Guidelines for Selection of Routes of Opioid Administration
Disadvantages of the Oral Route
Selected Oral Opioid Formulations
Trend in Oral Analgesics for Postoperative Pain
Tamper-Resistant and Abuse-Deterrent Oral Opioid Formulations
REGARDLESS of the type of pain being treated, opioids should be administered by the least invasive and safest route capable of producing satisfactory analgesia. The oral route is the most common and generally is selected before other routes because it is relatively safe, convenient, and inexpensive (Coyle, Cherny, Portenoy, 1995; Hanks, Cherny, Fallon, 2004; Stevens, Ghazi, 2000). The transdermal route is an accepted alternative for long-term therapy. If pain is severe and a rapid onset of analgesia is desired, the IV route can be used for rapid titration with close monitoring; the patient can be transitioned to the oral route when pain is under control.
Over time, it is often necessary to switch routes of administration (Hanks, Cherny, Fallon, 2004). For example, in a postoperative patient, the process of transitioning from IV or epidural to oral may require the use of both the old and the new routes to ensure continuous analgesia. For example, the patient may be started on the oral formulation while receiving PRN boluses by the IV route. When the oral route is established and at the approximate required dose, the IV route is discontinued.
In a survey of patients with cancer pain, more than half required more than one route of administration to maintain pain control during the last 4 weeks of life. This occurred usually when patients were unable to swallow. The routes used included rectal, SC, IV, and epidural. Sometimes patients required more than one route at a time (Coyle, Adelhardt, Foley, et al., 1990). Table 14-1 summarizes the advantages and disadvantages to some of the routes of opioid administration. This chapter presents most of the routes by which opioids are administered. The intraspinal routes are discussed separately in Chapter 15.
Table 14-1
Routes of Opioid Administration

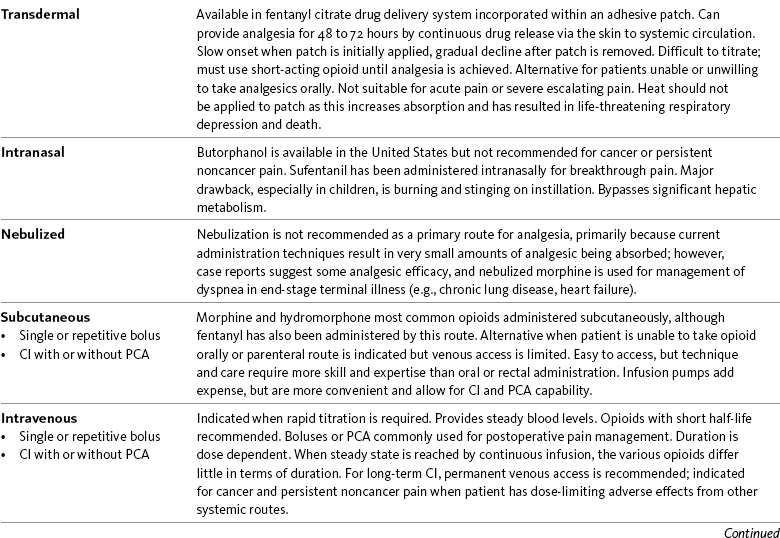

ATC, Around-the-clock; CI, continuous infusion; FDA, Food and Drug Administration; PCA, patient-controlled analgesia.
Opioid analgesics can be administered by a wide variety of routes. This table summarizes the advantages and disadvantages to some of these. See text for detailed discussion.
From Pasero, C., & McCaffery, M. (2011). Pain assessment and pharmacologic management, pp. 369-370, St. Louis, Mosby. Data from Buxton, I. L. O. (2006). Pharmacokinetics and pharmacodynamics. The dynamics of drug absorption, distribution, action, and elimination. In L. L. Brunton, J. S. Lazo, & K. L. Parker KL (Eds.), Goodman & Gilman’s the pharmacological basis of therapeutics, ed 11, New York, mcgraw-Hill; Darwish, M., Kirby, M., Jiang, J. G., et al. (2008). Bioequivalence following buccal and sublingual placement of fentanyl buccal tablet 400 microg in healthy subjects. Clin Drug Investig, 28(1), 1-7.; Darwish, M., Kirby, M., Robertson, P. Jr, et al. (2007). Absolute and relative bioavailability of fentanyl buccal tablet and oral transmucosal fentanyl citrate. J Clin Pharmacol, 47(3), 343-350; Dale, O., Hjortkjær, R., & Kharasch, E. D. (2002). Nasal administration of opioids for pain management in adults. Acta Anaesthesiol Scand, 46(7), 759-770; Gordon, D. B. (2008). New opioid formulations and delivery systems. Pain Manage Nurs, 8(3, Suppl 1), S6-S13; Gutstein, H. B., & Akil, H. (2006). Opioid analgesics. In L. L. Brunton, J. S. Lazo, & K. L. Parker (Eds.), Goodman & Gilman’s the pharmacological basis of therapeutics, ed 11, New York, mcgraw-Hill; Hanks, G., Cherny, N. I., & Fallon, M. (2004). Opioid analgesic therapy. In D. Doyle, G. Hanks, N. I. Cherny, et al. (Eds.), Oxford textbook of palliative medicine, ed 3, New York, Oxford Press; Holmquist, G. (2009). Opioid metabolism and effects of cytochrome P450. Pain Med, 10(Suppl 1), S20-S29; Shelley, K., & Paech, M. J. (2008). The clinical applications of intranasal opioids. Curr Drug Deliv, 5(1), 55-58; Smith, H. S. (2003). Drugs for pain. Philadelphia, Hanley & Belfus; Stevens, R. A., & Ghazi, S. M. (2000). Routes of opioid analgesic therapy in the management of cancer pain. www.medscape.com/viewarticle/408974. Accessed January 9, 2009; Swarm, R. A., Karanikolas, M., & Cousins, M. J. (2004). In D. Doyle, G. Hanks, N. I. Cherny, et al. (Eds.), Oxford textbook of palliative medicine, ed 3, New York, Oxford Press; Vascello, L., & McQuillan, R. J. (2006). Opioid analgesics and routes of administration. In O. A. de Leon-Casasola (Ed.), Cancer pain. Pharmacological, interventional and palliative care approaches, Philadelphia, Saunders. Pasero C, McCaffery M. May be duplicated for use in clinical practice.
Oral
The oral route is the most commonly employed for patients with continuous cancer or noncancer pain, or mild to moderate acute pain (Fine, Portenoy, 2007; Hanks, Cherny, Fallon, 2004; Menefee, Katz, Zacharoff, 2007). The many oral formulations available provide convenience and flexibility (American Pain Society [APS], 2003). Given the potential to titrate the oral dose to whatever level is necessary, the most common reason for failure to achieve analgesia by this route is insufficient dose administration. There is wide interindividual variation in response to opioids, however, and if attempts to escalate the oral dose of opioid do not achieve the desired response or result in unacceptable adverse effects, switching to a different oral opioid should be considered, as unresponsiveness to one opioid does not predict response to others (Fine, Portenoy, 2007).
Most first-line mu agonists and dual-mechanism drugs are available in oral form; fentanyl is not. Of the agonist-antagonist opioids, only pentazocine is available orally. Oral opioids in tablet form can be taken by most patients. Capsules and liquids are available for some opioids. Liquids should be carefully measured using an oral medication syringe or graduated liquid measuring spoon. Both of these measuring devices are available free from most pharmacies. When prescribing, dispensing, and administering opioids, care should be taken to ensure that the correct concentration and milligram amount are specified. Liquids should be ordered by milligram amount, not just volume, to avoid serious dosing errors (Institute for Safe Medication Practices, 2007). Non–modified-release tablet formulations can be crushed and taken with soft foods or put into suspensions (APS, 2003). Modified-release tablets; however, should not be cut, crushed, or chewed because this destroys the release mechanisms and risks severe overdose by releasing the half-day or full-day dose all at once. Modified-release medications in capsules may be opened and sprinkled on soft food, such as applesauce, but should not be chewed or allowed to dissolve (APS, 2003). Small amounts of food should be used to ensure consumption of the entire dose.
Disadvantages of the Oral Route
Two major disadvantages of the oral route are that it has a slow onset of action (typically 30 to 45 minutes) (APS, 2003) and a relatively delayed peak time (60 to 120 minutes after ingestion, and longer in the case of some of the modified-release tablets) (Hanks, Cherny, Fallon, 2004). As a result of these kinetics, the oral route is not ideal when it is imperative to get severe pain under control quickly, such as for pain crisis related to malignancy or myocardial infarction (Moryl, Coyle, Foley, 2008).
Although opioids tend to have a longer duration of action orally than parenterally, intervals between doses of short-acting preparations are relatively brief—commonly 4 hours. This requires the patient to take six to eight doses a day, a regimen that can interfere with patient activities, such as sleeping. The patient must remember to take all doses to maintain a constant level of analgesia.
A potential disadvantage for those opioids with active metabolites is that the ratio between the metabolite concentration and the parent compound is much higher when the drug is given orally and subjected to a larger first-pass effect through the liver than when it is given parenterally. This is true for morphine and its glucuronidated metabolites, for example (Lotsch, 2005). In most patients, this difference between oral and other routes is not clinically significant, but for some, particularly those with renal insufficiency, the relative concentration of the metabolites could be high enough to cause adverse effects.
The oral route is not an option for patients who are NPO (nothing by mouth), such as immediately after surgery. Some patients cannot tolerate the oral route because of GI obstruction or difficulty swallowing (Fine, Portenoy, 2007). Absorption by the oral route can be altered by a number of factors, including presence of food, gastric emptying time, and GI motility. Modified-release preparations appear to be less affected by the presence of food than short-acting preparations (see individual drugs later in this chapter, and see Chapter 13 for more).
The effectiveness of the oral route depends on patient compliance. Patients who must self-administer their medications but cannot adhere to the dosing regimen necessary to maintain stable effects are not good candidates for the oral route unless they are able to take formulations designed for once-a-day dosing (Lehne, 2004).
Finally, some oral medications, especially those in tablet or liquid form, have a bitter or unpleasant taste, to which most patients object. After administration, “chasers” of applesauce or lemon drops may be helpful in reducing the bitterness (Gardner-Nix, 1996).
Selected Oral Opioid Formulations
As mentioned, all of the first-line mu opioids except fentanyl are available in short-acting oral formulations, and several modified-release formulations exist. Opioids available in oral modified-release formulations in the United States include morphine (MS Contin, Oramorph SR, Kadian, Avinza, and generics), oxycodone (OxyContin), oxymorphone (Opana ER), hydromorphone (Exalgo), and tramadol (Ultram ER). Codeine is available as a modified-release product outside of the United States. Of the modified-release preparations, MS Contin has the smallest tablet, which is an important consideration in patients who have difficulty swallowing. MS Contin tablets are color-coded according to dose, as are Kadian and Avinza capsules, which may help prevent errors in dosing.
The modified-release opioid preparations have rendered the oral route more convenient than in the past by requiring only once- or twice-daily dosing. This may improve patient adherence to medication regimens and may also decrease the patient’s sense of being sick (APS, 2003; Fine, Portenoy, 2007; Gallagher, Welz-Bosna, Gammaitoni, 2007). These preparations simplify the regimen necessary to maintain relatively stable blood levels of the drug, potentially increasing their effectiveness for continuous pain (APS, 2003). Although unproven, some experts recommend modified-release products as one of the treatment strategies for patients at risk of opioid abuse because they may be less reinforcing of some drug-related behaviors and may be less likely to cause euphoria (Webster, Dove, 2007).
Patients should be observed closely for the need to shorten the recommended dosing interval of the modified-release agent. For example, although MS Contin and Oramorph were designed for 12-hour dosing, it is not unusual for patients to experience some end-of-dose failure (pain at the end of the dosing interval), which can be eliminated by switching to 8-hour dosing (Argoff, 2007). An observational cohort study found that 86% and 91% of patients taking modified-release morphine or oxycodone, respectively, required dosing more frequently than that recommended by the product’s manufacturers (Gallagher, Welz-Bosna, Gammaitoni, 2007). This underscores the importance of systematic assessment to determine the optimal dose interval as well as a need to develop modified-release formulations that provide satisfactory and sustained analgesia throughout the recommended dosing interval.
Although further research is warranted, the time of day that patients take their once-daily opioid dose does not appear to matter. A multicenter, randomized, placebo-controlled, cross-over study of patients with advanced cancer found essentially the same pain intensities when the once-daily dose of modified-release morphine was taken in the morning as when it was taken in the evening (Currow, Plummer, Cooney, et al., 2007). Patients can be told to find the time that works best to keep their pain under control.
Following is a discussion of the various oral formulations of morphine, oxycodone, oxymorphone, and hydromorphone. Formulations designed to deter abuse are also discussed.
Oral Morphine
Morphine is available in 15 and 30 mg short-acting tablets, and in 2, 4, 10, and 20 mg/mL solutions. These dose forms are used primarily when opioid therapy is initiated and for breakthrough pain (see Chapter 12 for more on breakthrough pain). MS Contin and Oramorph SR, modified-release formulations of morphine, are available in 15, 30, 60, and 100 mg tablets; MS Contin also is available in a 200 mg tablet. There are also generic products available at varying strengths. The recommended dosing interval is every 12 hours and no less than every 8 hours. There are two other modified-release morphine formulations, Kadian and Avinza, supplied as capsules that contain pellets which release drug at different rates. Kadian is available in 10, 20, 30, 50, 60, 80, 100, and 200 mg capsules and can be given every 12 or 24 hours. Avinza is available in 30, 45, 60, 75, 90, and 120 mg capsules and is approved for once-daily dosing. The Avinza prescribing information contains a black box warning that alcohol is not to be ingested while taking Avinza, as there is a risk of the pellets dissolving and the full daily dose of morphine being released at once (King Pharmaceuticals, 2008a). Kadian does not require such a warning (Alpharma Pharmaceuticals, 2008; Johnson, Wagner, Sun, et al., 2008), but alcohol can have additive CNS effects when ingested by a person taking any opioid and should be used with great caution. See Patient Education Form IV-9 (pp. 562-563) on short-acting morphine (includes concentrate), Form IV-10 (pp. 564-565) on modified-release 12-hour morphine, and Form IV-11 (pp. 566-567) on modified-release 24-hour morphine at the end of Section IV.
