13 INFLAMMATION AND FEVER
INTRODUCTION
Inflammation has been recognised for millennia. The earliest known reference to inflammation can be found in ancient Egyptian writings associating the clinical features with a flame. The Greeks also documented inflammation and used a word that meant ‘to burn’ to describe the heat associated with inflammatory responses, but it was the Roman writer Cornelius Celsus in the first century AD who listed the four cardinal signs of inflammation: redness (rubor), swelling (tumor), heat (calor) and pain (dolor).1 However, this lists only the clinical manifestations of inflammation, not the pathophysiology. Like other aspects of the immune system, it is only in the last 50 years that our understanding of cellular and chemical properties and how these are coordinated to instigate inflammatory responses has been elucidated. In fact, new aspects are still being discovered and so are contributing to complete the whole pathophysiological picture.
Inflammation can arise from many conditions. It is often observed in hospitalised patients — for instance, patients who have undergone surgery have inflammatory responses in the form of tissue repair and wound healing. This is an entirely normal anatomical and physiological response to injury. Alternatively, individuals may be hospitalised due to inflammatory processes, such as those that occur with atherosclerosis in coronary heart disease. Therefore, inflammation can be viewed either as providing restoration for the body — that is, homeostasis — or as a destructive process that significantly contributes to the disease process.
Inflammation is one of the primary responses of the innate immune system when a foreign substance passes the first line of defence. In Chapter 12 we explored innate immunity, and in this chapter we cover in detail aspects important to inflammation. It is essential that you have an understanding of the innate and adaptive immune responses to explain how inflammation fits in.
Inflammation can be divided into several phases: acute, chronic and granulomatosus inflammation. The acute inflammatory response is self-limiting — that is, it continues only until the threat to the host is eliminated. This usually takes 8–10 days from onset to healing. This is the most commonly encountered inflammatory response and you will have observed this many times in your own body. For instance, a superficial graze of the skin results in swelling, redness and warmth around the site. However, if the acute inflammatory response proves inadequate, chronic inflammation may develop and persist for weeks or months. If a continued response is necessary, inflammation may progress to a granulomatosus response that is designed to contain the infection or damaged site so it no longer poses any harm to the individual. Granulomatosus inflammation results in a mass of immune cells, mainly macrophages and other cells that are organised in a fashion to ward off further foreign threats. The characteristics of the early (i.e. acute) inflammatory response differ from those of the later (i.e. chronic) response and each phase involves different biochemical mediators and cells that function together. The acute and chronic phases may lead to healing without progression to the next phase.
The word inflammation is derived from the Latin word inflammare meaning to set on fire. This nicely explains one of the main manifestations of inflammation: heat, which often manifests as fever. Accordingly, we devote the second half of this chapter to thermoregulation and fever, as this is often present in individuals with an inflammatory response and the pathophysiological processes causing fever should be known.
We start by exploring the most common type of inflammatory response: acute inflammation.
ACUTE INFLAMMATION
As part of innate immunity, inflammation does not have the ability to identify antigens. But inflammatory responses are rapid, occurring within seconds to minutes — depending on the location of the tissue injury. Inflammation forms one of the primary defence mechanisms of the immune system in response to tissue injury and infection.2 When cells and tissues are injured, inflammation will result — it occurs when injury to vascularised tissues (having a blood supply) activates mechanisms that coordinate the inflammatory response. The classic symptoms of inflammation include redness, heat, swelling, pain and loss of function. The following characteristic microscopic changes also occur within seconds of the injury (see Figure 13-1):
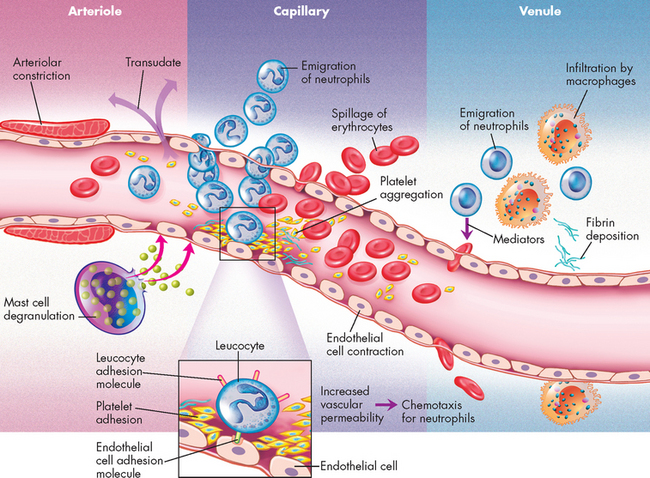
These three primary events are characterised in Figure 13-2.
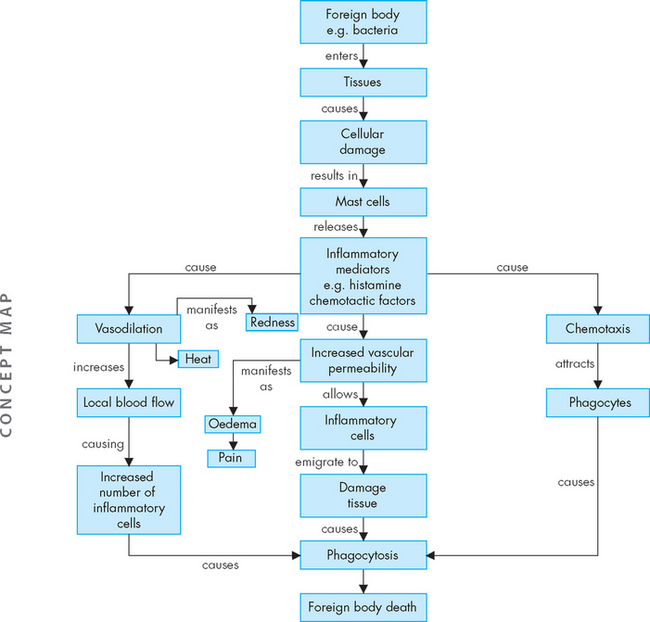
FIGURE 13-2 The inflammatory response.
A foreign substance, commonly bacteria, enters the body and causes tissue damage. This stimulates mast cells to release their intracellular granules (inflammatory mediators), which triggers three main actions: vasodilation, increased vascular permeability (cells of the capillary membrane move apart to allow access from the blood to the tissues) and chemotaxis (chemical attractor of inflammatory cells). Collectively, this allows inflammatory cells to move to the site and stimulates the release of other pathways (complement, kinin and clotting) that limit the effect of the foreign substance, allowing time for the immune response.
Each of the characteristic changes associated with inflammation is the direct result of the activation and interaction of a host of chemicals and cellular components found in the blood and tissues. The vascular changes deliver leucocytes, plasma proteins and other biochemical mediators to the site of injury, where they act in concert (see Figure 13-3). There are several benefits of inflammation:
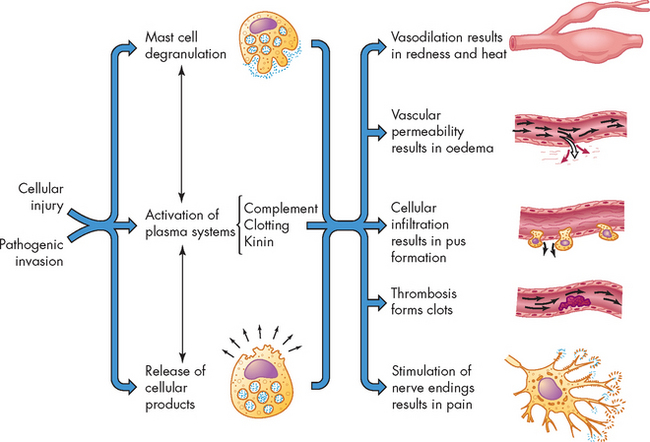
FIGURE 13-3 Acute inflammation.
Mast cell degranulation, the activation of three plasma systems and the release of subcellular components from the damaged cells occur as a consequence of cellular injury. These systems are interdependent, so that induction of one (e.g. mast cell degranulation) can result in the induction of the other two. The result is the development of the characteristic microscopic and clinical hallmarks of inflammation.
We now turn to an exploration of the cells and chemicals that are primarily involved in inflammation. We introduced the main components that affect innate immunity in Chapter 12, but here we provide a more complete picture of inflammatory responses to injury and infection.
CELLULAR COMPONENTS OF INFLAMMATION
Several types of cells participate in the inflammatory response. The primary circulating white blood cells are granulocytes, so-called because of the many enzyme-containing granules in their cytoplasm. These are the neutrophils, eosinophils and basophils. Other blood components include platelets, monocytes (precursors of macrophages that are found in the tissues) and various forms of lymphocytes. Lymphocyte-like natural killer cells are found in both the circulation and tissues. Under normal conditions, these circulating blood cells move unimpeded with the flow of blood. However, many of the biochemical mediators responsible for the initiation of inflammation are produced by mast cells from the activation of plasma protein systems, or are released from dying cells.
Mast cells
The mast cell is probably the most important activator of the inflammatory response (see Figure 13-4).3 Mast cells are filled with granules and are located in the loose connective tissues close to blood vessels near the body’s outer surfaces (i.e. in the skin and lining the gastrointestinal and respiratory tracts). Basophils are found in the blood and probably function in the same way as tissue mast cells (see Figure 13-5). A great number of stimuli activate mast cells to release potent soluble inducers of inflammation. These are released by (1) degranulation and (2) synthesis.
