Drug therapy in special situations
Prescribing in pregnancy
Guidelines for prescribing during pregnancy are set out in the British National Formulary (BNF). Pregnancy can be associated with medical problems that require treatment (Ch. 45), but exposure of the fetus to any unnecessary drugs is undesirable, particularly in the first trimester between the third and eleventh weeks of pregnancy, because of the risk of teratogenicity. In the second and third trimesters drugs may affect the growth or functional development of the fetus, whereas drugs given at full term may influence labour or affect the neonate after delivery. The magnitude of the potential problem is illustrated by the fact that about 50% of women take prescribed medication at some stage during pregnancy, and an unrecorded number will take over-the-counter medications, including herbal and homeopathic remedies, without guidance from a medical practitioner or a pharmacist.
Unequivocal teratogenic activity of drugs in humans is limited to a relatively small number of compounds, but the effects are irreversible and affect the whole life of the offspring. The potential catastrophic consequences of the administration of a teratogenic drug were highlighted by the thalidomide tragedy in the 1960s. Thalidomide was introduced as a sedative and hypnotic and used for the treatment of pregnancy-associated morning sickness. Following its introduction there was a dramatic increase in the incidence of phocomelia (abnormal or absent development of limb buds). The drug was banned once the association had been recognised, and this resulted in the incidence of phocomelia decreasing to previous levels (Fig. 56.1). Thalidomide is not teratogenic in rodents, and teratogenic effects are seen in rabbits only at doses about 100 times higher than those in humans or other primates; this observation resulted in the requirement for two or more animal species in preclinical testing for teratogenicity. The thalidomide tragedy led to a significant reduction in the proportion of women taking any prescription drug during pregnancy, particularly in the first trimester. The recent use of thalidomide as an unlicensed treatment for leprosy raises the spectre of teratogenicity; the drug should never be given to women with child-bearing potential.
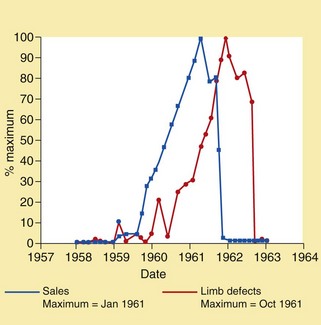
Fig. 56.1 Sales of thalidomide and the incidence of phocomelia.
Thalidomide sales and phocomelia incidence are each expressed as a percentage of the reported maximum.
The list of drugs known to be teratogenic is relatively short but includes thalidomide, many anticonvulsants, some chemotherapeutic drugs (e.g. alkylating agents and antimetabolites), warfarin, androgens, danazol, diethylstilbestrol, lithium and retinoids (Table 56.1). Because of their long half-lives, some retinoids can result in teratogenesis even if treatment for the mother is stopped before pregnancy occurs. Although teratogenesis is commonly thought of in terms of structural abnormalities or dysfunctional growth in utero, it also refers to long-term functional defects. For example, maternal consumption of alcohol during pregnancy may cause behavioural and cognitive abnormalities in childhood, despite the birth of a seemingly unaffected infant. Some drugs may initially appear harmless yet exhibit a long latency period. Diethylstilbestrol, which was given during pregnancy between the 1940s and early 1970s in the mistaken belief that it reduced the risk of miscarriage, resulted in abnormalities in the offspring when they reached adulthood, including hypogonadism in males and vaginal adenocarcinoma in females.
Table 56.1
Examples of drug-induced teratogenicity and fetal/neonatal toxicity
| Therapeutic drug | Teratogenic and adverse effects in fetus and neonate |
| ACE inhibitors | Affect fetal and neonatal blood pressure control and renal function; oligohydramnios |
| Alcohol | Fetal alcohol syndrome; growth restriction (Ch. 54) |
| Aminoglycosides | Auditory or vestibular nerve damage |
| Amiodarone | Neonatal goitre |
| Androgens | Virilisation of female fetus |
| Anti-cancer drugs | Carcinogenic and teratogenic effects (also avoid before pregnancy) |
| Barbiturates | Fetal abnormalities; withdrawal effects in neonates |
| Benzodiazepines | Withdrawal effects in neonates |
| Beta-adrenoceptor antagonists | Intra-uterine growth restriction, neonatal hypoglycaemia and bradycardia |
| Carbamazepine | Neural tube defects |
| Carbimazole | Neonatal goitre |
| Corticosteroids | Intra-uterine growth suppression (with prolonged treatment) |
| Dapsone | Neonatal haemolysis and methaemoglobinaemia |
| Diethylstilbestrol | Hypogonadism in male offspring and vaginal cancer in female offspring |
| Fibrinolytics | Premature separation of placenta in first 18 weeks |
| Lamotrigine | Teratogenicity |
| Leflunomide | Teratogenic in animals; effective contraception necessary for at least 2 years after end of treatment for women and 3 months for men |
| Lithium salts | Teratogenicity; cardiac abnormalities |
| NSAIDs | Premature closure of ductus arteriosus; pulmonary hypertension |
| Opioids | Neonatal respiratory depression and risk of withdrawal syndrome if the mother is habituated |
| Oral anticoagulants | Malformations; fetal or neonatal haemorrhage |
| Oxcarbazepine | Neural tube defects |
| Phenytoin | Congenital malformations; risk of neonatal haemorrhage due to vitamin K deficiency |
| Primaquine | Neonatal haemolysis and methaemoglobinaemia |
| Retinoids and retinoid-like drugs | Teratogenic, craniofacial malformations; some have long half-lives and effective contraception is essential for prolonged periods after stopping treatment and before pregnancy |
| Ribavirin | Teratogenic in animals; effective contraception necessary for at least 6 months after treatment for both women and men |
| Statins | Decreased cholesterol synthesis affects fetal development |
| Sulphonamides | Neonatal haemolysis and methaemoglobinaemia |
| Sulfonylureas | Neonatal hypoglycaemia |
| Thiazide diuretics | Growth retardation; electrolyte disturbance |
| Valproate | Congenital malformations and developmental delay in offspring |
Manufacturers of most drugs advise that they should be taken in pregnancy only if the potential benefit outweighs the possible risk; also, many recommend that prescribing to women of childbearing age should be carried out with pregnancy in mind and contraception should be adequate before, during and after treatment. ACE, angiotensin-converting enzyme; NSAIDs, non-steroidal anti-inflammatory drugs.
