These organisms are now recognized as a cause of diarrhea in children and adults in developed as well as developing countries. These can also be a cause of traveler’s diarrhea.81 EAEC attach to enterocytes via adherence fimbriae and are aided by dispersin, a bacterial surface protein that neutralizes the negative surface charge of lipopolysaccharide. While the bacteria do produce enterotoxins related to Shigella enterotoxin and ETEC ST toxin, histologic damage is minimal and the characteristic adherence lesions are only visible by electron microscopy.82 EAEC organisms cause nonbloody diarrhea that may be prolonged in individuals with the acquired immunodeficiency syndrome (AIDS).
PSEUDOMEMBRANOUS COLITIS
Pseudomembranous colitis, generally caused by Clostridium difficile, is also known as antibiotic-associated colitis or antibiotic-associated diarrhea. The latter terms apply to diarrhea developing during or after a course of antibiotic therapy and may be due to C. difficile as well as Salmonella, C. perfringens type A, or Staphylococcus aureus. The latter two organisms produce enterotoxins and are common agents of food poisoning.
Pathogenesis.
It is likely that disruption of the normal colonic flora by antibiotics allows C. difficile overgrowth. Although almost any antibiotic may be responsible, third-generation cephalosporins are implicated most frequently. Immunosuppression is also a predisposing factor for C. difficile colitis. Toxins released by C. difficile cause the ribosylation of small GTPases, such as Rho, and lead to disruption of the epithelial cytoskeleton, tight junction barrier loss, cytokine release, and apoptosis.83 The mechanisms by which these processes lead to the characteristic morphology of pseudomembranous colitis are incompletely understood.
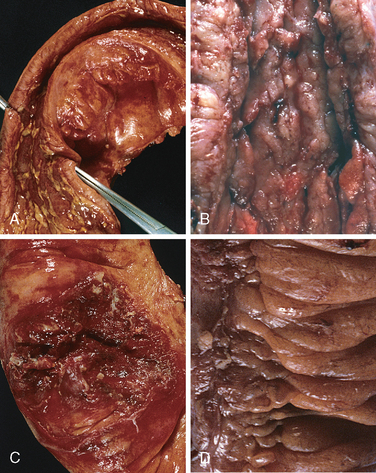
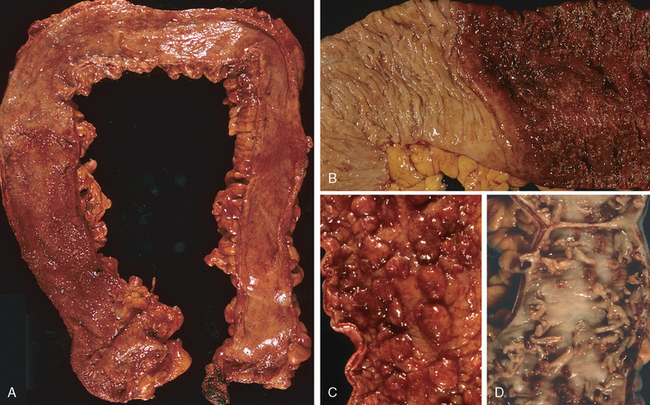
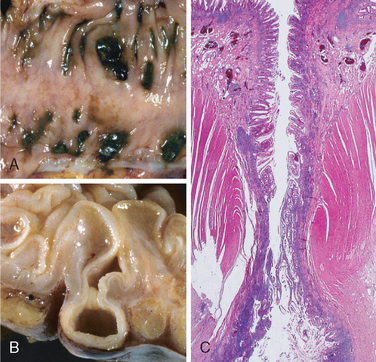
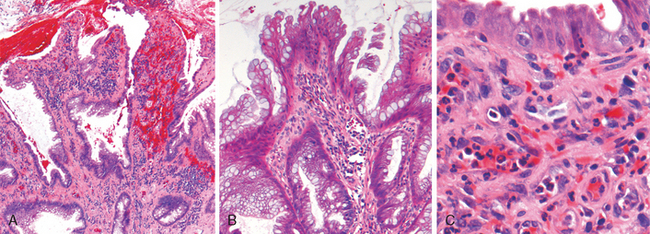
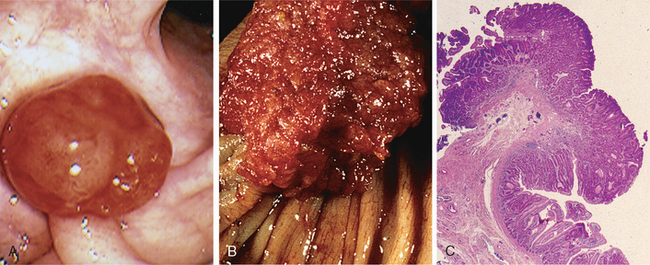
Morphology. Fully developed C. difficile–associated colitis is accompanied by formation of pseudomembranes (Fig. 17-29A, B), made up of an adherent layer of inflammatory cells and debris at sites of colonic mucosal injury. While pseudomembranes are not specific and may occur in ischemia and necrotizing infections, the histopathology of C. difficile–associated colitis is striking. The surface epithelium is denuded, and the superficial lamina propria contains a dense infiltrate of neutrophils and occasional fibrin thrombi within capillaries. Superficially damaged crypts are distended by a mucopurulent exudate that forms an eruption reminiscent of a volcano (Fig. 17-29C). These exudates coalesce to form the pseudomembranes.