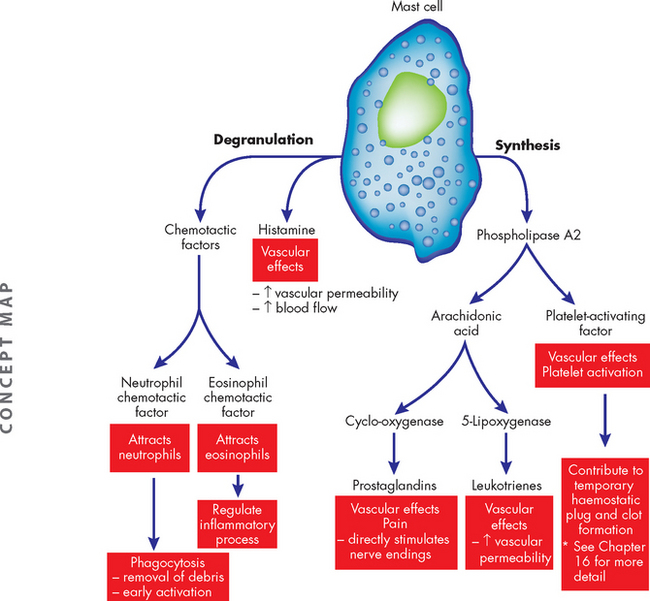
FIGURE 13-4 Mast cells and the degranulation and synthesis of inflammatory mediators upon activation.
Mast cells are filled with darkly staining granules that contain a large number of biologically active substances. Among these are histamine, which is a major initiator of vascular changes, and a variety of chemotactic factors. These substances are released immediately after stimulation of mast cells. Other substances are synthesised in response to mast cell stimulation. These include lipid-based molecules that originate from plasma membrane phospholipids as a result of the action of phospholipase A2, including platelet-activating factor and a variety of prostaglandins and leukotrienes.
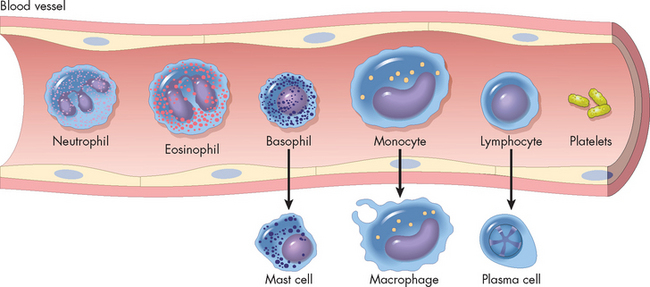
FIGURE 13-5 Inflammatory cells and their differentiation when migrating into the tissues.
Source: Based on Damjanov I. Pathology for the health professions. 3rd edn. Philadelphia: Saunders; 2006.
In response to a stimulus, biologically active molecules are released from the mast cell granules within seconds and exert their effects immediately. This process is called degranulation, literally meaning that the granules inside the cell are released into the extracellular space. These granules contain molecules that are important to propagating the inflammatory response, called mediators. These molecules include histamine, chemotactic factors and heparin.
In addition to the degranulation of inflammatory mediators, mast cells once activated begin new synthesis (production) of other mediators of inflammation, which are released later than those in the granules. These include leukotrienes, prostaglandins and platelet-activating factor, which are produced from lipids in the plasma membrane.
All of these molecules either released or synthesised by the mast cell are crucial to the inflammatory response, and at the site of tissue damage they are responsible for the hallmarks of inflammation. These chemical mediators are discussed later in the chapter.
Neutrophils
Neutrophils are produced by the bone marrow and are the most prevalent white blood cells, accounting for 50–60% of all white blood cells. They are sometimes referred to as polymorphonuclear neutrophils (see Figure 13-5) due to the multilobed nucleus.4 Neutrophils are also the most prominent member of the granulocytes, referring to the granules that are observed when stained. They are also in the group of cells called phagocytes, referring to the process of phagocytosis, which literally means ingesting foreign substances (see later in the chapter for more on phagocytosis). Neutrophils are the predominant phagocytes in the early inflammatory site, arriving 6–12 hours after the initial injury. Several inflammatory mediators specifically and rapidly attract neutrophils from the circulation and activate them.
Neutrophils are short-lived at the inflammatory site as they are mature cells and become a component of the purulent exudate or pus, which is removed from the body through the epithelium or via the lymphatic system (the lymphatic system is described in Chapter 22). The primary roles of neutrophils are the removal of debris and dead cells and phagocytosis of bacteria.
Monocytes and macrophages
Monocytes (the immature form of this white blood cell in the blood) and macrophages (the mature cell in the tissues) have fewer and larger lysosomes (organelles containing digestive enzymes; see Chapter 3) in their cytoplasm than do granulocytes.5 Monocytes are the largest normal blood cells and have a nucleus that is often indented or horseshoe-shaped. Monocytes are produced in the bone marrow, enter the circulation and migrate to the inflammatory site, where they develop into macrophages. Monocytes also appear to be the precursors of macrophages that are fixed in tissues (tissue macrophages), including Kupffer cells in the liver, alveolar macrophages in the lungs and microglia in the brain. Macrophages are generally larger and more active as phagocytes than their monocytic precursors (see Figure 13-5).
Macrophages enter the site after 24 hours or later, and they gradually replace the neutrophils. They migrate to the site after the neutrophils because they move more sluggishly and because many of the chemotactic factors that attract them, such as macrophage chemotactic factor, must first be released by neutrophils. Macrophages are better suited than neutrophils to long-term defence against infectious agents because macrophages can survive and divide in the acidic inflammatory site.
The bactericidal activity (ability to kill bacteria) of macrophages can increase markedly with the help of inflammatory cytokines produced by cells of the acquired immune system (subsets of T lymphocytes; see Chapter 12). (Cytokines are a family of proteins that are secreted and activate other inflammatory cells. They are discussed in detail later in this chapter.) Macrophages have cell surface receptors for these cytokines and are further activated to become more effective killers of infectious microorganisms.
Eosinophils
Although eosinophils (see Figure 13-5) are only mildly phagocytic, they have two specific functions: (1) they serve as the body’s primary defence against parasites; and (2) they help regulate vascular mediators released from mast cells.6
The regulation of mast cell-derived inflammatory mediators is a critical function of eosinophils and helps limit inflammation. Mast cells produce eosinophil chemotactic factor-A, which attracts eosinophils to the site of inflammation. Eosinophil lysosomes contain several enzymes that degrade vasoactive molecules, thereby controlling the vascular effects of inflammation. This role is very important, because if inflammation is not limited to the site of injury or infection, it may damage normal tissue.7
Platelets
Platelets are cytoplasmic fragments formed from megakaryocytes (large cells in the bone marrow responsible for platelet production) (see Figure 13-5). They circulate in the bloodstream until vascular injury occurs. After injury, platelets are activated by many products of tissue destruction and inflammation, including collagen, thrombin and platelet-activating factor. Activation results in: (1) their interaction with components of the coagulation cascade to stop bleeding; and (2) degranulation, releasing biochemical mediators such as serotonin, which has vascular effects similar to those of histamine. (Platelet function is described in detail in Chapter 16.)
Phagocytosis
Phagocytosis is the process by which a cell ingests and disposes of foreign material, including microorganisms. This process resembles endocytosis (refer to Chapter 3). Cells that perform this process are called phagocytes. The two most important phagocytes are neutrophils and macrophages. Both cells are circulating in the blood and must first leave the circulation and migrate to the site of inflammation before initiating phagocytosis. During inflammation the endothelial cells of the capillaries move apart (endothelial cell contraction). In addition, the phagocytes produce the surface molecules that increase the adhesion, or stickiness, between leucocytes and endothelial cells, causing the leucocytes to adhere to the walls of the capillaries and venules in a process called margination, or pavementing.8 The surface adhesion molecules assist the phagocytes to undergo diapedesis, or emigration of the cells through the endothelial junctions that have retracted in response to inflammatory mediators (see Figure 13-6).
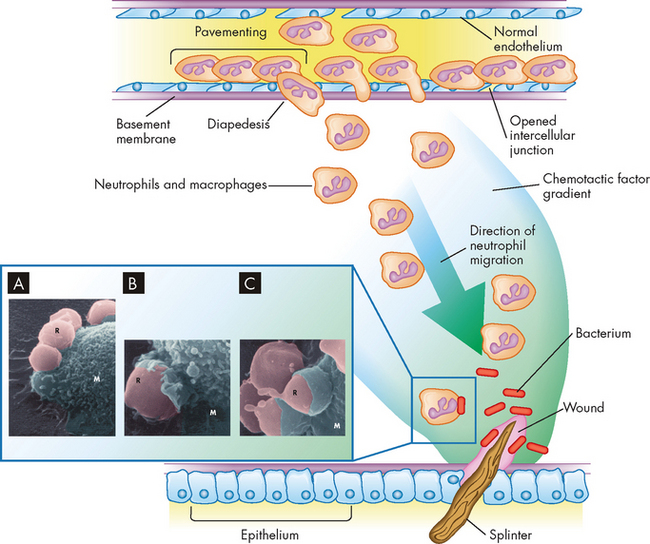
Phagocytosis is a multistep process that involves diffusion of chemotactic factors from a site of injury. Many additional factors affect the blood vessels and increase adhesion molecules on endothelial cells and phagocytes, resulting in adherence of the neutrophils to the vessel wall (pavementing), retraction of endothelial cells (vascular permeability), and movement of the neutrophils through the opened intercellular junctions (diapedesis) and into the tissue. The cells move along the gradient towards the highest concentration of chemotactic factors (chemotaxis). At the site of injury, phagocytes begin phagocytosis of contaminating bacteria. The actual process of phagocytosis involves several steps (see enlargement). The electron micrographs shows phagocytosis of damaged red blood cells by a macrophage: A Red blood cells (R) attach to the macrophage (M). B The plasma membrane of the macrophage begins to enclose the red blood cells. C The red blood cells are almost totally ingested by the macrophage.
Source: Inset Electron micrograph from Thibodeau GA. The human body in health & disease. 4th edn. St Louis: Mosby; 2005.
Once inside the tissue, leucocytes are attracted to the inflammatory site by chemotactic factors. At the inflammatory site, the process of phagocytosis involves five steps: (1) adherence of the phagocyte to its target; (2) engulfment (ingestion or endocytosis); (3) formation of a phagosome (a vacuole or membrane-bound ‘compartment’ that contains the foreign body); (4) fusion of the phagosome with lysosomal granules within the phagocyte; and (5) destruction of the target (see Figure 13-6) (lysosomes are described in Chapter 3). Throughout the process, both the target and the digestive enzymes are isolated within membrane-bound vesicles. Isolation protects the phagocyte itself from the harmful effects of the target microorganisms, as well as its own enzymes.