Notwithstanding the limited list of drugs that are known to cause teratogenesis, there is a much larger number that should be avoided or used with caution in pregnancy because of their potential to produce detrimental effects in the fetus. Examples include warfarin-induced anticoagulation (Ch. 11), which may predispose to cerebral haemorrhage in the fetus during delivery; in contrast, heparin is an effective anticoagulant in the mother and does not cross the placenta. Non-steroidal anti-inflammatory drugs (NSAIDs; Ch. 29) can produce premature closure of the ductus arteriosus before delivery. Adverse effects produced at therapeutic doses, such as tachycardia with tricyclic antidepressants and growth restriction with corticosteroids, may also affect the fetus or neonate.
Whenever a drug is given to a pregnant woman, or a woman who has child-bearing potential, an assessment should be made, taking into account any risk to the fetus balanced against the benefit to the mother and any risk associated with not treating the mother. For example, treatment with anticonvulsants or antimalarials may be essential for the mother, despite the possible risk to the fetus/neonate. The risk to the fetus/neonate should be minimised whenever possible by selecting the drug with the least potential for teratogenicity, by prescribing the lowest effective dose and minimizing the use of multiple drugs. Absence of evidence of teratogenicity does not imply that a risk does not exist. Drugs which have been used extensively in pregnancy and appear to be safe should nevertheless be preferred to new or untried drugs. The BNF provides detailed information on potential adverse drug effects on the fetus and neonate, and guidance is also available from the UK Teratology Information Service (www.uktis.org).
Pharmacokinetics in pregnancy
The placenta provides a potential barrier to the transfer of macromolecules from the maternal circulation, but low-molecular-weight drugs will cross the placenta, particularly if they are lipid-soluble. Some metabolism of drugs can occur in the placenta, which may further restrict fetal exposure, although the placenta does not have a high drug-metabolising capacity. The fetal liver and kidneys have only modest abilities to eliminate drugs, so drugs in the fetal circulation are usually cleared by the maternal routes of elimination. The fetus therefore represents a slowly equilibrating maternal kinetic compartment, with transfer across the placenta being determined by the concentration gradient between fetal and maternal circulations.
Maternal pharmacokinetics are affected by a number of physiological changes, especially in late pregnancy. Compared to non-pregnant women, these include:
These changes mean that maternal drug concentrations are often lower than those in a non-pregnant woman given the same dose, so drug doses may need to be increased in pregnancy to compensate.
Drugs and breastfeeding
Almost any compound present in the maternal circulation will enter breast milk and be ingested by the suckling baby. However, with a few exceptions there is little evidence that drug intake via breastfeeding is of concern because most drugs enter breast milk in quantities too small to affect the baby. In general, drugs licensed for use in children can be safely given to the nursing mother, whereas drugs known to have serious toxic effects in adults, or known to affect lactation, such as bromocriptine, should be avoided.
The American Academy of Pediatrics (2001) divides drugs into:
i. cytotoxic drugs that may interfere with cellular metabolism of the nursing infant (e.g. cyclophosphamide, ciclosporin, doxorubicin, methotrexate),
ii. drugs of abuse for which adverse effects on the infant during breastfeeding have been reported (e.g. amfetamine, cocaine, heroin, marijuana),
iii. radioactive compounds that require temporary cessation of breastfeeding (e.g. radioiodine),
iv. drugs for which the effect on nursing infants is unknown but may be of concern (a list of about 40 miscellaneous drugs),
v. drugs that have been associated with significant effects on some nursing infants and should be given to nursing mothers with close monitoring (e.g. acebutolol, atenolol, bromocriptine, aspirin, ergotamine, lithium, phenindione, phenobarbital/primidone),
vi. maternal medication usually compatible with breastfeeding (the vast majority of drugs),
The reader should refer to the up-to-date information in the BNF for detailed advice. The absence of safety information for many drugs in lactation means that only essential drugs should be given to mothers during breastfeeding.
Pharmacokinetics in lactation
Several factors influence drug transfer from the maternal circulation into breast milk, including the characteristics of the milk, the physicochemical properties of the drug and the amount of drug in the maternal circulation. The concentrations of drugs in breast milk are in equilibrium with those in the maternal circulation. At equilibrium, the free concentration in milk and plasma will be the same, but the total concentration will be influenced by the extent of protein binding and uptake into the lipid phase (see Fig. 2.3). Water-soluble drugs diffuse from plasma into milk, and the concentration in breast milk is similar to the non-protein-bound fraction in the maternal plasma. Lipid-soluble compounds diffuse into breast milk and may concentrate because of the high fat content in milk.
