Opioid analgesics and the management of pain
Pain and pain perception
Pain is a complex phenomenon that involves the person's awareness of, and response to, a noxious stimulus. Pain is highly subjective to the individual, and psychological factors will determine to what extent the individual experiences suffering or distress (Fig. 19.1). Pain can be acute, lasting only until the initiating trauma resolves, which is sometimes described as nociceptive pain. Pain can also become protracted and chronic, outlasting the original trauma, and can in some cases become intractable as a result of persistent pathological change in the way that the nociceptive (pain-carrying) neuronal pathways function (neuropathic pain).
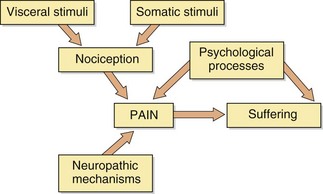
Nociceptive pain is a defensive response to a variety of stimuli (e.g. mechanical, thermal or chemical) that activate nociceptor sensory units on nerve endings. It is defensive as it induces behaviour that avoids exacerbation of the pain, and allows us to protect a damaged part of the body while it heals. Painful stimuli are transmitted to the central nervous system (CNS) by two types of fibre. Fast fibres in the neospinothalamic pathways (mechanical and thermal stimuli) carry pain that is appreciated as sharp and localised, while slow fibres in the paleospinothalamic pathways (chemical stimuli) produce poorly localised aching, throbbing or burning pain (Fig. 19.2).
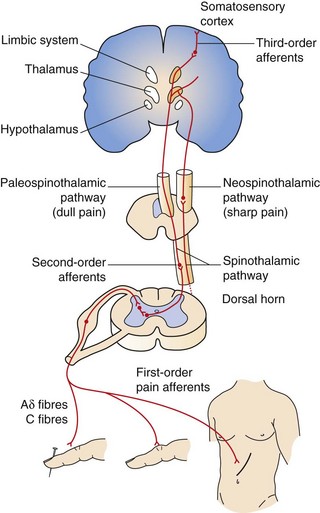
Fig. 19.2 Ascending pathways of pain perception.
Ascending pathways are activated following stimulation of afferent sensory nociceptive nerve terminals. Many mediators and neurotransmitters are involved during afferent stimulation of the nociceptive pathway. Mediator release (bradykinin, serotonin, prostaglandins) and thermal and mechanical influences can stimulate and sensitise the sensory nerve terminals of pain fibres, resulting in increased cation influx, depolarisation and generation of action potentials (see Fig. 19.3). Onward afferent transmission of ascending nerve impulses at the synapses in the dorsal horn involves transmitters such as substance P, glutamate and calcitonin gene-related peptide (CGRP) (Fig. 19.5). Hyperexcitability of pain fibres can also be promoted by other mediators. Prolonged activation of the nociceptive pathways can produce pathophysiological and phenotypic changes resulting in neuropathic pain that persists when the original pathological cause of the pain has resolved, and generation of nociceptive signals can occur at low levels of axonal stimulation.
Neuropathic pain may result from abnormal neuronal activity that persists beyond the time expected for healing of the injury. Examples include phantom limb pain following amputation and shingles causing pain well beyond the healing of the injury. There are multiple pathophysiological mechanisms underlying neuropathic pain. Neuropathic pain can be spontaneous (stimulus-independent), when it is usually described by the sufferer as shooting or lancinating sensations, electric shock-like pain or an abnormal unpleasant sensation (dysaesthesia). Alternatively, it can be an exaggerated response to a painful stimulus (hyperalgesia) or a painful response to a trivial stimulus (allodynia).
Some pain states have mixed nociceptive and neuropathic elements, for example mechanical spinal pain with local nerve damage such as radiculopathy or myelopathy.
The pathophysiological and molecular explanations for nociceptive and neuropathic pain and the endogenous responses that modulate pain are complex and incompletely understood. In brief, the genesis of nociceptive pain results from initial stimulation of afferent sensory neurons (nociceptors) by thermal, mechanical or chemical stimuli sufficient to stimulate the nociceptors (free nerve endings responsive to high-threshold noxious stimuli) on Aδ and C axons (Fig. 19.3). Nociceptors carry many surface receptors that modulate their sensitivity to stimulation, such as those for γ-aminobutyric acid (GABA), opioids, bradykinin, histamine, serotonin (5-hydroxytryptamine, 5-HT) and capsaicin, and mechanosensitive ion channels (responsive to touch, pressure, etc.). The particular type of painful stimulus may determine which receptors or ion channels are activated. Activation of the ion channels or receptors results in an influx of Na+/Ca2+ sufficient to generate action potentials in the nerve (Fig. 19.3). Most nociceptors are dormant (silent), but when tissue damage occurs nociceptors that were not normally active can be recruited and sensitise the area to painful stimuli. The afferent fibres from these nociceptors enter both the dorsal and ventral horns of the spinal cord, and link to ascending pathways. Chemical transmission of the afferent impulse travels across synapses in the spinal cord and in the projections of the ascending pathways to the thalamus, and from there to the reticular formation and the cortex. Transmitters in these central pathways include excitatory substances such as glutamate, tachykinins (neurokinins and substance P) and calcitonin gene-related peptide (CGRP). Synaptic transmission responsible for nociceptive pain can be reduced by local inhibitory neurons, mediated by GABA. There are also important descending modulatory pathways arising from the locus coeruleus that inhibit ascending pain pathways, with a further host of neurotransmitters including endogenous opioids, noradrenaline (acting on α2-adrenoceptors) and serotonin. Activation of these systems produces hyperpolarisation of neurons in the pain pathways (by inhibiting Na+/Ca2+ influx and enhancing K+ efflux) and prevents generation of nociceptive action potentials (Figs 19.4 and 19.5). The endogenous cannabinoids (anandamide and 2-arachidonyl glycerol) may also have modulatory functions in the descending pathways.
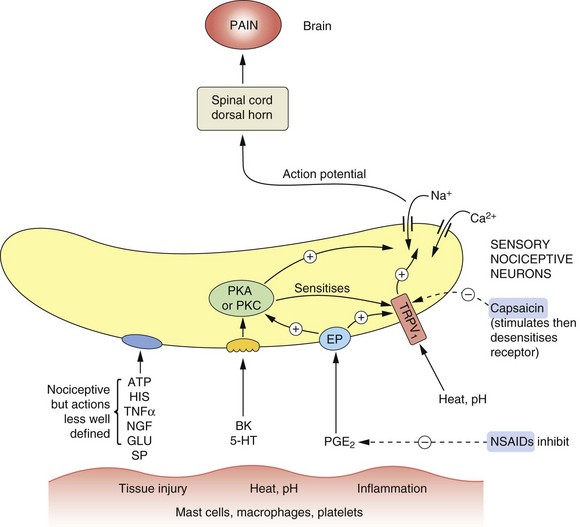
Fig. 19.3 Mediators involved in the genesis and modulation of pain.