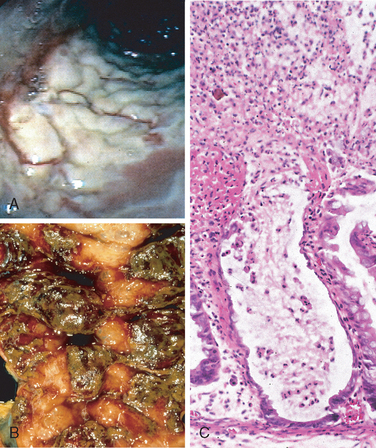
FIGURE 17-29 Clostridium difficile colitis. A, The colon is coated by tan pseudomembranes composed of neutrophils, dead epithelial cells, and inflammatory debris (endoscopic view). B, Pseudomembranes are easily appreciated on gross examination. C, Typical pattern of neutrophils emanating from a crypt is reminiscent of a volcanic eruption.
Clinical Features.
Risk factors for C. difficile–associated colitis include advanced age, hospitalization, and antibiotic treatment. The organism is particularly prevalent in hospitals; as many as 30% of hospitalized adults are colonized with C. difficile (a rate tenfold greater than the general population), but most colonized patients are free of disease. Individuals with C. difficile–associated colitis present with fever, leukocytosis, abdominal pain, cramps, hypoalbuminemia, watery diarrhea, and dehydration. Fecal leukocytes and occult blood may be present, but grossly bloody diarrhea is rare. Diagnosis of C. difficile–associated colitis is usually accomplished by detection of C. difficile toxin, rather than culture, and is supported by the characteristic histopathology. Metronidazole or vancomycin are generally effective therapies, but antibiotic-resistant and hypervirulent C. difficile strains as well as recurrent disease are increasingly common.84
WHIPPLE DISEASE
Whipple disease is a rare, multivisceral chronic disease first described as intestinal lipodystrophy in 1907 by George Hoyt Whipple. A mere 27 years later the pathologist went on to win the Nobel Prize for his work on pernicious anemia. He was a contemporary, but not a relative, of Allen Oldfather Whipple, the surgeon who pioneered the pancreatoduodenectomy.
Pathogenesis.
Whipple’s original case report described an individual with malabsorption, lymphadenopathy, and arthritis of undefined origin. Postmortem examination demonstrated the presence of foamy macrophages and large numbers of argyrophilic rods in the lymph nodes, providing evidence that the disease was infectious.85 The Gram-positive actinomycete, named Tropheryma whippelii, that is responsible for Whipple disease was identified by PCR in 1992 and finally cultured in 2000.86 Clinical symptoms occur because organism-laden macrophages accumulate within the small intestinal lamina propria and mesenteric lymph nodes, causing lymphatic obstruction. Thus, the malabsorptive diarrhea of Whipple disease is due to impaired lymphatic transport.
