Chapter 29 The Eye
Although this chapter comes at the end of the book, it is not the least important. Vision is a major quality-of-life issue. Before the public awareness of AIDS and Alzheimer disease, the most feared disease among Americans was cancer, and the second most feared disease was blindness. So great is the fear of blindness that even today, people often tell their physicians, “Doctor, I’d rather be dead than be blind!”
In general, diseases that produce loss of vision do not attract as much of our attention as do many of the life-threatening conditions described in this book. For example, age-related macular degeneration (ARMD) is the most common cause of irreversible visual loss in the United States. Most individuals with ARMD do not even suffer from a total loss of vision—an immersion into total darkness. The histopathology is unspectacular: small scars develop in the macula. But consider the effect of these tiny scars perhaps in a retired schoolteacher with ARMD. The small macular scars make it impossible for this person to see anything clearly in the central portion of her or his vision. The faces of spouse or grandchildren are not visible. He or she cannot read a book or newspaper. Once a model of independence, this teacher can no longer drive a car and must be chauffered everywhere. True, his or her life is not threatened by the small scars in the eyes’ macula, but the quality of life declines as this person is robbed of the common joys that most of us take for granted until they are lost.
To study the eye, one needs to comprehend all that has come before. For example, the pathology of the eyelids builds on knowledge of dermatopathology (Chapter 25), and the pathology of the retina and optic nerve extends what was learned in Chapter 28 about the brain and central nervous system. However, the study of ocular pathology does not merely repeat what has been presented thus far. The eye provides the only site in which a physician can easily visualize a variety of pathophysiologic disturbances in the microcirculation ranging from arteriosclerosis to angiogenesis in a clinical setting. Although there are conditions that are unique to the eye (such as cataract and glaucoma), many ocular conditions share similarities with disease processes elsewhere in the body that are modified by the unique structure and function of the eye (Fig. 29-1). Moreover, the eye has much to teach us about important mechanisms of disease that extend far beyond the visual system. For example, the tumor suppressor gene, RB, was described in retinoblastoma,1 a quite uncommon ocular tumor of infants and very young children, but the discovery of RB opened an important pathway to the understanding of the regulation of cellular replication.
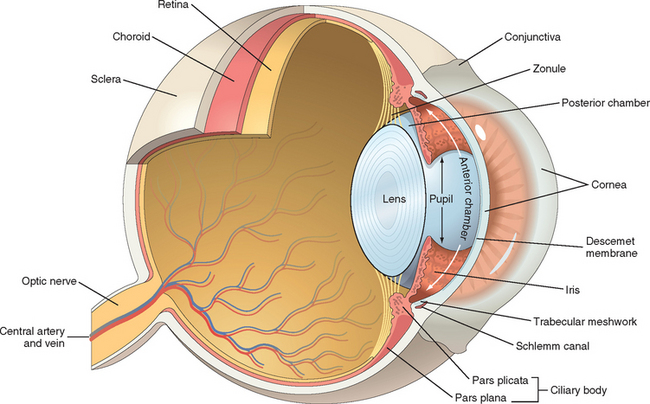
This chapter is organized on the basis of ocular anatomy. The discussion of each region of the eye begins with anatomic and functional considerations, and their impact on the understanding of ocular diseases.
Orbit
FUNCTIONAL ANATOMY AND PROPTOSIS
The orbit is a compartment that is closed medially, laterally, and posteriorly. Diseases that increase orbital contents therefore displace the eye forward, a condition known as proptosis. Aside from the obvious cosmetic concerns, the proptotic eye might not be covered completely by the eyelids, and the tear film might not be distributed evenly across the cornea. Corneal exposure is painful and can predispose to corneal ulceration and infection. Proptosis may be axial (directly forward) or positional. For example, any enlargement of the lacrimal gland from inflammation (e.g., sarcoidosis) or neoplasm (e.g., lymphoma, pleomorphic adenoma, or adenoid cystic carcinoma) produces a proptosis that displaces the eye inferiorly and medially, because the lacrimal gland is positioned superotemporally within the orbit.
Masses contained within the cone formed by the horizontal rectus muscles generate axial proptosis: the eye bulges straight forward. The two most common primary tumors of the optic nerve (a tract of the central nervous system and not a peripheral nerve), glioma and meningioma, produce axial proptosis because the optic nerve is positioned within the muscle cone. The orbital contents are subject to the same disease processes that affect other tissues. Representative inflammatory conditions and neoplasms of the orbit are discussed briefly next.
THYROID OPHTHALMOPATHY (GRAVES DISEASE)
In the chapter on endocrine disorders (Chapter 24) it was noted that axial proptosis is an important clinical manifestation of Graves disease. Proptosis is caused by the accumulation of extracellular matrix proteins and variable degrees of fibrosis in the rectus muscles (Fig. 29-2). The development of thyroid ophthalmopathy may be independent of the status of thyroid function.2
OTHER ORBITAL INFLAMMATORY CONDITIONS
The floor of the orbit is the roof of the maxillary sinus, and the medial wall of the orbit—the lamina papyracea—separates the orbit from the ethmoidal sinuses. Thus, uncontrolled sinus infection may spread to the orbit either as an acute infection (orbital cellulitis) or as a component of a fungal infection (mucormycosis) in immunosuppressed individuals, in patients with diabetic ketoacidosis, or, rarely, in persons without any predisposition. Systemic conditions such as Wegener granulomatosis (Chapter 11) may present first in the orbit and may be confined there for prolonged periods of time,3 or alternatively, it may involve the orbit secondarily by extension from the sinuses.
Idiopathic orbital inflammation, also known as orbital inflammatory pseudotumor (Fig. 29-3), is another inflammatory condition affecting the orbit. This condition may be unilateral or bilateral, and may affect all orbital tissue elements or may be confined to the lacrimal gland (sclerosing dacryoadenitis), the extra-ocular muscles (orbital myositis), or the Tenon’s capsule, the fascial layer that wraps around the eye (posterior scleritis). In long-term follow-up a subset of individuals with idiopathic orbital inflammation may show evidence of systemic vasculitis or other forms of connective tissue diseases.
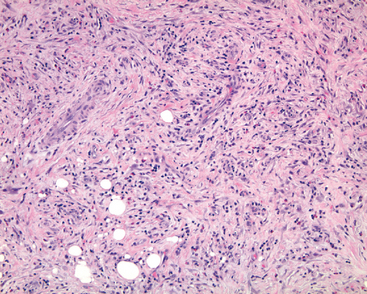
FIGURE 29-3 In idiopathic orbital inflammation (orbital inflammatory pseudotumor) the orbital fat is replaced by fibrosis. Note the chronic inflammation, accompanied in this case by eosinophils.
Morphology. Idiopathic orbital inflammation is characterized histologically by chronic inflammation and variable degrees of fibrosis. The inflammatory infiltrate typically includes lymphocytes and plasma cells and occasionally eosinophils. Germinal centers, when present, raise the suspicion of a reactive lymphoid hyperplasia. Vasculitis may be present, suggesting an underlying systemic condition. The presence of necrotic collagen along with vasculitis should raise the suspicion of Wegener granulomatosis. Idiopathic orbital inflammation is typically confined to the orbit but may develop concomitantly with sclerosing inflammation in the retroperitoneum, the mediastinum, and the thyroid.
NEOPLASMS
The most frequently encountered primary neoplasms of the orbit are vascular in origin: the capillary hemangioma of infancy and early childhood and the lymphangioma (both of which are unencapsulated) and the encapsulated cavernous hemangioma found typically in adults. These are described in other chapters. Only a handful of orbital masses are encapsulated (e.g., pleomorphic adenoma of the lacrimal gland, dermoid cyst, neurilemmoma), and the recognition of encapsulation on imaging studies allows the surgeon to anticipate pathologic findings.
Non-Hodgkin lymphoma, like idiopathic orbital inflammation, can affect the entire orbit or can be confined to compartments of the orbit such as the lacrimal gland. Orbital lymphomas are classified according to the WHO classification system (Chapter 13).
Primary orbital malignancies may arise from any of the orbital tissues and are classified according to the scheme used for the parent tissue. For example, the lacrimal gland may be considered a minor salivary gland, and tumors of the lacrimal gland are classified as salivary gland tumors are classified.
Metastases to the orbit may present with distinctive signs and symptoms that point to the origin of the tumor. For example, metastatic prostatic carcinoma may present clinically like idiopathic orbital inflammation; metastatic neuroblastoma and Wilms tumor—richly vascular neoplasms—may produce characteristic periocular ecchymoses. Neoplasms may also invade from the sinuses into the orbit.
Eyelid
FUNCTIONAL ANATOMY
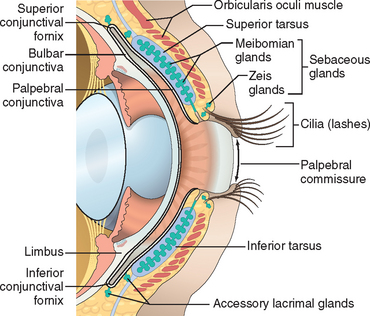
The eyelid is composed of skin externally and mucosa (the conjunctiva) on the surface apposed to the eye (Fig. 29-4). In addition to covering and protecting the eye, elements within the eyelid generate critical components of the tear film. If the drainage system of the sebaceous glands is obstructed by chronic inflammation at the eyelid margin (blepharitis) or, less commonly, by neoplasm, then lipid may extravasate into surrounding tissue and provoke a granulomatous response producing a lipogranuloma, or chalazion.
NEOPLASMS
The most common malignancy of the eyelid is basal cell carcinoma. The second most common malignancy is sebaceous carcinoma, followed by squamous cell carcinoma. Surprisingly, primary melanomas of the eyelid skin are extremely rare. Regardless of histogenesis, eyelid neoplasms may distort tissue and prevent the eyelids from closing completely. Exposure of the cornea is not only painful but predisposes the individual to corneal ulceration. Therefore, prompt treatment of locally invasive basal cell carcinomas, which are typically not a threat to the affected individual’s life, is imperative to preserve vision. Basal cell carcinoma has a distinct predilection for the lower eyelid and the medial canthus.
Sebaceous carcinoma may form a local mass that mimics chalazion or may diffusely thicken the eyelid. This neoplasm may also resemble inflammatory processes such as blepharitis or ocular cicatricial pemphigoid because of intraepithelial spread as occurs in Paget disease of the nipple (Chapter 23) or vulva. Sebaceous carcinoma tends to spread first to the parotid and submandibular nodes. The overall mortality rate can be as high as 22%.4
Morphology. In moderately differentiated or welldifferentiated sebaceous carcinoma, vacuolization of the cytoplasm is present and helps in the diagnosis. This cancer may, however, resemble a variety of other malignancies histologically, including basal cell carcinoma, and establishing the correct diag-nosis can be difficult. Pagetoid spread (Fig. 29-5) may mimic Bowenoid actinic keratosis in the eyelid and carcinoma in situ in the conjunctiva. Sebaceous carcinoma may spread through the conjunctival epithelium and the epidermis to the lacrimal drainage system and the nasopharynx. It may also extend into the lacrimal gland ductules and thereby into the main lacrimal gland.
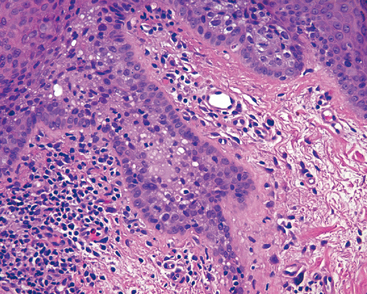
In individuals with AIDS, Kaposi sarcoma may develop in either the eyelid or the conjunctiva. In the eyelid the lesion may appear clinically to have a purple hue because the vascular lesion is embedded in the dermis, but in the thin mucous membrane of the conjunctiva, Kaposi sarcoma appears bright red and may be confused clinically with a subconjunctival hemorrhage.
Conjunctiva
FUNCTIONAL ANATOMY
The conjunctiva is divided into zones (see Fig. 29-4), each with distinctive histologic features and responses to disease. The conjunctiva lining the interior of the eyelid, the palpebral conjunctiva, is tightly tethered to the tarsus and may respond to inflammation by being thrown into minute papillary folds as may occur in allergic conjunctivitis and bacterial conjunctivitis. The conjunctiva in the fornix is a pseudostratified columnar epithelium rich in goblet cells. The fornix also contains accessory lacrimal tissue, and the ductules of the main lacrimal gland pierce through the conjunctiva in the fornix superiorly and laterally. The lymphoid population of the conjunctiva is most noticeable in the fornix, and in viral conjunctivitis, lymphoid follicles may enlarge sufficiently to be visualized clinically by slit-lamp examination. Granulomas associated with systemic sarcoidosis may be detected in the conjunctival fornix, and the yield of granulomas from a nondirected conjunctival biopsy in individuals suspected of having sarcoid may be as high as 50%.5 Primary lymphoma of the conjunctiva (typically indolent marginal zone B-cell lymphoma) is most likely to develop in the fornix. The bulbar conjunctiva—the conjunctiva that covers the surface of the eye—is a nonkeratinizing stratified squamous epithelium.