Numerous mediators are able to stimulate or sensitise primary sensory neurons (nociceptors), leading to activation of nociceptive fibres. Tissue injury and other noxious stimuli such as heat or extremes of pH can stimulate the release of substances that act to promote pain, such as bradykinin (BK) and serotonin (5-hydroxytryptamine, 5-HT). Prostaglandin E2 (PGE2) acting at prostaglandin E receptors (EP) sensitises the nerve endings to the actions of nociceptive mediators including BK and 5-HT. ATP and histamine (HIS) also have nociceptive actions, acting in poorly defined ways. Heat and H+ stimulate the transient receptor potential vanilloid 1 (TRPV1) receptor, producing pain. The TRPV1 receptor is also sensitised by many other mediators. Capsaicin and other TRPV1 receptor stimulants desensitise the receptor on persistent stimulation, resulting in an analgesic effect. Non-steroidal anti-inflammatory drugs (NSAIDs) inhibit the production of PGE2. Overall the effect of nociceptive stimuli is to depolarise the neuron, setting up action potentials in the fibres to the dorsal horn and pain-perceiving areas of the brain. Substance P (SP) and calcitonin gene-related peptide (CGRP) may also be involved in nociception. GLU, glutamate; NGF, nerve growth factor; PKA, protein kinase A; PKC, protein kinase C; TNFα, tumour necrosis factor α.
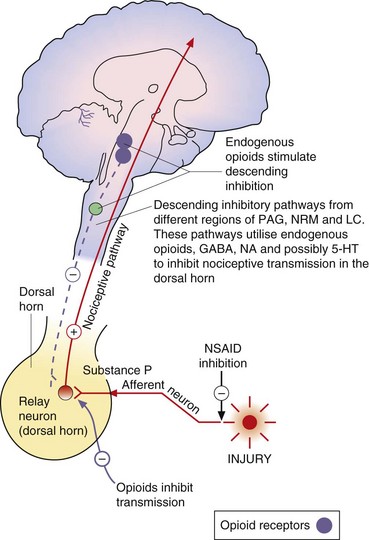
Fig. 19.4 Transmitters and receptors for pain perception and control.
The afferent nociceptive pathways are subject to inhibitory control. Opioids act at opioid receptor-rich sites in the periaqueductal grey matter (PAG), the nucleus raphe magnus (NRM) and other midbrain sites to stimulate descending inhibitory fibres that inhibit nociceptive transmission in the dorsal horn. Descending pathways from the locus coeruleus (LC) that are noradrenergic are also involved. Opioids also act at a local level in the dorsal horn (Fig. 19.5). Inhibitory modulation of nociceptive transmission via local nerve networks also results from actions of other agents (Fig. 19.5). 5-HT, 5-hydroxytryptamine (serotonin); GABA, γ-aminobutyric acid; NA, noradrenaline; NSAID, non-steroidal anti-inflammatory drug.
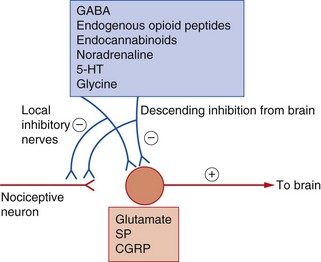
Fig. 19.5 Neurotransmitter substances involved in the genesis and modulation of pain.
Inhibitory modulation of nociception can occur via activation of the descending regulatory pathways and also by actions of local interneurons. 5-HT, 5-hydroxytryptamine (serotonin); CGRP, calcitonin gene-related peptide; GABA, γ-aminobutyric acid; SP, substance P.
Activation of pain pathways generates intracellular protein synthesis through stimulation of the proto-oncogene c-Fos and various inducible transcription factors in CNS neurons. These mediate the many changes in neuronal structure and the neuronal proliferation that occur with chronic pain.
Neuropathic pain may result from chronic hypersensitisation and phenotypic change in neurons. These changes can occur both peripherally and centrally, probably as a result of repetitive stimulation of synaptic transmission. In neuropathic and other chronic pain states, the following mechanisms may be important.
 Sensitisation of afferent inputs: this may include recruitment of silent nociceptors and a lower threshold for generation of action potentials in the spinal cord (through the action of cytokines released from glial cells), leading to enhanced action potential generation (Figs 19.3 and 19.5). Central sensitisation is also important with excessive neurotransmitter release. Abnormal voltage-gated Na+ channel expression may also be important in both peripheral and central sensitisation.
Sensitisation of afferent inputs: this may include recruitment of silent nociceptors and a lower threshold for generation of action potentials in the spinal cord (through the action of cytokines released from glial cells), leading to enhanced action potential generation (Figs 19.3 and 19.5). Central sensitisation is also important with excessive neurotransmitter release. Abnormal voltage-gated Na+ channel expression may also be important in both peripheral and central sensitisation.
Dysfunctional descending pain-modulatory and -facilitatory pathways: the functioning of the descending modulatory pathway (Fig. 19.4) may become inadequate as a result of persistent nociceptive pain. Persistent painful stimuli may be related to the overactivity of endogenous pain facilitatory pathways that run in tracts from the medulla to the spinal dorsal horn. The imbalance in the pain pathways then maintains neuropathic pain states.
Loss of inhibitory neurons and generation of new synapses that act as nociceptive neurons (neuronal plasticity): in neuropathic pain, Aβ nerve fibres that normally transmit tactile stimuli can sometimes undergo phenotypic change and take on the properties of a nociceptive neuron.
Analgesic drugs
Non-steroidal anti-inflammatory drugs (NSAIDs; Ch. 29) and opioids are the major classes of pain-relieving (analgesic) drugs. They act at different levels in the pain-transmitting pathways to influence the production and recognition of pain as indicated in Figures 19.3 and 19.4.
Non-steroidal anti-inflammatory drugs
These act mainly by blocking the peripheral generation of the nociceptive impulses. Prostaglandins enhance nociceptive impulses in peripheral afferent neurons by enhancing the ability of thermal, mechanical or chemical stimuli to increase Na+/Ca2+ influx and thereby to generate action potentials in nociceptive afferent neurons. NSAIDs inhibit the production of prostaglandins by the cyclo-oxygenase type 1 and type 2 (COX-1, COX-2) isoenzymes and thereby reduce the sensitivity of sensory nociceptive nerve endings to agents released by injured tissue that initiate pain, such as bradykinin and substance P. NSAIDs may also act on pain pathways in the CNS. Paracetamol, although not usually considered to be an NSAID, may also work in part through the same pathways. These drugs are considered fully in Chapter 29.
Opioids
These act on the spinal cord and limbic system, and stimulate the long descending inhibitory pathways from the midbrain to the dorsal horn. They produce their effects via specific receptors that are closely associated with the neuronal pathways which transmit pain from the periphery to the CNS.
Non-opioid, non-NSAID analgesics
A variety of drugs that were developed for other purposes are being used increasingly for their analgesic actions when NSAIDs and opioids are less effective, for example in neuropathic pain (see Table 19.2, below).
Opioid analgesics
Opioid is a term used for both naturally occurring and synthetic molecules that produce their effects by an agonist action at opioid receptors. The terms opiate analgesic (specifically, a drug derived from the juice of the opium poppy, Papaver somniferum) and narcotic analgesic (which literally means a ‘stupor-inducing pain killer’) are no longer used.
Mechanism of action
The brain produces several endogenous opioid peptides, which are neurotransmitters that act via specific opioid receptors. Among these are the two pentapeptide enkephalins. These each contain the amino acid sequence Tyr-Gly-Gly-Phe as the message domain, linked to either leucine or methionine, and are called leu-enkephalin and met-enkephalin. Other agonists incorporating the same amino acid sequence are dynorphins A and B and the most potent agonist β-endorphin, a 31-amino acid peptide with met-enkephalin at its carboxyl end. Two further peptides, endomorphins 1 and 2, have been identified that have Tyr-Pro-Phe and Tyr-Pro-Trp message domain sequences. All opioid peptides are derived by selective cleavage of larger precursor molecules.