Phagocytosis using innate receptors (phagocyte surface receptors that recognise bacteria) is a relatively slow process. Opsonisation (the coating of foreign bodies with opsonin to attract phagocytes) greatly enhances adherence by acting as a glue to increase the adherence between the phagocyte and the target cell. The most efficient opsonins are antibodies and C3b produced by the complement system. Antibodies are made against antigens on the surface of bacteria and are highly specific to that particular microorganism (see Chapter 12). Alternatively, the complement system is activated during inflammation and deposits C3b on the bacterial surface, which increases phagocytosis (see complement systems later in this chapter). The surface of phagocytes contains a variety of specific receptors that strongly bind to opsonins.
Engulfment (endocytosis) is carried out by small pseudopods that extend from the plasma membrane and surround the adherent microorganism (see Figure 13-6) forming an intracellular phagosome. After the formation of the phagosome, lysosomes converge, fuse with the phagosome and discharge their contents. Destruction of the bacterium (foreign body) takes place inside this compartment.9
Often the process of phagocytosis results in the death of the phagocyte. When a phagocyte dies at an inflammatory site, it frequently lyses (breaks open) and releases its cytoplasmic contents, including the lysosomal enzymes, into the tissue. These can be very destructive and digest the surrounding tissue, causing much of the tissue destruction associated with inflammation. The destructive effects of many enzymes released by dying phagocytes are minimised by natural inhibitors found in the blood, such as α1-antitrypsin, a plasma protein produced by the liver. Released lysosomal products also may contribute to inflammation by increasing vascular permeability, attracting additional monocytes, and activating the complement and kinin systems (see the section on plasma proteins later in the chapter).
INFLAMMATORY MEDIATORS
Inflammatory mediators are substances that promote inflammation. Literally a flood of these inflammatory substances is released by cells at the site of injury or infection. The mast cell releases several substances when stimulated. Alternatively, some inflammatory substances are synthesised by the cell and act later in the inflammatory period. In addition, some inflammatory mediators circulate in the plasma in an inactive form and are activated during inflammation. Furthermore, for inflammation to occur, many different kinds of cells must cooperate. That cooperation is achieved by the secretion of a variety of proteins that work locally — these are cytokines. Therefore, these chemical substances are crucial to inflammation and immunity, and we explore the main mediators to provide an overview of their functions and how they interact during inflammation.
Histamine
Histamine is a small weight molecule with potent effects on many other cells, particularly those that control the circulation. Histamine is a vasoactive amine, meaning that its action is on blood vessels. Histamine causes temporary, rapid constriction of smooth muscle and dilation of the post-capillary venules, which results in increased blood flow into the microcirculation. Histamine also causes increased vascular permeability resulting from retraction of endothelial cells lining the capillaries (see Figures 13-1 and 13-7) and increased adherence of leucocytes to the endothelium. Antihistamines are drugs that block the binding of histamine to its receptors, resulting in decreased inflammation.
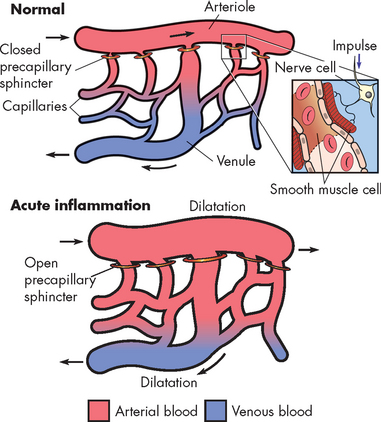
FIGURE 13-7 Vasodilation of the capillary beds during inflammation induced by histamine and nitric oxide.
Relaxation of the pre-capillary sphincter in the arterioles results in flooding of the capillary network and dilatation of the capillaries and post-capillary venules.
Source: Damjanov I. Pathology for the health professions. 3rd edn. Philadelphia: Saunders; 2006.
Chemotactic factors
Chemotactic factors are released from mast cell granules. Chemotactic factors diffuse from a site of inflammation forming a gradient and causing the directional movement (chemotaxis) of cells towards the inflammation (see Figure 13-8). Mast cells contain two specific chemotactic factors: neutrophil chemotactic factor and eosinophil chemotactic factor of anaphylaxis. These factors provide a strong attraction for neutrophils and eosinophils, which are vital to inflammation. Neutrophils are the predominant cell needed to kill bacteria in the early stages of inflammation, and eosinophils help regulate the inflammatory response.
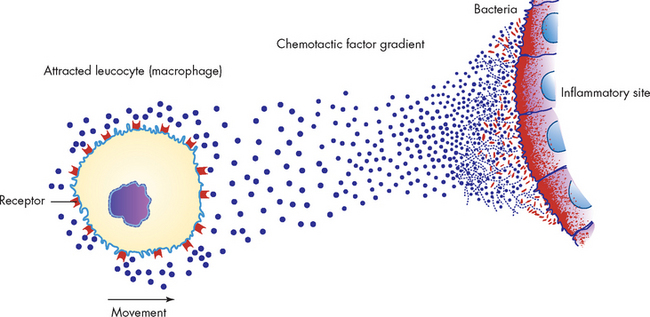
This occurs when chemotactic factors are released, which attracts inflammatory cells to site of inflammation. Multiple receptors on the leucocyte’s plasma membrane sense the area of highest concentration of chemotactic factor (dots), and the leucocyte (usually a phagocyte) moves towards this area.
Source: McCance KL, Huether SE. Pathophysiology: the biologic basis for disease in adults and children. 5th edn. St Louis: Mosby; 2006.
Leukotrienes
Leukotrienes are sulfur-containing lipids that produce histamine-like effects: smooth muscle contraction and increased vascular permeability. Leukotrienes appear to be important in the later stages of the inflammatory response because they stimulate slower and more prolonged responses than do histamines.
Nitric oxide
Endothelial cells release nitric oxide (not to be confused with nitrous oxide, or laughing gas), which has at least two effects on inflammation. First, nitric oxide causes vasodilation by inducing relaxation of vascular smooth muscle, a response that is local and short-lived (see Figure 13-7). Second, nitric oxide may suppress mast cell release of inflammatory molecules and decrease platelet adhesion and aggregation. Therefore, nitric oxide has both pro- and anti-inflammatory aspects.
Prostaglandins
Prostaglandins cause increased vascular permeability, neutrophil chemotaxis and pain by direct effects on nerves. They are long-chain, unsaturated fatty acids and are produced when the enzyme cyclo-oxygenase converts arachidonic acid to prostaglandin. Two particular types of prostaglandins, E1 and E2, cause increased vascular permeability and smooth muscle contraction. The enzyme cyclo-oxygenase (commonly abbreviated to COX) and arachidonic acid are important to remember because aspirin and other non-steroidal anti-inflammatory drugs (NSAIDs), such as ibuprofen, block the synthesis of prostaglandins by inhibiting cyclo-oxygenase, thereby inhibiting inflammation.
Platelet-activating factor
Platelet-activating factor is produced from a lipid of the phospholipid plasma membrane. Although mast cells are a major source of platelet-activating factor, this molecule can also be produced during inflammation by neutrophils, monocytes, endothelial cells and platelets. The biological activity of platelet-activating factor is virtually identical to that of leukotrienes, namely causing endothelial cell retraction to increase vascular permeability, leucocyte adhesion to endothelial cells and platelet activation.
Cytokines
Cytokines are a large group of proteins that provide a means of communication for the inflammatory and immune cells. Cytokines can be pro-inflammatory or anti-inflammatory, depending on whether they tend to induce or inhibit the inflammatory response. These molecules diffuse over short distances, bind to the appropriate target cells and affect the function of the target cells. Some effects occur over long distances, such as the systemic induction of fever by some cytokines (i.e. endogenous pyrogens) away from where they are produced at an inflammatory site (see the section on fever later in the chapter). Cytokine effects on other cells are mediated through specific cell-surface receptors. The binding of cytokines to a target cell often induces synthesis of additional cellular products. These are detailed in Table 13-1. Below we list some of the cytokines that strongly influence inflammation.
Table 13-1 THE ACTIONS OF THE PRIMARY INFLAMMATORY MEDIATORS
| MEDIATOR | PRINCIPAL SOURCES | ACTION |
|---|---|---|
| CELL-DERIVED | ||
| Histamine | Mast cells, basophils, platelets | Vasodilation, increased vascular permeability, endothelial activation |
| Serotonin | Platelets | Vasodilation, increased vascular permeability |
| Prostaglandins | Mast cells, leucocytes | Vasodilation, pain, fever |
| Leukotrienes | Mast cells, leucocytes | Increased vascular permeability, chemotaxis, leucocyte adhesion and activation |
| Platelet-activating factor | Leucocytes, mast cells | Vasodilation, increased vascular permeability, leucocyte adhesion, chemotaxis, degranulation |
| Nitric oxide | Endothelium, macrophages | Vascular smooth muscle relaxation, killing of microbes |
| Cytokines (TNF-α, IL-1, IL-6) | Macrophages, endothelial cells, mast cells | Local endothelial activation, fever, pain, hypotension, decreased vascular resistance (shock) |
| Chemokines | Leucocytes, activated macrophages | Chemotaxis, recruitment of leucocytes to sites of inflammation, migration of cells to normal tissues |
| PLASMA PROTEIN–DERIVED | ||
| Complement products (C5a, C3a, C4a) | Plasma (produced in liver) | Leucocyte chemotaxis and activation, vasodilation (mast cell stimulation) |
| Kinins | Plasma (produced in liver) | Increased vascular permeability, smooth muscle contraction, vasodilation, pain |
| Proteases activated during coagulation | Plasma (produced in liver) | Endothelial activation, leucocyte recruitment |
| IL-1 = interleukin-1; IL-6 = interleukin-6; TNF-α = tumour necrosis factor-alpha. | ||
Source: Based on Kumar V. Robbins & Cotran pathologic basis of disease. 8th edn. Philadelphia: Saunders; 2009.