The effects of drugs in breast milk depend not only on maternal pharmacokinetics but also on the extent of absorption, distribution and elimination of the drug in the neonate or infant (see below). Drugs may also have different pharmacodynamic properties in neonates or infants compared to older children and adults. If drugs are given during breast feeding, compounds with short half-lives are preferred because they are less likely to accumulate in neonates, who have lower drug clearance. Neonatal exposure can also be minimised if the feed is timed to coincide with the trough blood concentration in the mother, which is just before a dose is taken. The World Health Organization (WHO) nevertheless recommends that the health and developmental benefits of breastfeeding are usually greater than any likely risk from drugs in breast milk.
Prescribing for children
Both the pharmacokinetics and responses to drugs may differ in neonates, infants and children compared with adults. There are considerable differences among neonates (<1 month), infants (1–12 months) and children, because many metabolic and physiological processes are immature at birth and develop rapidly in the first months of life. These differences may affect the absorption and distribution of drugs and the rate of elimination of the drug from the body, and also the sensitivity of tissues to its actions or adverse effects. Particular care is needed in prescribing drugs that may affect growing or maturing organ systems such as the bones and teeth and the reproductive system. Box 56.1 shows some of the differences between the young and adults.
Although medicines should usually be used within the terms of the product license, many drugs given to children have not undergone formal clinical evaluation in this age group, and are not specifically licensed for paediatric use. It is recognised that ‘off-label’ prescribing may be necessary and the UK Medicines Act (1968) does not prohibit such unlicensed use. There is an increasing recognition of the need for formal clinical trials in the paediatric population, but such studies raise significant ethical issues. The BNF for Children (www.bnf.org/bnf/index.htm) gives specific guidance on prescribing for children in the UK and further information on the regulation of paediatric medicines is available at the following places.
UK: www.mhra.gov.uk/Howweregulate/Medicines/Medicinesforchildren/index.htm
EU: http://ec.europa.eu/health/human-use/paediatric-medicines/index_en.htm
Pharmacokinetics in neonates and children
In neonates, inefficient metabolism and renal clearance mean that lower doses of some drugs are needed after allowing for body weight, and doses need to be calculated with special care. The processes of drug elimination are largely mature by a few weeks of age, after which drug clearance (adjusted to body weight) is similar to or higher than that in adults (see below). However, children may be more susceptible to effects on growing or maturing tissues and organs. Generalisations are difficult and each drug needs to be considered in its own right.
Absorption
Slow rates of gastric emptying and intestinal transit may reduce the rate of drug absorption in neonates, but total absorption of poorly absorbed drugs may eventually be more complete because of longer contact with the intestinal mucosa. In the neonate, gastric pH is neutral and this can reduce the absorption of weak acids but increase the absorption of weak bases.
Distribution
Neonates and young children have a lower body fat content and higher total body water than adults; this influences the distribution of both lipid- and water-soluble drugs. Neonates have a lower plasma albumin concentration, which also has a lower affinity for drug binding. In addition, the higher plasma concentrations of free fatty acids and bilirubin compete with drugs for plasma protein-binding sites. The overall effect is reduced plasma protein binding, which not only increases the apparent volume of distribution of the drug but also increases the proportion of drug able to cross the blood–brain barrier; it also increases the amounts diffusing into the liver and therefore available for metabolism. Drugs that are strongly bound to albumin should not be used during neonatal jaundice because the drugs may displace bilirubin (which is mostly in the unconjugated form) from protein-binding sites and increase the risk of kernicterus.
Metabolism
The drug-metabolising enzyme systems are immature in the neonatal liver, and first-pass metabolism and hepatic drug clearance are low, especially for substrates of CYP1A2, CYP3A4 and glucuronidation. The clearances for substrates for these enzymes are two to six times lower in neonates compared with adults. When the enzyme systems mature, drug metabolism processes become more extensive. Plasma drug clearance is often higher in young children than in adults, because of their higher relative liver mass and greater hepatic blood flow per kilogram of body weight; hepatic blood flow is the rate-limiting step in the elimination of high-clearance drugs.
Renal elimination
Renal function in the neonate and infant is much less developed than in children or adults. The glomerular filtration rate in the newborn is about 40% of the adult level, and tubular secretory processes are poorly developed. Elimination of drugs such as digoxin, gentamicin and penicillin will therefore be slower until about 6–8 months of age.
In children, the larger volume of distribution and faster hepatic elimination mean that doses of metabolised drugs need to be higher than in adults after correcting for the difference in body weight. Prescribed doses are most accurately judged by considering both age and body surface area. In children, body surface area is a better guide to appropriate drug dosage than body weight. It can be estimated from a nomogram or by using the Du Bois formula:
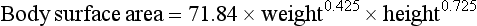
where body surface area is in square metres, weight in kilograms and height in metres. The drug dose for a child can be then approximated as:
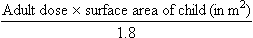
where 1.8 m2 is the average body surface area of a 70 kg adult.
Prescribing for the elderly
The elderly (usually taken to mean those over 70 years old) comprise a heterogeneous group who show considerable variation in ‘biological’ age. Changes occur in both the pharmacodynamics and pharmacokinetics of drugs with increasing age.
The density or numbers of receptors may be reduced with age; for example, β-adrenoceptors decrease in number, reducing the response to agonist drugs. The elderly are often more susceptible to sedatives, hypnotics and antipsychotic drugs, possibly because of changes in receptor numbers and/or reduced efficiency of the blood–brain barrier. They are also more susceptible to the adverse effects of NSAIDs on the gut.
Altered structure and function of target organs can also influence the effects of drugs. For example, baroreceptor function is impaired in the elderly and vasodilator drugs are more likely to provoke postural hypotension. The high peripheral resistance and less distensible arterial tree found with increasing age also respond less well to arterial vasodilators.
