DISORDERS OF THE ACCESSORY ORGANS OF DIGESTION
The accessory organs of digestion (liver, gallbladder, pancreas) secrete substances necessary for digestion and, in the case of the liver, carry out metabolic functions needed to maintain life. Inflammatory disease is a common cause of accessory organ dysfunction. Inflammation disrupts secretory function and prevents secretions from flowing into the duodenum. Lack of accessory organ secretions is a major cause of maldigestion and malabsorption in the small intestine. Other causes of accessory organ dysfunction are obstruction of ducts by aggregates in the secretions themselves (e.g., obstruction of bile flow by gallstones) or by tumors. (Cancers of the digestive tract are described at the end of this chapter.)
Clinical Manifestations of Liver Disorders
Of all the accessory organ disorders, acute or chronic liver disease leads to the most systemic, life-threatening complications. These complications include portal hypertension, ascites, hepatic encephalopathy, jaundice, and hepatorenal syndrome.
Portal Hypertension
Portal hypertension is abnormally high blood pressure in the portal venous system primarily caused by resistance to portal blood flow. Pressure in this system is normally 3 mmHg; portal hypertension is an increase to at least 10 mmHg. The portal veins carry blood from the gastrointestinal tract, pancreas, and spleen to the liver. In the liver the blood flows through the sinusoids and empties into the hepatic veins, which carry it into the inferior vena cava. The inferior vena cava delivers blood to the right atrium. The portal veins, sinusoids, and hepatic veins comprise the portal venous system (see Chapter 38).
PATHOPHYSIOLOGY Portal hypertension is caused by disorders that obstruct or impede blood flow through any component of the portal venous system or vena cava. Intrahepatic causes result from vascular remodeling with intrahepatic shunts, thrombosis, inflammation, or fibrosis of the sinusoids, as occurs in cirrhosis of the liver, viral hepatitis, or schistosomiasis (a parasitic infection). Posthepatic causes occur from hepatic vein thrombosis or cardiac disorders—such as failure of the right side of the heart or constrictive pericarditis—that impair the pumping ability of the right heart. This causes blood to back up and increase pressure in the portal system. Thrombosis or narrowing of the portal vein is the major prehepatic cause. The most common cause of portal hypertension is obstruction caused by cirrhosis of the liver (see p. 1491), the formation of intrahepatic arteriovenous shunts, and increased portal blood flow from anterior splanchnic vasodilation.229
High pressure in the portal veins causes collateral vessels to open between the portal veins and the systemic veins, in which blood pressure is considerably lower (Figure 39-16). This enables blood to bypass the obstructed portal vessels. The collateral veins develop in the esophagus, anterior abdominal wall, and rectum. High pressure and increased flow volume are transmitted through these veins from the portal to the systemic venous circulation.
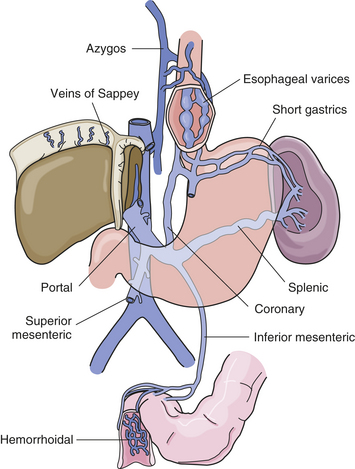
Figure 39-16 Varices related to portal hypertension. Portal vein, its major tributaries, and the most important shunts (collateral veins) between the portal and caval systems. (From Monahan FD et al: Phippss’ Medical-surgical nursing: concepts and clinical practice, ed 8, St Louis, 2007, Mosby.)
Hepatopulmonary syndrome and portopulmonary hypertension are common complications of portal hypertension. Arterial pulmonary hypertension develops as a result of microvascular intrapulmonary vasodilation caused by release of nitric oxide and carbon monoxide in the presence of liver injury.230 There are no specific clinical manifestations, although dyspnea, cyanosis, and digital clubbing may occur. Diagnosis is made by contrast echocardiography and right heart catheterization when pulmonary artery pressure is 730 mmHg by echocardiography. Treatment may include systemic vasodilators and endothelin receptor antagonists, which can reduce pulmonary and portal hypertension231 and improve outcome of liver transplant.
Long-term portal hypertension causes several problems that are difficult to treat and can be fatal:
1. Varices (distended, tortuous, collateral veins): prolonged elevation of pressure in collateral veins causes their transformation into varices, particularly in the lower esophagus and stomach but also in the rectum.
2. Splenomegaly (enlargement of the spleen): caused by increased pressure in the splenic vein, which branches from the portal vein.
3. Ascites (the accumulation of fluid in the peritoneal cavity, which is the space between the visceral peritoneum and the parietal peritoneum): caused in part by increased pressure in the mesenteric tributaries of the portal vein. Hydrostatic pressure forces water out of these vessels and into the peritoneal cavity. (This process, termed transudative effusion, is described in Chapter 33.)
4. Hepatic encephalopathy (also called portosystemic encephalopathy): characterized by central nervous system disturbances with astrocyte changes that lead to alterations of consciousness.
CLINICAL MANIFESTATIONS The vomiting of blood from bleeding esophageal varices is the most common clinical manifestation of portal hypertension.232 Slow, chronic bleeding from varices causes anemia or melena. Usually the bleeding is from varices that have developed slowly over a period of years.
Rupture of esophageal varices causes hemorrhage and voluminous vomiting of dark blood. The ruptured varices are usually painless. Rupture is caused by a combination of erosion by gastric acid and elevated venous pressure. Hemorrhoidal varices present as hematochezia and copious rectal bleeding. Mortality from ruptured esophageal varices ranges from 30% to 60%. Recurrent bleeding of esophageal or gastric varices indicates a poor prognosis. Most individuals die within 1 year.
EVALUATION AND TREATMENT Diagnosis of portal hypertension is often made at the time of variceal bleeding and confirmed by endoscopy and evaluation of portal venous pressure. Distended collateral veins may radiate over the abdomen, giving rise to caput medusae (Medusa’s head) from opening of the paraumbilical veins. The individual usually has a history of jaundice, hepatitis, or alcoholism.
Beta-blockers can be effective in preventing variceal bleeding. Emergency management of bleeding varices includes fluid resuscitation, prophylactic antibiotics, vasoactive drugs, endoscopic variceal band ligation, compression of the varices with an inflatable tube or balloon, and injection of a sclerosing agent.233,234 Surgical construction of transjugular intrahepatic portosystemic shunts (TIPS) (anastomosis of the portal vein to the inferior vena cava) may decompress the varices, but this treatment can precipitate encephalopathy or liver failure resulting from reduced hepatic blood flow. There is no effective, definitive treatment for portal hypertension. Liver transplant is the most successful option for liver failure.235
Splenomegaly
Splenomegaly is an enlargement of the spleen. Portal hypertension contributes to congestive splenomegaly by increasing intrasplenic blood pressure. Thrombocytopenia (decreased platelet count) is the most common manifestation of congestive splenomegaly and can contribute to an increased bleeding tendency. Splenomegaly also can be predictive of esophageal varices severity.236
Ascites
Ascites is the accumulation of fluid in the peritoneal cavity. Ascites traps body fluid in a “third space” from which it cannot escape. The effect reduces the amount of fluid available for normal physiologic functions. Cirrhosis is the most common cause of ascites; ascites is the most common complication of cirrhosis. Other diseases associated with ascites include heart failure, constrictive pericarditis, abdominal malignancies, nephrotic syndrome, and malnutrition. Twenty-five percent of individuals who develop ascites caused by cirrhosis die within 1 year. Continued heavy drinking of alcohol is associated with this mortality rate.
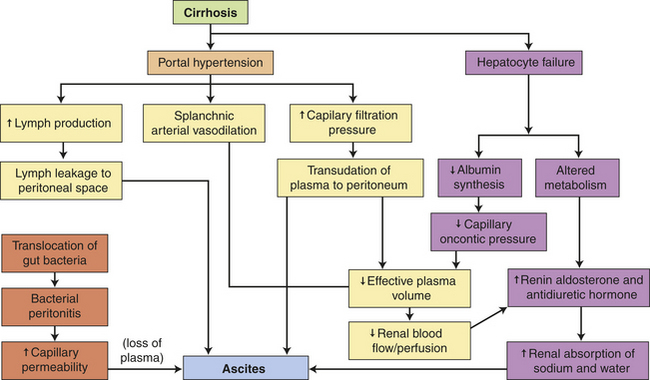
PATHOPHYSIOLOGY Several factors contribute to the development of ascites, including portal hypertension, splanchnic vasodilation, hepatocyte failure, and sodium retention. Impaired excretion of sodium by the kidneys promotes water retention, but the initiating event is not clear. The overflow theory proposes that renal sodium retention is stimulated by portal hypertension with intravascular hypervolemia and overflow into the peritoneal cavity. Portal hypertension also increases the production of hepatic lymph, which “weeps” into the peritoneal cavity. The underfill theory proposes an increase in hepatic sinusoidal hydrostatic pressure and decreased plasma oncotic pressure with weeping of lymph fluid from the surface of the liver. There is a decrease in effective circulating plasma volume activating the renin-angiotensin-aldosterone system and stimulating the kidney to retain more sodium and water, leading to intravascular volume overload.237 The arterial vasodilation theory proposes that circulating nitric oxide or release of endotoxin from translocation of intestinal bacteria triggers arterial vasodilation of the splanchnic organs early in the course of cirrhosis and stimulates renal sodium retention through renin-angiotensin-aldosterone, increased sympathetic tone, and changes in the intrarenal blood flow.238,239
In cases of cirrhosis, both portal hypertension and decreased production of albumin by hepatocytes contribute to the ascites. Deranged liver metabolism also permits the accumulation of hormones that regulate sodium and water balance. Excessive amounts of aldosterone and antidiuretic hormone remain in the blood, stimulating the kidneys to retain sodium and water. High aldosterone levels can be attributed also to increased secretion mediated by excessive plasma renin activity. The increased plasma renin activity may develop because of decreased metabolic function of the liver, increased renal secretion stimulated by low blood flow, or both.
As ascites sequesters more and more body fluid, the kidneys respond by retaining sodium and water in amounts exceeding intake. Retention of sodium and water expands plasma volume, thereby accelerating portal hypertension and ascites formation.
Ascites can be complicated by bacterial peritonitis. Peritonitis involves an inflammatory response that worsens ascites by increasing mesenteric capillary permeability. As plasma seeps out of the permeable mesenteric capillaries, it adds to the volume of ascitic fluid. Figure 39-17 summarizes the mechanisms by which cirrhosis of the liver causes ascites.
CLINICAL MANIFESTATIONS The accumulation of ascitic fluid causes weight gain, abdominal distention, and increased abdominal girth (Figure 39-18). Large volumes of fluid (10 to 20 L) displace the diaphragm and cause dyspnea by decreasing lung capacity. Respiratory rate increases, and the individual assumes a semi-Fowler position to relieve the dyspnea. Some peripheral edema is usually present. Dilutional hyponatremia is a consequence of excess fluid volume. Approximately 10% of individuals with ascites develop bacterial peritonitis, either spontaneously or as a result of paracentesis (needle aspiration of ascitic fluid). Peritonitis causes fever, chills, abdominal pain, decreased bowel sounds, and cloudy ascitic fluid.
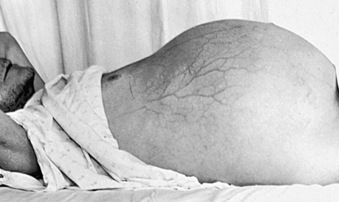
Figure 39-18 Massive ascites in an individual with cirrhosis. Distended abdomen, dilated upper abdominal veins, and inverted umbilicus are classic manifestations. (From Prior JA, Silberstein JS, Stang JM: Physical diagnosis: the history and examination of the patient, ed 6, St Louis, 1981, Mosby.)
EVALUATION AND TREATMENT Diagnosis of ascites is usually based on clinical manifestations and identification of liver disease. The serum-ascites albumin gradient (SAAG) is the most specific diagnostic indicator for portal hypertension-related ascites.240 Paracentesis is used to aspirate ascitic fluid for bacterial culture, biochemical analysis, and microscopic examination. The goal of treatment is to relieve discomfort. If the restoration of liver function is possible (e.g., in ascites caused by viral hepatitis), the ascites diminishes spontaneously. In the meantime, dietary salt restriction and potassium-sparing diuretics can reduce ascites. Strong diuretics, such as furosemide or ethacrynic acid, may be used and vasopressin receptor 2 antagonists are effective for dilutional hyponatremia.241 Albumin may be given.242 Serum electrolytes are monitored carefully because the individual is at risk for hyponatremia and hypokalemia.
Palliative measures include paracentesis to remove 1 or 2 L of ascitic fluid and relieve respiratory distress. This procedure can have serious complications, however. The removal of too much fluid too fast relieves pressure on blood vessels causing arteriolar vasodilation and carries the risk of hypotension, shock, or death.243 Despite repeated paracentesis, ascitic fluid reaccumulates in individuals with irreversible disease, drawing more albumin and electrolytes out of the vascular compartment. Paracentesis is also likely to cause peritonitis. Peritonitis is treated with long-term antibiotics. A transjugular intrahepatic portosystemic shunt or peritoneovenous shunt may be used to treat refractory ascites.237 Individuals with ascites and portal hypertension have a poor prognosis, and liver transplant is the best treatment option.
Hepatic Encephalopathy
Hepatic encephalopathy (portosystemic encephalopathy) is a complex neurologic syndrome characterized by impaired cognitive function, flapping tremor (asterixis), and electroencephalogram (EEG) changes. The syndrome may develop rapidly during acute fulminant hepatitis or slowly during the course of chronic liver disease. Risk factors in the presence of advanced liver disease include gastrointestinal bleeding, increased dietary protein, electrolyte imbalance, and hypoxia.
PATHOPHYSIOLOGY Hepatic encephalopathy probably results from a combination of biochemical alterations that affect neurotransmission. Liver dysfunction and collateral vessels that shunt blood around the liver to the systemic circulation permit neurotoxins and other harmful substances absorbed from the gastrointestinal tract to circulate freely to the brain. Also, permeability of the blood-brain barrier may be increased. The most hazardous substances are end products of intestinal protein digestion, particularly ammonia.244 The digestion of blood from leaking or ruptured varices adds to the amount of ammonia present in systemic blood, as does the action of ammonia-forming bacteria in the colon. The astrocyte is the most vulnerable because it is the site of ammonia detoxification.245 Ammonia that reaches the brain is metabolized to glutamine with osmotic disturbances and alterations in cerebral blood flow that interfere with neurotransmitters, cause astrocyte edema and oxidation, and can result in brain herniation and death.246,247
Blood levels of ammonia do not account for all symptoms associated with hepatic encephalopathy. The accumulation of short-chain fatty acids, serotonin, tryptophan, and false neurotransmitters probably contributes to neural derangement. Excessive gamma-aminobutyric acid (GABA), an inhibitory neurotransmitter, may contribute to reduced levels of consciousness.248 Infection, hemorrhage, inflammation, electrolyte imbalance, sedatives, and analgesics also can precipitate stupor and coma in the presence of liver disease.
CLINICAL MANIFESTATIONS Subtle changes in personality, memory loss, irritability, lethargy, and sleep disturbances are common initial manifestations of hepatic encephalopathy. Symptoms then can progress to confusion, flapping tremor of the hands, stupor, convulsions, and coma. Coma is usually a sign of liver failure and ultimately results in death.
EVALUATION AND TREATMENT Diagnosis of hepatic encephalopathy is based on a history of liver disease and clinical manifestations. Electroencephalography and blood chemistry tests, including blood ammonia levels, provide supportive data. There is no specific diagnostic test.
Correction of fluid and electrolyte imbalances and withdrawal of depressant drugs metabolized by the liver are the first steps in the treatment of hepatic encephalopathy. Reduction of blood ammonia levels is a major objective. This is accomplished by restricting dietary protein intake and eliminating intestinal bacteria. Neomycin is effective in sterilizing the bowel, but it can be nephrotoxic. Lactulose may be administered to prevent ammonia absorption in the colon.244 Antibiotics appear superior to nonabsorbable disaccharides in improving hepatic encephalopathy by reducing bacterial production of ammonia but adverse effects are a concern.249 Sodium benzoate and L-ornithine-L-aspartate also detoxify ammonia.
Jaundice
Jaundice (icterus) is a yellow or greenish pigmentation of the skin caused by hyperbilirubinemia (total plasma bilirubin concentrations greater than 2.5 to 3 mg/dl). Hyperbilirubinemia and jaundice can result from (1) extrahepatic obstruction to bile flow (gallstones), (2) intrahepatic obstruction (hepatocellular disease such as cirrhosis or hepatitis), or (3) excessive production of bilirubin (excessive hemolysis of red blood cells) (Figure 39-19). Jaundice in newborns is caused by impaired bilirubin uptake and conjugation (see Chapter 40).
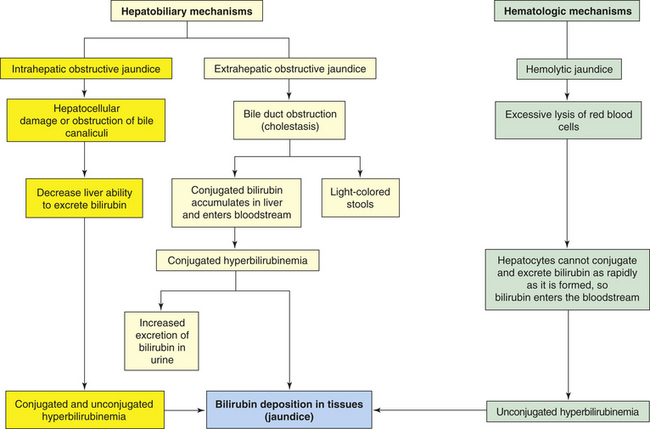
PATHOPHYSIOLOGY Obstructive jaundice can result from extrahepatic or intrahepatic obstruction.250 Extrahepatic obstructive jaundice develops if the common bile duct is occluded by a gallstone, tumor, or compression from edema of pancreatitis. Because the bile duct is obstructed, bilirubin is conjugated by the hepatocytes but cannot flow into the duodenum. (Conjugated bilirubin is soluble in water and is then soluble in aqueous bile.) Therefore, it accumulates in the liver and enters the bloodstream, causing hyperbilirubinemia. Because conjugated bilirubin is water soluble, it appears in the urine. The stools may be light colored or clay colored because they lack bile pigments. The stools also lack urobilinogen because bile is not available for conversion to urobilinogen.
Intrahepatic obstructive jaundice involves disturbances in hepatocyte function and obstruction of bile canaliculi. The uptake, conjugation, and excretion of bilirubin are affected with elevated levels of both conjugated and unconjugated bilirubin. Hepatocellular damage increases plasma concentrations of unconjugated bilirubin. The major disorder, however, is obstruction of bile canaliculi, which diminishes flow of conjugated bilirubin into the common bile duct with elevations in the plasma. In mild cases, some of the bile canaliculi open. Consequently, the amount of bilirubin in the intestinal tract may be only slightly decreased. The stools may appear normal or light colored.
Excessive hemolysis (breakdown) of red blood cells or absorption of hematoma can cause hemolytic jaundice (prehepatic or nonobstructive jaundice). An increased amount of unconjugated bilirubin is formed through metabolism of the heme component of destroyed red blood cells. The extra amount of unconjugated bilirubin exceeds the conjugation ability of the liver, causing blood levels of unconjugated bilirubin to rise. Unconjugated hyperbilirubinemia is the major cause of hemolytic jaundice. Because unconjugated bilirubin is not water soluble, it is not excreted in the urine. The reserve conjugation ability of the liver usually prevents long-term unconjugated hyperbilirubinemia greater than 4 to 5 mg/dl. Severe hemolytic crisis, as occurs with sickle cell disease (see Chapter 28), is a cause of hemolytic jaundice. Hemolytic drugs also can cause jaundice. If unconjugated hyperbilirubinemia exceeds 5 mg/dl, both hemolytic and liver disorders are indicated.
Hyperbilirubinemia and jaundice can be caused also by metabolic defects that impair the uptake or conjugation of unconjugated bilirubin in the liver. Gilbert disease, for example, causes an elevation of unconjugated bilirubin in the plasma but no other symptoms of liver disease. Gilbert disease is probably caused by an inherited deficiency of glucuronyl transferase enzyme, which is required for the hepatic uptake of unconjugated bilirubin. The causes of jaundice are summarized in Table 39-7.
Table 39-7
Three Common Types of Jaundice
| Type | Mechanism | Causes |
| Hemolytic jaundice (predominantly unconjugated bilirubin) | Excessive destruction of erythrocytes | Membrane defect of erythrocytesHemolytic anemias |
| Immune reaction | ||
| Severe infection | ||
| Toxic substances in the circulation (e.g., snake venom) | ||
| Transfusion of incompatible blood | ||
| Obstructive (cholestatic) jaundice (predominantly conjugated bilirubin) | Obstruction to passage of conjugated bilirubin from liver to intestine | Obstruction of bile duct by gallstones or tumor (extrahepatic obstructive jaundice) |
| Obstruction of bile flow through the liver (intrahepatic obstructive jaundice) | ||
| Drugs | ||
| Hepatocellular jaundice (both conjugated and unconjugated bilirubin) | Failure of liver cells (hepatocytes) to conjugate bilirubin and of bilirubin to pass from liver to intestine | Genetic defect of hepatocyte (decreased enzymes), such as occurs in premature infants (see Chapter 40) |
| Hepatitis or biliary cirrhosis |
CLINICAL MANIFESTATIONS The clinical manifestations of jaundice vary and are related to the underlying pathology. Conjugated hyperbilirubinemia may cause the urine to darken several days before the onset of jaundice. The complete obstruction of bile flow from the liver to the duodenum causes light-colored stools. With partial obstruction, stool color is normal and bilirubin is present in the urine. Extrahepatic biliary obstruction is associated with increased intestinal permeability and bacterial translocation, and may contribute to the pathogenesis of sepsis and renal failure.251
Fever, chills, and pain often accompany jaundice resulting from viral or bacterial inflammation of the liver (e.g., viral hepatitis). Manifestations of liver injury from any cause commonly include anorexia, malaise, and fatigue. Yellow discoloration may first occur in the sclera of the eye and then progress to the skin. Skin xanthomas (cholesterol deposits) and pruritus commonly accompany jaundice with an elevation of serum alkaline phosphatase.252
EVALUATION AND TREATMENT Laboratory evaluation of serum establishes whether elevated plasma bilirubin is conjugated or unconjugated or both. Unconjugated bilirubinemia results from hemolysis or hereditary disorders of bilirubin metabolism. Elevations of conjugated bilirubin indicate liver injury or extrahepatic obstruction. The history, physical examination and laboratory tests identify underlying disorders, such as alcoholism, exposure to hepatitis virus, or gallbladder disease. The treatment for jaundice consists of correcting the cause.