Interleukins
The interleukins are produced predominantly by macrophages and lymphocytes in response to their recognition of a pathogen or stimulation by other products of inflammation. Interleukin-1 (IL-1) is a pro-inflammatory cytokine (which means that it encourages the inflammatory process) produced mainly by macrophages. It is endogenous, meaning that it is produced from within the body. This endogenous pyrogen (i.e. fever-causing cytokine) reacts with receptors on cells of the hypothalamus and affects the body’s thermostat, resulting in fever. It also activates phagocytes and lymphocytes, thereby enhancing both innate and adaptive immunity, and acts as a growth factor for many cells. It has several effects on neutrophils, including induction of proliferation (resulting in an increase in the number of circulating neutrophils), chemotaxis, increased cellular respiration and increased lysosomal enzyme activity.
Interleukin-6 (IL-6) is produced by macrophages, lymphocytes, fibroblasts and other cells. It directly induces hepatocytes (liver cells) to produce many of the proteins needed in inflammation (acute-phase reactants, discussed later in this chapter). IL-6 also stimulates growth and differentiation of precursors of blood cells in the bone marrow and the growth of fibroblasts.
Interleukin-10 (IL-10) is an example of an anti-inflammatory cytokine and is primarily produced by lymphocytes to slow both the inflammatory and the acquired immune responses. It achieves this through suppressing the growth of lymphocytes and the production of pro-inflammatory cytokines by macrophages.
More than 30 interleukins have been identified. Their varied effects include the following:
Tumour necrosis factor-alpha
Macrophages secrete tumour necrosis factor-alpha (TNF-α) in response to recognition of foreign materials. Other cells, such as mast cells, are additional and crucial sources of this pro-inflammatory cytokine. TNF-α induces a multitude of pro-inflammatory effects, including enhancement of endothelial cell adhesion molecule expression, which results in increased adherence of neutrophils. When secreted in large amounts, TNF-α has the following systemic effects:
 causing muscle wasting (cachexia) and intravascular thrombosis in cases of severe infection and cancer.
causing muscle wasting (cachexia) and intravascular thrombosis in cases of severe infection and cancer.Very high levels of TNF-α can be lethal and may be responsible for fatalities from shock caused by gram-negative bacterial infections. (The distinction between gram-positive and gram-negative is discussed in Chapter 14.)
PLASMA PROTEIN SYSTEMS
Three key plasma protein systems are essential to an effective inflammatory response: the complement system, the coagulation system and the kinin system (see Figure 13-9). Although each system has a unique role in inflammation, the systems have many similarities. Each system consists of multiple proteins in the blood. To prevent activation in unnecessary situations, each protein is normally in an inactive form. Each system contains a few proteins that can be activated by products of tissue damage or infection. Activation of the first component results in sequential activation of other components of the system, leading to a biological function that helps protect the individual. This sequential activation is referred to as a cascade. Thus, we refer to the complement cascade, the coagulation cascade or the kinin cascade.
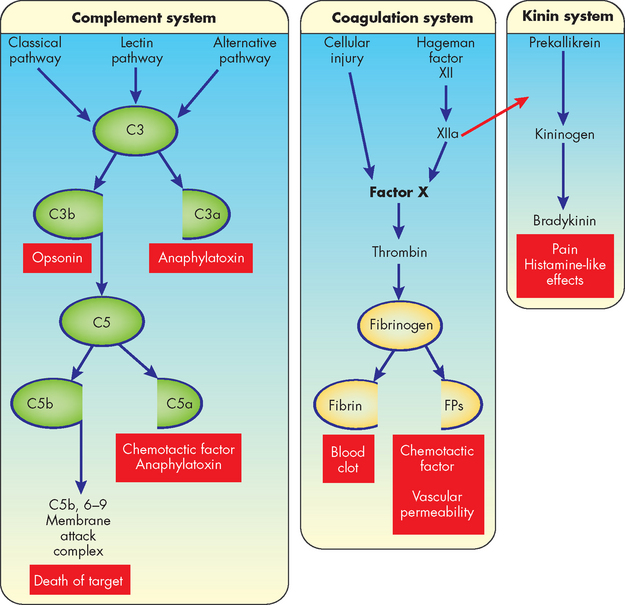
FIGURE 13-9 Plasma protein systems in inflammation: complement, coagulation and kinin systems.
See the text for more detailed information. In this scheme some systems are activated by multiple pathways that come together (at C3 for the complement system and factor X for the coagulation system). Some of the components of the pathways are activated by being split into two active components (C3 and C5 of the complement system and fibrinogen of the coagulation system). The larger of the components usually activates the next component of the pathway (as do C3b and C5b of the complement system). Hageman factor participates in both the coagulation and kinin pathways (i.e. activated factor XII [XIIa] helps activate factor X and prekallikrein). Many of the components of each pathway have potent biological activities (in red-coloured boxes). Many other components of each pathway are not shown in this drawing but play very important roles in activation of the pathway. FP = fibrinopeptides.
The complement system
The complement system consists of a large number of proteins (sometimes called complement components) that together constitute about 10% of the total circulating serum protein.10 The complement system is extremely important because activated components can destroy pathogens directly and can activate or collaborate with virtually every other component of the inflammatory response. For these reasons, proteins of the complement system are among the body’s most potent defenders against bacterial infection.
The most important portion of the complement cascade is activation of C3 and C5, which results in a variety of subunits that are opsonins, chemotactic factors or anaphylatoxins. Opsonins are molecules that coat bacteria and increase their susceptibility to being eaten and killed by inflammatory cells, such as neutrophils and macrophages. Anaphylatoxins are molecules that induce rapid degranulation of mast cells, thus increasing inflammation. The most potent complement products are C3b (opsonin), C3a (anaphylatoxin) and C5a (anaphylatoxin, chemotactic factor).
Complement components C5b to C9 (membrane attack complex, or MAC) form a complex that creates pores in the outer membranes of cells or bacteria. The pores disrupt the cell’s membrane and permit water to enter, causing lysis and death of the cell. This is a particularly effective mechanism for destroying foreign cells that have entered the body.
Complement activation can be accomplished in three different ways (see Figure 13-9):
The classical pathway is primarily activated by proteins of the acquired immune system, antibodies.11 When antibodies bind to antigens they activate the first component of complement, C1, which through other complement components leads to activation of C3 and C5. Thus, the acquired immune response can use the complement system to kill bacteria and activate inflammation.
The lectin pathway is similar to the classic pathway but is independent of antibody. Thus, infectious agents that do not activate the alternative pathway may be susceptible to complement through the lectin pathway.
The alternative pathway is activated by several substances found on the surface of infectious organisms. This pathway uses unique proteins to form a complex that activates C3, which leads to C5 activation and convergence with the classical pathway. Thus, the complement system can be directly activated by certain infectious organisms without antibody being present.
In summary, the complement cascades can be activated by at least three different means and its products have four functions: (1) opsonisation, (2) anaphylatoxic activity resulting in mast cell degranulation, (3) leucocyte chemotaxis and (4) cell lysis.
The coagulation system
The coagulation (clotting) system is a group of plasma proteins that, when activated sequentially, form a fibrinous meshwork at an injured or inflamed site.12 This: (1) forms a clot that stops bleeding; (2) traps infectious organisms and prevents their spread to adjacent tissues; (3) keeps microorganisms and foreign substances at the site of greatest inflammatory cell activity; and (4) provides a framework for future repair and healing. The main substance in this fibrinous mesh is fibrin, an insoluble protein produced by the coagulation cascade.
Like the complement cascade, the coagulation cascade can be activated through different pathways that converge and result in the formation of a clot (see Figure 13-9). The coagulation cascade converges at factor X. From that point on, a common pathway results in fibrin formation. The coagulation cascade is discussed further in Chapter 16.
Many substances that are released during tissue destruction and infection, such as bacterial products (e.g. endotoxin), collagen and cellular proteases, can activate the coagulation system. Activation of some clotting factors produces fragments that enhance the inflammatory response.
The kinin system
The third plasma protein system, the kinin system, interacts closely with the coagulation system (see Figure 13-9). Both the coagulation and kinin systems are activated through activated factor XII (factor XIIa). Another name for factor XIIa is prekallikrein activator because it enzymatically activates the first component of the kinin system, prekallikrein. The final product of the kinin system is a small-molecular-weight molecule, bradykinin. At low doses, bradykinin causes dilation of blood vessels, acts with prostaglandins to induce pain, causes smooth muscle cell contraction and increases vascular permeability (see Figure 13-3). Bradykinin induces smooth muscle contraction more slowly than histamine and may be more important during the later phases of inflammation.
Interaction of the plasma protein systems
The three plasma protein systems are highly interactive so that activation of one results in secondary activation of the other two. It is beneficial to the individual to activate all three systems, but it would be detrimental if the systems continued producing potent pro-inflammatory molecules indefinitely. Therefore, the systems are tightly regulated to control and localise inflammation to the appropriate sites.
CHRONIC INFLAMMATION
Superficially, the difference between acute and chronic inflammation is duration; chronic inflammation lasts 2 weeks or longer, regardless of cause. Chronic inflammation is sometimes preceded by an unsuccessful acute inflammatory response (see Figure 13-10). For example, if bacterial contamination or foreign objects (e.g. dirt, wood splinters, glass) persist in a wound, an acute response may be prolonged beyond 2 weeks. Pus formation, purulent discharge and incomplete wound healing may characterise this type of chronic inflammation.