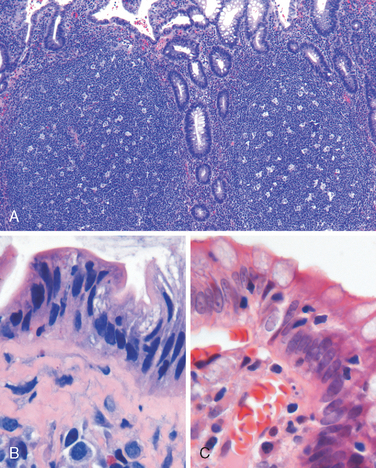
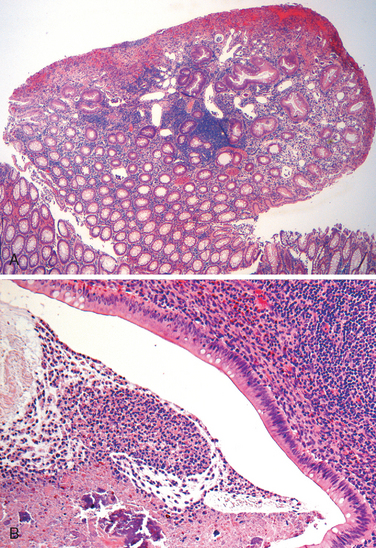
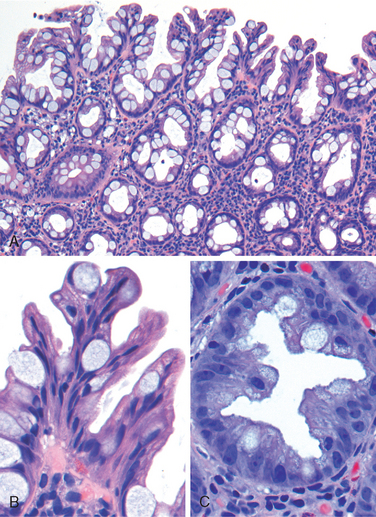
Morphology. The morphologic hallmark of Whipple disease is a dense accumulation of distended, foamy macrophages in the small intestinal lamina propria (Fig. 17-30A). The macrophages contain periodic acid–Schiff (PAS)-positive, diastase-resistant granules that represent lysosomes stuffed with partially digested bacteria (Fig. 17-30B). Intact rod-shaped bacilli can also be identified by electron microscopy (Fig. 17-30C). A similar infiltrate of foamy macrophages is present in intestinal tuberculosis (Fig. 17-30D), and in both diseases the organisms are PAS-positive. However, the acid-fast stain can be used to distinguish between these, since mycobacteria stain positively (Fig. 17-30E) while T. whippelii do not.
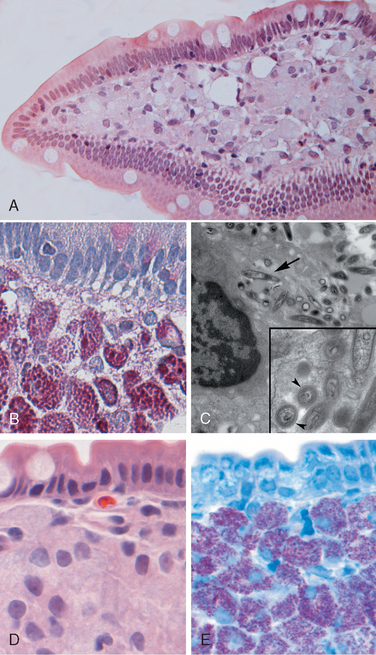
FIGURE 17-30 Whipple disease and mycobacterial infection. A, H&E staining shows effacement of normal lamina propria by a sheet of swollen macrophages. B, PAS stain highlights macrophage lysosomes full of bacilli. Note the positive staining of mucous vacuoles in the overlying goblet cells. C, An electron micrograph of one lamina propria macrophage shows these bacilli within the cell (top) and at higher magnification (inset). D, The morphology of mycobacterial infection can be similar to Whipple disease, particularly in the immunocompromised host. Compare with A. E, Mycobacteria are positive with stains for acid-fast bacteria.
