Biosynthesis of Cholesterol and Steroids
Learning objectives
Introduction
Cholesterol is essential for cellular structure and function
Cholesterol is an essential component of mammalian cell membranes. It is also a precursor of important compounds such as the steroid hormones, vitamin D, and the bile acids. Humans synthesize approximately 1 g of cholesterol each day. The rate of endogenous cholesterol synthesis and its dietary intake determine its plasma concentration. Excess of dietary cholesterol reduces its endogenous synthesis.
A typical Western diet contains approximately 500 mg (1.2 mmol) of cholesterol daily, mainly in meat, eggs, and dairy products (Chapter 22). Under normal circumstances, 30–60% of this is absorbed during passage through the gut. Following intestinal absorption, cholesterol is transported to the liver and to peripheral tissues as a component of lipoprotein particles, the chylomicrons.
In the cell, all cholesterol is present within membranes. The regulatory events occur either within membranes or in their close vicinity
Cellular cholesterol synthesis, and cholesterol uptake from the extracellular fluid (primarily as a component of the low-density lipoprotein [LDL] particles) are precisely regulated at both translational and post-translational level – and the membrane concentration of the free cholesterol is the main regulator of cholesterol synthesis. The pathway of cholesterol synthesis in its early stages, provides substrates for the synthesis of compounds important for cell proliferation the electron transport and for combating oxidative stress. The key intermediates in cholesterol synthesis are mevalonate, farnesyl pyrophosphate, squalene and lanosterol (Fig. 17.1). The oxysterols generated at the later stages of the pathway also contribute to the regulation of cholesterol homeostasis. Finally, range of genetically controlled transporters regulate cholesterol absorption and its intestinal and biliary transport.
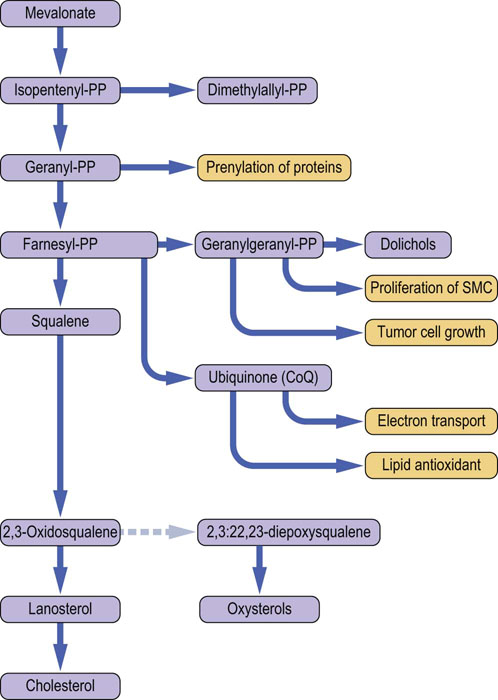
Fig. 17.1 Cholesterol synthesis and related pathways. The pathway of cholesterol synthesis is also a source of compounds that participate in a variety of cell functions. These are shown in orange-colored boxes. (modified from Charlton-Menys V, Durrington PN. Exp Physiol 2007; 93:27–42, with permission). SMC, smooth muscle cells; CoQ, coenzyme Q; PP, pyrophosphate.
The liver repackages cholesterol and triglycerides into very low-density lipoproteins (VLDL). After peripheral hydrolysis of VLDL by lipoprotein lipase, the VLDL remnants and LDL deliver cholesterol back to the liver. Chapter 18 describes lipoprotein metabolism in detail.
Humans cannot metabolize the sterol ring of cholesterol
Cholesterol is excreted by the liver in bile either in the form of bile acids or as free cholesterol. The so called primary bile acids are synthesized in the liver, and another ‘set’ known as the secondary bile acids is produced from them by the intestinal bacteria. Most bile acids are reabsorbed in the terminal ileum and recycled back to the liver.
Clinical significance
Excess supply of cholesterol is important in the etiology of atherosclerosis (Chapter 18). Cholesterol is also a major component of gallstones. Another cause of clinical disorders are the inherited deficiencies of enzymes that participate in the synthesis of steroid hormones from cholesterol. The latter are important in neonatal medicine.
The Cholesterol Molecule
The structure of cholesterol is shown in Figure 17.2. It has a molecular weight of 386 Da and contains 27 carbon atoms, of which 17 are incorporated into four fused rings (the cyclopentanoperhydrophenanthrene nucleus). Two further carbons are in the methyl groups at the junctions of rings AB and CD, and eight are in the side chain. Cholesterol is almost entirely composed of carbon and hydrogen atoms; there is a solitary hydroxyl group is attached to carbon 3. Cholesterol is also almost completely saturated structure, having just one double bond between carbon atoms 5 and 6.
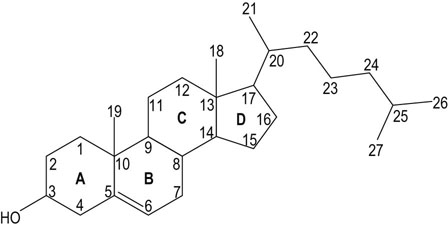
Fig. 17.2 Structure of cholesterol.
A–D is the conventional notation used to describe the four rings. Numbers 1–27 describe the carbon atoms.
Cholesterol changes membrane fluidity
Cholesterol is an essential component of cell membranes. It is held in the lipid bilayer by physical interactions between the planar steroid ring and the fatty acid chains. The absence of covalent bonding means that it may easily transfer in and out of the membrane. Membranes are fluid structures where both the lipid and protein molecules move and undergo conformational change (Chapter 8). The more fluid the phospholipid bilayer becomes, the more permeable is the membrane. At body temperature, the long hydrocarbon chains of the lipid bilayer are capable of considerable motion. Cholesterol is located between these hydrocarbon chains. Cholesterol increases fluidity of membranes rich in phospholipids and sphingolipids that contain saturated fatty acids.
Cholesterol is clustered in regions within the lipid bilayer. In a such a cluster, there may be 1 mole of cholesterol per mole of phospholipid, while in adjacent areas there may be no cholesterol. Thus, the membrane contains cholesterol-rich impermeable patches and more permeable cholesterol-free regions. Different cell organelles may differ in cholesterol content by the factor of 10. It is, for instance, virtually absent from the inner mitochondrial membrane.
Free and esterified cholesterol
Cholesterol is poorly soluble in water. Only about 30% of circulating cholesterol occurs in the free form, the majority forms esters with long-chain fatty acids such as oleic and linoleic acids. Cholesteryl esters (CE) are even less soluble in water than free cholesterol.
Cholesterol is esterified in the plasma by the enzyme cholesterol-lecithin acyltransferase and in the cells by the acyl-CoA : cholesterol acyltransferase (ACAT). In the plasma, it is incorporated into a range of lipoproteins (Chapter 18) where it is present mostly as CE. CE are stored in lipid droplets in the endoplasmic reticulum.