The pharmacokinetics of the modified-release formulations are complex because the time to steady state is determined by the half-life of absorption rather than the terminal elimination half-life. All of the modified-release formulations approach steady state in a 2- to 3-day timeframe. Like other oral formulations, the drugs are not preferable for rapid titration to address severe pain. Although patients can be titrated using the modified-release drug, an accepted alternative approach is to titrate first to a stable dose of short-acting morphine, then switch to the modified-release formulation (APS, 2003).
Co-administration of a short-acting opioid for breakthrough pain is conventional practice during the treatment of patients with pain related to active cancer or other types of serious illnesses; it is implemented on a case-by-case basis during the treatment of persistent noncancer pain based on a separate analysis of risk and benefit (Fine, Portenoy, 2007). Availability of a short-acting drug during dose titration of the modified-release formulation may facilitate dose finding and the comfort of the patient; the modified-release dose can be titrated every 24 to 48 hours (Twycross, Wilcock, 2007). The short-acting dose may or may not be continued after a stable dose of the modified-release drug is found. One study randomized 40 patients with uncontrolled cancer pain to titration with short-acting oral morphine given every 4 hours or titration with modified-release morphine (Kapanol, Kadian) given once daily (Klepstad, Kaasa, Jystad, et al., 2003). The mean time to achieve adequate pain control was 2.1 days with short-acting morphine and 1.7 days with modified-release morphine, and those taking the latter reported feeling less tired at the end of titration. No other differences in adverse effects, health-related quality of life functions, or satisfaction with treatment were noted.
Although the various forms of modified-release morphine contain the same drug and are of the same dose strength, they may or may not be bioequivalent. MS Contin and Oramorph SR, for example, are pharmaceutically equivalent because they contain the same drug, have the same dose form, can deliver the same amount of drug, are both available in the same dose strengths, and are given by the same route; however, the two are not necessarily therapeutically equivalent because they use different modified-release mechanisms. This means that the same dose of each product may not affect the patient in the same way (McCaffery, Lochman, 1996). The FDA does not consider any of the modified-release dose forms to be therapeutically equivalent unless bioequivalence data have been submitted. Given the many choices now available, it is best not to assume that very similar products will behave the same in a given individual. It should be recognized, however, that the FDA and some state laws have allowed pharmacists, physicians and other prescribers, institutions, and health care plans to consider drugs containing the molecule to be therapeutically equivalent, even in the absence of confirmatory clinical data. If patients report a change in the outcomes associated with stable drug therapy, the clinician should assess whether the formulation may have been changed by the pharmacist (McCaffery, Lochman, 1996). Box 14-1 provides recommendations when switching from one pharmaceutically equivalent product to another.
The first modified-release morphine formulations were Oramorph and MS Contin. Oramorph contains a simple matrix system; GI fluid penetrates the tablet, hydrates the matrix, and forms a gel layer that breaks down and dissolves gradually over the dosing period (Amabile, Bowman, 2006). MS Contin tablets contain morphine in a dual-control (hydrophilic and hydrophobic) polymer matrix that controls the release of morphine (Amabile, Bowman, 2006). This sophisticated hydrophilic/hydrophobic relationship is reported to provide a more constant and predictable release of morphine from the system than is possible with the simpler Oramorph release mechanism (Amabile, Bowman, 2006).
Avinza is a capsule containing both fast-acting and modified-release beads of morphine. The primary advantage of this combination is that the fast-acting component allows the morphine concentration to plateau rapidly (within 30 minutes), and the modified-release component maintains the plasma concentration throughout the dosing interval (Amabile, Bowman, 2006). Most patients obtain adequate pain relief with either 12- or 24-hour dosing of the drug (Argoff, 2007). A comparative steady-state analysis of once-daily Avinza and twice-daily modified-release morphine (MS Contin) found that the two formulations provide similar total systemic exposure of morphine and its metabolites throughout a 24-hour period but have distinct pharmacokinetic profiles due to divergent technologies (Portenoy, Sciberras, Eliot, et al., 2002). Avinza maintains morphine concentrations at or greater than 50% and 75% for a longer duration of time than MS Contin. This may help to explain why studies have shown that the two drugs appear to have similar efficacy, but that Avinza may offer some additional benefits. For example, Avinza produced analgesia and adverse effects comparable to MS Contin but with greater improvements in sleep in patients with osteoarthritis (OA) pain (Caldwell, Rapoport, Davis, et al., 2002; Rosenthal, Moore, Groves, et al., 2007). Another study in patients with a variety of types of noncancer pain showed improvements in pain relief and both sleep and physical functioning over the 3-month study period with Avinza (Adams, Chwiecko, Ace-Wagoner, et al., 2006). Patients with noncancer pain of various origins (including neuropathic) that was unresponsive to short-acting opioid regimens experienced reduced pain and improvements in depressive symptoms and cognitive functioning after taking Avinza for 4 weeks (Panjabi, Panjabi, Shepherd, et al., 2008).
Once-daily Avinza has also been shown to produce more consistent opioid plasma concentrations with less frequent dosing compared with twice-daily modified-release oxycodone (OxyContin, see the paragraphs that follow) in healthy volunteers (Eliot, Geiser, Loewen, 2001). A multicenter study randomized 392 patients with persistent back pain (including some with neuropathic pain) to receive either Avinza every 24 hours or modified-release oxycodone every 12 hours; supplemental ibuprofen was allowed (Rauck, Bookbinder, Bunker, et al., 2006). Following a titration period, 174 patients took their study drug at a fixed dose for four weeks followed by a four-week period during which the dose could be changed as needed. Those taking Avinza experienced better pain control with a lower daily opioid dose, consumed fewer breakthrough doses, and had better sleep quality than those taking modified-release oxycodone. Adverse effects were similar. Patient surveys were used to evaluate physical function and revealed improvements in both groups but no significant differences between the two; however, fewer patients were unable to work due to illness or treatment in the Avinza group than in the modified-release oxycodone group (Rauck, Bookbinder, Bunker, et al., 2007).
Kadian, the other once-daily morphine formulation, is available in a capsule containing polymer-coated modified-release pellets of morphine (Alpharma Pharmaceuticals, 2008; Amabile, Bowman, 2006). If differs from Avinza in that it does not contain a fast-acting component. Its time to maximum serum level (tmax) is the longest (approximately 9.5 hours) of any of the modified-release morphine formulations (Rosielle, 2007). An analysis of data on nearly 1042 patients with noncancer pain who were started on Kadian once daily and could switch after two weeks to 12-hour dosing if necessary (Nicholson, Ross, Weil, et al., 2006) was undertaken to determine factors that influenced the patients’ choice of dosing interval (Weil, Nicholson, Sasaki, 2009). At the end of the study, 56.8% were taking the drug once daily and 43.2% were taking it every 12 hours. Race and gender did not influence dosing, but those with higher baseline and 2-week pain intensities were more likely to switch to a 12-hour dosing schedule, and older patients were more likely to remain on the 24-hour dosing schedule. Those who switched to 12-hour dosing experienced improved pain control, and by week 4, efficacy was comparable between the two dosing schedules. A review of the data of 68 patients taking Kadian for persistent pain showed the drug to be safe and effective for long-term opioid therapy (mean treatment = 12 months) (Chao, 2005). Patients in the review had a variety of pain conditions including radiculopathy and neck, head, and back pain. The median daily dose was 60 mg (range 20 to 400 mg); pain intensity (0 to 10) was reduced from a mean baseline of 7.8 to 5.2; 29% were considered non-responders, but over one-third of the patients experienced a reduction to 2.9; and, as in the previous study, over one-half of the patients were maintained on once-daily dosing.
In summary, some studies have shown patient preference for one morphine product or another, but as long as one product is used consistently and titrated to effect, they should all provide equally effective analgesia with the same adverse effect profile (Rosielle, 2007). Key to this is ensuring that the optimal dosing interval is prescribed. As mentioned, a high percentage of patients require more frequent dosing intervals than are recommended by the manufacturer (Argoff, 2007; Gallagher, Welz-Bosna, Gammaitoni, 2007).
Oral Oxycodone
Oxycodone is used extensively by the oral route and is available in both short-acting and modified-release formulations. It is also available alone or in combination (2.5 to 10 mg) with varying amounts of acetaminophen, aspirin, or ibuprofen (see Table 13-9 on p. 351). Single-entity oxycodone is available in 5, 15, and 30 mg tablets (capsules in 5 mg) and in two solution strengths, 5 mg/5mL and a 20 mg/mL concentrate (see the safety considerations for liquid opioids earlier in the chapter). The short-acting dose forms typically are used for short-term acute pain and for breakthrough pain.
With the varying doses and dose types (tablet, capsule, liquid) available, there is potential for confusion on the part of both clinician and patient. Prescriptions must be carefully written for each individual, and it is prudent to have the patient bring in the prescription bottle if refills are needed.
A drawback to the use of oxycodone combinations is that the clinician must carefully monitor the dose of acetaminophen, aspirin, or ibuprofen to ensure that maximum safe levels are not exceeded. Increases in the dose of oxycodone for inadequate pain relief are limited by acetaminophen’s and aspirin’s recommended maximum daily dose of 4000 mg and ibuprofen’s limit of 3200 mg (see Section III). At the time of publication, the United States Food and Drug Administration (U.S. FDA) was considering the need to restrict the availability of a maximum dose/tablet of acetaminophen to 325 mg and eliminate analgesics with fixed combinations of opioids-nonopioids (e.g., oxycodone plus acetaminophen [Percocet, Vicodin]) because of concerns of overdose and resultant liver failure (U.S. FDA, 2009b; Harris, 2008).
Single-entity preparations have allowed broader use of oxycodone. Oxycodone is one of four opioid analgesics that are available in the United States in 12-hour modified-release form (OxyContin) for twice daily dosing; it is also approved for 8-hour dosing for patients who do not maintain pain relief for 12 hours (see Tamper-Resistant and Abuse-Deferrent Oral Opioid Formations on p. 378). OxyContin is available in 10, 15, 20, 30, 40, 60, 80, and 160 mg tablets. Doses of 60, 80 and 160 mg or any single dose of greater than 40 mg are approved for opioid-tolerant patients only. These are small tablets that are color-coded according to dose. See Patient Education Form IV-14 (pp. 572-573) on oxycodone with acetaminophen, Form IV-12 (pp. 568-569) on short-acting oxycodone, and Form IV-13 (pp. 570-571) on modified-release oxycodone at the end of Section IV.
OxyContin exhibits a biphasic release profile, with an initial peak at approximately 0.6 hours and a second peak at approximately 6.9 hours (Purdue Pharma, 2007). Analgesic onset occurs in most patients within 1 hour of administration, sooner than that produced by the delivery system in MS Contin (Kalso, 2005). The rapid-release phase of OxyContin has a half-life of 37 minutes and releases 38% of the total dose; the slower phase has a half-life of 6.2 hours and accounts for the remaining 62% of the dose (De Pinto, Dunbar, Edwards, 2006). The rationale for this formulation was to provide an extended duration of analgesia without significantly compromising the brisk onset of analgesia inherent in conventional short-acting products (Davis, Varga, Dickerson, et al., 2003). Early clinical trials demonstrated that patients with cancer pain or noncancer pain could be converted from other opioids and titrated to comfort using modified-release oxycodone as readily as with a short-acting opioid (Salzman, Roberts, Wild, et al., 1999).
The bioavailability of modified-release oxycodone is similar to short-acting oxycodone, and it is as effective at 12-hour dosing as the equivalent dose of short- acting oxycodone taken at 4-hour intervals (Davis, Varga, Dickerson, et al., 2003). A morphine to oxycodone ratio of 1.5:1 is considered equianalgesic.
Modified-release oxycodone has been found to be effective for a wide variety of types of pain (Riley, Eisenberg, Muller-Schwefe, et al., 2008) (see research below). Like modified-release morphine, it is used for continuous cancer pain and non–cancer-related pain of all types. It also has been used for treatment of some types of acute pain. The extensive literature on OxyContin includes the following types of pain:
• Acute postoperative pain (Blumenthal, Min, Marquardt, et al., 2007; Cheville, Chen, Oster, et al., 2001; de Beer, Winemaker, Donnelly, et al., 2005; Dorr, Raya, Long, et al., 2008; Ginsberg, Sinatra, Adler, et al., 2003; Kampe, Warm, Kaufmann, et al., 2004) (See also Trend in Oral Analgesics for Postoperative Pain in paragraphs that follow.)
• Cancer pain (Gralow, 2002; Pan, Zhang, Zhang, et al., 2007; Reid, Martin, Sterne, et al., 2006).
• Persistent noncancer pain (Portenoy, Farrar, Backonja, et al., 2007; Roth, Fleischmann, Burch, et al., 2000)
• Acute exacerbation of noncancer pain (Ma, Jiang, Zhou, et al., 2008)
• Neuropathic pain (Eisenberg, McNicol, Carr, 2006; Furlan, Sandoval, Mailis-Gagnon, et al., 2006; Gimbel, Richards, Portenoy, 2003; Watson, Moulin, Watt-Watson, et al., 2003).