Opioid receptors are found on the presynaptic and postsynaptic membranes of neurons in CNS pain pathways, and also in the peripheral nervous system. Three major classes of opioid receptor have been identified, which mediate distinct effects: the µ (mu), κ (kappa) and δ (delta) receptors, also termed MOP, KOP and DOP, respectively (Box 19.1). There is a distinctive regional distribution of opioid peptides and their receptors in the CNS, with high concentrations in the limbic system and spinal cord. These regions also contain high concentrations of a neutral endopeptidase (enkephalinase), which rapidly hydrolyses the pentapeptides into fragments. The various endogenous opioid peptides show preferential receptor-binding affinities: β-endorphin binds equally to µ- and δ-receptors, endomorphins bind mainly to µ-receptors, dynorphins bind preferentially to κ-receptors and the enkephalins bind preferentially to δ-receptors. The different physiological effects produced by these receptors are due to their specific neuronal distributions. An ‘orphan’ opioid receptor has been identified that does not bind the major opioid peptides, which has been named the nociceptin receptor after its specific endogenous agonist. It is widely distributed in the brain and facilitates descending enkephalinergic pathways in the spinal cord.
Opioid receptors are found on both presynaptic and postsynaptic neurons. The postsynaptic actions inhibit neuronal depolarisation, and the presynaptic effect inhibits neurotransmitter release. All opioid receptors are coupled to inhibitory G-proteins (Gi/G0), and receptor activation has many intracellular consequences:
inhibition of adenylyl cyclase with reduced intracellular generation of cAMP, which reduces neurotransmitter release,
phosphorylation of extracellular signal-regulated kinase (ERK) may be important in suppression of the immune system,
activation of voltage-gated inwardly rectifying K+ channels, which hyperpolarises the target cells, making them less responsive to depolarising impulses,
inhibition of voltage-gated N-type Ca2+ channels in target neurons, which reduces neurotransmitter release.
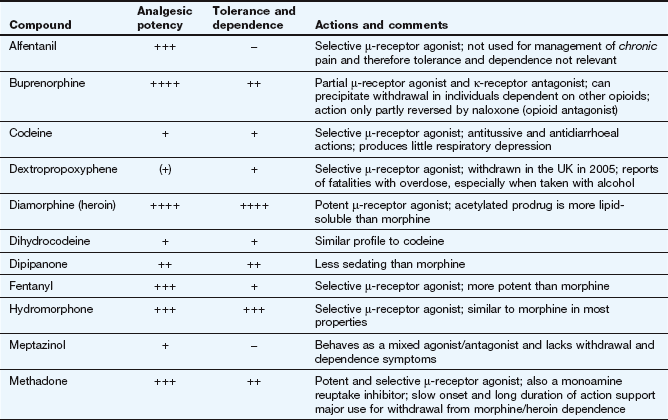
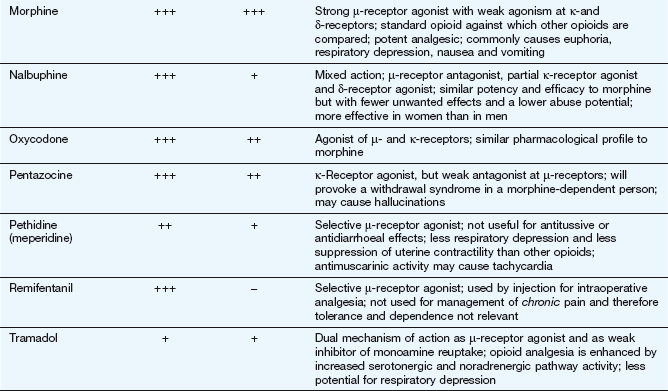
Morphine and synthetic opioid analgesics produce their effects largely by acting as agonists at specific opioid receptors in the CNS. Opioid drugs show receptor selectivity and can have agonist, partial agonist or antagonist properties at various opioid receptor types (Table 19.1). The analgesic action of opioids is the result of a complex series of neuronal interactions. In the nucleus raphe magnus of the brain, µ-receptor stimulation decreases activity in inhibitory GABA neurons that project to descending inhibitory serotonergic neurons in the brainstem. These neurons in turn connect presynaptically with afferent nociceptive fibres in the dorsal horn of the spinal cord. Inhibition of the GABA neurons permits increased firing of the descending inhibitory serotonergic neurons. Analgesia is produced by inhibition of the release of the pain pathway mediators, substance P, glutamate and nitric oxide from the afferent nociceptive neurons (Fig. 19.5). Activation of κ-receptors antagonises the analgesia produced by µ-receptor stimulation, by inhibiting the descending (pain-modulating) serotonergic neurons in the pain pathway. However, there is a paradoxical spinal analgesic effect from unopposed κ-receptor activation.
Opioid receptors are also present on peripheral nerves in the pain pathways, and a µ-receptor agonist reduces the sensitivity of peripheral nociceptive neurons to pain stimuli, particularly in inflamed tissues. Non-neuronal κ-receptors are involved in the inflammatory response, and are found on endothelial cells, T-lymphocytes and macrophages; κ-receptor agonists are being developed to modulate the inflammatory response orchestrated by these cells.
Full agonists: these act principally at µ-receptors and include morphine, diamorphine, fentanyl, pethidine, codeine and dihydrocodeine. They also have weak agonist activity at δ- and κ-receptors.
Mixed agonist–antagonist: pentazocine has agonist effects at the κ-receptor (and, to a lesser extent, the δ-receptor) and is a weak µ-receptor antagonist.
Mixed partial agonist–antagonist: buprenorphine is a potent partial agonist at the µ-receptor and has antagonist activity at κ-receptors. The latter action will enhance the analgesic action produced via the µ-receptors.
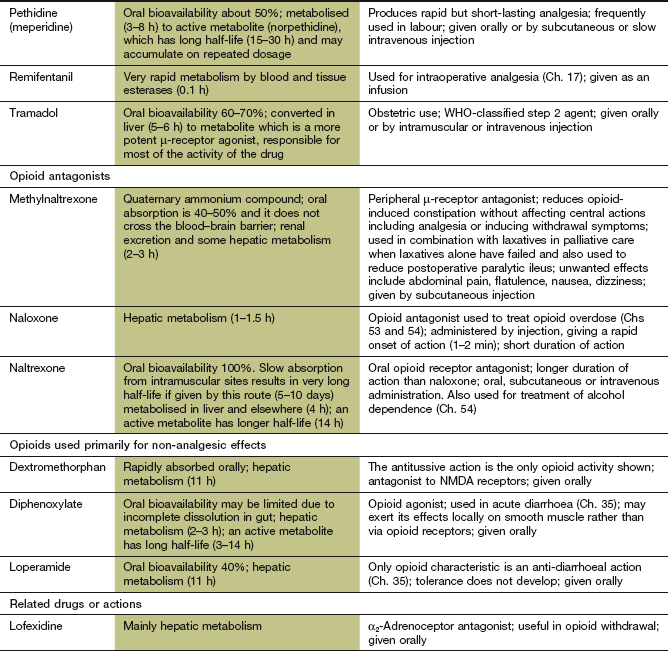
Some opioids have additional properties: meptazinol is a µ-receptor agonist with muscarinic receptor agonist activity; tramadol and methadone are µ-receptor agonists that also inhibit neuronal noradrenaline and serotonin uptake. These supplementary actions of tramadol and methadone contribute to their analgesic actions, since amine-mediated neurotransmission potentiates descending inhibitory pain pathways (Fig. 19.4). Methadone is also an antagonist at glutamate N-methyl-D-aspartate (NMDA) receptors, an action which can also inhibit pain transmission (Fig. 19.5).