Hepatorenal Syndrome
Hepatorenal syndrome (HRS) is a complication of advanced liver disease characterized by functional renal failure with oliguria, sodium and water retention (with or without ascites and peripheral edema), hypotension, and peripheral vasodilation. There are two types of HRS: type 1 involves rapid and progressive renal failure, and type 2 is more chronic and stable accompanied by refractory ascites.253 Renal disorders associated with liver disease can have numerous causes, but HRS is usually associated with alcoholic cirrhosis and fulminant hepatitis. The renal failure is not caused by primary renal disease or other extrinsic factors, but rather by arterial vasodilation of the splanchnic vasculature and renal vasoconstriction with decreased blood flow (prerenal renal failure) and it is reversible.254
PATHOPHYSIOLOGY Oliguric hepatic failure generally accompanies a sudden decrease in blood volume secondary to massive gastrointestinal bleeding or hypotension caused by failing liver function. Hypotension also can be caused by the excessive use of diuretics to treat ascites with decreased renal blood flow, decreased glomerular filtration rate, and oliguria. The hypotension results in decreased glomerular filtration; however, the ability to concentrate and dilute urine is usually maintained. A significant number of individuals with advanced liver disease develop oliguria unrelated to any precipitating event. Inappropriate constriction of renal arterioles is proposed as the causative mechanism. Intrarenal vasoconstriction may result from the selective effects of vasoactive substances that accumulate in the blood because of liver failure. The diseased liver fails to remove excessive angiotensin, vasopressin, prostaglandins, and catecholamines from the blood. These substances travel to the kidneys and cause vasoconstriction. Vasoconstriction also may be a compensatory response to portal hypotension and vasodilation in the splanchnic circulation. The exact reason for the renal vasoconstriction is unknown but is related to vasoconstrictive mediators and sympathetic nerve stimulation. Systemic vasodilation caused by increases in nitric oxide and other substances also may contribute to vascular alterations and renal failure in advanced liver disease.255
CLINICAL MANIFESTATIONS The onset of hepatorenal manifestations may be gradual or acute. Oliguria and complications of advanced liver disease, including jaundice, ascites, and gastrointestinal bleeding, are usually present. Systolic blood pressure is usually less than 100 mmHg. Nonspecific symptoms of hepatorenal syndrome include anorexia, weakness, and fatigue.
EVALUATION AND TREATMENT Diagnosis of HRS is made by excluding all other causes of renal failure. Despite oliguria, serum potassium levels do not become dangerously elevated until the terminal stages of the hepatorenal syndrome. Blood urea increases, followed by an increase in serum creatinine concentration. Urine osmolality is increased, but urine sodium concentrations are below normal (unlike acute tubular necrosis). Urine specific gravity is greater than 1.015. The prognosis for hepatorenal syndrome is usually poor and is related to liver function. Secondary problems, including fluid and electrolyte disorders, bleeding, infections, and encephalopathy, are vigorously treated. Treatment may include systemic vasoconstrictors (α-adrenergic agonists). Pentoxifylline is used to treat HRS in severe alcoholic hepatitis. Symptoms improve with TIPS and extracorporeal albumin dialysis (ECAD). Liver transplant reverses symptoms.256
Disorders of the Liver
Viral hepatitis is a relatively common systemic disease that affects primarily the liver. Six strains of viruses cause various types of hepatitis: hepatitis A virus (HAV), hepatitis B virus (HBV), hepatitis D virus (HDV), hepatitis C virus (HCV), and hepatitis E virus (HEV). Hepatitis A was previously known as infectious hepatitis, and hepatitis B as serum hepatitis. Coinfection of HBV, HCV, HDV, and HIV occurs because these viruses share the same routes of transmission with more rapid progression of liver disease.257 Hepatitis G virus is a blood-borne virus and is usually not a significant cause of liver disease.258 Characteristics of the various types are presented in Table 39-8.
Table 39-8
Characteristics of Viral Hepatitis
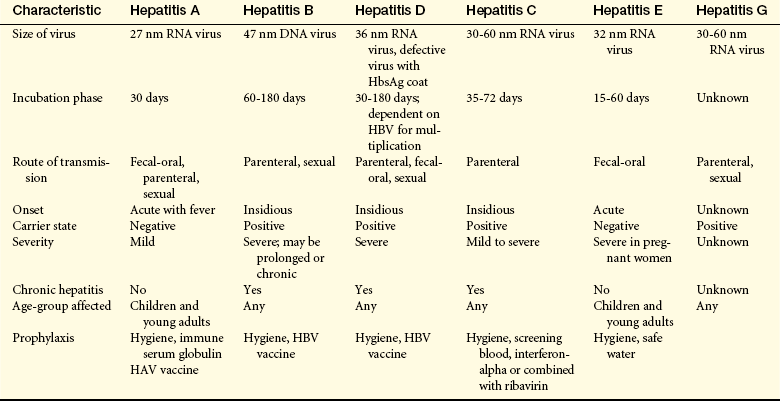
DNA, Deoxyribonucleic acid; HbsAg, hepatitis B surface antigen; HBV, hepatitis B virus; RNA, ribonucleic acid.
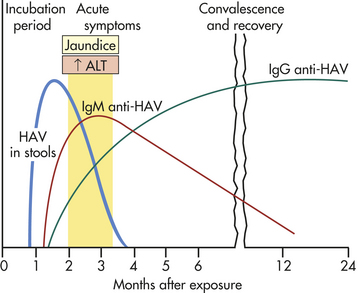
Hepatitis A: HAV can be recovered from the feces, bile, and sera of infected individuals. The usual mode of transmission is the fecal-oral route (contaminated food or water), but the virus can be spread also by the transfusion of infected blood. Approximately 45% of adults in urban areas have HAV antibodies in their blood. The disease spreads readily in crowded, unsanitary conditions, usually through contaminated food or water. Person-to-person spread is more likely to occur in settings such as daycare centers or institutions for the mentally retarded, where there is contact between clients and caregivers who are not vaccinated.259
The incubation period (the time between exposure and onset of symptoms) for HAV is 4 to 6 weeks (Figure 39-20). Fecal shedding of the virus is greatest for 10 to 14 days before the onset of symptoms and during the first week of symptoms and up to 3 months after onset of symptoms. The disease is most contagious during this time. Antibodies to HAV (anti-HAV) develop about 4 weeks after infection. The serum immunoglobulin M (IgM) concentration increases initially and is followed by an increase of serum IgG, whose levels remain elevated for several years after infection, creating immunity to the disease. (See Chapters 6 and 7 for a description of immune functions.) Immunization is effective in preventing the disease and confers long-term immunity.260 A combined HAV and HBV vaccine is available and effective.261
Hepatitis B: Hepatitis B is transmitted through contact with infected blood, body fluids, or contaminated needles. Hepatitis B is also a sexually transmitted disease (see Chapter 24). Transmission among homosexual men may be by oral or genital contact with bleeding lesions in the rectal mucosa. People receiving hemodialysis, multiple blood transfusions, or immunosuppressive drugs have a greater risk of exposure or less resistance to HBV. Coinfection with HCV, HDV, and HIV is common because these viruses share the same routes of transmission.257,262 Mother-infant transmission of HBV occurs if the mother becomes infected during the third trimester of pregnancy. Approximately 0.3% of adults in the United States and up to 400 million worldwide carry the hepatitis B surface antigen (HBsAg) marker for active HBV.263 HBV is a major cause of chronic hepatitis, cirrhosis, and hepatocellular carcinoma.264
Three types of viral particles are involved in HBV infection. The larger (47 nm) Dane particle probably represents the intact HBV. The Dane particle has a double-layered outer coat and carries HBsAg, which was originally called the Australia antigen. HBsAg can be identified in the serum by radioimmunoassay. Hepatitis B core antigen (HBcAg) usually is not detected in the serum. The HBeAg is a derivative of HBcAg and is a marker of HBV replication. The HBV has an incubation period of 6 to 8 weeks. The initial serologic change is a transient increase in IgM. Levels of IgG antibodies to HBsAg rise more slowly and remain elevated for years (Figure 39-21). Chronic infection develops in 15% to 30% of those with acute infection with increased risk for cirrhosis and hepatocellular carcinoma.265 Antiviral and immunomodulatory treatment for chronic hepatitis B includes combination therapy and prevention of drug resistance.266 Vaccine prevents transmission of hepatitis B and the development of acute or chronic hepatitis B, particularly in high-risk populations.267
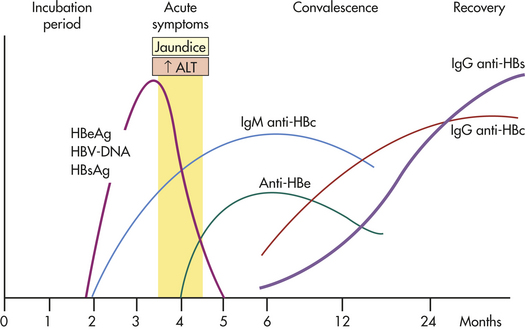
Figure 39-21 Course of infection with the hepatitis B virus (HBV). HbsAg, Hepatitis B surface antigen; anti-HBs, antibody to HBsAg; HbeAg, hepatitis B e-antigen; anti-Hbe, antibody to HBeAg; anti-HBc, antibody to hepatitis B core antigen. The antibody to HBs (anti-HBs) is IgG, the immunoglobulin that creates immunity. ALT, Alanine transaminase; DNA, deoxyribonucleic acid; IgG, immunoglobulin G; IgM, immunoglobulin M.
Hepatitis C: HCV (previously known as non-A, non-B hepatitis) is a parenterally transmitted flavivirus with six genotypes. About 40% of HCV cases involve intravenous drug users, who also have a high incidence of HIV infection.268 Coinfection with HBV also is prevalent.269 It is the most common cause of chronic liver disease and HCV in the Western world. The variants of HCV make vaccine development difficult and resistance to drug therapy is common.270 Persistent infection with recurring acute symptoms and elevated aminotransferase levels represent the clinical presentation. Half of infected individuals have a viral response to treatment.271
Hepatitis D: HDV occurs in individuals with hepatitis B. The delta virus depends on the HBV for its replication because the coat of the delta virus consists of HBsAg molecules that are on the surface of HBV. Hepatitis D has been shown to suppress replication of HBV.272 Parenteral drug users have a high incidence of HDV infection. The clinical course of HDV is similar to that of hepatitis A and B, although it is sometimes more severe. Treatment for chronic HDV may best be treated with antiviral drugs (i.e., pegylated interferon alpha and ribavirin).273
Hepatitis E: Hepatitis E is most common in developing countries and is transmitted by the fecal-oral route, usually by way of contaminated water. It is also found in developed countries and must be differentiated from drug-induced liver injury.274 Animal reservoirs of HEV include domestic pigs, wild boars, and deer.275 It is more prevalent among adults and has the highest mortality in pregnant women.276 Clinically, it resembles HAV. No vaccine for HEV is available but development is in progress.277
PATHOPHYSIOLOGY The pathologic lesions of hepatitis are similar to those caused by other viral infection. Hepatic cell necrosis, scarring, Kupffer cell hyperplasia, and infiltration by mononuclear phagocytes occur with varying severity. Cellular injury is promoted by cell-mediated immune mechanisms (i.e., cytotoxic T cells, T regulatory cells, and natural killer cells).281 Regeneration of hepatic cells begins within 48 hours of injury. The inflammatory process can damage and obstruct bile canaliculi, leading to cholestasis and obstructive jaundice. In milder cases the liver parenchyma is not damaged. Damage tends to be most severe in cases of hepatitis B and hepatitis C. Hepatitis B is also associated with acute fulminating hepatitis, a rare form of the disease that is characterized by massive hepatic necrosis (see p. 1491). Acute fulminating hepatitis causes severe encephalopathy, which is manifested as confusion, stupor, and coma. Liver failure can occur, leading to intestinal bleeding, cardiorespiratory insufficiency, and renal failure. Mortality is high, but recovery can be complete.
CLINICAL MANIFESTATIONS The clinical manifestations of the various types of hepatitis are very similar. The spectrum of manifestations ranges from absence of symptoms to fulminating hepatitis, with rapid onset of liver failure and coma. Acute viral hepatitis causes abnormal liver function test results. The serum aminotransferase values, aspartate transaminase (AST) and alanine transaminase (ALT), are elevated, but their elevation may not be consistent with the extent of cellular damage. The clinical course of hepatitis usually consists of four phases: incubation, prodromal, icteric, and recovery phases. The incubation phase is reviewed in Table 39-8.
Prodromal Phase: The prodromal (preicteric) phase of hepatitis begins about 2 weeks after exposure and ends with the appearance of jaundice. Fatigue, anorexia, malaise, nausea, vomiting, headache, hyperalgia, cough, and low-grade fever are prodromal symptoms that precede the onset of jaundice. About 10% of individuals may develop extrahepatic symptoms including rash, arthralgias, and purpura. HBV and HCV may cause nephritis related to glomerular immune complex deposition.282 Food odors often cause nausea, and changes in taste suppress the desire to smoke and drink alcohol. Right upper abdominal pain is common, and a weight loss of 2 to 4 kg is not unusual. The infection is highly transmissible during this phase.
Icteric Phase (Jaundice): The icteric phase begins about 1 to 2 weeks after the prodromal phase and lasts 2 to 6 weeks. Hepatocellular destruction and intrahepatic bile stasis cause jaundice (icterus). The urine may be dark and the stools clay colored before the onset of jaundice from conjugated hyperbilirubinemia. The icteric phase is the actual phase of illness. The liver is enlarged, smooth, and tender, and percussion over the liver causes pain. During the icteric phase, gastrointestinal and respiratory symptoms subside, but fatigue and abdominal pain may persist or become more severe. The stools may be lighter in color as a result of cholestasis. Serum bilirubin levels range from 5 to 10 mg/dl, with conjugated bilirubin fraction increasing. The jaundice may last 2 to 6 weeks or longer. Mild and transient itching often accompanies jaundice. The prothrombin time may be prolonged in individuals with more serious forms of the disease.
Recovery Phase: The posticteric or recovery phase begins with resolution of jaundice, about 6 to 8 weeks after exposure. Although the liver may still be enlarged and tender, symptoms diminish. In most cases, liver function test results return to normal within 2 to 12 weeks after the onset of jaundice.
Chronic hepatitis may begin at this point and is associated with HBV and HCV infection. Chronic active hepatitis is the persistence of clinical manifestations and liver inflammation after acute stages of HBV, HCV, and HDV. Liver function tests remain abnormal for longer than 6 months, and HBsAg persists. Chronic, active HBV is a predisposition to cirrhosis and primary hepatocellular carcinoma. Chronic active hepatitis constitutes a carrier state, and hepatitis C can be transmitted from mothers to infants.283
EVALUATION AND TREATMENT Diagnosis of type A hepatitis is based on the presence of anti-HAV, as is the diagnosis of HCV. The most specific diagnostic test for HBV is serologic analysis for HBsAg, which is the marker for HBV. The assay for HDV is the total antibody to HDV and antigen (anti-HDV). A test for HEV has not been developed. Liver enzyme levels and function tests also can indicate other viral liver diseases, drug toxicity, or alcoholic hepatitis.
Specific treatments are previously described for the different types of hepatitis viruses. Physical activity may be restricted. A low-fat, high-carbohydrate diet is beneficial if bile flow is obstructed.
To prevent transmission of hepatitis A, handwashing and use of gloves for disposing of fecal matter are imperative. Molecular procedures are available for direct surveillance of HAV in food and environmental samples should be used for the prevention of food-borne infections.284 There should be no direct contact with blood or body fluids of individuals with hepatitis B or hepatitis C. The administration of immunoglobulin before exposure or early in the incubation period can prevent hepatitis A. A combined vaccine for HAV and HBV is available. Hepatitis B immunoglobulin provides passive prophylactic immunity against HBV. Prophylaxis is recommended for healthcare workers, liver transplant recipients, and others who are at risk for contact with infected body fluids.285
Fulminant Hepatitis
Fulminant hepatitis is a clinical syndrome resulting in severe impairment or necrosis of liver cells and potential liver failure. The disorder may occur as a complication of hepatitis C or hepatitis B, particularly HBV infection compounded by infection with the delta virus. Toxic reactions to drugs and congenital metabolic disorders also can cause fulminant hepatitis.286 Acetaminophen overdose is the leading cause of acute liver failure in the United States (see What’s New? Acetaminophen and Acute Liver Failure).
Causative mechanisms of fulminant hepatic failure are poorly understood. Hepatocytes become edematous, and patchy areas of necrosis and inflammatory cell infiltrates disrupt the parenchyma. The death of hepatocytes may be caused by toxic, viral, or immunologic damage.
Fulminant hepatitis usually develops 6 to 8 weeks after the initial symptoms of viral hepatitis or a metabolic liver disorder. Anorexia, vomiting, abdominal pain, and progressive jaundice are initial signs, followed by ascites and gastrointestinal bleeding. Hepatic encephalopathy is manifested as lethargy, altered motor functions, and coma. Liver function tests show elevations of both direct and indirect serum bilirubin, serum transaminases, and blood ammonia. Prothrombin time is prolonged.
Treatment of fulminant hepatitis is supportive. The hepatic necrosis is irreversible, and 60% to 90% of affected children die. Liver transplantation may be lifesaving.287 Survivors usually do not develop cirrhosis or chronic liver disease. Artificial liver support systems are continuing to be developed for use as a bridge to transplant or recovery from acute or acute on chronic liver failure288 (see What’s New? Artificial Liver Support).
Cirrhosis
Cirrhosis is an irreversible inflammatory disease that disrupts liver structure and function and is a leading cause of death in the United States. Disorganization of hepatic tissues is caused
by nodular regeneration forming fibrous bands, giving the liver a cobbly appearance.289 The liver may be larger or smaller than normal, and usually it is firm or hard when palpated. A variety of disorders can cause cirrhosis. Therefore, it is often classified by cause (Table 39-9).
Table 39-9
| Type and Disease Name | Causal Mechanisms | Pathophysiology |
| Alcoholic cirrhosis, Laennec cirrhosis, portal cirrhosis, fatty cirrhosis | Toxic effects of chronic, excessive alcohol intake; acetaldehyde formed by alcohol metabolism damages hepatocytes | Fatty liver, inflammation (alcoholic steatohepatitis), and derangement of the lobular architecture by necrosis and fibrosis (cirrhosis) with obstruction of biliary and vascular channels |
| Biliary cirrhosis (intrahepatic or extra hepatic obstruction of bile flow) | ||
| Primary biliary cirrhosis | Unknown; possible an autoimmune mechanism | Inflammation and scarring of lobular bile ducts |
| Secondary biliary cirrhosis | Obstruction by neoplasms, strictures, or gallstones | Inflammation and scarring of bile ducts proximal to the obstruction |
| Postnecrotic cirrhosis | Viral hepatitis caused by hepatitis A, B, or C virus; drugs or other toxins; autoimmune destruction | Replacement of necrotic tissue with cirrhotic tissue, particularly fibrous, nodular scar tissue |
| Metabolic cirrhosis | Metabolic defects and storage disease, such as α1-antitrypsin deficiency, glycogen storage disease, hemochromatosis, Wilson disease, galactosemia | Inflammation and scarring with specific morphologic changes related to cause |
The precise process of cellular injury depends on the cause of cirrhosis, and the causes are not all clearly understood. Structural changes result from fibrosis, which is a consequence of leukocyte release, inflammatory cytokines, and chemokines with activation of fibrogenic fibroblasts.290 The parenchyma of the liver becomes distorted, and biliary channels may be altered or obstructed, producing jaundice. Obstruction caused by cirrhosis can cause portal hypertension (see p. 1482). New vascular channels can form shunts, and blood from the portal vein bypasses the liver. These vascular changes compromise liver function further, and the process of regeneration is replaced by hypoxia; necrosis; atrophy; and ultimately, liver failure.
Cirrhosis develops slowly over a period of years. Its severity and rate of progression depend on the cause. If toxins, such as alcohol, are involved, the rate of cell death and the severity of inflammation depend on the amount of toxin present.291
Alcoholic Liver Disease: Deaths from alcohol-related liver disease have increased over the past decade and the amount and duration of alcohol consumption are positively related to the extent of liver damage. Abuse of any type of alcoholic beverage can cause cirrhosis. Malnutrition may add to the risk of cirrhosis in alcohol abusers. The incidence of alcoholic cirrhosis is greatest in middle-aged men. In the United States, mortality resulting from cirrhosis is highest among non-white individuals. Although alcoholic cirrhosis is the most prevalent of the various types of cirrhosis, the occurrence of cirrhosis among persons with alcoholism is relatively low (approximately 25%).