The conjunctiva, like the eyelid, is richly invested with lymphatic channels. Malignant neoplasms arising in the eyelid and conjunctiva tend to spread to regional lymph nodes (parotid and submandibular node groups).
CONJUNCTIVAL SCARRING
Many cases of bacterial or viral conjunctivitis cause redness and itching, but most heal without sequelae. However, infection with Chlamydia trachomatis (trachoma) may produce significant conjunctival scarring. Conjunctival scarring is also seen after exposure of the ocular surface to caustic alkalis or as a sequela to ocular cicatricial pemphigoid (Chapter 25). A reduction in the number of goblet cells due to conjunctival scarring leads to a decrease in surface mucin, which is essential for the adherence of the aqueous component of tears to the corneal epithelium. Thus, even if the aqueous component of the tear film is adequate, the affected individual will suffer from a dry eye, a condition that, when severe, can be painful and can predispose to corneal opacification and ulceration. More commonly, however, dry eye results from a deficiency in the aqueous component of the tear film generated by the accessory lacrimal glands embedded within the eyelid and fornix.
The conjunctiva may be scarred iatrogenically through reaction to drugs or as a consequence of surgery. In other parts of the body, cancer surgery requires excision of the lesion with a margin of normal tissue to ensure complete removal. However, extensive surgical excision of even diseased conjunctiva can remove a large number of goblet cells or compromise lacrimal gland ductules that traverse the conjunctiva. Thus, removal of a conjunctival neoplasm or a precursor lesion may leave the affected individual with a painful dry eye that can compromise vision. Therefore, surgeons often remove only the invasive components of conjunctival neoplasms, and treat the intraepithelial components with tissue-sparing modalities such as cryotherapy or topical chemotherapy delivered as eyedrops.
PINGUECULA AND PTERYGIUM
Both pinguecula and pterygium appear as submucosal elevations on the conjunctiva. They result from actinic damage and are therefore located in the sun-exposed regions of the conjunctiva (i.e., in the fissure between both the upper and lower eyelids—the interpalpebral fissure). Pterygium typically originates in the conjunctiva astride the limbus. It is formed by a submucosal growth of fibrovascular connective tissue that migrates onto the cornea, dissecting into the plane occupied normally by the Bowman layer. Pterygium does not cross the pupillary axis and, aside from the possible induction of mild astigmatism, does not pose a threat to vision. Although most pterygia are entirely benign, it is worthwhile submitting the excised tissue for pathologic examination because, on occasion, precursors of actinic-induced neoplasms—squamous cell carcinoma and melanoma—are detected in these lesions.
Pinguecula, which, like pterygium, appears astride the limbus, is a small, yellowish submucosal elevation. Although the pinguecula does not invade the cornea as pterygium does, the presence of a focal conjunctival elevation near the limbus can result in an uneven distribution of the tear film over the adjacent cornea. As a consequence of focal dehydration, a saucer-like depression in the corneal tissue—a dellen—may develop.
NEOPLASMS
Both squamous neoplasms and melanocytic neoplasms and their precursors tend to develop at the limbus. Conjunctival squamous cell carcinoma may be preceded by intraepithelial neoplastic changes analogous to those seen in the evolution of cervical squamous cell carcinoma. In the conjunctiva the spectrum of changes from mild dysplasia through carcinoma in situ is designated as conjunctival intraepithelial neoplasia. Squamous papillomas and conjunctival intraepithelial neoplasia may be associated with the presence of human papillomavirus types 16 and 18.6 Although conjunctival squamous cell carcinoma tends to follow an indolent course, mucoepidermoid carcinoma of the conjunctiva (reflecting the ability of conjunctival stem cells to differentiate into squamous epithelium and goblet cells) follows a much more aggressive course.
Conjunctival nevi are encountered commonly in clinical practice but seldom invade the cornea or appear in the fornix or over the palpebral conjunctiva.7 Pigmented lesions in these zones of the conjunctiva most likely represent melanomas or melanoma precursors. Compound nevi of the conjunctiva characteristically contain subepithelial cysts lined by surface epithelium (Fig. 29-6A, B). In late childhood or adolescence, conjunctival nevi may acquire an inflammatory component rich in lymphocytes, plasma cells, and eosinophils. The resultant inflamed juvenile nevus is completely benign and not associated with vitiligo or halo nevus.
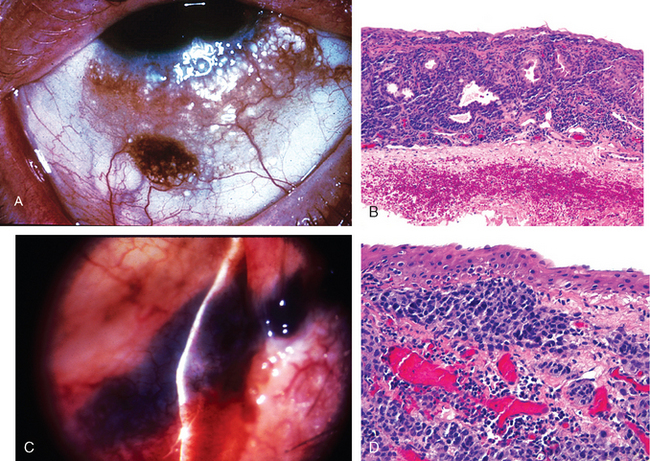
FIGURE 29-6 A, B, Cystic compound nevus of the conjunctiva. C, D, Conjunctival malignant melanoma. In C, note the deflection of the beam of the slit lamp over the surface of the lesion, indicative of invasion.
(A, B, From Folberg R et al.: Benign conjunctival melanocytic lesions: clinicopathologic features. Ophthalmology 96:436, 1989.)
Conjunctival melanomas are unilateral neoplasms, typically affecting fair-complexioned individuals in middle age8 (Fig. 29-6C, D). Most cases of conjunctival melanoma develop through a phase of intraepithelial growth termed primary acquired melanosis with atypia, which is roughly analogous to melanoma in situ but does not correspond neatly to the radial growth phase of cutaneous melanoma. Between 50% and 90% of individuals with incompletely treated primary acquired melanosis with atypia will develop conjunctival melanoma; the best treatment of conjunctival melanoma is its prevention through extirpation of its precursor lesion. The lesions tend to spread first to the parotid or submandibular lymph nodes. The mortality for conjunctival melanoma is 25%.
Sclera
The sclera consists mainly of collagen and contains few blood vessels and fibroblasts; hence, wounds and surgical incisions tend to heal poorly. Immune complex deposits within the sclera, such as in rheumatoid arthritis, may produce a necrotizing scleritis.
The sclera may appear “blue” in a variety of conditions. It may become thin following episodes of scleritis, and the normally brown color of the uvea may appear blue clinically because of the optical Tyndall effect. Sclera may also be thinned in eyes with exceptionally high intraocular pressure and because this zone of scleral ectasia is lined by uveal tissue, the resulting lesion, known as a staphyloma, also appears blue. The sclera may appear blue in osteogenesis imperfecta. Finally, the sclera may appear blue because of a heavily pigmented congenital nevus of the underlying uvea, a condition known as congenital melanosis oculi. When accompanied by periocular cutaneous pigmentation, this condition is known as nevus of Ota.
Cornea
FUNCTIONAL ANATOMY
The cornea and its overlying tear film—not the lens—make up the major refractive surface of the eye (Fig. 29-7). Parenthetically, myopia typically develops because the eye is too long for its refractive power, and hyperopia results from an eye that is too short. The popularity of procedures such as laser-assisted in situ keratomileusis (LASIK) to sculpt the cornea and change its refractive properties attests to the importance of corneal shape in contributing to the refractive power of the eye.
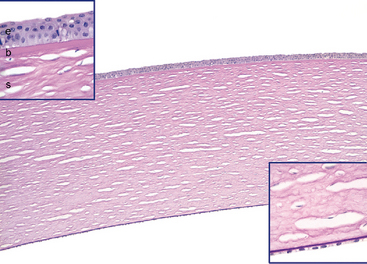
FIGURE 29-7 Normal corneal microarchitecture. The corneal tissue is stained by periodic acid–Schiff (PAS) to highlight basement membranes. The inset at the upper left is a high magnification of the anterior layers of the cornea: the epithelium (e), Bowman layer (b), and the stroma (s). A very thin PAS-positive basement membrane separates the epithelium from the Bowman layer. Note that the Bowman layer is acellular. The inset at the lower right is a high magnification of the PAS-positive Descemet membrane and the corneal endothelium. The “holes” in the stroma are artifactitious spaces between parallel collagenous stromal lamellae.
Anteriorly, the cornea is covered by epithelium that rests on a basement membrane. The Bowman layer, situated just beneath the epithelial basement membrane, is acellular and forms an efficient barrier against the penetration of malignant cells from the epithelium into the underlying stroma.
The corneal stroma lacks blood vessels and lymphatics, a feature that contributes not only to the transparency of the cornea, but also to high rate of success of corneal transplantation. Indeed, non-immunological graft failure (associated with loss of endothelial cells and subsequent corneal edema) is seen more commonly than is immunological graft rejection. The risk of corneal graft rejection increases with stromal vascularization and inflammation. A precise alignment of collagen in the corneal stroma also contributes to transparency. Scarring and edema both disrupt the spatial alignment of stromal collagen and contribute to corneal opacification. Scars may result from trauma or inflammation. Normally, the corneal stroma is in a state of relative deturgescence (dehydration), maintained in large part by active pumping of fluid from the stroma back into the anterior chamber by the corneal endothelium.
The corneal endothelium is derived from neural crest and is not related to vascular endothelium. It rests on its basement membrane, Descemet membrane. A decrease in endothelial cells or a malfunction of endothelium results in stromal edema, which may be complicated by bullous separation of the epithelium (bullous keratopathy). Descemet membrane increases in thickness with age. It is the site of copper deposition in the Kayser-Fleischer ring of Wilson disease (Chapter 18).
KERATITIS AND ULCERS
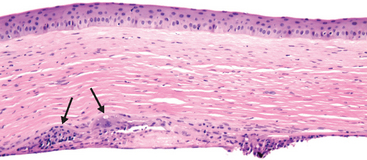
Various pathogens—bacterial, fungal, viral (especially herpes simplex and herpes zoster), and protozoal (Acanthamoeba)—can cause corneal ulceration. In all forms of keratitis, dissolution of the corneal stroma may be accelerated by activation of collagenases within corneal epithelium and stromal fibroblasts (also known as keratocytes). Exudate and cells leaking from iris and ciliary body vessels into the anterior chamber may be visible by slit-lamp examination and may accumulate in sufficient quantity to become visible even by a penlight examination (hypopyon). Although the corneal ulcer may be infectious, the hypopyon seldom contains organisms and is an example par excellence of the vascular response to acute inflammation. The specific forms of keratitis may have certain distinctive features. For example, chronic herpes simplex keratitis may be associated with a granulomatous reaction involving the Descemet membrane (Fig. 29-8).
CORNEAL DEGENERATIONS AND DYSTROPHIES
Ophthalmologists have traditionally divided many corneal disorders into degenerations and dystrophies. Corneal degenerations may be either unilateral or bilateral and are typically nonfamilial. By contrast, corneal dystrophies are typically bilateral and are hereditary. Corneal dystrophies may affect selective corneal layers (e.g., Reis-Bückler dystrophy affects Bowman layer, and posterior polymorphous dystrophy affects the endothelium), or the changes may be distributed throughout multiple layers.
Band Keratopathies
Two types of band keratopathy serve as examples of corneal degenerations. Calcific band keratopathy is characterized by deposition of calcium in the Bowman layer. This condition may complicate chronic uveitis, especially in individuals with chronic juvenile rheumatoid arthritis. Actinic band keratopathy develops in individuals who are exposed chronically to high levels of ultraviolet light. In this condition, extensive solar elastosis develops in the superficial layers of corneal collagen in the sun-exposed interpalpebral fissure, hence the horizontally distributed band of pathology. Similar to pinguecula, the sun-damaged collagen of the cornea appears clinically to be yellow to the point that this condition is sometimes erroneously called “oil-droplet keratopathy.”
