Digestion and Absorption of Nutrients
The Gastrointestinal Tract
Learning objectives
After reading this chapter you should be able to:
 Describe the main stages of digestion.
Describe the main stages of digestion.
Discuss mechanisms involved in the absorption of nutrients from the digestive tract.
Discuss the role of digestive enzymes.
Discuss digestion of the main classes of nutrients: carbohydrates, proteins and fats.
Identify compounds arising from the digestion of carbohydrates, proteins and fats that become substrates for further metabolism.
Introduction
The gastrointestinal tract and the organs functionally associated with it, are responsible for digestion and absorption of food
All organisms require sources of energy and other materials to enable function and growth. Their survival depends on the ability to extract and assimilate these resources from the ingested food. Also, the intestinal epithelium and the tight junctions between the enterocytes form the most important barrier between the organism and its external environment. The barrier has selective absorption and secretion functions and also may become a scene of immune (or autoimmune) response.
The gastrointestinal (GI) tract and the organs functionally associated with it, principally the liver and pancreas, are responsible for digestion and absorption of food. Digestion is the process by which food is broken down into components simple enough to be absorbed in the intestine. Absorption is the uptake of the products of digestion by intestinal cells (enterocytes) and their delivery to blood or lymph. Digestion and absorption of nutrients are closely linked and are regulated by the nervous system, hormones and paracrine factors. The physical presence of food particles in the GI tract also stimulates these processes.
Importantly, the absorption and secretion of ions such as sodium, chloride potassium, and bicarbonate, and the absorption of water, are also essential functions of the gastrointestinal tract. Therefore many clinical problems associated with digestion and absorption are closely linked to fluid and electrolyte disorders (Chapter 24). It would be a mistake to regard these two fields as separate: a clinician must see them in an integrated way.
Impairment of digestion and absorption results in maldigestion and malabsorption syndromes, respectively. Maldigestion denotes impaired breakdown of nutrients to their absorbable products. Malabsorption is the defective absorption, uptake and transport of nutrient products that were adequately digested.
The key clinical signs of signs of malabsorption and/or maldigestion are diarrhea, steatorrhea and loss of weight. In children there is failure to thrive. While acute diarrhea carries a risk of fast dehydration and electrolyte depletion, chronic diarrhea is associated with progressive malnutrition. Worldwide, diarrheal disease is, according to the WHO data (2011), a 5th leading cause of death. Malabsorption and maldigestion may also develop as consequences of surgical intervention such as gastrectomy, small bowel resection or colectomy.
The overall function of the GI tract is to break down food into components that can be absorbed and utilized by the body (Fig. 10.1), and then to excrete the nonabsorbable material. Its different anatomical segments have specific functions relating to digestion and absorption:
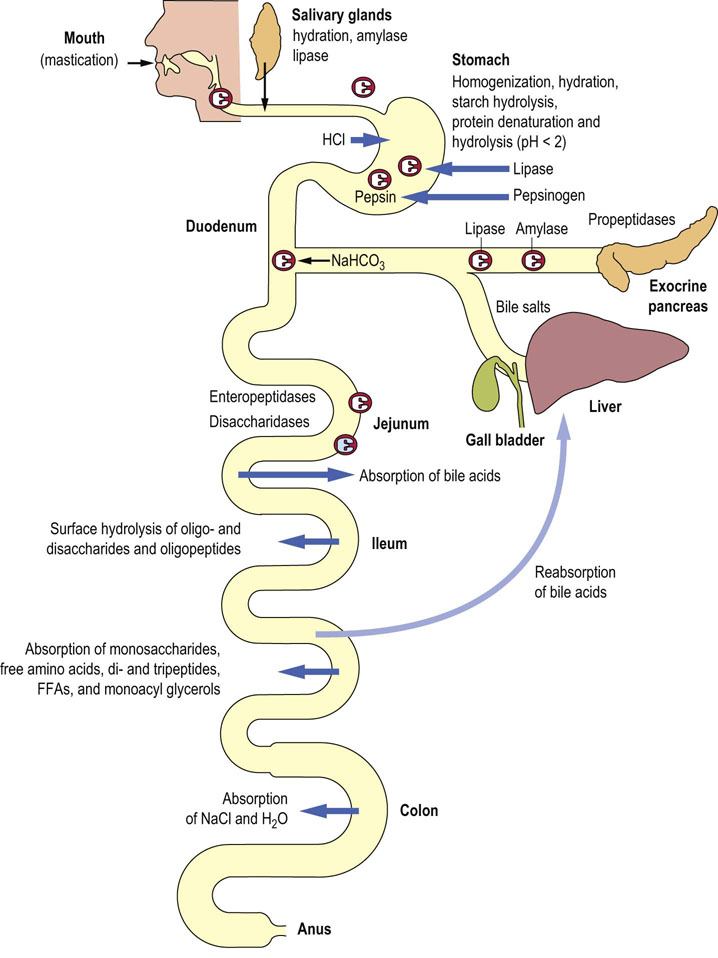
Fig. 10.1 The gastrointestinal (GI) tract.
Digestion and absorption of nutrients require integrated function of several organs. Mixing of food and initiation of digestion take place in the stomach. The absorptive processes start in the jejunum. However, the bulk of nutrients are absorbed in the ileum. The large intestine is involved in the absorption of water and electrolytes, and participates in the recirculation of the bile acids to the liver (Chapter 30). Taking into account all intake and secretions, a large amount of fluid (approximately 10 liters) passes through the GI tract every day. FFA, free fatty acids. Reproduced from Dominiczak MH. Medical Biochemistry Flash Cards. London: Elsevier, 2012, Card 34.
The mouth, stomach and duodenum deal with the initial process of mixing ingested food and initiating digestion.
In the duodenum, bile and pancreatic secretions enter through the common bile duct.
The small intestine is the main digestive area: in the jejunum digestive processes continue and absorption is initiated; it continues in the ileum.
The large intestine (cecum, colon and rectum; primarily the colon) is involved in the absorption and secretion of electrolytes and water.
Water and electrolyte handling in the gastrointestinal tract
Handling of electrolytes and water by the GI tract is one of its main functions
Handling of electrolytes and water by the GI tract is one of its main functions, and includes not only their absorption and secretion but also cell volume maintenance, and affects cell proliferation and differentiation, as well as apoptosis and carcinogenesis.
Large volume of fluid is secreted and reabsorbed by the GI tract
In a 24 h period, around 10.0 L of fluid enter and leave the GI tract. One liter of saliva is secreted per day; it contains electrolytes, protein and mucus. Intestinal secretions total about 7.0 liters every day, over and above an average water intake of about 2.0 liters. Most of this fluid is reabsorbed by the small intestine. The colon is particularly active in reabsorption: it absorbs around 90% of the fluid that passes through it, and only about 150–250 mL of water are normally contained in the stool.
Electrolytes are secreted by the salivary glands, stomach and the pancreas
Several secretory processes take place in the GI tract. Salivary glands, stomach and pancreas secrete digestive enzymes in the form of inactive zymogens. There is the hydrogen ion secretion in the stomach. Secretion of the bicarbonate ion takes place throughout the GI tract, with particularly large amounts being secreted in the pancreatic juice. Potassium secretion occurs predominantly in the colon and is regulated by aldosterone.
Impaired intestinal function leads to fluid-electrolyte and acid–base disorders
All this has clinical implications: diseases of the GI tract, and surgical removal of large segments of small and large intestine, carry a risk of major water and electrolyte disorders. Before treatment for cholera was known, a person with fulminant diarrhea caused by Vibrio cholerae infection could die of dehydration within a few hours. Acidosis due to bicarbonate loss can also be a feature of bowel disease (Chapter 25).