Opioid receptor antagonists such as naloxone have no analgesic actions and are used in the treatment of opioid overdose (Ch. 53).
Effects and clinical uses
Effects on the central nervous system:
Analgesia: The analgesia produced by morphine is most effective for chronic visceral pain, but can still be helpful for some types of neuropathic pain. In addition to its antinociceptive effect, morphine alters the perception of pain, making it less unpleasant. This supraspinal effect, possibly at the limbic system, is less marked with some opioids such as pentazocine. Opioid analgesics have no anti-inflammatory effect. In fact, morphine can release the inflammatory mediator histamine locally at the site of an injection. Full µ-receptor agonists are the most powerful opioid analgesics (see Table 19.1 for list of full and partial agonists). However, some full µ-receptor agonists, for example codeine, have a low affinity for the receptors and have a limited analgesic effect. There is growing evidence that the antagonist action of methadone at NMDA receptors can produce effective analgesia in people who have become tolerant to high doses of morphine (see below). Short-acting opioids such as fentanyl and remifentanil are used for analgesia during general anaesthesia (Ch. 17).
The ceiling analgesic effect of a µ-receptor partial agonist is lower than that of a full agonist. If a person receiving high doses of a potent full µ-receptor agonist is given a µ-receptor partial agonist (e.g. buprenorphine) or a µ-receptor antagonist (e.g. pentazocine), then some of the full agonist molecules will be displaced from receptor sites by the less effective partial agonist molecules or antagonist molecules. The degree of analgesia may then be reduced, and in dependent individuals withdrawal symptoms can be produced (see below and Table 19.1).
Euphoria: The use of morphine is often associated with an elevated sense of well-being (euphoria, mediated by µ-receptors), an action that contributes considerably to its analgesic efficacy. The opposite effect (dysphoria, mediated by agonist activity at κ-receptors) counteracts the euphoric action, and the degree of euphoria produced will depend on the receptor-binding characteristics of the drug.
Respiratory depression: The sensitivity of the respiratory centre to stimulation by carbon dioxide is reduced by morphine at doses that produce analgesia. Respiratory depression is a common cause of death in opioid overdose. Occasionally, the effect on respiratory rate can be of clinical benefit; for example, intravenous morphine relieves the dyspnoea associated with acute pulmonary oedema, and morphine is used orally or by subcutaneous infusion for the treatment of breathlessness in palliative care. Meptazinol and tramadol are claimed to cause less respiratory depression than other opioids.
Suppression of the cough centre: Opioids possess an antitussive action. Compounds such as codeine and dextromethorphan are effective for cough suppression (Ch. 13), despite having relatively weak analgesic effects.
Vomiting: Opioids stimulate the chemoreceptor trigger zone and cause vomiting in up to 30% of people. Tolerance to the nausea and vomiting can occur with repeated doses. Powerful opioids such as morphine are usually given with an anti-emetic (Ch. 32), particularly when used for acute pain.
Miosis: Stimulation of the third-nerve nucleus results in pupillary constriction. Pinpoint pupils, together with coma and slow respiration, are signs of opioid overdose (Ch. 53).
Endocrine effects: Opioids inhibit the hypothalamic–pituitary–adrenal axis, leading to a progressive decline in plasma cortisol levels (Ch. 44). They also increase prolactin and decrease luteinizing hormone release, which leads to testosterone deficiency in men and a reduction in oestrogen synthesis in women (Chs 45 and 46). Men usually benefit from testosterone replacement during long-term opioid use (Ch. 46).
Gastrointestinal tract: There is a general increase in resting tone of the gut wall and sphincters. These effects arise from stimulation of µ- and κ-receptors on neuronal plexuses in the gut wall. An increase in biliary pressure caused by opioid-induced spasm at the sphincter of Oddi can exacerbate biliary colic. In the stomach, a decrease in motility and pyloric tone can produce anorexia, nausea and vomiting. In the small and large intestines there is increased segmenting activity and decreased propulsive activity. Thus, opioid administration is associated with constipation, and up to 80% of people who take opioids in the long term will need a laxative. Methylnaltrexone, a specific antagonist of peripheral opioid receptors, is used to treat opioid-induced constipation during palliative care when laxatives are inadequate. The effects of opioids on gastrointestinal motility make them useful in the treatment of diarrhoea (Ch. 35). Pethidine and tramadol have less effect on the gastrointestinal tract than do equi-analgesic doses of morphine.
Cardiovascular system: Opioids have little effect on the heart or circulation except at high doses that can depress the medullary vasomotor centre. Hypotension can occur with parenteral use of morphine, possibly because of histamine release.
Other systems: Opioids have minor effects on other systems. For example, there is an increase in tone of the bladder wall and sphincter, which can lead to urinary retention. There is increasing evidence that long-term use of opioids suppresses immune function by inhibiting the development and differentiation of many types of immune cells. The clinical relevance of this is not known.
Tolerance and dependence: Tolerance and dependence result from changes in the functioning of opioid receptors during continuous opioid administration. As a consequence of adaptive changes, more of the drug is necessary to produce the same effect (tolerance) and withdrawal of the drug produces adverse physiological effects until the compensatory changes are reset (dependence).
Tolerance to opioids occurs in two ways. Associative (learned) tolerance has a major psychological component. Non-associative (adaptive) tolerance involves downregulation or desensitisation of opioid receptors. This arises in response to increased firing of neurons in the noradrenergic pathways of the locus coeruleus (an area of the brain involved in the physiological response to stress, which is rich in inhibitory opioid receptors), and activation of the reward pathway in the brain (see Ch. 54). It may also involve increased activity at NMDA receptors for excitatory glutamate-mediated neurotransmission in spinal and supraspinal circuits. Tolerance to the analgesia, euphoria, respiratory depression and emesis develops rapidly during regular opioid administration, but much less to the constipation or miosis. A high degree of cross-tolerance is shown by many opioids; consequently, individuals who develop tolerance to one opioid are often (but not invariably) tolerant to another. Opioid-induced NMDA receptor activation can also produce abnormal pain sensitivity at spinal cord dorsal horn cells. This sensitisation process can be confused with tolerance and lead to opioid dose escalation. Methadone may be useful in this situation (see above).
Dependence manifests itself as a withdrawal syndrome, which can be precipitated when individuals who have taken the drug for a long period of time have their intake stopped or are given an opioid antagonist or partial agonist (Ch. 54). This is most often a problem for people who abuse the drug, but can occur from long-term intake of a prescribed opioid.
During the first 12 h after opioid withdrawal, effects such as nervousness, sweating and craving are largely psychological, because they can be alleviated by the administration of a placebo. Following this period the effects of physiological dependence manifest themselves; for example, dilated pupils, anorexia, weakness, depression, insomnia, gastrointestinal and skeletal muscle cramps, increased respiratory rate, pyrexia, piloerection with goose-pimples, and diarrhoea. The time course for the development and resolution of these symptoms varies among the opioids. In the case of morphine, the maximum intensity of withdrawal effects occurs quickly (after about 1–2 days) and subside rapidly (about 5–10 days), and often the intensity of the symptoms is intolerable.