Keratoconus
With an incidence of 1 in 2000, keratoconus is a fairly common disorder characterized by progressive thinning and ectasia of the cornea without evidence of inflammation or vascularization. Such thinning results in a cornea that has a conical rather than spherical shape. This abnormal shape generates irregular astigmatism that is difficult to correct with spectacles. Rigid contact lenses generate a smooth, spherical surface to the cornea and may provide refractive relief for individuals with keratoconus. Patients whose vision cannot be corrected with spectacles or contact lenses are excellent candidates for corneal transplantation, which has a high degree of success in this condition. The etiology of keratoconus is unknown. Unlike many degenerations, it is typically bilateral. There is an association between keratoconus, Down syndrome, and Marfan syndrome, as well as with atopic disorders. Activation of collagenases, gelatinases, and matrix metalloproteinases has been implicated in the pathogenesis of this condition.
Morphology. Thinning of the cornea with breaks in the Bowman layer are the histologic hallmarks of keratoconus (Fig. 29-9). In some patients the Descemet membrane may rupture precipitously, allowing the aqueous humor in the anterior chamber to gain access to the corneal stroma. The sudden effusion of aqueous humor through a gap in the Descemet membrane—corneal hydrops—may also cause vision to worsen suddenly. An episode of hydrops may be followed by corneal scarring that can also contribute to visual loss. Acute corneal hydrops can complicate Descemet membrane ruptures that develop secondary to extraordinary elevations of intraocular pressure in infantile glaucoma (Haab’s striae) or following the now uncommon obstetric forceps injury to the eye.
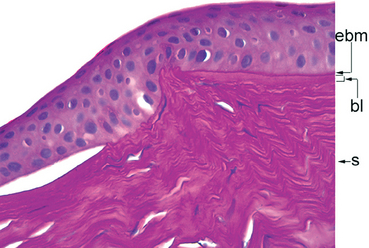
FIGURE 29-9 Keratoconus. The tissue section is stained by PAS to highlight the epithelial basement membrane (ebm), which is intact, the Bowman layer (bl), situated between the epithelial basement membrane, and the stroma (s). Following the Bowman layer from the right side of the photomicrograph toward the center, there is a discontinuity, diagnostic of keratoconus. The epithelial separation just to the left of the Bowman layer discontinuity resulted from an episode of corneal hydrops, caused by a break in the Descemet membrane (not shown).
Fuchs Endothelial Dystrophy
This condition, one of several dystrophies affecting the endothelium, is one of the principal indications for corneal transplantation in the United States. The two major clinical manifestations of Fuchs endothelial dystrophy—stromal edema and bullous keratopathy—are both related to a primary loss of endothelial cells. Early in the course of the disease endothelial cells produce droplike deposits of abnormal basement membrane material (guttata) that resemble the fetal component of the Descemet membrane ultrastructurally. Guttata can be visualized clinically by slit-lamp examination. With disease progression, there is a decrease in the total number of endothelial cells, and the residual cells are incapable of maintaining stromal deturgescence. Consequently the stroma becomes edematous and thickens; it acquires a ground-glass appearance clinically, and vision is blurred (Fig. 29-10). Because of chronic edema, the stroma may eventually become vascularized. On occasion the number of endothelial cells may decrease following cataract surgery even in individuals who do not have early forms of Fuchs dystrophy, and the condition is then known as pseudophakic bullous keratopathy.

FIGURE 29-10 Fuchs dystrophy. This tissue section is stained by PAS to highlight the Descemet membrane, which is thick. Numerous droplike excrescences—guttata—protrude downward from the Descemet membrane. Endothelial cell nuclei are not seen. Epithelial bullae, not shown in this micrograph, were present, reflecting corneal edema.
With increasing stromal edema, the epithelium undergoes hydropic change, and the detachment of the epithelium from the Bowman layer produces epithelial bullae that may eventually rupture. Fibrous connective tissue may be deposited between the epithelium and Bowman layer (degenerative pannus) either by ingrowth from the limbus or perhaps through fibrous metaplasia of the corneal epithelium.
Stromal Dystrophies
In these conditions the stromal deposits generate discrete opacities in the cornea may eventually compromise vision. Deposits in the vicinity of the epithelium, its basement membrane, and Bowman layer may result in painful epithelial erosions. Scarring in the vicinity of Bowman layer may generate an irregular corneal surface, further compromising vision. Macular corneal dystrophy is so named because early in the disease, small nummular (macular) deposits of keratan sulfate develop in the corneal stroma. Later in the course of this autosomal recessive dystrophy, keratan sulfate is distributed diffusely throughout the stroma and may affect the endothelium.
The identification of specific mutations responsible for various stromal dystrophies is generating a new molecular classification of these disorders.9 One example involves an autosomal dominant form of stromal dystrophy associated with mutations in the TGFB1 gene, which encodes an extracellular matrix protein called keratoepithelin. Diverse mutations in TGFB1 disrupt the folding of keratoepithelin, and depending on the exact mutation, lead to the deposition of various types of proteinaceous deposits in the cornea. These include needle-shaped deposits of amyloid (lattice dystrophy); chunky deposits of hyalin (granular dystrophy); and combinations of these opacities in the same person (Avelino dystrophy, named for the location of the first families with this condition).
Anterior Segment
FUNCTIONAL ANATOMY
The anterior chamber is bounded anteriorly by the cornea, laterally by the trabecular meshwork, and posteriorly by the iris (Fig. 29-11). Aqueous humor, formed by the pars plicata of the ciliary body, enters the posterior chamber, bathes the lens, and circulates through the pupil to gain access to the anterior chamber.
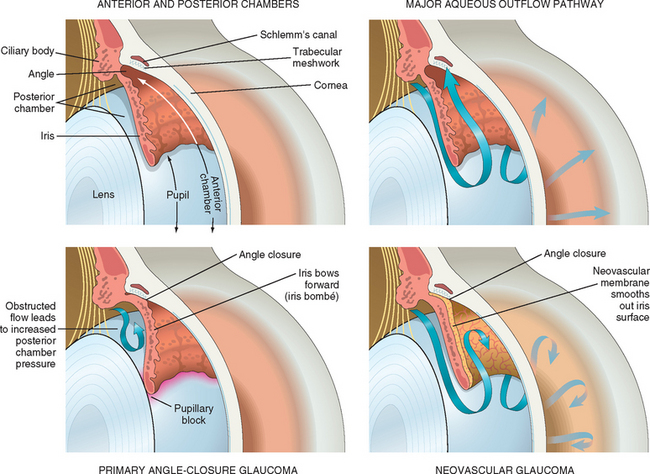
FIGURE 29-11 Upper left, The normal eye. Note that the surface of the iris is highly textured with crypts and folds. Upper right, The normal flow of aqueous humor. Aqueous humor, produced in the posterior chamber, flows through the pupil into the anterior chamber. The major pathway for the egress of aqueous humor is through the trabecular meshwork, into the Schlemm canal. Minor outflow pathways (uveoscleral and iris, not depicted) contribute to a limited extent to aqueous outflow. Lower left, Primary angle-closure glaucoma. In anatomically predisposed eyes, transient apposition of the iris at the pupillary margin to the lens blocks the passage of aqueous humor from the posterior chamber to the anterior chamber. Pressure builds in the posterior chamber, bowing the iris forward (iris bombé) and occluding the trabecular meshwork. Lower right, A neovascular membrane has grown over the surface of the iris, smoothing the iris folds and crypts. Myofibroblasts within the neovascular membrane cause the membrane to contract and to become apposed to the trabecular meshwork (peripheral anterior synechiae). Outflow of aqueous humor is blocked, and the intra-ocular pressure becomes elevated.
The lens is a closed epithelial system; the basement membrane of the lens epithelium (known as the lens capsule) totally envelops the lens. Thus, the lens epithelium does not exfoliate like the epidermis or a mucosal epithelium. Instead, the lens epithelium and its derivative fibers accumulate within the confines of the lens capsule, thus “infoliating.” With aging, therefore, the size of the lens increases. Neoplasms of the lens have not been described.
CATARACT
The term cataract describes lenticular opacities that may be congenital or acquired. Systemic diseases (such as galactosemia, diabetes mellitus, Wilson disease, and atopic dermatitis), drugs (especially corticosteroids), radiation, trauma, and many intraocular disorders are associated with cataract. Age-related cataract typically results from opacification of the lens nucleus (nuclear sclerosis). The accumulation of urochrome pigment may render the lens nucleus brown, thus distorting the individual’s perception of blue color (the predominance of yellow hues in Rembrandt’s paintings later in life might have been a consequence of nuclear sclerotic cataracts). Other physical changes in the lens may generate opacities. For example, the lens cortex may liquefy. Migration of the lens epithelium posterior to the lens equator may result in posterior subcapsular cataract secondary to enlargement of abnormally positioned lens epithelium. The technique that is most commonly used to remove opacified lenses extracts the lens contents, leaving the lens capsule intact (extracapsular cataract extraction). A prosthetic intra-ocular lens may be inserted into the eye. Residual lens epithelial cells may migrate over the lens capsule, contributing to opacification of the capsule and reduction in vision after surgery.
Inflammatory reactions to lens material may develop following exposure of the intact lens cortex caused by rupture of the capsule due to trauma or as a result of cataract extraction. It has been suggested that antigen-antibody complexes containing lens cortical material develop especially in the presence of Propionibacterium acnes (which acts as an adjuvant), generating a lens-induced uveitis.
Occasionally, the lens cortex may liquefy nearly entirely, a condition known as hypermature or morgagnian cataract. High-molecular-weight proteins from liquefied lens cortex may leak through the lens capsule (phacolysis). This phacolytic protein—either free or contained within macrophages—may clog the trabecular meshwork and contribute to elevation in intra-ocular pressure and optic nerve damage; phacolytic glaucoma is an example of secondary open-angle glaucoma.
THE ANTERIOR SEGMENT AND GLAUCOMA
The term glaucoma refers to a collection of diseases characterized by distinctive changes in the visual field and in the cup of the optic nerve. Most of the glaucomas are associated with elevated intra-ocular pressure, although some individuals with normal intra-ocular pressure may develop characteristic optic nerve and visual field changes (normal or low-tension glaucoma). The relationship between intra-ocular pressure and optic nerve damage is discussed later under “Optic Nerve.”
To understand the pathophysiology of glaucoma it is useful to consider the formation and drainage of aqueous humor. As Figure 29-11 illustrates, aqueous humor is produced in the ciliary body and passes from the posterior chamber through the pupil into the anterior chamber. Although there are multiple pathways for the egress of fluid from the anterior chamber, most of the aqueous humor drains through the trabecular meshwork, situated in the angle formed by the intersection between the corneal periphery and the anterior surface of the iris. With this background, glaucoma can be classified into two major categories. In open-angle glaucoma the aqueous humor has complete physical access to the trabecular meshwork, and the elevation in intra-ocular pressure results from an increased resistance to aqueous outflow in the open angle. In angle-closure glaucoma the peripheral zone of the iris adheres to the trabecular meshwork and physically impedes the egress of aqueous humor from the eye. Both open-angle and angle-closure glaucoma can be subclassified into primary and secondary types.
In primary open-angle glaucoma, the most common form of glaucoma, the angle is open, and few changes are apparent structurally. Mutations in the MYOC gene have been associated with a subset of individuals with juvenile and adult primary open-angle glaucoma. The function of the gene product, myocilin, is unclear. MYOC is present in the tracetabular meshwork, in other anterior segment tissues, and in the optic nerve. The pathogenesis of primary open-angle glaucoma may be related to several genes, but the genes identified thus far account for a small percentage of affected individuals with this condition.10 The role of these genes in the pathogenesis of glaucoma is not clear.
There are multiple causes of secondary open-angle glaucoma. Particulate material such as high-molecular-weight lens proteins produced by phacolysis, senescent red cells after trauma (ghost cell glaucoma), iris epithelial pigment granules (pigmentary glaucoma), fragments of oxytalan fibers (exfoliation glaucoma), and necrotic tumors (melanomalytic glaucoma) can clog the trabecular meshwork in the presence of an open angle. Elevations in the pressure on the surface of the eye (episcleral venous pressure) in the presence of an open angle also contributes to secondary open-angle glaucoma. This type of glaucoma is associated with surface ocular vascular malformations seen in Sturge-Weber syndrome or as a consequence of arterialization of the episcleral veins following a spontaneous or traumatic carotid-cavernous fistula.