 Clinical box Causes of fluid and electrolyte loss from the gastrointestinal tract
Clinical box Causes of fluid and electrolyte loss from the gastrointestinal tract
Prolonged vomiting causes loss of water, hydrogen and chloride ions, and a further loss of potassium due to the body's compensatory mechanisms. Diarrhea may be caused by increased intestinal secretion due to, for instance, inflammation, or may be caused by malabsorption of nutrients. Severe diarrhea, leading to the loss of alkaline intestinal contents may lead to dehydration and metabolic acidosis. It also results in the loss of sodium, potassium and other minerals (Chapters 11, 23 and 24). Patients with a short gut syndrome resulting from extensive small bowel resection, may develop severe fluid balance problems due to inability to reabsorb water.
Mechanisms of water and electrolyte transport in the intestine
Sodium-potassium ATPase is the driving force for transport processes in the enterocytes
Enterocytes possess an array of transporters and ion channels (Fig. 10.2). The sodium-potassium ATPase, described in more detail in Chapter 24, is located on the basolateral membrane (the ‘blood side’) and transports the sodium ion outside the cell in exchange for the potassium (3Na+ to every 2K+ ions). This creates a sodium concentration gradient, and hyperpolarizes the membrane, increasing the intracellular negative potential and driving the passive transport systems (and thus transcellular ion transport). Moreover, transport of sodium (and chloride) is accompanied by passive transport of water, which is both paracellular (through the tight junctions) and transcellular (by membrane water transporters, the aquaporins).
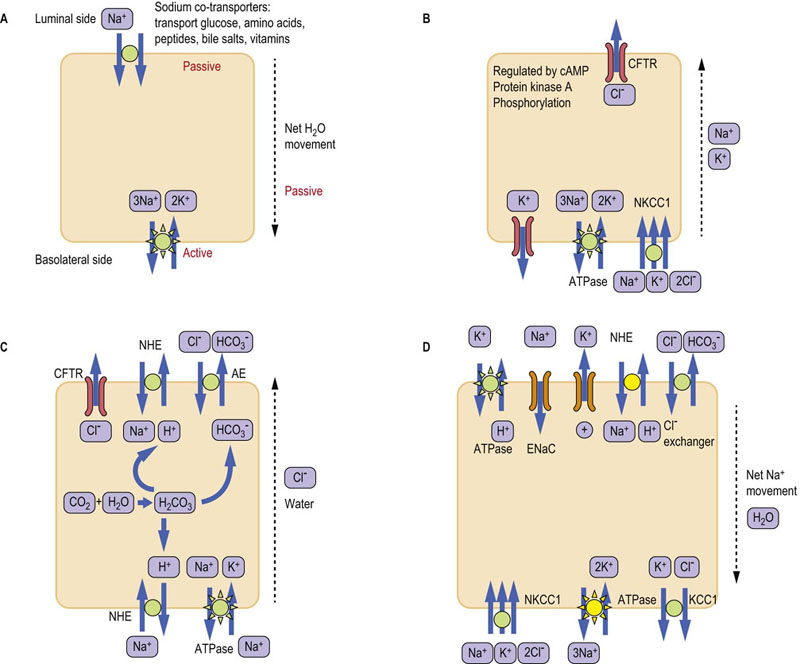
Fig. 10.2 Intestinal electrolyte transport systems.
(A) Sodium co-transporters transport a wide range of substrates, including glucose. Sodium gradient is created by the Na+/K+-ATPase located in the basolateral membrane. (B) CFTR transporter secretes chloride ion and is regulated by the cAMP-PKA signaling cascade. Sodium and potassium may also be secreted as counterions. Note the potassium ‘leak’ channel in the basolateral membrane. The NKCC1 transporter supplies chloride to the cell. (C) The electroneutral sodium absorption and the secretion of bicarbonate. (D) Electrogenic sodium absorption and potassium secretion in the distal colon. Transporters marked yellow are regulated by aldosterone in the distal colon. See text for details. CFTR, cystic fibrosis transmembrane conductance regulator; NHE, sodium/hydrogen exchanger; ENaC, epithelial sodium channel; AE, anion exchanger (chloride/bicarbonate exchanger); NKCC1, Na+ K+ Cl− co-transporter; KCC1, K+ Cl− co-transporter.
Sodium co-transporters are a common mode of intestinal transport
Sodium co-transporters transport the sodium ion together with another molecule (Fig. 10.2A). For instance, glucose is absorbed together with sodium by the sodium-glucose co-transporter present in the luminal membrane, known as SGLT-1 (Sodium/Glucose Linked Transport-1). Glucose is subsequently extruded into plasma at the basolateral membrane by the GLUT2 transporter (Fig. 10.7 below). The discovery of the link between intestinal transport of sodium and glucose had enormous clinical consequences. It was during a cholera epidemic in Manila, in the late 1960s, that researchers observed that patients who had been dehydrated because of diarrhea did not absorb oral sodium chloride well during attempts at oral rehydration. However, they started to do so when glucose was also provided. This observation led to the formulation of the WHO oral rehydration solution, which subsequently saved the lives of millions of children affected by severe diarrhea worldwide.
Other modes of sodium transport are the electroneutral (Fig. 10.2B) and electrogenic transport
The electroneutral sodium transport is through sodium/hydrogen exchanger (NHE), usually combined with chloride transport via the chloride/bicarbonate exchanger known as AE exchanger (Fig. 10.2C). The exchangers are present on both luminal and basolateral membranes. This type of transport is responsible for most of the sodium chloride reabsorption in the colon. In the distal colon the NHE exchanger is upregulated by glucocorticoids.
The electrogenic absorption of sodium occurs through the epithelial sodium channels (ENaCs, also known as amiloride-sensitive sodium channels), which are present on the luminal side of the epithelium (Fig. 10.2D). ENaCs are regulated by steroids (aldosterone) and are important particularly in the distal colon. Absorption of Na+ is accompanied by Cl− following through a chloride channel (which could be the CFTR – see below). Aldosterone also upregulates the Na+/K+-ATPase.
Chloride transport: the cystic fibrosis transmembrane conductance regulator (CFTR)
Luminal secretion of chloride occurs via the cystic fibrosis transmembrane conductance regulator (CFTR; Fig. 10.2B). The CFTR is a single-polypeptide membrane ion channel. It is also present in the epithelia of the lung and sweat glands. Its function is controlled by the G-protein–cAMP-protein kinase A (PKA) signaling cascade (Chapter 40). Because the CFTR is activated by cAMP, prostaglandin E2 (PGE2), serotonin, as well as the cholera toxin and the E. coli heat-stable enterotoxin, all activate chloride secretion. On the other hand, the loss-of-function mutations of CFTR are the cause of cystic fibrosis, where the chloride transport is impaired or inhibited. CFTR also has regulatory function: its phosphorylation inhibits the NHE exchanger, thus decreasing Na+ absorption. Interestingly CFTR is also able to transport chloride in the opposite direction, aiding chloride reabsorption (above).
The basolateral C1− uptake occurs through the Na+ K+ C1− co-transporter (known as NKCC1) and through chloride/bicarbonate exchangers.
Clinical box Cystic fibrosis
Cystic fibrosis, a monogenic autosomal recessive disorder, involves inhibition of chloride transport due to the absence of the CFTR. Different mutations of the CFTR gene lead to either complete absence of the transporter or impair its functionality.