PATHOPHYSIOLOGY Chronic alcoholic hepatitis is a precursor of cirrhosis characterized by inflammation, degeneration, and necrosis of hepatocytes, infiltration of polymorphonuclear leukocytes and lymphocytes, immunologic alterations, and lipid peroxidation. The injured hepatocytes contain Mallory bodies (hyaline endoplasmic reticulum). The presence of Mallory bodies indicates the onset of fibrosis. Neutrophils infiltrate and surround degenerating hepatocytes. The mechanism of hepatocyte injury is not clearly understood, but inflammatory mediators, acetaldehyde, reactive oxygen and nitrogen species, and genetic factors are involved.292,293 Serum IgA is often elevated in individuals with alcoholic hepatitis, and liver antigens and antibodies have been identified in persons with progressive alcoholic liver disease. The inflammation and necrosis caused by alcoholic hepatitis stimulate the fibrosis characteristic of the cirrhotic stage of disease.294
Alcoholic cirrhosis is a complex process that begins with fatty infiltration (hepatic steatosis). Fatty infiltration can occur without subsequent hepatitis or cirrhosis. Fat deposition (deposition of triglycerides) within the liver hepatocytes is caused primarily by increased lipogenesis and decreased fatty acid oxidation by hepatocytes. Cessation of alcohol intake reverses fat accumulation. Lipids mobilized from adipose tissue or dietary fat intake may contribute to fat accumulation.
Alcoholic cirrhosis is caused by the toxic effects of alcohol metabolism on the liver, immunologic alterations, lipid peroxidation, and malnutrition.295 Alcoholic cirrhosis is more severe when associated with HCV.296 The oxidative metabolism of alcohol occurs primarily in the liver (see Chapter 2). Alcohol is transformed to acetaldehyde. Excessive amounts of acetaldehyde induce lipid peroxidation, oxidative stress, and disrupt cytoskeleton and membrane function. Acetaldehyde inhibits export of proteins from the liver, alters metabolism of vitamins and minerals, promotes liver fibrosis, and induces malnutrition.297 Mitochondrial function is impaired, decreasing oxidation of fatty acid. Enzyme and protein synthesis may be depressed or altered, and hormone and ammonia degradation is diminished. Alcohol also may stimulate the formation of autoantibodies specific to hepatic cells. Bacterial endotoxin from the intestine contributes to progressive injury and inflammation.298,299 Cellular damage initiates an inflammatory response. Inflammatory cytokines, including TNF-α and IL-6, IL-8, and IL-18, are associated with alcoholic liver disease. Inflammation and necrosis result in excessive collagen formation. Transforming growth factor-beta (TGF-β) contributes to fibrosis and is produced in part by activated Kupffer cells.300 TGF-β activates hepatic stellate cells to produce excess collagen.301 Dense bands of fibrosis surround regenerative hepatocellular nodules. Fibrosis and scarring alter the structure of the liver and obstruct biliary and vascular channels.302 Examples of liver damage are shown in Figure 39-22.
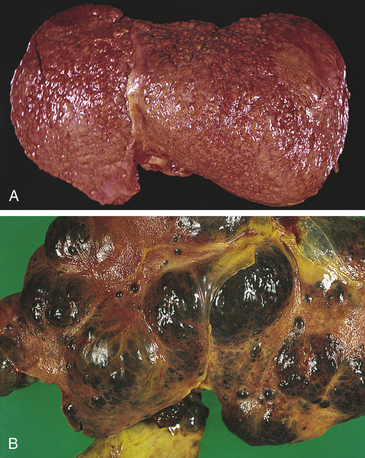
Figure 39-22 Cirrhosis. A, Micronodular cirrhosis. The nodular appearance develops from regeneration of hepatocytes projecting through fibrous bands of tissue. B, Macronodular cirrhosis. (From Damjanov I, Linder J, editors: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
CLINICAL MANIFESTATIONS Fatty infiltration causes no specific symptoms or abnormal liver function test results. The liver is usually enlarged, however, and the individual has a history of continuous alcohol intake during the previous weeks or months. Anorexia, nausea, jaundice, and edema develop with advanced fatty infiltration or the onset of alcoholic hepatitis (Figure 39-23).
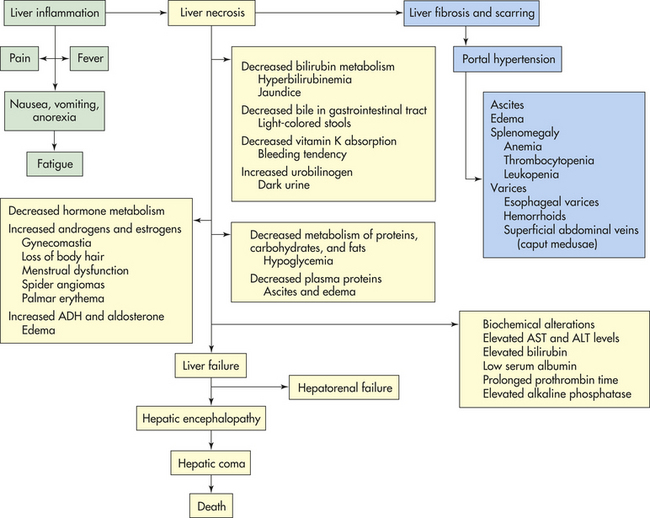
Figure 39-23 Clinical manifestations of cirrhosis. ADH, Antidiuretic hormone; ALT, alanine transaminase; AST, aspartate transaminase.
The clinical manifestations of alcoholic hepatitis can be mild or severe. Nonspecific symptoms include fatigue, weight loss, and anorexia.303 Manifestations of acute illness include nausea, anorexia, fever, abdominal pain, and jaundice. Toxic effects of alcohol also can cause testicular atrophy, reduced libido, azoospermia, and decreased testosterone in men.304 Cirrhosis is a multiple-system disease and causes hepatomegaly, splenomegaly, ascites, gastrointestinal hemorrhage, portal hypertension, hepatic encephalopathy, and esophageal varices. Anemia results from blood loss, poor nutrition, and hypersplenism. Risk for infection is greater, in part because of altered macrophage function.305 The presence of numerous and severe manifestations increases the risk of death. The clinical features of alcoholic cirrhosis depend on the duration of the disease and the severity of liver damage.
EVALUATION AND TREATMENT The diagnosis of alcoholic hepatitis is based on the individual’s history and clinical manifestations. The results of liver function tests are abnormal, and serologic studies show elevated serum enzymes and bilirubin and decreased serum albumin. Prolonged prothrombin time cannot easily be corrected with vitamin K therapy. Liver biopsy can confirm the diagnosis of cirrhosis, but biopsy is not necessary if clinical manifestations of cirrhosis are evident.
There is no specific treatment for alcoholic cirrhosis, but many of the complications are treatable. Rest, a nutritious diet, corticosteroids, antioxidants, drugs that slow fibrosis, and management of complications such as ascites, gastrointestinal bleeding, infection, and encephalopathy slow disease progression. Cessation of alcohol consumption slows the progression of liver damage, improves clinical symptoms, and prolongs life. Although the liver damage is irreversible, measures that halt the inflammation and destruction of liver cells prolong life. Liver transplant is the treatment for liver failure289,306 and artificial liver support systems are being developed (see What’s New? Artificial Liver Support).
Biliary Cirrhosis: Biliary cirrhosis differs from alcoholic cirrhosis in that the damage and inflammation leading to cirrhosis begin in bile canaliculi and bile ducts, rather than in the hepatocytes. The two types of biliary cirrhosis are primary and secondary. Although both involve bile duct pathology, they differ with respect to cause, risk factors, and mechanisms of obstruction and inflammation.
Primary Biliary Cirrhosis: Primary biliary cirrhosis is an autoimmune disease of unknown etiology leading to destruction of small intrahepatic bile ducts. Mitochondrial autoantibodies are a hallmark of the disease and may be triggered by xenobviotics of infectious agents in genetically susceptible individuals.307 The disease is characterized by inflammation and destruction of small intrahepatic bile ducts with portal inflammation and, ultimately, fibrosis. Women are affected more commonly (90%) than men. Symptoms rarely develop before the age of 30 years. Primary biliary cirrhosis often accompanies the autoimmune diseases.308
Primary biliary cirrhosis develops insidiously. It begins with inflammation, destruction, fibrosis, and obstruction of the intrahepatic bile ducts. Nodular regeneration and cirrhosis follow. Portal hypertension develops during the later stages of the disease.309
Individuals with primary biliary cirrhosis may be asymptomatic or symptomatic at diagnosis.308 The earliest manifestations are pruritus, fatigue, and abdominal pain. Jaundice and light-colored stools are later symptoms. These symptoms are caused by intrahepatic obstruction of bile flow. Steatorrhea and fat-soluble vitamin deficiencies are present in some cases. The malabsorption can lead to osteomalacia and osteoporosis. Cirrhosis, symptoms of portal hypertension and encephalopathy, and ultimately liver failure develop.
Serologic tests show elevated alkaline phosphatase levels, hyperbilirubinemia, and hyperlipidemia, with or without other clinical manifestations. Most individuals have a circulating IgG antimitochondrial antibody that is not found in other types of liver disease. Evaluation involves ruling out biliary obstruction caused by gallstones, tumor, or inflammation of the common bile duct (i.e., secondary biliary cirrhosis). The presence of positive cholestatic liver test for 6 months’ duration, positive serum antimitochondrial antibody, and liver biopsy confirms the diagnosis of primary biliary cirrhosis.310
Corticosteroids or azathioprine may be used to suppress the immune response. No specific treatment is available. The distressing pruritus may be relieved by cholestyramine, which binds bile salts in the intestine. Intramuscular injections of vitamins D and K alleviate the vitamin deficiency. The other symptoms of cirrhosis are managed as they develop. Long-term treatment with ursodeoxycholic acid slows disease progression.308 Life expectancy is about 8 to 10 years after symptom onset.311 Liver transplant is the only definitive therapy.
Secondary Biliary Cirrhosis: Secondary biliary cirrhosis develops when there is prolonged partial or complete obstruction of the common bile duct or its branches. The obstruction may be caused by gallstones, tumors, fibrotic strictures, or chronic pancreatitis. Biliary atresia and cystic fibrosis cause secondary biliary cirrhosis in children.
Chronic obstruction to bile flow increases pressure in the hepatic bile duct and results in the accumulation of bile in the centrilobular spaces. Necrotic areas develop and are followed by proliferation and inflammation of the portal ducts that result in edema and fibrosis. Pools of bile form when the portal ducts rupture into surrounding necrotic areas. Injury is accompanied by regeneration of hepatic cells with the development of finely nodular cirrhosis.
Clinical manifestations are similar to those of primary biliary cirrhosis, with jaundice and pruritus the most distressing symptoms. Right upper quadrant pain is common, and a low-grade fever may be present from bile duct inflammation (cholangitis).
Cholangiography provides the most definitive diagnosis. Laboratory tests usually show elevated conjugated bilirubin and alkaline phosphatase levels. Aminotransferase increases if there is an accompanying cholangitis. Surgery or endoscopy relieves obstruction, prolongs survival, and diminishes or resolves symptoms. Continued obstruction leads to advanced cirrhosis and liver failure.
Disorders of the Gallbladder
Obstruction and inflammation are the most common disorders of the gallbladder. Obstruction is caused by gallstones (cholelithiasis), which are aggregates of substances in the bile. The gallstones may remain in the gallbladder or be ejected, with bile, into the cystic duct. Gallstones that become lodged in the cystic duct obstruct the flow of bile into and out of the gallbladder and cause inflammation. Gallstone formation is termed cholelithiasis. Inflammation of the gallbladder or cystic duct is known as cholecystitis.
Cholelithiasis (Gallstones)
Cholelithiasis is a prevalent disorder in developed countries, where incidence is 10% to 20%. The actual incidence is unknown because many individuals who have gallstones are asymptomatic. Risk factors include obesity; rapid weight loss in obese individuals; middle age; female gender; oral contraceptives; American Indian ancestry; gallbladder, pancreatic, or ileal disease; high dietary cholesterol; and gene-environmental interactions.312
PATHOPHYSIOLOGY Gallstones are commonly of two types: cholesterol and pigmented.313 Cholesterol stones are the most common. Pigmented stones, which are less common, occur later in life and are associated with cirrhosis. Cholesterol gallstones form in bile that is supersaturated with cholesterol produced by the liver. Supersaturation sets the stage for cholesterol crystal formation, or the formation of “microstones.” More crystals then aggregate on the microstones, which grow to form “macrostones.” This process usually occurs in the gallbladder, which may have decreased motility. The stones may lie “silent” or become lodged in the cystic or common duct, causing pain and cholecystitis. Gallstone formation may be such that the stones accumulate and fill the entire gallbladder (Figure 39-24). Impaired gallbladder motility and gallbladder stasis also may contribute to stone formation.314
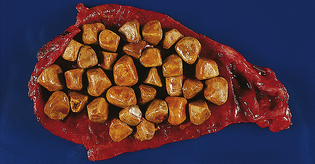
Figure 39-24 Resected gallbladder containing mixed gallstones. (From Kissane JM, ed: Anderson’s pathology, ed 9, St Louis, 1990, Mosby.)
It is not known why the hepatocytes secrete bile that is supersaturated with cholesterol. Proposed mechanisms include (1) an enzymatic defect that increases the hepatocytes’ synthesis of cholesterol; (2) diminished secretion of bile acids, which normally promote cholesterol solubility; (3) decreased resorption of bile salts from the ileum, which decrease the bile acid pool; (4) gallbladder smooth muscle hypomotility and stasis; (5) genetic predisposition; and (6) some combination of these mechanisms.315 In obese individuals the mechanism appears to involve cholesterol synthesis, whereas in nonobese individuals, it appears to involve decreased secretion of bile acids.
Pigmented stones are created by cholesterol, calcium bilirubinate, or pigmented polymers. The formation of pigmented stones is associated with biliary tract obstruction and bacterial degradation and precipitation of biliary lipids.316
CLINICAL MANIFESTATIONS Epigastric and right hypochondrium pain and intolerance to fatty foods are the cardinal manifestations of cholelithiasis. Vague symptoms include heartburn, flatulence, epigastric discomfort, pruritus, jaundice, and food intolerances, particularly to fats and cabbage. The pain, often called biliary colic, is most characteristic and is caused by the lodging of one or more gallstones in the cystic or common duct.317 The pain can be intermittent or steady. It usually is located in the right upper quadrant and radiates to the mid-upper back. Jaundice indicates that the stone is located in the common bile duct. Abdominal tenderness and fever indicate cholecystitis. Complications can include pancreatitis.
EVALUATION AND TREATMENT Diagnosis is based on the individual’s medical history, physical examination, and radiographic evaluation. An oral cholecystogram usually outlines the stones. Intravenous cholangiography is used to differentiate cholelithiasis from other causes of extrahepatic biliary obstruction if the cholecystogram is negative. Endoscopic or percutaneous cholangiography and endoscopic or transabdominal ultrasonography are diagnostic options.318
Laparoscopic cholecystectomy is the preferred treatment for gallstones that cause obstruction or inflammation. Use of transluminal endoscopic surgery is advancing rapidly.319 Endoscopic retrograde cholangiopancreatography and sphincterotomy with stone retrieval is used for the treatment of bile duct stones. Large stones may be managed with lithotripsy.320 An alternative treatment is the administration of drugs that dissolve smaller stones. For example, the bile acid chenodeoxycholic acid (CDCA) can completely or partially dissolve cholesterol gallstones. Ursodeoxycholic acid (UDCA), which is structurally similar to CDCA, is also effective, is less toxic to hepatocytes, and does not cause fatty diarrhea, as does CDCA.
Cholecystitis
Cholecystitis can be acute or chronic. Both forms are almost always caused by the lodging of a gallstone in the cystic duct. Obstruction causes the gallbladder to become distended and inflamed. The pain is similar to that caused by gallstones. Pressure against the distended wall of the gallbladder decreases blood flow. Ischemia, necrosis, and perforation of the gallbladder are possible. Fever, leukocytosis, rebound tenderness, and abdominal muscle guarding are common findings. Serum bilirubin and alkaline phosphatase levels may be elevated. Nevertheless, the acute abdominal pain of cholecystitis must be differentiated from the pain caused by other disorders, such as pancreatitis, myocardial infarction, and acute pyelonephritis of the right kidney. Cholangiography or radioactive scan can confirm a diagnosis of cholecystitis.
Treatment includes pain control, replacement of fluid and electrolytes, and fasting. Antibiotics are often prescribed to manage bacterial infection in severe cases. Immediate cholecystectomy is required for complications such as peritonitis from gallbladder perforation. Persistent symptoms or development of chronic cholecystitis punctuated by recurrent acute attacks usually requires cholecystectomy. If pancreatic abscesses develop, they usually are resected.321
Disorders of the Pancreas
Pancreatitis, or inflammation of the pancreas, is a relatively rare and potentially serious disorder. Incidence is about equal in men and women and is more common between 50 and 60 years of age. Pancreatitis can be acute or chronic. It is associated with several other clinical conditions, including alcoholism, obstructive biliary tract disease (particularly cholelithiasis), peptic ulcers, trauma, hyperlipidemia, and certain drugs. The cause is unknown in 15% to 25% of cases.322
Acute Pancreatitis
PATHOPHYSIOLOGY Acute pancreatitis (acute hemorrhagic pancreatitis) is initiated by intrapancreatic activation of proteases (Figure 39-25). It is usually a mild disease, but about 20% of those afflicted develop a severe pancreatic inflammation requiring hospital care. Although the precise pathogenic mechanism or sequence of events often is unknown, alcoholism and biliary tract obstruction are commonly associated. Bile reflux into the pancreas occurs if gallstones obstruct thecommon bile duct and bile contributes to attacks of acute pancreatitis.323 The pancreatic acinar cell metabolizes ethanol with the generation of toxic metabolites.324 The most common theory is that pancreatitis develops because of an injury or disruption of pancreatic acinar cells, which permit leakage of pancreatic enzymes (trypsin, chymotrypsin, and elastase) into pancreatic tissue.325 Activated proteolases (trypsin and elastase) and lipases break down tissue and cell membranes, causing inflammation, edema, vascular damage, hemorrhage, necrosis, and fibrosis (Figure 39-26).326 (Fatty necrosis is described in Chapter 2.) Toxic enzymes, infiltration of macrophages and leukocytes with release of inflammatory mediators (TNF-α, IL-1β, IL-6, IL-8, IL-10, C5a, intercellular-adhesion molecule [ICAM], and substance P) into the bloodstream cause injury to vessels and other organs, such as the lungs, heart, and kidneys. Translocation of bacteria occurs with gut barrier dysfunction resulting in local or systemic infection.327
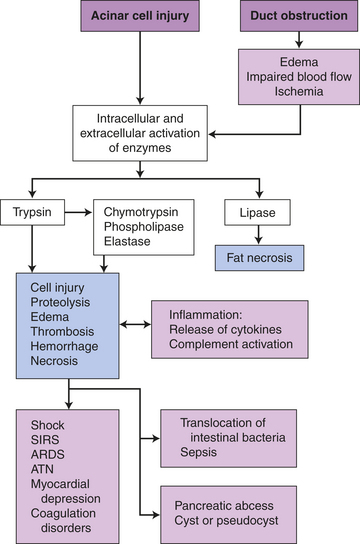
Figure 39-25 Pathophysiology of acute pancreatitis. ARDS, Acute respiratory distress syndrome; ATN, acute tubular necrosis; SIRS, systemic inflammatory response syndrome.
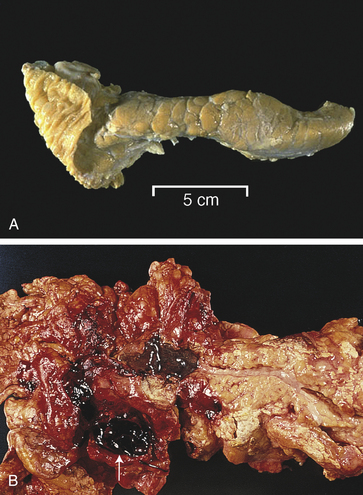
Figure 39-26 Normal and acute hemorrhagic pancreatitis. A, Normal pancreas. B, Acute hemorrhagic pancreatitis. The pancreas has hemorrhage, fat necrosis (white patches), and a pseudocyst filled with blood (white arrow). (A from Klatt EC, editor: Robbins and Cotran atlas of pathology, Philadelphia, 2006, Saunders. B from Damjanov I, Linder J, editors: Pathology: a color atlas, St Louis, 2000, Mosby.)
CLINICAL MANIFESTATIONS Epigastric or midabdominal pain is the cardinal symptom of acute pancreatitis. The pain may radiate to the back because of the retroperitoneal location of the pancreas. The pain is caused by edema, which distends the pancreatic ducts and capsule; chemical irritation and inflammation of the peritoneum; and irritation or obstruction of the biliary tract. Fever and leukocytosis accompany the inflammatory response. Nausea and vomiting are caused by hypermotility or paralytic ileus secondary to the pancreatitis or peritonitis.
Abdominal distention accompanies bowel hypermotility and the accumulation of fluids in the peritoneal cavity. Hypotension and shock occur frequently because enzymes and kinins released into the circulation increase vascular permeability and dilate vessels. Hypovolemia, hypotension, and myocardial insufficiency result. A small percentage of individuals develop tachypnea and hypoxemia secondary to pulmonary edema, atelectasis, or pleural effusions caused by circulating pancreatic enzymes and inflammatory mediators. In severe cases, hypovolemia decreases renal blood flow sufficiently to impair renal function.328 Pancreatic encephalopathy and coagulation abnormalities are complications of severe pancreatitis and often occur with multiple organ failure.329 Tetany may develop as a result of calcium deposition in areas of fat necrosis or as a decreased response to parathyroid hormone. Transient hyperglycemia also can occur if glucagon is released from damaged A cells in the pancreatic islets. A systemic inflammatory response and multiple organ failure account for most deaths with severe pancreatitis.330 In hemorrhagic pancreatitis, some individuals develop flank or periumbilical ecchymosis, a sign of poor prognosis.