In contrast to morphine, withdrawal from methadone is a slow process because of its very long half-life, but the effects are far less intense (peak effect at almost 1 week and symptoms persist for about 3 weeks). Therefore, morphine- or heroin-dependent individuals who have been abusing the drug are often transferred from their drug of abuse to methadone prior to withdrawal (opioid-substitution therapy). Methadone also produces less euphoria than morphine or heroin. After a period of treatment with methadone, the methadone dosage is gradually reduced and the person undergoes a more tolerable withdrawal. Buprenorphine is used as an alternative to methadone, and produces less sedation and a low intensity of withdrawal symptoms. It can be given for 6 days in a rapid detoxification programme, or for long-term maintenance for reducing relapse in people who are addicted to opioids, since its partial agonist activity blocks the ‘high’ from illicit opioid use.
Rapid in-hospital tapering of opioids over 2 weeks has an early 80% success rate. On an outpatient basis, slow tapering over 6 months is more successful than rapid withdrawal, but still leads to only a 40% success rate. Long-term buprenorphine therapy, combined with high-intensity psychosocial group therapy, has achieved up to 75% withdrawal rates after 1 year. Detoxification from opioids can be helped by the presynaptic α2-adrenoceptor agonists clonidine or lofexidine (a clonidine analogue with fewer unwanted effects). These inhibit the excessive sympathetic nervous system activity associated with opioid withdrawal, such as lacrimation, rhinorrhoea, muscle pain, joint pain and gastrointestinal symptoms. However, the lethargy, insomnia and restlessness persist.
Naltrexone is a µ- and κ-receptor antagonist and a weak agonist at the δ-receptor. It blocks the effects of opioid agonists, and is used for prevention of relapse in people who were dependent on opioids, and who have withdrawn for at least 7–10 days.
Pharmacokinetics
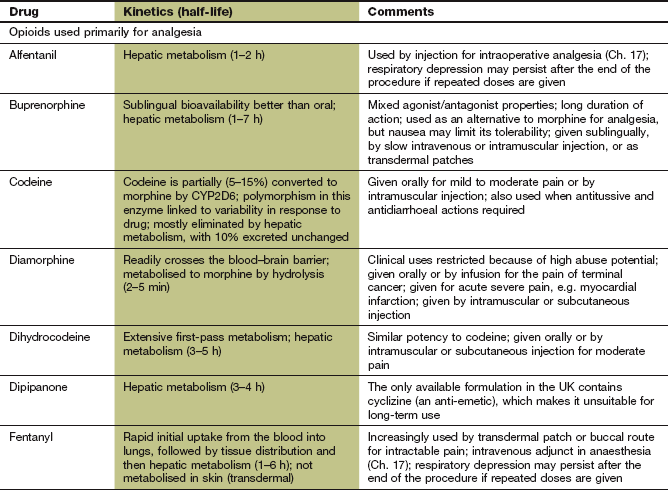
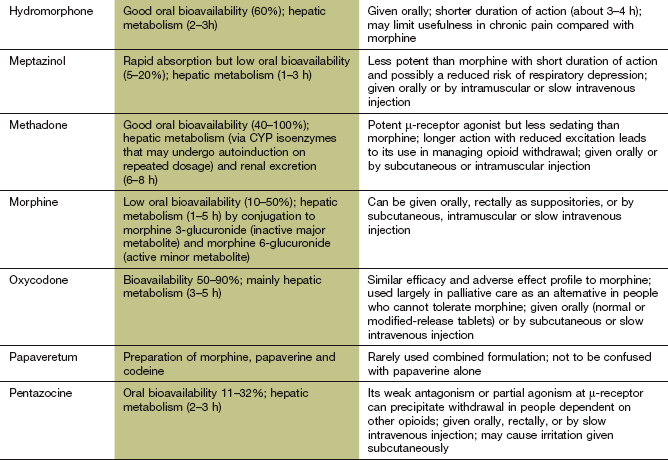
The pharmacokinetic properties of individual opioid analgesics are summarised in the Compendium at the end of this chapter. Most opioids are available for oral use. However, buprenorphine is formulated for sublingual absorption, and fentanyl as a sublingual or buccal tablet or a nasal spray for rapid pain relief. Some opioids, such as morphine, buprenorphine, meptazinol, methadone, oxycodone and tramadol can be given by intravenous or intramuscular injection. Morphine and oxycodone can also be given by subcutaneous infusion in palliative care. Diamorphine is more potent than morphine and can be given by subcutaneous infusion in smaller volumes, which can be useful for emaciated people. Morphine and pentazocine are available as suppositories. Fentanyl and buprenorphine can be delivered transdermally via self-adhesive patches for prolonged analgesia.
Some opioids, such as morphine, have a low and variable absorption across the gut wall, and the dose should be reduced when given parenterally to achieve equivalent analgesia and avoid unwanted effects. Others, such as dihydrocodeine, have a low oral bioavailability due to extensive first-pass metabolism. Opioids are eliminated by hepatic metabolism. A metabolite of morphine, morphine 6-glucuronide, has more analgesic activity than the parent compound and is excreted by the kidney. The dose of morphine must therefore be reduced in renal failure. Diamorphine is an acetylated morphine derivative that is converted to morphine by hydrolysis in plasma and in most tissues, including the brain. Codeine is a prodrug that is metabolised by CYP2D6, which shows genetic polymorphism, to several active metabolites. Codeine 6-glucuronide is responsible for most of the analgesic activity, while about 5% is converted to morphine. About 10% of people have low CYP2D6 activity, and have a reduced analgesic response to codeine.
Most opioid analgesics have half-lives in the range of 1–6 h. For long-term pain control, morphine, and some other powerful opioids, is often given as a modified-release formulation to prolong the duration of action. Fentanyl has a short half-life (1–6 h) but can be given as a transdermal delivery patch, when pain relief lasts 12–24 h due to slow drug delivery (Ch. 2). In addition, the effect persists for several hours after removing the patch owing to build-up of a subcutaneous drug reservoir at the site of application. Care is necessary both to maintain analgesia and to avoid unwanted opioid effects if the dose of fentanyl is increased or fentanyl patches are changed for another opioid.
Unwanted effects
The unwanted effects of opioids are caused by their actions on those opioid receptors that are not the primary site for therapeutic benefit. For example, respiratory depression and constipation are unwanted effects when an opioid is used as an analgesic, but the same effects are therapeutically beneficial in the treatment of breathlessness or diarrhoea. Tolerance and dependence can also be regarded as unwanted problems associated with long-term use.
Non-opioid, non-NSAID agents used for analgesia
A diverse group of drugs are now being used for pain control in circumstances where opioids and NSAIDs are inadequate (Table 19.2). These are dealt with individually in the next section and the detailed pharmacology of the drugs is given in the chapters describing their main clinical uses.
Table 19.2
The mechanisms of action of some non-opioid, non-NSAID analgesicsa
| Drug | Mechanismb |
| Gabapentin, pregabalin | GABA concentrations increased |
| Carbamazepine, lamotrigine | Membrane stabilisation; Na+ channel blockade; attenuation of glutamate-related synaptic transmission |
| Baclofen | GABAB receptor agonist; attenuation of glutamate-related synaptic transmission |
| Clonidine | α2-Adrenoceptor agonist |
| Tricyclic antidepressants (imipramine, amitriptyline) | Increase noradrenaline availability |
| Ketamine, dextromethorphan | Glutamate NMDA receptor antagonists |
| Local anaesthetics | Neuronal transmission (Na+ channel block) |
| Capsaicin | Substance P depletion, stimulation of vanilloid (TRPV1) receptor followed by desensitisation |
| Cannabinoids | Stimulation of cannabinoid (CB) receptors |
GABA, γ-aminobutyric acid; NMDA, N-methyl-D-aspartate; TRPV1, transient receptor potential vanilloid 1.
aThe mechanisms of pain control by these classes of drugs are imperfectly understood. Some are selective in their actions. For example, pain related to diabetic neuropathy and post-herpetic neuralgia (shingles), but not lower back pain, responds to tricyclic antidepressants.
bSee also Fig. 19.3 for details about mechanism.