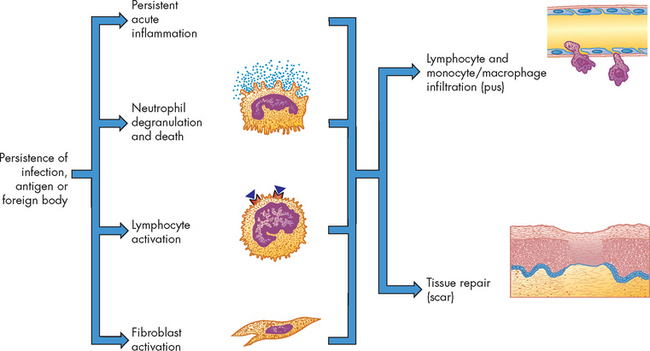
FIGURE 13-10 Chronic inflammation.
Inflammation usually becomes chronic because of the persistence of an infection, an antigen or a foreign body in the wound. Chronic inflammation is characterised by the persistence of many of the processes of acute inflammation. In addition, large amounts of neutrophil degranulation and death, the activation of lymphocytes and the concurrent activation of fibroblasts result in the release of mediators that induce the infiltration of more lymphocytes and monocytes/macrophages and the beginning of wound healing and tissue repair.
Chronic inflammation can occur also as a distinct process without previous acute inflammation. Some microorganisms, chemicals or physical irritants (e.g. inhaled dusts, wood splinters and suture material) can cause a prolonged inflammatory response (see Figure 13-11).
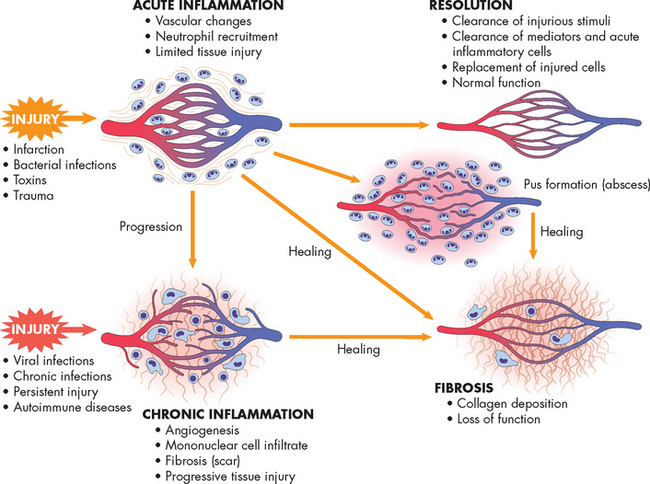
FIGURE 13-11 The differences between acute and chronic inflammation.
Acute and chronic inflammation can lead to fibrosis and loss of tissue function.
Source: Kumar V et al. Robbins & Cotran pathologic basis of disease. 7th edn. Philadelphia: Saunders; 2005.
Chronic inflammation is characterised by a dense infiltration of lymphocytes and macrophages. If macrophages are unable to protect the host from tissue damage, the body attempts to wall off and isolate the infected area, thus forming a granuloma. The process of granuloma formation begins when some macrophages differentiate into large epithelial-like cells, which are incapable of phagocytosing large bacteria but are capable of taking up debris and other small particles. Other macrophages fuse into multinucleated giant cells, which are active phagocytes that can engulf very large particles — larger than can be engulfed by a single macrophage. These two types of differentiated macrophages form the centre of the granuloma, which is surrounded by a wall of lymphocytes. The granuloma itself is often encapsulated by fibrous deposits of collagen and may become cartilaginous, or possibly calcified, which results in inflexible, somewhat hardened tissue that replaces the normal tissue.
CLINICAL MANIFESTATIONS OF INFLAMMATION
As you will now be aware, inflammation produces a distinct clinical picture. Individuals with inflammation often experience local and systemic signs and symptoms. This is because the inflammatory cells, plasma protein systems and especially the chemical mediators interact to produce all the characteristics of inflammation. Local inflammation accompanies all types of cellular and tissue injury, whether infected or sterile, and is responsible for initiating healing.
All the local characteristics of acute inflammation (i.e. swelling, pain, heat and redness) result from vascular changes and the subsequent leakage of circulating components into the tissue. Heat and redness are the result of vasodilation and increased blood flow through the injured site. Swelling (oedema) occurs as exudate (fluid and cells) accumulates in the tissues. Swelling is usually accompanied by pain caused by pressure exerted by exudate accumulation, as well as the presence of soluble biochemical mediators such as prostaglandins and bradykinin.
Exudate varies in composition, depending on the stage of the inflammatory response. In early or mild inflammation, the exudate is watery (called serous exudate) with very few plasma proteins or leucocytes. An example of serous exudate is the fluid in a blister. In more severe or advanced inflammation, the exudate may be thick and clotted (called fibrinous exudate), such as in the lungs of individuals with pneumonia. If a large number of leucocytes accumulate, as in persistent bacterial infections, the exudate consists of pus and is called a purulent exudate. Purulent exudate is characteristic of walled-off lesions (cysts or abscesses).
There are three primary systemic changes associated with the acute inflammatory response: fever, leucocytosis and an increased level of circulating plasma proteins. Fever is one of the most common clinical manifestations of inflammation. It is an important clinical sign and you should have a thorough understanding of the pathogenesis and patterns of fever, as many individuals in healthcare facilities will experience fever. We discuss fever in detail in the following section.
Leucocytosis is an increase in the number of circulating white blood cells. Often the leucocytosis causes production of an immature form of neutrophils such that they are present in relatively greater than normal proportions; however, because they are immature, they may not function as normal neutrophils.
The synthesis of many plasma proteins, mostly products of the liver, is increased during inflammation. These proteins, which can be either pro-inflammatory or anti-inflammatory in nature, are referred to as acute-phase reactants. Acute-phase reactants reach maximal circulating levels 10–40 hours after the start of inflammation. IL-1 indirectly induces the synthesis of acute-phase reactants by increasing production of IL-6, which directly stimulates synthesis of acute-phase reactants by liver cells. Common laboratory tests for inflammation measure levels of acute-phase reactants. For example, an increase in blood levels of acute-phase reactants such as C-reactive protein and fibrinogen is considered a good indicator of an acute inflammatory response.
Fat is ‘inflammatory’
Obesity has increased dramatically in recent years and is a significant health problem in Western countries like Australia and New Zealand. The many health-related problems linked to obesity appear to be secondary to a chronic systemic low-grade inflammatory response mediated by the adipose tissue itself. Pro-inflammatory molecules produced by adipose tissue include tumour necrosis factor-alpha (TNF-α) and interleukin 6 (IL-6), and several other cytokines. These lead to increases in circulating C-reactive protein (a protein produced in the liver) and other inflammatory mediators that place the individual at increased risk for cardiovascular disease and other complications.
Source: Fantuzzi G. Adipose tissue, adipokines, and inflammation. J Allergy Clin Immunol 2005; 115:911–909; Schaffler A et al. Role of adipose tissue as an inflammatory organ in human diseases. Endocr Rev 2006; 27(5):449–467.
FEVER
Fever is a fundamental sign of almost all infectious diseases and many non-infectious conditions that arise as a consequence of inflammation. However, to appreciate the mechanisms of fever you need to understand normal thermoregulation to provide a distinction. Therefore, we first explore temperature deviations so that they can be distinguished from fever.
Temperature regulation
We regulate body temperature within a relatively narrow range using both autonomic and behavioural mechanisms. Behavioural responses are very powerful — for instance, when we are cold, we can apply more clothing or move towards a heat source. This increases skin temperature, which in turn increases core temperature. In contrast, autonomic responses (those we do not have conscious control over) are mediated through our nervous system and act to maintain homeostasis.
In all homeothermic animals (animals with the ability to maintain a stable core temperature independent of environmental temperature), temperature regulation is achieved through precise balancing of heat production and heat loss. Maintenance of body temperature within the normal range is necessary for life. In humans, core temperature is maintained in a range around 37°C — the normal range being about 36.2 to 37.5°C — and rarely exceeds 41°C. However, this is dependent on many factors — for instance, stages in the menstrual cycle and time of day both influence core temperature. In addition, an individual’s body parts will vary in temperature, which means that finding a representative core temperature is difficult. Skin temperature is strongly associated with air temperature and varies according to anatomical location: the extremities are generally cooler than the trunk and the temperature at the core of the body (as measured by the rectal temperature) is generally 0.5°C higher than at the surface (as measured by oral temperature). The daily fluctuating temperature in both sexes peaks at around 6 pm and is at its lowest during sleep.
Hypothalamic control of temperature
Temperature regulation is mediated primarily by the hypothalamus, specifically the pre-optic region of the anterior hypothalamus. Peripheral thermoreceptors in the skin and central thermoreceptors in the hypothalamus, spinal cord, abdominal organs and other central locations provide the hypothalamus with information about the body temperatures. Simply, if these temperatures are low, the hypothalamus triggers heat-production mechanisms, such as shivering. The hypothalamus also stimulates the sympathetic nervous system, which stimulates the adrenal cortex, increasing skeletal muscle tone, initiating the muscle shivering response and producing vasoconstriction. The hypothalamus relays information to the cerebral cortex about cold and voluntary responses result, such as increased body movement. Alternatively, if body temperatures are high, the hypothalamus will initiate heat-loss mechanisms. These include vasodilation, an increase in skin blood flow to dissipate heat to the environment, and sweating. In this way, core temperature is defended and homeostasis is maintained.
Mechanisms of heat production and loss
Body heat is produced by the chemical reactions of metabolism, skeletal muscle tone and contraction, and chemical thermogenesis (heat producing). Heat is distributed by the circulatory system. Heat loss is achieved through: (1) radiation; (2) conduction; (3) convection; (4) vasodilation; (5) decreased muscle tone; (6) evaporation; (7) increased respiration; (8) voluntary measures; and (9) adaptation to warmer climates. These mechanisms are summarised in Table 13-2.