These changes reflect the ageing process itself, but they are often complicated by the presence of chronic disease (frequently involving multiple pathological processes) and variation due to both genetic and environmental influences. The risks of unwanted effects are higher in the elderly as a consequence of these changes. Significant numbers of hospital admissions in the elderly are due to adverse drug reactions, most of which are the more predictable type A effects (see Ch. 53). In addition, drug interactions are more common in the elderly because of the coexistence of different treatable conditions requiring the simultaneous use of several drugs (polypharmacy). For these reasons, it is usual to start drug treatment in the elderly with the smallest effective dose. Rational prescribers should also seek to minimise the numbers of drugs used, with clear explanations of usage instructions and regular review of drug regimens.
Pharmacokinetics in the elderly
Drug absorption across the gut wall is not greatly affected by ageing, although bioavailability may be increased due to reduced first-pass metabolism.
Distribution
Older people tend to have a lower lean body mass and a relative increase in body fat compared with young adults. The apparent volume of distribution (Vd) of water-soluble drugs such as digoxin may therefore be lower in the elderly and a smaller loading dose would be needed. Conversely, lipid-soluble drugs may be eliminated more slowly because of their increased Vd resulting from increased body fat and reduced hepatic metabolism.
Metabolism
The size of the liver and its blood flow decrease with age. Although enzyme activity per hepatocyte probably shows little change, the overall capacity for drug metabolism, particularly phase 1 metabolic reactions (Ch. 2), is reduced. This is particularly important for lipid-soluble drugs, such as nifedipine or propranolol, which undergo extensive first-pass metabolism, because lower hepatic metabolism increases bioavailability and reduces systemic clearance.
Renal elimination
Increasing age is also associated with a progressive reduction in glomerular filtration rate (GFR), so the elimination of polar drugs and metabolites is slower. This can produce toxicity when renally eliminated drugs with a low therapeutic index are prescribed in the elderly, for example lithium, digoxin or gentamicin. Creatinine clearance, which is an estimate of GFR, usually correlates well with the clearance of drugs that are eliminated in the urine unchanged as the parent drug. Reduced muscle mass in elderly people results in reduced creatinine production. Plasma creatinine in the elderly therefore frequently remains within the normal laboratory reference range even when renal function is substantially reduced, because the decreased creatinine production balances its reduced elimination. The Cockcroft and Gault equation, which relates plasma creatinine to creatinine clearance, contains elements reflecting sex- and age-dependent differences in muscle mass.
Creatinine clearance (mL·min–1) for males equals:
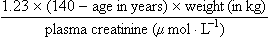
and for females it equals:
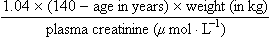
As an alternative, the eGFR (see below) is used to approximate GFR.
Prescribing in renal failure
Individuals with renal failure show increased responses to many drugs, especially when the drug, or its active metabolite, is eliminated in the urine. The extent to which dose adjustment is necessary depends on the extent of renal impairment and on the proportion of total plasma clearance that is due to renal clearance. The estimated glomerular filtration rate (eGFR) is often reported with laboratory estimations of serum creatinine. This is a useful guide to renal function, but does not consider weight as a variable that affects GFR. Nevertheless, for most drugs that are excreted by the kidney it is an adequate guide for dosage adjustment.
There are also pharmacodynamic changes in renal failure; for example, there are altered responses to drugs in people with uraemia, and drugs acting on the central nervous system (CNS) in particular produce enhanced responses, possibly because of increased permeability of the blood–brain barrier.
The BNF gives advice on drug prescribing to those with renal impairment. People with renal impairment may show an abnormal drug response due to one or more of the following factors:
 failure to excrete the drug or its metabolites may produce toxicity,
failure to excrete the drug or its metabolites may produce toxicity,
there may be increased sensitivity, even if elimination is unaltered,
many unwanted effects are poorly tolerated in such individuals,
Pharmacokinetics in renal failure
The activity of most drugs is not affected by impaired renal function, because most drugs are cleared by hepatic metabolism, but the kidneys provide the major route of elimination for water-soluble drugs and water-soluble metabolites (see Ch. 2). Renal elimination of drugs can be affected indirectly by abnormal renal perfusion, such as might occur in shock, or directly by changes in the kidney, for example renal tubular necrosis. Reduced renal function may increase the risk of toxicity from the parent drug or its metabolites due to their accumulation in the body, or toxicity may arise due to increased sensitivity in renal failure without an obvious impairment in drug elimination.
There are several other ways in which renal impairment may influence the handling of drugs.
Metabolism in the liver can be impaired in people with uraemia, particularly metabolic reactions involving reduction, acetylation and ester hydrolysis.
The kidney has important metabolic activities, such as the 1-α-hydroxylation of vitamin D and the degradation of insulin, both of which can be impaired in renal failure.
The distribution of drugs can be affected by changes in fluid balance in renal failure, and more importantly by altered protein binding (see below). Circulating concentrations of albumin are decreased in severe renal failure with proteinuria. In addition, retained endogenous metabolites, such as the tryptophan metabolite indican, may compete for drug-binding sites on plasma proteins. The increased concentrations of free drug can lead to an enhanced response or to its increased elimination by glomerular filtration or metabolism.
Tissue binding of digoxin is reduced in renal failure, so a lower loading dose should be given to compensate for the reduced volume of distribution.
The elimination of drugs by the kidney is significantly impaired only when the glomerular filtration rate is reduced below 50 mL⋅min−1. For some drugs, clinically important accumulation does not occur until much lower filtration rates. Changes in renal tubular secretion of drugs in renal disease are less well understood.