The prevalence of CF is 1 : 3000 live births in the USA and northern Europe. In the USA, cystic fibrosis is the No. 1 cause of malabsorption. It manifests itself predominantly in childhood. The main problems are usually respiratory. Chloride secretion is decreased and the Na+ reabsorption is accelerated. This results in decreased hydration of epithelial secretions. In the respiratory tract there is decreased hydration of the airway mucus and thus failure of its clearance, with ensuing bacterial infections. Gastrointestinal problems include meconium ileus and intestinal obstruction. The absence of the CFTR also affects functioning of the Cl−/HCO3− exchanger (and thus the passive secretion of Na+) – this impairs pancreatic enzyme secretion. Thickened biliary secretions may be a cause of focal biliary cirrhosis and chronic cholelithiasis. There also is impairment of mucus secretion in the colonic crypts, with enhanced Na+ reabsorption through Na+ channels and Na+/H+ transporters.
Potassium absorption and potassium secretion in the colon is aided by a several potassium channels
Potassium absorption is mediated by H+/K+ ATPases (belonging to the family of P-type ATPases) in the luminal membrane. On the other hand, the basolateral potassium transport is by potassium channels and the K+ and Cl− cotransporter (KCC1). Both basolateral and luminal K+ channels are necessary to hyperpolarize the membrane to establish a driving force for the ENaC transporter.
K+ secretion through luminal K+ channels parallels the Cl− secretion through CFTR, and is similarly stimulated by cAMP, cGMP and protein kinase C (PKC). The expression of luminal K+ channels is also stimulated by aldosterone and glucocorticoids.
Reabsorption of short-chain fatty acids occurs together with bicarbonate secretion
The colon reabsorbs short-chain fatty acids (SCFAs) derived from bacterial fermentation of fiber, and this is combined with secretion of bicarbonate. Bicarbonate is secreted using luminal anion exchangers SCFA/HCO3− or C1−/HCO3−.
Aquaporins control colonic water reabsorption
Water reabsorption in the colon is mediated by ion channels known as aquaporins (AQP). AQP1,3 and 4 are located on basolateral and AQP8 on luminal membranes. The intestinal electrolyte transport systems are summarized in Figure 10.2.
The pH of intestinal secretions vary
Hydrogen ion concentration varies widely in different parts of the GI tract. This is essential for the digestive process and for protection of the underlying tissues in the stomach and intestines. Saliva secreted into the mouth is alkaline due to its bicarbonate content. On the other hand, the contents of the stomach are strongly acidic, but the mucus protecting its walls is alkaline. Thus while the parietal cells of the stomach secrete large amounts of hydrogen ion (principally through the action of the luminal H+/K+-ATPase), the gastric surface cells secrete mucus containing bicarbonate ion; they employ the Cl−/HCO3− exchanger. On entry to the duodenum the acidic content of the stomach is neutralized by strongly alkaline (due to the bicarbonate content) pancreatic secretions.
Digestion
There is a significant mechanical component to digestion. Chewing breaks down food to enable it to be swallowed, whilst the addition of saliva in the mouth begins the digestive process and acts as lubrication to facilitate swallowing. The food is then moved into the esophagus by a process driven by the esophageal reflex. As it transfers into the stomach, it is broken down into smaller particles. The presence of the digest triggers peristalsis, which further helps mixing and stimulates digestive secretions. Major stimuli to peristalsis are mediated through the parasympathetic nervous system. Absorption of nutrients depends on the rate of transit: thus, increased motility may lead to malabsorption.
The stomach and intestines are lined by epithelium, which has an invaginated surface that greatly increases its area. The small intestine is lined by enterocytes arranged in intestinal villi, and in addition each cell contains microvilli. The total absorptive surface area of the intestine is approximately 250 m2, roughly the area of a junior basketball court.
 Advanced concept box Digestive function of the stomach
Advanced concept box Digestive function of the stomach
There are different cell types in the mucosal wall of the stomach, performing different digestive functions. Cells called ‘chief cells’ secrete pepsinogen, which is a precursor of pepsin. Pepsinogen is activated to pepsin in the acidic environment of the stomach lumen. Parietal cells generate hydrogen ions through the action of carbonic anhydrase and then pump them into the lumen by an ATP-dependent proton pump on the luminal membrane. The H+ secretion is dependent on the parallel export of K+ through luminal K+ channel.
Parietal cell activity is stimulated by the action of histamine acting on H2 receptors. (Chapter 41), produced by histamine-secreting cells. The hormone gastrin is secreted by G-cells, and is triggered by food entering the stomach. Stomach cells also secrete the intrinsic factor (IF), which facilitates absorption of vitamin B12 in the intestine (Chapter 11). Last, but not least, epithelial cells secrete alkaline mucus, which protects the stomach lining from the effects of the strong acid.
Damage to the lining of the stomach or duodenum leads to ulceration. Treatment of acid-related symptoms such as dyspepsia or gastroesophageal reflux, can be achieved with antacids which simply neutralize the pH, H2 antagonists, e.g. cimetidine or ranitidine, which prevent histamine release, or proton pump inhibitors, e.g. omeprazole, which block H+ secretion by the parietal cells.
Digestion is a sequential, ordered series of processes
The carbohydrates, proteins and fats are split to absorbable products. Some ingested material, such as complex carbohydrates of plant origin, is indigestible and constitutes fiber.
The process of digestion is characterized by a number of stages that occur in a sequence, allowing the interaction of fluid, pH, emulsifying agents and enzymes. This, in turn, requires concerted secretory action from the salivary glands, liver and gall bladder, the pancreas, and the intestinal mucosa. The processes involved are outlined in Figure 10.1 and can be summarized as follows:
Lubrication and homogenization of food with fluids secreted by glands of the intestinal tract, starting in the mouth.
Secretion of enzymes that break down macromolecules to a mixture of oligomers, dimers and monomers.
Secretion of electrolytes, hydrogen ions and bicarbonate within different parts of the GI tract to optimize the conditions for enzymic hydrolysis.
Secretion of bile acids to emulsify dietary lipid, facilitating enzymic hydrolysis and absorption.
Further hydrolysis of oligomers and dimers by membrane-bound enzymes (jejunum).
Specific transport of digested material into enterocytes and thence to blood or lymph.
Recycling of bile acids. Absorption of the SCFAs produced by colonic bacterial fermentation.
Spare capacity in the intestinal tract
There is considerable functional reserve in all aspects of digestion and absorption
Because if this, minor functional loss may go unnoticed, allowing pathology to progress for some time before being diagnosed. A considerable impairment of structure/function needs to be present before signs and symptoms of GI maldigestion or malabsorption occur. Each of the organs involved in digestion and absorption has the capacity to increase its activity several fold in response to specific stimulation; this adds to the reserve capacity. For example, pancreatic disease manifests itself only after 90% of the pancreatic function is destroyed.
Note also that digestion of a particular nutrient takes place at several points in the GI tract. Lipids, carbohydrates and proteins can be digested at multiple points along the GI tract. Therefore disruption of digestive mechanisms at a single point is unlikely to cause a complete inability to digest a nutrient group (Fig. 10.3).
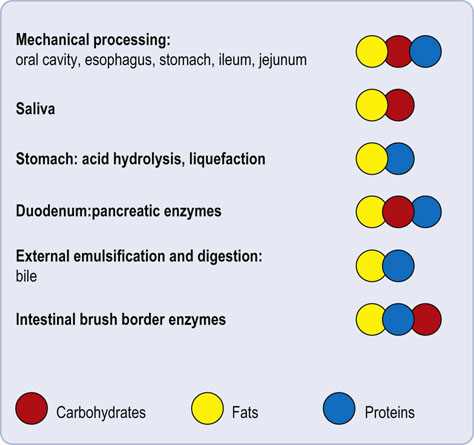
Fig. 10.3 Digestion as a multiorgan process.