Among the controlled trials conducted with OxyContin have been several demonstrating the potential utility of combination therapy. One randomized study of 338 patients with painful diabetic neuropathy, for example, found that those who took a combination of modified-release oxycodone and gabapentin required less rescue medication, experienced significantly better pain relief and sleep, and were less likely to discontinue treatment due to lack of therapeutic effectiveness than those who took gabapentin plus placebo (Hanna, O’Brien, Wilson, 2008). This finding was not surprising given data from another controlled trial (N = 87) demonstrating that modified-release oxycodone was more effective than gabapentin for relief of acute pain of herpes zoster (Dworkin, Barbano, Tyring, et al., 2009). Similarly, oxycodone has been included in multimodal postoperative pain treatment plans (see later in this chapter for trends in the use of oral opioids for postoperative pain).
Oral Oxymorphone
Oxymorphone is available in short-acting (Opana) and modified-release (Opana ER) oral formulations. Short-acting oxymorphone is available in 5 and 10 mg tablets, and modified-release oxymorphone is available in 5, 7.5, 10, 15, 20, 30, and 40 mg tablets. The tablets are color-coded according to dose.
Oxymorphone is more lipophilic than morphine, which may account for the slightly faster onset of action of its short-acting formulation (30 to 45 minutes) (Smith, 2009). A mean time to peak effect of 30 minutes has been associated with all doses of short-acting oxymorphone (Smith, 2009). The oral bioavailability of oxymorphone is 10% (Prommer, 2006b), and consumption of food at the time of dosing, particularly food with a high fat content, can increase the plasma concentration of oral oxymorphone (short-acting and modified-release) by as much as 50%. Oxymorphone is extensively metabolized in the liver and produces clinically inert metabolites (Smith, 2009). Oxymorphone’s half-life (7 to 11 hours) is longer than morphine’s (2 to 4 hours) (Chamberlin, Cottle, Neville, 2007), and, as a consequence, the time required to approach steady state is longer (Smith, 2009). Oxymorphone is more potent than morphine and oxycodone, and has suggested oral conversion ratios of 3:1 and 2:1, respectively (Smith, 2009).
Depending on pain severity, the initial dose of short-acting oxymorphone usually is 5 to 10 mg in opioid-naïve patients (Endo, 2006). Because food can increase the plasma concentration of oral oxymorphone, the drug should be taken on an empty stomach (1 hour before or 2 hours after a meal), and alcohol should be avoided as co-ingestion can increase serum levels up to 270% (Chamberlin, Cottle, Neville, 2007; Guay, 2007; Smith, 2009). These are important considerations when selecting an opioid; oral oxymorphone would not be a good choice in those who are unable to follow these dosing restrictions.
The use of short-acting oral oxymorphone is similar to morphine, hydromorphone, and oxycodone formulations. It is most appropriate for treatment of acute pain, such as postoperative pain (Gimbel, Ahdieh, 2004), and cancer- and non–cancer-related breakthrough pain (Sloan, Slatkin, Ahdieh, 2005). A randomized, placebo- controlled, parallel-group trial in 331 patients following abdominal surgery demonstrated that oxymorphone 5 mg or 10 mg provided comparable pain relief to oxycodone 10 mg (Aqua, Gimbel, Singla, et al., 2007). Another randomized controlled trial administered 5 mg of short-acting oxymorphone or placebo hourly as needed for up to 8 hours to 122 patients with mostly moderate-intensity pain following outpatient knee arthroscopy (Gimbel, Walker, Ma, et al., 2005). Patients in the oxymorphone group had significantly better pain relief, required less rescue medication, and were more likely to rate their pain relief as very good or excellent.
Modified-release oxymorphone (Opana ER) was approved for use in the United States in 2006 for the treatment of moderate to severe persistent pain. Its unique formulation allows the release of oxymorphone dependent on the rate of penetration of water into a hydrophilic matrix. Modified-release oxymorphone is dosed every 12 hours, and, like short-acting oxymorphone, steady state is reached after 3 days of every-12-hour dosing (Smith, 2009). As mentioned, oxymorphone is more potent than oxycodone. One study explored the dose equivalency of modified-release formulations of oxymorphone and oxycodone and established an equianalgesic dose ratio of 2:1 (oxymorphone was twice as potent as oxycodone) (Gabrail, Dvergsten, Ahdieh, 2004). See Patient Education Form IV-15 (pp. 574-575) on short-acting oxymorphone and Form IV-16 (pp. 576-577) on modified-release oxymorphone at the end of Section IV.
Although it is common to titrate the opioid dose using the short-acting formulation, and then switch to the modified-release formulation, one study demonstrated that it is safe to start with the lowest modified-release oxymorphone dose (5 mg every 12 hours) in opioid-naïve patients with moderate to severe noncancer pain and titrate from there (Rauck, Ma, Kerwin, et al., 2008). Similar to other opioids, research has shown that fixed dosing and rapid titration resulted in a higher incidence of adverse effects than gradual titration of modified-release oxymorphone (Brennan, 2009).
Modified-release oxymorphone has been found to be effective in the treatment of a variety of types of persistent cancer and noncancer pain (Brennan, 2009; Prager, Rauk, 2004; Sloan, Barkin, 2008; Sloan, Slatkin, Ahdieh, 2005; Slatkin, Tormo, Ahdieh, 2004). One study that evaluated modified-release oxymorphone in patients with persistent low back pain found that positive effects were less profound for those aspects of the pain likely to be neuropathic in origin (and described as cold, itchy, sensitive, tingling, and numb) than pains that were inferred to be nociceptive (and were described as sharp, aching, and deep) (Gould, Jensen, Victor, et al., 2009). This study lends support to the conclusion that is applied to all opioid drugs, i.e., that opioids are effective for neuropathic pain but may be relatively less effective for some pains of this type than pains conventionally considered to be nociceptive. (See Section I for more on nociceptive pain and neuropathic pain.)
The number of clinical trials that evaluate modified-release oxymorphone in diverse types of acute and persistent pain has been increasing. Studies have appeared in the following types:
• Cancer pain (Sloan, Slatkin, Ahdieh, 2005; Gabrail, Dvergsten, Ahdieh, 2004)
• Persistent low back pain (Gould, Jensen, Victor, et al., 2009; Hale, Ahdieh, Ma, et al., 2007; Hale, Dvergsten, Gimbel, 2005; Katz, Rauck, Ahdieh, et al., 2007; Penniston, Gould, 2009; Rauck, Ma, Kerwin, et al., 2008)
• OA pain (Kivitz, Ma, Ahdieh, et al., 2006; Matsumoto, Babul, Ahdieh, 2005; McIlwain, Ahdieh 2005; Rauck, Ma, Kerwin, et al., 2008)
Oxymorphone has been shown to be safe for long-term therapy (McIlwain, Ahdieh, 2005; Prager, Rauck, 2004; Rauck, Ma, Kerwin, et al., 2008). A multicenter, open-label, nonrandomized study (N = 126) evaluated opioid-naïve patients with noncancer pain during a 6-month gradual dose-titration and stabilization phase followed by a 5-month maintenance phase and found that modified-release oxymorphone provided effective, well-tolerated, and stable analgesia in 75% of the patients (Rauck, Ma, Kerwin, et al., 2008).
Although further research in special populations is needed, plasma concentrations of the drug and its metabolites have been shown to be 40% (mean) higher in older adults; therefore, initial low doses should be used in these patients, and titration should proceed cautiously (Smith, 2009). As with short-acting oxymorphone, Guay (2007) recommends beginning with the lowest dose of modified-release oxymorphone, 5 mg every 12 hours. Dose adjustments of oxymorphone are also likely to be necessary in patients with moderate renal and hepatic disease (Smith, 2009). Guay (2007) recommends avoiding oxymorphone entirely in patients with moderate to severe hepatic impairment. The drug was shown in one study to be removed by hemodialysis (Smith, 2009). There appears to be a low risk for interaction with concurrent medications that are metabolized by the CYP450 enzyme system, which may be a significant benefit in patients who are poor metabolizers or those who take multiple medications that rely on this enzyme system for metabolism, such as some antidepressants, beta blockers, antipsychotics, chemotherapeutic agents, and some other opioids (Adams, Pieniaszek, Gammaitoni, et al., 2005; Chamberlin, Cottle, Neville, 2007; McIlwain, Ahdieh 2005; Smith, 2009) (see Chapter 11 for more on cytochrome P450 enzymes and drug-drug interactions). The reader is referred to a 2009 journal supplement devoted to content on oxymorphone: Pain Med 10(Suppl 1).
Oral Hydromorphone
Oral short-acting hydromorphone is available in 2, 4, and 8 mg tablets and in a 1 mg/mL oral solution. Modified-release formulations of oral hydromorphone are available in Canada and Europe, and a once-daily formulation (Exalgo) (8, 12, and 16 mg tablets) was approved in the United States in 2010 (Gupta, Sathyan, 2007). The formulation uses a novel bilayer tablet system, the OROS® Push-Pull™ technology, to release hydromorphone at a relatively constant rate during a 24-hour period (Gardner-Nix, Mercadante, 2010). The bilayer core within the semipermeable tablet consists of a single drug layer (the “pull” layer) and a hydrophilic expanding compartment (the “push” layer). After tablet ingestion, fluid from the gastrointestinal (GI) tract forms a drug suspension and causes the push layer to expand. This exerts force on the pull layer and pushes the suspended drug out of the tablet through a laser-drilled orifice in the tablet shell membrane (Gardner-Nix, Mercadante, 2010).
Research in 31 healthy volunteers demonstrated that the pharmacokinetics of modified-release hydromorphone are linear and dose proportional (Sathyan, Xu, Thipphawong, et al., 2007a). Median peak concentration was noted between 12 and 16 hours with a mean terminal half-life of approximately 11 hours, both independent of dose. Steady state is reached after 48 hours of dosing (Gupta, Sathyan, 2007). The presence of food has little effect on the bioavailability of the drug (Sathyan, Xu, Thipphawong, et al., 2007b), and alcohol does not cause immediate release (“dose dumping”) of the drug (Sathyan, Sivakumar, Thipphawong, et al., 2008). Drug release is independent of pH and agitation (Gupta, Sathyan, 2007).
A study of opioid-tolerant patients with persistent cancer pain (N = 73) or persistent noncancer pain (N = 331) stabilized the patients on their previous opioid, converted this dose to modified-release hydromorphone, then titrated to optimal dose using a stepwise approach, which was then maintained for 2 weeks (Palangio, Northfelt, Portenoy, et al., 2002). The majority of patients reached a stable dose of modified-release hydromorphone quickly (mean 12.1 days), and most required no or few steps to achieve it. The most common adverse effects were nausea and constipation. A morphine to hydromorphone conversion ratio of 5:1 was used, but the researchers reinforced the principle of decreasing the equianalgesic dose of the new opioid by 25% to 50% until research establishes differently (see Chapter 18). This study also suggested that direct conversion from another opioid to modified-release hydromorphone could be done without the intermediate step of titration with short-acting hydromorphone first. In other clinical trials, the once-daily hydromorphone has also been well tolerated with an adverse effect profile similar to other short- and modified-release opioid analgesics, such as morphine and oxycodone (Cousins, 2007; Gardner-Nix, Mercadante, 2010; Gupta, Sathyan, 2007; Hale, Tudor, Khanna, et al., 2007; Hanna, Thipphawong, 118 Study Group, 2008; Wallace, Thipphawong, 2007; Wirz, Wartenberg, Elsen, et al., 2006).
Long-term use of modified-release hydromorphone has been shown to be effective and safe. A multicenter open-label study (N = 388) administered modified-release hydromorphone for 274 days to patients with persistent cancer or noncancer pain (Wallace, Moulin, Rauck, et al., 2009). The median daily dose was 48 mg at 6, 9, and 12 months, with 75.9% of patients reporting overall treatment as good to excellent at 12 months. The most common adverse effects were nausea and constipation. A ratio of 5:1 (5 mg of morphine equivalents to 1 mg of hydromorphone) was used to convert opioid-tolerant patients with persistent noncancer pain from other oral opioids to modified-release hydromorphone without loss of efficacy or increase in adverse effects (Wallace, Rauck, Moulin, et al., 2007). (See Exalgo package insert for patient medication guide.)
Trend in Oral Analgesics for Postoperative Pain
With the current trend toward early discharge of patients after relatively major surgical procedures, consideration must be given to more aggressive pain treatment in the home setting than is possible with the traditional fixed combination opioid/nonopioid analgesics. The fixed dose of the nonopioid in these preparations limits the number of tablets that patients may take in a 24-hour period without exceeding the maximum safe daily dose (e.g., 4000 mg of acetaminophen). Single-entity opioids, such as morphine, oxycodone, and oxymorphone, are better choices if the anticipated severity or persistence of the pain increases the likelihood that dose titration will be needed.
Research has addressed the safety of early postoperative oral analgesia. A randomized controlled trial (N = 227) showed that early oral analgesia (first postoperative day) with scheduled 20 mg doses of short-acting morphine every 4 hours and an additional 10 mg dose every 2 hours PRN was safe and effective, producing similar analgesia as IV PCA with a basal rate after intraabdominal surgery (Pearl, McCauley, Thompson, et al., 2002). Others have found similar positive results with this approach following orthopedic surgery (Zaslansky, Eisenberg, Peskin, et al., 2006).
Modified-release opioids, used most often for patients with persistent cancer or noncancer pain, are increasingly prescribed in selected patients in the postoperative setting (Holt, Viscusi, Wordell, 2007; Pasero, McCaffery, 2007). The modified-release opioid formulations, e.g., MS Contin, OxyContin, and Opana ER, are FDA-approved for postoperative pain treatment in patients who were taking the particular opioid prior to surgery. They should also be considered for some opioid-naïve patients who are undergoing major surgeries that are associated with moderate to severe pain and are likely to require repeated doses of analgesics over several days. Modified-release opioids are not appropriate for pain that is mild or not expected to persist for an extended period of time.