Primary angle-closure glaucoma typically develops in eyes with shallow anterior chambers, often found in individuals with hyperopia. Transient apposition of the pupillary margin of the iris to the anterior surface of the lens may result in obstruction to the flow of aqueous humor through the pupillary aperture (pupillary block). Continued production of aqueous humor by the ciliary body thus elevates pressure in the posterior chamber and may bow the iris periphery forward (iris bombé), apposing it to the trabecular meshwork. These anatomic changes provoke a marked elevation in intra-ocular pressure (see Fig. 29-11). Since the crystalline lens is avascular and the lens epithelium receives its nutrition from the aqueous humor, unremitting elevation in intra-ocular pressure in primary angle-closure glaucoma can damage the lens epithelium. This leads to minute anterior subcapsular opacities that are visible by slit-lamp examination (glaukomflecken). Although the affected individual might have a normal complement of healthy corneal endothelial cells, sustained elevated intra-ocular pressure can produce corneal edema and bullous keratopathy.
There are many causes of secondary angle-closure glaucoma. Contraction of various types of pathologic membranes that form over the surface of the iris can draw the iris over the trabecular meshwork, occluding aqueous outflow. For example, chronic retinal ischemia is associated with the up-regulation of VEGF and other pro-angiogenic factors. The appearance of VEGF in the aqueous humor is thought to induce the development of thin, clinically transparent fibrovascular membranes over the surface of the iris. Contraction of myofibroblastic elements in these membranes leads to occlusion of the trabecular meshwork by the iris: neovascular glaucoma (see Fig. 29-11). Necrotic tumors, especially retinoblastomas, can also induce iris neovascularization and glaucoma. Secondary angle-closure glaucoma may be caused by other mechanisms as well; for example, tumors in the ciliary body can mechanically compress the iris onto the trabecular meshwork, closing off the major pathway of aqueous outflow.
ENDOPHTHALMITIS AND PANOPHTHALMITIS
In intra-ocular inflammation, vessels in the ciliary body and iris become leaky, allowing cells and exudate to accumulate in the anterior chamber. These changes can be visualized with a slit lamp; at times the inflammatory cells may adhere to the corneal endothelium, forming clinically visible keratic precipitates. The size and shape of these precipitates can provide clues to the underlying cause of the inflammation. For example, aggregates of macrophages on the endothelium in sarcoid produce characteristic “mutton-fat” keratic precipitates.
Just as pleural exudate in acute bronchopneumonia can lead to adhesions between the visceral and parietal pleura, the presence of exudate in the anterior chamber can facilitate the formation of adhesions between the iris and the trabecular meshwork or cornea (anterior synechiae) or between the iris and anterior surface of the lens (posterior synechiae). Anterior synechiae can lead to elevation in intra-ocular pressure, which may lead to optic nerve damage. Prolonged contact between the iris and the anterior surface of the lens can deprive lens epithelium of contact with aqueous humor and can induce fibrous metaplasia of the lens epithelium: anterior subcapsular cataract (Fig. 29-12). The pharmacologic induction of pupillary dilation and cycloplegia in individuals with intra-ocular inflammation is intended in part to prevent the formation of synechiae and their sequelae.
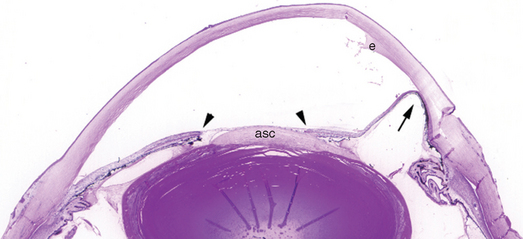
FIGURE 29-12 Sequelae of anterior segment inflammation. This eye was removed for complications of chronic corneal inflammation (not visible at this magnification). The exudate (e) present in the anterior chamber would have been visualized with a slit lamp as an optical “flare.” The iris is adherent focally to the cornea, obstructing the trabecular meshwork (anterior synechia, arrow), and to the lens (posterior synechiae, arrowheads). An anterior subcapsular cataract (asc) has formed. The radial folds in the lens are artifacts.
Although inflammation confined to the anterior segment is technically intra-ocular inflammation, the term endophthalmitis is not applied clinically unless there is inflammation within the vitreous humor. The retina lines the vitreous cavity, and suppurative inflammation in the vitreous humor (endophthalmitis) is poorly tolerated by the retina; after only a few hours of exposure to acute inflammation, the retina may be irreversibly damaged. Endophthalmitis is classified as exogenous (originating in the environment and gaining access to the interior of the eye through a wound) or endogenous (delivered to the eye hematogenously). The term panophthalmitis is applied to inflammation within the eye that involves the retina, choroid, and sclera and extends into the orbit (Fig. 29-13).
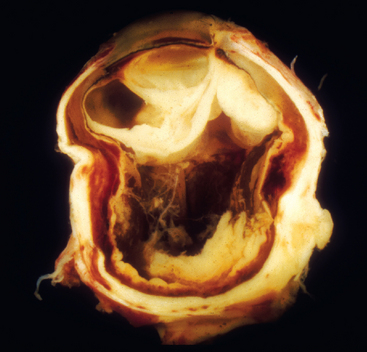
FIGURE 29-13 Exogenous panophthalmitis. This eye was removed after a foreign body injury. Note the suppurative inflammation behind the lens that is drawn up to the right of the lens to the cornea, the site of the wound. The central portion of the vitreous humor was extracted surgically (by vitrectomy). Note the adhesions to the surface of the eye at the eight o’clock position, indicating that the intra-ocular inflammation has spread through the sclera into the orbit: panophthalmitis.
(From Folberg R: The eye. In Spencer WH (ed): Ophthalmic Pathology—An Atlas and Textbook, 4th ed. Philadelphia, WB Saunders, 1985.)
Uvea
Together with the iris, the choroid and ciliary body constitute the uvea. The choroid is among the most richly vascularized sites in the body. As in the retina, there are no lymphatics within the uvea.
UVEITIS
The term uveitis can be applied to any type of inflammation in one or more of the tissues that compose the uvea. Thus, the iritis that develops after blunt trauma to the eye or that accompanies a corneal ulcer is technically a form of uveitis. However, in clinical practice the term uveitis is restricted to a diverse group of chronic diseases that may be either components of a systemic process or localized to the eye. Uveal inflammation may be manifest principally in the anterior segment (e.g., in juvenile rheumatoid arthritis) or may affect both the anterior and posterior segments. The complications of chronic anterior segment inflammation were discussed earlier; the remainder of this discussion therefore focuses on the effects of uveal inflammation on the posterior segment of the eye. As will be described briefly, uveitis is frequently accompanied by retinal pathology. Uveitis may be caused by infectious agents (e.g., Pneumocystis carinii), may be idiopathic (e.g., sarcoidosis), or may be autoimmune in origin (sympathetic ophthalmia). Some examples are described below.
Granulomatous uveitis is a common complication of sarcoidosis (Chapter 15). In the anterior segment it gives rise to an exudate that evolves into “mutton-fat” keratic precipitates described earlier. In the posterior segment, sarcoid may involve the choroid and retina. Thus, granulomas may be seen in the choroid. Retinal pathology is characterized by perivascular inflammation; this is responsible for the well-known ophthalmoscopic sign of “candle wax drippings.” Conjunctival biopsy can be used to detect granulomatous inflammation and confirm the diagnosis of ocular sarcoid.
Numerous infectious processes can affect the choroid or the retina. Inflammation in one compartment is typically associated with inflammation in the other. Retinal toxoplasmosis is usually accompanied by uveitis and even scleritis. Individuals with AIDS may develop cytomegalovirus retinitis and uveal infection such as pneumocystis or mycobacterial choroiditis.11-13
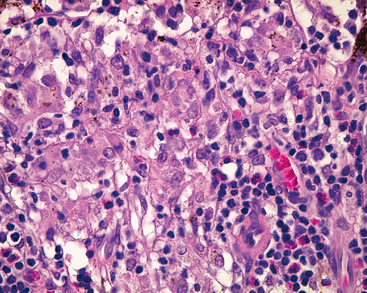
Sympathetic ophthalmia is an example of noninfectious uveitis limited to the eye. This condition is characterized by bilateral granulomatous inflammation typically affecting all components of the uvea: a panuveitis. Sympathetic ophthalmia, which blinded young Louis Braille, may complicate a penetrating injury of the eye. In the injured eye, retinal antigens sequestered from the immune system may gain access to lymphatics in the conjunctiva and thus set up a delayed hypersensitivity reaction that affects not only the injured eye but also the contralateral, noninjured eye.14 The condition may develop from 2 weeks to many years after injury. Enucleation of a blind eye (which can be the sympathizing eye rather than the directly injured eye) may yield diagnostic findings. Sympathetic ophthalmia is treated by the administration of systemic immunosuppressive agents. It is characterized by diffuse granulomatous inflammation of the uvea (choroid, ciliary body, and iris). Plasma cells are typically absent, but eosinophils may be identified in the infiltrate (Fig. 29-14).
NEOPLASMS
The most common intra-ocular malignancy of adults is metastasis to the uvea, typically to the choroid. The occurrence of metastases to the eye is associated with an extremely short survival, and treatment of ocular metastases, usually by radiotherapy, is palliative.
Uveal Nevi and Melanomas
Uveal melanoma is the most common primary intra-ocular malignancy of adults. Although it was formerly thought to be rare (7 per million per year), the incidence of this tumor increases with age, and by the seventh decade the incidence is more than 20 per million per year. Unlike cutaneous melanoma, the occurrence of uveal melanoma has remained stable over many years.15 Although excessive childhood exposure to ultraviolet radiation has been implicated, the link between ultraviolet light and uveal melanoma is not nearly as clear as it is for cutaneous melanoma. Therefore the etiology of uveal melanoma is still unresolved at this time. Uveal nevi, especially choroidal nevi, are rather common, affecting an estimated 10% of the Caucasian population but their progression to melanoma is exceptionally uncommon.
There are no lymphatics within the eye; hence, uveal melanomas, with very rare exception, spread exclusively by a hematogenous route (the only exception being the rare case of melanoma that spreads through the sclera and invades the conjunctiva, thereby gaining access to conjunctival lymphatics). Most uveal melanomas spread first to the liver, thereby providing an excellent example of organ-specific metastasis. Although the 5-year survival rate is approximately 80%,16 the cumulative melanoma mortality rate is 40% at 10 years, increasing 1% per year thereafter.17 Examples of metastases appearing many years after treatment are well known, making uveal melanoma a prime candidate for the investigation of tumor dormancy.
Morphology. Histologically, uveal melanomas may contain two types of cells, spindle and epithelioid, in various proportions (Fig. 29-15). Spindle cells are fusiform in shape and have little atypia, whereas epithelioid cells are spherical and have greater cytologic atypicality. Melanomas situated exclusively in the iris tend to follow a relatively indolent course, whereas melanomas of the ciliary body and choroid are more aggressive. The prognosis of choroidal and ciliary body melanomas is related to (1) size (in contrast to cutaneous melanoma, the lateral extent of the tumor rather than tumor depth is the size dimension related to adverse outcome); (2) cell type (tumors containing epithelioid cells have a worse prognosis than do those containing exclusively spindle cells); (3) and proliferative index. In contrast to cutaneous melanomas, large numbers of tumor-infiltrating lymphocytes are associated with an adverse outcome.18 Extra-ocular extension is related to poor prognosis. Other features associated with poor prognosis include monosomy 3 and trisomy 8, and the presence of looping patterns rich in laminin that surround packets of tumor cells.19 These “spaces” (which are not blood vessels) connect to blood vessels and serve as extravascular conduits for the transport of plasma and possibly blood.20 In vitro studies and examination of human tissues suggest that these patterns are formed by aggressive tumor cells in a process termed vasculogenic mimicry.21,22
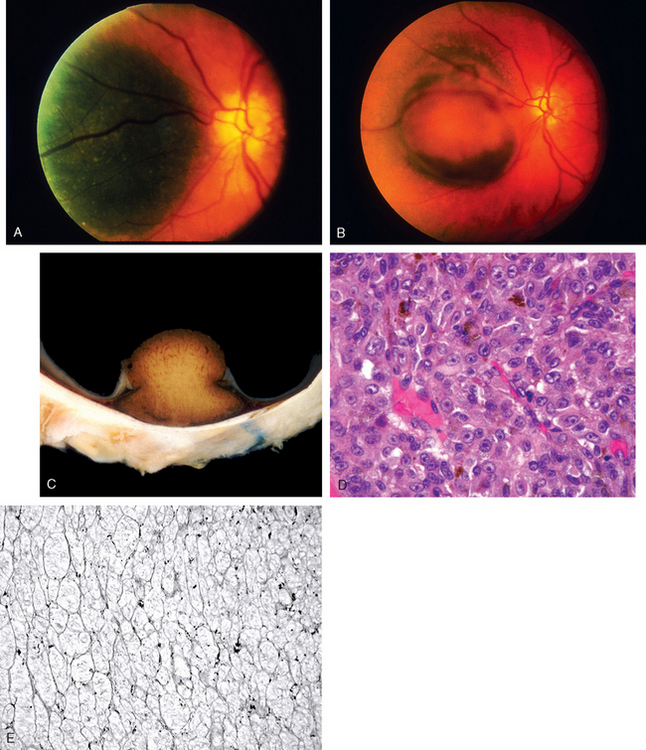
FIGURE 29-15 Uveal melanoma. A, Fundus photograph from an individual with a relatively flat pigmented lesion of the choroid near the optic disc. B, Fundus photograph of the same individual several years later; the tumor has grown and has ruptured through the Bruch membrane. C, Gross photograph of a choroidal melanoma that has ruptured the Bruch membrane. The overlying retina is detached. D, Epithelioid melanoma cells associated with an adverse outcome. E, Patterns rich in laminin (PAS positive) surround aggregates of melanoma cells; these patterns form a “fluid-conducting meshwork” in uveal melanoma and are associated with an adverse outcome.