(C, Courtesy of George Kasnic and Dr. William Clapp, University of Florida, Gainesville, FL.)
The villous expansion caused by the dense macrophage infiltrate imparts a shaggy gross appearance to the mucosal surface. Lymphatic dilatation and mucosal lipid deposition account for the common endoscopic detection of white to yellow mucosal plaques. In Whipple disease, bacteria-laden macrophages can accumulate within mesenteric lymph nodes, synovial membranes of affected joints, cardiac valves, the brain, and other sites.
The clinical presentation of Whipple disease is usually a triad of diarrhea, weight loss, and malabsorption. Extraintestinal symptoms, which can exist for months or years before malabsorption, include arthritis, arthralgia, fever, lymphadenopathy, and neurologic, cardiac, or pulmonary disease.
Mycobacterial infections are considered in detail in Chapter 8.
VIRAL GASTROENTERITIS
Symptomatic human infection is caused by several distinct groups of viruses. The most common are discussed here.
Norovirus.
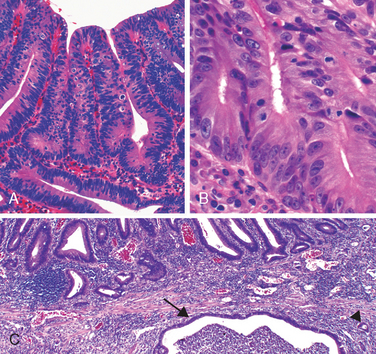
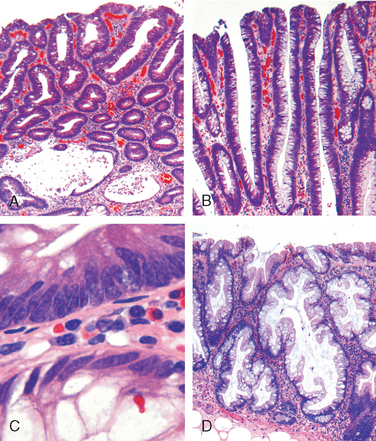
This was previously known as norwalk-like virus and is a common cause of nonbacterial infectious gastroenteritis.87 These are small icosahedral viruses with a single-stranded RNA genome that forms a genus within the Caliciviridae family. Norovirus causes approximately half of all gastroenteritis outbreaks worldwide and is a common cause of sporadic gastroenteritis in developed countries. Local outbreaks are usually related to contaminated food or water, but person-to-person transmission underlies most sporadic cases. Infections spread easily within schools, hospitals, nursing homes, and, most recently, cruise ships. Following a short incubation period, affected individuals develop nausea, vomiting, watery diarrhea, and abdominal pain. Biopsy morphology is nonspecific. When detected, abnormalities are most evident in the small intestine and include mild villous shortening, epithelial vacuolization, loss of the microvillus brush border, crypt hypertrophy, and lamina propria infiltration by lymphocytes (Fig. 17-31A). The disease is self-limited.
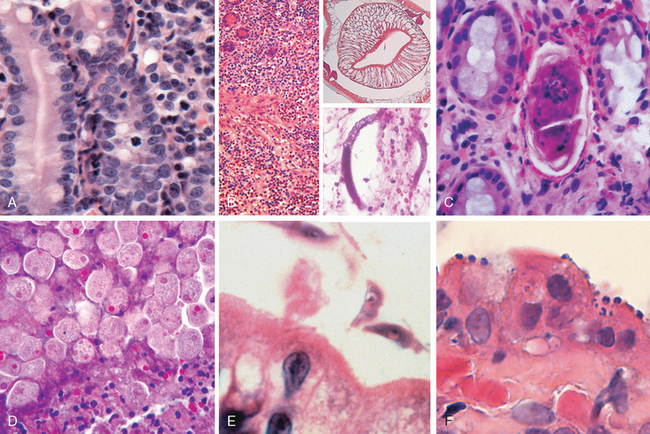
FIGURE 17-31 Infectious enteritis. A, Histologic features of viral enteritis include increased numbers of intraepithelial and lamina propria lymphocytes and crypt hypertrophy. B, Diffuse eosinophilic infiltrates in parasitic infection. This case was caused by Ascaris (upper inset), but a similar tissue reaction could be caused by Strongyloides (lower inset). C, Schistosomiasis can induce an inflammatory reaction to eggs trapped within the lamina propria. D, Entamoeba histolytica in a colon biopsy specimen. Note some organisms ingesting red blood cells. E, Giardia lamblia, which are present in the luminal space over nearly normal-appearing villi, are easily overlooked. F, Cryptosporidia organisms are seen as small blue spheres that appear to lay on top of the brush border but are actually enveloped by a thin layer of host cell cytoplasm.