Pain management
Appropriate management of pain depends on its origin and severity. The analgesic ladder developed by the World Health Organization (WHO) is useful for choosing drug therapy appropriate to the level of pain (Box 19.2).
Step 1 drugs
Paracetamol (Ch. 29) is suitable for mild pain. However, an NSAID (Ch. 29) may be more appropriate if there is local inflammation. The choice will be determined by the balance of benefits and risks of NSAIDs. Examples of pain that respond better to an NSAID are soft-tissue injury, tissue compression, visceral pain caused by pleural or peritoneal irritation and bone pain caused by metastatic deposits. Bone metastases cause local secretion of prostaglandins, and NSAIDs can be particularly effective. Individual responses to an NSAID vary; about 60% of people will respond to an alternative drug even if the first was ineffective.
Step 2 drugs
A weak opioid should be added to a step 1 drug for moderate pain, or when the response to a step 1 drug is inadequate. Opioids suitable for moderate pain include codeine and dihydrocodeine. These are often used in combination with paracetamol, such as the compound formulation co-codamol (codeine and paracetamol), although the dose of opioid in some combinations is too low to produce useful additional analgesia. Tramadol has been advocated at this stage, but it is no more effective than co-codamol, and claims that it has less effect on respiration and gastrointestinal motility are of uncertain clinical importance.
Step 3 drugs
The drug of choice for moderate to severe pain is morphine. It is usually effective orally, using a rapid-onset formulation for initial pain control. Doctors are often unwilling to give adequate doses of strong opioid because of concern about addiction. In severe chronic pain this is not an issue, and it should not be used as a reason for avoiding the use of morphine in non-cancer pain. Opioid addiction is not a problem when opioids are given appropriately for relief of pain.
Acute pain
Acute pain usually has an obvious cause and is often accompanied by anxiety. For rapid pain relief in a self-limiting condition, for example migraine, a readily absorbed, short-acting drug will be appropriate. For more protracted conditions, for example sprains, a long-acting drug may be helpful to improve adherence by reducing the frequency of administration.
The principles of the WHO analgesic ladder should be followed. Very severe acute pain, for example with myocardial infarction, will require a powerful opioid such as morphine given intravenously for rapid effect. Intramuscular injection should be avoided if possible, since severe pain is often accompanied by sympathetic nervous system stimulation which produces peripheral vasoconstriction and thereby delays drug absorption. Some acute severe pain, such as that arising postoperatively or from trauma, renal colic, cholecystitis, pancreatitis or sickle cell crisis, should be treated initially with a powerful analgesic, then less powerful analgesics as the condition resolves. This involves applying the principles of the WHO analgesic ladder in reverse.
Postoperative pain has many components, including hyperalgesia in the area of the incision, local ischaemia in the wound and central neuronal sensitisation in addition to the inflammatory response to surgery. Many studies have shown that the treatment of postoperative pain is often inadequate. There is some evidence that intraoperative and postoperative opioid use can increase the risk of hyperalgesia (acute opioid-induced hyperalgesia), an effect that can be modulated by the use of α2-adrenoceptor agonists (such as clonidine), cyclo-oxygenase (COX)-2 inhibitors (Ch. 29) or glutamate NMDA receptor antagonists (such as ketamine or methadone).
Persistent postsurgical pain is common, and is usually considered to be present if symptoms continue for more than 2 months after the procedure. There are many surgical, psychosocial, genetic and environmental factors that predict the development of this condition. Postoperative regional anaesthesia and combination analgesic regimens can reduce the risk of developing the syndrome. Although the principles of the WHO pain ladder are still important for management of postoperative pain, it is clear that combinations of regional anaesthetic techniques (Ch. 18) and analgesics (including those more commonly used for neuropathic pain) after surgery may be important to ensure optimal outcomes.
Chronic pain
Chronic pain is usually defined as pain lasting for at least 3 months, or at least until acute tissue pathology that initiated the pain would have been expected to heal. It can be a result of persistent nociceptive stimulation or can have a neuropathic origin. There are numerous psychosocial contributors to the development of chronic pain as well as physical pathology, and therefore management of much chronic pain is more effective if facilitated by multidisciplinary pain teams. Drug therapy is not the only solution for chronic pain, and, depending on the cause, non-pharmacological or local treatments are often appropriate. Examples include:
surgery for neoplastic, structural or ischaemic disorders,
physical methods such as acupuncture, transcutaneous electrical nerve stimulation (TENS; which activates spinal inhibitory neurons by acting as a counter-irritant) and local anaesthetic nerve block (Ch. 18),
behavioural modification, for example biofeedback, relaxation techniques, hypnosis,
a corticosteroid (Ch. 44) for raised intracranial pressure or spinal cord compression, or to reduce inflammation which can be associated with a cancer.
As for neuropathic pain, many forms of chronic non-cancer pain, such as fibromyalgia, osteoarthritis or low-back pain respond better to adjuvant analgesics such as tricyclic antidepressants or anticonvulsant drugs (see below) than to conventional analgesics. Combinations of both adjuvant and conventional analgesics are often used. If conventional analgesia is needed (especially for nociceptive pain and several types of cancer-related pain) then the principles of escalation of analgesia described by the WHO analgesic ladder should be followed.
For most severe chronic nociceptive pain morphine is the treatment of choice. If pain remains severe with a low initial dosage of morphine, the dosage should be increased by 50–100% every 24 h. Once the pain is moderate in intensity, increments of 25–50% daily are usually sufficient to achieve control without excessive unwanted effects. A modified-release formulation of morphine can be substituted once a stable dosage has been determined, although a rapid-acting formulation may still be required to treat breakthrough pain. Oral administration may not be possible if there is vomiting, dysphagia or intestinal obstruction. In these circumstances, rectal administration of morphine or subcutaneous infusion of morphine using a syringe driver can be used. Diamorphine can also be given by epidural or intrathecal injection for intractable pain. Transdermal delivery of an opioid such as fentanyl from an adhesive patch is an alternative to modified-release oral morphine. There is substantial evidence that if a person is unable to tolerate a particular opioid then an alternative opioid may be better tolerated. The factors that predict intolerance to a particular agent are poorly understood, although it is now recognised that intolerance to morphine may be associated with a mutation in the multidrug resistance 1 transporter protein (Chs 2 and 52). Oxycodone is increasingly used as an alternative to morphine, and there is evidence that the response to methadone can persist when other µ-receptor agonists are ineffective.
Neuropathic pain
Neuropathic pain, such as trigeminal neuralgia, post-herpetic neuralgia and phantom limb pain after an amputation, often responds less well than nociceptive pain to conventional analgesia. Although the mechanisms of neuropathic pain are now better understood there is still little agreement on how to use this knowledge to direct treatment. As with all forms of chronic pain a multidisciplinary approach that considers the psychosocial contributors to the pain and promotes appropriate physical activity is often necessary.