Each of the main group of nutrients (carbohydrates, proteins and fats) undergoes digestion at multiple points.
The GI tract can also accommodate loss of function of one particular constituent organ. For example, if the stomach is surgically removed, the pancreas and small intestine can compensate for the loss of gastric digestion. On the other hand, in pancreatic disease, lingual lipases can accommodate, some loss of pancreatic lipase.
Digestive enzymes and zymogens
Most digestive enzymes are secreted as inactive precursors
With the exception of salivary amylase and lingual (associated with the tongue; hence oral) lipases, digestive enzymes are secreted into the gut lumen as inactive precursors termed zymogens (Chapter 6). Secretion of enzymes is similar in the salivary glands, gastric mucosa and pancreas. These organs contain specialized cells for the synthesis, packaging and transport of zymogen granules to the cell surface and thence to the intestinal lumen. These secretions are termed exocrine, i.e. ‘secreting to the outside’, as opposed to the endocrine secretion of hormones.
Enzymes involved in digestion of protein (proteases) and fat (lipase: phospholipase A2) are synthesized as inactive zymogens and are only activated on their release to the gut lumen. In general, these enzymes, once in their active form, can activate their own precursors. Activation of the precursors can also occur by change in pH (e.g. pepsinogen in the stomach is converted at pH below 4.0 into pepsin) or by the action of specific enteropeptidases bound to the mucosal membrane of the duodenum (Fig. 10.1).
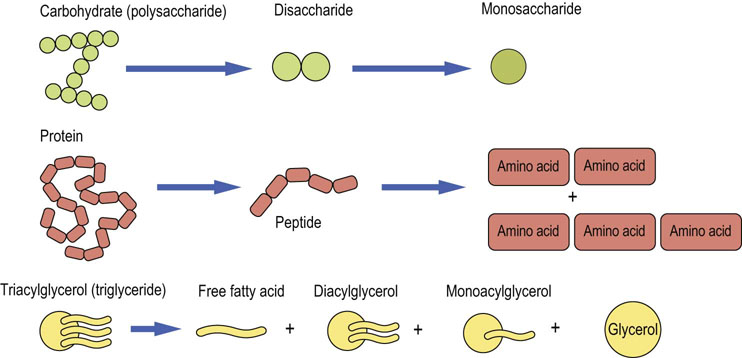
All digestive enzymes are hydrolases
All digestive enzymes hydrolyze their substrates. The products of such hydrolytic procedures are oligomers, dimers and monomers of the parent macromolecule. Thus, carbohydrates are hydrolyzed into a mixture of dissacharides and monosaccharides. Proteins are broken down to a mixture of di- and tripeptides and amino acids. Lipids are broken down to a mixture of fatty acids (FAs), glycerol and mono- and diacylglycerols (Fig. 10.4).
Digestion and absorption of carbohydrates
Dietary carbohydrates enter the GI tract as mono-, di- and polysaccharides
Dietary carbohydrates consist of mainly plant and animal starches (polysaccharides), the disaccharides sucrose and lactose, and the monosaccharides (Fig. 10.5). Monosaccharides include glucose, fructose and galactose, which are either present in the diet or are generated by digestion of di- and polysaccharides. Lactose, for instance, is a disaccharide derived from dairy products, and is hydrolyzed to the monosaccharides glucose and galactose by lactase and β-galactosidase. These sugar monomers can then be absorbed from the GI tract.
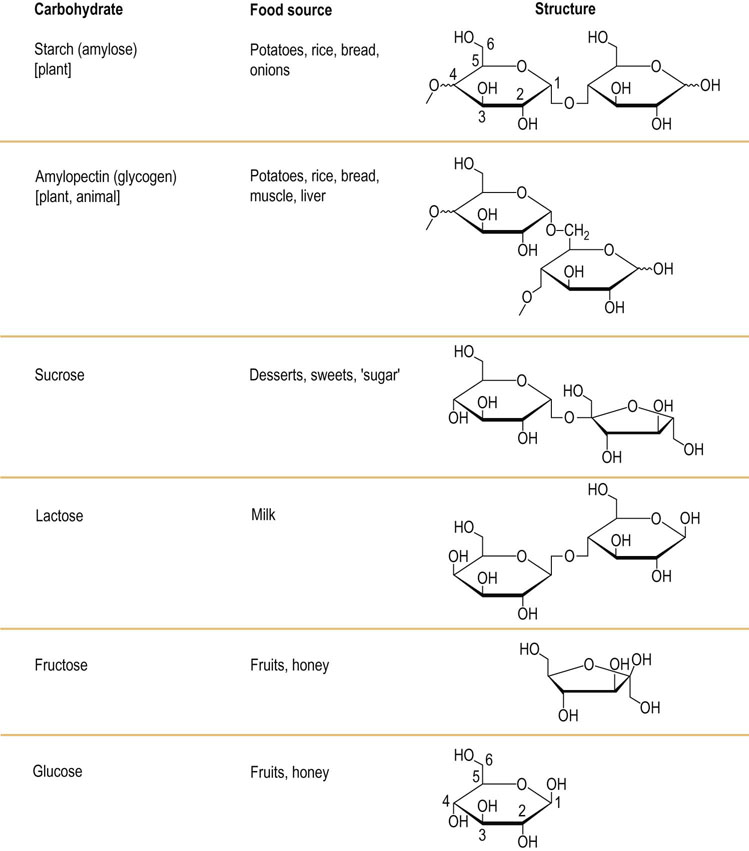
Fig. 10.5 Structure of the key dietary carbohydrates.
Starch and amylopectin are polysaccharides, and only two component sugar molecules are shown for each to illustrate the intermolecular linkages. Sucrose and lactose are the most common disaccharides, and fructose and glucose the most common monosaccharides. Refer to the glucose molecule for the standard numbering of carbon atoms (see also Chapter 3).
Advanced concept box Role of amylase, α-glucosidases and isomaltase in polysaccharide digestion
During eating, homogenization of food occurs by chewing. It is aided by contractions of the stomach wall muscles and gastric folds. One consequence of this is that dietary polysaccharides become hydrated. This is necessary for the action of amylase, which is specific for internal α-1,4-glycosidic linkages and not the α-1,6 linkages. Amylase also does not act on α-1,4 linkages of glycosyl residues serving as branching units. Thus, the cleaved units formed by its action are the disaccharide maltose, the trisaccharide maltotriose, and an oligosaccharide with one or more α-1,6 branches and containing on average eight glycosyl units, termed the ‘α-limit dextrin’. These compounds are further cleaved to glucose by oligosaccharidase and α-glucosidase, the latter removing single glucose residues from α-1,4-linked oligosaccharides (including maltose). A sucrase–isomaltase complex is secreted as a single polypeptide precursor molecule and is activated into two separate enzymes, one of which (isomaltase) is responsible for the hydrolytic cleavage of α-1,6-glycosidic bonds. Thus the final product of digestion of starches is glucose. The amylase occurs free in the lumen, whereas α-glucosidases and isomaltase are attached to the membrane of the enterocyte.
Disaccharides, and polysaccharides such as starch and glycogen, require hydrolytic cleavage into monosaccharides before absorption
Disaccharides are broken down by membrane-bound disaccharidases present on the intestinal mucosal surface. Starch and glycogen require additional hydrolytic capacity of the enzyme amylase found in the secretions of the salivary glands and pancreas (Fig. 10.6).