Rotavirus.
This encapsulated virus with a segmented double-stranded RNA genome infects 140 million people and causes 1 million deaths each year, making rotavirus the most common cause of severe childhood diarrhea and diarrheal mortality worldwide. Children between 6 and 24 months of age are most vulnerable. Protection in the first 6 months of life is probably due to the presence of antibodies to rotavirus in breast milk, while protection beyond 2 years is due to immunity that develops following the first infection.88 Outbreaks in hospitals and daycare centers are common, and infection spreads easily; the estimated minimal infective inoculum is only 10 viral particles. Rotavirus selectively infects and destroys mature enterocytes in the small intestine, and the villus surface is repopulated by immature secretory cells. This results in loss of absorptive function and net secretion of water and electrolytes that is compounded by an osmotic diarrhea from incompletely absorbed nutrients. Like norovirus, rotavirus has a short incubation period followed by several days of vomiting and watery diarrhea. Vaccines are now available, and their use will probably change the epidemiology of rotavirus infection.
Adenovirus.
The second most common cause of pediatric diarrhea (after rotavirus), adenovirus also affects immunocompromised patients.89 Small intestinal biopsy specimens can show epithelial degeneration but more often exhibit nonspecific villous atrophy and compensatory crypt hyperplasia. Viral nuclear inclusions are uncommon. Disease typically presents after an incubation period of 1 week with nonspecific symptoms that include diarrhea, vomiting, and abdominal pain. Fever and weight loss may also be present. Symptoms generally resolve within 10 days.
PARASITIC ENTEROCOLITIS
Although viruses and bacteria are the predominant enteric pathogens in the United States, parasitic disease and protozoal infections affect over one half of the world’s population on a chronic or recurrent basis. The small intestine can harbor as many as 20 species of parasites, including nematodes, such as the roundworms Ascaris and Strongyloides; hookworms and pinworms; cestodes, including flatworms and tapeworms; trematodes, or flukes; and protozoa. Parasitic infections are covered in Chapter 8, and we will briefly discuss only those that are common in the intestinal tract.
Ascaris lumbricoides.
This nematode infects over a billion individuals worldwide as a result of human fecal-oral contamination. Ingested eggs hatch in the intestine and larvae penetrate the intestinal mucosa, but disease is caused when larvae migrate from the splanchnic circulation to the systemic circulation and create hepatic abscess or Ascaris pneumonitis. In the latter case, larvae migrate up the trachea, are swallowed, and arrive again in the intestine to mature into adult worms. Adult worm masses induce an eosinophil-rich inflammatory reaction (Fig. 17-31B) that can physically obstruct the intestine or biliary tree. Diagnosis is usually made by detection of eggs in stool samples.
Strongyloides.
The larvae of Strongyloides live in fecally contaminated ground soil and can penetrate unbroken skin. They migrate through the lungs, where they induce inflammatory infiltrates, and then reside in the intestine while maturing into adult worms. Unlike other intestinal worms, which require an ova or larval stage outside the human, the eggs of Strongyloides can hatch within the intestine and release larvae that penetrate the mucosa, causing autoinfection (see Fig. 17-31B). Hence, Strongyloides infection can persist for life, and immunosuppressed individuals can develop overwhelming autoinfection. Strongyloides incite a strong tissue reaction and induce peripheral eosinophilia.
Necator duodenale and Ancylostoma duodenale.
These hookworms infect 1 billion people worldwide and cause significant morbidity. Infection is initiated by larval penetration through the skin and, after further development in the lungs the larvae migrate up the trachea and are swallowed. Once in the duodenum the worms attach to the mucosa, suck blood, and reproduce. This causes multiple superficial erosions, focal hemorrhage, and inflammatory infiltrates and, in chronic infection, iron deficiency anemia. Diagnosis can be made by detection of the eggs in fecal smears.