First-line treatment is usually with an anticonvulsant that binds to voltage-gated Ca2+ channels (such as gabapentin or pregabalin) (Ch. 23). Tricyclic antidepressants (Ch. 22) that increase synaptic noradrenaline and serotonin concentrations in the descending spinal inhibitory pathways are an alternative first-line approach (Figs 19.4 and 19.5). The importance of noradrenaline in modulating pain is shown by the lack of efficacy of selective serotonin reuptake inhibitor (SSRI) antidepressants for neuropathic pain. However, selective serotonin and noradrenaline reuptake inhibitors (SNRI) such as duloxetine (Ch. 22), which have fewer unwanted effects than tricyclic antidepressants, can be effective.
Opioids are useful in some cases of stimulus-independent pain when central mechanisms may be important. Other pharmacological treatments that are sometimes considered include baclofen (Ch. 24) and the NMDA receptor antagonist ketamine (Ch. 17), which modulate spinal transmission of the pain signal. The use of cannabinoids (the active components of cannabis; Ch. 54) for relief of hyperalgesia in conditions such as multiple sclerosis is receiving considerable attention. Stimulation of cannabinoid receptors produces an antinociceptive action and inhibits pain transmission in the spinal cord.
Stimulus-evoked pain may respond to topical treatment if it is localised and arises from peripheral mechanisms. Lidocaine cream or topical patches can be effective through its local anaesthetic actions (Ch. 18) for conditions such as mechanical allodynia in post-herpetic neuralgia. Alternatively, capsaicin, a derivative of red chilli peppers that stimulates C fibres in the afferent nociceptive pathway, can be applied topically as a counter-irritant. This releases substance P and stimulates the transient receptor potential vanilloid 1 (TRPV1) receptor and initially provokes hyperalgesia by promoting depolarisation and action potential generation. Subsequent depletion of substance P and even nerve terminal degeneration then blocks nerve function (Fig. 19.3).
Trigeminal neuralgia is distinct from other forms of neuropathic pain. It can be treated surgically, but also responds well to carbamazepine (Ch. 23), which can reduce the frequency and intensity of attacks. Alternative treatments include phenytoin and lacosamide (Ch. 23).
True/false questions
1. Opioids inhibit pain transmission in the dorsal horn of the spinal cord.
2. Opioids can cause euphoria or dysphoria.
3. Tolerance develops uniformly to all of the biological effects of the opioids
4. Morphine can cause itching and vasodilation at injection sites.
5. Methadone has a rapid onset of action and a short half-life.
6. Meptazinol is a pure µ-receptor agonist.
7. Naloxone is a short-acting opioid agonist.
8. Drugs that inhibit the reuptake of noradrenaline can be effective analgesics in neuropathic pain.
9. Anticonvulsants should not be used in the treatment of neuropathic pain.
10. Buprenorphine is a partial agonist at µ-opioid receptors.
11. Pentazocine can precipitate withdrawal symptoms in morphine addicts.
12. Pethidine (meperidine) is used in childbirth because of its short half-life.
One-best-answer (OBA) questions
1. Which of the following is the most appropriate statement about opioid analgesics?
A In the elderly, tolerance rapidly develops to the constipating effects of morphine.
B An opioid analgesic is the drug of choice for phantom limb pain following a below-knee amputation after a road traffic accident.
C The analgesic potency of codeine and dihydrocodeine are similar to that of morphine.
D Tolerance does not develop to the miotic effect of opioids.
E Fentanyl can be used for opioid withdrawal and maintenance of the chronically relapsing heroin addict.
Case-based questions
Pain control in terminal cancer: a 60-year-old man was admitted to a hospice. He had previously had a left nephrectomy for renal cell carcinoma and now had intense metastatic bone pain in his ankles, right iliac crest and left upper arm. He was also having periods of dyspnoea. Prior to admission, his medication was co-codamol three times daily and diclofenac (150 mg) at night. He was also taking cimetidine (400 mg) twice daily. His pain was not well controlled on admission. After a week of assessment and optimisation of drug therapy, his treatment comprised the following drugs.

1. Was this man taking an opioid analgesic before admission to the hospice?
2. How does morphine exert its pharmacological action as an analgesic?
3. Why was oral morphine solution (an immediate-release form) made available in addition to the slow-release formulation?
4. Was the dependence potential of morphine likely to present a problem in this man?
5. What alternative opioids might you consider as an immediate replacement for morphine?
6. How does diclofenac control inflammation and why was it useful in this man?
7. What was the rationale for the use of dexamethasone?
8. Metoclopramide is an anti-emetic. Why do you think that this man was likely to suffer from nausea and possibly vomiting?
9. How does metoclopramide act to alleviate nausea and what other anti-emetic drugs could be used?
10. Why might gastric or duodenal ulceration be a problem in this man?
11. Why was cimetidine given before admission and why was it replaced with lansoprazole?
12. Why was constipation likely to be a problem in this man?
13. What is the mechanism of action of docusate sodium and what alternative laxative agents could have been used?
1. True. Opioids act at opioid receptors to block transmitter release and postsynaptic trasmission in the dorsal horn.
2. True. In people who have pain, analgesia is often associated with well-being (µ-receptors), whereas in pain-free people dysphoria can occur (κ-receptors).
3. False. Much less tolerance to miosis and constipation develops than to the other biological effects of opioids, including analgesia and respiratory depression.
4. True. Morphine is an alkaloid which in parenteral doses may release histamine from mast cells, leading to itch, vasodilation and hypotension.
5. False. Methadone has good oral absorption and less potential to cause euphoria, so it is used in controlled withdrawal in people with opioid dependence. Its slow onset of action and long half-life mean that withdrawal symptoms are more gradual and less intense than with morphine or heroin.
6. True. Because the µ-opioid receptors produce analgesia and the κ-receptors are involved in respiratory depression, it is claimed that meptazinol has less respiratory depressant action.
7. False. Naloxone is a short-acting opioid antagonist, blocking µ-, κ- and δ-receptors. It is used in opioid overdose, although severe withdrawal symptoms can occur in people addicted to opioids following naloxone administration.
8. True. Tricyclic antidepressants can be effective for the treatment of some cases of neuropathic pain. They may act by enhancing amine levels in the descending inhibitory pathways that control the pain gate mechanism (see Table 19.2).
9. False. Anticonvulsants such as carbamazepine and lamotrigine can be of use in the treatment of neuropathic pain, probably by stabilising neuronal membranes and inhibiting release of excitatory neurotransmitters, while gabapentin and pregabalin increase levels of the inhibitory transmitter γ-aminobutyric acid (GABA).
10. True. The partial agonism of buprenorphine at µ-opioid receptors is associated with lower abuse potential than morphine.
11. True. A partial agonist (such as pentazocine) can reduce the effects of a full agonist (such as morphine) at the same receptor.
12. True. The short half-life of pethidine reduces respiratory depression in the neonate and allows rapid adjustment of dosage. Pethidine also has less suppressive effect on the uterine muscles than morphine.
OBA answers
A Constipation continues to be a problem with long-term morphine treatment, and laxatives are often required.
B Chronic pain is usually less responsive to opioids, and non-opioid treatments (e.g. anticonvulsants) may be required.
C Codeine and dihydrocodeine are less potent analgesics than morphine.
D This is correct. Miosis is one of the signs of opioid abuse.
E Fentanyl is not suitable. Methadone can be used as a substitute in the detoxification process.
A Unlike the street drug, heroin for medical use is not contaminated and can be effective for severe pain in terminal cancer and acute myocardial infarction.
B Opioid analgesia occurs by central action and not by effects on histamine release in inflamed tissues.
C Fentanyl can be administered by transdermal patch due to its high potency and lipid solubility.