EVALUATION AND TREATMENT Diagnosis of pancreatitis is based on clinical findings, identification of associated disorders, and laboratory studies. Elevated serum amylase and lipase are characteristic diagnostic features and serum lipase is more specific and sensitive. The amylase level usually rises within 12 hours after the onset of symptoms and returns to normal within 3 to 5 days in most cases. Serum lipase levels increase within 4 to 8 hours of clinical symptom onset and decrease within 8 to 14 days. Serum trypsin levels are very specific for pancreatitis but may not be readily available. Urine trypsinogen-2 and urine amylase also are elevated. C-reactive protein elevates within 48 hours and is a marker of severity.331,332 The ratio of amylase clearance to creatinine clearance by the kidney can be diagnostic because, in cases of pancreatitis, amylase clearance increases significantly compared with creatinine clearance. Serum transferrin is a diagnostic marker for alcoholic acute pancreatitis.333 Acute pancreatitis is difficult to diagnose because several other disorders can cause similar clinical and laboratory findings. These disorders include perforating duodenal ulcer, acute cholecystitis, small-bowel obstruction, and kidney stones. Ultrasound and CT scan are used in more severe cases to evaluate extent of involvement and complications. Intra-abdominal pressure monitoring assesses risk for abdominal compartment syndrome334 (see What’s New? Abdominal Compartment Syndrome).
The goal of treatment for acute pancreatitis is to stop the process of autodigestion and prevent systemic complications. Narcotic medications may be needed to relieve pain. Meperidine hydrochloride (Demerol) is used instead of morphine because it causes less spasm of the sphincter of Oddi than morphine. Nasogastric suction may not be necessary with mild pancreatitis but may help relieve pain and prevent paralytic ileus in individuals who are nauseated and vomiting. Enteral nutrition with use of nasogastric or jejunal tube feedings is often effective, but an effort is made to maintain normal enteral nutrition.335 Probiotics may be helpful. Parenteral fluids are essential to restore blood volume and prevent hypotension and shock. Parenteral hyperalimentation should be initiated
when enteral feeding is not tolerated. Drugs that decrease gastric acid production (e.g., cimetidine) can decrease stimulation of the pancreas by secretin. Necrotizing pancreatitis requires surgical resection, and antibiotics may control infection. The risk of mortality increases significantly with the development of pulmonary, cardiac, and renal complications.
Chronic Pancreatitis
Chronic alcohol abuse is the most common cause of chronic pancreatitis. Obstruction from gallstones, autoimmune disease, gene mutations, smoking, and obesity can be contributing factors.336 The disease is idiopathic in about 25% of cases.337 Toxic metabolites and chronic release of inflammatory cytokines contribute to the destruction of acinar cells and islets of Langerhans. Fibrosis, strictures, calcification, ductal obstruction, and pancreatic cysts are the common lesions of chronic pancreatitis. The cysts are walled-off areas or pockets of pancreatic juice, necrotic debris, or blood within or adjacent to the pancreas. Continuous or intermittent abdominal pain, weight loss, and less commonly, steatorrhea and diabetes mellitus accompany disease progression. Pain is associated with increased intraductal pressure, increased tissue pressure, ischemia, neuritis, ongoing injury, and changes in central pain perception.338 Preventing disease progression includes lifestyle modification to stop alcohol use and smoking. Pain management is complex with use of analgesics. A fat-free diet and oral enzyme replacements prevent malabsorption and weight loss. Surgical drainage or partial resection of the pancreas may be required to relieve pain and prevent cystic rupture.339 Chronic pancreatitis is a risk factor for pancreatic cancer.340
CANCER OF THE DIGESTIVE SYSTEM
Cancer occurs throughout the alimentary tract and the accessory organs of digestion (liver, gallbladder, and pancreas) (Table 39-10). A genetic predisposition is being evaluated.341
Table 39-10
Cancer of the Gut, Liver, and Pancreas
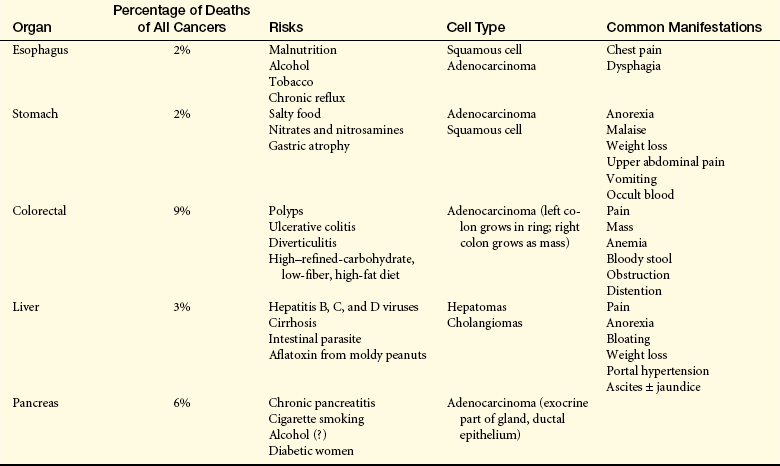
NOTE: Esophageal (men), colorectal, liver, and pancreatic cancers are within the top 10 causes of death from cancer.
From American Cancer Society, Inc., Surveillance and Health Policy Research: Estimated New Cancer Cases and Deaths by Sex. U.S., 2009 available at: www.cancer.org/downloads/stt/CFF2009_EstCD_3.pdf.
Cancer of the Gastrointestinal Tract
Carcinoma of the esophagus is a rare disease with 16,470 new cases and 14,530 deaths each year. Adenocarcinoma is increasing in white men and women in the United States.342 Squamous cell carcinoma is more prevalent in China, Iran, South America, and South Africa and is more common in black men in the United States.343 The incidence in the United States and Europe is less than 1% of new cancers per year.344 The U.S. incidence is higher in blacks than in whites and peaks at about 60 years of age.
Squamous cell carcinoma is associated with malnutrition caused by poor economic conditions, dietary habits, alcoholism, tobacco use, obesity, radiation exposure, and chronic gastroesophageal reflux.345 Adenocarcinoma is associated with reflux esophagitis and sliding hiatal hernia. Both of these conditions can cause erosive esophagitis and ulceration that can eventually lead to metaplasia (Barrett esophagus) and neoplastic changes.
PATHOGENESIS Carcinomas of the esophagus are often secondary to infiltration by a gastric carcinoma or to the presence of Barrett (dysplastic) epithelium (columnar rather than squamous epithelium in the lower esophagus), which is associated with chronic gastroesophageal reflux. The disease progresses from Barrett metaplasia, to dysplasia, to adenocarcinoma, and then metastasis. Carcinomas can occur at any level of the esophageal tract but are most common at the gastroesophageal junction.346
The pathogenesis of esophageal carcinoma is facilitated by (1) alterations of esophageal structure and function that permit food and drink to remain in the esophagus for prolonged periods; (2) ulceration and metaplasia caused by esophageal reflux; and (3) long-term exposure to irritants, such as alcohol and tobacco, that cause neoplastic transformation (see Chapter 11). H. pylori do not colonize the intestinal epithelium of Barrett esophagus and may be a protection against esophageal adenocarcinoma.347 Chronic inadequate nutrition can impair structure and function of the esophagus. Mutation of the TP53 gene is an early event in Barrett adenocarcinoma.348
CLINICAL MANIFESTATIONS Early stages of esophageal carcinoma are asymptomatic. The two main manifestations of esophageal carcinoma are chest pain and dysphagia. The most common type of pain is heartburn (pyrosis). It is initiated by eating spicy or highly seasoned foods and by lying down. Dysphagia (pain on swallowing), another common symptom, is usually pressure-like and may radiate posteriorly between the scapulae. Some individuals with esophageal cancer complain of a constant retrosternal pain that radiates to the back. Dysphagia usually progresses rapidly.
EVALUATION AND TREATMENT Individuals who present with dysphagia undergo endoscopy so that specimens can be obtained and examined for neoplastic change and type of carcinoma. CT studies of the thorax also are used for diagnosis. Prevention and treatment of gastroesophageal reflux are essential to the management of Barrett esophagus. Untreated esophageal cancer metastasizes rapidly and has a poor prognosis. The cancer has often metastasized by the time of diagnosis. The lymphatic vessels of the esophagus are continuous with vital mediastinal structures and drain to the lymph nodes from the neck of the celiac axis, making it impossible to remove all the lymph nodes with the tumor. Removal of the primary lesion and the local lymph nodes, however, can benefit the individual with esophageal cancer and cure is likely if there is not malignancy. If spread has occurred, treatment is combined radiation, chemotherapy, and palliative care.346
Cancer of the Stomach
Although the incidence of gastric cancer has declined in the United States, it still represents about 2% (21,130 cases) of all new cancer cases and 10,620 deaths annually.344 The incidence of gastric cancer is greater in men than in women. In countries such as Japan, the British Isles, and Iceland, the incidence of stomach cancer has remained high consistently. Gastric cancer is the second most common cause of death from cancer in Asia.349 These data illustrate the importance of environmental factors, such as diet, to carcinogenesis.
Nonenvironmental risk factors include a family history of gastric adenocarcinoma; blood type (blood group A); type A atrophic gastritis; and pernicious anemia, which is associated with atrophy of the gastric mucosa in the same locations where gastric tumors arise.350
The most important environmental risk factors in causing gastric cancer are (1) infection with H. pylori that carries the CagA gene product cytotoxin-associated antigen A; (2) salt added to food; (3) food additives (e.g., nitrates) in pickled or salted foods (e.g., bacon); and (4) low intake of fruits and vegetables. Infection with H. pylori and severe chronic gastritis change the mucosal cell proliferation pattern, increasing the risk for gastric and duodenal carcinoma.351,352 H. pylori also is causatively linked to mucosa-associated lymphoid tissue (MALT) lymphoma (a low-grade B-cell lymphoma) that originates in the stomach.353 Vitamin C and carotenoids are possible protective factors.354 Dietary salt enhances the conversion of nitrates to carcinogenic nitrosamines in the stomach. Salt is also caustic to the stomach and can cause chronic atrophic gastritis. Finally, hypertonic salt solutions delay gastric emptying. Delayed emptying increases the time during which carcinogenic nitrosamines can exert their effects on the stomach mucosa.
The metabolism of nitrates and nitrites is very complex. Nitrates interact with amino acids in the stomach to form nitrosamines. The conversion of these carcinogenic nitrosamines is enhanced at a low pH by iodides and thiocyanates. Nitrates are thought to be active only when converted to nitrites and to cause stomach cancer once atrophic gastritis has occurred.
PATHOGENESIS Gastric cancer usually begins in the glands of the stomach mucosa. Approximately 50% of all gastric cancers develop in the prepyloric antrum (Figure 39-27). Atrophic gastritis and intestinal metaplasia are strongly linked to the development of gastric cancer.355 Insufficient acid secretion by the atrophic mucosa creates a relatively alkaline environment that permits bacteria to multiply and act on nitrates. The resulting increase in nitrosoamines damages the deoxyribonucleic acid (DNA) of mucosal cells further, promoting metaplasia and neoplasia. Duodenal reflux also may contribute to intestinal metaplasia. The reflux contains caustic bile salts that destroy the mucosal barrier that normally protects the stomach. Alterations in TP53 and p21 gene expression and DNA ploidy occur in gastric carcinomas.356
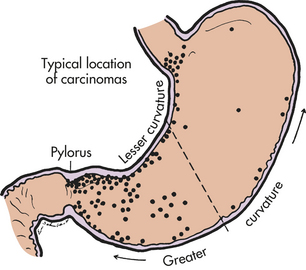
Figure 39-27 Typical sites of stomach cancer. (From del Regato JA, Spjut HJ, Cox JD: Cancer: diagnosis, treatment, and prognosis, ed 2, St Louis, 1985, Mosby.)
CLINICAL MANIFESTATIONS The early stages of gastric cancer are generally asymptomatic or produce vague symptoms such as loss of appetite (especially for meat), malaise, and “indigestion.” Later manifestations include unexplained weight loss, upper abdominal pain, vomiting, change in bowel habits, and anemia caused by persistent occult bleeding. The prognosis is poor because symptoms usually do not occur until the tumor has penetrated the muscle layers of the stomach; spread to surrounding tissues; and entered the draining lymph nodes and veins, causing distant metastases. Generally the first manifestations of carcinoma are caused by distant metastases.
EVALUATION AND TREATMENT The choice of diagnostic tests depends on the clinical manifestations at the time of presentation. Most symptoms suggest a problem in the upper gastrointestinal tract. Direct endoscopic visualization and biopsy usually establish the diagnosis, or microscopic examination of exfoliated cells obtained by lavage during endoscopy.
Surgery is the treatment for gastric cancer. Staging is determined by pathologic findings after resection. Chemotherapy combined with chemoradiotherapy may provide the best postoperative outcomes. Screening and treatment for H. pylori infection is the best preventive approach to gastric cancer, and there are increasing challenges with antibiotic resistance.357
Cancer of the Colon and Rectum
Cancer of the lower intestinal tract (colon [78%] rectum [28%]) is the third most common cause of cancer death in the United States for men and women. Colorectal cancer (CRC) accounts for about 9% of all cancer deaths; 49,920 deaths in 2009.339 Cancer of the colon tends to occur in individuals older than 50 years with 146,970 new cases in 2009. It is more common in African Americans and is rare in children.344 Worldwide, the prevalence of colorectal cancer is highest in populations with high socioeconomic standards, possibly because of dietary and lifestyle habits.358,359
Small intestine carcinoma is rare and represents less than 1% of gastrointestinal cancers (6230 new cases in 2009).344,360 Carcinoma occurs more frequently in familial adenomatous polyposus and Crohn disease. Long term management includes frequent screening and endoscopic surveillance.361 Anal carcinioma is rare (5290 cases in 2009).344 The most common risk factor is infection with human papillomavirus (93%), and less commonly, anal involvment in Crohn disease.361
PATHOGENESIS Genetic and environmental factors are associated with the development of CRC (Figure 39-28). Alterations in the tumor-suppressor TP53 gene are present in 75% to 85% of colorectal cancers.362,363 Allelic deletion on chromosomes 5, 17, and 18 appears to promote transition from normal to malignant colon mucosa. Progression from adenomas to colon cancer involves a multistep cascade of genetic mutations involving the DCC (deleted in colorectal cancer gene) and TP53 (protein 53) tumor-suppressor genes; k-ras (retrovirus-associated DNA sequence) gene mutations that are more common in hyperplastic polyps; loss of heterozygosity in which cells lose one allele of some genes with loss of genetic stability and tumor suppressor function; and DNA methylation that can silence DNA expression and inactivate tumor suppression. Cyclooxygenase-2 (COX-2) is variably expressed in right-sided and left-sided colorectal cancer, and COX-2 may participate in regulation of apoptosis, angiogenesis, and invasiveness of adenomatous polyposis coli.364 Numerous environmental factors can increase the risk of colon cancer, presumably by modulating these molecular pathways.365
The adenomatous polyposis coli (APC) gene was first identified as the gene mutated (chromosome 5q) in an autosomal dominant inherited syndrome of colon cancer known as familial adenomatous polyposis (FAP) coli. These cancers are more common in the left colon. Hereditary nonpolyposis colon cancer (HNPCC), also known as Lynch syndrome, is less common and caused by an inherited mutation in a DNA mismatch repair (MMR) gene.366 These tumors are more common in the right colon and occur in other organs.367 Hereditary colon cancers have a high risk of occurrence and represent about 3% to 5% of all colon cancers368; most colon cancers are sporadic and caused by somatic mutations.
Most CRC develops from polyps. A polyp is a finger-like projection arising from the mucosal epithelium. Most polyps are benign. The two types of neoplastic polyps are hypoplastic and adenomatous. Most colon cancers arise from adenomatous polyps (Figure 39-29). Hypoplastic polyps generally lack atypia but when they are large (greater than 1 cm), numerous (more than 20), and located in the right colon they are associated with cancer. The two major types of adenomatous polyps are pedunculated (stalk or tubular), the most common, and sessile (papillary, villous or serrated) (Figure 39-30). Once the adenoma traverses the muscularis mucosae, it becomes invasive and highly malignant. Adenomas can be detected early, however, and the submucosa may not be penetrated for several years. The larger the polyp is, the greater the risk of colorectal cancer. Although lesions larger than 1.5 cm occur less often, they are more likely to be malignant than those smaller than 1 cm. The adenomatous polyp forms in an area of epithelial cell hyperproliferation and crypt dysplasia. Table 39-11 gives other conditions commonly confused with colorectal cancer.
Table 39-11
Conditions Commonly Confused with Colorectal Cancer
| Condition | Significant Characteristics |
| Diverticulitis | Left-sided pain similar to that of appendicitis; tender lower left quadrant. Associated findings: nausea, vomiting, fever, obstruction, anorexia, and leukocytosis; mucosa is intact, and perforation, peritonitis, and abscesses occur more often than in cancer; ultrasound, CT scan, MRI, and proctosigmoidoscopy are used to distinguish from cancer |
| Ulcerative colitis | Younger people with chronic attacks of bloody diarrhea, crampy abdominal pain, fever, malnutrition, and dehydration; usually involves the left colon and rectum; endoscopy, barium enema, and biopsy performed for definitive diagnosis |
| Crohn disease (granulomatous colitis) | Generally involves the right colon; chronic diarrhea with abdominal cramps, fever, weight loss, and often a palpable abdominal mass; difficult at times to distinguish Crohn disease from ulcerative colitis; endoscopic examination and CT scan used to distinguish from cancer |
| Appendicitis | Vague abdominal symptoms, often with a tender or nontender mass in the lower right quadrant; associated symptoms: mild fever and leukocytosis; CT scan used to distinguish cancer of the cecum from appendiceal abscess |
| Thrombosed hemorrhoids | Examination shows a tender, swollen, bluish painful mass in the anus; individual has a history of hemorrhoids |
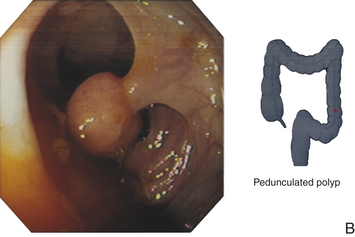
Figure 39-29 Development of cancer of the colon from adenomatous polyps. A, The tumor becomes invasive if it penetrates the muscularis mucosae and enters the submucosal layer. B, Endoscopic image of pedunculated polyp in descending colon. (A from del Regato JA, Spjut HJ, Cox JD: Cancer: diagnosis, treatment, and prognosis, ed 2, St Louis, 1985, Mosby. B courtesy David Bjorkman, MD, University of Utah School of Medicine, Department of Gastroenterology, Salt Lake City.)
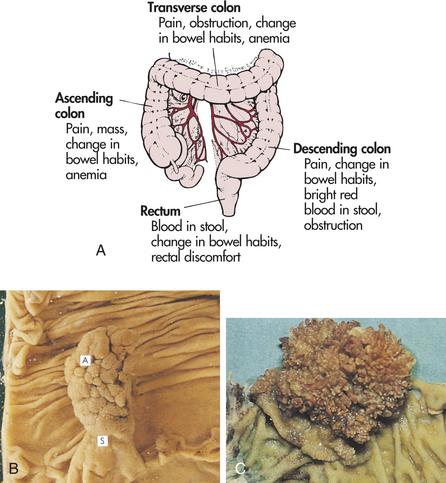
Figure 39-30 Signs and symptoms of colorectal cancer by location of primary lesion. A, Clinical manifestations are listed in order of frequency for each region (lymphatics of colon also shown). B, Tubular adenomata (A) are rounded lesions 0.5 to 2 cm in size that are generally red and sit on a stalk (S) of normal mucosa that has been dragged up by traction of the polyp in the bowel lumen. C, Villous adenomata are frondlike lesions about 0.6 cm thick that occupy a broad area of mucosa generally 1 to 5 cm in diameter. (B and C from Stevens A, Lowe J: Pathology, ed 2, London, 2000, Mosby.)
Most colorectal cancers are moderately differentiated adenocarcinomas. These tumors have a long preinvasive phase, and when they invade they tend to grow slowly. Because the lymphatic channels are located underneath the muscularis mucosae, the lesions must traverse this layer before metastasis can occur. Systemic lymphatic spread occurs along the aorta to the mesenteric and pancreatic lymph nodes. Liver metastasis follows invasion of the mesenteric veins (left colon) or superior veins (right colon), which drain into the portal circulation.
Lower risk for colon cancer may be associated with diets high in cereal grains, vegetables, folic acid, calcium, and vitamin D; hormone replacement therapy; physical activity; and use of NSAIDs.369,370 Cancer-promoting mechanisms are related to prolonged contact of the fecal mass with colon mucosa, including processed red meats, and substances produced by microflora of the sigmoid colon.371,372
CLINICAL MANIFESTATIONS Tumors of the right (ascending) and left (descending) colon evolve into two distinct types. On the right side the lesions are polypoid and extend along one wall of the cecum and ascending colon. Clinical manifestations include pain, a palpable mass in the lower right quadrant, anemia, and dark red or mahogany-colored blood mixed with the stool (see Figure 39-30). These large, bulky tumors become necrotic and ulcerated, contributing to persistent blood loss and anemia. Obstruction is unusual because the feces are more liquid.
Tumors of the left, or descending, colon start as small, elevated, button-like masses. They grow circumferentially and spread along the entire bowel wall, eventually ulcerating in the middle as the tumor penetrates the blood supply. Obstruction is common but occurs slowly. Manifestations include progressive abdominal distention, pain, vomiting, constipation, need for laxatives, cramps, and bright red blood on the surface of the stool.