Intestinal absorption of cholesterol
Cholesterol is absorbed (and secreted) in the intestine by specific transporters
Dietary cholesterol is absorbed from the intestine via a membrane transporter known as the Nieman–Pick C1-like (NPC1L1) protein. Another transporter present in the apical (luminal) side of enterocytes is the ATP-binding cassette G5/G8, comprising two half-transporters – ABCG5 and ABCG8. These transport cholesterol in the ‘reverse’ direction, back to the intestine, and are also involved in secretion of other sterols into the bile. These transporters are upregulated by the nuclear transcription factor, the liver X receptor (see below). The genes coding for these transporters have a sterol response element in their promoter regions. Mutations in these genes result in tissue accumulation of plant sterols (sitosterolemia). The drug ezetimibe suppresses the NPC1L1-mediated cholesterol transport and has been used in the treatment of hypercholesterolemia.
Biosynthesis of cholesterol
Cholesterol is synthesized from acetyl coenzyme A
Virtually all human cells have the capacity to make cholesterol. Liver is the major site of cholesterol synthesis and smaller amounts are synthesized in the intestine, adrenal cortex and gonads. The synthesis of cholesterol molecule requires a source of carbon atoms, a source of reducing power and significant amounts of energy. The acetyl-coenzyme A (acetyl-CoA) provides a high-energy starting point. Acetyl-CoA may be provided from the β-oxidation of long-chain fatty acids, the dehydrogenation of pyruvate and the oxidation of ketogenic amino acids such as leucine and isoleucine. The reducing power is provided by reduced nicotinamide a dinucleotide phosphate (NADPH) generated in the pentose phosphate pathway (Chapter 12).
Energy is provided by the breakdown of adenosine triphosphate (ATP). Overall, the synthesis of 1 mole of cholesterol requires 18 moles of acetyl-CoA, 36 moles of ATP and 16 moles of NADPH. All the biosynthetic reactions occur within the cytoplasm, although some of the required enzymes are bound to the ER membranes.
Mevalonic acid is the first unique compound in the pathway of cholesterol synthesis
Three molecules of acetyl-CoA are converted into the 6-carbon mevalonic acid (Fig. 17.3). The first two steps occur in the cytoplasm and are condensation reactions leading to the formation of the 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA). These reactions, catalyzed by acetoacetyl-CoA thiolase and HMG-CoA synthase, are the same as the ones in the synthesis of ketone bodies, although the latter process occurs within mitochondria.
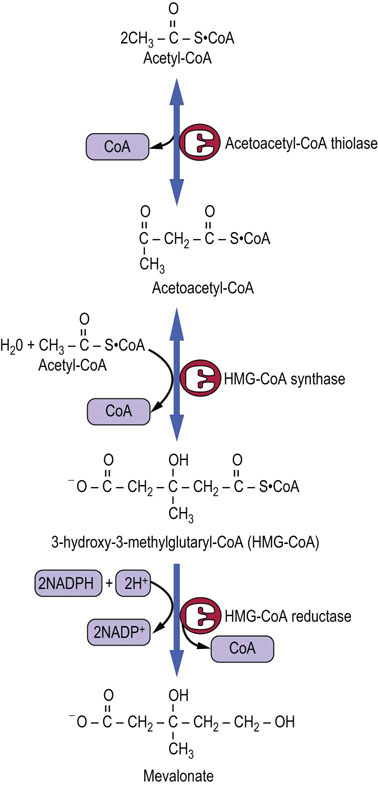
HMG-CoA reductase is the rate-limiting enzyme in the pathway
The rate-limiting reaction in the pathway of cholesterol synthesis is that catalyzed by the HMG-CoA reductase (HMGR), which leads to the irreversible formation of mevalonic acid. The reaction uses two molecules of NADPH.
HMGR is embedded in the ER. It is controlled at multiple levels: by feedback inhibition, by the rate of its degradation, by phosphorylation (it is active in a nonphosphorylated state) and by changes in gene expression. It is also affected by several hormones: insulin and tri-iodothyronine increase its activity, while glucagon and cortisol inhibit it. HMGR can also be phosphorylated (and thus inhibited) by the ‘energy sensor’ enzyme, the AMP-dependent kinase (AMPK, Chapter 22). Oxysterols also regulate the HMGR.
Farnesyl pyrophosphate is made up of three isoprene units
Three molecules of mevalonic acid are phosphorylated in two reactions requiring kinases and ATP. Subsequent decarboxylation yields the isomeric 5-carbon isoprene units, isopentenyl pyrophosphate and dimethylallyl pyrophosphate, which condense together to form geranyl pyrophosphate. The geranyl pyrophosphate is enlarged to geranyl-geranyl pyrophosphate. Further condensation with isopentenyl pyrophosphate produces the 15-carbon atom molecule, farnesyl pyrophosphate (Fig. 17.4). As well as being an intermediate in cholesterol biosynthesis, farnesyl pyrophosphate is the branching point for the synthesis of dolichol (a substrate in glycoprotein synthesis) and ubiquinone (Fig. 17.1).
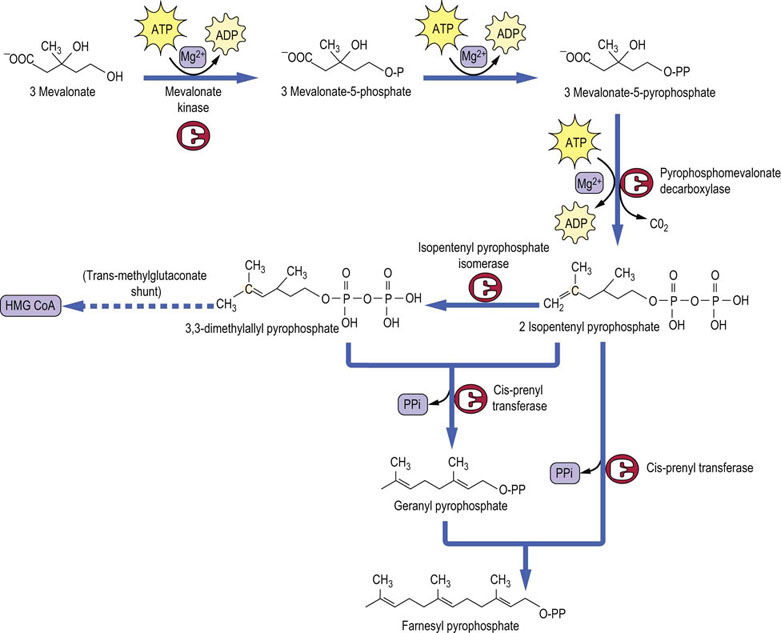
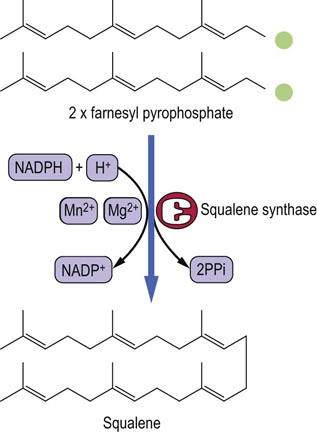
Squalene is a linear molecule capable of a ring formation
Squalene synthase condenses two molecules of farnesyl pyrophosphate to form squalene, a 30-carbon hydrocarbon containing six double bonds (Fig. 17.5), which later enable it to fold into a ring similar to the steroid nucleus. Several intermediates are involved at this stage.
Squalene cyclizes to lanosterol
Before the closure of the ring, squalene is converted to squalene 2,3-oxide by squalene monooxygenase. This NADPH-dependent monooxygenase inserts oxygen molecule into the structure. Thereafter, cyclization is catalyzed by oxidosqualene cyclase, yielding lanosterol (Fig. 17.6).