Oxycodone (OxyContin) is the most widely studied modified-release opioid for treatment of postoperative pain. One study randomized 40 patients to receive either 20 mg of modified-release oxycodone or placebo preoperatively and every 12 hours postoperatively, in addition to IV morphine via PCA and IV acetaminophen (1 g) for 2 days following lumbar discectomy (Blumenthal, Min, Marquardt, et al., 2007). Those who received oxycodone consumed significantly less morphine; had significantly lower pain scores during rest, coughing, and with movement; experienced less nausea and vomiting and earlier recovery of bowel function; and reported higher satisfaction with their pain treatment than those who received placebo. Another study (N = 59) demonstrated that, compared with placebo, modified-release oxycodone given preoperative and every 12 hours postoperatively produced better pain relief, greater range of motion and quadriceps strength during physical therapy, and a shorter length of hospital stay by 2.3 days in patients following total knee arthroplasty (Cheville, Chen, Oster, et al., 2001). Others have found similar superior pain relief, reduced supplemental analgesic requirements, and cost savings with a range of doses (10 to 30 mg) of pre- and postoperative modified-release oxycodone following knee or hip replacement (de Beer, Winemaker, Donnelly, et al., 2005; Dorr, Raya, Long, et al., 2006) and breast surgery (Kampe, Warm, Kaufmann, et al., 2004).
Patients have been rapidly converted from IV opioids to modified-release oxycodone following major surgery. A multicenter, open-label study of 189 patients who were receiving IV PCA opioid for 12 to 24 hours following abdominal, orthopedic, or gynecologic surgery were given an initial dose of modified-release oxycodone at 12 hours postoperatively (Ginsberg, Sinatra, Adler, et al., 2003). The initial dose of oxycodone was calculated by multiplying the amount of IV morphine used in the previous 24 hours by a conversion factor of 1.2 to determine the daily dose of oxycodone, which was then divided by 2 to determine the every-12-hour dose of oxycodone (matched with available tablet strengths). This calculated amount of modified-release oxycodone plus breakthrough doses of NSAIDs or short-acting oxycodone every 4 hours PRN was well tolerated and provided satisfactory pain control for seven days postoperatively.
Though further research is needed, positive results were found with the use of modified-release oxymorphone for postoperative pain. One study randomized patients to receive modified-release oxymorphone 20 mg every 12 hours or placebo following knee arthroplasty (Ahdieh, Ma, Babul, et al., 2004). IV oxymorphone PCA was used for rescue analgesia. Patients who received modified-release oxymorphone had significantly better pain control and used significantly less rescue analgesia than those who received placebo. Treatment was well tolerated.
Tamper-Resistant and Abuse-Deterrent Oral Opioid Formulations
Several oral opioid formulations designed to be tamper-resistant and deter abuse were in various phases of investigation, development, and approval at the time of publication (Fleming, Noonan, Wheeler, et al., 2008; Jones, Johnson, Wagner, et al., 2008; Katz, Adams, Chilcoat, et al., 2007; Katz, Sun, Fox, et al., 2008; King Pharmaceuticals, 2008b; Medical News Today, 2008). A unique modified-release formulation (ALO-01, EMBEDA) contains pellets of morphine and sequestered naltrexone, an opioid antagonist (Johnson, Sun, Stuaffer, et al., 2007). Embeda was approved in 2009 and is available in capsules containing morphine/naltrexone in the following strengths: 20 mg/0.8 mg, 30 mg/1.2 mg, 50 mg/2 mg, 60 mg/2.4 mg, 80 mg/3.2 mg, and 100 mg/4 mg (King Pharmaceuticals, 2009). When taken as directed, the naltrexone remains sequestered in a pellet core and passes through the GI tract without significant absorption; however, if the product is crushed, chewed, or dissolved, the naltrexone is released and free to reverse opioid effects. A phase II multicenter, randomized-controlled, cross-over trial of 113 patients with moderate to severe OA pain was conducted to compare ALO-01 and modified-release morphine (Kadian) (Katz, Sun, Fox, et al., 2008). After a washout period to induce pain flare, patients were titrated to comfort with Kadian then randomized to receive either Kadian or ALO-01 for 14 days. Patients were then treated with Kadian for 7 days followed by a cross-over to the other study medication (either Kadian or ALO-01) for 14 days. Most of the patients (Kadian, 80% and ALO-01, 92%) rated the analgesics as good to excellent. Morphine exposure at steady state was similar, and plasma naltrexone levels were below quantification and had no effect on analgesia in those who took ALO-01. Adverse effects were similar and typical of opioids. A crossover study randomized 113 patients with moderate-to-severe OA pain to receive modified-release morphine (Kadian) or ALO-01 and demonstrated similar morphine exposure at steady state with the two formulations, and again the sequestered naltrexone had no effect on pain scores (Katz, Sun, Johnson, et al., 2009). When tested in recreational opioid users, crushed ALO-01 reduced euphoria due to naltrexone absorption and was no more desirable than intact morphine (Jones, Johnson, Wagner, et al., 2008).
OxyContin (12-hour modified-release oxycodone) is available in a novel abuse-deterrent formulation. The drug is contained within a hard gelatin capsule designed to resist tampering, such as crushing or dissolving in alcohol; other similar oxycodone formulations are in development (Fleming, Noonan, Wheeler, et al., 2008; Gordon, 2008; King Pharmaceuticals, 2008b). Research in healthy volunteers demonstrated that the technology successfully protected the drug from rapid release in various tampering simulations, such as chewing (Fleming, Noonan, Wheeler, et al., 2008). Plasma levels were lower when the drug was taken in a fasted state.
Oral Transmucosal
The oral mucosa functions similar to the skin as a barrier to dangerous substances. It differs from the skin in that it is significantly more vascular, more permeable to drugs with similar properties, and has a lower drug depot (storage) effect than the skin. Although mucosal drug absorption also involves hydrophilic pathways, absorption is optimized with drugs that are lipid soluble, such as fentanyl, buprenorphine, and methadone (Reisfield, Wilson, 2007). Three areas within the mouth can be used for oral transmucosal drug delivery: the sublingual, buccal, and gingival areas. These areas usually are regarded as separate routes of administration and are studied and discussed separately. Drug development for oral transmucosal routes of delivery has focused on acute pain management, primarily breakthrough pain, because a relatively rapid onset of effect can be achieved using these formulations.
Sublingual
Use of the sublingual route involves placing the drug under the tongue for absorption through the oral mucosa into the systemic circulation. Because the drug is absorbed directly into systemic circulation, the first-pass effect is avoided (Zhang, Zhang, Streisand, 2002). Of all the areas within the mouth for oral transmucosal drug administration, the sublingual area appears to be the highest in drug permeability. It is only 25% as thick as the buccal mucosa and, unlike the gingival mucosa, is nonkeratinized (Reisfield, Wilson, 2007). The sublingual route is an alternative when oral, parenteral, and rectal routes are unavailable or impractical (Reisfield, Wilson, 2007).
Few opioids have been administered by the sublingual route. Hospice nurses report success with morphine by the sublingual route (Robinson, Wilkie, Campbell, 1995), but it is thought that this is related to the fact that the drug is eventually swallowed; sublingual absorption of morphine and other hydrophilic drugs is poor (Hanks, Cherny, Fallon, 2004; Reisfield, Wilson, 2007).
The effects of lipid solubility, oral cavity pH, and drug contact time on sublingual absorption of various opioids and naloxone have been studied. Normal saliva pH is 6.5, but can vary with mouth breathing, nutritional status, food or beverage consumption, vomiting, stomatitis, and decreased salivary flow. Salivary pH also varies by region of the mouth (Reisfield, Wilson, 2007). Absorption of drugs is improved with high lipid solubility and an alkaline environment. Compared with morphine at pH 6.5 (18% absorption), the more lipophilic opioids—buprenorphine (55%), fentanyl (51%), and methadone (34%)—were absorbed to a significantly greater extent, whereas levorphanol, hydromorphone, oxycodone, heroin, and the opioid antagonist naloxone were not (Weinberg, Inturrisi, Reidenberg, et al., 1988). At a pH of 8.5, methadone absorption increased to 75%. Drug absorption was not affected by concentration, but was affected by contact time. Sixty percent of the maximum methadone and fentanyl absorption at 10 minutes was seen at 2.5 minutes of contact time; maximum buprenorphine absorption was complete by 2.5 minutes of contact time. Adverse effects were minor (bitter taste, burning sensation, lightheadedness), with fentanyl and buprenorphine associated with the lowest incidence.
Sublingual oxycodone and hydromorphone have been studied to a very limited degree (Reisfield, Wilson, 2007). An alkalinized oxycodone sublingual spray had a bioavailability of 70% in an animal model. Hydromorphone in healthy volunteers had a bioavailability of only 25%. The injectable forms of sufentanil and alfentanil have been used sublingually for breakthrough pain, but have not been studied (Gardner-Nix, 2001a; Hanks, Cherny, Fallon, 2004); with their high lipophilicity and potency (which exceeds fentanyl), they are a good theoretical choice, but the lack of commercially available products makes them impractical (Reisfield, Wilson, 2007). Fentanyl and methadone are well absorbed sublingually, but no preparations are commercially available (see the following discussion on oral transmucosal fentanyl) (Hanks, Cherny, Fallon, 2004); a sublingual fentanyl tablet now is available in some countries (Lennernas, Hedner, Holmberg, et al., 2005). A sublingual buprenorphine wafer has been approved for use in treatment of opioid addiction (Heit, Gourlay, 2008). A buprenorphine liquid product is available in other countries and may provide relief of mild to moderate pain. Sublingual buprenorphine absorption occurs within 3 to 5 minutes, bioavailability is 51%, and peak plasma concentrations generally occur at approximately 60 minutes (Johnson, Fudala, Payne, 2005).
An advantage of the sublingual route is that administration requires little expertise, preparation, or supervision (Reisfeld, Wilson, 2007; Stevens, Ghazi, 2000). Unfortunately, the sublingual route currently has limited value for the administration of most opioids because formulations are lacking, absorption is poor for most opioids, and high doses cannot be given. In addition, proper administration is seldom possible because the drug must be in contact with the oral mucosa at least 5 minutes, a length of time most patients find intolerable (Reisfield, Wilson, 2007).
Buccal and Gingival
The buccal route of administration involves placement of the drug, usually in tablet form, inside the mouth between the mucosal surface of the cheek and the gum of the upper molars (see buccal fentanyl in following paragraphs). The gingival route involves placing the tablet form between the upper lip and the gum of the incisors. Of all areas in the mouth for oral transmucosal drug administration, the gingival route appears to be the lowest in drug permeability (Reisfield, Wilson, 2007). If the buccal or gingival routes must be used, the site should be rinsed with water to remove residues of the drug after absorption. If these routes are used repeatedly, the site should be rotated because irritation of the mucous membrane can occur.
Oral Transmucosal Fentanyl for Breakthrough Pain
Breakthrough pain (sometimes called pain flare, episodic pain, or transient pain) is defined as a transitory exacerbation of pain in a patient who has relatively stable and adequately controlled baseline pain (Portenoy, Forbes, Lussier, et al., 2004) (see Chapter 12 for a detailed discussion of breakthrough pain). The ideal medication for breakthrough pain has been described as one with a fast onset, relatively short duration of action, and minimal adverse effects (Zeppetella, 2008). Transmucosal delivery of a lipophilic and potent drug such as fentanyl meets these characteristics. Fentanyl had been incorporated into three products approved in the United States for the treatment of breakthrough cancer pain at the time of publication; numerous others are in development. Although indicated for cancer-related breakthrough pain, these products are widely used for breakthrough pain in noncancer pain syndromes as well (Prime Therapeutics, 2007).
Oral transmucosal fentanyl citrate (OTFC, Actiq®) is provided as a solid matrix or lozenge on a plastic stick (Figure 14-1, A). The medication is intended to be dissolved by saliva and absorbed through all oral mucosal surfaces. An effervescent fentanyl buccal tablet (FBT, Fentora®) is designed for placement against the buccal mucosa—between the upper gum and cheek—until it is dissolved (Figure 14-1, B). The newest formulation, BEMA (BioErodible MucoAdhesive; Onsolis) is a microadhesive polymer disk, about the size of a nickel, that contains fentanyl and is designed to stick to the oral mucosa (inside of the cheek) and dissolve within 15 to 30 minutes (Blum, Breithaupt, Hackett, et al., 2008).
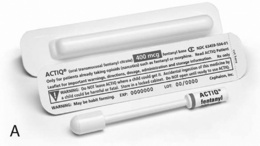
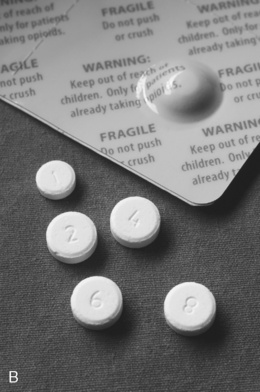
Figure 14-1 A, Oral transmucosal fentanyl citrate (OTFC). B, Fentanyl buccal tablet (FTB). Cephalon, Inc.
The safety guidelines for the oral transmucosal products are strict, and the titration recommendations are conservative (see the following). Although safe use in opioid-naïve patients with severe persistent noncancer pain has been described (Collado, Torres, 2008), these products have been approved for treatment of breakthrough pain in opioid-tolerant cancer patients only (Cephalon, 2007, 2008), and they should not be prescribed to patients with minimal or no existing opioid treatment unless appropriate monitoring is available. Titration usually should begin at the lowest dose (Cephalon, 2007, 2008). Despite using the same drug by a similar route, OTFC and FBT are not equivalent or interchangeable. These are important drugs for the treatment of breakthrough pain and are safe when used as directed; however, their potency must be respected, and clinicians, patients, and families must be well-versed in safe utilization.