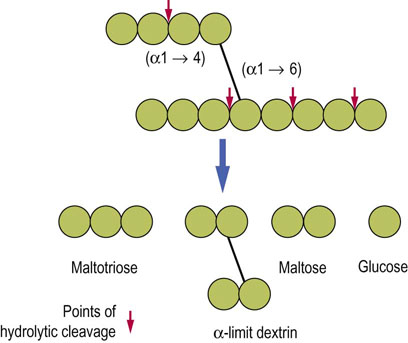
Fig. 10.6 Hydrolytic cleavage of polysaccharides.
Enzymatic hydrolysis is the mechanism of digestion of polysaccharides and disaccharides. The arrows illustrate points of cleavage and the type of hydrolyzed bond. Note that α-limit dextrin still contains both α-1,4 and α-1,6 bonds.
Starch is a plant polysaccharide and glycogen is its animal equivalent. Both contain a mixture of linear chains of glucose molecules linked by α-1,4-glycosidic bonds (amylose) and by branched glucose chains with α-1,6 linkages (amylopectin). Glycogen has a far more branched structure than starch. Digestion of these polysaccharides is promoted by the endosaccharidases, and amylase.
The products of hydrolysis of starch are the disaccharide maltose, the trisaccharide maltotriose and a branched unit, termed the α-limit dextrin. These products are further hydrolyzed by enzymes bound to the enterocytes, finally yielding the monosaccharide glucose (Fig. 10.7A).
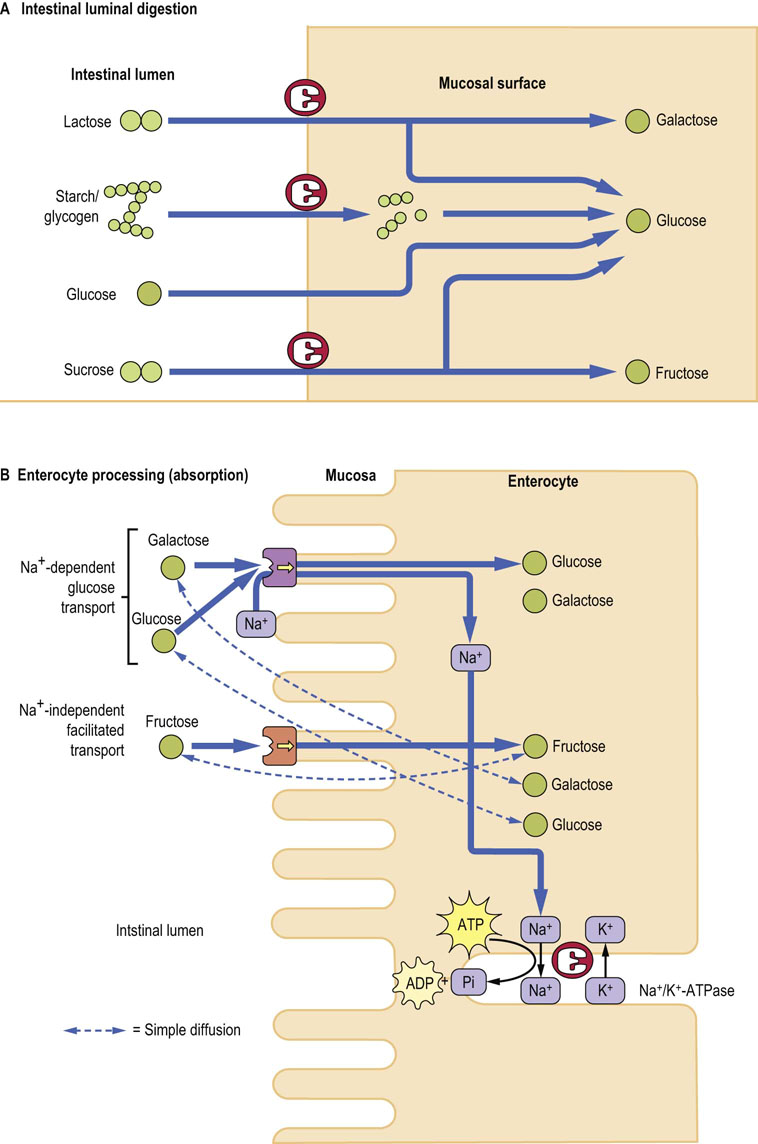
Fig. 10.7 Digestion and absorption of dietary carbohydrates.
(A) Monosaccharides are released as a result of hydrolysis of different polysaccharides. Note that preliminary digestion occurs in the gut lumen, and the final stage takes place on the mucosal surface. (B) Links between absorption of monosaccharides and sodium, and their relationship to the activity of Na+/K+-ATPase. ADP, adenosine diphosphate; ATP, adenosine triphosphate; Pi, inorganic phosphate. Compare with Figure 8.5.
Dietary disaccharides such as lactose, sucrose and trehalose (a disaccharide made up of two glucose molecules joined by an α-1,1 linkage) are hydrolyzed to their constituent monosaccharides by specific disaccharidases attached to the small intestinal brush border membrane. The catalytic domains of these enzymes project into the gut lumen to react with their specific substrates, whilst their noncatalytic, structural domain(s) are attached to the enterocyte membrane.
Disaccharidases are inducible, with the exception of lactase
The greater the amount of a disaccharide (e.g. sucrose) present in the diet or produced by digestion, the greater is the amount of the relevant specific disaccharidase (e.g. sucrase) produced by the enterocytes. The rate-limiting step in the absorption of dietary disaccharides is thus the transport of the resultant monosaccharides. Lactose, on the other hand, is a noninducible brush border disaccharidase, and therefore the rate-limiting factor in lactose absorption is its hydrolysis.
Clinical box Types of diarrhea
Diarrhea can be caused by the nonabsorbable solutes present in the gut (osmotic diarrhea), by the failure to either digest or absorb nutrients, and also by the secretory agonists (secretory diarrhea).
Osmotic diarrhea may be caused by malabsorption, digestive enzyme deficiencies or short bowel, and inflammatory disease.
Secretory diarrhea may be caused by infections, malabsorption of bile salts, malabsorption of fat or by endocrine causes such as carcinoid syndrome or Zollinger–Ellison syndrome. Main causes of inflammatory diarrhea are Crohn's disease, ulcerative colitis, and irritable bowel syndrome.
Absorption can be impaired, and secretion increased, in conditions that lead to the inflammation of the bowel (inflammatory diarrhea). Characteristically, the secretory diarrhea but not osmotic one, persists on fasting.
Active and passive transport systems transfer monosaccharides across the brush border membrane
The process of digestion results in a large increase in the number of osmotically active monosaccharide particles within the gut lumen. This leads to water being drawn into the lumen from the GI tract mucosa and vascular compartment. Increased brush border hydrolysis increases the osmotic load, while increased monosaccharide transport across the brush border enterocyte decreases it. For most oligo- and disaccharidases, the transport of the resulting monomers is rate limiting. As monomeric sugar concentrations (and osmolality) increase in the gut lumen, there is a compensatory decrease in the activity of brush border disaccharidases. This controls the osmotic load and prevents fluid shifts.
Glucose, fructose and galactose are the primary monosaccharides resulting from the digestion of dietary carbohydrates. Absorption of these sugars and other minor monosaccharides occurs by means of specific carrier-mediated mechanisms (Fig. 10.7B), all of which demonstrate substrate specificity and stereospecificity, show saturation kinetics and can be specifically inhibited. In addition, all monosaccharides can cross the brush border membrane by a simple diffusion, although this is extremely slow.