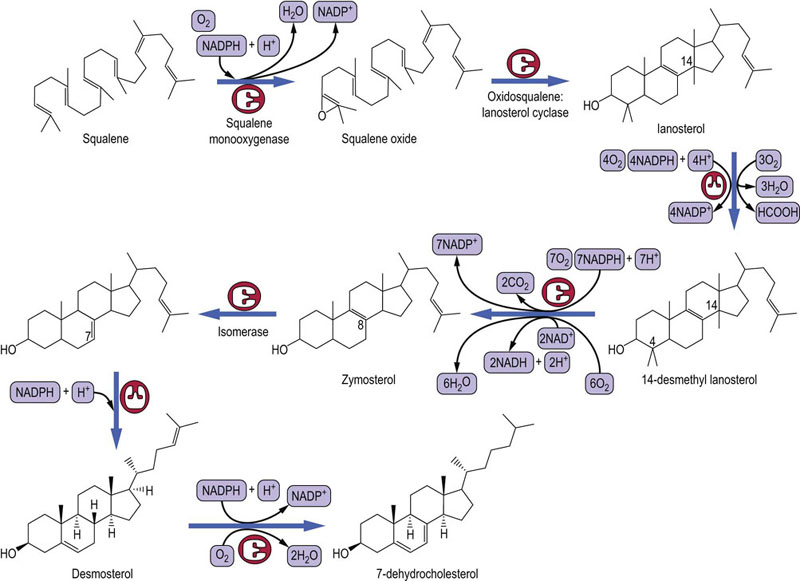
Fig. 17.6 Later stages of cholesterol synthesis.
These reactions occur while bound to a squalene- and sterol-binding proteins. FAD, flavin adenine dinucleotide; NADH, reduced nicotinamide adenine dinucleotide.
In plants, there is a different product of squalene cyclization, known as cycloartenol, which is further metabolized to a range of phytosterols, including sitosterol, rather than to cholesterol.
Final stages of cholesterol biosynthesis occur on a carrier protein
Squalene, lanosterol and all the subsequent intermediates in cholesterol synthesis are hydrophobic molecules. In order for the final steps of the pathway to occur in an aqueous medium, these intermediates react while bound to a squalene- and sterol-binding protein. The conversion from the 30-carbon lanosterol to the 27-carbon cholesterol involves decarboxylation reactions, isomerization and a reduction (Fig. 17.6).
Cholesterol can be converted to oxysterols by oxidation of its side chain. Oxysterols are important in cholesterol metabolism in the brain
This is performed by a cytochrome P450 enzyme, cholesterol 24-hydroxylase (CYP46A1, present in the brain) and 27-hydroxylase (CYP27A1, present in other tissues). The 27-hydroxycholesterol can cross the blood–brain barrier without the need for an energy-requiring transporter. The 25-hydroxycholesterol regulates the liver X receptors (LXRs). In the brain, also through LXR, it regulates the expression of apolipoprotein E (an important transporter of cholesterol in the brain) and the expression of transporters ABCA1, ABCG1 and ABCG4 present on astrocyte membranes.
Measurements of plant sterols and cholesterol precursors are used as markers of cholesterol absorption and metabolism
In the studies of cholesterol metabolism, the plant sterols campesterol, sitosterol and biliary sterol 5α-cholestanol have been used as markers of cholesterol absorption. On the other hand, the measurements of cholesterol precursors such as mevalonic acid, squalene or lanosterol have been used as markers of cholesterol synthesis.
 Advanced concept box PCSK9 protease regulates degradation of LDL receptors
Advanced concept box PCSK9 protease regulates degradation of LDL receptors
Serine protease PCSK9 (proprotein convertase subtilisin/kexin type 9) is a regulator of LDL receptors. PCSK9 is secreted from the liver, is present in plasma, and binds to the extracellular domain of the LDL receptor. After the LDL-receptor complex is internalized, PCSK9 prevents it from recycling to the membrane and channels it towards degradation. PSK9 overexpression in transgenic mice lowers the LDL receptor levels. In hypercholesterolemic individuals with a gain-of-function mutation, the PCSK9 has increased affinity for the LDL receptor. On the other hand, the loss-of-function mutation results in lower plasma cholesterol concentrations.
Regulation of cholesterol synthesis
At an organism level, the supply of cholesterol is either through diet or through de novo synthesis
Many factors are involved in the regulation of the intracellular concentration of cholesterol (Table 17.1). Under normal circumstances, there is an inverse relationship between dietary cholesterol intake and the rate of cholesterol biosynthesis. This ensures a relatively constant supply of cholesterol. It also explains why dietary restriction is only likely to achieve a moderate reduction in the plasma cholesterol concentration.
Table 17.1
Regulation of intracellular cholesterol concentration
Processes that increase the free cholesterol concentration
De novo synthesis
Hydrolysis of intracellular cholesteryl esters by cholesterol ester hydrolase
Dietary intake of cholesterol
Receptor-mediated uptake of LDL: upregulation of the LDL receptors
Processes that decrease intracellular free cholesterol concentration
Inhibition of de novo cholesterol synthesis
Downregulation of the LDL receptors
Esterification of cholesterol by acyl-coenzyme A : cholesterol acyl transferase
Release of cholesterol from the cell to high-density lipoproteins (HDL)
Conversion of cholesterol to bile acids or to steroid hormones
Factors that affect the activity of HMG-CoA reductase
Intracellular concentration of HMG-CoA
Membrane concentration of cholesterol
Hormones: insulin, tri-iodothyronine, glucagon, cortisol
The cell acquires cholesterol from both de novo synthesis and the external supply
Note that the ‘external supply’ in the case of a cell does not necessarily equal dietary source. The exogenous cholesterol reaches cells predominantly within lipoproteins: as a component of chylomicron remnants, VLDL remnants, or LDL. These lipoproteins bind to the apoB/E receptors present on the plasma membranes and the lipoprotein/receptor complexes are taken up into the cell (Chapter 18). In the cytoplasm, vesicles carrying the internalized complexes are acted upon by lysosomal enzymes, which separate the LDL from the receptor and hydrolyze cholesterol esters. Free cholesterol is released to the membrane. The LDL apoprotein B is degraded.
Regulation of intracellular cholesterol concentration involves HMG-CoA reductase, LDL receptor, 7α-hydroxylase and a network of nuclear receptors
The two sources of cholesterol supply, synthesis de novo and external delivery by lipoproteins, are reciprocally related. The intracellular (intramembrane) cholesterol concentration is a key factor regulating both cellular cholesterol synthesis and the expression of LDL receptors. Thus, an increase in the free cholesterol concentration results in the following (Fig. 17.7):
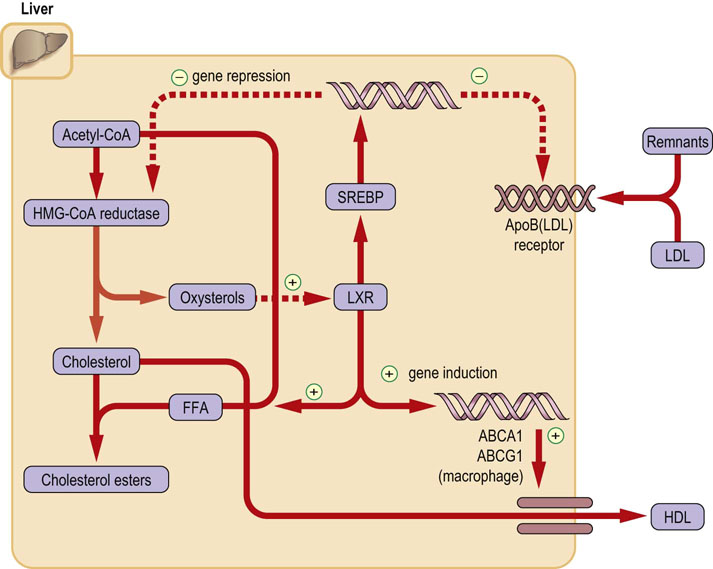
Fig. 17.7 Regulation of intracellular cholesterol concentration.