A reduction in drug dosage in renal failure is usually necessary only if a high proportion of the drug is eliminated by the kidney and when the compound has a low therapeutic index. Maintenance dosage may be lowered by either reducing the dose or increasing the dose interval (see Ch. 2, equation 2.24). Loading doses do not usually require any modification. Large dose modifications are rarely needed for drugs that do not have dose-related unwanted effects. For the purposes of prescribing and dosage adjustment in renal impairment, the BNF uses the classification of Chronic Kidney Disease based on eGFR values (in mL/min/1.73 m2):
stage 1 (normal): eGFR >90 (with other evidence of kidney damage),
stage 2 (mild): eGFR 60–89 (with other evidence of kidney damage),
For some drugs, only established renal failure (stage 5) needs to be considered (for example, a reduction in dosage is recommended for ampicillin), while for other drugs even mild impairment (stage 2) may be important (for example, dosage reduction is recommended for carboplatin, whereas cisplatin should be avoided).
A further important consideration is the avoidance of drugs that have toxic effects on the kidney. Use of these in renal impairment can sometimes produce an irreversible decline in renal function.
Prescribing in liver disease
Changes in both drug responses and pharmacokinetics can occur in liver disease. The BNF lists six main potential problems in prescribing for individuals with liver disease:
The severity of the liver disease is important, as is whether the disease is decompensated and includes jaundice, hypoproteinaemia or encephalopathy. Many of the pharmacodynamic and pharmacokinetic changes in liver failure arise from decreased hepatic synthesis of proteins that perform essential functions within the hepatocyte, or which are released into the blood, such as albumin and clotting factors.
CNS-depressant drugs such as morphine and chlorpromazine have an enhanced effect in people with liver failure. This is caused by increased sensitivity of neuronal tissue and can provoke encephalopathy in susceptible people. Decreased plasma protein binding may contribute to this greater sensitivity by increasing the percentage of free drug so that more drug crosses the blood–brain barrier. Benzodiazepines used during investigational procedures in individuals with liver failure can produce profound and long-lasting effects, which may require reversal by the administration of the benzodiazepine antagonist flumazenil.
Encephalopathy may be triggered by drugs that cause constipation (which increases the formation of potentially toxic metabolites, such as ammonia, by the intestinal bacteria). Diuretics that produce hypokalaemia can also precipitate hepatic encephalopathy in chronic liver disease. Therefore, potassium-sparing diuretics such as spironolactone are usually preferred to diuretics such as furosemide; an additional advantage of spironolactone is that it blocks the effects of circulating aldosterone, which is often increased in decompensated liver disease.
The reduced ability to synthesise vitamin K-dependent clotting factors makes people with chronic liver disease prone to clotting problems; they would be very sensitive to anticoagulant drugs, which are clearly contraindicated.
People with pre-existing liver disease are likely to be more susceptible to hepatotoxic drugs. This raises a problem for pain relief, since paracetamol is hepatotoxic at high doses, whereas NSAIDs can increase the risk of gastrointestinal bleeding and cause fluid retention, while opioids can precipitate encephalopathy. In practice, lower doses of paracetamol are usually given, taking care that the amounts do not exceed the reduced threshold for hepatotoxicity shown by such individuals (see Ch. 53).
Pharmacokinetics in liver disease
The rate of absorption of drugs from the gut lumen is not greatly affected in liver disease, but other aspects of drug handling may be altered. Distribution of drugs may be affected if synthesis of albumin is reduced, resulting in a higher percentage of free drug in plasma and possibly a greater risk of toxicity; examples of highly protein-bound drugs are phenytoin and prednisolone. Free drug concentrations may also be increased by elevated plasma bilirubin, which can displace drugs such as lidocaine and propranolol from their plasma protein-binding sites.
The liver has characteristics that facilitate the rapid and extensive uptake and metabolism of lipid-soluble drugs (Fig. 56.2). These include:
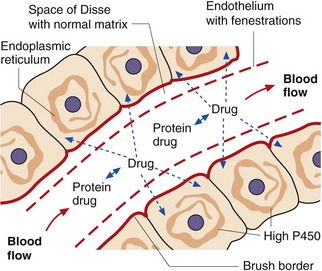
fenestrations in the endothelium, allowing ready access to extracellular fluid,
rapid diffusion across the space of Disse (which is a matrix consisting primarily of type 4 collagen),
a brush border on hepatocytes, allowing rapid uptake,
high intracellular enzyme activity for both phase 1 and phase 2 metabolism.
During chronic liver disease, a number of changes may occur that reduce the capacity of the liver to metabolise drugs (Fig. 56.3):
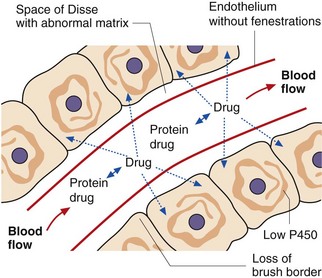
fenestrations in the endothelium are lost,
diffusion across the space of Disse may be reduced in fibrosis/cirrhosis as type 4 collagen is replaced by type 1 and type 3 collagen (which can form dense fibrils),
the brush border on hepatocytes is lost,
intracellular enzyme activity is reduced,
intrahepatic vascular shunts may reduce the perfusion of hepatocytes.
Reduced hepatic uptake and metabolism or decreased biliary excretion of drugs may result in a greater proportion of the drug or its metabolites being eliminated by other routes, such as the urine.