Table 13-2 MECHANISMS OF HEAT PRODUCTION AND LOSS
| CONDITION | DESCRIPTION |
|---|---|
| Heat production | |
| Chemical reactions of metabolism | Occur during ingestion and metabolism of food and while maintaining the body at rest (basal metabolism); occur in body core (liver). |
| Skeletal muscle contraction | Gradual increase in muscle tone or rapid muscle oscillations (shivering). |
| Chemical thermogenesis (heat production) | Adrenaline is released and produces a rapid, transient increase in heat production by raising basal metabolic rate; quick, brief effect that counters heat lost through conduction and convection; involves brown adipose tissue, which decreases markedly in older adults. Thyroid hormone increases metabolism. |
| Heat loss | |
| Radiation | Heat loss through electromagnetic waves emanating from surfaces with temperature higher than the surrounding air. |
| Conduction | Heat loss by direct molecule-to-molecule transfer from one surface to another, so that warmer surface loses heat to cooler surface. |
| Convection | Transfer of heat through currents of gases or liquids; exchanges warmer air at body’s surface with cooler air in surrounding space. |
| Vasodilation | Diverts core-warmed blood to surface of body, with heat transferred by conduction to skin surface and from there to the surrounding environment; occurs in response to autonomic stimulation under control of hypothalamus. |
| Decreased muscle tone | Washed-out feeling caused by moderately reduced muscle tone and curtailed voluntary muscle activity. |
| Evaporation | Body water evaporates from surface of skin and linings of mucous membranes; major source of heat reduction connected with increased sweating in warmer surroundings. |
| Increased ventilation | Air is exchanged with environment through normal process; minimal effect. |
| Voluntary mechanisms | ‘Stretching out’ and ‘slowing down’ in response to high body temperatures, increasing the body surface area available for heat loss; dressing in light-coloured, loose-fitting garments. |
| Adaptation to warmer climates | Gradual process beginning with lassitude, weakness and faintness, proceeding through increased sweating, lowered sodium content, decreased heart rate, increased stroke volume and extracellular fluid volume, and terminating in improved warm weather functioning and decreased symptoms of heat intolerance (work output, endurance and coordination increase, and subjective feelings of discomfort decrease). |
Disorders of temperature regulation
Hyperthermia
Hyperthermia (an increase in core temperature above normal) can produce nerve damage, coagulation of cell proteins and death (see Table 13-3). In core temperatures above 41°C, nerve damage can produce convulsions in the adult, and death is imminent in core temperatures above 43°C. Hyperthermia is not mediated by pyrogens.
Table 13-3 CLINICALLY SIGNIFICANT CORE AND SKIN TEMPERATURES
| CORE TEMPERATURES | |
|---|---|
| > 45°C | Possible death without treatment |
| > 40.5°C | Profound clinical hyperthermia (heatstroke) |
| > 39.5°C | Profound hyperthermia |
| 38.5–39.5°C | Moderate hyperthermia (heat exhaustion) |
| 37.2–38.5°C | Mild hyperthermia |
| 36.5–37°C | Normothermia |
| SKIN TEMPERATURES | |
|---|---|
| > 50°C | Second-degree burn |
| > 45°C | Tissue damage |
| 41–43°C | Burning pain |
| 39–41°C | Pain |
| 33–39°C | Skin warmth through to discomfort (hot) |
| 28–33°C | Thermal comfort |
Source: Taylor NAS, Groeller H (eds). Physiological bases of human performance during work and exercise. Sydney: Elsevier; 2008.
Hyperthermia may be therapeutic, accidental or associated with stroke or head trauma. Therapeutic hyperthermia, a controversial therapy, is a form of local or general body-induced hyperthermia used to destroy pathological microorganisms or tumour cells by facilitating the host’s natural immune process or tumour blood flow.13
The four forms of accidental hyperthermia are summarised as follows:14
Hypothermia
Hypothermia (a decrease in core temperature below the normal range) produces depression of the central nervous and respiratory systems, vasoconstriction, alterations in microcirculation, coagulation and ischaemic tissue damage. Most tissues can tolerate low temperatures in controlled situations, such as surgery. However, in severe hypothermia, ice crystals form on the inside of the cells, causing the cells to rupture and die. Tissue hypothermia slows cell metabolism, increases blood viscosity, slows microcirculatory blood flow, facilitates blood coagulation and stimulates profound vasoconstriction.
The pathogenesis of fever
Fever (also called febrile response) is a temporary ‘resetting of the hypothalamic thermostat’ to a higher level. Usually, thermoregulatory mechanisms adjust heat production and heat loss to maintain body core temperature within a narrow range. During fever, this is re-set to a higher level, so that the thermoregulatory centre in the hypothalamus adjusts heat production and loss to maintain the core temperature at the new, higher temperature.16 Fever is usually characterised by an increase in core temperature of 1–4°C. However, the primary difference between fever and hyperthermia is that in fever the increase in temperature is a response to an endogenous or exogenous pyrogen. A pyrogen is a substance that causes fever. There are two broad classifications of pyrogens: exogenous pyrogens arise from outside of the body; and endogenous pyrogens arise from inside the body.
Exogenous pyrogens, or endotoxins produced by pathogens, stimulate inflammatory cells, mainly macrophages and neutrophils, to release substances such as TNF-α, IL-1, IL-6 and interferon. These pyrogenic cytokines induce the synthesis of prostaglandins in the hypothalamus and this increases the level of hypothalamic temperature. In reality, the hypothalamus does not regulate body temperature from a ‘set-point’ temperature, but rather a range of temperatures called the interthreshold zone.17 This is elevated in fever. In response, the hypothalamus signals an increase in heat production and decreases heat-loss avenues to raise body temperature (see Figure 13-12).18 The individual feels colder, dresses more warmly, decreases body surface area by curling up and may go to bed in an effort to get warm. Body temperature is maintained at the new higher level until the fever ‘breaks’, when the interthreshold zone begins to return to normal. This response is mediated in part by cytokines associated with the inflammatory response.19 There are decreased heat-production and increased heat-loss mechanisms. The individual feels very warm, dons cooler clothes, throws off the covers and stretches out. Once the body has returned to a normal temperature the individual feels more comfortable and the hypothalamus adjusts thermoregulatory mechanisms to maintain the new temperature. Therefore, fever is mediated from exogenous and endogenous pyrogens and elevates the core temperature as a defence mechanism.
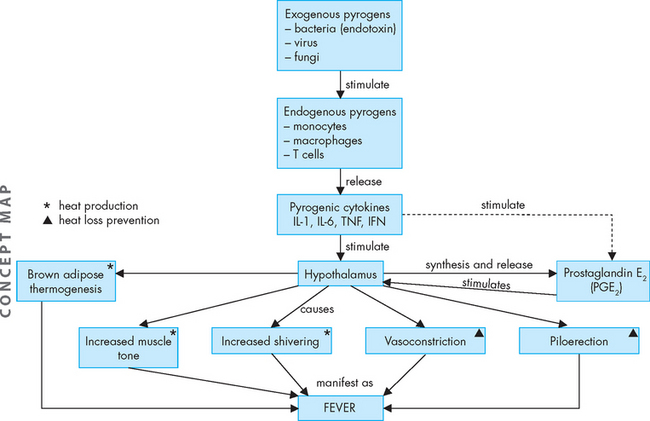
FIGURE 13-12 The pathogenesis of fever.
Exogenous pyrogens stimulate cells that release cytokines that stimulate fever (exogenous pyrogens). These pyrogenic cytokines stimulate the hypothalamus (pre-optic of anterior hypothalamus) to synthesise and release prostaglandin E2, which acts on the cells locally in the hypothalamus to increase heat production area and decrease heat-loss avenues. This causes an increase in core temperature, which is manifest as fever.
The benefits of fever
Fever is usually beneficial and assists the immune system to respond to pathogenic microorganisms.
It does this through several mechanisms:20,21
Conversely, fever can also be harmful to the individual as it may augment the body’s susceptibility to the effects of endotoxins associated with gram-negative bacterial infections (bacterial toxins are described in Chapter 14).
Suppression of fever by pharmacological treatment with antipyretic medications, such as aspirin or paracetamol, occurs via inhibition of the cyclo-oxygenase enzyme, which is required for the production of prostaglandin. Therefore, prostaglandin does not influence the hypothalamus and an increase in heat production does not occur. However, antipyretics should be used only if a fever (> 38.5°C) produces or is high enough to produce serious side effects, such as cardiovascular stress, nerve damage or convulsion.22
Clinical patterns of fever
Although fever is accompanied by an increase in core temperature, the patterns can be varied. Four categorised patterns are seen in clinical practice (see Figure 13-13).
During sustained fever, the core temperature remains elevated, often for days, and there is very little variation. Remittent fever occurs when there are minor fluctuations in body temperature but the fever is sustained. Relapsing fever is associated with days of normal temperature following fever, with an increase in core temperature, often associated with increasing severity of illness.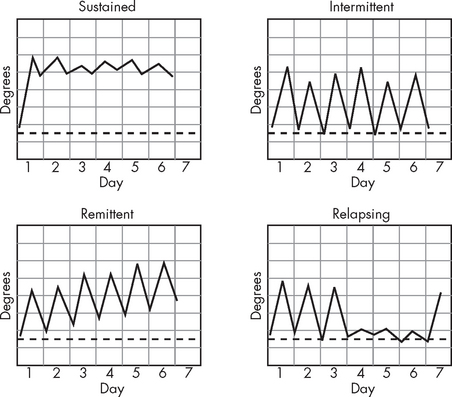
FIGURE 13-13 Different patterns of fever.
Source: McGee S. Evidence-based physical diagnosis. 2nd edn. St Louis: Saunders; 2007.
Unfortunately, despite differing patterns, these fever patterns are not usually associated with any particular condition or disease. They represent fever mediated through pyrogens from both infectious and non-infectious agents.
WOUND HEALING
Tissue damage is followed by a period of healing that begins during acute inflammation and may last for as long as 2 years (see Figure 13-14).23 The most favourable outcome is a return to normal structure and function. The repaired tissues may be close to normal if damage is minor, no complications occur and destroyed tissues are capable of regeneration. However, regeneration may not be possible if extensive damage is present, the tissue is not capable of regeneration, infection results in abscess or granuloma formation, or fibrin persists in the lesion. In those cases, destroyed tissue is replaced with scar tissue. Scar tissue is composed primarily of collagen that fills in the lesion. The tensile strength of scar tissue is not the same as the initial tissue and it cannot carry out the physiological functions of the destroyed tissue.