Rectal carcinomas are defined as tumors occurring up to 15 cm from the anal opening. Tumors of the rectum can spread through the rectal wall to nearby structures: the prostate in men and the vagina in women. Penetration occurs more readily in the lower third of the rectum because it has no serosal covering. Systemic and pulmonary metastases occur through the hemorrhoidal plexus, which drains into the vena cava.
EVALUATION AND TREATMENT Individuals with hereditary polyposis should begin screening at an early age (10 to 12 years) using colonoscopy with a consideration of prophylactic surgery.367 Genetic markers in stool and blood are being developed.373 Screening for nonhereditary CRC in asymptomatic individuals older than age 50 includes fecal occult blood and immunochemical tests, and sigmoidoscopy or colonoscopy.374,375 A diet rich in vegetables, grains, fruit, and calcium and low in fat and red meat may modify cancer risk. Uncertainties remain in the dietary modulation of colon cancer.376,377 In 8% to 29% of cases, bowel obstruction is the primary symptom at diagnosis.378
The staging of colorectal cancer involves preoperative testing and operative exploration. Preoperative testing begins with physical examination of the abdomen to detect liver enlargement and ascites and palpation of appropriate lymph nodes. Elevations of carcinoembryonic antigen (CEA) are often detected in the sera of individuals with colorectal carcinoma. The amount of CEA in the serum is a function of the stage of the disease and the type of tumor. Operative staging consists of careful exploration during surgery and biopsy of possible metastases. The National Cancer Institute379 classification is widely used for staging of colorectal cancer and is as follows:
Stage 0 (carcinoma in situ): involves only the mucosal lining, also known as carcinoma in-situ
Stage I: extension of cancer to the middle layers of the colon wall, no spread to lymph nodes. Stage I colon cancer is sometimes called Dukes’A colon cancer.
Stage II: extension beyond the colon wall to nearby tissues around the colon or rectum, and/or through the peritoneum. Stage II colon cancer is sometimes called Dukes’ B colon cancer.
Stage III: spread beyond the colon into lymph nodes and nearby organs and/or through the peritoneum. Stage III colon cancer is sometimes called Dukes’ C colon cancer.
Stage IV: spread to nearby lymph nodes and has spread to other parts of the body, such as the liver or lungs. Stage IV colon cancer is sometimes called Dukes’D colon cancer.
Treatment for cancer of the colon is always surgical. The location and amount of colon resected depend on the site of the cancer. Resection and anastomosis can be performed for cancer of the ascending, transverse, descending, or sigmoid colon and upper rectum. These surgeries are performed through abdominal incisions, and natural defecation is preserved.
Growths in the lower portion of the rectum require removal of the entire rectum with formation of a permanent colostomy. Prognosis after surgery depends on the stage and location of the tumor.
Radiation therapy is often given before surgery in the hope that it will shrink the tumor, alter the malignant cells, or both so that these cells will not survive after surgery. Adjuvant chemotherapy is used to treat metastatic disease and cases with a high risk of recurrence. New chemotherapeutic agents are improving first line treatment.380 Immunotherapy can boost the immune response.381 Recombinant vaccines for colon cancer are in clinical trials.382,383 Molecular prognostic markers are being developed.384
Cancer of the Accessory Organs of Digestion
Cancer of the liver is the fifth most common cause of cancer and the third leading cause of cancer death worldwide; however, the incidence is increasing as a result of chronic hepatitis B and C infection.385 Cancer in the liver is usually caused by metastatic spread from a primary site elsewhere in the body. Liver cancer is relatively rare in the United States but is common in densely populated parts of the Far East, southern Africa, China, and Greece. The number of deaths in the United States from liver cancer is greatest among Asians and Pacific Islanders (the third leading cause of cancer deaths) and is higher in blacks and Hispanics compared with whites. For reasons not understood, incidence is higher in men than in women.386 Primary liver cancer is rare before the age of 40 years and most common during the sixth decade. Together, primary and secondary liver cancer accounts for about 3% of all cancer deaths in the United States.344 The incidence of hepatocellular carcinoma is increasing from dissemination of hepatitis B and, particularly, C infection.387
Risk factors for primary liver cancer include the following:
1. Infection with HBV, HCV, and HDV, particularly in conjunction with cirrhosis, acts either as a carcinogen or as a co-carcinogen in chronically infected hepatocytes.388
2. Chronic liver disease, especially cirrhosis.389
3. Exposure to mycotoxins. The most significant mycotoxins are the aflatoxins, particularly those produced by Aspergillus flavus, a mold found on spoiled corn, peanuts, and grain. Aflatoxins cause mutation of the TP53 suppressor gene and activation of WNT signal transduction pathway.390
4. Heavy smoking and heavy drinking of alcohol.391,392
5. The presence of liver flukes (ingestion of raw fish) in Southeast Asia.393
PATHOGENESIS Primary carcinomas of the liver are hepatocellular or cholangiocellular. Hepatocellular carcinoma develops in the hepatocytes, whereas cholangiocellular carcinoma (cholangiocarcinoma) develops in the bile ducts. Hepatocellular carcinoma (hepatocarcinoma) (HCC) can be nodular (consisting of multiple, discrete nodules), massive (consisting of a large tumor mass having satellite nodules), or diffuse (consisting of very small nodules distributed throughout most of the liver). HCC is the type of primary liver cancer that is closely associated with cirrhosis (Figure 39-31). Chronic hepatitis and cirrhosis give rise to HCC repetitive cellular proliferation that occurs in the inflamed liver in response to growth factor and cytokine stimulation. Numerous genetic and epigenetic alterations, including failure of tumor-suppressor genes and signaling pathways, combine to promote carcinogenesis.394,395 Because carcinoma of the liver invades the hepatic and portal veins, it often spreads to the heart and lungs. Other sites of metastases are the brain, kidney, and spleen.
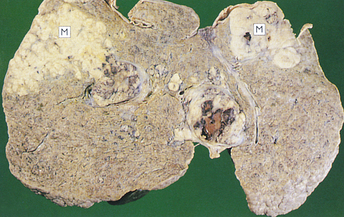
Figure 39-31 Hepatocellular carcinoma. Macroscopically, hepatocellular carcinomas may be single or multifocal. They usually develop in a liver already affected by cirrhosis. Tumor appears as an abnormal mass (M) within the liver. (From Stevens A, Lowe J: Pathology, ed 2, London, 2000, Mosby.)
Cholangiocellular carcinomas (cholangiocarcinoma) occur less often than hepatocellular carcinomas in the United States. This type of primary liver cancer is most common in areas where liver fluke infestation is prevalent in many parts of Southeast Asia. The mechanism by which fluke infestation causes cholangiocellular carcinoma is multifactorial and includes parasite secretions, immunopathology, and mechanical damage.388 Other risk factors include primary sclerosing cholangitis, hepatolithiasis, and choledochal cysts. However, most individuals diagnosed with cholangiocellular carcinoma do not have an identifiable risk factor.396 Cholangiocellular carcinoma can occur anywhere along the bile duct and extend directly into the liver, usually as a solitary lesion. It is difficult to distinguish an invasion of cholangiocellular carcinoma from a metastatic adenocarcinoma except by neoplastic changes found in nearby ducts.
CLINICAL MANIFESTATIONS The clinical presentation of liver cancer in adults is characterized by vague abdominal symptoms, such as nausea and vomiting, fullness, pressure, dull ache in the right hypochondrium, and weight loss. Manifestations of hepatocellular carcinoma can occur slowly or abruptly. In individuals with cirrhosis, deepening jaundice or abrupt lack of appetite is a sign of hepatocellular carcinoma. Obstruction by the tumor can cause sudden worsening of portal hypertension and development of ascites. As the tumor enlarges, it causes pain. Cholangiocellular carcinoma more commonly presents insidiously as pain, loss of appetite, weight loss, and gradual onset of jaundice. Some carcinomas of the liver rupture spontaneously, causing hemorrhage. Others are discovered accidentally during evaluation of a bone fracture or surgical exploration.
EVALUATION AND TREATMENT There is no specific test for the diagnosis of liver cancer. For high-risk individuals alpha fetoprotein and abdominal ultrasound are common screening tools. The diagnosis is based on additional laboratory findings, radiologic examination, biopsy findings, and exploratory laparotomy.397 Serum levels of alkaline phosphatase, aspartate aminotransferate (AST), and alanine aminotransferase (ALT) are commonly elevated in individuals with hepatocellular carcinoma. Excessive levels of alpha fetoprotein (>400 ng/ml) have been correlated with hepatitis B antigen, rapid tumor growth, and poor tumor differentiation. Erythrocytosis is secondary to erythropoietin production. Hepatitis B vaccination and antiviral treatment of hepatitis C will advance prevention of HCC.398
Staging of the tumor is important to guiding treatment but does not accurately predict postoperative outcome.399 Surgical resection is possible in noncirrhotic individuals if the tumor is localized to a removable lobe. Tumors of the posterior segment of the right lobe are not resectable, because this segment contains the right hepatic vein. Radiofrequency (thermal) ablation has emerged as the most effective method for local tumor destruction.400 Tyrosine kinase inhibitors and monoclonal antibodies are improving survival. Transarterial embolization and radiation is used for management of pain and to reduce tumor size. Surgical resection or liver transplant is the only alternative for cure.401
Cancer of the Gallbladder
Cancer of the gallbladder and biliary tract is a rare but lethal disease. It is more common in women than in men by a ratio of about 2 to 1.344 It occurs rarely before age 40 and is most common between the ages of 50 and 60 years. Obesity is a risk factor. Native populations in North and South America have greater risk of gallbladder cancer, and it is more common in Chile, Poland, India, Japan, and Israel.402,403 Most gallbladder cancer is caused by metastasis. Primary carcinoma of the gallbladder is rare and is usually associated with chronic cholecystitis and cholelithiasis.
PATHOGENESIS Most primary carcinomas of the gallbladder are adenocarcinomas. A few are squamous cell carcinomas; TP53 gene mutation, altered expression of P-glycoprotein, COX-2, epidermal growth factor receptor, and K-ras gene mutation occur.404 Invasion of the liver occurs early. Spreading progresses to the cystic and periportal lymph nodes with invasion of the pancreas and retroperitoneal lymph nodes. Direct invasion of the stomach and the duodenum can cause pyloric obstruction. Infection often accompanies cancer of the gallbladder. Generalized peritonitis, gangrene, perforation, and liver abscesses are potential complications of infection.
CLINICAL MANIFESTATIONS A typical presentation of carcinoma of the gallbladder is steady upper-right-quadrant pain for about 2 months. Other manifestations include diarrhea, belching, weakness, loss of appetite, weight loss, and vomiting. Obstructive jaundice can occur if an enlarging tumor presses on the extrahepatic ducts.
EVALUATION AND TREATMENT Early diagnosis of cancer of the gallbladder is not possible because of lack of symptoms, and individuals present with an advanced stage of disease. Individuals with gallstones, especially older women, are evaluated carefully. Inflammatory disorders, such as cholangitis (bile duct inflammation) and peritonitis, often obscure an underlying malignancy. The most specific diagnostic procedures include ultrasonography, CT, and MRI.
Complete surgical resection of the gallbladder is the only effective treatment. Because advanced malignancies cannot be resected, gallbladders containing stones are removed as a preventive measure. Palliative chemotherapy provides symptom improvement but does not improve survival.405 The prognosis of gallbladder cancer is extremely poor; most individuals die within 1 to 2 years after surgery.406
Cancer of the Pancreas
Pancreatic cancer now ranks fourth in men in as a cause of cancer deaths in the United States. The incidence of pancreatic cancer rises steadily with age. Men are affected slightly more often than women and blacks more often than whites. Pancreatic cancer accounts for about 35,240 deaths annually in the United States.344 Mortality is about 95% within 12 months. Pancreatic cancer is a disease of inherited and acquired mutation in cancer-related genes.407 With the exception of an association of risk with cigarette smoking, no external risk factors have been identified.408 Pancreatitis is associated with 50% of pancreatic cancers.409
PATHOGENESIS Cancer of the pancreas can arise from exocrine or endocrine cells. Most pancreatic tumors arise from exocrine cells in the ducts and are called ductal adenocarcinomas. Tumors arising in small ducts invade nearby glandular tissue, penetrate the covering of the pancreas, and extend into surrounding tissues.410 A K-ras mutation is the most common genetic alteration; tumor suppressor gene alterations are also found, including TP53, p16, and DCC. Growth factors are overexpressed in ductal cancer.411
Ductal adenocarcinomas can occur in the head, body, or tail of the pancreas. Tumors of the head quickly spread to obstruct the common bile duct and portal vein (Figure 39-32). These tumors can then infiltrate the superior mesenteric artery, the vena cava, and the aorta. Cancer cells that enter the blood vessels can form emboli. Tumors of the body and tail infiltrate the posterior abdominal wall. Lymphatic invasion occurs early and rapidly and involves local and regional lymph nodes. Venous invasion causes metastases to the liver. Tumor implants on the peritoneal surface can obstruct veins and promote development of ascites.
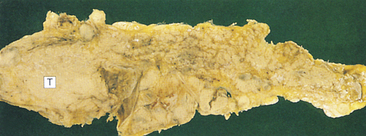
Figure 39-32 Pancreatic carcinoma. Tumors appear as gritty, gray, hard nodules (T) irregularly invading the adjacent gland and local structures. (From Stevens A, Lowe J: Pathology, ed 2, London, 2000, Mosby.)
Ductal adenocarcinomas arising in the head of the pancreas cause biliary obstruction somewhat early in the disease. Individuals with such tumors survive slightly longer than those with cancer of the body and tail, presumably because they seek medical attention earlier.
Tumors of the endocrine pancreas are rare neoplasms of the islets of Langerhans known as apudomas. The first four letters in apudoma derive from amine precursor uptake and decarboxylation. The apudomas are so named because they contain neurosecretory granules. Endocrine neoplasms secrete abnormal amounts of hormones, such as insulin.
CLINICAL MANIFESTATIONS Cancer of the body and tail of the pancreas is generally asymptomatic until there is intraductal obstruction or the tumor invades adjacent tissue. Often vague back pain is an initial symptom.412 Jaundice develops in most cases, usually caused by obstruction of the bile duct. Because obstruction impairs enzyme secretion and flow to the duodenum, pancreatic cancer causes fat and protein malabsorption, resulting in weight loss. Distant metastases are found in the neck nodes, the lungs, and the brain. Most individuals die of hepatic failure, malnutrition, or systemic diseases.
EVALUATION AND TREATMENT Pancreatic cancer is usually at an advanced stage at the time of diagnosis and has a poor prognosis.413 A laparotomy is often performed, particularly if jaundice is present. Ultrasonography, CT, or endodoscopic retrograde cholangiopancreatography may be needed to confirm the need for a laparotomy, especially in individuals without jaundice. Laparotomy is used to establish a definitive diagnosis, evaluate the extent of disease, and determine whether palliative bypass surgery (i.e., cholecystojejunostomy and gastrojejunostomy) is needed. Most individuals require palliative double bypass of the blocked bile ducts, as well as gastrojejunostomy to prevent duodenal obstruction.
Many surgeons recommend a total pancreatectomy because cancer of the pancreas seldom consists of a single lesion.414 Adjuvant chemotherapy and new systemic agents are improving survival.415 Radiochemotherapy may produce favorable controls in locally advanced cancer.416
Achalasia 1457
Acute gastritis 1463
Acute occlusion of mesenteric artery blood flow (acute mesenteric ischemia) 1477
Acute pancreatitis (acute hemorrhagic pancreatitis) 1496
Afferent loop obstruction 1470
Alcoholic cirrhosis 1492
Alcoholic hepatitis 1492
Alkaline reflux gastritis 1469
Anal carcinoma 1500
Anorexia 1452
Anorexia nervosa (AN) 1480
Appendicitis 1475
Ascites 1483
Biliary cirrhosis 1493
Bulimia nervosa 1481
Cholangiocellular carcinoma (cholangiocarcinoma) 1503
Cholecystitis 1495
Cholelithiasis 1494
Chronic active hepatitis 1490
Chronic alcoholic hepatitis 1492
Chronic gastritis 1464
Chronic mesenteric insufficiency 1477
Chronic pancreatitis 1497
Cirrhosis 1491
Constipation 1453
Crohn disease (CD) 1473
Curling ulcer 1467
Cushing ulcer 1467
Diarrhea 1454
Diverticula (sing., diverticulum) 1474
Diverticulitis 1474
Diverticulosis 1474
Dumping syndrome 1468
Duodenal ulcer 1465
Dysphagia 1456
Eosinophilic esophagitis 1459
Esophageal varices 1483
Fulminant hepatitis 1491
Gallstone 1494
Gastric ulcer 1467
Gastritis 1463
Gastroesophageal reflux disease (GERD) 1458
Gluconeogenesis 1481
Glycogenolysis 1481
Hematemesis 1456
Hematochezia 1456
Hemolytic jaundice (prehepatic jaundice, nonobstructive jaundice) 1486
Hepatic encephalopathy 1485
Hepatocellular carcinoma (hepatocarcinoma; HCC) 1503
Hepatopulmonary syndrome 1483
Hepatorenal syndrome (HRS) 1487
Hiatal hernia 1459
Hyperbilirubinemia 1485
Icteric phase (jaundice) 1490
Incubation phase of hepatitis 1490
Intestinal obstruction 1460
Irritable bowel syndrome (IBS) 1476
Ischemic ulcer 1467
Jaundice (icterus) 1485
Kwashiorkor 1481
Lactase deficiency 1470
Large bowel obstruction 1461
Leptin resistance 1479
Long-term starvation 1481
Lower gastrointestinal bleeding 1456
Malabsorption 1470
Maldigestion 1470
Marasmus 1481
Melena 1456
Mesenteric venous thrombosis 1477
Motility diarrhea 1455
Mucosa-associated lymphoid tissue (MALT) lymphoma 1499
Nausea 1453
Obesity 1477
Obstructive jaundice 1485
Occult bleeding 1456
Osmotic diarrhea 1454
Polyp 1500
Pancreatic insufficiency 1470
Pancreatitis 1495
Paraesophageal hiatal hernia 1459
Paralytic ileus 1460
Parietal pain 1455
Peptic ulcer 1464
Polyp 1500
Portal hypertension 1482
Primary biliary cirrhosis 1493
Prodromal (preicteric) phase 1490
Projectile vomiting 1453
Pyloric obstruction 1460
Recovery phase 1490
Rectal carcinoma 1501
Referred pain 1456
Reflux esophagitis 1458
Retching 1453
Secondary biliary cirrhosis 1493
Secretory diarrhea 1454
Short bowel syndrome 1474
Short-term starvation 1481
Sliding hiatal hernia 1459
Small intestine carcinoma 1500
Small intestine obstruction 1461
Splenomegaly 1483
Starvation 1481
Stress ulcer (stress-related mucosal disease) 1467
Ulcerative colitis (UC) 1471
Upper gastrointestinal bleeding 1456
Viral hepatitis 1488
Visceral pain 1455
Vomiting 1452
Zollinger-Ellison syndrome 1467
REFERENCES
1. Meadows, N. The central control of vomiting. J Pediatr Gastroenterol Nutr. 1995;21(Suppl 1):S20–S21.
2. Navari, R.M. Prevention of emesis from multiple-day and high-dose chemotherapy regimens. J Natl Compr Canc Netw. 2007;5(1):51–59.
3. Santucci, G., Mack, J.W. Common gastrointestinal symptoms in pediatric palliative care: nausea, vomiting, constipation, anorexia, cachexia. Pediatr Clin North Am. 2007;54(5):673–689.
4. Talley, N.J. Definitions, epidemiology, and impact of chronic constipation. Rev Gastroenterol Disord. 2004;4(Suppl 2):S3–S10.
5. Loening-Baucke, V. Prevalence rates for constipation and faecal and urinary incontinence. Arch Dis Child. 2007;92(6):486–489.
6. Mitolo-Chieppa, D., et al. Cholinergic stimulation and nonadrenergic, noncholinergic relaxation of human colonic circular muscle in idiopathic chronic constipation. Dig Dis Sci. 1998;43(12):2719–2726.
7. Spinzi, G.C. Bowel care in the elderly. Dig Dis. 2007;25(2):160–165.
8. Panchal, S.J., Muller-Schwefe, P., Wurzelmann, J.I. Opioid-induced bowel dysfunction: prevalence, pathophysiology and burden. Int J Clin Pract. 2007;61(17):1181–1187.
9. Rao, S.S. Constipation: evaluation and treatment of colonic and anorectal motility disorders. Gastroenterol Clin North Am. 2007;36(3):686–711.
10. Chiarioni, G., Heymen, S., Whitehead, W.E. Biofeedback therapy for dyssnergic defecation. World J Gastroenterol. 2006;12(44):7069–7074.
11. Feldman, M., Friedman, L.S., Brandt, L.J. ed 8. Sleisenger & Fordtran’s Gastrointestinal and Liver Diseases. Philadelphia: Saunders; 2006;vol. 1.
12. Spiller, R. Role of motility in chronic diarrhoea. Neurogastroenterol Motil. 2006;18(12):1045–1055.
13. Sellin, J.H. A practical approach to treating patients with chronic diarrhea. Rev Gastroenterol Disord. 2007;7(Supp l3):S19–S26.
14. Al-Chaer, E.D., Traub, R.J. Biological basis of visceral pain: recent developments. Pain. 2002;96(3):221–225.
15. Vergnolle, N. Postinflammatory visceral sensitivity and pain mechanisms. Neurogastroenterol Motil. 2008;20(Suppl 1):73–80.