OTFC is available in 200, 400, 600, 800, 1200, and 1600 mcg strengths (Cephalon, 2007). Individual titration, usually beginning with the lowest dose (200 mcg), is necessary. Dose adjustment on the basis of age alone is not required (Kharasch, Hoffer, Whittington, 2004), although elimination in older adults is prolonged (Gordon, 2006). To administer OTFC, the patient is instructed to hold onto the stick and place the lozenge between the gum and cheek. The stick is used to move and twirl the lozenge around the oral mucosa, particularly between the gums and cheek and above and below the tongue, so that it dissolves in the saliva. Patients should be told not to suck on the lozenge as one would suck on a candy lollipop; this will result in much of the drug being swallowed (oral route), negating the benefits of exposure to direct systemic circulation that the oral transmucosal route offers. Further, clinicians should not refer to OTFC as a lollipop, sucker, or popsicle, as this is not only misleading but can result in family members, particularly children in the home, misunderstanding that this is a medication (not candy) that should be consumed by the patient only. Biting or chewing will cause a greater proportion to be swallowed, also resulting in decreased effectiveness. It has been suggested that the patient swish the fentanyl-containing saliva around the mouth prior to swallowing to enhance oral mucosal absorption (Gordon, 2006). If pain relief is insufficient after 15 to 25 minutes and the entire lozenge has been consumed, a second lozenge may be used; there are no published data about the use of more than two lozenges successively, and this is not recommended (Cephalon, 2007) (see Box 14-2 for complete dosing recommendations). See Patient Education Form IV-4 (pp. 551-552) on oral transmucosal fentanyl at the end of Section IV.
Guidelines
Box 14-2
Dosing of Oral Transmucosal Fentanyl Citrate (Actiq)
1. The initial dose of Actiq to treat episodes of breakthrough pain is 200 mcg.
2. Instruct the patient to place the Actiq unit in the mouth between the cheek and lower gum and to occasionally move the drug matrix from one side to the other using the handle. The Actiq unit should be sucked and not chewed. Chewing the matrix could result in lower peak concentrations and efficacy.
3. The Actiq unit should be consumed over a 15-minute period. Longer or shorter consumption times may produce less efficacy than reported in clinical trials.
4. Tell the patient that if pain relief occurs or signs of excessive opioid effects such as sedation appear before the unit is consumed, the dose unit should be removed from the patient’s mouth immediately, disposed of properly (see below), and subsequent doses should be decreased.
5. From this initial dose, closely follow the patient and change the dose level until the patient reaches a dose that provides adequate analgesia using a single Actiq dose unit per breakthrough episode.
6. Ask patients to record their use of Actiq over several episodes of breakthrough pain and review their experience with their prescriber to determine if a dose adjustment is warranted.
Redosing within a single episode
Until the appropriate dose is reached, patients may find it necessary to use an additional Actiq unit during a single episode.
1. Redosing may start 15 minutes after the previous unit has been completed (30 minutes after the start of the previous unit).
2. While patients are in the titration phase and consuming units that individually may be subtherapeutic, no more than 2 units should be taken for each individual breakthrough pain episode.
If treatment of several consecutive breakthrough pain episodes requires more than 1 Actiq per episode, consider an increase in dose to the next higher available strength.
At each new dose of Actiq during titration, it is recommended that 6 units of the titration dose be prescribed.
Evaluate each new dose of Actiq used in the titration period over several episodes of breakthrough pain (generally 1 to 2 days) to determine whether it provides adequate efficacy with acceptable adverse effects.
The incidence of adverse effects is likely to be greater during this initial titration period compared with later, after the effective dose is determined.
1. Increase the dose of Actiq when patients require more than 1 dose unit per breakthrough pain episode for several consecutive episodes.
2. When titrating to an appropriate dose, prescribe small quantities (6 units) at each titration step.
3. Once a successful dose has been found (i.e., average episode is treated with a single unit), patients should limit consumption to 4 or fewer units per day.
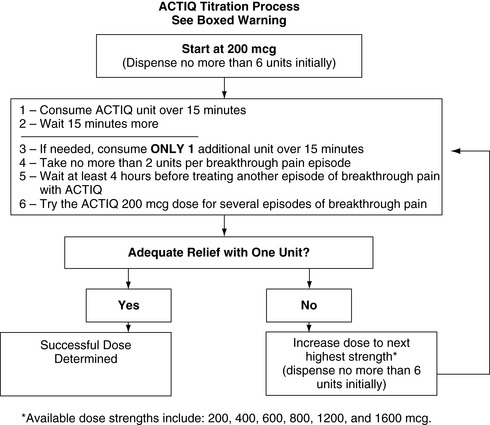
4. Consider increasing the around-the-clock opioid dose used for persistent pain in patients experiencing more than four breakthrough pain episodes daily.
Clinicians should not refer to OTFC as a lollipop, sucker, or popsicle as this is not only misleading but can result in family members, particularly children in the home, misunderstanding that this is a medication that should be consumed by the patient only.
This box describes the initial dosing, titration, and subsequent dosing of oral transmucosal fentanyl citrate (Actiq). See prescription information insert for other information, such as boxed warnings, contraindications, and storage.
From Pasero, C., & McCaffery, M. (2011). Pain assessment and pharmacologic management, pp. 382-383, St. Louis, Mosby. Data from Cephalon. (2007). Actiq prescribing information. Cephalon Inc, Frazer, PA. Available at http://www.actiq.com/pdf/actiq_package_insert_4_5_07.pdf. Accessed April 13, 2008. Pasero C, McCaffery M. May be duplicated for use in clinical practice.
After administration, a portion of the fentanyl diffuses across the oral mucosa (25%) and the rest is swallowed and partially absorbed through the stomach and the intestine (75%). In total, OTFC has about 50% bioavailability (Mystakidou, Katsouda, Parpa, et al., 2005). The fentanyl absorbed from the mucosa rapidly crosses the blood-brain barrier to the CNS, its primary site of action (Mystakidou, Katsouda, Parpa, et al., 2005).
There is no predictable dose relationship between background opioid dose for persistent pain and an effective OTFC dose, and it may take several days to determine the optimal dose (Coluzzi, Schwartzberg, Conroy, et al., 2001). This observation, based on safety and efficacy trials, contradicts the usual assumption that an effective dose of breakthrough medication is a percentage of the total daily dose (see Chapter 12) (Mercadante, Villari, Ferrera, et al., 2007). A small study (N = 25) tested this conclusion with a fixed dose of OTFC based on the daily morphine dose. The results suggested that patients receiving more than 180 mg of oral morphine equivalents can be safely started at 600 mcg of OTFC (Mercadante, Villari, Ferrera, et al., 2007). It should be emphasized that this was a small, nonblinded study, but it is the first clinical trial to directly confront the issue of prolonged conventional titration.
Compared with oral (i.e., swallowed) fentanyl administration, OTFC yields higher and more rapidly attained plasma concentrations and greater bioavailability. These characteristics provide evidence that OTFC passes by mucosal transport directly into the systemic circulation without undergoing first-pass metabolism in the liver. Based on studies that show similar pharmacokinetics in single vs. multiple doses (Gordon, 2006), it appears that there is no depot effect in the mouth, in contrast to transdermally administered fentanyl. The clinical implications of this are that the onset of analgesia will be more consistent and rapid than by the oral or transdermal route of administration and adverse effects will dissipate quickly when administration is discontinued.
Although there is considerable variation in the time-action relationship following administration of a dose, OTFC usually has a more rapid onset, earlier peak effect, and shorter duration of action than a conventional short-acting oral opioid. With a typical onset of 30 to 45 minutes and a peak effect of 60 minutes or more, the oral drug has a profile that is likely to be poorly matched to the usual timing of a breakthrough pain episode (Zeppetella, 2008). Ashburn, Fine, and Stanley (1989) first reported effectiveness in the use of OTFC to manage breakthrough pain in a patient with metastatic carcinoma of the lung. They cited fentanyl’s short duration (considered a drawback in the management of continuous pain) as an advantage in treating breakthrough pain because it allowed patients to avoid excessive sedation and other adverse effects associated with longer-acting oral opioids typically used for breakthrough cancer pain.
In clinical trials for OTFC in breakthrough pain, approximately 75% of patients were able to titrate to an effective transmucosal fentanyl dose, and these patients reported a faster onset than their usual oral breakthrough pain medication, with equal or better effectiveness, and acceptable adverse effects (Mystakidou, Katsouda, Parpa, et al., 2005; Mercadante, Villari, Ferrera, et al., 2007; Coluzzi, Schwartzberg, Conroy, et al., 2001). Analysis of the breakthrough pain experience of patients using OTFC concluded that OTFC had a positive impact on quality of life, particularly enjoyment of life and improvements in mood and ability to work (Taylor, Webster, Chun, et al., 2007). Another small study demonstrated that OTFC is an effective analgesic with no increase in adverse effects compared with placebo for burn patients during painful dressing changes (MacIntyre, Margetts, Larsen, et al., 2007).
Adverse effects include sedation, dizziness, nausea, constipation, and itching, all with an incidence of less than 15% (Bennett, Burton, Fishman, et al., 2005b). OTFC contains sugar and has been associated with dental caries. Patients should be instructed in good oral care, and diabetics should be informed that each OTFC lozenge contains 2 grams of sugar (Laverty, 2007; Gordon, 2006).
Potential cost benefits have been reported with the use of OTFC. Patients with non–cancer-related pain states who had a history of emergency department (ED) visits or hospitalizations for pain-control issues substituted OTFC for their usual breakthrough pain medication (Tennant, Herman, 2002). After 3 months, over 78% of the patients (N = 90) estimated that, based on previous experience, they had avoided at least one ED visit for pain control. Similar results have been found for cancer patients (Burton, Driver, Mendoza, et al., 2004) and sickle cell patients (Shaiova, Wallenstein, 2004).
Fentanyl Buccal Tablet (FBT)
An effervescent fentanyl tablet designed to rapidly dissolve when placed between the upper rear molar and cheek (buccal mucosa) approved for breakthrough cancer pain (fentanyl buccal tablet; FBT; Fentora) (see Figure 14-1, B) is available in 100, 200, 300, 400, 600, and 800 mcg tablets. Clinicians should not refer to FBT as a candy” as this is not only misleading but can result in family members, particularly children in the home, misunderstanding that this is a medication (not a candy) that should be consumed by the patient only.
FBT is clinically distinct from OTFC. Absorption through the oral mucosa is slightly faster, and the amount of fentanyl reaching the systemic circulation is higher with FBT. The effervescent design of the tablet increases pH, which enhances tablet dissolution and membrane permeability (Taylor, 2007). Fentora is twice the potency of OTFC, and titration of FBT usually should begin at the lowest dose (100 mcg). If pain relief is insufficient after 15 minutes and the entire tablet has been consumed, a second tablet may be used (Cephalon, 2008). Similar to OTFC, there are no data concerning a sequencing of more than 2 tablets, and it is not recommended. If a patient requires a higher dose, two tablets may be placed on each side of the mouth. It is estimated that a 30% smaller dose of FBT achieves systemic exposure equivalent to OTFC (Darwish, Kirby, Robertson, et al., 2007). It is, therefore, recommended that when switching from OTFC to FBT, the starting dose of FBT should be adjusted (Cephalon 2008) (see Box 14-3 for complete dosing recommendations and see Patient Education Form IV-3, pp. 549-550, on fentanyl buccal tablets).
Clinical trials with cancer patients with breakthrough pain have shown FBT to be superior to placebo in both pain intensity difference (pain score before and after intervention) and pain relief (Slatkin, Xie, Messina, et al., 2007; Blick, Wagstaff, 2006). One study evaluating FBT administered sublingually demonstrated nearly equivalent pharmacokinetics compared with buccal administration (Darwish, Kirby, Jiang, et al., 2008). It has not been subjected to a clinical trial, but sublingual may be an alternative route for some patients.
Adverse effects of FBT are similar to OTFC. The importance of correct prescribing and teaching patients proper use is stressed. The United States FDA issued an advisory about unsafe prescribing and use of Fentora® after serious toxicity and deaths had been reported (U.S. FDA 2007b).
BioErodible MucoAdhesive (BEMA) Patch
At the time of publication, the most recently approved oral transmucosal fentanyl formulation in the United States was the BEMA patch. BioErodible MucoAdhesive (BEMA; Onsolis), is a bilayer polymer disk, about the size of a nickel, which contains fentanyl and is designed to stick to the oral mucosa (inside of the cheek) and dissolve within 15 to 30 minutes and provide rapid analgesia via systemic circulation (Blum, Breithaupt, Hackett, et al., 2008). The formulation was approved in July 2009 for the treatment of breakthrough cancer pain in opioid-tolerant individuals only (U.S. FDA, 2009a). It is available in 200, 400, 600, 800, and 1200 mcg strengths.
One benefit of this formulation is that the mucoadhesive polymer delivery system helps to control the mucosal surface application area and time in contact with the mucosa to optimize drug delivery. Two phase 1 open-label, randomized, crossover studies in 12 healthy volunteers evaluated the pharmacokinetics of BEMA (Vasisht, Stark, Finn, 2008). Absolute bioavailability was greater than 70% for both a single- and multi-unit (4 × 200 mcg) regimen, of which a high percentage (51%) of the fentanyl was absorbed through the oral mucosa.
Several clinical trials have demonstrated efficacy and safety of BEMA in patients with cancer pain (Blum, Breithaupt, Hackett, et al., 2008; Blum, Finn, 2008; North, Kapoor, Bull, et al., 2008; Slatkin, Hill, Finn, 2008). One study evaluated the breakthrough pain experience in 80 cancer patients on stable opioid doses who had participated in a previous double-blind, cross-over study in which their optimal BEMA dose for breakthrough pain treatment was established (North, Kapoor, Bull, et al., 2008). After treatment with their optimal BEMA dose or placebo, patients recorded their pain intensity at multiple intervals for 60 minutes after administration during up to nine breakthrough pain episodes. BEMA demonstrated significantly greater pain relief than placebo at all time intervals and through 60 minutes. Other studies of patients with breakthrough pain have found only 10% of patients required additional rescue medication and 85% rated their breakthrough treatment with BEMA as “good” or better (North, Kapoor, Bull, et al., 2008). A double-blind, randomized, placebo-controlled, multiple cross-over study and an open-label study showed that titration to optimal BEMA dose for breakthrough pain was well tolerated in cancer patients receiving concomitant long-term opioid therapy (Blum, Breithaupt, Hackett, et al., 2008). Adverse effects were typical of opioids: somnolence (6%), nausea (5.3%), dizziness (4.6%), and vomiting (4%). A multicenter, open-label study reported similar incidences of adverse effects (Slatkin, Hill, Finn, 2008). Three patients (1.4%) experienced mild stomatitis, which did not necessitate discontinuation of treatment. An abstract describing over 60,000 doses of BEMA taken for breakthrough pain in three different clinical trials reported a low incidence (4.6%) of application site reactions, including stomatitis (1.6%) (Blum, Finn, 2008).