Clinical box A boy with abdominal discomfort, bloating and diarrhea: lactose intolerance
A 15-year-old African-American boy came across to the UK on an exchange visit for 2 months. After 2 weeks in the UK, he complained of abdominal discomfort, a feeling of being bloated, increased passage of urine and, more recently, the development of diarrhea. His only change in diet noted at the time was the introduction of milk. He had developed a considerable liking for milk and was consuming 1–2 large cartons per day. A lactose tolerance test was performed, whereby the young man was given 50 g lactose in an aqueous vehicle to drink. Plasma glucose levels did not rise by more than 1 mmol/L (18 mg/dL) over the next 2 hours, with sampling at 30-minute intervals. A diagnosis of lactose intolerance was made.
Lactose intolerance results from acquired lactase deficiency. Lactase activity decreases with increasing age in children, but the extent of the decline in activity is genetically determined and demonstrates ethnic variation. Lactase deficiency in the adult black population varies from 45% to 95%. If symptoms of malabsorption occur after the introduction of milk to adult diets, the diagnosis of acquired lactase deficiency should be considered. The diagnosis is made by challenging the small bowel with lactose and monitoring the rise in plasma glucose. An increase of more than 1.7 mmol/L (30 mg/dL) is considered normal. A rise of less than 1.1 mmol/L (20 mg/dL) is diagnostic of lactase deficiency. A rise of 1.1–1.7 mmol/L (20–30 mg/dL) is inconclusive.
There are at least two carrier-mediated transport mechanisms for monosaccharides
At the brush border membrane both glucose and galactose are transported by the sodium-dependent glucose transporter mentioned above. This membrane-linked protein binds with glucose (or galactose) and Na+ at separate sites, and transports both into the enterocyte cytosol. Na+ is transported down its concentration gradient (the concentration within the gut lumen exceeding that inside cells), and carries glucose along against the glucose concentration gradient. This transport is linked to Na+/K+-ATPase). The transport of glucose or galactose is thus an indirect active process.
Fructose is transported across the brush border membrane by a sodium-independent facilitated diffusion involving the membrane-associated glucose transporter GLUT-5 present on the brush border side of the enterocyte, and GLUT-2, which transfers monosaccharides out of the enterocyte into the circulation (Table 8.2).
An incomplete digestion of carbohydrates (the components of fiber) leads to their conversion to short-chain fatty acids (acetate, propionate, butyrate) by the colonic bacteria.
Clinical box A young man with weight loss, diarrhea, abdominal bloating and anemia: celiac disease
A 22-year-old man presented with a history of weight loss, diarrhea, abdominal bloating and anemia. He described his stools as pale and bulky. Laboratory features included hemoglobin of 90 g/L (9 g/dL) (reference range 130–180 g/L; 13–18 g/dL). Biopsy of his small bowel demonstrated flattening of the mucosal surface, villous atrophy and disappearance of microvilli. A diagnosis of gluten-induced enteropathy (celiac disease) was made. All wheat products were removed from the patient's diet and the symptoms resolved.
Celiac disease is an autoimmune condition precipitated by sensitivity to gluten resulting in inflammation of the small bowel mucosa. Gluten is a storage protein of wheat, barley and rye. It is actually a mixture of proteins, which includes the gliadins (the alcohol-soluble fraction of gluten) and glutelins. The gliadins pass through the intestinal barrier during, for instance, infections, and trigger the immune response. The inflammatory reaction ensues. The result is villous athrophy and hyperplasia of the crypts. Since the absorptive surface is markedly reduced, the resulting malabsorption can be severe.
Circulating antibodies to wheat gluten and its fractions are frequently present in cases of celiac disease. The diagnosis involves duodenal biopsy and testing the response to a gluten-free diet. The autoantibodies are tested for are antigliadin antibodies and/or tissue transglutaminase antibodies (transglutaminase is an enzyme that deamidates gliadin in the intestinal wall). Celiac disease is underdiagnosed, especially in patients with unexplained anemia.
For hematology reference values, refer to Table 5.2.
Advanced concept box Short-chain fatty acids are produced in the large bowel from undigested carbohydrates
Decreased absorption of dietary starch leads to the bacterial production of short-chain fatty acids (SHFAs) by the colonic bacteria.
SHFAs can be produced from fermentable oligosaccharides, disaccharides, monosaccharides and polyols (FODMAPs). Animal studies showed the presence of the short chain fatty acids receptor 2 (FFA2), a G-protein-coupled receptor present on intestinal endocrine cells., Binding of SCFA releases serotonin, leading to the increase in intestinal motility.
Digestion and absorption of lipids
Approximately 90% of fat in the diet is triacylglycerol (TAG; also termed triglyceride). The remainder consists of cholesterol, cholesteryl esters, phospholipids and nonesterified fatty acids (NEFAs).
Fats need to be emulsified before digestion
The hydrophobic nature of fats prevents the access of water-soluble digestive enzymes. Furthermore, fat globules present only a limited surface area for enzyme action. These issues are overcome by the emulsification process. The change in the physical nature of lipids begins in the stomach: the core body temperature helps to liquefy dietary lipids, and the peristaltic movements of the stomach facilitate formation of a lipid emulsion. The emulsification process is also aided by the acid-stable salivary and gastric lipases. The initial rate of hydrolysis is slow, due to the separate aqueous and lipid phases and limited lipid–water interface. Once hydrolysis begins, however, the water-immiscible TAGs are degraded to fatty acids, which act as surfactants. They confer a hydrophilic surface to lipid droplets and break them down into smaller particles, thus increasing the lipid–water interface and facilitating hydrolysis. The lipid phase disperses throughout the aqueous phase as an emulsion. Dietary phospholipids, fatty acids and monoacyl glycerols also act as surfactants.
Bile salts and pancreatic enzymes act on the lipid emulsion in the duodenum
The lipid emulsion passes from the stomach into the duodenum where the further digestion occurs, driven by enzymes secreted by the pancreas. Solubilization is aided by the release of bile salts from the gall bladder, stimulated by the hormone cholecystokinin.
The major enzyme secreted by the pancreas is pancreatic lipase. Lipase, however, remains inactive in the presence of bile salts normally secreted into the small intestine. This inhibition is overcome by the concomitant secretion of co-lipase by the pancreas. Co-lipase binds to both the water–lipid interface and to pancreatic lipase, simultaneously anchoring and activating the enzyme. As shown in Figure 10.8, only a small proportion of dietary TAGs become completely hydrolyzed to glycerol and fatty acids. The ‘second’ and ‘third’ fatty acids in TAGs are hydrolyzed with increasing difficulty: the pancreatic lipase produces mainly 2-monoacyl glycerols (2-MAGs), which are absorbed into enterocytes.
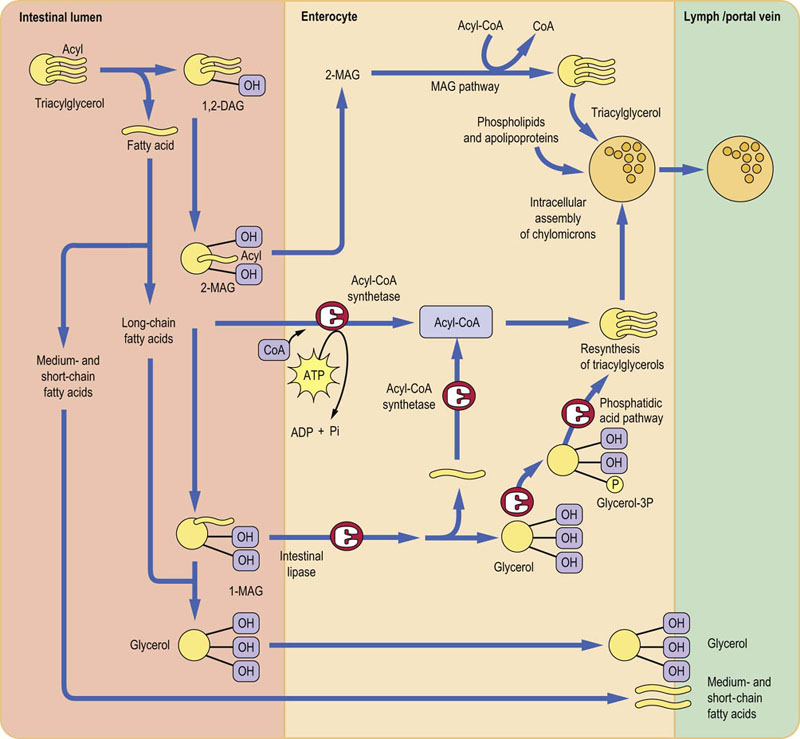
Fig. 10.8 Digestion and absorption of dietary lipids.