Free membrane cholesterol (and oxysterols) regulate intracellular cholesterol concentration by inducing or suppressing gene expression. Note that increase in intracellular cholesterol concentration will suppress synthesis of HMG-CoA reductase and ApoB/E receptor and at the same time increase cholesterol esterification and its transport from cells. See text for details. FFA: free fatty acid, Acetyl-CoA: acetyl coenzyme A, LXR: liver X receptor, SREBP: sterol regulatory element-binding protein.
 A reduction in both the activity and expression of HMG-CoA reductase. This limits cholesterol synthesis.
A reduction in both the activity and expression of HMG-CoA reductase. This limits cholesterol synthesis.
Downregulation of LDL receptors. This limits the cellular uptake of cholesterol.
An increase in cholesterol and phospholipid efflux from cell to apoproteins A (i.e. the high-density lipoprotein [HDL]).
An increase in the rate of conversion of cholesterol to bile acids. This increases cholesterol excretion.
Advanced concept box Trans-methylglutaconate shunt
The dimethylallyl pyrophosphate, one of the isoprene units formed from mevalonate (Fig. 17.4), can be dephosphorylated and broken down into acetoacetate and acetyl-CoA, which may then be diverted into other pathways, such as fatty acid biosynthesis. This mechanism is known as the trans-methylglutaconate shunt. Thus, high-energy compounds once destined to be converted into cholesterol may be redeployed to meet a higher priority need. Note that an increase in fatty acid synthesis will increase the amount of substrate for cholesterol esterification.
Sterol regulatory element-binding proteins (SREBPs) regulate the genes coding for enzymes involved in cholesterol synthesis
SREBPs are synthesized as 120 kDa inactive precursors, which are integral part of the ER membrane. They bind to the ER protein known as the SREBP cleavage-activating protein (SCAP). The SCAP/SREBP complex transfers from the ER to the Golgi apparatus, where SREBPs are cleaved by a protease, releasing the active transcription factors, which in turn translocate to the nucleus and activate all the genes in the cholesterol synthetic pathway (Fig. 17.7). This process is subject to ingenuous regulatory mechanism.
Cholesterol molecules bind to sterol-sensing intermembrane domains (cholesterol ‘receptors’) present on the SCAP protein. This allows binding of the SCAP/SREBP complex to another ER protein, Insig-1 (Insig stands for ‘insulin-induced gene’). The stability of the SCAP/SREBP/Insig-1 complex is a key regulatory event. This is how it works:
When cholesterol is depleted, the SCAP/SREBP complex dissociates from Insig-1 and travels to the Golgi apparatus. However, when cholesterol concentration in the membrane is high, cholesterol binding to SCAP induces conformational change which stabilizes the SCAP/SREBP/Insig-1 complex, blocking its movement to the Golgi. Consequently, there is a decrease in nuclear SREBP and the transcription of genes associated with cholesterol synthesis remains repressed. The synthesis of cholesterol is inhibited. Cholesterol excess also decreases the level of Insig-1 mRNA (Fig. 17.8).
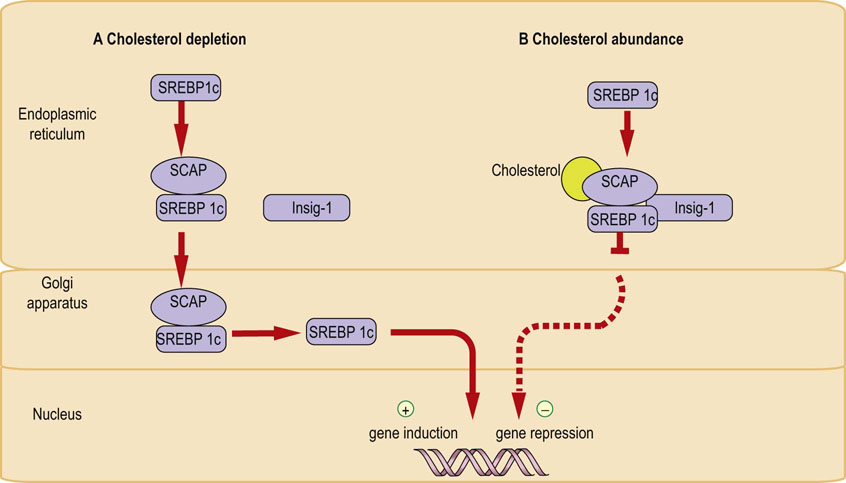
Fig. 17.8 Control of gene transcription by sterol-regulated transcription factors (SREBPs).
(A) When the membrane concentration of free cholesterol is low, the concentration SCAP/SREBP complex transfers from the ER to Golgi apparatus. Proteolysis takes place and the active transcription factor enters nucleus and initiates gene transcription. (B) When the membrane concentration of free cholesterol is high, cholesterol-binding-induced conformational change in the SCAP protein stabilizes its binding to the Insig-1. The complex remains in the ER with SREBP in inactive form, and gene transcription is repressed. See text for details. ER, endoplasmic reticulum; SREBP, sterol regulatory element binding protein; SCAP, SREBP-cleavage-activating protein.
When cells become again deprived of cholesterol, SCAP/SREBP dissociates from Insig-1, and the complex is again free to move to the Golgi apparatus, restoring nuclear SREBP level and restarting cholesterol synthesis. The SREBPs also induce Insig-1 synthesis.
HMG-CoA reductase is regulated by cholesterol in a mechanism involving its degradation
HMGR also possesses the sterol-sensing domain. When cholesterol level is high, HMGR, similarly to the SCAP/SREBP complex, binds to the Insig-1 protein. Here, however, the effect of such binding is different: it increases the ubiquination of the enzyme and channels it towards degradation. The overall effect is inhibition of cholesterol synthesis.
SREBPs have wide-ranging effects on the synthesis of cholesterol and fatty acids
In addition to the effect on cholesterol synthesis, SREBPs increase the expression of LDL receptor gene and affect fatty acid synthesis. In mammals, there are two closely related SREBPs: SREBP 1a and 1c. They are produced by the same gene, by alternative splicing. SREBP2 regulates cholesterol synthesis and LDL-receptor gene expression, while SREBP1c controls fatty acid synthesis. SREBP 1a induces all SREBP responsive genes.