Intranasal
The intranasal route has been used for centuries to administer a number of different drugs. It may be attractive as a noninvasive, rapid-onset, short-acting, and often convenient alternative to parenteral and oral opioids (Shelley, Paech, 2008).
The nasal mucosa has a surface area of 150 to 180 square centimeters of thin, permeable membrane with blood flow greater per cubic centimeter than muscle, brain, and liver (Shelley, Paech, 2008). This large surface area has uniform temperature and high permeability, and it provides easy access to extensive vasculature for rapid systemic absorption that eliminates the hepatic first-pass effect, potentially providing greater bioavailability compared with oral administration (Stoker, Reber, Waltzman, et al., 2008). In addition, there is evidence from animal studies that intranasal opioids may bypass the blood-brain barrier by a direct route to the brain via the olfactory nerve (Westin, Bostrom, Grasjo, et al., 2006).
Although the nasal mucosal surface provides a relatively large surface area, the volume of drug-containing fluid that can be administered per dose is limited (0.15 mL on each side). The drug needs to remain in contact with the mucosal surface long enough for absorption to occur. Chitosan, a naturally occurring substance that adheres to the mucosal surface and may enhance absorption, has been suggested as a vehicle for intranasal drug administration (Charlton, Davis, Illum, 2007; Pavis, Wilcock, Edgecombe, et al., 2002). A newer proprietary formulation uses pectin for the same purpose. Drug concentration and pH of the nasal environment also affect absorption (Dale, Hjortkjaer, Kharasch, 2002; Wolfe, 2007).
The only intranasal opioid approved in the United States at the time of publication is the mixed agonist-antagonist butorphanol (Stadol NS), which has been used for migraine (Rapoport, Bigal, Tepper, et al., 2004) and for postoperative pain (Dale, Hjortkjaer, Kharasch, 2002). Other opioids have been used by this route for procedural (Finn, Wright, Fong, et al., 2004; Wolfe, 2007), dental (Christensen, Cohen, Mermelstein, et al., 2008; Wermeling, Grant, Lee, et al., 2005), postoperative (Mathieu, Cnudde, Engelman, et al., 2006; Stoker, Reber, Waltzman, et al., 2008), and breakthrough pain (Jackson, Ashby, Keech, 2002; Fitzgibbon, Morgan, Dockter, et al., 2003) and for prehospital use by emergency services (Rickard, O’Meara, mcgrail, et al., 2007; Wolfe, 2007). It has also been suggested as an alternative to IV PCA (Miaskowski, 2005; Shelley, Paech, 2008). Intranasal opioids in use and under investigation include morphine, hydromorphone, butorphanol, sufentanil, alfentanil, and fentanyl.
As with other routes of administration, drug selection and proper technique are required for safety and efficacy. In clinical trials, nasal opioids have been administered using a simple mechanical device that delivers a metered-dose spray. In general, the more lipophilic opioids are better absorbed across mucosal surfaces, but this characteristic does not seem to apply to the nasal mucosa (Dale, Hjortkjaer, Kharasch, 2002).
Both morphine and hydromorphone have shown rapid absorption when administered intranasally. Hydromorphone has a peak time to effect of 20 minutes and is approximately 55% bioavailable (Cashman, 2008). A multicenter, open-label study compared single doses (2 mg, 4 mg, 6 mg, 8 mg, and 10 mg) of intranasal hydromorphone in the treatment of acute trauma pain in the ED and reported a mean decrease in pain intensity from baseline at 30 minutes of 24% in those who received 2 mg and 39% to 44% for those who received the higher doses (Wermeling, Clinch, Rudy, et al., 2009). Adverse effects were typical of opioids and of mild to moderate intensity. A randomized controlled clinical trial (N = 187) using an intranasal morphine-chitosan formulation (Rylomine) postoperatively following bunionectomy in 187 opioid-naïve patients demonstrated that the intranasal analgesic was comparable to IV morphine for both pain relief and systemic adverse effects (Stoker, Reber, Waltzman, et al., 2008). The minimally effective morphine dose was 7.5 mg, and 30 mg produced unacceptable adverse effects. The optimal dosing interval for 7.5 mg was 1 to 2 hours and for 15 mg was 2 to 3 hours. The researchers described the capacity of each nostril to hold only 150 to 200 microliters as a safety mechanism of this route of administration; it requires approximately 15 minutes for the drug to clear the nasal passages, and any attempt to instill additional drug would result in it being swallowed or dripping out of the nose.
A pilot study of a different morphine formulation administered intranasally (40 mg single dose) for breakthrough pain in cancer patients demonstrated meaningful pain relief at approximately 7 minutes (Fitzgibbon, Morgan, Dockter, et al., 2003). Intranasal fentanyl was shown to be comparable to IV morphine in a randomized controlled trial in a prehospital emergency setting (Rickard, O’Meara, mcgrail, et al., 2007) and to produce the same or better analgesia than oral morphine in 75% of hospice patients in a pilot study (Zeppetella, 2000a). A randomized crossover study found that intranasal fentanyl produced equivalent pain relief as oral morphine for burn dressing changes in children (Borland, Bergesio, Pascoe, et al., 2005). Intranasal sufentanil has also been demonstrated in a randomized controlled clinical trial to provide effective postoperative pain relief (Mathieu, Cnudde, Engelman, et al., 2006). Intranasal hydromorphone has not been studied clinically, but pharmacokinetic studies have shown rapid absorption and good bioavailability (Rudy, Coda, Archer, et al., 2004). Similar results were found with methadone, but current formulations are probably too irritating for clinical applications (Dale, Hoffer, Sheffels, et al., 2002). Preliminary study of intranasal oxycodone suggests that it may have lower bioavailability than the oral oxycodone (Lugo, Kern, 2004). Anecdotal reports of intranasal alfentanil suggest it is effective for breakthrough pain but that patients may prefer the buccal route (Duncan, 2002).
Although studies of intranasal opioid delivery have produced promising results, many questions remain unanswered. Optimal formulations and delivery devices have yet to be determined. Patient selection, drug selection for specific indications, and safety and acceptability issues are still being defined (Dale, Hjortkjær, Kharasch, 2002). Bitter taste and transient nasal irritation are the most common local effects (Stoker, Reber, Waltzman, et al., 2008), which may be unacceptable to some patients.
Nebulized
Nebulized opioids, particularly morphine and fentanyl, have been used to treat dyspnea (Shirk, Donahue, Shirvani, 2006; Coyne, Viswanathan, Smith, 2002; Chandler, 1999) (see Chapter 20). Although the effectiveness of this route remains controversial, there has been interest in expanding its role to pain control. Published efficacy data are limited, but promising, and there are a few case studies. A study comparing placebo, IV morphine (4 mg) via 2 minute infusion, and either one (2.2 mg) or three (6.6 mg) inhaled doses of morphine over a period of 30 seconds to 2 minutes following bunionectomy showed that three doses of inhaled morphine compared favorably to IV morphine (Thipphawong, Babul, Morishige, et al., 2003). Nebulized morphine (20 mg in 3 to 5 mL buffered saline) was used to treat acute chest wall pain in 2 patients with sickle cell disease (Ballas, Viscusi, Epstein, 2004). Both patients reported significant pain relief, and treatment was continued for 10 days until the pain subsided. A prospective, cohort study, however, found that 0.2 mg/kg nebulized morphine was ineffective in treating acute pain in the emergency setting (Bounes, Ducasse, Bona, et al., 2009). Zeppetella (2000b) reported two cases of patients with advanced cancer. One patient, who was taking modified-release morphine for his pain, initially was started on inhaled fentanyl 25 mcg for dyspnea, but found that it relieved his pain more than his dyspnea. He subsequently used the same dose for breakthrough pain with significant relief within 15 minutes. A second patient, maintained on transdermal fentanyl, started on inhaled fentanyl 25 mcg and was titrated to good relief for pain on movement to 125 mcg.
The use of nebulized opioids for dyspnea is premised on the potential for local effects from drug binding to opioid receptors located in the airways or lungs, which may augment effects produced centrally following absorption through the lungs and into the systemic circulation (Shirk, Donahue, Shirvani, 2006). When used for pain, presumably the systemic effects are most relevant, unless the pain relates specifically to pulmonary pathology.
Morphine is absorbed rapidly by inhalation and reaches peak concentration in 10 minutes or less. Bioavailability is variable and is apparently a function of the efficiency of the delivery system rather than the capacity for absorption by the lung. Of the drug deposited in the lung, near 100% bioavailability of morphine and fentanyl are reached (Shirk, Donahue, Shirvani, 2006).
Adverse effects of nebulized opioids are typical of opioids in general: sedation, dizziness, nausea, and vomiting (Shirk, Donahue, Shirvani, 2006; Coyne, Viswanathan, Smith, 2002; Chandler, 1999). Airway irritation manifested by cough or bronchospasm have not been major problems but presumably can occur. Fentanyl is believed to be less likely to cause local irritation (Chandler, 1999). Clinicians sometimes cite concerns about the use of nebulized opioids in opioid-naïve patients with dyspnea; however, one prospective, nonrandomized study used transcutaneous carbon dioxide measurement and pulse oximetry and found that there was no higher risk of respiratory depression in opioid-naïve compared with opioid-tolerant palliative patients treated with nebulized morphine (Clemens, Quednauk, Klaschik, 2008).
Nebulized opioid administration is somewhat cumbersome because of the equipment required; some patients object to the mask needed to deliver the nebulized drug (Zeppettella, 2000a, 2000b), and some complain of a bitter taste (Chandler, 1999). Nonetheless, it is considered a possible alternative to IV administration because it is less expensive and invasive, and could be used more readily in the home than IV opioids (Shirk, Donahue, Shirvani, 2006). Significant improvements in equipment efficiency and portability may make the inhalation of opioids a viable alternative in the future.
Rectal
The rectal route offers an excellent, but frequently overlooked, alternative to the oral route. The rectum provides a large mucosal surface (although less than the upper GI tract) (Lugo, Kern, 2004) that permits passive diffusion of medications and absorption into the systemic circulation. Nurses and caregivers who care for the terminally ill often use the rectal route to administer medications. It is inexpensive and does not involve technical equipment or expertise to the extent that is required for parenteral administration. The rectal route is an alternative to the oral and parenteral routes for patients unable to swallow, experiencing nausea or vomiting, with a bowel obstruction, with a mental status change, or near death (Davis, Walsh, LeGrand, et al., 2002). (See Chapter 8 for rectal nonopioid analgesic administration in the perioperative setting.) It is particularly useful when other routes are unavailable and a delay is expected before the patient can be assessed for alternative treatment (Warren, 1996). The rectal route may be contraindicated in patients who are neutropenic or thrombocytopenic (platelet count of 50,000 per microliter or less) because of potential rectal bleeding from insertion of the suppository. Diarrhea, perianal abscess or fistula, and abdominoperineal resection are also relative contraindications (Davis, Walsh, LeGrand, et al., 2002).
Morphine, hydromorphone, and oxymorphone suppositories are commercially available. Custom-made capsules and suppositories containing methadone have been reported (Watanabe, Belzile, Kuehn, et al., 1996), as has the rectal administration of commercially available liquid preparations and modified-release formulations (Wiffen, McQuay, 2007; Mercadante, Arcuri, Fusco, et al., 2005; Davis, Walsh, LeGrand, et al., 2002; Walsh, Tropiano, 2002). All opioids may be compounded for rectal administration (Stevens, Ghazi, 2000). Oral tablets, oral solutions and suspensions, and injectable products have been administered rectally, sometimes without alteration of the medication, because of the lack of commercially available rectal preparations of opioids in various strengths. For example, intact oral tablets may be administered rectally, or they may be placed in an empty gelatin capsule for rectal insertion. From a pharmacoeconomic, convenience, and patient management perspective, modified-release preparations developed for oral administration are less expensive, provide for flexible dosing, and may be easier to use than suppositories (Walsh, Tropiano, 2002).
Rectal Anatomy and Drug Absorption
Understanding the anatomy of the rectum is important for effective use of the rectal route. The rectum makes up the last 15 to 19 cm of the large intestine. Minimal migration of rectal preparations occurs through the length of the rectum. Thus, the total surface area for drug absorption consists of a region approximately 6 to 8 cm long with no digestive enzymes. Three veins are responsible for blood return in the rectum. The middle and inferior rectal veins drain the remainder of the rectum into the inferior vena cava, bypassing the liver. The superior rectal vein drains the upper rectum into the portal vein, which transports the drug to the liver. However, there is no precise anatomic division that differentiates the rectal regions that drain to the different veins. Most drugs will, therefore, be absorbed by both systemic and hepatic circulation, with some degree of hepatic metabolism (Davis, Walsh, LeGrand, et al., 2002).