Enterobius vermicularis.
Also known as pinworms, these parasites infect people in industrialized and developing countries; in the United States more than 60 million people have pinworms. Because they do not invade host tissue and live their entire life within the intestinal lumen, they rarely cause serious illness. Infection by E. vermicularis, or enterobiasis, is primarily by the fecal-oral route. Adult worms living in the intestine migrate to the anal orifice at night, where the female deposits eggs on the perirectal mucosa. The eggs cause intense irritation. Rectal and perineal pruritus ensues and leads to contamination of the fingers, which promotes human-to-human transmission. Both eggs and adult pinworms remain viable outside the body, and repeat infection is common. Diagnosis can be made by applying cellophane tape to the perianal skin and examining the tape for eggs under a microscope.
Trichuris trichiura.
Whipworms primarily infect young children. Similar to E. vermicularis, Trichuris trichiura does not penetrate the intestinal mucosa and rarely cause serious disease. Heavy infections, however, may cause bloody diarrhea and rectal prolapse.
Schistosomiasis.
This disease involving the intestines most commonly takes the form of adult worms residing within the mesenteric veins. Symptoms of intestinal schistosomiasis are caused by trapping of eggs within the mucosa and submucosa (Fig. 17-31C). The resulting immune reaction is often granulomatous and can cause bleeding and even obstruction. More details are presented in Chapter 8.
Intestinal cestodes.
The three primary species of cestodes that affect humans are Diphyllobothrium latum, fish tapeworms; Taenia solium, pork tapeworms; and Hymenolepsis nana, dwarf tapeworms. They reside exclusively within the intestinal lumen and are transmitted by ingestion of raw or undercooked fish, meat, or pork that contain encysted larvae. Release of the larvae allows attachment to the intestinal mucosa through its head, or scolex. The worm derives its nutrients from the food stream and enlarges by formation of egg-filled segments termed proglottids. Humans are usually infected by a single worm, and, since the worm does not penetrate the intestinal mucosa, peripheral eosinophilia does not generally occur. Nevertheless, the parasite burden can be staggering, since adult worms can grow to many meters in length. Large numbers of proglottids or individual eggs are shed in the feces. Clinical symptoms include abdominal pain, diarrhea, and nausea. Diagnosis is established by stool examination.
Entamoeba histolytica.
This protozoan that causes amebiasis is spread by fecal-oral transmission. E. histolytica infects approximately 500 million people in developing countries such as India, Mexico, and Colombia, and causes 40 million cases of dysentery and liver abscess annually. E. histolytica cysts, which have a chitin wall and four nuclei, are resistant to gastric acid, a characteristic that allows them to pass through the stomach without harm. Cysts then colonize the epithelial surface of the colon and release trophozoites, ameboid forms that reproduce under anaerobic conditions.
Amebiasis is seen most frequently in the cecum and ascending colon, although the sigmoid colon, rectum, and appendix can also be involved. Dysentery develops when the amebae attach to the colonic epithelium, induce apoptosis, invade crypts, and burrow laterally into the lamina propria. This recruits neutrophils, causes tissue damage, and creates a flask-shaped ulcer with a narrow neck and broad base. Histologic diagnosis can be difficult, since amebae are similar to macrophages in size and general appearance (see Fig. 17-31D). Parasites may penetrate splanchnic vessels and embolize to the liver to produce abscesses in about 40% of patients with amebic dysentery. Amebic liver abscesses, which can exceed 10 cm in diameter, have a scant inflammatory reaction at their margins and a shaggy fibrin lining. The abscesses persist after the acute intestinal illness has passed and may, rarely, reach the lung and the heart by direct extension from the liver. Amebae may also spread via the bloodstream into the kidneys and brain.
Individuals with amebiasis may present with abdominal pain, bloody diarrhea, or weight loss. Occasionally, acute necrotizing colitis and megacolon occur, and both are associated with significant mortality. The parasites lack mitochondria or Krebs cycle enzymes and are thus obligate fermenters of glucose. Therefore, metronidazole, which inhibits the enzyme pyruvate oxidoreductase that is required for fermentation, is the most effective treatment.