(A to C, From Folberg R: Pathology of the Eye—an Interactive CD-ROM Program. Philadelphia, Mosby, 1996; E, from Maniotis AJ et al.: Control of melanoma morphogenesis, endothelial survival, and perfusion by extracellular matrix. Lab Invest 82:1031, 2002.)
Uveal melanomas can have an adverse effect on vision, producing changes ranging from retinal detachment to glaucoma. There seems to be no difference in survival between tumors treated by removal of the eye (enucleation) and those treated by radiation treatment. It should be noted that patients treated by radiation and other vision-sparing modalities are diagnosed clinically without tissue removal for pathological examination. There is currently no effective treatment for metastatic uveal melanoma.
Retina and Vitreous
FUNCTIONAL ANATOMY
The neurosensory retina, like the optic nerve, is an embryologic derivative of the diencephalon. The retina therefore responds to injury by means of gliosis. As in the brain, there are no lymphatics. The architecture of the retina explains the ophthalmoscopic appearance of a variety of ocular disorders. Hemorrhages in the nerve fiber layer of the retina are oriented horizontally and appear as streaks or “flames;” the external retinal layers are oriented perpendicular to the retinal surface, and hemorrhages in these outer layers appear as dots (cross-sections of cylinders). Exudates tend to accumulate in the outer plexiform layer of the retina, especially in the macula (Fig. 29-16).
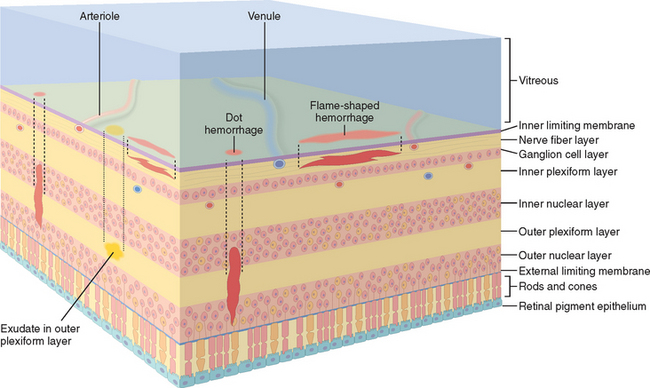
FIGURE 29-16 Clinicopathologic correlations of retinal hemorrhages and exudates. The location of the hemorrhage within the retina determines its appearance by ophthalmoscopy. The retinal nerve fiber layer is oriented parallel to the internal limiting membrane, and hemorrhages of this layer appear to be flame-shaped ophthalmoscopically. The deeper retinal layers are oriented perpendicular to the internal limiting membrane and hemorrhages in this location appear as cross-sections of a cylinder or “dot” hemorrhages. Exudates that originate from leaky retinal vessels accumulate in the outer plexiform layer.
The retinal pigment epithelium (RPE), like the retina, is derived embryologically from the primary optic vesicle, an outpouching of the brain. Separation of the neurosensory retina from the RPE defines a retinal detachment. The RPE has an important role physiologically in the maintenance of the outer segments of the photoreceptors. Disturbances in the RPE-photoreceptor interface may play important roles in hereditary retinal degenerations such as retinitis pigmentosa.
The adult vitreous humor is avascular. Incomplete regression of fetal vasculature running through the vitreous humor can produce significant pathology as a retrolental mass (persistent hyperplastic primary vitreous). The vitreous humor can be opacified by hemorrhage from trauma or retinal neovascularization. With age the vitreous humor may liquefy and collapse, creating the visual sensation of “floaters.” Also, with aging, the posterior face of the vitreous humor—the posterior hyaloid—may separate from the neurosensory retina (posterior vitreous detachment). The relationship between the posterior hyaloid and the neurosensory retina has a key role in the pathogenesis of retinal neovascularization and in some forms of retinal detachment.
RETINAL DETACHMENT
Retinal detachment (separation of the neurosensory retina from the RPE) is broadly classified by etiology based on the presence or absence of a break in the retina. Rhegmatogenous retinal detachment is associated with a full-thickness retinal defect. Retinal tears may develop after the vitreous collapses structurally, and the posterior hyaloid exerts traction on points of abnormally strong adhesion to the retinal internal limiting membrane. Liquefied vitreous humor then seeps through the tear and gains access to the potential space between the neurosensory retina and the RPE (Fig. 29-17). Re-attachment of the retina to the RPE generally requires relief of vitreous traction through indenting of the sclera by surgical procedures. This can be accomplished by the application of strips of silicon to the surface of the eye (scleral buckling) and possibly by removal of vitreous material (vitrectomy). Rhegmatogenous retinal detachment may be complicated by proliferative vitreoretinopathy, the formation of epiretinal or subretinal membranes by retinal glial cells (Müller cells) or RPE cells.
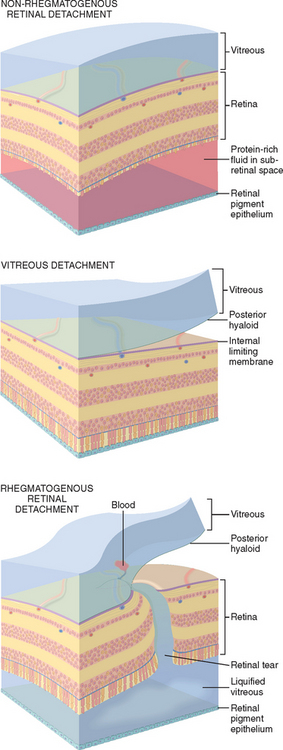
FIGURE 29-17 Retinal detachment is defined as the separation of the neurosensory retina from the RPE. Retinal detachments are classified broadly into non-rhegmatogenous (without a retinal break) and rhegmatogenous (with a retinal break) types. Top, In non-rhegmatogenous retinal detachment the subretinal space is filled with protein-rich exudate. Note that the outer segments of the photoreceptors are missing (see Fig. 29-16 for orientation of layers). This indicates a chronic retinal detachment, a finding that can be seen in both non-rhegmatogenous and rhegmatogenous detachments. Middle, Posterior vitreous detachment involves the separation of the posterior hyaloid from the internal limiting membrane of the retina and is a normal occurrence in the aging eye. Bottom, If, during a posterior vitreous detachment, the posterior hyaloid does not separate cleanly from the internal limiting membrane of the retina, the vitreous humor will exert traction on the retina, which will be torn at this point. Liquefied vitreous humor seeps through the retinal defect, and the retina is separated from the RPE. The photoreceptor outer segments are intact, illustrating an acute detachment.
Non-rhegmatogenous retinal detachment (retinal detach-ment without retinal break) may complicate retinal vascular disorders associated with significant exudation and any condition that damages the RPE and permits fluid to leak from the choroidal circulation under the retina. Retinal detachments associated with choroidal tumors and malignant hypertension are examples of non-rhegmatogenous retinal detachment.
RETINAL VASCULAR DISEASE
Hypertension
Normally, the thin walls of retinal arterioles permit a direct visualization of the circulating blood by ophthalmoscopy. In retinal arteriolosclerosis the thickened arteriolar wall changes the ophthalmic perception of circulating blood: vessels may appear narrowed, and the color of the blood column may change from bright red to copper and to silver depending on the degree of vascular wall thickness (Fig. 29-18A). Retinal arterioles and veins share a common adventitial sheath. Therefore, in pronounced retinal arteriolosclerosis the arteriole may compress the vein at points where both vessels cross (Fig. 29-18B). Venous stasis distal to arteriolar-venous crossing may precipitate occlusions of the retinal vein branches.
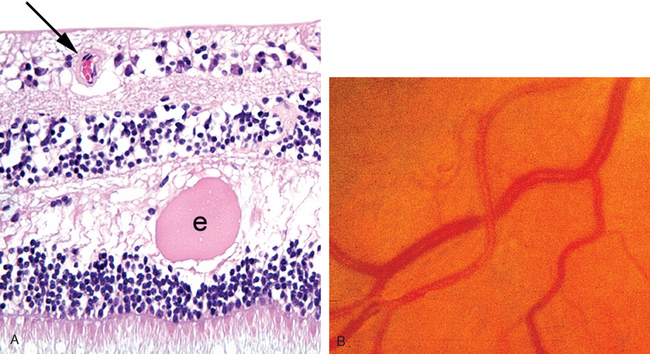
FIGURE 29-18 The retina in hypertension. A, The wall of the retinal arteriole (arrow) is thick. Note the exudate (e) in the retinal outer plexiform layer. B, The fundus in hypertension. The diameter of the arterioles is reduced, and the color of the blood column appears to be less saturated (copper wire–like). If the wall of the vessel were thicker still, the degree of red color would diminish such that the vessels might appear clinically to have a “silver-wire” appearance. In this fundus photograph, note that the vein is compressed where the sclerotic arteriole crosses over it.
(B, Courtesy of Dr. Thomas A. Weingeist, Department of Ophthalmology and Visual Science, University of Iowa, Iowa City, IA.)
In malignant hypertension vessels in the retina and choroid may be damaged. Damage to choroidal vessels may produce focal choroidal infarcts, seen clinically as Elschnig pearls. Damage to the choriocapillaris, the internal layer of the choroidal vasculature, may, in turn, damage the overlying RPE and permit the exudate to accumulate in the potential space between the neurosensory retina and the RPE, thereby producing a retinal detachment. Exudate from damaged retinal arterioles typically accumulates in the outer plexiform layer of the retina (see Fig. 29-18A). The ophthalmoscopic finding of a macular star—a spokelike arrangement of exudate in the macula in malignant hypertension—results from exudate accumulating in the outer plexiform layer of the macula that is oriented obliquely instead of perpendicular to the retinal surface.
Occlusion of retinal arterioles may produce infarcts of the nerve fiber layer of the retina (axons of the retinal ganglion cell layer populate the nerve fiber layer). Axoplasmic transport in the nerve fiber layer is interrupted at the point of axonal damage, and accumulation of mitochondria at the swollen ends of damaged axons creates the histologic illusion of cells (cytoid bodies). Collections of cytoid bodies populate the nerve fiber layer infarct, seen ophthalmoscopically as “cotton-wool spots” (Fig. 29-19). Although nerve fiber layer infarcts are described here in the context of hypertension, they may be detected in a variety of retinal occlusive vasculopathies. For example, retinal nerve fiber layer infarcts may develop in individuals with AIDS due to a retinal vasculopathy that is similar to the brain vasculopathy that may develop in this condition.
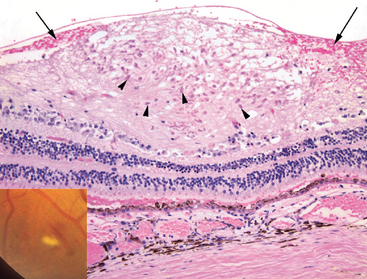
FIGURE 29-19 Nerve fiber layer infarct. A “cotton-wool spot” is illustrated in the inset, adjacent to a flame-shaped (nerve fiber layer) hemorrhage. The histology of a cotton-wool spot—an infarct of the nerve fiber layer of the retina—is illustrated in the photomicrograph. A focal swelling of the nerve fiber layer is occupied by numerous red to pink cytoid bodies (arrowheads), bulbous ends of severed axons. Hemorrhage (arrows) surrounding the nerve fiber layer infarct as illustrated here is a variable and inconsistent finding.