The bioavailability of rectal medications is affected by drug placement, rectal contents and tissue health, and degree of ionization of the medication (Fine, Portenoy, 2007; Davis, Rudy, Archer, et al., 2004). Morphine, including morphine in a modified-release delivery system (MS Contin) has 30% to 40% bioavailability (Stevens, Ghazi, 2000). Tradition and customary manufacturer recommendations are to insert rectal suppositories blunt end first to maximize absorption; however, a systematic review of the literature found that there is no research to support this recommendation or to guide correct suppository insertion procedure (Bradshaw, Price, 2007). Suppositories placed lower in the rectum may be better absorbed into the systemic circulation, while those higher in the rectum are absorbed into the portal circulation (Fine, Portenoy, 2007).
Contact time with the mucosal epithelium and moisture content of the rectum also influence absorption. The drug must dissolve in 1 to 3 mL of rectal fluid before being absorbed by the rectal mucosa. In addition, it may be necessary to remove rectal contents so that the medication can be placed against the rectal wall (Davis, Walsh, LeGrand, et al., 2002).
Given the variable influence of all these factors, it is not surprising that interindividual variability in response has been reported in relation to rectal drug administration (Davis, Rudy, Archer, et al., 2004; Lugo, Kern, 2004). Within-patient, dose-to-dose variability is a concern because of the difficulty in ensuring that tablet placement, contact time, and moisture level remain stable over time.
When administering a solution or suspension, the likelihood of spontaneous expulsion by the patient before the drug is absorbed can be prevented by administering volumes no greater than 60 mL (Warren, 1996). Even with caution, however, local irritation can develop over time, limiting the use of this route (Lugo, Kern, 2004). See Box 14-4 for guidelines on maximizing the rate and amount of drug absorption by the rectal route of administration and for minimizing complications.
Rectal Dosing
Acknowledging the concern about variability related to anatomy and other factors, the effective dose of opioids by the rectal route is, overall, approximately equal to oral dosing (Mercadante, Arcuri, Fusco, et al., 2005; Davis, Walsh, LeGrand, et al., 2002; Walsh, Tropiano, 2002). Nonetheless, when switching from the oral to the rectal routes, the starting dose typically is reduced by approximately 25%. In one study, 39 patients who were receiving MS Contin orally were switched to the same dose by the rectal route (Maloney, Kesner, Klein, 1989). All but 1 patient achieved adequate analgesia, but 28% required a decrease in the rectal dose because of excessive drowsiness.
A major disadvantage of the rectal route is that many individuals find it objectionable and are reluctant to use it. This can be especially problematic when family members must administer rectal medications and when medications must be administered several times a day. The need for frequent dosing can be mitigated by the use of a modified-release oral formulation. These oral formulations can be used rectally until the modified-release suppositories under development are commercially available (Walsh, Tropiano, 2002). The modified-release oral drugs demonstrate good rectal absorption and slow steady release compared with the rapid absorption and high peak effect of short-acting preparations. Compared with modified-release opioids given orally, studies show equal or greater systemic absorption, lower peak concentration, and a more prolonged time to peak by the rectal route. The apparent equivalence between the oral and rectal routes may be the result of a balance between slower rectal absorption but less first-pass effect (Davis, Walsh, LeGrand, et al., 2002). Modified-release oral formulations must not be crushed or dissolved.
In an early study, modified-release oral morphine tablets (Oramorph SR) were found to be safe and effective when administered every 12 hours by the rectal or vaginal routes to 8 patients with cancer pain (Grauer, Bass, Wenzel, et al., 1992). No consistent changes were found from the oral route in the frequency of adverse experiences, morphine requirements, or pain intensity ratings. Three of the eight patients reported better pain relief, and five reported the same relief as achieved with the oral route. Two small studies comparing the oral and rectal routes using modified-release morphine showed no differences in pain relief or pharmacokinetics between the groups (Wiffen, McQuay, 2007). In a study comparing liquid morphine administered orally versus rectally, pain relief was much faster in the rectal group and pain scores were better through 180 minutes (Wiffen, McQuay, 2007). Oxycodone tablets administered orally and rectally had similar bioavailability, but with great interindividual variability (Lugo, Kern, 2004).
Opioids that require extensive hepatic metabolism, such as codeine and tramadol, appear to be as effective rectally as when given by the oral route. This suggests a significant degree of absorption from the rectum into the hepatic circulation. These are drugs that would be at a disadvantage if not subjected to the first-pass effect (Davis, Walsh, LeGrand, et al., 2002).
Just as with the oral route, underdosing can occur with the rectal route. The most common reason for failure to achieve adequate analgesia is insufficient dose administration. It is best to control severe escalating pain rapidly with parenteral opioids, and then switch to rectal administration with scheduled ATC doses. Even severe pain that is stable can be managed by the rectal route if the principles of good pain management are followed (McCaffery, Martin, Ferrell, 1992).
Stomal
Treatment of a variety of cancers may include creation of an ostomy. Sigmoid colostomies (left-sided), which produce formed stool, are more likely than other ostomies to be an effective alternative route for administering opioids. Ostomies that are constructed of jejunum, ileum, ascending, transverse, and high descending colon (produce wet, liquid, or semisolid effluent) generally have rapid transit times. The constant pressure of the watery effluent from these ostomies will push out the drug form before it can be dissolved and absorbed (McCaffery, Martin, Ferrell, 1992).
Colostomies usually are created in areas of the colon that are drained by vessels that go directly to the portal vein, subjecting drugs that are present to hepatic metabolism. Thus ostomy administration cannot be equated to rectal administration because it does not avoid the first-pass effect (Warren, 1996); however, the starting dose usually is the same as that for oral or rectal administration. Very limited information is available about the efficacy of stomal administration of opioids, but this route may be an effective alternative for some patients, at least temporarily. Box 14-5 presents guidelines for the administration of medications through a stoma.
Vaginal
Although opioid preparations for vaginal administration are not commercially available, opioids are absorbed by this route (Fisher, Stiles, Heim, et al., 2006). Anecdotally, many patients have reported using the vaginal route for opioid administration. Experience with the administration of other medications (i.e., misoprostol) suggests that vaginal pH may affect efficacy; the lower the pH (acidic), the faster the absorption and the lower the dose required to achieve effect (Abd-El-Maeboud, Ghazy, Nadeem, et al., 2008).
An early study showed that modified-release morphine (Oramorph SR) was safe and effective when administered every 12 hours by the vaginal route to patients with cancer pain (Grauer, Bass, Wenzel, et al., 1992). Patients in this study reported no consistent changes from the oral route in the frequency of adverse experiences, morphine requirements, or pain intensity ratings. In a case report, a patient achieved adequate analgesia using modified-release and short-acting morphine as well as morphine suppositories via the vaginal route as an alternative to IV PCA (Ostrop, Lamb, Reid, 1998). Unpredictable bioavailability was reported as a concern.
A more recent study examined the use of vaginal fentanyl in 4 patients with stable cancer pain (Fisher, Stiles, Heim, et al., 2006). The researchers compounded 50 mcg of fentanyl into a suppository with a water-soluble base and administered two suppositories 1.5 to 2 hours prior to the patients’ regularly scheduled oral opioid. No plasma fentanyl was detected, and patients reported no discernable pain relief with this dose, so the researchers increased the dose to 200 mcg in the fourth patient. This patient reported transient mild dizziness 1 hour after administration, and although her pain intensity did not change, she required less breakthrough analgesia on that day. The researchers concluded that although their study showed that fentanyl is absorbed vaginally, a 200 mcg dose of fentanyl vaginally was at the low end of what would be needed for analgesic activity. As with any route or drug that has not been fully researched, other routes known to be safe and effective are preferred and recommended.
Transdermal
The oral and transdermal routes are generally the preferred routes of administration for long-term use of opioids. They are noninvasive and usually have high acceptability among patients. The transdermal route is particularly attractive for patients who are nauseated or have difficulty swallowing or for whom it is difficult to adhere to an oral regimen (Fine, Portenoy, 2007). The transdermal route avoids hepatic first-pass metabolism, which may pose an advantage for some patients. Drug is delivered at a relatively constant rate, which in some patients may reduce fluctuating effects associated with peak and trough plasma concentrations following oral administration. For some, adherence to the treatment regimen may be improved by the relatively long dosing interval possible with this route. The primary disadvantages of transdermal delivery compared with oral administration are that the skin and other tissues serve as both a barrier and a reservoir: there is significant lag time before the effects of the drug are felt after transdermal application (see the section that follows), and the drug continues to enter the systemic circulation for a variable period after the patch is removed (Kaestli, Wasilewski-Rasca, Bonnabry, et al., 2008).
Two opioids are commercially available for transdermal administration: buprenorphine (Sittl, Likar, Nautrup, 2005; Skaer, 2004) (not released in the United States at time of publication) and fentanyl. Transdermal fentanyl patches are available in two delivery systems: a drug reservoir patch (e.g., Duragesic) and a matrix patch (e.g., Mylan transdermal system). Both these transdermal systems provide analgesia by passive diffusion of the fentanyl across the skin and into the systemic circulation (see discussion below). The equianalgesic ratio between transdermal buprenorphine and transdermal fentanyl has not been established. A retrospective study using a prescription database suggested a ratio of fentanyl 110 to 115:1 (fentanyl:buprenorphine) (Sittl, Likar, Nautrup, 2005).
A distinction should be made between transdermal and topical drug delivery. Transdermal medications are absorbed through the skin, enter the bloodstream, and then are transported throughout the body. They provide systemic analgesia and are an alternative to oral and IV administration. Topical medications may also be absorbed through the skin, but a much smaller percentage of the drug enters the circulation. Topical medications work locally (see Chapter 24 and Figure 24-1 on p. 685).
The outer layer of the skin, the stratum corneum, is the major barrier to absorption of medications through the skin. A lipid-rich intracellular pathway permits passive diffusion of highly lipophilic drugs through this and other layers. In addition to lipophilicity, other characteristics that enhance absorption include low molecular weight, solubility in water as well as oil, and a low melting point. Fentanyl meets all of these requirements. In contrast, morphine is very hydrophilic and is not absorbed across intact skin. Even when applied in a medium intended to increase absorption, quantifiable levels of morphine are not detectable in plasma (Paice, Von Roenn, Hudgins, et al., 2008).
Transdermal Fentanyl
The fentanyl transdermal patch (available in 12, 25, 50, and 100 mcg/h doses) is typically applied to the back or chest and left in place 48 to 72 hours (see application instructions below). The availability of a 12 mcg/hour-dose patch provides a better option than in the past for initiating therapy in patients who are opioid-naïve or have minimal prior opioid exposure; the availability of a lower starting dose in these patients reduces the risk of adverse effects when starting therapy (Mercadante, Villari, 2001).
Because of its lipophilicity, fentanyl is widely distributed in the body (Figure 14-2). A subcutaneous depot or reservoir is established in the skin near the patch, but following systemic uptake, the drug also redistributes into fat and muscle. At steady state, the level of drug in the blood represents the end result of absorption from the patch, movement into and out of these storage sites in various tissues, and drug elimination processes.
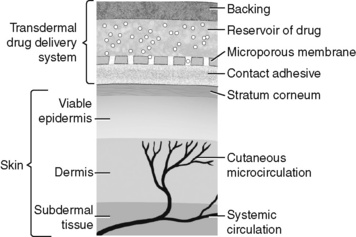
Figure 14-2 Pathway of absorption from transdermal fentanyl. This image illustrates the pathway of absorption from transdermal fentanyl. The skin under the drug reservoir absorbs fentanyl, and depots of the drugs concentrate in the upper skin layers, fat, and skeletal muscles. From these depots, fentanyl is gradually released into systemic circulation. From Pasero, C., & McCaffery, M. (2011). Pain assessment and pharmacologic management, p. 391, Mosby, Inc. May be duplicated for use in clinical practice.
Conversion to transdermal fentanyl from another opioid is a multistep process that takes several days and requires close attention by the clinician. The starting dose of the patch may be determined using the dose conversion tables in the package inserts provided by the manufacturer or one of the conventional strategies developed by clinicians (see the paragraphs that follow). When the first patch is applied, 12 to 18 hours are required for clinically relevant serum levels to be reached. A common approach is to provide a supplemental short-acting opioid along with the patch, at least during this titration period. This reduces the risk that both pain and withdrawal might occur in patients who are being switched from another opioid to transdermal fentanyl. This supplemental analgesia can be scheduled (not PRN) for the first 12 to 16 hours, with each dose equivalent to 10% to 15% of the total daily dose of the previous opioid (Skaer, 2006); a well-educated patient can be instructed to use the supplemental dose PRN, self-administering it in response to worsening pain or symptoms consistent with withdrawal.
As an alternative approach to transitioning from another drug and route (e.g., an oral modified-release opioid) to transdermal fentanyl, the patient can be instructed to overlap the administration of the last dose of the drug that is being discontinued with the placement of the first patch. Although it is still prudent to make available a supplemental short-acting opioid dose, at least during the transition period, there is less risk of pain flare or withdrawal if this overlap is instituted.
During the initial titration period, at least 24 hours should elapse before the fentanyl dose is increased; a 48-hour or 72-hour period reduces the risk of treatment-emergent adverse effects, but may be too long to wait if it is apparent that there are insufficient effects by the end of the first day. The increment in the dose depends on the existing dose, pain severity, and an assessment of medical factors associated with the risk of adverse effects. Similar to oral dosing, the usual increment is between 33% and 50%, and sometimes as high as 100%; when the dose is very low (e.g., 12 to 25 mcg/h), the dose is often doubled.
As noted, transdermal fentanyl patches are available in two delivery systems: a drug reservoir patch (e.g., Duragesic) (Figure 14-3, A) and a matrix patch (e.g., Mylan transdermal system) (see Figure 14-3, B). As the name implies, the reservoir patch has a fentanyl-gel reservoir that diffuses through a membrane to the skin. The fentanyl is incorporated into the adhesive of the matrix patch (Kaestli, Wasilewski-Rasca, Bonnabry, et al., 2008). There is the potential for leakage of the drug from a damaged reservoir patch; there is no such risk with the matrix patch. Although some patients report differences between the matrix and reservoir patches in terms of efficacy and patch adhesiveness, studies have shown they are bioequivalent and can be expected to produce similar analgesic efficacy and adverse effects (Hair, Keating, McKeage, 2008; Kress, Von der Laage, Hoerauf, et al., 2008; Marier, Lor, Morin, et al., 2006; Marier, Lor, Potvin, et al., 2006). A randomized controlled study also established similar efficacy and adverse effects when both transdermal fentanyl products and oral opioid formulations were compared (Kress, Von der Laage, Hoerauf, et al., 2008). Patients should be monitored closely when switching from one formulation to another to ensure ease of use, satisfactory analgesia, and tolerable and manageable adverse effects.