SREBP1c can be activated by liver X receptors
SREBP 1c is upregulated by the liver X receptors (LXRs). LXRs are ligand-activated transcription factors that are members of the nuclear receptor superfamily (Chapter 40). They form heterodimers with other similar molecules, such as the retinoid X receptors (RXRs) and the farnesyl X receptors (FXRs). Resultant complexes bind to the LXR response elements on the DNA, regulating gene expression. LXRs also sense intracellular cholesterol concentration and contribute to regulating both its synthesis and its efflux from cells. However, it is not the cholesterol that binds to the LXR but oxysterols, such as 25-hydroxycholesterol or 27-hydroxycholesterol (Fig. 17.1).
SREBP 1c regulates cholesterol efflux from cells
High concentration of cholesterol in the hepatocyte induces, also through LXR-SREBP1c mechanism, genes coding for cholesterol transporters that control its efflux from cells to HDL particles: the expression of ABCA1 (the transporter that controls efflux of cholesterol from cells to nascent HDL) and ABCG1 (the transporter that stimulates efflux of cholesterol to more mature HDL2 and HDL3). Note that another transcription factor, PPARα, also regulates cholesterol efflux, acting through LXR (Chapter 18). PPARa is affected by a group of lipid-lowering drugs known as fibrates (derivatives of the fibric acid).
SREBP 1c regulates fatty acid synthesis
High intracellular cholesterol concentration also induces (through SREBP1c) genes coding for all the enzymes catalyzing fatty acid synthesis. The increased supply of fatty acids provides substrates for cholesterol esterification. Thus an increase in cholesterol concentration increases the concentration of oxysterols, and this in turn stimulates fatty acid synthesis, providing the substrate for cholesterol esterification. SREBP1c also induces genes involved in NADPH generation and the synthesis of triglycerides and phospholipids.
Statins are drugs that inhibit HMG-CoA reductase
The HMGR inhibitors, known as statins, lower cholesterol by binding to the site of HMG-CoA binding on the enzyme and competitively inhibiting its activity. This results in a decrease in the intracellular cholesterol concentration. The decrease in free cholesterol stimulates the expression of LDL receptors. LDL clearance increases and the plasma LDL-cholesterol decreases. The hepatic HMGR exhibits a diurnal rhythm: its activity is at a peak about 6 hours after dark and at a minimum some 6 hours after exposure to light. Therefore, statins are usually taken at night to ensure maximum effect.
 Clinical box A 50-year-old man with hypercholesterolemia treated with statin
Clinical box A 50-year-old man with hypercholesterolemia treated with statin
Despite a strict low cholesterol diet, a 50-year-old man, who had a family history of early cardiovascular disease, had a serum cholesterol concentration of 8.0 mmol/L (309 mg/dL); desirable concentration is 4.0 mmol/L (<155 mg/dL). He also smoked 15 cigarettes per day. He was given smoking cessation advice and was prescribed a statin. He tolerated the therapy well and 3 months later his cholesterol was 5.5 mmol/L (212 mg/dL). The dose of the statin was increased and after a further 3 months his plasma cholesterol concentration was 4.1 mmol/L (158 mg/dL).
Partial inhibition of the HMGR brings about a lowering of total plasma cholesterol by 30–50% and LDL-cholesterol by 30–60%. A range of statins is now available: this follows the original discovery that compactin (later renamed mevastatin), a fungal metabolite isolated from Penicillium citrinum, had HMGR-inhibiting properties. The inhibition of HMGR activity leads to a decrease in intracellular free cholesterol concentration, and to consequent increased expression of the cell membrane LDL receptor (Chapter 18). The end result is the lowering of the plasma total cholesterol and LDL-cholesterol.
Bile acids
The liver removes cholesterol either in a free form or as bile acids
The bile acids are quantitatively the most abundant metabolic products of cholesterol. In man, there are four main bile acids (Fig. 17.9). They all possess 24 carbon atoms: the terminal three carbons of the cholesterol side chain are removed during synthesis. They also have a saturated steroid nucleus and differ only in the number and position of the additional hydroxyl groups. All these hydroxyl groups have the α-configuration (i.e. are located below the plane of the nucleus), This means that the isomerization of the 3β-hydroxyl group of cholesterol must occur.
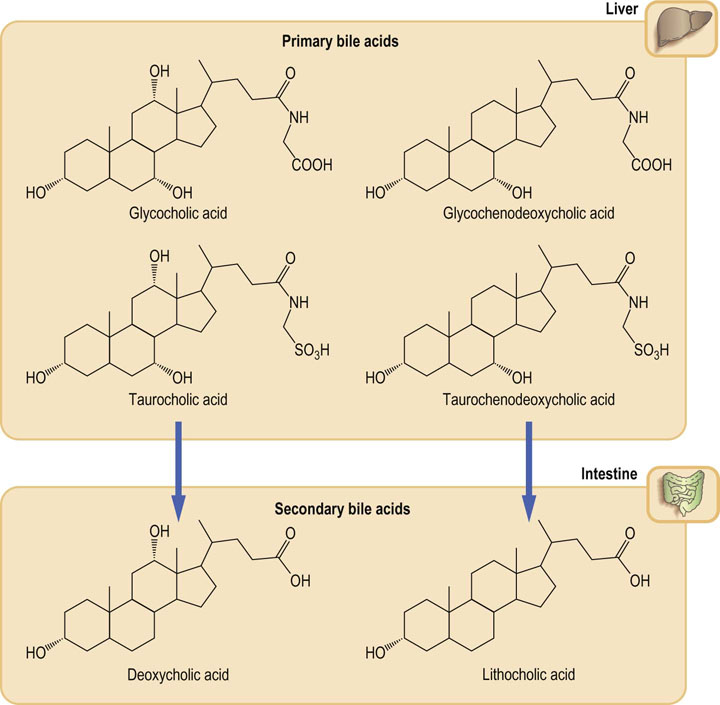
Primary bile acids are synthesized in the liver
Biosynthesis of the bile acids occurs in liver parenchymal cells, where cholic and chenodeoxycholic acids are produced. They are known as the primary bile acids. The rate-limiting step in the biosynthesis is a reaction catalyzed by the the microsomal 7α-hydroxylase (designated also CYP7A1), which introduces a hydroxyl group at 7α position of cholesterol ring. It is another microsomal monooxygenase (compare the ones involved in cholesterol synthesis).
Prior to their secretion, the primary bile acids are conjugated through the carboxyl group, forming amide linkages with either glycine or taurine. In man, there is a 3 : 1 ratio in favor of glycine conjugates. The secreted products are thus principally glycocholic, glycochenodeoxycholic, taurocholic and taurochenodeoxycholic acids. At physiologic pH, the bile acids are mainly ionized, therefore they are present as sodium or potassium salts. The terms ‘bile acids’ and ‘bile salts’ are used interchangeably. As their name suggests, these compounds are either directly secreted into the duodenum or are stored in the gall bladder. They form an important component of bile, together with water, phospholipids, cholesterol and excretory products such as bilirubin.
Liver X receptors participate in bile synthesis and secretion
Cholesterol is pumped into bile by ABCG5 and ABCG8 transport proteins, the expression of which is regulated by the LXR. Importantly, bile supersaturated with cholesterol facilitates formation of cholesterol gallstones. The LXR coordinate expression of several genes relevant to cholesterol excretion, including the cholesterol 7α-hydroxylase. Cholesterol excretion into the bile is also regulated by other nuclear receptors: the farnesyl X receptor (FXR) heterodimerizes with retinoic X receptor and binds to the bile acid response elements on the DNA. FXR acts as the cellular bile acid sensor by binding bile acids and suppressing their synthesis. FXR also induces bile acid export pump ABCB11, which removes bile acids from the hepatocyte into the bile.