16. DiMaio, C.J., Stevens, P.D. Nonvariceal upper gastrointestinal bleeding. Gastrointest Endosc Clin North Am. 2007;17(2):253–272. [v, 2007].
17. Edelman, D.A., Sugawa, C. Lower gastrointestinal bleeding: a review. Surg Endosc. 2007;21(4):514–520.
18. Killip, S., Bennett, J.M., Chambers, M.D. Iron deficiency anemia. Am Fam Physician. 2007;75(5):671–678.
19. Vela, M.F., Vaezi, M.F. Cost-assessment of alternative management strategies for achalasia. Expert Opin Pharmacother. 2003;4(11):2019–2025.
20. Boeckxstaens, G.E. Achalasia. Best Pract Res Clin Gastroenterol. 2007;21(4):595–608.
21. Nellemann, H., et al. Bread and barium: diagnostic value in patients with suspected primary esophageal motility disorders. Acta Radiol. 2000;41(2):145–150.
22. Smout, A.J. Advances in esophageal motor disorders. Curr Opin Gastroenterol. 2008;24(4):485–489.
23. Pohl, D., Tutuian, R. Achalasia: an overview of diagnosis and treatment. J Gastrointestin Liver Dis. 2007;16(3):297–303.
24. Long, J.D., Orlando, R.C. Nonerosive reflux disease. Minerva Gastroenterol Dietol. 2007;53(2):127–141.
25. Richter, J.E. Gastrooesophageal reflux disease. Best Pract Res Clin Gastroenterol. 2007;21(4):609–631.
26. Havemann, B.D., Henderson, C.A., El-Serag, H.B. The association between gastro-oesphageal reflux disease and asthma: a systematic review,. Gut. 2007;56(12):1654–1664.
27. Johanson, J.F. Epidemiology of esophageal and supraesophageal reflux injuries. Am J Med. 2000;108(Suppl 41):99S.
28. Orlando, R.C. Mechanisms of reflux-induced epithelial injuries in the esophagus. Am J Med. 2000;108(Suppl 4a):104S.
29. Rajendra, S., et al. Helicobacter pylori, ethnicity, and the gastroesophageal reflux disease spectrum: a study from the East. Helicobacter. 2007;12(2):177–183.
30. Holmes, R.S., Vaughan, T.L. Epidemiology and pathogenesis of esophageal cancer. Semin Radiat Oncol. 2007;17(1):2–9.
31. Anderson, L.A., et al. Relationship between Helicobacter pylori infection and gastric atrophy and the stages of the oesophageal inflammation, metaplasia, adenocarcinoma sequence: results from the FINAR case-control study. Gut. 2008;57(6):734–739.
32. Sqouros, S.N., Mantides, A. Refractory heartburn to proton pump inhibitors: epidemiology, etiology and management. Digestion. 2006;73(4):218–227.
33. Haggitt, R.C. Histopathology of reflux-induced esophageal and supraesophageal injuries. Am J Med. 2000;108(Suppl 4a):109S.
34. Hornick, J.L., Odze, R.D. Neoplastic precursor lesions in Barrett’s esophagus. Gastroenterol Clin North Am. 2007;36(4):775–796.
35. Holmes, R.L., Fadden, C.T. Evaluation of the patient with chronic cough. Am Fam Physician. 2004;69(9):2159–2166.
36. Vaezi, M.F. Extraesophageal manifestations of gastroesophageal reflux disease. Clin Cornerstone. 2003;5(4):32–38. [discussion 39-40, 2003].
37. Levine, M.S., Rubesin, S.E., Laufer, I. Barium esophagography: a study for all seasons. Clin Gastroenterol Hepatol. 2008;6(1):11–25.
38. Tytgat, G.N., et al. New algorithm for the treatment of gastro-oesphageal reflux disease. Aliment Pharmacol Ther. 2008;27(3):249–256.
39. Vakil, N. Review article: the role of surgery in gastro-oesophageal reflux disease. Aliment Pharmacol Ther. 2007;25(12):1365–1372.
40. Blanchard, C., Rothenberg, M.E. Basic pathogenesis of eosinophilic esophagitis. Gastrointest Endosc Clin North Am. 2008;18(1):133–143.
41. Straumann, A. The natural history and complications of eosinophilic esophagitis,. Gastrointest Endosc Clin North Am. 2008;18(1):99–118.
42. Boushey, R.P., et al. Laparoscopic repair of paraesophageal hernias: a Canadian experience. Can J Surg. 2008;51(5):355–360.
43. Karmali, S., et al. Primary laparoscopic and open repair of paraesophageal hernias: a comparison of short-term outcomes. Dis Esophagus. 2008;21(1):63–68.
44. Stawowy, M., et al. Endoscopic stenting for malignant gastric outlet obstruction. Surg Laparosc Endosc Percutan Tech. 2007;17(1):5–9.
45. Maron, D.F., Fry, R.D. New therapies in the treatment of postoperative ileus after gastrointestinal surgery. Am J Ther. 2008;15(1):59–65.
46. Attard, J.A., MacLean, A.R. Adhesive small bowel obstruction: epidemiology, biology and prevention. Can J Surg. 2007;50(4):291–300.
47. Tumage, R.H., Heldmann, M., Cole, P. Intestinal obstruction and ileus. In Feldman M., Friedman L.S., Lawrence B.L., eds.: Sleisenger & Fordtran’s gastrointestinal and liver disease, ed 8, Philadelphia: Saunders, 2006.
48. Cappell, M.S., Batke, M. Mechanical obstruction of the small bowel and colon. Med Clin North Am. 2008;92(3):575–577. [viii, 2008].
49. Cerro, P., et al. Sonographic diagnosis of intussusceptions in adults. Abdom Imaging. 2000;25(1):45.
50. Maglinte, D.D., et al. Small-bowel obstruction: state-of-the-art imaging and its role in clinical management. Clin Gastroenterol Hepatol. 2008;6(2):130–139.
51. McNamara, R., Mihalakis, M.J. Acute colonic pseudo-obstruction: rapid correction with neostigmine in the emergency department. J Emerg Med. 2008;35(2):167–170.
52. Parfitt, J.R., Driman, D.K. Pathological effects of drugs on the gastrointestinal tract: a review. Hum Pathol. 2007;38(4):527–536.
53. Yardley, J.H., Hendrix, T.R. Gastritis and gastropathy. In Yamada T., Alpers D.H., Laine L., eds.: Atlas of gastroenterology, ed 3, Philadelphia: Lippincott Williams & Wilkins, 1999.
54. Neesse, A., et al. Multifiocal early gastric cancer in a patient with autoimmune atrophic gastritis and iron defciency anaemia. Z Gastroenterol. 2009;47(2):223–227.
55. Kapadia, C.R. Gastric atrophy, metaplasia, and dysplasia: a clinical perspective. J Clin Gastroenterol. 2003;36(5 Suppl):S29–S36. [discussion S61-S62, 2003].
56. D’Elios, M.M., et al. Gastric autoimmunity: the role of Helicobacter pylori and molecular mimicry. Trends Mol Med. 2004;10(7):316–323.
57. Oksanen, A., et al. Atrophic gastritis and Helicobacter pylori infection in outpatients referred for gastroscopy. Gut. 2000;46(4):460.
58. Presotto, F., et al. Helicobacter pylori infection and gastric autoimmune disease: is there a link? Helicobacter. 2003;8(6):578–584.
59. Annibale, B., Laqhner, E. Assessing the severity of atrophic gastritis. Eur J Gastroenterol Hepatol. 2007;19(12):1059–1063.
60. Boyanova, L., et al. Prevalence and evolution of Helicobacter pylori resistance to 6 antibacterial agents over 12 years and correlation between susceptibility testing methods. Diagn Microbiol Infect Dis. 2008;60(4):409–415.
61. Erkurt, M.A., et al. Effects of cyanocobalamin on immunity in patients with pernicious anemia. Med Princ Pract. 2008;17(2):131–135.
62. Lethbridge-Cejku, M., Vickerie, J. Summary health statistics for U.S. adults: National Health Interview Survey, 2003. Vital Health Stat. 10(225), 2005.
63. Goodwin, R.D., Stein, M.B. Generalized anxiety disorder and peptic ulcer disease among adults in the United States. Psychosom Med. 2002;64:862–866.
64. Razvodovsky, Y.E. Suicide and stomach ulcer mortality rate in Russia. Psychiatr Danub. 2007;19(1-2):35–41.
65. Levenstein, S. The very model of a modern etiology: a biopsychosocial view of peptic ulcer [review],. Psychosom Med. 2000;62(2):176.
66. Hobsley, M., Tovey, F.I., Holton, J. Precise role of H. pylori in duodenal ulceration. World J Gastroenterol. 2006;12(40):6413–6419.
67. Ji, K.Y., Hu, F.L. Interaction or relationship between Helicobacter pylori and non-steroidal anti-inflammatory drugs in upper gastrointestinal diseases. World J Gastroenterol. 2006;12(24):3789–3792.
68. Kurata, J.H. Epidemiology of peptic ulcer disease. In: Swabb E.A., Szabo S., eds. Investigation and basis for therapy. New York: Marcel Dekker, 1991.
69. Schineller, B.A., Ramchandani, D. Psychologic factors associated with peptic ulcer disease. Med Clin North Am. 1991;75(4):865.
70. Kuyvenhoven, J.P., Veenendaal, R.A., Vandenbroucke, J.P. Peptic ulcer bleeding: interaction between non-steroidal anti-inflammatory drugs, Helicobacter pylori infection, and the ABO blood group system. Scand J Gastroenterol. 1999;34(11):1082–1086.
71. Sharara, A.I., et al. Association of gastroduodenal disease phenotype with ABO blood group and Helicobacterp pylori virulence-specific serotypes. Dig Liver Dis. 2006;38(11):829–833.
72. Gisbert, J.P., et al. H. pylori-negative duodenal ulcer prevalence and causes in 774 patients. Dig Dis Sci. 1999;44(11):2295.
73. Tovey, F.I., Hobsley, M. Is Helicobacter pylori the primary cause of duodenal ulceration? J Gastroenterol Hepatol. 1999;14(11):1053.
74. Axon, A.T. Relationship between Helicobacter pylori gastritis, gastric cancer and gastric acid secretion. Adv Med Sci. 2007;52:55–60.
75. Hirschi, A.M., Makristathis, A. Methods to detect Helicobacter pylori: from culture to molecular biology. Helicobacter. 2007;12(Suppl 2):6–11.
76. Chuang, C.H., et al. Adjuvant effect of vitamin C on omeprazole-amoxicillin-clarithromycin triple therapy for Helicobacter pylori eradication. Hepatogastroenterology. 2007;54(73):320–324.
77. Ramakrishna, K., Salinas, R.C. Peptic ulcer disease. Am Fam Physician. 2007;76(7):1005–1012.
78. Aldoori, W.H., et al. Prospective study of diet and the risk of duodenal ulcer in men. Am J Epidemiol. 1997;145(1):42.
79. Agarwal, K., Agarwal, S. Helicobacter pylori vaccine: from past to future. Mayo Clin Proc. 2008;83(2):169–175.
80. Kamada, T., et al. endoscopic characteristics and Helicobacter pylori infection in NSAID-associated gastric ulcer. J Gastroenterol Hepatol. 2006;21(1 Pt 1):98–102.
81. Vere, C.C., et al. Endoscopical and histological features in bile reflux gastritis. Rom J Morphol Embryol. 2005;46(4):269–274.
82. Anlauf, M., et al. Sporadic versus hereditary gastrinomas of the duodenum and pancreas: distinct clinico-pathological and epidemiological features. World J Gastroenterol. 2005;12(34):5440–5446.
83. Campana, D., et al. Zollinger-Ellison syndrome: Diagnosis and therapy. Minerva Med. 2005;96(3):187–206.
84. Stollman, N., Metz, D.C. Pathophysiology and prophylaxis of stress ulcer in intensive care unit patients. J Crit Care. 2005;20(1):35–45.
85. Yang, Y.X., Lewis, J.D. Prevention and treatment of stress ulcers in critically ill patients. Semin Gastroentest Dis. 2003;14(1):11–19.
86. Klebl, F.H., Scholmerich, J. Therapy insight: prophylaxis of stress-induced gastrointestinal bleeding in critically ill patients. Nat Clin Pract Gasteroenterol Hepatol. 2007;4(10):562–570.
87. Lipof, T., Shapiro, D., Kozol, R.A. Surgical perspectives in peptic ulcer disease and gastritis. World J Gastroenterol. 2006;12(20):3248–3252.
88. Martin, R.F. Surgical management of ulcer disease. Surg Clin North Am. 2005;85(5):907–929.
89. Tack, J. Gastric motor disorders. Best Pract Res Clin Gastroenterol. 2007;21(4):633–644.
90. Ukleja, A. Dumping syndrome: pathophysiology and treatment. Nutr Clin Pract. 2005;20(5):517–525.
91. Carvajal, S.H., Mulvihill, S.J. Postgastrectomy syndromes: dumping and diarrhea. Gastroenterol Clin North Am. 1994;23(2):261.
92. Karamanolis, G., Tack, J. Nutrition and motility disorders. Best Pract Res Clin Gastroenterol. 2006;20(3):485–505.
93. Pedrazzani, C., et al. Postoperative complications and functional results after subtotal gastrectomy with Billroth II reconstruction for primary gastric cancer. Dig Dis Sci. 2007;52(8):1757–1763.
94. Didden, P., Penning, C., Masclee, A.A. Octreotide therapy in dumping syndrome: analysis of long-term results. Aliment Pharmacol Ther. 2006;24(9):1367–1375.
95. Zobolas, B., et al. Alkaline reflux gastritis: early and late results of surgery. World J Surg. 2006;30(6):1043–1049.
96. Kim, H.C., et al. Afferent loop obstruction after gastric cancer surgery: helical CT findings. Abdom Imaging. 2003;28(5):624–630.
97. Beyan, C., et al. Post-gastrectomy anemia: evaluation of 72 cases with post-gastrectomy anemia. Hematology. 2007;12(1):81–84.
98. Ferrone, M., Raimondo, M., Scolapio, J.S. Pancreatic enzyme pharmacotherapy. Pharmacotherapy. 2007;27(6):910–920.
99. Kuokkanen, M., et al. Mutation in the translated region of the lactase gene (LCT) underlie congenital lactase deficiency. Am J Hum Genet. 2006;78(2):339–344.
100. Troelsen, J.T. Adult-type hypolactasia and regulation of lactase expression. Biochem Biophys Acta. 2005;1723(1-3):19–32.
101. Heyman, M.B., et al. Lactose intolerance in infants, children, and adolescents. Pediatrics. 2006;118(3):1279–1286.
102. Obermayer-Pietsch, B.M., et al. Adult-type hypolactasia and calcium availability: decreased calcium intake or impaired calcium absorption? Osteoporosis Int. 2007;18(4):445–451.
103. Norlin, M., Wikvall, K. Enzymes in the conversion of cholesterol into bile acids. Curr Mol Med. 2007;7(2):199–218.
104. Loftus, C.G., et al. Update on the incidence and prevalence of Crohn’s disease in ulcerative colitis in Olmstead County, Minnesota, 1940-2000. Inflamm Bowel Dis. 2007;13(3):254–261.
105. Baumgart, D.C. What’s new in inflammatory bowel disease in 2008? World J Gastroenterol. 2008;14(3):329–330.
106. Scaldaferri, F., Fiocchi, C. Inflammatory bowel disease: progress and current concepts of etiopathogenesis. J Dig Dis. 2007;8(4):171–178.
107. Karban, A., Eliakim, R. Effect of smoking on inflammatory bowel disease: is it disease or organ specific? World J Gastroenterol. 2007;13(15):2150–2152.
108. Packey, C.D., Sartor, R.B. Interplay of commensal and pathogenic bacteria, genetic mutations, and immunoregulatory defects in the pathogenesis of inflammatory bowel diseases. J Intern Med. 2008;263(6):597–606.
109. Zhong, W., et al. Chemokines orchestrate leukocyte trafficking in inflammatory bowel disease. Front Biosci. 2008;13:1654–1664.
110. Thoreson, R., Cullen, J.J. Pathophysiology of inflammatory bowel disease. Surg Clin North Am. 2007;87(3):575–585.
111. Baumgart, D.C., Sandborn, W.J. Inflammatory bowel disease: clinical aspects and established and evolving therapies. Lancet. 2007;369(9573):1641–1657.
112. Xie, J., Itzkowitz, S.H. Cancer in inflammatory bowel disease. World J Gastroenterol. 2008;14(3):378–389.
113. Agrawal, D., Rukkannagari, S., Kethu, S. Pathogenesis and clinical approach to extraintestinal manifestations of inflammatory bowel disease. Minerva Gastroenterol Dietol. 2007;53(3):233–248.
114. Denese, S., et al. Inflammation and coagulation in inflammatory bowel disease: the clot thickens. Am J Gastroenterol. 2007;102(1):174–186.
115. Freeman, H.J. Venous thromboembolism with inflammatory bowel disease. World J Gastroenterol. 2008;14(7):991–993.
116. Nikolaus, S., Schreiber, S. Diagnostics of inflammatory bowel disease. Gastroenterology. 2007;133(5):1670–1689.
117. Hanauer, S.G. Risks and benefits of combining immunosuppressives and biological agents in inflammatory bowel disease: is the synergy worth the risk? Gut. 2007;56(9):1181–1183.
118. Lakatos, P.L., Szamosi, T., Lakatos, L. Smoking in inflammatory bowel disease: good, bad or ugly? World J Gastroenterol. 2007;13(46):6134–6139.
119. McGilligan, V.E., et al. Hypothesis about mechanisms through which nicotine might exert its effect on the interdependence of inflammation and gut barrier function in ulcerative colitis. Inflamm Bowel Dis. 2007;13(1):108–115.
120. Ba’ath, M.E., et al. Surgical management of inflammatory bowel disease. Arch Dis Child. 2007;92(4):312–316.
121. Caprilli, R., Viscido, A., Latella, G. Current management of severe ulcerative disease. Nat Clin Pract Gastroenterol Hepatol. 2007;4(2):92–101.
122. Brant, S.R., et al. A population-based case-control study of CARD15 and other risk factors in Crohn’s disease and ulcerative colitis,. Am J Gastroenterol. 2007;102(2):313–323.
123. de Mesquita, M.B., Cvitelli, F. Levine A: Epidemiology, genes and inflammatory bowel diseases in childhood. Dig Liver Dis. 2007;40(1):3011.
124. Ng, A.Z.-V.S.-S., et al. Associate of the T allele of an intronic single nucleotide polymorphism in the colony stimulating factor 1 receptor with Crohn’s disease: a case-control study. J Immune Based Ther Vaccines. 2004;2:6.
125. Bruzzese, E., et al. Microflora in inflammatory bowel diseases: a pediatric perspective. J Clin Gastroenterol. 2004;38(6 Suppl):S91–S93.
126. Darfeuille-Michaud, A., et al. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology. 2004;127(2):412–421.
127. Shinzaki, S., et al. IgG oligosaccharide alterations are a novel diagnostic marker for disease activity and the clinical course of inflammatory bowel disease. Am J Gastroenterol. 2008;103(5):1173–1181.
128. Shah, S.B., Hanauer, S.B. Treatment of diarrhea in patients with inflammatory bowel disease: concepts and cautions. Rev Gastroenterol Disord. 2007;7(Suppl 3):S3–S10.
129. Allison, M.C., et al. Inflammatory bowel disease. St Louis: Mosby; 1998.
130. Friedman, S. Cancer in Crohn’s disease. Gastroenterol Clin North Am. 2006;35(3):621–639.
131. Behm, B.W., Bickston, S.J. Tumor necrosis factor-alpha for maintenance of remission in Crohn’s disease. Cochrane Database Syst Rev. (1):2008. [CD006893].
132. Casillas, S., Delaney, C.P. Larparoscopic surgery for inflammatory bowel disease. Dig Surg. 2005;22(3):135–142.
133. Tursi, A. New physiopathological and therapeutic approaches to diverticular disease of the colon. Expert Opin Pharmacother. 2007;8(3):299–307.
134. Petruzziello, L., et al. Review article: uncomplicated diverticular disease of the colon. Aliment Pharmacol Ther. 2006;23(10):1379–1391.
135. Colecchia, A., et al. Diverticular disease of the colon: new perspectives in symptom development and treatment. World J Gastroenterol. 2003;9(7):1385–1389.
136. Floch, M.H., Bina, O. The natural history of diverticulitis: fact and theory,. J Clin Gastroenterol. 2004;38(5 Suppl):S2–S7.
137. Bogardus, S.T. Jr: What do we know about diverticular disease? A brief overview,. Clin Gastroenterol. 2006;40(7 Suppl 3):S108–S111.
138. Szojda, M.M., et al. Review article: management of diverticulitis. Aliment Pharmacol Ther. 2007;226(Suppl 2):67–76.
139. Addiss, D.G., et al. The epidemiology of appendicitis and appendectomy in the United States,. Am J Epidemiol. 1990;132(5):910–925.
140. Brennan, G.D. Pediatric appendicitis: pathophysiology and appropriate use of diagnostic imaging. CJEM. 2006;8(6):425–432.
141. Birchley, D. Patients with clinical acute appendicitis should have pre-operative full blood count and C-reactive protein assays. Ann R Coll Surg Engl. 2006;88(1):27–32.
142. Kosaka, N., et al. Difficulties in the diagnosis of appendicitis: review of CT and US images. Emerg Radiol. 2007;14(5):289–295.
143. Ditillo, M.F., Dziura, J.D., Rabinovici, R. Is it safe to delay appendectomy in adults with acute appendicitis? Ann Surg. 2006;244(5):656–660.
144. Parkes, G.C., et al. Gastrointestinal microbiota in irritable bowel syndrome: their role in its pathogenesis and treatment. Am J Gastroenterol. 2008;103(6):1557–1567.