Dietary triglycerides undergo variable degrees of hydrolysis in the intestinal lumen. Subsequently, medium- and short-chain fatty acids are absorbed into the portal blood. However, long-chain fatty acids are resynthesized into triacylglycerols and then are incorporated into chylomicrons. The fatty acids are activated by acetyl-CoA before the synthesis of acylglycerols can take place. Note that enterocytes do not possess glycerol kinase: formation of glycerol phosphate requires the presence of glucose. TAG, triacylglycerol; DAG, diacylglycerol; MAG, monoacylglycerol; CoA, coenzyme A. Reproduced from Dominiczak MH. Medical Biochemistry Flash Cards. London: Elsevier, 2012, Card 38.
Bile salts are essential for solubilizing lipids during the digestive process
Bile acids (which are bile salts at the alkaline pH of the intestine) act as detergents and reversibly form lipid aggregates (micelles). Micelles are considerably smaller than lipid emulsion droplets. Their size depends on the bile acid concentration and the ratio of bile acids to lipids.
Thus, the lipid digest changes from fat emulsion droplets into micellar structures. The micelles transport the lipids to the brush border of the enterocyte.
The absorption of lipids into the epithelial cells lining the small intestine occurs by diffusion through the plasma membrane. Almost all the fatty acids and 2-MAGs are absorbed, as both are water soluble. Water-insoluble lipids are poorly absorbed: only 30–40% of dietary cholesterol is absorbed. The bile salts pass into the ileum, where they themselves are reabsorbed and transferred back to the liver. This is called the enterohepatic circulation (Chapter 17).
Clinical box An alcoholic man with central abdominal pain: pancreatitis
A 56-year-old man with a long history of alcohol abuse presented with chronic central abdominal pain, weight loss and diarrhea. He described his bowel motions as pale and greasy, and difficult to flush away. The abdominal radiograph revealed epigastric calcification in the area of the pancreas, and CT scanning revealed an atrophic calcified pancreas. The stool sample sent for fecal elastase quantification revealed this to be significantly reduced. Treatment was initiated with pancreatic enzyme supplements, resulting in resolution of his diarrhea and weight gain.
Acute pancreatitis is a serious and life-threatening illness, caused by gallstones blocking the pancreatic duct, alcohol abuse, or more rarely by drugs such as azathioprine, viruses such as mumps, or hypertriglyceridemia. Patients present with severe abdominal pain, nausea and vomiting. The most important biochemical marker of pancreatitis is increased amylase activity in serum. Increased activity of lipase and a decrease in serum calcium can also occur.
Chronic pancreatitis is a consequence of long-term inflammation and leads to malnutrition and steatorrhea (excessive fecal fat). It is also associated with failure of endocrine pancreatic function, leading to hyperglycemia.
Advanced concept box Exocrine function of the pancreas
The pancreas has two distinct functional roles: an exocrine function, i.e secretion of digestive enzymes via the pancreatic duct; and an endocrine function, i.e secretion of insulin, glucagon and other hormones by the islets of Langerhans. (Chapter 21) These hormones are responsible for glycemic control and aspects of gastrointestinal function.
Exocrine secretions flow into the pancreatic duct, which empties into the duodenum along with the common bile duct from the liver and the gall bladder. Food entering the duodenum stimulates the secretion of cholecystokinin, and this in turn stimulates pancreatic enzyme production and secretion. The acidity of the stomach contents entering the duodenum stimulates the release of another hormone, secretin, which triggers the secretion of bicarbonate-rich pancreatic fluid, which neutralizes the acidity in the duodenum.
The pancreas secretes enzymes which digest carbohydrates, lipids and proteins. Pancreatic amylase digests carbohydrates to oligo- and monosaccharides; lipase digests triacylglycerols while cholesteryl esterase yields free cholesterol and fatty acids; finally, proteases and peptidases break down proteins and peptides. To prevent the powerful proteases breaking down the pancreas itself (autodigestion), they are secreted as proenzymes (Chapter 6) and are activated in the intestinal lumen.
The fate of fatty acids depends on their chain length
Medium- and short-chain fatty acids (less than 10 carbon atoms) pass directly through the enterocytes into the hepatic portal system. In contrast, fatty acids containing more than 12 carbon atoms are bound to a fatty acid-binding protein within the cell, and are transferred to the rough endoplasmic reticulum for resynthesis into TAGs. The glycerol required for this process is obtained from the absorbed 2-MAGs (the MAG pathway; Fig. 10.8), from the hydrolysis of 1-MAG (which yields free glycerol), or from the glycerol-3-phosphate obtained from glycolysis (the phosphatidic acid pathway). Glycerol produced in the intestinal lumen is not used in the enterocyte for TAG synthesis, and passes directly to the portal vein.
Triacylglycerol synthesis requires activation of fatty acids
All absorbed long-chain fatty acids are reutilized to form TAG before being transferred to chylomicrons. Fatty acid activation is accomplished by the acyl-CoA synthase. Chylomicrons are assembled within the rough endoplasmic reticulum before being released by exocytosis into the intercellular space. They leave the intestine via lymph (Chapter 18).
Digestion and absorption of proteins
The gut receives 70–100 g dietary proteins per day and 35–200 g of endogenous proteins. The latter (mostly enzymes) are either secreted into the gut or shed from the epithelium as a result of cell turnover. The digestion and absorption of protein is extremely efficient: of this large load, only 1–2 g of nitrogen, equivalent to 6–12 g of protein, are lost in the feces daily.
Proteins are hydrolyzed by peptidases
Proteins are broken down by hydrolysis of peptide bonds by peptidases. These enzymes can either cleave internal peptide bonds (endopeptidases) or cleave off one amino acid at a time from either the –COOH (carboxypeptidases) or –NH2 (aminopeptidases) terminal of the polypeptide. Endopeptidases break down large polypeptides to smaller oligopeptides, which can subsequently be acted upon by the exopeptidases to produce amino acids and di- and tripeptides, the final products of protein digestion which are absorbed by the enterocytes. Depending on the source of the peptidases, the protein digestion can be divided into gastric, pancreatic and intestinal phases (Fig. 10.9).
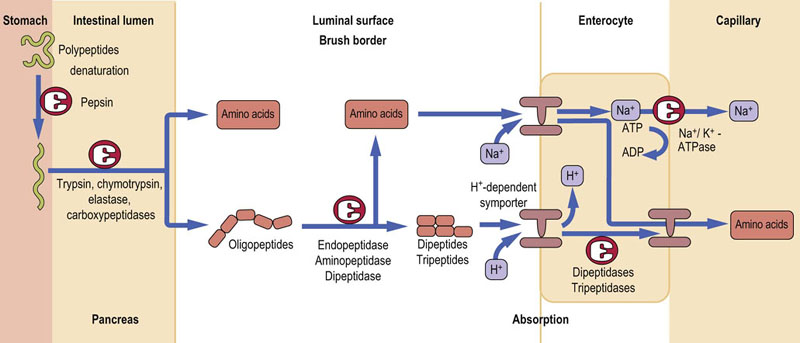
Fig. 10.9 Digestion and absorption of dietary proteins.