(Fundus photograph, Courtesy of Dr. Thomas A. Weingeist, Department of Ophthalmology and Visual Science, University of Iowa, Iowa City, IA.)
Diabetes Mellitus
The eye is profoundly affected by diabetes mellitus. The effects of hyperglycemia on the lens and iris have already been mentioned. Thickening of the basement membrane of the epithelium of the pars plicata of the ciliary body is a reliable histologic marker of diabetes mellitus in the eye (Fig. 29-20) and is reminiscent of similar changes in the glomerular mesangium. This discussion focuses on the retinal microangiopathy associated with diabetes mellitus, a prototype for the consideration of other retinal microangiopathies.
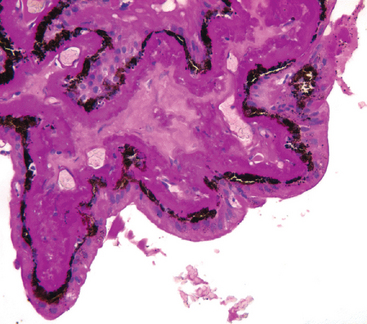
FIGURE 29-20 The ciliary body in chronic diabetes mellitus, PAS stain. Note the massive thickening of the basement membrane of the ciliary body epithelia, reminiscent of changes in the mesangium of the renal glomerulus.
The retinal vasculopathy of diabetes mellitus can be classified into background (preproliferative) diabetic retinopathy and proliferative diabetic retinopathy.23
Background (preproliferative) diabetic retinopathy includes a spectrum of changes ranging from structural and functional abnormalities of angiogenesis located within the retina (i.e., confined beneath the internal limiting membrane of the retina). As with diabetic microangiopathy in general, the basement membrane of retinal blood vessels is thickened. In addition, the number of pericytes relative to endothelial cells diminishes. Microaneurysms are an important manifestation of diabetic microangiopathy. They are typically smaller than the resolution of direct ophthalmoscopes, and findings customarily described as microaneurysms by ophthalmoscopy may in fact be retinal microhemorrhages. Structural changes in the retinal microcirculation have been associated with a physiologic breakdown in the blood-retinal barrier. Thus, the retinal microcirculation in diabetics may be exceptionally leaky, giving rise to macular edema, a common cause of visual loss in these patients. The vascular changes may also produce exudates that accumulate in the outer plexiform layer. Although the retinal microcirculation is often hyperpermeable, it is also subject to the effects of micro-occlusion. Both vascular incompetence and vascular micro-occlusions can be visualized clinically after intravenous injection of fluorescein.
Nonperfusion of the retina due to the microcirculatory change described above is associated with up-regulation of VEGF and retinal angiogenesis.24 The development of intraretinal angiogenesis—new vessels confined within the retina beneath the internal limiting membrane—can be included with lesions termed intraretinal microangiopathy.
Clinically, proliferative diabetic retinopathy is defined by the appearance of new vessels that sprout from existing vessels—angiogenic vessels—on the surface of either the optic nerve head, which is termed neovascularization of the disc, or the surface of the retina, which is designated by the nebulous term neovascularization elsewhere. It is worth emphasizing that the term retinal neovascularization is not applied either clinically or pathologically unless the newly formed vessels breach the internal limiting membrane of the retina. The quantity and location of retinal neovascularization guide the ophthalmologist in the treatment of proliferative diabetic retinopathy. The web of newly formed vessels is called a neovascular membrane both clinically and histopathologically. It is composed of angiogenic vessels with or without a substantial supportive fibrous or glial stroma (Fig. 29-21).
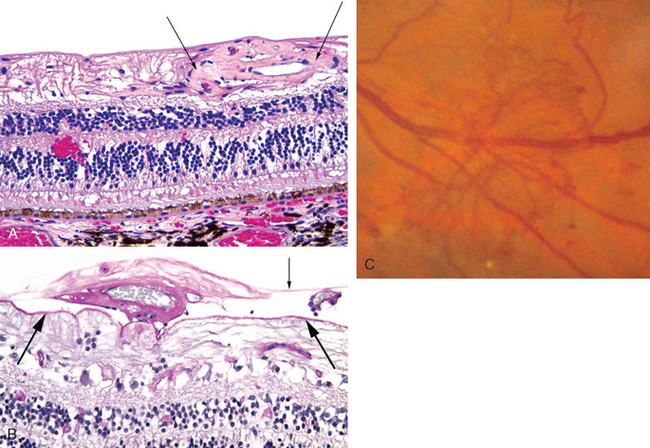
FIGURE 29-21 The retina in diabetes mellitus (see Fig. 29-16 for a schematic of retinal structure). A, A tangle of abnormal vessels lies just beneath the internal limiting membrane of the retina on the right half of the photomicrograph (between arrows). This is an example of intraretinal angiogenesis known as intraretinal microangiopathy (IRMA). Note the retinal hemorrhage in the outer plexiform layer in the left half. The ganglion cell layer and the nerve fiber layer—the axons of the ganglion cells—are absent. The rarefied space beneath internal limiting membrane to the left of the focus of IRMA consists largely of elements of retinal glial (Müller) cells. Absence of the ganglion cell and nerve fiber layers is a hallmark of glaucoma. The chronic diabetes mellitus in this individual was complicated by iris neovascularization and secondary angle-closure glaucoma (neovascular glaucoma). B, In this section stained by PAS, the internal limiting membrane is indicated by the thick arrows and the posterior hyaloid of the vitreous by the thin arrow. In the potential space between these two landmarks, the vessels to the left of the thin arrow are invested with a fibrous-glial stroma and would appear ophthalmoscopically as a white neovascular membrane. The thin-walled vessel to the right of the thin arrow is not invested with connective tissue. A posterior vitreous detachment in an eye such as this might exert traction on these new vessels and precipitate a massive vitreous hemorrhage. C, Ophthalmoscopic view of retinal neovascularization (known clinically as neovascularization “elsewhere” in contrast with neovascularization of the optic disc) creating a neovascular membrane.
If the vitreous humor has not detached and the posterior hyaloid is intact, neovascular membranes extend along the potential plane between the retinal internal limiting membrane and the posterior hyaloid. Thus, the separation of the vitreous humor from the internal limiting membrane of the retina (posterior vitreous detachment) after retinal neovascularization may precipitate massive hemorrhage from the disrupted neovascular membrane. Organization of the retinal neovascular membrane may wrinkle the retina, disrupting the orientation of retinal photoreceptors and producing visual distortion, and may exert traction on the retina, separating it from the RPE (retinal detachment). Traction retinal detachment may begin as a non-rhegmatogenous detachment, but severe traction may tear the retina, producing a traction rhegmatogenous detachment.
Retinal neovascularization may be accompanied by the development of a neovascular membrane on the iris surface, presumably secondary to increased levels of VEGF in the aqueous humor.25 Contraction of the iris neovascular membrane may lead to adhesions between the iris and trabecular meshwork (anterior synechiae), thus occluding a major pathway for aqueous outflow and thereby contributing to elevation of the intra-ocular pressure (neovascular glaucoma). Ablating nonperfused retina by laser photocoagulation or cryopexy triggers regression of both retinal and iris neovascularization.
Retinopathy of Prematurity (Retrolental Fibroplasia)
At term, the nasal (medial) aspect of the retina is vascularized, but the temporal (lateral) aspect of the retinal periphery is incompletely vascularized. In premature or low-birth-weight infants treated with oxygen, the immature retinal vessels in the temporal retinal periphery can constrict, rendering the retinal tissue distal to this zone ischemic. Retinal ischemia can result in up-regulation of pro-angiogenic factors such as VEGF and lead to retinal angiogenesis.26 Contraction of a peripheral retinal neovascular membrane may result in “dragging” of the temporal aspect of the retina toward the temporal peripheral zone such that the macula (situated temporal to the optic nerve) is displaced laterally. With significant contraction the retina can detach.
Sickle Retinopathy, Retinal Vasculitis, Radiation Retinopathy
Retinopathy affecting individuals with sickle hemoglobinopathies (Chapter 14) has been divided into two types that roughly parallel those used for diabetic retinopathy: nonproliferative (intraretinal angiopathic changes) and proliferative (retinal neovascularization). The final common pathway in both types is vascular occlusion.27 Low oxygen tension within the blood vessels in the retinal periphery results in sickling and red cell deformation causing microvascular occlusions. In the nonproliferative form (which occurs in individuals with SS, and SC hemoglobins), vascular occlusions are thought to contribute to preretinal, intraretinal, and subretinal hemorrhages. The resolution of these hemorrhages may give rise to a variety of ophthalmoscopically visible changes, known as salmon patches, iridescent spots, and black sunburst lesions. Organization of pre-retinal hemorrhage may result in retinal traction and retinal detachment. Vascular occlusions may also contribute to angiogenesis secondary to up-regulation of both VEGF and basic fibroblast growth factor.28 This can give rise to florid zones of retinal neovascularization in the periphery, described clinically as “sea-fans.”
Neovascularization also occurs in a variety of other clinical settings such as peripheral retinal vasculitis, and in irradiation used to treat intra-ocular tumors. The feature common to these conditions is damage to retinal vessels, producing zones of retinal ischemia that drive retinal angiogenesis and its complications, hemorrhage and traction.
Retinal Artery and Vein Occlusions
The central retinal artery or its branches can be occluded by disorders that affect the vessels in general. For example, the lumen of the central retinal artery can be narrowed significantly by atherosclerosis, thus predisposing to thrombosis. Emboli to the central retinal artery can originate from thrombi in the heart or on ulcerated atheromatous plaques in the carotid arteries. Fragments of atherosclerotic plaques can lodge within the retinal circulation (Hollenhorst plaques). Total occlusion of a branch retinal artery can produce a segmental infarct of the retina. With sudden cessation of blood supply, the retina (an embryologic derivative of brain tissue) swells acutely and becomes optically opaque. By ophthalmoscopy the fundus in the affected area appears white instead of red or orange, because the retinal opacity blocks the view of the richly vascular choroid.
Total occlusion of the central retinal artery can produce a diffuse infarct of the retina. Following an acute occlusion, the retina appears relatively opaque by ophthalmoscopy. The fovea and foveola are physiologically thin; therefore, the normal orange-red of the choroid is not only visible but highlighted by the surrounding opaque retina—the origin of the cherry-red spot of the central retinal artery occlusion. The cherry-red spots seen in rare storage diseases such as Tay-Sachs and Niemann-Pick diseases also have their basis in the anatomic variations of the macula. The storage material accumulates in retinal ganglion cells: the ganglion cell layer of the macula surrounding the fovea is thick, but there are no ganglion cells in the center of the macula, the fovea. Thus, the fovea is relatively transparent to the underlying choroidal vasculature but is rimmed by relatively opaque retina, the result of storage material accumulating in the perifoveal macular ganglion cells (Fig. 29-22).
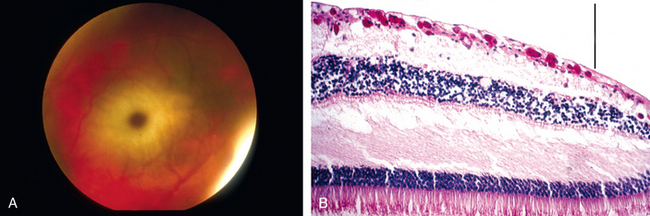
FIGURE 29-22 The cherry-red spot in Tay-Sachs disease. A, Fundus photograph of the cherry-red spot in Tay-Sachs disease. B, Photomicrograph of the macula in an individual with Tay-Sachs disease, stained with PAS to highlight the accumulation of ganglioside material in the retinal ganglion cells. The presence of ganglion cells filled with gangliosides outside the fovea blocks the transmission of the normal orange-red color of the choroid, but absence of ganglion cells within the fovea (to the right of the vertical bar) permits the normal orange-red color to be visualized, accounting for the so-called cherry-red spot.
(A, Courtesy of Dr. Thomas A. Weingeist, Department of Ophthalmology and Visual Science, University of Iowa, Iowa City, IA; B, from the teaching collection of the Armed Forces Institute of Pathology.)
Retinal arterial occlusions are typically sudden events; therefore, they are not often complicated by prolonged ischemia to allow for up-regulation of pro-angiogenic factors. Hence, retinal arterial occlusions are seldom complicated by either retinal or iris neovascularization.
Retinal vein occlusion may occur with or without ischemia.29 In ischemic retinal vein occlusion, VEGF and other proangiogenic factors are up-regulated in the retina, leading to neovascularization of the retina and surface of the optic nerve head as well as neovascularization of the iris and subsequent angle-closure glaucoma.30 Non-ischemic retinal vein occlusion may be complicated by hemorrhages, exudates, and macular edema but is seldom complicated by retinal or iris neovascularization.