First-pass metabolism may be considerably reduced in conditions such as liver cirrhosis; the consequences are most apparent with drugs that normally undergo extensive hepatic first-pass metabolism. In liver failure, their bioavailability may increase considerably from <20% to almost 100%.
Biliary excretion is impaired in conditions causing reduced formation of bile. A correlation between drug clearance and serum bilirubin would be expected for drugs eliminated unchanged in bile, such as rifampicin and fusidic acid. Reduced biliary elimination of drug metabolites could affect enterohepatic circulation (Ch. 2). Reduced bile production can affect the absorption of highly lipid-soluble molecules, such as the fat-soluble vitamins, that require micelle formation for effective absorption.
Systemic clearance may be reduced for drugs eliminated by hepatic metabolism. The changes that occur in liver disease affect both high-clearance drugs, where the elimination rate is dependent on effective liver blood flow, and low-clearance drugs, where it is dependent on hepatic extraction and enzyme activity.
Prescribing in liver disease should be undertaken with care, and drugs that are extensively metabolised by the liver should be given in smaller doses. The need for dose reduction arises primarily from an increase in bioavailability and a decrease in systemic clearance, both of which increase the average steady-state plasma concentration.
Prescribing in palliative care
Symptom relief in palliative care often presents challenges to the prescriber, and the evidence to guide choice of treatments is often derived from experience rather than controlled trials. Palliative care services produce comprehensive guidelines for symptom control, often advising use of drugs for unlicensed indications. Awareness of psychological, emotional and social contributors to symptoms will help to guide strategies for management. The following synopsis is not comprehensive, but covers an approach to some key symptoms.
Pain
Accurate diagnosis of the cause of pain is essential for a rational approach to therapy. The principles of the WHO analgesic ladder apply (Ch. 19). Analgesics should be given regularly, and preferably by mouth with additional methods of pain control considered in all cases. These may include co-analgesics for neuropathic pain (Ch. 19), surgery, radiotherapy, nerve blocks, TENS, acupuncture and addressing psychological problems.
If a strong opioid is needed, then immediate-release morphine every 4 h is preferred initially, increasing the dose by 30–50% every 2–3 days as required. When pain control is achieved, modified-release morphine every 12 h can be used (giving the same total daily dose), with immediate-release morphine for breakthrough pain. Continuing pain despite persisting unwanted effects such as drowsiness suggests that the pain is not fully opioid-responsive. A laxative should always be given with a strong opioid.
Alternative opioids for palliative care include oxycodone or hydromorphone (which have a slightly different unwanted effect profile), diamorphine for higher doses by subcutaneous infusion (can be given in a smaller volume of fluid), methadone (particularly for neuropathic pain) or fentanyl (for transcutaneous use or in severe chronic kidney disease). Transdermal delivery of an opioid can be helpful if there is vomiting, intractable constipation or other unwanted effects in the presence of opioid-responsive pain. Care must be taken to give equivalent doses when changing from one opioid to another.
Nausea and vomiting
It is important to identify the cause if possible, since this will guide treatment. Drug therapy should always be considered as a cause of vomiting, and the responsible drug stopped or the dose reduced if possible. Non-drug measures include psychotherapeutic techniques, acupuncture, acupressure and ginger.
If drug therapy is required, then the choice will depend on the predominant contributory causes (Ch. 32). Examples include dexamethasone for raised intracranial pressure, levomepromazine or benzodiazepines for anxiety, metoclopramide or prochlorperazine for drug-related vomiting, metoclopramide for gastric stasis, and metoclopramide, levomepromazine or cyclizine if the cause is not clear.
Anorexia/cachexia/fatigue syndrome
This syndrome arises in terminal cancer, heart failure and with chronic infection or inflammation. There is usually profound loss of weight and muscle bulk. It is often not possible to deal with causative factors, and management may include dexamethasone to suppress inflammation, methylphenidate, or modafanil (Ch. 22) for fatigue and encouraging exercise.
Constipation
Constipation may arise from drug therapy (e.g. opioids, antidepressants, antispasmodics, ondansetron), inactivity, dehydration, hypercalcaemia or concurrent disease. Adequate fluid intake is important and if the underlying cause is not amenable to treatment then symptomatic treatment should be given (Ch. 35). Macrogols are usually used for opioid-induced constipation, although a stimulant such as senna may also be needed.
Breathlessness
Breathlessness is often multifactorial, and specific treatments may be successful. If there is no treatable cause then nebulised saline can help to loosen secretions. Morphine, sometimes together with a benzodiazepine such as diazepam, can reduce the subjective sensation of breathlessness.
Hiccups
Hiccups can have a peripheral cause such as gastric distention, diaphragmatic irritation, liver enlargement or intrathoracic tumour. A variety of treatments have been advocated, indicating that all have limited efficacy. Options include metoclopramide, domperidone, a proton pump inhibitor, dexamethasone, baclofen and nifedipine. Central causes include raised intracranial pressure and uraemia, and treatment options include dexamathasone, levomepromazine, haloperidol and diazepam.
Use of a syringe driver
Near the end of life drugs may need to be given by subcutaneous infusion via a small battery-powered pump. Maintaining steady plasma drug concentrations may aid symptom control or give relief in someone who cannot swallow. Examples of drugs given by this route are:
cyclizine, haloperidol or metoclopramide for vomiting,
dexamethasone for neuropathic pain, raised intracranial pressure or vomiting,
morphine, oxycodone or diamorphine for pain control,
glycopyrronium to reduce respiratory secretions,
hyoscine to relieve intestinal colic and to reduce secretions,
Drug interactions
Many people take more than one drug during a course of treatment because:
combination therapy is preferable or necessary for producing an adequate effect or response; important examples are the chemotherapy of malignant disease and the treatment of hypertension,
a single condition or pathology may give rise to a variety of symptoms that are controlled by different drugs,
the person may suffer from more than one condition or pathology requiring treatment with drugs that are unrelated pharmacologically.