D The opioid antagonist naloxone can be used in opioid overdose but triggers severe withdrawal effects in opioid-addicted individuals.
E Although its oral bioavailability is relatively low, morphine can be administered by oral, rectal or parenteral routes.
Case-based answers
1. The simple analgesic paracetamol can be given in compound formulations with the opioid analgesic codeine (co-codamol) or with dihydrocodeine (co-dydramol). A non-steroidal anti-inflammatory drugs (NSAID) analgesic (diclofenac) was also given to this man. Although this regimen conforms to the WHO analgesic ladder principle of combining opioid, NSAID and simple analgesics, codeine lacks the efficacy of morphine and failed to provide adequate pain control before admission.
2. Morphine acts at specific opioid receptors at spinal and supraspinal sites to produce analgesia. Morphine is a strong agonist at µ-opioid receptors that produce analgesia, euphoria, sedation, respiratory depression, dependence and inhibition of gastrointestinal motility.
3. Oral morphine solution is used to control breakthrough exacerbations of pain on a patient-initiated basis. Repeated use of oral morphine solution should signal a reassessment of the dose of the long-acting morphine. When the person is unable to take oral medication because of weakness or vomiting, rectal or continuous subcutaneous infusion may be required. Normally, 80% of people require less than 200 mg morphine per day to control severe pain. With terminally ill people having persistent severe pain, the dose is gradually increased over a period of 1–2 weeks until appropriate control is achieved. The maximum dose may be as high as 2–3 g per day. Unwanted effects can occur so close monitoring is needed when an opioid is initiated or its dosage altered.
4. For reasons that are not easily explained, dependence seldom occurs in people with a high degree of pain. Possible reasons include high levels of endogenous opioids or catecholamines.
5. Diamorphine can be used instead of morphine. It is more potent but no more efficacious. Its major advantage is its high solubility, which reduces the volume of intramuscular injections or continuous subcutaneous infusion if these are required. Infrequently, an unusual response to morphine may require its replacement by other opioids. Fentanyl delivered via a transdermal patch has fewer unwanted effects.
6. Diclofenac is an aspirin-like NSAID often used in the treatment of arthritic conditions (Ch. 29). Unlike opioids, it has both analgesic and anti-inflammatory actions, due to its inhibition of the synthesis of prostaglandins by cyclo-oxygenase. The pain from bone metastases is compounded by local inflammation. Prostaglandins sensitise nociceptors to pain stimuli, and inflammatory swelling triggers pain due to increased pressure. Inflammation is reduced by diclofenac, thereby reducing the requirement for morphine.
7. Dexamethasone is a potent glucocorticoid (Ch. 44), reducing inflammation and swelling at metastatic sites.
8. Nausea is an unwanted effect of morphine, occurring particularly during the first week of use, but it may also be a consequence of the cancer itself or related complications such as hypercalcaemia. Tolerance to the nausea induced by morphine occurs.
9. Metoclopramide is a dopamine antagonist that acts in the chemoreceptor trigger zone (CTZ) to reduce nausea caused by opioid analgesics, chemotherapy and radiotherapy. It also has prokinetic activity on the gut that reduces the risk of nausea and vomiting. Other dopamine antagonists such as prochloperazine can also be used (Ch 32).
10. Gastric and/or duodenal inflammation (which may cause considerable discomfort) or even ulceration may occur with prolonged use of diclofenac and a corticosteroid, due to inhibition of the synthesis of protective prostaglandins in the gut wall.
11. Cimetidine is a histamine H2 receptor antagonist that reduces histamine-mediated gastric acid secretion by the parietal cells (Ch. 33), and was used twice-daily to reduce the risk of NSAID-induced gastric or duodenal ulceration. Cimetidine inhibits the enzymes that convert codeine into morphine and may reduce its analgesic effect. After admission, cimetidine was replaced with the proton pump inhibitor lansoprazole, which is more effective in blocking acid secretion and can be given once daily (Ch. 33).
12. Constipation is a feature of morphine therapy. Tolerance does not develop to opioid-induced constipation. Peristalsis is reduced, while the tone of the intestinal muscle is increased.
13. Docusate sodium has some faecal-softening properties and is a stimulant of intestinal smooth muscle, which restores peristalsis. In practice, terminally ill people are often given danthron, in combination with either docusate sodium (co-danthrusate) or poloxamer (co-danthramer). Danthron is a stimulant drug and stool softener and is particularly useful when ‘bowel movements must be by strain’. The irritant properties of danthron and its carcinogenic potential restrict its general use. The alternatives in use include senna preparations (stimulants) and magnesium sulphate (a bulk purgative).
14. Temazepam is a short-acting benzodiazepine anxiolytic sedative used to aid sleeping (Ch. 20).
Note. Pain control must also take note of the psychological, social and spiritual condition of the person. At all times, if pain control is inadequate, adjuvant treatments such as radiotherapy or transcutaneous electrical nerve stimulation (TENS) should be considered. Where neuropathic pain is evident, tricyclic antidepressants or anticonvulsants should also be considered.
Ballantyne, JC, Mao, J. Opioid therapy for chronic pain. N Engl J Med. 2003;349:1943–1953.
Bennetto, L, Patel, NK, Fuller, G. Trigeminal neuralgia and its management. BMJ. 2007;334:201–205.
Berde, CB, Sethna, NF. Analgesics for the treatment of pain in children. N Engl J Med. 2002;347:1094–1103.
Eisenberg, E, McNichol, ED, Carr, DB. Efficacy and safety of opioid agonists in the treatment of neuropathic pain of nonmalignant origin. JAMA. 2005;293:3043–3052.
Freynhagen, R, Bennett, MI. Diagnosis and management of neuropathic pain. BMJ. 2009;339:391–395.
Gonzalez, G, Oliveto, A, Kosten, TR. Treatment of heroin (diamorphine) addiction. Drugs. 2002;62:1331–1343.
Holdcroft, A, Power, I. Management of pain. BMJ. 2003;326:635–639.
Jensen, TS. Anticonvulsants in neuropathic pain: rationale and clinical evidence. Eur J Pain. 2002;6(Suppl A):61–68.
Johnson, RW, Dworkin, RH. Treatment of herpes zoster and postherpetic neuralgia. BMJ. 2003;326:748–750.
Lobmaier, P, Gossop, M, Waal, H, Bramness, J. The pharmacological treatment of opioid addiction: a clinical perspective. Eur J Clin Pharmacol. 2010;66:537–545.
Mendell, JR, Sahenk, Z. Painful sensory neuropathy. N Engl J Med. 2003;348:1243–1255.
O'Connor, AB, Dworkin, RH. Treatment of neuropathic pain: an overview of recent guidelines. Am J Med. 2009;122(Suppl 1):S22–S32.
Portenoy, RK. Treatment of cancer pain. Lancet. 2011;377:2236–2247.
Somogyi, AA, Barratt, DT, Coller, JK. Pharmacogenetics of opioids. Clin Pharm Ther. 2007;81:429–444.
Turk, DC, Wilson, HD, Cahana, A. Treatment of chronic non-cancer pain. Lancet. 2011;377:2226–2235.
Vadalouca, A, Siafaka, I, Argyra, E, et al. Therapeutic management of chronic neuropathic pain: an examination of pharmacologic treatment. Ann NY Acad Sci. 2007;1088:164–186.
Vanderah, TW. Pathophysiology of pain. Med Clin North Am. 2007;91:1–12.
Wu, CL, Raja, SN. Treatment of acute postoperative pain. Lancet. 2011;377:2215–2225.