145. De Giorgio, R., Barbara, G. Is irritable bowel syndrome an inflammatory disorder? Curr Gastroenterol Rep. 2008;10(4):385–390.
146. Spiller, R., Garsed, K. Postinfectious irritable bowel syndrome. Gastroenterology. 2009;136(6):1979–1988.
147. Hammerle, C.W., Surawicz, C.M. Updates on treatment of irritable bowel syndrome, World J of Gastroenterol. 2008;14(17):2639–2649.
148. American College of Gastroenterology Task Force on Irritable Bowel Syndrome: An evidence-based review on the management of irritable bowel syndrome. Am J Gastroenterol. 2009;104(Supplement 1):1–40.
149. Sreenarasimhaiah, J. Chronic mesenteric ischemia. Best Pract Res Clin Gastroenterol. 2005;19(2):283–295.
150. Herbert, G.S., Steele, S.R. Acute and chronic mesenteric ischemia. Surg Clin North Am. 2007;87(5):1115–1134. [ix, 2007].
151. Kvietys, P.R., Barrowmand, A., Granger, N.D. Pathophysiology of the splanchnic circulation. Boca Raton, FL: CRC Press; 1987.
152. Berland, T., Oldenburg, W.A. Acute mesenteric ischemia. Curr Gastroenterol Rep. 2008;10(3):341–346.
153. National Heart, Lung, and Blood Institute, National Institutes of Health: Clinical guidelines on the identification, evaluation, and treatment of overweight and obesity in adults—executive summary, 1998. Available at www.nhlbi.nih.gov/guidelines/obesity/sum_intr.htm.
154. James, W.P. The epidemiology of obesity: the size of the problem,. J Intern Med. 2008;263(4):336–352.
155. Wolin, K.Y., Colditz, G.A. Can weight loss prevent cancer? Br J Cancer. 2008;99(7):995–999.
156. Barness, L.A., Opitz, J.M., Gilbert-Barness, E. Obesity: genetic, molecular and environmental aspects. Am J Med Genet A. 2007;143A(24):3016–3034.
157. Korner, A., et al. Polygenic contribution to obesity: genome-wide strategies reveal new targets. Front Horm Res. 2008;36:12–36.
158. Farooqi, S., O’Rahilly, S. Genetics of obesity in humans. Endocr Rev. 2006;27(7):710–718.
159. Rosengren, A., Lissner, L. The sociology of obesity,. Front Horm Res. 2008;36:260–270.
160. Rocha, V.Z., Libby, P. The multiple facets of the fat tissue,. Thyroid. 2008;18(2):175–183.
161. Bays, H.E., et al. Pathogenic potential of adipose tissue and metabolic consequences of adipocyte hypertrophy and increased visceral adiposity. Expert Rev Cardiovasc Ther. 2008;6(3):343–368.
162. Coll, A.P., Farooqui, I.S., O’Rahilly, S. The hormonal control of food intake,. Cell. 2007;129(2):251–262.
163. Myers, M.G., Cowley, M.A., Munzberg, H. Mechanisms of leptin action and leptin resistance. Annu Rev Physiol. 2008;70:537–556.
164. Levin, B.E., Dunn-Meynell, A.A., Banks, W.A. Obesity-prone rats have normal blood-brain barrier transport but defective central leptin signaling before obesity onset. Am J Physiol Regul Integr Comp Physiol. 2004;286(1):R143–R150.
165. Banks, W.A. The blood-brain barrier as a cause of obesity,. Curr Pharm Des. 2008;14(16):1606–1614.
166. Pan, W. Modulation of feeding-related peptide/protein signals by the blood-brain barrier. J Neurochem. 2004;90(2):455–461.
167. Scarpace, P.J., Zhang, Y. Elevated leptin: consequence or cause of obesity? Front Biosci. 2007;12:3531–3544.
168. Martin, S.S., Qasim, A., Reilly, M.P. Leptin resistance: a possible interface of inflammation and metabolism in obesity-related cardiovascular disease. J Am Coll Cardiol. 2008;52(15):1201–1210.
169. Calabro, P., Yeh, E.T. Intra-abdominal obesity, inflammation and cardiovascular risk: new insight into global cardiometabolic risk. Curr Hypertens Rep. 2008;10(1):32–38.
170. Leite-Moreira, A.F., Rocha-Sousa, A., Henriques-Coelho, T. Cardiac, skeletal and smooth muscle regulation by ghrelin. Vitam Horm. 2008;77:207–238.
171. Wu, J.T., Kral, J.G. Ghrelin: Integrative neuroendocrine peptide in health and disease. Ann Surg. 2004;239(4):464–474.
172. Williams, J., Mobarhan, S. A critical interaction: leptin and ghrelin,. Nutr Rev. 2003;61(11):391–393.
173. Klok, M.D., Jakobsdottir, S., Drent, M.L. The role of leptin and ghrelin in the regulation of food intake and body weight in humans, a review,. Obes Rev. 2007;8(1):21–34.
174. Fantuzzi, G. Adiponectin and inflammation: consensus and controversy. J Allergy Clin Immunol. 2008;121(2):326–330.
175. Guerre-Millo, M. Adiponectin: an update. Diabetes Metab. 2008;34(1):12–18.
176. Zick, Y. Molecular basis of insulin action. Novartis Found Symp. 2004;262(36-50):265–268. [discussion 50-55, 2004].
177. Lazar, M.A. Resistin-and obesity-associated metabolic diseases. Horm Metab Res. 2007;39(10):710–716.
178. Hamdy, O., Porramatikul, S., Al-Ozairi, E. Metabolic obesity: the paradox between visceral and subcutaneous fat. C urr Diabetes Rev. 2006;2(4):367–373.
179. Kong, A.P., Chan, N.N., Chan, J.C. The role of adipocytokines and neurohormonal dysregulation in metabolic syndrome,. Curr Diabetes Rev. 2006;2(4):397–407.
180. Damjanovi, M., Barton, M. Fat intake and cardiovascular response. Curr Hypertens Rep. 2008;10(1):25–31.
181. Yanai, H., et al. The underlying mechanisms for development of hypertension in the metabolic syndrome. Nutr J. 2008;7:10.
182. Mokhlesi, B., Tulaimat, A. Recent advances in obesity hypoventilation syndrome. Chest. 2007;132(4):1322–1336.
183. Shore, S.A. Obesity and asthma: possible mechanisms. J Allergy Clin Immunol. 2008;121(5):1087–1093.
184. Shimura, R., et al. Fat accumulation, leptin, and hypercapnia in obstructive sleep apnea-hypopnea syndrome. Chest. 2005;127(2):543–549.
185. Wearing, S.C., et al. Musculoskeletal disorders associated with obesity: a biomechanical perspective. Obes Rev. 2006;7(3):239–250.
186. Pischon, T., Nothlings, U., Boeing, H. Obesity and cancer. Proc Nutr Soc. 2008;67(2):128–145.
187. Renehan, A.G., Roberts, D.L., Dive, C. Obesity and cancer: pathophysiological and biological mechanisms. Arch Physiol Biochem. 2008;114(1):71–83.
188. Sweeting, H.N. Measurement and definitions of obesity in childhood and adolescence: a field guide for the uninitiated. Nutr J. 2007;6:32.
189. Centers for Disease Control and Prevention: BMI—body mass index: BMI calculator for children and teens, 2004. Available at www.cdc.gov/growthcharts.
190. Jaski, C.B., Lustig, R.H. Adolescent obesity and puberty: the “perfect storm,”. Ann N Y Acad Sci. 2008;1135:265–279.
191. Hensrud, D.D. Diet and obesity. Curr Opin Gastroenterol. 2004;20(2):119–124.
192. Ruser, C.B., Federman, D.G., Kashaf, S.S. Whittling away at obesity and overweight: small lifestyle changes can have the biggest impact. Postgrad Med. 2005;117(1):37–40. [31-34, 2005].
193. Teixeira, P.J., et al. A review of psychosocial pre-treatment predictors of weight control,. Obes Rev. 2005;6(1):43–65.
194. Tsai, A.G., Wadden, T.A. Systematic review: an evaluation of major commercial weight loss programs in the United States. Ann Intern Med. 2005;142(1):56–66.
195. Volek, J.S., Vanheest, J.L., Forsythe, C.E. Diet and exercise for weight loss: a review of current issues. Sports Med. 2005;35(1):1–9.
196. Wynne, K., et al. Appetite control. J Endocrinol. 2005;184(2):291–318.
197. Kruger, J., Blanck, H.M., Gillespie, C. Dietary practices, dining out behavior, and physical activity correlates of weight loss maintenance. Prev Chronic Dis. 2008;5(1):A11.
198. Aylwin, S., Al-Zamin, Y. Emerging concepts in the medical and surgical treatment of obesity. Front Horm Res. 2008;36:229–259.
199. Powell, L.H., Calvin, J.E., 3rd., Calvin, J.E., Jr. Effective obesity treatments. Am Psychol. 2007;62(3):234–246.
200. Suastika, K. Update in the management of obesity. Acta Med Indones. 2006;38(4):231–237.
201. Waseem, T., et al. Pathophysiology of obesity: why surgery remains the most effective treatment. Obes Surg. 2007;17(10):1389–1398.
202. Kaye, W. Neurobiology of anorexia and bulimia nervosa. Physiol Behav. 2008;94(1):121–235.
203. National Institute of Mental Health. Eating disorders, NIH Pub 93-3477. Washington, DC: US Government Printing Office; 1993.
204. Berkman, N.D., Lohr, K.N., Bulik, C.M. Outcomes of eating disorders: a systematic review of the literature. Int J Eat Disord. 2007;40(4):293–309.
205. Law, S.A., Golding, J.M. Sexual assault history and eating disorder among White, Hispanic, and African-American women and men. Am J Public Health. 1996;86(14):579.
206. Emous, S.J. Eating disorders in adolescent girls. Pediatr Int. 2000;42(1):107.
207. Hebebrand, J., et al. The role of leptin in anorexia nervosa: clinical implications,. Mol Psychiatry. 2007;12(1):23–35.
208. Mantzoros, C., et al. Cerebrospinal fluid leptin in anorexia nervosa: correlation with nutritional status and potential role in resistance to weight gain. J Clin Endocrinol Metab. 1997;82(6):1845–1851.
209. Bulik, C.M., et al. The genetics of anorexia nervosa,. Annu Rev Nutr. 2007;27:263–275.
210. American Psychiatric Association. Diagnostic and statistical manual of mental disorders, ed 4. Washington, DC: Academic Press; 1994.
211. Seidenfeld, M.E., Sosin, E., Rickert, V.I. Nutrition and eating disorders in adolescents. Mt Sinai J Med. 2004;71(3):155–161.
212. Rayworth, B.B., Wise, L.A., Harlow, B.L. Childhood abuse and risk of eating disorders in women. Epidemiology. 2004;15(3):271–278.
213. Mitchell, J.E., Crow, S. Medical complications of anorexia nervosa and bulimia nervosa. Curr Opin Psychiatry. 2006;29(4):438–443.
214. Casiero, D., Frishman, W.H. Cardiovascular complications of eating disorders. Cardiol Rev. 2006;14(5):227–231.
215. Kohn, M., Golden, N.H. Eating disorders in children and adolescents: epidemiology, diagnosis, and treatment. Paediatr Drugs. 2001;3(2):91–99.
216. Gowers, S.G. management of eating disorders in children and adolescents. Arch Dis Child. 2008;93(4):331–334.
217. Guarda, A.S. Treatment of anorexia nervosa: insights and obstacles. Physiol Behav. 2008;94(1):113–120.
218. Williams, P.M., Goodie, J., Motsinger, C.D. Treating eating disorders in primary care. Am Fam Physician. 2008;77(2):187–195.
219. Yantis, M.A., Velander, R. How to recognize and respond to refeeding syndrome. Nursing. 2008;38(5):34–39.
220. Mehler, P.A. Eating disorders: bulimia nervosa. Hosp Pract (Off Ed). 1996;31(2):107.
221. Lam, R.W., Golder, E.M., Gerwal, A. Seasonality of symptoms in anorexia and bulimia nervosa. Int J Eat Disord. 1996;19(1):35.
222. Shapiro, J.R., et al. Bulimia nervosa treatment: a systematic review of randomized controlled trials. Int J Eat Disord. 2007;40(4):321–336.
223. Williamson, D.A., Martin, C.K., Stewart, T. Psychological aspects of eating disorders. Best Pract Res Clin Gastroenterol. 2004;18(6):1073–1088.
224. Incalzi, R.A., et al. Energy intake and in-hospital starvation. Arch Intern Med. 1996;156(4):425.
225. Finn, P.F., Dice, J.F. Proteolytic and lipolytic responses to starvation. Nutrition. 2006;22(7-8):830–844.
226. Jahoor, F., et al. Protein metabolism in severe childhood malnutrition. Ann Trop Paediatr. 2008;28(2):87–101.
227. Berkley, J.A., et al. Prognostic indicators of early and late death in children admitted to district hospital in Kenya: cohort study. BMJ. 2003;326(7385):361.
228. Lidder, P.G., Lewis, S. Perioperative and postoperative nutrition. Hosp Med. 2004;65(12):717–720.
229. Zipprich, A. Hemodynamics in the isolated cirrhotic liver. J Clin Gastroenterol. 2007;41(10 Suppl 3):S254–S258.
230. Varghese, J., et al. Hepatopulmonary syndrome—past to present. Ann Hepatol. 2007;6(3):135–142.
231. Colle, I., et al. Hepatopulmonary syndrome and portopulmonary hypertension: what’s new? Acta Gastroenterol Belg. 2007;70(2):203–209.
232. Habib, A., Sanyal, A.J. Acute variceal hemorrhage. Gastrointest Endosc Clin North Am. 2007;17(2):223–252. [v, 2007].
233. Abraldes, J.G., Bosch, J. The treatment of acute variceal bleeding. J Clin Gastroenterol. 2007;41(10 Suppl 3):S312–S317.
234. Thabut, D., Bernard-Chabert, B. Management of acute bleeding from portal hypertension. Best Pract Res Clin Gastroenterol. 2007;21(1):19–29.
235. Ravindra, K.V., Eng, M., Marvin, M. Current management of sinusoidal portal hypertension. Am Surg. 2008;74(1):4–10.
236. Poordad, F. Review article: thrombocytopenia in chronic disease. Aliment Pharmacol Ther. 2007;26(Suppl 1):5–11.
237. Kashani, A., et al. Fluid retention in cirrhosis: pathophysiology and management. QJM. 2008;101(2):71–85.
238. Iwakiri, Y. The molecules: mechanisms of arterial vasodilation observed in the splanchnic and systemic circulation in portal hypertension,. J Clin Gastroenterol. 2007;41(10 Suppl 3):S288–S294.
239. LaVilla, G., Gentillini, P. Hemodynamic alterations in liver cirrhosis. Mol Aspects Med. 2008;29(1-2):112–118.
240. McGibbon, A., et al. An evidence-based manual for abdominal paracentesis. Dig Dis Sci. 2007;52(12):3307–3315.
241. Gines, P., Cardenas, A. The management of ascites and hyponatremia in cirrhosis,. Semin Liver Dis. 2008;28(1):43–58.
242. Gentilini, P., et al. Albumin improves the response to diuretics in patients with cirrhosis and ascites: results of a randomized, controlled trial. J Hepatol. 1999;30(4):639.
243. Coll, S., et al. Mechanisms of early decrease in systemic vascular resistance after total paracentesis: influence of flow rate of ascites extraction. Eur J Gastroenterol Hepatol. 2004;16(3):347–353.
244. Av, S.P. Hepatic encephalopathy: pathophysiology and advances in therapy. Trop Gastroenterol. 2007;28(1):4–10.
245. Wright, G., Jalan, R. Management of hepatic encephalopathy in patients with cirrhosis. Best Pract Res Clin Gastroenterol. 2007;21(1):95–110.
246. Haussinger, D., Schliess, F. Pathogenetic mechanisms of hepatic encephalopathy. Gut. 2008;57(8):1156–1165.
247. Vaquero, J., Butterworth, R.F. Mechanisms of brain edema in acute liver failure and impact of novel therapeutic interventions. Neurol Res. 2007;29(7):683–690.
248. Ahboucha, S., Butterworth, R.F. The neurosteroid system: an emerging therapeutic target for hepatic encephalopathy,. Metab Brain Dis. 2007;22(3-4):291–308.
249. Bass, N.M. Review article: the current pharmacological therapies for hepatic encephalopathy. Aliment Pharmacol Ther. 2007;25(Suppl 1):23–31.
250. Roche, S.P., Kobos, R. Jaundice in the adult patient. Am Fam Physician. 2004;69(2):299–304.
251. Assimakopoulos, S.F., Scopa, C.D., Vagianos, C.E. Pathophysiology of increased intestinal permeability in obstructive jaundice. World J Gastroenterol. 2008;13(48):6458–6464.
252. Raiford, D.S. Pruritus of chronic cholestasis. QJM. 1995;88(9):603.
253. Fabrizi, F., Martin, P., Messa, P., Recent advances in the management of hepato-renal syndrome (HRS). Acta Clin Belg Suppl. 2007(2):393–396.
254. Turban, S., Thuluvath, P.J., Atta, M.G. Hepatorenal syndrome. World J Gastroenterol. 2007;13(30):4046–4055.
255. Angeli, P., Merkel, C. Pathogenesis and management of hepatorenal syndrome in patients with cirrhosis. J Hepatol. 2008;48(Suppl 1):S93–S103.
256. Barve, A., et al. Treatment of alcoholic liver disease. Ann Hepatol. 2008;7(1):5–15.
257. Sukowski, M.S. Viral hepatitis and HIV coinfection. J Hepatol. 2008;48(2):353–367.
258. Kumar, D., et al. Occurrence & nucleotide sequence analysis of hepatitis G virus in patients with acute viral hepatitis and fulminant hepatitis. Indian J Med Res. 2007;125(6):752–755.
259. Ciocca, M. Clinical course and consequences of hepatitis A infection. Vaccine. 2000;18(Suppl 1):S71.
260. Brundage, S.C., Fitzpatrick, A.N., Hepatitis, A. Am Fam Physician. 2006;73(12):2162–2168.
261. Keystone, J.S., Hershey, J.H. The underestimated risk of hepatitis A and hepatitis B: benefits of an accelerated vaccination schedule,. Int J Infect Dis. 2007;12(1):3–11.
262. McGovern, B.H. The epidemiology, natural history and prevention of hepatitis B: implications of HIV coinfection. Antivir Ther. 2007;12(Suppl 3):H3–H13.
263. Kumar, R., Agrawal, B. Novel treatment options for hepatitis B virus infection. Curr Opin Investig Drugs. 2004;5(2):171–178.
264. Lin, C.L., Kao, J.H. Hepatitis B viral factors and clinical outcomes of chronic hepatitis B. J Biomed Sci. 2008;15(2):137–145.
265. Ayoub, W.S., Keeffe, E.B. Review article: current antiviral therapy of chronic hepatitis B. Aliment Pharmacol Ther. 2008;28(2):167–177.
266. Balsano, C., Alisi, A. Viral hepatitis B: established and emerging therapies. Curr Med Chem. 2008;15(9):930–939.
267. Oldfield, E.C., 3rd., Keeffe, E.B. The A’s and B’s of vaccine-preventable hepatitis: improving prevention in high-risk adults,. Rev Gastroenterol Disord. 2007;7(1):1–21.
268. Balasubramanian, A., Groopman, J.E., Ganju, R.K. Underlying pathophysiology of HCV infection in HIV-positive drug users. J Addict Dis. 2008;27(2):75–82.
269. Cheruvu, S., Marks, K., Talal, A.H. Understanding the pathogenesis and management of hepatitis B/HIV and hepatitis B/hepatitis C virus coinfection. Clin Liv Dis. 2007;11(4):917–943. [ix-x, 2007].
270. Koev, G., Kati, W. The emerging field of HCV drug resistance. Expert Opin Investig Drugs. 2008;17(3):303–319.
271. Yuan, H.J., Lee, W.M. Nonresponse to treatment for hepatitis C: current management strategies. Drugs. 2008;68(1):27–42.
272. Tong, M.J., Terrault, N.A., Klintmalm, G. Hepatitis B transplantation: special conditions. Semin Liver Dis. 2000;20((Suppl 1):25.
273. Berg, T. Tailored treatment for hepatitis C. Clin Liver Dis. 2008;12(3):507–528.
274. Dalton, H.R., et al. The role of hepatitis E virus testing in drug-induced liver injury,. Aliment Pharmacol Ther. 2007;26(10):1429–1435.
275. Okamomo, H. Genetic variability and evolution of hepatitis E virus. Virus Res. 2007;127(2):216–228.
276. Panda, S.K., Thakral, D., Rehman, S. Hepatitis E virus. Rev Med Virol. 2007;17(3):151–180.
277. Mushahwar, I.K., Hepatitis, E. molecular virology, clinical features, diagnosis, transmission, epidemiology, and prevention. J Med Virol. 2008;80(4):646–658.
278. Baggio-Zappia, G.L., Hernandes Granato, C.L. HIV-GB virus C co-infection an overview. Clin Chem Lab Med. 2009;47(1):12–19.
279. Reshetnyak, V.I., et alKarlovich, T.I., Hehenko, L.U. Hepatitis G virus. World J Gastroenterol. 2008;14(30):4725–4734.
280. Leao-Filho, G.C. Hepatitis G virus infection in patients with hepatocellular carcinoma in Recife, Brazil. Jpo J Clin Oncol. 2007;37(8):632–636.
281. Billerbeck, E., Bottler, T., Thimme, R. Regulatory T cells in viral hepatitis. World J Gastroenterol. 2007;13(36):4858–4864.