Giardia lamblia.
These organisms, also referred to as G. duodenalis or G. intestinalis, were initially described by van Leeuwenhoek, the inventor of the microscope, who discovered the pathogen in his own stool. They are the most common pathogenic parasitic infection in humans and are spread by fecally contaminated water or food. Infection may occur after ingestion of as few as 10 cysts. Because cysts are resistant to chlorine, Giardia are endemic in unfiltered public water supplies. They are commonly present in rural streams, explaining infection in campers who use these as a water source. Infection may also occur by the fecal-oral route and, because the cysts are stable, they may be accidentally swallowed while swimming in contaminated water.
Giardia are flagellated protozoans that cause decreased expression of brush-border enzymes, including lactase; microvillous damage; and apoptosis of small intestinal epithelial cells. Secretory IgA and mucosal IL-6 responses are important for clearance of Giardia infections, and immunosuppressed, agammaglobulinemic, or malnourished individuals are often severely affected.90 Giardia can evade immune clearance through continuous modification of the major surface antigen, variant surface protein, and can persist for months or years while causing intermittent symptoms.
Giardia trophozoites can be identified in duodenal biopsies by their characteristic pear shape with two nuclei of equal size, each of which contains a complete copy of the genome. Despite large numbers of trophozoites, which are sickle-shaped in profile and tightly bound to the brush border of villous enterocytes, there is no invasion and small intestinal morphology may be normal by light microscopy (see Fig. 17-31E). Villous blunting with increased numbers of intraepithelial lymphocytes and mixed lamina propria inflammatory infiltrates may be present in patients with heavy infections.
Giardiasis may be subclinical or accompanied by acute or chronic diarrhea, malabsorption, and weight loss.90 Infection is usually documented by immunofluorescent detection of cysts in stool samples. Although oral antimicrobial therapy is effective, recurrence is common.
Cryptosporidium.
Like Giardia, Cryptosporidia are an important cause of diarrhea worldwide. Cryptosporidiosis was first discovered in the 1980s as an agent of chronic diarrhea in AIDS patients and is now recognized as a cause of acute, self-limited disease in immunologically normal hosts. Cryptosporidiosis causes traveler’s diarrhea as well as persistent diarrhea in residents of developing countries. The organisms are present worldwide, with the exception of Antarctica, perhaps because the oocysts are killed by freezing. The oocysts are resistant to chlorine and may, therefore, persist in treated, but unfiltered, water. Contaminated drinking water continues to be the most common means of transmission. The largest documented outbreak, a result of inadequate water purification, occurred in 1993 in Milwaukee, Wisconsin, and affected more than 400,000 people. Like giardiasis, cryptosporidiosis can be spread to water sport participants via contaminated water. Food-borne infection occurs less frequently.
Humans are infected by several different Cryptosporidium species, including C. hominis and C. parvum. All are able to go through an entire life cycle, with asexual and sexual reproductive phases, in a single host. The ingested encysted oocyte, of which 10 are sufficient to cause symptomatic infection, is activated by acid in the stomach to produce proteases that allow release of sporozoites from the oocysts. The sporozoites are motile and have a specialized organelle for attachment to the enterocyte brush border, where they induce actin polymerization. This drives extension of the epithelial cell membrane to engulf the parasite and form a vacuole within the microvilli. Sodium malabsorption, chloride secretion, and increased tight junction permeability are responsible for the nonbloody, watery diarrhea that ensues.
Mucosal histology is often only minimally altered, but persistent cryptosporidiosis in children is associated with villous atrophy. Heavy infection in immunosuppressed patients may be associated with villous atrophy, crypt hyperplasia, and variable inflammatory infiltrates. Although the sporozoite is intracellular, it appears, by light microscopy, to sit on top of the epithelial apical membrane (Fig. 17-31F). Organisms are typically most concentrated in the terminal ileum and proximal colon, but can be present throughout the gut, biliary tract, and even the respiratory tract of immunodeficient hosts. Diagnosis is based on finding oocysts in the stool.