AGE-RELATED MACULAR DEGENERATION
From the name of this disorder, it is clear that advancing age is a risk factor. The cumulative incidence of age-related macular degeneration (ARMD) in individuals 75 years of age and older is 8%, and with increasing longevity ARMD is becoming a major health problem.31
Nearly 71% of cases are estimated to be heritable, but the identification of genetic risk factors has not yet contributed to therapeutic strategies that modulate the clinical course.32 Attention is now focused on the roles of several genes, especially CFH (complement factor H) in the pathogenesis of this condition.33 Individuals with the CFH CC genotype who have smoked at least 10 pack-years (i.e., at least 20 cigarettes per day for 10 years) have a 144-fold increase in developing the neovascular form of ARMD than individuals with this genotype who have smoked for fewer than 10 pack-years.34
To understand the pathogenesis of ARMD it is important to appreciate the existence of a structural and functional unit composed of the retinal pigment epithelium (RPE), Bruch membrane (which contains the basement membrane of the RPE), and the innermost layer of the choroidal vasculature, the choriocapillaris. Disturbance in any component of this “unit” affects the health of the overlying photoreceptors, producing visual loss.
It is commonplace to describe ARMD as either nonneovascular (atrophic or dry) or neovascular (exudative or wet). Non-neovascular ARMD is identified ophthalmoscopically by diffuse or discrete deposits in the Bruch membrane (drusen) and geographic atrophy of the RPE. Approximately 10% to 20% of individuals with non-neovascular ARMD develop choroidal neovascular membranes. Loss of vision is substantially more severe in these individuals.
Choroidal neovascularization is defined by the presence of angiogenic vessels that presumably originate from the choriocapillaris and penetrate through the Bruch membrane beneath the RPE (Fig. 29-23). This neovascular membrane may also penetrate the RPE and become situated directly beneath the neurosensory retina. The vessels in this membrane may leak, and the exuded blood may be organized by RPE cells into macular scars. Occasionally, hemorrhage from these neovascular membranes can be massive, leading to the localized suffusion of blood that may be mistaken clinically for an intra-ocular neoplasm, or may produce diffuse vitreous hemorrhage. Currently the mainstay of treatment for neovascular ARMD is the injection of VEGF antagonists into the vitreous of the affected eye.35
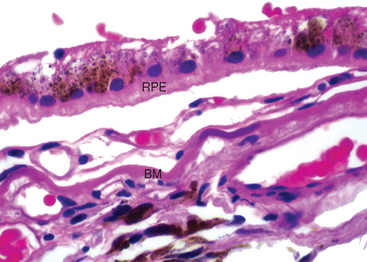
FIGURE 29-23 Age-related macular degeneration. A neovascular membrane is positioned between the RPE and Bruch membrane (BM). Note the blue discoloration of Bruch membrane to the right of the label, indicating focal calcification.
Choroidal neovascular membranes can develop in conditions that are unrelated to age, such as pathologic myopia (Fuchs spot), following traumatic disruption of the Bruch membrane, angioid streaks, or an immunological response to systemic histoplasmosis (presumed ocular histoplasmosis syndrome).
OTHER RETINAL DEGENERATIONS
Retinitis Pigmentosa
The term “retinitis” pigmentosa is an unfortunate relic that is used to describe a collection of inherited retinal disorders that were formerly incorrectly presumed to be inflammatory. The conditions that are grouped under the rubric of retinitis pigmentosa are fairly common and have an incidence of 1 in 3600. They may be inherited as X-linked recessive, autosomal recessive, or autosomal dominant (the age of onset correlates with the inheritance pattern, with autosomal dominant retinitis pigmentosa appearing later in life). Retinitis pigmentosa may be part of a syndrome such as Refsum disease or may develop in isolation (nonsystemic retinitis pigmentosa).
Retinitis pigmentosa is linked to mutations in genes that regulate the functions of either the photoreceptor cells or the RPE. These include genes that regulate the visual cascade and visual cycle, structural genes (transpanins), transcription factors, retinal catabolic pathways, and mitochondrial metabolism.36 Typically, both rods and cones are lost to apoptosis, though in varying proportions. Loss of rods may lead to early night blindness and constricted visual fields. As cones are lost, central visual acuity may be affected. Clinically, retinal atrophy is accompanied by constriction of retinal vessels and optic nerve head atrophy (“waxy pallor” of the optic disk) and the accumulation of retinal pigment around blood vessels, thus accounting for the “pigmentosa” in the disease’s name. The electroretinogram reveals abnormalities characteristic of this disease.
RETINITIS
A variety of pathogens can contribute to the development of infectious retinitis. For example, Candida may disseminate to the retina hematogenously, especially in the setting of intravenous drug abuse or in systemic candidemia from other causes. Hematogenous dissemination of pathogens to the retina typically results in multiple retinal abscesses. As was mentioned previously, cytomegalovirus retinitis is an important cause of visual morbidity in immunocompromised individuals, especially those with AIDS.
RETINAL NEOPLASMS
Retinoblastoma
Retinoblastoma is the most common primary intra-ocular malignancy of children. The molecular genetics of retinoblastoma has been discussed in detail (Chapter 7). Although the name retinoblastoma might suggest origin from a primitive retinal cell that is capable of differentiation into both glial and neuronal cells, it is now clear that the cell of origin of retinoblastoma is neuronal. Recall that in approximately 40% of cases, retinoblastoma occurs in individuals who inherit a germline mutation of one RB allele. Retinoblastomas arising in the context of germline mutations not only may be bilateral but also may be associated with pinealoblastoma (“trilateral” retinoblastoma), which is associated with a dismal outcome.37
Morphology. The pathology of retinoblastoma, both hereditary and sporadic types, is identical. Tumors may contain both undifferentiated and differentiated elements. The former appear as collections of small, round cells with hyperchromatic nuclei. In well-differentiated tumors there are FlexnerWintersteiner rosettes and fleurettes reflecting photoreceptor differentiation. It should be noted, however, that the degree of tumor differentiation does not appear to be associated with the prognosis. As seen in Figure 29-24, viable tumor cells are found encircling tumor blood vessels with zones of necrosis typically found in relatively avascular areas, illustrating the dependence of retinoblastoma on its blood supply. Focal zones of dystrophic calcification are characteristic of retinoblastoma.
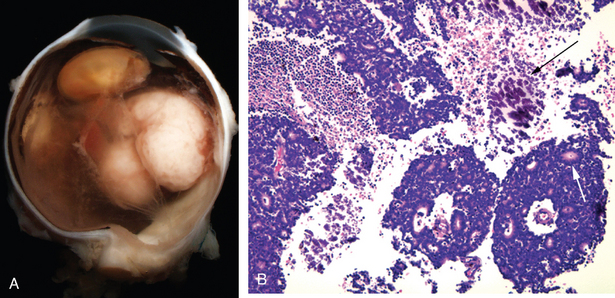
FIGURE 29-24 Retinoblastoma. A, Gross photograph of retinoblastoma. B, Tumor cells appear viable when in proximity to blood vessels, but necrosis is seen as the distance from the vessel increases. Dystrophic calcification (dark arrow) is present in the zones of tumor necrosis. Flexner-Wintersteiner rosettes—arrangements of a single layer of tumor cells around an apparent “lumen”—are seen throughout the tumor, and one such rosette is indicated by the white arrow.
In an effort to preserve vision and eradicate the tumor, many ophthalmic oncologists now attempt to reduce tumor burden by administration of chemotherapy; after chemoreduction, tumors may be obliterated by laser treatment or cryopexy. Retinoblastoma tends to spread to the brain and bone marrow and seldom disseminates to the lungs. Prognosis is adversely affected by extra-ocular extension and invasion along the optic nerve, and by choroidal invasion. A variant of retinoblastoma—retinocytoma or retinoma—has been reported and appears to be a pre-malignant lesion.38 The appearance of retinoblastoma in one eye and retinocytoma in the other eye is characteristic of heritable retinoblastoma.
Retinal Lymphoma
Primary retinal lymphoma is analogous to primary large-cell lymphoma of the brain; therefore, it involves the two retinal layers derived from brain: the neurosensory retina and the RPE. The underlying choroid is typically filled with a cytologically benign lymphoid infiltrate. Primary intra-ocular lymphoma tends to occur in older individuals and may mimic uveitis clinically. The diagnosis depends on a demonstration of lymphoma cells in vitreous aspirates.39
Optic Nerve
As a sensory tract of the central nervous system, the optic nerve is surrounded by meninges, and cerebrospinal fluid circulates around the nerve. The pathology of the optic nerve is similar to the pathology of the brain. For example, the most common primary neoplasms of the optic nerve are glioma (typically pilocytic astrocytomas) and meningioma.
ANTERIOR ISCHEMIC OPTIC NEUROPATHY
There are striking similarities between stroke and a condition known in ophthalmic terminology as anterior ischemic optic neuropathy (AION).40 As used clinically, the term AION includes a spectrum of injuries to the optic nerve varying from ischemia to infarction. Thus, transient partial interruptions in blood flow to the optic nerve can produce episodes of transient loss of vision, whereas total interruption in blood flow can produce an optic nerve infarct, either segmental or total. Zones of relative ischemia may surround segmental infarcts of the optic nerve. Optic nerve function in these poorly perfused but not infarcted zones may recover. The optic nerve does not regenerate, and visual loss from infarction is permanent.
Interruption in the blood supply to the optic nerve can result from inflammation of the vessels that supply the optic nerve, known as arteritic AION, or from embolic or thrombotic events, known as non-arteritic AION. Bilateral total infarcts of the optic nerve resulting in total blindness have been reported in temporal arteritis (arteritic AION), adding urgency to the treatment of this condition with high doses of corticosteroids.
PAPILLEDEMA
Edema of the head of the optic nerve may develop as a consequence of compression of the nerve (as in a primary neoplasm of the optic nerve) or from elevations of cerebrospinal fluid pressure surrounding the nerve. The concentric increase in pressure encircling the nerve contributes to venous stasis both at the nerve head and in axoplasmic transport, leading to nerve head swelling. Swelling of the optic nerve head in elevated intra-cranial pressure is typically bilateral (unless the affected individual has experienced previous unilateral optic atrophy) and is commonly termed papilledema. Typically, acute papilledema from increased intra-cranial pressure is not associated with visual loss. Ophthalmoscopically, the optic nerve head is swollen and hyperemic; by contrast, the optic nerve head in the relatively acute phases of anterior ischemic optic neuropathy appears swollen and pale because of decreased nerve perfusion (Fig. 29-25). In papilledema secondary to increased intra-cranial pressure, the optic nerve may remain congested for a prolonged period of time.
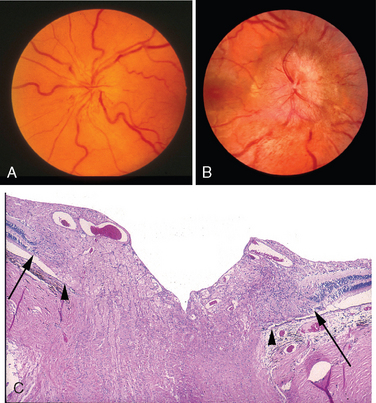
FIGURE 29-25 The optic nerve in anterior ischemic optic neuropathy (AION) and papilledema. A, In the acute phases of AION the optic nerve may be swollen, but it is relatively pale because of decreased perfusion. B, In papilledema secondary to increased intra-cranial pressure, the optic nerve is typically swollen and hyperemic. C, Normally, the termination of Bruch membrane (arrowhead) is aligned with the beginning of the neurosensory retina, as indicated by the presence of stratified nuclei (arrow), but in papilledema the optic nerve is swollen, and the retina is displaced laterally. This is the histologic explanation for the blurred margins of the optic nerve head seen clinically in this condition.
(A and B, Courtesy of Dr. Sohan S. Hayreh, Department of Ophthalmology and Visual Science, University of Iowa, Iowa City, IA; C, from the teaching collection of the Armed Forces Institute of Pathology.)