The preliminary stage is protein denaturation, which takes place in the stomach. The peptide bonds between amino acids are hydrolyzed by endo- and exopeptidases. Single amino acids and di- and tripeptides are absorbed using specific transport systems located in the enterocyte membrane.
Protein digestion begins in the stomach
In the stomach, the secreted HCl reduces the pH to 1–2, with consequent denaturation of dietary proteins. Denaturation unfolds polypeptide chains, making proteins more accessible to proteases. In addition, the chief cells of the gastric mucosa secrete pepsin. It is released as inactive precursor-, pepsinogen, and is activated by either an intramolecular reaction (autoactivation) at pH below 5.0 or by active pepsin (autocatalysis). At pH above 2.0 the liberated peptide remains bound to pepsin and acts as an inhibitor of its activity. This inhibition is removed by either a drop in pH below 2.0 or by further pepsin action. The products of digestion of proteins by pepsin are large peptide fragments and some free amino acids. They stimulate cholecystokinin release in the duodenum, in turn triggering the release of the main digestive enzymes by the pancreas, as well as the contraction of the gallbladder to release bile.
Proteolytic enzymes are released from the pancreas as inactive zymogens, in a manner similar to pepsinogen
Duodenal enteropeptidase converts trypsinogen to the active trypsin. This enzyme is then capable of autoactivation. It also activates all other pancreatic zymogens (chymotrypsin, elastase and carboxypeptides A and B). Because of this prime role of trypsin in activating other pancreatic enzymes, its activity is controlled within the pancreas and pancreatic ducts by a low-molecular-weight inhibitory peptide.
Pancreatic proteases cleave peptide bonds in different locations in a protein
Trypsin cleaves proteins at arginine and lysine residues, chymotrypsin at aromatic amino acids, and elastase at hydrophobic amino acids. The combined effect is to produce an abundance of free amino acids and low-molecular-weight peptides of 2–8 amino acids in length. Alongside protease secretion, the pancreas also produces copious amounts of sodium bicarbonate. This neutralizes the stomach as they pass into the duodenum, thus promoting pancreatic protease activity.
Final digestion of peptides is dependent on small intestinal peptidases
The final digestion of di- and oligopeptides is carried out in the small intestine by membrane-bound endopeptidases, dipeptidases and aminopeptidases. The end-products of this are free amino acids, and di- and tripeptides, which are then absorbed across the enterocyte membrane by specific carrier-mediated transport. Within the enterocyte, di- and tripeptides are further hydrolyzed to their constituent amino acids. The final step is the transfer of free amino acids out of the enterocyte into the portal blood.
Clinical box Diagnostic approaches to malabsorption
Malabsorption can be caused by cystic fibrosis, and lactase or other specific digestive enzyme deficiencies. The most common cause of carbohydrate malabsorption is lactose deficiency. Pancreatic insufficiency is also an important cause, as is inadequate amount of bile. Malabsorption can also result from the damage to the intestinal wall by, for instance, lymphoma, inflammatory bowel disease or radiotherapy. Important causes are surgical interventions: gastrectomy, pancreatectomy and resection of large fragments of the small bowel.
Rare endocrine causes include Zollinger–Ellison syndrome and abetalipoproteinemia (a rare disorder of lipoprotein metabolism in which chylomicron assembly is impaired).
The signs of malabsorption are chronic diarrhea, steatorrhea and loss of weight, and-in children – failure to thrive. Its complications result from the inadequate intake of nutrients, vitamins or trace metals (Chapter 11).
Diagnosis of malabsorption syndromes involves the conventional hematology and biochemistry tests and testing for active inflammatory processes (C-reactive protein), as well as stool examination and stool culture. Specialist tests include testing for vitamin deficiencies. Imaging-based investigations such as abdominal ultrasound and CT scan can be performed, and portions of the GI tract can be visualized through esophago-gastro-duodenoscopy. Biopsies can be taken from the stomach, duodenum and the small bowel.
The hydrogen breath tests are used in the diagnosis of carbohydrate malabsorption. Older tests for malabsorption include the xylose absorption test and lactose absorption test. If pancreatic insufficiency is suspected, the fecal excretion of enzymes such as elastase and lipase can be assessed, and endoscopic retrograde pancreatography (ERCP) performed.
Advanced concept box Active transport of amino acids into intestinal epithelial cells
Mechanisms of active transport of amino acids and di- or tripeptides into intestinal epithelial cells are similar to those described for glucose. At the brush border membrane, Na+-dependent symporters mediating amino acid uptake are linked to ATP-dependent pumping out of Na+ at the basolateral membrane. A similar H+-dependent symporter is present on the brush border surface for di- and tripeptide transport into the cell. Na+-independent transporters are present on the basolateral surface, allowing facilitated transport of amino acids into the portal vein.
At least six specific symporter systems have been identified for the uptake of L-amino acids from the intestinal lumen:
Neutral amino acid symporter for amino acids with short or polar side chains (Ser, Thr, Ala).
Neutral amino acid symporter for aromatic or hydrophobic side chains (Phe, Tyr, Met, Val, Leu, Ileu).
Imino acid symporter (Pro, OH-Pro).
Basic amino acid symporter (Lys, Arg, Cys).
These transport systems are also present in the renal tubules and defects in their molecular structure can lead to disease (e.g. Hartnup disease, an inherited disorder with defects of intestinal amino acid absorption and urinary loss of neutral amino acids described in the box on p. 84).
Summary
Digestion is a series of processes which prepare food for absorption.
Digestion and absorption of foods make the metabolic fuels available to the organism.
Carbohydrates are digested to simple sugars.
Fats are hydrolyzed to di- and monoglycerides.
Proteins are hydrolyzed to di- and tripeptides and free amino acids.
Defects in these mechanisms result in a variety of malabsorption and food intolerance syndromes.
Baumgart, DC, Sandborn, WJ. Crohn's disease. Lancet. 2012; 380:1590–1605.
Binder, HJ. Causes of chronic diarrhea. N Engl J Med. 2006; 355:236–239.
Broer, A, Cavanaugh, JA, Rasko, JEJ, et al. The molecular basis of neutral aminoacidurias. Pflugers Arch Eur J Physiol. 2006; 451:511–517.
Drozdowski, LA, Thomson, ABR. Intestinal sugar transport. World J Gastroenterol. 2006; 12:1657–1670.
Harris, JB, LaRocque, RC, Qadri, F, et al. Cholera. Lancet. 2012; 379:2466–2476.
Hou, W, Schubert, ML. Gastric secretion. Curr Opin Gastroenterol. 2006; 22:593–598.
Kunzelmann, K, Mall, M. Electrolyte transport in the mammalian colon: mechanisms and implications for disease. Physiol Rev. 2002; 82:245–289.
Diarrhoea: why children are still dying and what can be done, WHO 2009. www.who.int/maternal_child_adolescent/documents/9789241598415/en/index.html.
Johns Hopkins Children's Center. What is malabsorption. www.hopkinschildrens.org/tpl_rlinks_nav1up.aspx?id=5130.
Malabsorption. http://labtestsonline.org/understanding/conditions/malabsorption/.
MedlinePlus. Malabsorption. www.nlm.nih.gov/medlineplus/ency/article/000299.htm.
UNICEF rehydration solution 2006. www.unicef.org/media/media_31825.html.