The term ‘interaction’ implies that the response to the combination of drugs is different to that which could be predicted from a simple summation of the effects of each drug given singly.
The consequences of treatment with a combination of drugs can be divided into four different types:
dose-addition, where each drug produces the same response and the magnitude of response to the combination of both drugs is given by simple addition of the doses, after allowing for any difference in potency; this is the usual situation when more than one drug is used to treat a single condition,
response-addition, where each drug produces a different response and their combination gives each response as if the other drug were not present; this is the usual situation when two drugs are used to treat two different conditions,
synergism, where the magnitude of response to a drug combination is greater than would be predicted by simple addition of the separate drug doses, after allowing for any difference in potency; synergism is often produced when the drugs have different mechanisms or act at different steps in the process leading to the overall response,
antagonism, where the magnitude of response to a drug combination is lower than would be predicted by simple addition of the doses, after allowing for any difference in potency; this sort of interaction can occur if a partial receptor agonist is given with a full agonist at the same receptor and reduces the overall activity.
Interactions that result in antagonism or synergism may be beneficial, or potentially harmful because of a lack of clinical response or the risk of toxicity. Beneficial interactions are usually well characterised and have clear advantages – for example, combinations of different anti-cancer drugs – and are the basis of prescribing recommendations. This section therefore focuses on adverse interactions, which are of greatest importance for drugs that have a narrow therapeutic index and for groups of people at increased risk, such as the elderly.
Drug interactions may arise at the site of the mechanism of action (pharmacodynamic) or from altered delivery of the drug to its site of action (pharmacokinetic).
Pharmacodynamic interactions
Pharmacodynamic interactions are usually predictable based on the known mechanisms of action of the drugs. Interactions may relate to the principal site of action of the drug, or to secondary sites of action that are responsible for unwanted effects. In principle, drugs that are highly selective for a single site of action are less likely to produce pharmacodynamic interactions than drugs that show low selectivity. An example of a serious adverse synergistic interaction is between an angiotensin-converting enzyme (ACE) inhibitor, such as enalapril (Ch. 6) and spironolactone (Ch. 14); the ACE inhibitor reduces the production of aldosterone, thereby reducing the excretion of K+, an effect which is exaggerated by the action of spironolactone, and the combination can cause potentially life-threatening hyperkalaemia.
Pharmacokinetic interactions
Co-administration of two drugs could give an interaction if one drug affects the rate or extent of absorption of the other drug. Changes in the rate of absorption, for example by increasing or decreasing gastric emptying or intestinal motility, will affect the peak concentration, but not usually the extent of absorption. Interactions affecting the extent of absorption are usually more important; examples include the retention of drugs in the gut lumen (e.g. tetracycline antibiotics bind to divalent or trivalent metals, such as Ca2+ or Fe3+, to form complexes that are not absorbed) and the inhibition or induction of first-pass metabolism of drugs in the gut lumen, gut wall or liver.
Distribution
The main interactions affecting drug distribution arise from competition for the non-specific binding sites on plasma proteins, such as albumin (see Table 2.3). Interactions affecting plasma protein binding may be clinically relevant when:
the displaced drug is highly protein bound; for example, if competition for protein-binding sites reduces binding from 98 to 96%, this will double the free drug concentration in plasma (from 2 to 4%); in contrast, a 2% change in the binding of a drug that is only 50% bound would not be clinically or biologically significant,
the displaced drug has a narrow therapeutic index, so that a two- to three-fold change in free drug concentration greatly increases the risk of drug toxicity,
the displaced drug has a low apparent volume of distribution, such that the plasma contains a significant proportion of the total body load,
the displacing drug is of such low potency that large doses must be given and the number of protein-binding sites becomes limiting.
Metabolism
Perhaps surprisingly, the simultaneous administration of two drugs that share a common pathway of metabolism rarely causes an interaction. This is because therapeutic drug concentrations are usually far below the Km values of the metabolising enzymes, such as cytochrome P450. The enzymes therefore do not become saturated and first-order kinetics (Ch. 2) still apply. An exception is the zero-order (saturated) metabolism of ethanol and methanol by alcohol dehydrogenase; this allows ethanol to be used to slow the metabolism of methanol to its toxic products and reduce the risk of blindness (Ch. 53).
Important interactions can occur, however, when one drug induces or inhibits the enzymes involved in the metabolism of another drug. This is well recognised for drugs affecting the cytochrome P450 enzyme system because of the large number of P450 isoenzymes and their importance for the elimination of most drugs (Table 2.7). Induction or inhibition of hepatic enzymes can affect both systemic clearance and first-pass metabolism after oral dosage.
Enzyme inhibition occurs as soon as the inhibiting drug concentration is sufficiently high, and can occur after a single dose (e.g. cimetidine). In contrast, enzyme induction requires a few days as it results from gene transcription and translation of additional enzyme; the increased enzyme activity then reduces the concentrations of the other drug. This may decrease the clinical response to the second compound, if it is an active drug, or it could increase the bioactivation of a prodrug to an active metabolite. When dosage with an enzyme-inducing drug is stopped, the enzyme activity usually declines over a period of 2–3 weeks. If the dosage of the second drug has been optimised for the drug combination, its plasma concentration may then increase markedly, giving a risk of toxicity.