GLAUCOMATOUS OPTIC NERVE DAMAGE
As discussed already, the majority of individuals with glaucoma have elevated intra-ocular pressure. However, there is a small group that develops the visual field and optic nerve changes typical of glaucoma with normal intra-ocular pressure: so-called normal-tension glaucoma. Interestingly, mutations in the optineurin gene are seen in individuals with normal-tension glaucoma but are not seen in individuals with primary open-angle glaucoma, in which pressure is elevated chronically.41 Conversely, some individuals with elevated intra-ocular pressure who are followed over long periods of time never develop visual field changes or optic nerve cupping. Therefore, it is clear that whatever the mechanism of damage to the retinal ganglion cell—the axons of which populate the optic nerve—there is a spectrum of neuronal susceptibility to the effects of elevated intra-ocular pressure. Therefore, considerable research is now directed toward understanding mechanisms by which the optic nerve axons may be protected from injury.42
Morphology. Characteristically, there is a diffuse loss of ganglion cells and thinning of the retinal nerve fiber layer (Fig. 29-26), which can be measured by optical coherence tomography. In advanced cases, the optic nerve is both cupped and atrophic, a combination unique to glaucoma. Elevated intra-ocular pressure in infants and children can lead to diffuse enlargement of the eye (buphthalmos) or enlargement of the cornea (megalocornea). Several mutations have been associated with the development of infantile glaucoma, but the mechanisms by which these genes produce glaucoma is unclear. After the eye reaches its adult size, prolonged elevation of intra-ocular pressure can lead to focal thinning of the sclera, and uveal tissue may line ectatic sclera (staphyloma).
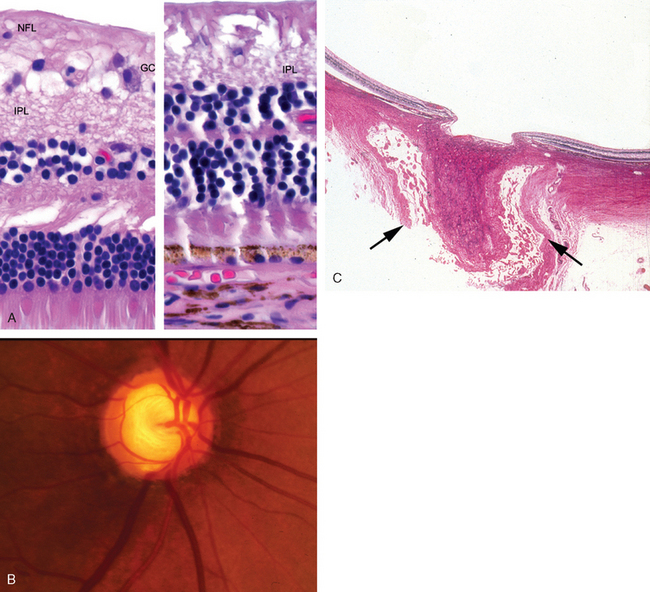
FIGURE 29-26 The retina and optic nerve in glaucoma. A, Left panel, normal retina; right panel, the retina in long-standing glaucoma (same magnification). The full thickness of the glaucomatous retina is captured (right), a reflection of the thinning of the retina in glaucoma. In the glaucomatous retina, the areas corresponding to the nerve fiber layer (NFL) and ganglion cell layer (GC) are atrophic; the inner plexiform layer (IPL) is labeled for reference. Note also that the outer nuclear layer of the glaucomatous retina is aligned with the inner nuclear layer of the normal retina due to the thinning of the retina in glaucoma. See Figure 29-16 for orientation. B, Glaucomatous optic nerve cupping results in part from loss of retinal ganglion cells, the axons of which populate the optic nerve. C, The arrows point to the dura of the optic nerve. Notice the wide subdural space, a result of atrophy of the optic nerve. There is a striking degree of cupping on the surface of the nerve as a consequence of long-standing glaucoma.
OTHER OPTIC NEUROPATHIES
Optic neuropathy may be inherited (as in Leber hereditary optic neuropathy) or may be secondary to nutritional deficiencies (as in so-called tobacco-alcohol amblyopia) or toxins such as methanol. Individuals may experience a severe visual disability if fibers in the optic nerve degenerate, especially if the central visual acuity is lost as a result of degeneration of the nerve fibers that originate from the macula.
The predilection for Leber hereditary optic neuropathy to develop in young men is explained by inheritance of mitochondrial gene mutations (Chapter 5). It is possible that these mutations provide for a genetic susceptibility to a variety of environmental exposures that constitute the final trigger for optic nerve degeneration.43 Since neuronal health is dependent on axoplasmic transport of mitochondria, mitochondrial dysfunctions give rise to neurologic disorders including optic neuropathy.44
OPTIC NEURITIS
Many unrelated conditions have historically been grouped under the heading of optic neuritis. Unfortunately, the term itself suggests optic nerve inflammation, which might not accurately describe the pathophysiologic changes. In common clinical usage the term optic neuritis is used to describe a loss of vision secondary to demyelinization of the optic nerve. One of the most important causes of optic neuritis is multiple sclerosis (Chapter 28). Indeed, optic neuritis may be the first manifestation of this disease. The 10-year risk of developing multiple sclerosis after the first attack of optic neuritis increases if the affected person has concomitant evidence of brain lesions as detected by magnetic resonance imaging. However, even when brain lesions are detectable, the risk of progression to multiple sclerosis is only 40%.45 Individuals with a single episode of optic nerve demyelinization may recover vision and remain disease free.
The End-Stage Eye: Phthisis Bulbi
Trauma, intra-ocular inflammation, chronic retinal detachment, and many other conditions can give rise to an eye that is both small (atrophic) and internally disorganized: phthisis bulbi. Congenitally small eyes—hypoplastic or microphthalmic eyes—are generally not disorganized internally. Phthisical eyes typically feature the following changes: the presence of exudate or blood between the ciliary body and sclera and the choroid and sclera (ciliochoroidal effusion); the presence of a membrane extending across the eye from one aspect of the ciliary body to the other (cyclitic membrane); chronic retinal detachment; optic nerve atrophy; the presence of intra-ocular bone, which is thought by many to originate from osseous metaplasia of the RPE; and a thickened sclera, especially posteriorly. Ciliochoroidal effusion is typically associated with the physiologic state of low intra-ocular pressure (hypotony). The normal pull of the extra-ocular muscles on a hypotonous eye may render the appearance of the eye as square rather than round.
1 Friend SH, et al. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature. 1986;323:643.
2 Hatton MP, Rubin PA. The pathophysiology of thyroid-associated ophthalmopathy. Ophthalmol Clin North Am. 2002;15:113.
3 Ahmed M, et al. Diagnosis of limited ophthalmic Wegener granulomatosis: distinctive pathologic features with ANCA test confirmation. Int Ophthalmol. 2008;28:35.
4 Song A, et al. Sebaceous cell carcinoma of the ocular adnexa: clinical presentations, histopathology, and outcomes. Ophthal Plast Reconstr Surg. 2008;24:194.
5 Rose AS, et al. Hepatic, ocular and cutaneous sarcoidosis. Clin Chest Med. 2008;29:509.
6 Scott IU, et al. Human papillomavirus 16 and 18 expression in conjunctival intraepithelial neoplasia. Ophthalmology. 2002;109:542.
7 Folberg R, et al. Benign conjunctival melanocytic lesions: clinicopathologic features. Ophthalmology. 1989;96:436.
8 Jakobiec FA, et al. Clinicopathologic characteristics of premalignant and malignant melanocytic lesions of the conjunctiva. Ophthalmology. 1989;96:147.
9 Vincent AL, et al. Inherited corneal disease: the evolving molecular, genetic and imaging revolution. Clin Experiment Ophthalmol. 2005;33:303.
10 Wiggs JL. Genetic etiologies of glaucoma. Arch Ophthalmol. 2007;125:30.
11 Holland GN. AIDS and ophthalmology: the first quarter century. Am J Ophthalmol. 2008;145:397.
12 Zamir E, et al. Massive mycobacterial choroiditis during highly active antiretroviral therapy: another immune-recovery uveitis? Ophthalmology. 2002;109:2144.
13 Vrabec TR. Posterior segment manifestations of HIV/AIDS. Surv Ophthalmol. 2004;49:131.
14 Chu DS, Foster CS. Sympathetic ophthalmia. Int Ophthalmol Clin. 2002;42:179.
15 Singh AD, Topham A. Incidence of uveal melanoma in the United States: 1973–1997. Ophthalmology. 2003;110:956.
16 Singh AD, Topham A. Survival rates with uveal melanoma in the United States: 1973–1997. Ophthalmology. 2003;110:962.
17 Seddon JM, et al. A prognostic factor study of disease-free interval and survival following enucleation for uveal melanoma. Arch Ophthalmol. 1983;101:1894.
18 Folberg R, et al. Recommendations for the reporting of tissues removed as part of the surgical treatment of common malignancies of the eye and its adnexa. The Association of Directors of Anatomic and Surgical Pathology. Hum Pathol. 2003;34:114.
19 Kilic E, et al. Clinical and cytogenetic analyses in uveal melanoma. Invest Ophthalmol Vis Sci. 2006;47:3703.
20 Clarijs R, et al. Presence of a fluid-conducting meshwork in xenografted cutaneous and primary human uveal melanoma. Invest Ophthalmol Vis Sci. 2002;43:912.
21 Maniotis AJ, et al. Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am J Pathol. 1999;155:739.
22 Folberg R, Maniotis AJ. Vasculogenic mimicry. APMIS. 2004;112:508.
23 Frank RN. Diabetic retinopathy. N Engl J Med. 2004;350:48.
24 Pe’er J, et al. Hypoxia-induced expression of vascular endothelial growth factor by retinal cells is a common factor in neovascularizing ocular diseases. Lab Invest. 1995;72:638.
25 Tolentino MJ, et al. Pathologic features of vascular endothelial growth factor–induced retinopathy in the nonhuman primate. Am J Ophthalmol. 2002;133:373.
26 Chen J, Smith LE. Retinopathy of prematurity. Angiogenesis. 2007;10:133.
27 Emerson GG, Lutty GA. Effects of sickle cell disease on the eye: clinical features and treatment. Hematol Oncol Clin North Am. 2005;19:957.
28 Mohan JS, et al. The angiopoietin/Tie-2 system in proliferative sickle retinopathy: relation to vascular endothelial growth factor, its soluble receptor Flt-1 and von Willebrand factor, and to the effects of laser treatment. Br J Ophthalmol. 2005;89:815.
29 Berker N, Batman C. Surgical treatment of central retinal vein occlusion. Acta Ophthalmol. 2008;86:245.
30 Funk M, et al. Intraocular Concentrations of Growth Factors and Cytokines in Retinal Vein Occlusion and the Effect of Therapy with Bevacizumab. Invest Ophthalmol Vis Sci. 2008. Epub.
31 Klein R, et al. Fifteen-year cumulative incidence of age-related macular degeneration: the Beaver Dam Eye Study. Ophthalmology. 2007;114:253.
32 Scholl HP, et al. An update on the genetics of age-related macular degeneration. Mol Vis. 2007;13:196.
33 Maller J, et al. Common variation in three genes, including a noncoding variant in CFH, strongly influences risk of age-related macular degeneration. Nat Genet. 2006;38:1055.
34 DeAngelis MM, et al. Cigarette smoking, CFH, APOE, ELOVL4, and risk of neovascular age-related macular degeneration. Arch Ophthalmol. 2007;125:49.
35 Jager RD, Mieler WF, Miller JW. Age-related macular degeneration. N Engl J Med. 2008;358:2606.
36 Daiger SP, et al. Perspective on genes and mutations causing retinitis pigmentosa. Arch Ophthalmol. 2007;125:151.
37 Balmer A, et al. Diagnosis and current management of retinoblastoma. Oncogene. 2006;25:5341.
38 Sampieri K, et al. Genomic differences between retinoma and retinoblastoma. Acta Oncol. 2008;47:1483.
39 Choi JY, et al. Primary intraocular lymphoma: a review. Semin Ophthalmol. 2006;21:125.
40 Hayreh SS. Ischemic optic neuropathy. Prog Retin Eye Res. 2008. Epub.
41 Wiggs JL, et al. Lack of association of mutations in optineurin with disease in patients with adult-onset primary open-angle glaucoma. Arch Ophthalmol. 2003;121:1181.
42 Levin LA. Neuroprotection and regeneration in glaucoma. Ophthalmol Clin North Am. 2005;18:585.
43 Yen MY, et al. Leber’s hereditary optic neuropathy: a multifactorial disease. Prog Retin Eye Res. 2006;25:381.
44 Mayorov VI, et al. Mitochondrial oxidative phosphorylation in autosomal dominant optic atrophy. BMC Biochem. 2008;9:22.
45 Beck RW, et al. High- and low-risk profiles for the development of multiple sclerosis within 10 years after optic neuritis: experience of the optic neuritis treatment trial. Arch Ophthalmol. 2003;121:944.