Secondary bile acids are synthesized in the intestine
Secondary bile acids form within the intestine through the action of the anaerobic bacteria (principally Bacteroides) on the primary bile acids. They are deoxycholic and lithocholic acids (Fig. 17.9). Only a proportion of primary bile acids is converted into secondary bile acids. This requires hydrolysis of the amide link to glycine or taurine prior to removal of the 7α-hydroxyl group.
Bile acids assist the digestion of dietary fat
The secretion of bile from the liver and the emptying of the gallbladder are controlled by the gastrointestinal hormones hepatocrinin and cholecystokinin, respectively. They are released when partially digested food passes from the stomach to the duodenum. Once secreted into the intestine, the bile acids act as detergents (they possess polar carboxyl and hydroxyl groups), assisting the emulsification of ingested lipids; this aids the enzymatic digestion and absorption of dietary fat (Chapter 10).
Bile acids recirculate via the enterohepatic circulation
Up to 30 g of bile acids pass from the bile duct into the intestine each day but only 2% of this (approximately 0.5 g) is lost with the feces. Most are deconjugated and reabsorbed. Their passive reabsorption occurs in the jejunum and colon but they are mostly taken up by active transport in the ileum. Reabsorbed bile acids are transported back to the liver via the portal vein, being noncovalently bound to albumin, and are re-secreted into the bile. The process is known as the enterohepatic circulation. This recirculation also explains why bile contains both primary and secondary bile acids. The total bile acid pool is only 3 g and therefore they have to recirculate 5–10 times a day.
The bile acid flux also contributes to the control of bile acid synthesis; 7α-hydroxylase is subject to feedback inhibition by the bile acids returning to the liver through the portal vein. Dietary bile acids also decrease the expression of 7α-hydroxylase. Bile acid metabolism is summarized in Figure 17.10.
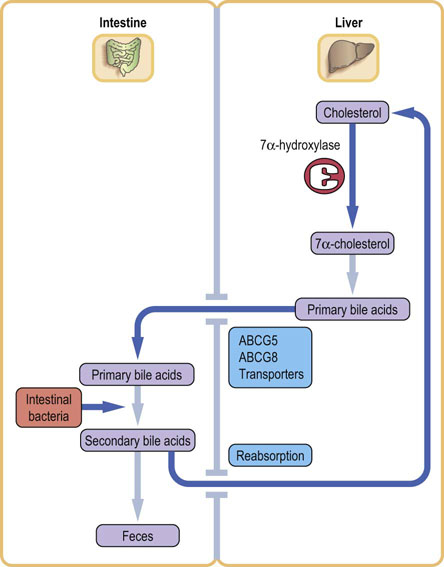
Cholesterol is excreted in the feces
Approximately 1 g of cholesterol is eliminated from the body each day through the feces: 50% of this is excreted as bile acids and the remainder as the isomeric saturated neutral sterols coprostanol (5β-) and cholestanol (5α-) produced by bacterial reduction of the cholesterol molecule.
Cholestyramine is a bile-acid binding resin which has been used to lower plasma cholesterol
Cholestyramine is a drug that interrupts enterohepatic circulation of the bile acids. It leads to an increase in 7α-hydroxylase activity, and thus to increased bile acid synthesis, and their increased excretion. This in turn leads to an increased cellular cholesterol synthesis and to increased expression of LDL receptors. Cholestyramine was one of the first cholesterol-lowering drugs but now has been superseded by the statins.
Clinical box A 45-year-old woman admitted with abdominal pain and vomiting: gallstones
A 50 years’ old woman woman complained of right upper quadrant abdominal pain and vomiting after eating fatty food. The only biochemical abnormality was a modestly raised alkaline phosphatase at 400 U/L (the upper reference limit is 260 U/L). An abdominal ultrasound was performed and it showed that the gall bladder contained gallstones. She was referred to the surgeons.
Gallstones occur in up to 20% of the population of Western countries. The condition results from the formation of cholesterol-rich stones within the gall bladder. Cholesterol is present in high concentrations in bile, being solubilized in micelles that also contain phospholipids and bile acids. When the liver secretes bile with a cholesterol-to-phospholipid ratio greater than 1 : 1, it is difficult to solubilize all the cholesterol in the micelles; thus there is a tendency for the excess to crystallize around any insoluble nuclei. This is compounded by further concentration of the bile in the gall bladder, which occurs as a result of reabsorption of water and electrolytes.
The condition may be managed conservatively by reducing dietary cholesterol, and by increasing availability of bile acids that will assist with cholesterol solubilization in the bile and its excretion via the gut. Alternative treatment includes disintegration of stones by shock waves (lithotripsy) and surgery. Elevated alkaline phosphatase is a marker of cholestasis (Chapter 30).
Steroid hormones
Cholesterol is the precursor of all steroid hormones
Mammals produce a wide range of steroid hormones, some of which differ only by a double bond or by the orientation of a hydroxyl group. Consequently, it has been necessary to develop a systematic nomenclature to detail their exact structures. There are three groups of steroid hormones (Fig. 17.11). The corticosteroids have 21 carbon atoms in the basic pregnane ring. The loss of the two carbon atoms from the cholesterol side chain produces the androstane ring and the hormones known as the androgens. Finally, the loss of the methyl group at carbon atom 19 as part of the aromatization of the A ring results in the estrane structure found in the estrogens. The presence and position of double bonds, and the position and orientation of the functional groups on the basic nucleus, are characteristics of individual hormones.
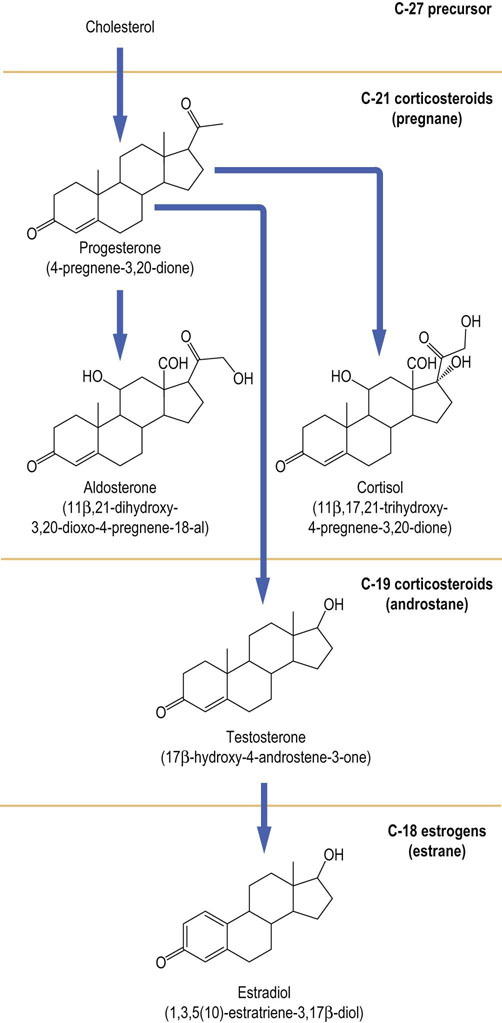
Fig. 17.11 Structure and nomenclature of the most important human steroid hormones.