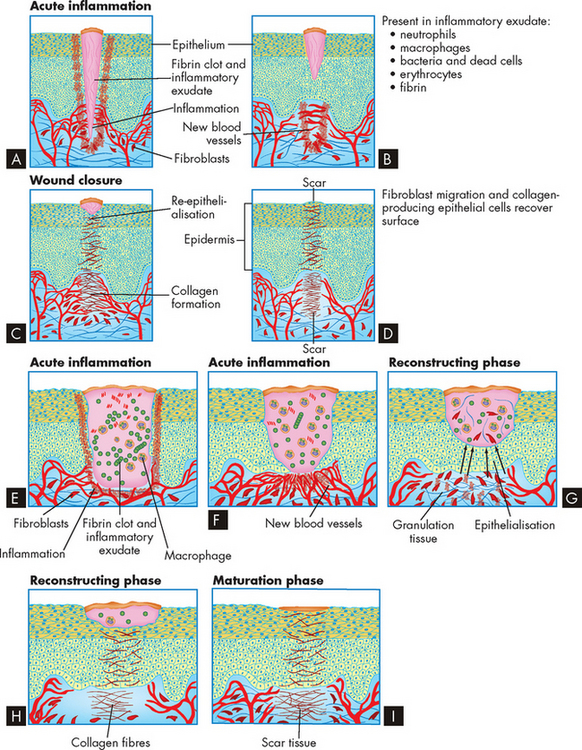
FIGURE 13-14 Wound healing by primary or secondary intention.
A to D Healing by primary intention. E to I Healing by secondary intention. See the text for more details.
Healing involves processes that (1) fill in, (2) seal and (3) shrink the wound. These characteristics of healing vary in importance and duration among different types of wounds. A clean incision, such as a paper cut or sutured surgical wound, heals primarily through the process of collagen synthesis. Because this type of wound has minimal tissue loss and close proximity of the wound edges, very little sealing (termed epithelialisation) and shrinkage (contraction) are required. Wounds that heal under conditions of minimal tissue loss are said to heal by primary intention.
Other wounds do not heal as easily. Healing of an open wound (one with extensive tissue loss), such as pressure ulcer (called a decubitus ulcer), requires a great deal of tissue replacement so that epithelialisation, scar formation and contraction take longer and healing occurs through secondary intention (see Figure 13-14). Healing by either primary or secondary intention may occur at different rates for different types of tissue injury.
Wound healing occurs in two overlapping phases: the reconstructive phase begins 3–4 days after injury and continues for as long as two weeks; and the maturation phase begins several weeks after injury and is normally complete within a few years.
The reconstructive phase
Surgical and penetrating wounds are useful models of both normal and abnormal (dysfunctional) healing. Bleeding is initially sealed off by a blood clot containing a cross-linked mesh of fibrin and trapped platelets. Most surgical wounds are completely sealed within hours after closure. This sealing helps unite the wound edges and creates a physical barrier to bacterial invasion. The fibrin mesh acts as a scaffold for fibroblasts and collagen deposition, which ultimately fills the wound.
For healing to proceed, the fibrin clot must be replaced by normal tissue or scar tissue.24 Macrophages invade the dissolving clot and clear away debris and dead cells. Macrophages also secrete biochemical mediators that promote healing. Granulation tissue grows into the wound from the surrounding healthy connective tissue. Granulation tissue is filled with new capillaries (called angiogenesis, meaning growing new blood vessels) derived from capillaries in the surrounding tissue, giving the granulation tissue a red, granular appearance. New lymphatic vessels also grow into the granulation tissue by a similar process.
During this process the healing wound must be protected. Epithelialisation is the process by which epithelial cells grow into the wound from surrounding healthy tissue. Epithelial cells migrate under the clot or scab to unravel collagen. Migrating epithelial cells contact similar cells from all sides of the wound and seal it, thereby halting migration and proliferation. The epithelial cells remain active, undergoing differentiation to give rise to the various epidermal layers (see Chapter 18). Epithelialisation of a skin wound can be hastened if the wound is kept moist (as opposed to allowing the wound to dry out), preventing the fibrin clot from becoming a scab.
Fibroblasts are the most important cells during healing because they secrete collagen and other connective tissue proteins, which are deposited in debrided areas about 6 days after the fibroblasts have entered the lesion. Collagen is the most abundant protein in the body. It contains high concentrations of the amino acids. As healing progresses, collagen undergoes chemical reactions to form collagen fibres. The complete process takes several months.
Wound contraction is necessary for closure to all wounds, especially those that heal by secondary intention. Contraction is noticeable 6–12 days after injury. The granulation tissue contains myofibroblasts — specialised cells that are responsible for wound contraction. Myofibroblasts have features of both smooth muscle cells and fibroblasts. They appear microscopically similar to fibroblasts but differ in that their cytoplasm contains bundles of parallel fibres similar to those found in smooth muscle cells. Wound contraction occurs as extensions from the plasma membrane of myofibroblasts establish connections between neighbouring cells, contract their fibres and exert tension on the neighbouring cells while anchoring themselves to the wound bed.
The maturation phase
Collagen matrix assembly, tissue regeneration and wound contraction continue into the maturation phase — a phase that can persist for years. Scar tissue is remodelled and capillaries disappear, leaving the scar avascular — that is, without a blood supply (the ‘a’ in front of the word vascular means without). Within 2 to 3 weeks after maturation has begun, the scar tissue has gained about two-thirds of its eventual maximum strength.
Epidermal wounds that heal by secondary intention and unsutured internal lesions are not completely restored by healing. At best, repaired tissue regains 80% of its original tensile strength. Essentially, this means that the scar tissue is never as strong as the original tissue. Only epithelial, hepatic (liver) and bone marrow cells are capable of the complete mitotic regeneration of the normal tissue known as compensatory hyperplasia (hyperplasia is described in Chapter 4). In fibrous connective tissue such as joints and ligaments, normal healing results in replacement of the original tissue with new tissue that does not have exactly the same structure or function as that of the original. In some cases this leads to recurrent injury, such as that experienced by athletes. For instance, an athlete sustains a knee injury, undergoes rehabilitation, but re-injures the same knee when competing again. This may occur because the new tissue is not quite as effective as the original tissue, leading to future problems. Some tissues heal without the replacement of cells. For example, damage resulting from myocardial infarction heals with a scar composed of fibrous tissue rather than with cardiac muscle, which can lead to cardiac conduction abnormalities as action potentials cannot propagate through the cardiac muscle tissue.
Dysfunctional wound healing
Dysfunctional healing may occur during any phase of the process and may involve insufficient repair, excessive repair or infection. The cause of dysfunctional healing can be related to a predisposing disorder, such as diabetes mellitus; to an acquired condition, such as hypoxaemia (insufficient oxygen in arterial blood); or to numerous drugs and nutrients. Wound repair delays healing by reactivating inflammatory processes.
Dysfunction during the inflammatory response
Healing is prolonged if bleeding is not stopped during acute inflammation. Large clots increase the amount of space that granulation tissue must fill and serve as mechanical barriers to oxygen diffusion. Excess blood cells resulting from haemorrhage must be cleared before repair and prolong the process. Accumulated blood is also an excellent culture medium for bacteria and promotes infection, thereby prolonging inflammation by increasing exudation and pus formation.
Excessive fibrin deposition is detrimental to healing. Fibrin released in response to injury must eventually be reabsorbed to prevent organisation into fibrous adhesions. Adhesions formed in the pleural, pericardial or abdominal cavities can bind organs together by fibrous bands and distort or strangulate the affected organ.
Decreased blood volume also inhibits inflammation because of vessel constriction rather than the dilation required to deliver inflammatory cells to the site of injury. Anti-inflammatory steroids prevent macrophages from migrating to the site of injury and they also inhibit fibroblast migration into the wound during the reconstructive phase.
Optimal nutrition is important during all phases of healing because metabolic needs increase. The substances most needed are glucose, oxygen and protein. Leucocytes need glucose for chemotaxis, phagocytosis and intercellular killing. Therefore, the wounds of individuals with diabetes mellitus who receive insufficient insulin usually heal poorly. People with diabetes mellitus are also at an increased risk for ischaemic wounds because they are likely to have both small-vessel diseases that impair the microcirculation and altered (glycosylated) haemoglobin (see Chapter 35), which has an increased affinity for oxygen and thus does not readily release oxygen in tissues. Oxygen-deprived (ischaemic) tissue is susceptible to infection, which prolongs inflammation. Hypoproteinaemia (a low level of protein in the blood, usually due to nutritional deficiency, liver or renal disease or burns) also prolongs inflammation because it impairs fibroblast proliferation.
Dysfunction during the reconstructive phase
Impaired collagen synthesis
Most of the factors that interfere with the production of collagen in healing tissues are nutritional.24 Protein and other nutrients are required for collagen synthesis. These include iron, oxygen, copper and calcium. Usually such minute amounts of these substances are required as cofactors that deficiencies are not clinically significant.
Dysfunctional collagen synthesis also may involve excessive production of collagen, causing surface overhealing leading to abnormal scarring. Hypertrophic scars are large, red scars that are often hard and raised compared to normal scars. They may form when excessive collagen tissue is produced but the defining characteristic is that the tissue remains within the wound edges. Hypertrophic scars tend to regress over time, usually years. Keloid scars, on the other hand, are raised scars that extend beyond the original boundaries of the wound and invade surrounding tissue. They are permanent and are often associated with tenderness and pain. Figure 13-15 shows hypertrophic and keloid scars.