282. Naicker, S., et al. Infection and glomerulonephritis. Semin Immunopathol. 2007;29(4):397–414.
283. Ohto, H., et al. Transmission of hepatitis C from mothers to infants. N Engl J Med. 1994;330(11):744.
284. Sánchez, G., Bosch, A., Pintó, R.M. Hepatitis A virus detection in food: current and future prospects. Lett Appl Microbiol. 2007;45(1):1–5.
285. Habib, S., Shaikh, O.S. Hepatitis B immune globulin. Drugs Today (Barc). 2007;43(6):379–394.
286. Gotthardt, D., et al. Fulminant hepatic failure: etiology and indications for liver transplant. Nephrol Dial Transplant. 2007;22(Suppl 8):viii5–viii8.
287. Khashab, M., Tector, A.J., Kwo, P.Y. Epidemiology of acute liver failure. Gastroenterol Rep. 2007;9(1):66–73.
288. Phau, J., Lee, K.H. Liver support devices. Curr Opin Crit Care. 2008;14(2):208–215.
289. Schuppan, D., Afdhal, N.H. Liver cirrhosis. Lancet. 2008;371(9615):838–851.
290. Wallace, K., Burt, A.D., Wright, M.C. Liver fibrosis. Biochem J. 2008;411(1):1–18.
291. Hill, D.B., Kugelmas, M. Alcoholic liver disease: treatment strategies for the potentially reversible stages. Postgrad Med. 1998;103(4):261.
292. Arteel, G., et al. Advances in alcoholic liver disease. Best Pract Res Clin Gastroenterol. 2003;17(4):625–647.
293. Bradbury, M.W., Berk, P.D. Lipid metabolism in hepatic steatosis. Clin Liver Dis. 2004;8(3):639–671.
294. Savolainen, V., et al. Early perivenular fibrosis —precirrhotic lesions among moderate alcohol consumers and chronic alcoholics. J Hepatol. 1995;23(5):524.
295. Zetterman, R.Z. Gitnick, G., eds. Current hepatology, vol. 16. St Louis: Mosby, 1996.
296. Singal, A.K., Anand, B.S. Mechanisms of synergy between alcohol and hepatitis C virus. J Clin Gastroenterol. 2007;41(8):761–772.
297. Greenwel, P. Acetaldehyde-mediated collagen regulation in hepatic stellate cells. Alcohol Clin Exp Res. 1999;23(5):930.
298. Nagata, K., Suzuki, H., Sakaguchi, S. Common pathogenic mechanism in development progression of liver injury caused by non-alcoholic or alcoholic steatohepatitis. J Toxicol Sci. 2007;32(5):453–468.
299. Paik, Y.H., et al. Toll-like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology. 2003;37(5):1043–1055.
300. Kolios, G., Valatas, V., Kouromalis, E. Role of Kupffer cells in the pathogenesis of liver disease. World J Gastroenterol. 2006;12(46):7413–7420.
301. Parsons, C.J., Takashima, M., Rippe, R.A. Molecular mechanisms of hepatic fibrogenesis. J Gastroenterol Hepatol. 2007;22(Suppl 1):S79–S84.
302. Yip, W.W., Burt, A.D. Alcoholic liver disease. Semin Diagn Pathol. 2006;23(3-4):149–160.
303. O’Shea, R.S., McCullough, A.J. Treatment of alcoholic hepatitis. Clin Liver Dis. 2005;9(1):103–134.
304. Gavaler, J.S., van Thiel, D.H. Ethanol: its adverse effects upon the hypothalamic pituitary-gonadal axis. J Lab Clin Med. 1983;101:21.
305. Gomez, F., Ruiz, P., Schreiber, A.D. Impaired function of macrophage Fc gamma receptors and bacterial infection in alcoholic cirrhosis. N Engl J Med. 1994;331(17):1122–1128.
306. Brave, A., et al. Treatment of alcoholic liver disease. Ann Hepatol. 2008;7(1):5–15.
307. Gershwin, M.E., Mackay, I.R. The causes of primary biliary cirrhosis: convenient and inconvenient truths,. Hepatology. 2008;47(2):737–745.
308. Kumagi, T., Heathcote, E.J. Primary biliary cirrhosis. Orphanet J Rare Dis. 2008;23(3):1.
309. Heathcote, E.J. Management of primary biliary cirrhosis. The American Association of the Study of Liver Disease practice guidelines,. Hepatology. 2000;31(4):1005.
310. Lazaridis, K.N., Talwalkar, J.A. Clinical epidemiology of primary biliary cirrhosis: incidence, prevalence, and impact of therapy. J Clin Gastroenterol. 2007;41(5):494–500.
311. Prince, M.I., et al. Asymptomatic primary biliary cirrhosis: clinical features, prognosis, and symptom progression in a large population based cohort. Gut. 2004;53(6):865–870.
312. Attasaranya, S., Fogel, E.L., Lehman, G.A. Choledocholithiasis, ascending cholangitis, and gallstone pancreatitis. Med Clin North Am. 2008;92(4):925–960.
313. Carey, M.C. Pathogenesis of gallstones. Am J Surg. 1993;165(4):410.
314. Portincasa, P., Di Ciaula, A., vanBerge-Henegouwen, G.P. Smooth muscle function and dysfunction in gallbladder disease. Curr Gastroenterol Rep. 2004;6(2):151–162.
315. Wang, H.H., Portincasa, P., Wang, D.Q. Molecular pathophysiology and physical chemistry of cholesterol gallstones. Front Biosci. 2008;13:401–423.
316. Donovan, J.M. Physical and metabolic factors in gallstone pathogenesis. Gastroenterol Clin North Am. 1999;28(1):75.
317. Berger, M.Y., et al. Abdominal symptoms: do they predict gallstones? A systemic review,. Scand J Gastroenterol. 2000;35(1):70.
318. Williams, E.J., et al. Guidelines on the management of common bile duct stones (CBDS). Gut. 2008;57(7):1004–1021.
319. Lammert, F., Miquel, J.F. Gallstone disease: from genes to evidence-based therapy. J Hepatol. 2008;48(Suppl 1):S124–S135.
320. Hochberger, J., et al. Management of difficult common bile duct stones. Gastrointest Endosc Clin North Am. 2003;13(4):623–634.
321. Venu, R.P., et al. Endoscopic transpapillary drainage of pancreatic abscess: technique and results. Gastrointest Endosc. 2000;51(4 Pt 1):391.
322. Frossard, J.L., Steer, M.L., Pastor, C.M. Acute pancreatitis. Lancet. 2008;37(9618):1072.
323. Arendt, T., et al. Gallstones, the choledochoduodenal junction and initiation of acute pancreatitis: are two stones the culprit rather than one stone? Med Hypotheses. 2000;54(4):570.
324. Vonlaufen, A., et al. Role of alcoholic metabolism in chronic pancreatitis. Alcohol Res Health. 2007;30(1):48–54.
325. Bhatia, M. Apoptosis versus necrosis in acute pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2004;286(2):G189.
326. Apte, M.V., Wilson, J.S. Alcohol-induced pancreatic injury. Best Pract Res Clin Gastroenterol. 2003;17(4):593–612.
327. Elfar, M., et al. The inflammatory cascade in acute pancreatitis: relevance to clinical disease,. Surg Clin North Am. 2007;87(6):1325–1340. [vii, 2007].
328. Zhang, X.P., Wang, L., Zhou, Y.F. The pathogenic mechanism of severe acute pancreatitis complicated with renal injury: a review of current knowledge,. Dig Dis Sci. 2008;53(2):297–306.
329. Kakafika, A., et al. Coagulation, platelets, and acute pancreatitis. Pancreas. 2007;34(1):15–20.
330. Lytras, D., et al. Persistent early organ failure: defining the high-risk group of patients with severe acute pancreatitis? Pancreas. 2008;36(3):249–254.
331. Kemppainen, E.A., et al. Advances in the laboratory diagnostics of acute pancreatitis. Ann Med. 1998;30(2):169.
332. Papachristou, G.I., Whitcomb, D.C. Predictors of severity and necrosis in acute pancreatitis. Gastroenterol Clin North Am. 2004;33(4):871–890.
333. Methuen, T., et al. Disialotransferrin, determined by capillary electrophoresis, is an accurate biomarker for alcoholic cause of acute pancreatitis. Pancreas. 2007;34(4):405–409.
334. Rosas, J.M., et al. Intra-abdominal pressure as a marker of severity in acute pancreatitis. Surgery. 2007;141(2):173–178.
335. Curtis, C.S., Kudska, K.A. Nutrition support in pancreatitis. Surg Clin North Am. 2007;87(6):1403–1415. [viii, 2007].
336. Spanier, B.W., Dijkgraaf, M.G., Bruno, M.J. Epidemiology, aetiology and outcome of acute and chronic pancreatitis: an update. Best Pract Res Clin Gastroenterol. 2008;22(1):45–63.
337. Keller, J., Layer, P. Idiopathic chronic pancreatitis. Best Pract Res Clin Gastroenterol. 2008;22(1):105–113.
338. Fregni, F., Pascual-Leone, A., Freedman, S.D. Pain in chronic pancreatitis: a salutogenic mechanism or a maladaptive brain response? Pancreatology. 2007;7(5-6):409–410.
339. Gachago, C., Draganov, P.V. Pain management in chronic pancreatitis. World J Gastroenterol. 2008;14(2):3137–3148.
340. Martin, R.F., Marion, M.D. Resectional therapy for chronic pancreatitis. Surg Clin North Am. 2007;87(6):1461–1475. [ix, 2007].
341. Robertson, E.V., Jankowski, J.A. Genetics of gastroesophageal cancer: paradigms, paradoxes, and prognostic utility. Am J Gastroenterol. 2008;103(2):443–449.
342. Brown, L.M., Devesa, S.S., Chow, W.H. Incidence of adenocarcinoma of the esophagus among white American by sex, stage and age. J Natl Cancer Inst. 2008;100(16):1184–1187.
343. Shimizu, M., Ban, S., Odze, R.D. Squamous dysplasia and other precursor lesions related to esophagesl squamous cell carcinoma. Gastroenterol Clin North Am. 2007;36(4):787–811. [v-vi, 2007].
344. American Cancer Society, Inc., Surveillance and Health Policy Research: Estimated New Cancer Cases and Deaths by Sex, U.S., 2009 Available at: http://www.cancer.org/downloads/stt/CFF2009_EstCD_3.pdf
345. Holmes, R.S., Vaughan, T.L. Epidemiology and pathogenesis of esophageal cancer. Semin Radiat Oncol. 2007;17(1):2–9.
346. Gee, D.W., Rattner, D.W. Management of gastroesophageal tumors. Oncologist. 2007;19(1):79–88.
347. Rokkas, T., et al. Relationship between Helicobacter pylori infection and esophageal neoplasia: a meta-analysis. Clin Gastroenterol Hepatol. 2007;5(12):1413–1417. [e1–2, 2007].
348. Lin, J., Beerm, D.C. Molecular biology of upper gastrointestinal malignancies. Semin Oncol. 2004;31(4):476–486.
349. Leung, W.K., et al. Screening for gastric cancer in Asia: current evidence and practice. Lancet Oncol. 2008;9(3):279–287.
350. Ye, W., Nyren, O. Risk of cancers of the oesophagus and stomach by histology or subsite in patients hospitalized for pernicious anemia. Gut. 2003;52(7):938–941.
351. Cheung, T.K., Xia, H.H., Wong, B.C. Helicobacter pylori eradication for gastric cancer prevention. J Gastroenterol. 2007;42(Suppl 17):10–15.
352. Lochhead, P., El-Omar, E.M. Helicobacter pylori infection and gastric cancer. Best Pract Res Clin Gastroenterol. 2007;21(2):281–297.
353. Du, Mq. MALT lymphoma: recent advances in aetiology and molecular genetics. J Clin Exp Hematop. 2007;47(2):31–42.
354. Tsugane, S., Sasazuki, S. Diet and risk of gastric cancer: review of epidemiological evidence. Gastric Cancer. 2007;10(2):75–83.
355. Houben, G.M., Stockbrugger, R.W. Bacteria in the aetio-pathogenesis of gastric cancer: a review. Scand J Gastroenterol. 1995;212(Suppl):13.
356. Wilksten, J.P., et al. Comparison of the prognostic value of a panel of tissue tumor markers and established clinicopathological factors in patients with gastric cancer. Anticancer Res. 2008;28(4C):2279–2287.
357. Fuccio, L., et al. Systematic review: Helicobacter pylori eradication for the prevention of gastric cancer. Aliment Pharmacol Ther. 2007;25(2):133–141.
358. Pufulete, M. Intake of diary products and risk of colorectal neoplasia. Nutr Res Rev. 2008;21(1):56–67.
359. Sharma, S., O’Keefe, S.J. Environmental influences on the high mortality from colorectal cancer in African Americans. Postgrad Med J. 2007;83(983):583–589.
360. Kam, M.H., et al. Small bowel malignancies: a review of 20 patients at a single centre. Colorectal Dis. 2004;6(3):195–197.
361. Will, O.C., et al. Familial adenomatous polyposis and the small bowel: a loco-regional review and current management strategies. Pathol Res Pract. 2008;204(7):449–458.
362. Ahnen, J.D. The genetic basis of colorectal cancer risk,. Adv Intern Med. 1996;41:531–532.
363. Jen, J., et al. Allelic loss of chromosome 18q and prognosis in colorectal cancer. N Engl J Med. 1994;331(4):213–221.
364. Eisinger, A.L., et al. The role of cyclooxygenase-2 and prostaglandins in colon cancer,. Prostaglandins Other Lipid Mediat. 2007;82(1-4):147–154.
365. Cappell, M.S. Pathophysiology: Clinical presentation and management of colon cancer. Gastroenterol Clin North Am. 2008;37(1):1–24. [v, 2008].
366. Evans, D.G., et al. Strategies for identifying hereditary nonpolyposis colon cancer. Semin Oncol. 2007;34(5):411–417.
367. Al-Sukhni, W., Aronson, M., Gallinger, S. Hereditary colorectal cancer syndromes: familial adenomatous polyposis and Lynch syndrome. Surg Clin North Am. 2008;88(4):819–844.
368. Rustgi, A.K. The genetics of hereditary colon cancer,. Genes Dev. 2007;21(20):2525–2538.
369. Antonakopoulos, N. The role of NSAIDs in colon cancer prevention,. Hepatogastroenterology. 2007;54(78):1694–1700.
370. Ryan-Harshman, M., Aldoori, W. Diet and colorectal cancer: Review of the evidence. Can Fam Physician. 2007;53(11):1913–1920.
371. Kuhnle, G.G., Bingham, S.A. Dietary meat, endogenous nitrosation and colorectal cancer. Biochem Soc Trans. 2007;36(Pt 5):1355–1357.
372. Rescigno, M. The pathogenic role of intestinal flora in IBD and colon cancer,. Curr Drug Targets. 2008;9(5):395–403.
373. Gupta, A.K., Brenner, D.E., Turgeon, D.K. Early detection of colon cancer: new tests on the horizon. Mol Diagn Ther. 2008;12(2):77–85.
374. Bonanno, E., et al. Stool test for colorectal cancer screening: what is going on? Surg Oncol. 2007;16(Suppl 1):S43–S45.
375. Cappell, M.S. From colonic polyps to colon cancer: pathophysiology, clinical presentation, screening and colonoscopic therapy. Minerva Gastroenterol Dietol. 2007;53(4):351–373.
376. Kim, Y.S., Milner, J.A. Dietary modulation of colon cancer risk. J Nutr. 2007;137(Suppl 11):2576S–2579S.
377. Weingarten, M.A., Zalmanovici, A., Yaphe, J. Dietary calcium supplementation for preventing colorectal cancer and adenomatous polyps. Cochrane Database Syst Rev. (1):2008. [CD003548].
378. De Salvo, G.L., et al. Curative surgery for obstruction from primary left colorectal carcinoma: primary or staged resection? Cochrane Database Syst Rev. (2):2004. [CD002101].
379. National Cancer Institute: Stages of colon cancer, 4/28/2008. Available at: http://cancerweb.ncl.ac.uk/cancernet/200008.html#2_STAGESOFCOLONCANCER
380. Braun, A.H., et al. New systemic frontline treatment for metastatic colorectal carcinoma. Cancer. 2004;100(8):1558–1577.
381. Midgley, R.S., Kerr, D.J. Immunotherapy for colorectal cancer. Expert Rev Anticancer Ther. 2003;3(1):63–78.
382. Schlom, J., et al. Strategies in the development of recombinant vaccines for colon cancer. Semin Oncol. 1999;26(6):672–682.
383. Sobrero, A., et al. New directions in the treatment of colon cancer: a look to the future. Eur J Cancer. 2000;36(5):559–566.
384. Lurje, G., Zhang, W., Lenz, H.J. Molecular prognostic markers in locally advanced colon cancer. Clin Colorectal Cancer. 2007;6(10):683–690.
385. El-Serag, H.B., Rudolph, K.L. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132(7):2557–2576.
386. Wong, R., Corley, D.A. Racial and ethnic variations in hepatocellular carcinoma incidence within the United States. Am J Med. 2008;121(6):525–531.
387. But, D.Y., Lai, C.L., Yuen, M.F. Natural history of hepatitis-related hepatocellular carcinoma. World J Gastroenterol. 2008;14(11):1652–1656.
388. Barazani, Y., et al. Chronic viral hepatitis and hepatocellular carcinoma. World J Surg. 2007;31(6):1243–1248.
389. Okuda, H. Hepatocellular carcinoma development in cirrhosis. Best Pract Res Clin Gastroentrol. 2007;21(1):161–173.
390. Hussain, S.P., et al. TP53 mutations and hepatocellular carcinoma: insights into the etiology and pathogenesis of liver cancer. Oncogene. 2007;26(15):2166–2176.
391. Hara, M., et al. Case-control study on cigarette smoking and the risk of hepatocellular carcinoma among Japanese. Cancer Sci. 2008;99(1):93–97.
392. Zhu, K., et al. Cigarette smoking and primary liver cancer: a population-based case control study in US men. Cancer Causes Control. 2007;18(3):315–321.
393. Kaewpitoon, N., et al. Opisthorchis viverrini: the carcinogenic human liver fluke. World J Gastroenterol. 2008;14(5):666–674.
394. Rougier, P., et al. Hepatocellular carcinoma (HCC): an update. Semin Oncol. 2007;34(2 Suppl 1):S12–S20.
395. Tommasi, S., et al. Molecular pathways and related target therapies in liver carcinoma. Curr Pharm Des. 2007;13(32):3279–3287.
396. Ben-Menachem, T. Risk factors for cholangiocarcinoma. Eur J Gastroenterol Hepatol. 2007;19(8):615–617.
397. El-Serag, H.B., et al. Diagnosis and treatment of hepatocellular carcinoma. Gastroenterology. 2008;134(6):1752–1763.
398. Tan, A., et al. Viral hepatocarcinogenesis: from infection to cancer. Liver Int. 2008;28(2):175–188.
399. Cho, C.S., et al. A novel prognostic nomogram is more accurate than conventional staging systems for predicting survival after resection of hepatocellular carcinoma,. J Am Coll Surg. 2008;206(2):281–291.
400. Lencioni, R., Crocetti, L. Image-guided thermal ablation of hepatocellular carcinoma. Crit Rev Oncol Hematol. 2008;66(3):200–207.
401. Volk, M.L., Marrero, J.A. Early detection of liver cancer: diagnosis and management. Curr Gastroenterol Rep. 2008;10(1):60–66.
402. Lowenfels, A.B., et al. Epidemiology of gallbladder cancer. Hepatogastroenterology. 1999;46(27):1529.
403. Pandy, M. Risk factors for gallbladder cancer: a reappraisal. Eur J Cancer Prev. 2003;12(1):15–24.
404. Malik, I.A. Gallbladder cancer: current status. Expert Opin Pharmacother. 2004;5(6):1271–1277.
405. Thomas, M.B. Targeted therapies for cancer of the gallbladder. Curr Opin Gastroenterol. 2008;24(3):372–376.
406. Foster, J.M., et al. Gallbladder cancer: defining the indications for primary radical resection and radical re-resection. Ann Surg Oncol. 14(2), 2007. [833–340].
407. Koorstra, J.B., et al. Pancreatic carcinogenesis. Pancreatology. 2008;8(2):110–125.
408. Hart, A.R., Kennedy, H., Harvey, I. Pancreatic cancer: a review of the evidence on causation. Clin Gastroenterol Hepatol. 2008;6(3):275–282.
409. Lowenfels, A.B., Maisonneuve, P., Lankisch, P.G. Chronic pancreatitis and other risk factors for pancreatic cancer. Gastroenterol Clin North Am. 1999;28(3):673–685.
410. Kloppel, G., Luttges, J. The pathology of ductal-type pancreatic carcinomas and pancreatic intraepithelial neoplasia: insights for clinicians,. Curr Gastroenterol Rep. 2004;6(2):111–118.
411. Pryczynicz, A., et al. Expression of EGF and EGFR strongly correlates with metastasis of pancreatic ductal carcinoma. Anticancer Res. 2008;28(2B):1399–1404.
412. Lomberk, G. Pain management. Pancreatology. 2008;8(6):542–543.
413. Chari, S.T. Detecting early pancreatic cancer: problems and prospects. Semin Oncol. 2007;34(4):284–294.
414. Koliopanos, A., et al. Radical resection of pancreatic cancer. Hepatobiliary Pancreat Dis Int. 2008;7(1):11–18.
415. von Wichert, G., Seufferlein, T., Adler, G. Palliative treatment of pancreatic cancer. J Dig Dis. 2008;9(1):1–7.
416. Wilkowski, R., Wolf, M., Heinemann, V. Primary advanced unresectable pancreatic cancer. Recent Results Cancer Res. 2008;177:79–93.