Their trivial and systematic names (in parentheses) are shown. For numbering of the atoms in a steroid molecule, see Fig. 17.1; see also Chapter 39.
Biosynthesis of the steroid hormones
Conversion of cholesterol into steroid hormones occurs in only three organs: the adrenal cortex, the testis in men, and the ovary in women
A simplification used in practice is to consider the corticosteroids as the products of the adrenal cortex, the androgens as the products of the testis and the estrogens as the products of the ovary. Such simplified pathway of steroid synthesis is shown in Figure 17.12 (see also Chapter 39). However, this is not absolute, and all three organs are capable of secreting small amounts of steroids belonging to other groups. In pathologic situations, such as a defect in steroidogenesis or a steroid-secreting tumor, a very abnormal pattern of steroid secretion may emerge.
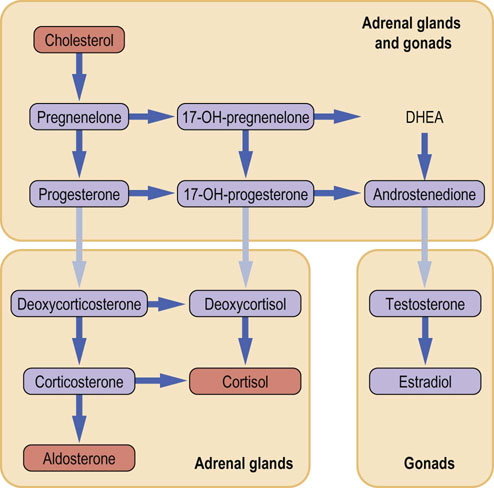
Cytochrome P450 monooxygenases control steroidogenesis
Most of the enzymes involved in converting cholesterol into steroid hormones are cytochrome P450 proteins that require oxygen and NADPH. In its simplest form, such an enzyme complex catalyzes the replacement of a carbon–hydrogen bond with a carbon–hydroxyl bond; hence, the term monooxygenase (compare cholesterol synthesis pathway above). Hydroxylation of the adjacent carbon atoms is the forerunner to cleavage of the carbon–carbon bond. Comparison of the structure of cholesterol (Fig. 17.2) with those of the steroid hormones (Fig. 17.11) demonstrates that the biosynthetic pathway largely consists of cleavage of carbon–carbon bonds and hydroxylation reactions. The enzymes involved have their own nomenclature in which the symbol CYP is followed by a specific suffix. Thus, CYP21A2 refers to the enzyme that hydroxylates carbon atom 21.
Clinical box Smith–lemli–opitz syndrome: a defect in 7-dehydrocholesterol reductase
The syndrome presents at birth with microencephaly, short nasal root, small chin, high arched palate, and often with midline cleft. There are often accompanying central nervous system (CNS) defects, polydactyly, and in males, ambiguous genitalia.
Despite the pathway of cholesterol synthesis and metabolism being well understood, a defect in 7-dehydrocholesterol reductase was only identified in 1993. The pathophysiology involves incomplete processing of embryonic signaling proteins (HH proteins), resulting in variable defects in different tissues.
While some of these children die in infancy, the rest, if assisted in feeding, survive with severe mental retardation (IQ 20–40). Most also develop growth retardation. Treatment involves giving additional cholesterol to the child. This improves growth but it appears to have no CNS benefits.
Corticosteroids
In the adrenal glands, zona fasciculata and zona reticularis are places of synthesis of the cortisol and the adrenal androgens. The aldosterone is synthesized in the outer layer (zona glomerulosa)
Cellular substructure of the adrenal cortex consists of three layers. The inner two layers (zona fasciculata and zona reticularis) are places of synthesis of cortisol, the main glucocorticoid, and the adrenal androgens. The cells of the outer layer (zona glomerulosa) synthesize aldosterone, the main mineralocorticoid (Chapter 23). Although many of the steps are similar, they are controlled by very different mechanisms.
Biosynthesis of the cortisol depends on stimulation by pituitary adrenocorticotropic hormone (ACTH) which binds to its plasma membrane receptor and triggers a range of intracellular events, which cause hydrolysis of cholesteryl esters stored in lipid droplets and the activation of the cholesterol 20,22-desmolase enzyme, which converts C-27 cholesterol into pregnenolone, the first of the C-21 pregnane family of corticosteroids. This is the rate-limiting step of steroidogenesis. Thereafter, conversion to cortisol requires a dehydrogenation – isomerization and three sequential hydroxylation reactions at C-17, C-21 and C-11, catalyzed by the CYP enzymes. The pathway is regulated by negative feedback control of ACTH secretion by cortisol (Chapter 39).
In case of aldosterone, the main stimulus to its synthesis is not ACTH but angiotensin II (Chapter 24), and potassium is an important secondary stimulus. Angiotensin II, by binding to its receptor, and potassium, work cooperatively to activate the first step in the pathway: the conversion of cholesterol into pregnenolone. Zona glomerulosa lacks the 17α-hydroxylase but has abundant amounts of 18-hydroxylase, which is the first step of a two-stage reaction forming the 18-aldehyde group of aldosterone.
Androgens
Conversion of corticosteroids into androgens requires the 17–20 lyase/desmolase and a substrate that contains a 17α-hydroxyl group
The 17α-hydroxyl group is added prior to breaking the C17–C20 bond to yield the androstane ring structure. The enzyme is abundant in the Leydig cells of the testis and in the granulosa cells of the ovary. However, in these two tissues the same biosynthetic step is controlled by two different hormones. In the testis the rate-limiting cholesterol side-chain cleavage step is stimulated by the luteinizing hormone (LH), whereas in the ovary it is stimulated by the follicle-stimulating hormone (FSH).
Estrogens
Conversion of androgens into estrogens involves removal of the methyl group at C-19 by the 19-aromatase
The A ring undergoes two dehydrogenations, yielding the characteristic 1,3,5(10)-estratriene nucleus. This aromatase is most abundant in the granulosa cells of the ovary, although the enzyme in adipose tissue can also convert some testosterone into estradiol. Biological actions of the steroid hormones are diverse and are best considered as belonging to the trophic hormone system (Chapter 39). Many genetic defects have been identified in the structures of the CYP enzymes. These defects lead to abnormal steroid biosynthesis and to clinical disorders such as congenital adrenal hyperplasia.
Advanced concept box Abnormalities in steroid synthesis are revealed by ALTERED pattern of urinary steroid metabolites
Steroid metabolites are excreted in urine mostly as water-soluble sulfate or glucuronic acid conjugates. The procedure used for their identification is gas chromatography – mass spectrometry (GCMS); it is very similar to methods adopted for the identification of anabolic steroids in sport. The first step in the analysis involves enzymatic release of the steroids from these conjugates; this is followed by chemical derivatization to increase their stability and improve separation, which is carried out by gas chromatography on capillary columns at high temperatures. Final detection is by mass fragmentation: for each steroid metabolite, a unique ion fragmentation ‘fingerprint’ is achieved, which allows positive identification and quantitation.