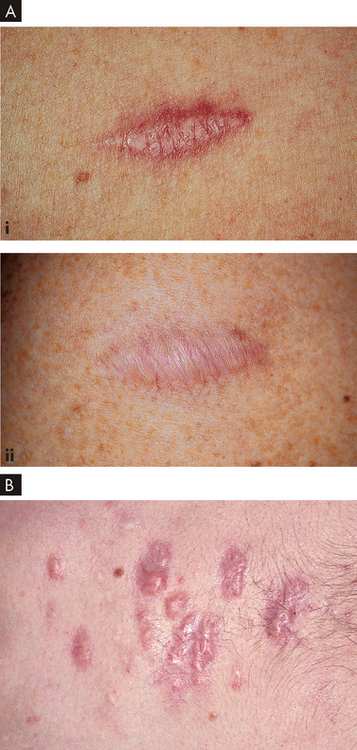
FIGURE 13-15 Different types of scars.
A Hypertrophic scars: (i) a 5-month-old scar that is still pink; and (ii) a 1-year-old scar that has reduced in size and lost colour (hypopigmented). B A keloid scar. Keloids on the chest and extremities are raised with a flat surface, and the base is wider than the top.
Source: A Courtesy of Jean L Bolognia MD. Bolognia JL. Dermatology. 2nd edn. St Louis: Mosby; 2008. B Habif TP. Clinical dermatology: a color guide to diagnosis and therapy. 5th edn. St Louis: Mosby; 2010.
Impaired epithelialisation
Epithelialisation is suppressed by anti-inflammatory steroids and hypoxaemia. Wound care technique may greatly influence epithelial cell migration. External wounds that are draining or healing by secondary intention often are debrided and protected with dressings. The ideal dressing absorbs some drainage without being incorporated into the clot or granulation tissue. Because epithelial cells must migrate across the wound during healing, dressings that debride healthy epithelial cells along with necrotic tissue prolong epithelialisation.
Many solutions that traditionally have been used to clean or irrigate wounds are deleterious to the fragile new cells in the wound bed. Normal saline and potable tap water are the most innocuous solutions that can be used to cleanse or irrigate a wound that is healing primarily by epithelialisation. Solutions such as iodine and hydrogen peroxide are very drying and subsequently inhibit rather than promote epithelial cell migration.
Wound disruption
A potential complication of wounds that are sutured closed is dehiscence, in which the wound pulls apart at the suture line. Dehiscence generally occurs 5–12 days after suturing, when collagen synthesis is at its peak. Approximately half of dehiscence occurrences are associated with wound infection, but they also may be the result of sutures breaking because of excessive strain. Obesity increases the risk for dehiscence because adipose tissue is difficult to suture. Wound dehiscence usually is heralded by increased serous drainage from the wound and a feeling that ‘something gave way’. Prompt surgical attention is required.
Impaired contraction
Wound contraction, although necessary for healing, may become pathological when contraction is excessive, resulting in a deformity or contracture. Burns are especially susceptible to contracture development. Internal contraction deformities include duodenal strictures caused by dysfunctional healing of an ulcer and oesophageal strictures caused by lye burns. Contracture may occur in cirrhosis of the liver. Scar tissue that becomes contracted constricts vascular flow and contributes to the development of portal hypertension and oesophageal varices. Proper positioning and range-of-motion exercises and surgery are among the physical means used to overcome myofibroblast pull and prevent contractures.
 PAEDIATRICS
PAEDIATRICSPAEDIATRICS AND FACTORS AFFECTING INFLAMMATION AND TEMPERATURE REGULATION
Newborns have a transiently depressed inflammatory response. The neutrophils are incapable of chemotaxis as they are lacking fluidity in the cell membrane. There is a tendency for infections associated with chemotactic defects — for example, cutaneous abscesses caused by Staphylococci and cutaneous candidiasis. There is also a partial deficiency in complement, especially components of alternative pathways.
Infants produce sufficient body heat, primarily through metabolism of brown fat (called brown fat thermogenesis), but cannot retain heat. This is due to many factors, including their small body size, a greater ratio of body surface to body weight, and an inability to shiver coupled with a small amount of subcutaneous fat.25
AGEING AND FACTORS AFFECTING INFLAMMATION AND TEMPERATURE REGULATION
Elderly individuals are at risk for impaired wound healing. This is often associated with chronic illness — for example, diabetes mellitus or cardiovascular disease — which is prevalent in the elderly population in Australia and New Zealand. In addition, medications such as anti-inflammatory steroids, commonly prescribed in the elderly population, may interfere with healing.
The elderly are also at risk for sustaining wounds because of impaired sensation or mobility and physiological changes in skin. There is a loss of subcutaneous fat associated with ageing, which diminishes the layer of protection, and thickened and less elastic collagen fibres, which contribute to less protection. The atrophied epidermis, including the underlying capillaries, decreases perfusion and increases the risk of hypoxia in the wound.
A number of factors affect the thermoregulatory capacity of elderly individuals. These factors are associated with ageing and can contribute to an elderly individual’s ability to respond to large changes in environmental temperature, both high and low. The factors include: sluggish blood circulation, with a diminished vasoconstrictor response; structural and functional skin changes, which change the perception of heat and cold, and mean that the elderly do not sweat as efficiently as younger people; a decreased shivering response (delayed onset and decreased effectiveness), which overall reduces heat-producing capacity. Moreover, elderly people often have a decreased thirst coupled with undernutrition, which affects thermoregulation.26–28 4 6
Acute inflammation
Inflammation is a rapid and nonspecific protective response to cellular injury from any cause. It can occur only in vascularised tissue.Cellular components of inflammation
Many different types of cells are involved in the inflammatory process, including neutrophils, monocytes and macrophages, eosinophils and platelets. The most important activator of the inflammatory response is the mast cell, which initiates inflammation by releasing biochemical mediators (histamine, chemotactic factors) from preformed cytoplasmic granules and synthesising other mediators (prostaglandins, leukotrienes) in response to a stimulus. Phagocytic cells (neutrophils and macrophages) engulf and destroy microorganisms by enclosing them in phagocytic vacuoles, within which toxic products and degradative lysosomal enzymes kill and digest the microorganisms. Polymorphonuclear neutrophils, the predominant phagocytic cells in the early inflammatory response, exit the circulation by diapedesis through the retracted endothelial cell junctions and move to the inflammatory site by chemotaxis. The macrophage, the predominant phagocytic cell in the late inflammatory response, is highly phagocytic, is responsive to cytokines and promotes wound healing. Eosinophils release products that control the inflammatory response and are the principal cell that kills parasitic organisms.Inflammatory mediators
Histamine is the major vasoactive amine released from mast cells. It causes constriction of vascular smooth muscles, dilation of capillaries and retraction of endothelial cells lining the capillaries, which increases vascular permeability. Chemotactic factors are released from mast cell granules, forming a chemical gradient to attract inflammatory cells towards the site of inflammation. Leukotrienes produce histamine-like effects, smooth muscle contraction and increased vascular permeability. The biological activity of platelet-activating factor is virtually identical to that of leukotrienes. Nitric oxide is released from the endothelium and causes vasodilation by inducing relaxation of vascular smooth muscle. It may suppress mast cell release of inflammatory molecules. Prostaglandins cause increased vascular permeability, neutrophil chemotaxis and pain by direct effects on nerves.Plasma protein systems
Inflammation is mediated by three key plasma protein systems: the complement system, the coagulation system and the kinin system. The components of all three systems are a series of inactive proteins that are activated sequentially. The complement system can be activated by antigen-antibody reactions (through the classical pathway) or by other products, especially bacterial polysaccharides (through the lectin pathway or the alternative pathway), resulting in the production of biologically active fragments that cause cell lysis. The most biologically potent products of the complement system are C3b (opsonin), C3a (anaphylatoxin) and C5a (anaphylatoxin, chemotactic factor). Opsonins, such as antibody and complement component C3b, coat microorganisms and make them more susceptible to phagocytosis by binding them more tightly to the phagocyte.Chronic inflammation
Clinical manifestations of inflammation
Local manifestations of inflammation are the result of the vascular changes associated with the inflammatory process, including vasodilation and increased capillary permeability. The symptoms include redness, heat, swelling and pain.Fever
Temperature regulation is achieved through precise balancing of heat-production and heat-loss mechanisms. Body temperature is maintained in a narrow range between approximately 36.2 and 37.5°C. Temperature regulation is mediated by the hypothalamus through thermoreceptors in the skin, hypothalamus, spinal cord and abdominal organs. Heat is produced through chemical reactions of metabolism, skeletal muscle contraction, chemical thermogenesis and vasoconstriction. Heat is lost through radiation, conduction, convection, vasodilation, decreased muscle tone, evaporation of sweat, increased respiration and voluntary mechanisms. Hyperthermia (an increase in core temperature above normal) can produce nerve damage, coagulation of cell proteins and death. Hypothermia (a decrease in core temperature below normal) slows the rate of chemical reaction (tissue metabolism), increases the viscosity of the blood, slows blood flow through the microcirculation, facilitates blood coagulation and stimulates profound vasoconstriction. Fever is triggered by the release of pyrogens from bacteria, leucocytes and other cells involved in the immune response. Fever is both a normal immunological mechanism and a symptom of a disease.Wound healing
Damaged tissue proceeds to restoration of the original tissue structure and function if little tissue has been lost or the injured tissue is capable of regeneration. This is called healing by primary intention. Tissues that sustained extensive damage or those incapable of regeneration heal by the process of repair resulting in the formation of a scar. This is called healing by secondary intention.Paediatrics and factors affecting inflammation and temperature regulation
Athena is 54 years old and married with 4 adult-age children. She works part-time in an after-school care centre during the school semesters. A few days ago she tripped over a toy in the playground and hurt her ankle. Her ankle was swollen, red and tender to touch. It was also painful and so she took several days off work. Athena noticed that when she walked after a few minutes her ankle became painful and more swollen than usual. She also had a fever up to 38.2°C. Her local doctor diagnosed a grade II ankle sprain (moderate tearing of ligaments, internal and external bruising, joint instability and difficulty walking). He prescribed rest, elevation, ice and antipyretics.