Excretion
Each of the three processes involved in the renal elimination of drugs – that is, glomerular filtration, pH-dependent reabsorption and renal tubular secretion – can be a site for drug interactions.
Glomerular filtration depends on renal perfusion and only removes free drug (not protein-bound). In consequence, drugs affecting renal perfusion or plasma protein binding can give rise to interactions.
pH-dependent reabsorption could be altered by drugs that affect urine pH, either directly or via metabolic effects; for example, the pH changes associated with aspirin overdose can affect the excretion of drugs taken concurrently.
Renal tubular secretion can give rise to interactions when there is competition for the transporter system. Aspirin can interfere with the transport of both endogenous compounds (e.g. uric acid) and drugs (e.g. methotrexate).
The biliary excretion of drugs is not an important site for drug interactions, but the enterohepatic cycling of drugs can be affected by the co-administration of poorly absorbed broad-spectrum antibacterials, which affect the hydrolysis of drug conjugates in the lower bowel (Fig. 2.13).
True/false questions
1. The highest risk of teratogenicity is during the final trimester of pregnancy.
2. Drug doses may need to be increased in pregnancy.
3. After correction for body weight, drug doses in children are the same as for adults.
4. The bioavailability of lipid-soluble drugs is increased in the elderly.
5. Most drugs are not affected by impaired renal function.
6. Drugs that cause constipation can trigger encephalopathy in people with chronic liver disease.
7. Drugs that delay gastric emptying reduce the extent of absorption of other drugs.
8. Enzyme inducers can enhance clinical responses to prodrugs.
1. False. The greatest risk of teratogenicity is during organogenesis in the first trimester.
2. True. While only the lowest effective doses of essential drugs should be used in pregnancy, these may need to be higher than in non-pregnant women, due to increased volume of distribution and higher clearance.
3. False. Correction for body weight may underestimate drug doses in children due to their relatively high hepatic clearance; correction by body surface area is a better guide.
4. True. Lipid-soluble drugs are typically cleared by hepatic metabolism; lower hepatic blood flow in the elderly can reduce first-pass metabolism and increase oral bioavailability.
5. True. Most drugs are cleared by hepatic metabolism, so the plasma concentration of the parent drug is not affected by impaired renal function.
6. True. In chronic liver disease, constipation can increase the risk of encephalopathy from the generation of ammonia and other toxic products by the gut flora.
7. False. Delayed gastric emptying will slow the rate of absorption of most drugs and reduce their peak plasma concentrations, but the extent of absorption is not usually affected.
8. True. Bioconversion of a prodrug to its active derivative may be enhanced by an enzyme inducer.
American Academy of Pediatrics. Committee on Drugs. The transfer of drugs and other chemicals into human milk. Pediatrics. 2001;108:776–789.
Bressler, R, Bahl, JJ. Principles of drug therapy for the elderly patient. Mayo Clin Proc. 2003;78:1564–1577.
Briggs, GG, Freeman, RK, Yaffe, SJ. Drugs in Pregnancy and Lactation. A Reference Guide to Fetal and Neonatal Risk, 5th edn. Baltimore: Williams and Wilkins; 1998.
Cresswell, KM, Fernando, B, McKinstry, B, Sheikh, A. Adverse drug events in the elderly. Br Med Bull. 2007;83:259–274.
Dickinson, BD, Altman, RD, Nielsen, NH, Sterling, ML, Council on Scientific Affairs, American Medical Association. Drug interactions between oral contraceptives and antibiotics. Obstet Gynecol. 2001;98:853–860.
Dorne, JLCM, Walton, K, Renwick, AG. Human variability in xenobiotic metabolism and pathway-related uncertainty factors for chemical risk assessment: a review. Food Chem Toxicol. 2005;43:203–216.
Ito, S. Drug therapy for breast-feeding women. N Engl J Med. 2000;343:118–126.
Johnson, TN. The development of drug metabolising enzymes and their influence on the susceptibility to adverse drug reactions in children. Toxicology. 2003;192:37–48.
Koren, G, Pastuszak, A, Ito, S. Drugs in pregnancy. N Engl J Med. 2000;338:1128–1137.
Mallet, L, Spinewine, A, Huang, A. Prescribing in elderly people 2. The challenge of managing drug interactions in elderly people. Lancet. 2007;370:185–191.
Nunn, T, Williams, J. Formulation of medicines for children. Br J Clin Pharmacol. 2005;59:674–676.
O'Mahony, D, Gallagher, PF. Inappropriate prescribing in the older population: need for new criteria. Age Ageing. 2008;37:138–141.
Patsalos, PN, Perucca, E. Clinically important drug interactions in epilepsy: interactions between antiepileptic drugs and other drugs. Lancet Neurol. 2003;2:473–481.
Routledge, PA, O'Mahony, MS, Woodhouse, KW. Adverse drug reactions in elderly patients. Br J Clin Pharmacol. 2004;57:121–126.
Spina, E, Scordo, MG, D'Arrigo, C. Metabolic drug interactions with new psychotropic agents. Fundam Clin Pharmacol. 2003;17:517–538.
Spinewine, A, Schmader, KE, Barber, N, et al. Prescribing in elderly people 1. Appropriate prescribing in elderly people: how well can it be measured and optimised? Lancet. 2007;370:173–184.
Stephenson, T. The medicines for children agenda in the UK. Br J Clin Pharmacol. 2006;61:716–719.
Turnheim, K. When drug therapy gets old: pharmacokinetics and pharmacodynamics in the elderly. Exp Gerontol. 2003;38:843–853.