CLINICAL BOX A neonate born with ambiguous genitalia
Congenital adrenal hyperplasia
A neonate is born with ambiguous genitalia. Within 48 hours the infant becomes distressed and hypotensive. Biochemical investigations reveal:
Na+ 115 mmol/L (135–145 mmol/L)
K+ 7.0 mmol/L (3.5–5.0 mmol/L)
17-hydroxyprogesterone 550 nmol/L (upper reference limit 50 nmol/L)
This baby has a severe form of steroid 21-hydroxylase deficiency, the commonest of a range of conditions known as congenital adrenal hyperplasia, which are characterized by defects in activity of one of the enzymes in the steroidogenic pathway. The condition has a genetic basis, and it leads to a failure to produce cortisol (and also possibly aldosterone). This results in reduced negative feedback inhibition of the pituitary production of ACTH. ACTH thus continues to stimulate the adrenal gland to produce steroids upstream of the enzyme block. The accumulating steroids include 17-hydroxyprogesterone, which is further metabolized to testosterone (Figs 17.12 and 17.13). This results in androgenization of a female neonate. Mineralocorticoid deficiency causes renal salt wasting and requires urgent treatment with steroids and fluids. Long-term maintenance therapy with hydrocortisone and a mineralocorticoid suppresses ACTH and androgen production.
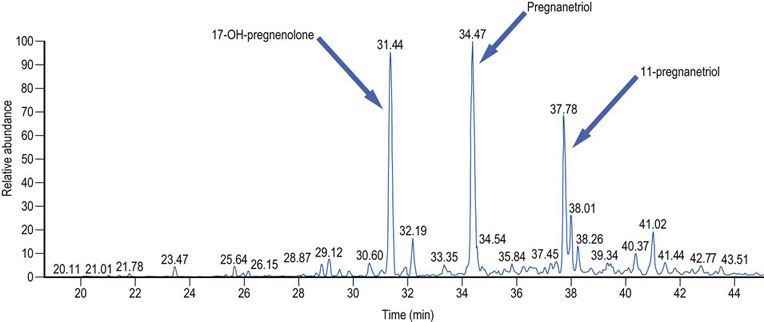
Fig. 17.13 Separation of urinary steroids performed by mass spectrometry.
In the clinical laboratory, the measurement of urinary steroid metabolites aids the diagnosis of a number of inherited disorders of the synthesis and metabolism of adrenal steroids, and steroid-producing tumors. It is particularly valuable in identifying the site of the defect in congenital adrenal hyperplasia. These investigations are most often performed in neonates with ambiguous genitalia, children with precocious puberty and in patients with suspected Cushing’s syndrome (Chapter 39). This is a urinary steroid metabolite pattern from a patient with 21-hydroxylase deficiency variant of congenital adrenal hyperplasia. The most prominent steroid metabolites are 17-hydroxypregnenolone, pregnanetriol and 11-oxo-pregnanetriol.
Total ion chromatogram of a urinary steroid metabolite pattern from a patient with 21-hydroxylase deficiency variant of congenital adrenal hyperplasia. In this condition the most prominent steroid metabolites are 17-hydroxypregnanolone, pregnanetriol and 11-oxo-preganetriol.
X-axis: time at which the chromatographically separated steroid metabolites are detected by the mass spectrometer. Y-axis: relative abundance (quantity of ions).
A less severe form of this condition is a result of partial enzyme deficiency. It occurs in young women who present with menstrual irregularity and hirsutism as a consequence of excess of adrenal androgens.
Mechanism of action of steroid hormones
Steroid hormones act via nuclear receptors
All steroid hormones act by binding to ligand-activated nuclear receptors. The superfamily of hormone receptors also includes receptors for the thyroid hormone T3 and the active forms of vitamins A and D (Chapter 40). Adjacent to the hormone-binding domain is a highly conserved DNA-binding domain, which is characterized by the presence of two zinc fingers (Fig. 34.3). Binding of the steroid ligand facilitates translocation of the activated receptor to the nucleus and its binding to a specific steroid response element in the promoter regions of target genes. This leads to gene transcription (Chapter 34). Genetic variability of steroid receptors structure may be associated with a variable degree of hormone resistance and diverse clinical presentations. See also discussion of the steroid receptor in Chapter 35.
Elimination of steroid hormones
Most steroid hormones are excreted in urine. There are two main steps in this process. Firstly, the biological potency of the steroid must be removed and this is achieved by a series of reduction reactions. Secondly, the steroid structure must be rendered water soluble: this is achieved by conjugation to a glucuronide or sulfate, usually through the hydroxyl group at C-3. As a result, many different steroid hormone conjugates are present in urine, some of them in high concentrations. Urinary steroid profiling by gas chromatography – mass spectrometry typically identifies more than 30 such steroids. Their relative concentrations may be used to pinpoint specific defects in the steroidogenic pathway (Fig. 17.13).
Vitamin D3
Vitamin D3 (cholecalciferol) is derived from cholesterol and plays a key role in calcium metabolism. The actions and metabolism of vitamin D are described in Chapter 26.
Summary
Cholesterol is an essential constituent of cell membranes and the precursor molecule for bile acids, steroid hormones and vitamin D.
Cholesterol is both supplied with the diet and synthesized de novo from the acetyl-CoA.
Cholesterol biosynthesis is precisely regulated. The rate-limiting enzyme in the synthetic pathway is the HMG-CoA reductase.
The transformation of cholesterol into bile acids and steroid hormones involves several hydroxylation reactions catalyzed by cytochrome P450 monooxygenases.
Charlton-Menys, V, Durrington, PN. Human cholesterol metabolism and therapeutic molecules. Exp Physiol. 2007; 93:27–42.
Goldstein, J, DeBose Boyd, RA, Brown, MS. Protein sensors for membrane sterols. Cell. 2006; 124:35–46.
Janowski, BA, Willy, PJ, Devi, TR, et al. An oxysterol signaling pathway mediated by the nuclear receptor LXRa. Nature. 1996; 383:728–731.
Marcil, M, Brooks-Wilson, A, Clee, SM, et al. Mutations in the ABC1 gene in familial HDL deficiency with defective cholesterol efflux. Lancet. 1999; 354:1341–1346.
Maxfield, FR, Van Meer, G. Cholesterol, the central lipid of mammalian cells. Curr Opin Cell Biol. 2010; 22:422–429.
Ory, DS. Nuclear receptor signaling in the control of cholesterol homeostasis. Circ Res. 2004; 95:660–670.
Sakai, J, Rawson, RB. The sterol regulatory element-binding protein pathway: control of lipid homeostasis through regulated intracellular transport. Curr Opin Lipidol. 2001; 12:261–266.
Vegiopoulos, A, Herzig, S. Glucocorticoids, metabolism and metabolic diseases. Mol Cell Endocrinol. 2007; 275:43–61.
Young, SG, Fong, LG. Lowering plasma cholesterol by raising LDL receptors – revisited. N Engl J Med. 2012; 366:1154–1155.
Cholesterol biosynthetic pathway – Rat Genome Database. http://rgd.mcw.edu/rgdweb/pathway/pathwayRecord.html?acc_id=PW:0000454.
Herman, GE, Disorders of cholesterol biosynthesis: prototypic metabolic malformation syndromes. Hum Mol Genet. 2003;12(suppl 1):R75–R8. http://hmg.oxfordjournals.org/content/12/suppl_1/R75.full
KEGG Pathway Database. Primary bile acid biosynthesis – Reference pathway. www.genome.jp/kegg/pathway/map/map00120.html.
King, M. http://themedicalbiochemistrypage.org/steroid-hormones.php