Figure 14-3 Transdermal fentanyl drug delivery systems. A, Reservoir. B, Matrix. From Marier, J. F., Lor, M., Potvin, D., et al. (2006). Pharmacokinetics, tolerability, and performance of a novel matrix transdermal delivery system of fentanyl relative to the commercially available reservoir formulation in healthy subjects. J Clin Pharmacol, 46(6), 642-653. As appears in Pasero, C., & McCaffery, M. (2011). Pain assessment and pharmacologic management, p. 391, Mosby, Inc. May be duplicated for use in clinical practice.
As with any modified-release opioid, concurrent administration of a supplemental, as-needed opioid may be provided for breakthrough pain (see Chapter 12) (Fine, Portenoy, 2007; Skaer, 2006). This is conventional practice for populations with pain due to active cancer or other serious illness; it is a strategy to be considered based on a separate assessment of potential benefit and risk in the population with persistent noncancer pain. If a decision is made to provide a supplement dose, this therapy can be transitioned simply from a supplemental dose given at the time of transitioning to the patch.
In the absence of an oral form of fentanyl, the typical breakthrough pain medication during treatment with transdermal fentanyl is one of the common oral opioids—morphine, hydromorphone, or oxycodone; alternatively, an oral or buccal transmucosal fentanyl formulation can be used. Some clinicians suggest an increase in the dose of the transdermal patch if a patient experiences more than three breakthrough episodes per day (Breitbart, Chandler, Eagle, et al., 2000), while others point out that the number of breakthrough doses that suggest inadequately controlled baseline pain varies widely and the decision to increase the dose of transdermal fentanyl (or any opioid) should be individualized to prevent an increase in unwanted adverse effects (Hewitt, 2000).
If pain consistently returns, or if more breakthrough medication is required on the third day of dosing, reducing the patch-application interval to 48 hours should be considered (Breitbart, Chandler, Eagel, et al., 2000). An observational cohort study found that 50% of patients taking transdermal fentanyl required dosing more frequently than the 72-hour interval recommended by the product’s manufacturers (i.e., every 24 to 48 hours) (Gallagher, Welz-Bosna, Gammaitoni, 2007). Others have described a similar need for a shorter interval in some patients (Sasson, Shvartzman, 2006). Patch changes more frequent than every 48 hours are not recommended, however (Breitbart, Chandler, Eagel, et al., 2000). Interindividual variation underscores the importance of systematic assessment to determine both the optimal dose and dose interval.
Converting to the fentanyl patch from another opioid requires calculating a safe starting dose (Fine, Portenoy, et al. 2009). The manufacturer provides a table of conversion values from oral morphine, but the morphine values constitute a fairly wide range at each dose interval, which can be confusing for the clinician. In addition, clinical experience has shown the values to be conservative, putting the patient at risk for pain and withdrawal and requiring a prolonged titration period (Skaer, 2006; Breitbart, Chandler, Eagel, et al., 2000). A more straightforward alternative method was proposed by Breitbart and colleagues (2000) using a morphine to fentanyl conversion ratio of approximately 2 mg/24 h:1 mcg/h (e.g., 60 mg/day of oral morphine provides analgesia approximately equal to a 25 mcg/h transdermal fentanyl patch). See Box 14-6 for calculation when switching from oral morphine to transdermal fentanyl. Clinical trials using this ratio have shown it to be safe and effective (Donner, Zenz, Tryba, et al., 1996; Mystakidou, Parpa, Tsilika, et al., 2004). Extensive clinical experience supports this approach (Carr, Goudas, 2000; Hewitt, 2000), or a somewhat more flexible guideline indicating that 100 mcg/h of transdermal fentanyl is equianalgesic to 2 to 4 mg/h of IV morphine or equivalent (Hanks, Cherny, Fallon, 2004; Fine, Portenoy, the Ad Hoc Expert Panel on Evidence Review and Guidelines for Opioid Rotation, 2009).
The use of parenteral fentanyl as an intermediate step in converting a patient to transdermal fentanyl is not recommended (Breitbart, Chandler, Eagle, et al., 2000); however, IV fentanyl may be helpful in controlling acute cancer-related pain. Patients (N = 9) experiencing an acute exacerbation of their continuous cancer pain were switched from transdermal fentanyl to IV fentanyl using a 1:1 conversion ratio (Kornick, Santiago-Palma, Schulman, et al., 2003). The median time to achieve mild levels of pain at rest was 1.5 days. Others have also used a 1:1 transdermal fentanyl-IV fentanyl conversion ratio during dose titration (Mercadante, Ferrera, Villari, et al., 2006).
Indications
Improper prescribing and use of transdermal fentanyl patches has resulted in serious toxicity and deaths (U.S. FDA, 2005, 2007c; Ross, Quigley, 2003). Transdermal patches should be used only by patients who have persistent cancer or noncancer pain and are able to follow application instructions (see the paragraphs that follow). It should not be used for acute or postoperative pain. In the United States, transdermal fentanyl is approved for opioid-tolerant patients only (see discussion on use in opioid-naïve patients in the paragraphs that follow). Despite a “black box” warning that reinforces its use in opioid tolerant patients only, it is sometimes prescribed as an initial drug. For example, a review of pharmacy data revealed that transdermal fentanyl was the most commonly prescribed formulation when opioid-naïve nursing home residents were started on a long-acting opioid (Dosa, Dore, Mor, et al., 2009).
Transdermal fentanyl has been shown to be effective for persistent cancer-related pain and various types of noncancer pain (see research below). Although a prospective study found no difference in the incidence of GI adverse effects with transdermal fentanyl compared with oral modified-release opioids (Wirz, Whittmann, Schenk, et al., 2009), several of the studies listed below demonstrated a lower incidence of opioid-induced adverse effects, particularly constipation (see Chapter 19). This and significant improvements in many of the indicators of quality of life (e.g., sleep, mood) may be primary reasons patients frequently express a preference for transdermal fentanyl over other modified-release formulations (Allan, Hays, Jensen, et al., 2001; Berliner, Giesecke, Bornhovd, 2007; Kornick, Santiago-Palma, Moryl, et al., 2003). The research that has demonstrated efficacy and effectiveness of transdermal fentanyl includes the following:
• Persistent cancer-related pain (Breitbart, Chandler, Eagel, et al., 2000; Mercadante, Porzio, Ferrera, et al., 2008; Mystakidou, Tsilika, Parpa, et al., 2003; Payne, Mathias, Pasta, et al., 1998; Vielvoye-Kerkmeer, Mattern, Uitendaal, 2000)
• Acute exacerbation of cancer pain (Kornick, Santiago-Palma, Schulman, et al., 2003)
• AIDS-related pain (Newshan, Lefkowitz, 2001)
• Pain associated with oral mucositis from chemotherapy (Cai, Huang, Sun, et al., 2008)
• Persistent postsurgical pain (Mystakidou, Parpa, Tsilika, et al., 2003)
• Osteoporosis (Mystakidou, Parpa, Tsilika, et al., 2003)
• OA pain (Collado, Torres, 2008; Langford, McKenna, Ratcliffe, et al., 2006)
• Rheumatoid arthritis (Berliner, Giesecke, Bornhovd, 2007; Collado, Torres, 2008)
• Persistent low back pain (Allan, Richarz, Simpson, et al., 2005; Collado, Torres, 2008)
• Persistent neck pain (Mystakidou, Parpa, Tsilika, et al., 2003)
• Multiple sclerosis (Mystakidou, Parpa, Tsilika, et al., 2003)
• Complex regional pain syndrome (Agarwal, Polydefkis, Block, et al., 2007; Mystakidou, Parpa, Tsilika, et al., 2003)
• Postamputation limb pain (Agarwal, Polydefkis, Block, et al., 2007)
• Trigeminal neuralgia (Collado, Torres, 2008)
• Ischemic vascular disease (Collado, Torres, 2008)
• Central pain (Collado, Torres, 2008)
• Neuropathic pain (variety) (Agarwal, Polydefkis, Block, et al., 2007; Collado, Torres, 2008; Mystakidou, Parpa, Tsilika, et al., 2003; Noble, Tregear, Treadwell, et al., 2008)
Transdermal Fentanyl in Opioid-Naïve Patients
Although transdermal fentanyl is approved in the United States as a second-line option in patients who require opioid analgesia for persistent pain (i.e., in opioid-tolerant patients only) (Kornick, Santiago-Palma, Moryl, et al., 2003), it has been used safely in opioid-naïve patients (Breitbart, Chandler, Eagel, et al., 2000). A large multicenter study randomized 680 patients with persistent low back pain to receive oral modified-release morphine (30 mg every 12 hours) or transdermal fentanyl (25 mcg/h every 72 hours) (Allan, Richarz, Simpson, et al., 2005). Starting doses were adjusted based on response. Pain relief was similar among the two groups, but those taking transdermal fentanyl experienced greater pain relief at rest and at night and less constipation. An open-label, prospective trial of 268 opioid-naïve and 321 opioid-tolerant (transferring from oral morphine) patients with cancer-related pain demonstrated improvements in pain and quality of life, high patient satisfaction, and no discontinuations due to adverse effects associated with the use of transdermal fentanyl for the 12-month study period (Mystakidou, Tsilika, Parpa, et al., 2003). An observational study of 215 opioid-naïve patients with noncancer pain found that transdermal fentanyl (starting dose, 12 mcg/h) administered for maintenance analgesia and OTFC (starting dose, 400 mcg) for breakthrough pain was well tolerated and produced sustained pain relief over the 6-month study period (Collado, Torres, 2008). Others have found similar results in patients with persistent cancer-related pain (Vielvoye-Kerkmeer, Mattern, Uitendaal, 2000). Breitbart and colleagues (2000) recommend beginning therapy in opioid-naïve patients with moderate to severe persistent pain with the lowest available dose of transdermal fentanyl (12 mcg/h) and providing a low-dose opioid-nonopioid (e.g., 5 mg oxycodone or hydrocodone plus acetaminophen) PRN every 4 hours for breakthrough pain.
Safety of Long-Term Use
Transdermal fentanyl has been shown to be safe for long-term opioid therapy. An observational study followed 73 cancer patients taking transdermal fentanyl for as long as 29 months after participating in a randomized clinical trial (Nugent, Davis, Brooks, et al., 2001). The median starting dose was 75 mcg/h, and the median final dose was 100 mcg/h. Treatment was well tolerated with a low incidence of adverse effects. Studies in populations with a variety of noncancer pain syndromes produced similar results (Milligan, Lanteri-Minet, Borchert, et al., 2001; Mystakidou, Parpa, Tsilika, et al., 2003). A review of randomized controlled trials of individuals with long-standing low back pain could not identify a pattern of baseline pain (e.g., patient characteristics and severity of pain) that could be used to predict who would or would not respond to long-term opioid (transdermal fentanyl or morphine) therapy (Kalso, Simpson, Slappendel, et al., 2007). The researchers suggested that a one-month trial of opioid treatment would be sufficient to determine response and tolerability in most patients.
Patch Application and Disposal
The transdermal fentanyl patch may be placed on any intact, flat skin surface where adhesion can be maintained for 3 days; flank, back, chest, and upper arms are typically used. For example, skin folds should be avoided (see Patient Education Form IV-5 [pp. 554-555] on fentanyl transdermal system at the end of Section IV). Hair may be clipped but not shaved, and inflamed or irradiated skin should be avoided. The skin must be clean and dry, and soaps, oils, and alcohol should not be used on skin where a patch will be placed. Once in place, firmly rubbing the edges and the body of the patch will help to ensure that the patch sticks to the skin. Skin chemistry varies among patients, and some patches may need additional reinforcement with tape or an occlusive dressing (Skaer, 2004). Folding or cutting the patch in an attempt to reduce the dose of fentanyl will damage the drug-releasing mechanism of the reservoir system and is not recommended in either patch formulation, although a creative method for dose reduction by limiting the area of absorption using an occlusive dressing was proposed by a group of clinicians (Peng, Sun, Mok, 2005). Research is needed to confirm the safety and effectiveness of this approach. Patches are kept in place for 48 to 72 hours, and sites are rotated with each patch change to minimize skin irritation.
Nurses and caregivers are advised to wear plastic gloves prior to applying transdermal fentanyl patches to patients to avoid absorbing fentanyl. A case report described somnolence, weight loss, and depression in a woman who, without wearing gloves, applied transdermal fentanyl patches every 36 hours to her daughter who had persistent pain (Gardner-Nix, 2001b). After the use of gloves to change the patches was instituted, the mother experienced withdrawal symptoms, gradual resolution of somnolence, and regained most of her lost weight. The clinician who reported the case suggested that the situation was exacerbated by the concurrent use of steroid spray and cream to decrease the patient’s skin irritation, which may have promoted absorption.
Proper disposal of medications has become a controversial topic because of the risk of misuse of prescription medications and environmental concerns about medications entering soil and drinking water. The U.S. federal government has issued medication disposal guidelines (Office of Drug Control Policy, 2007). These guidelines confirm the Duragesic manufacturer’s (Janssen, Ortho McNeil) recommendation that fentanyl patches should be folded in half (adhesive to adhesive) and disposed in the toilet. Some pharmacies and health systems and community household hazardous waste programs will also take used and unused medications for safe disposal.
Adverse Effects
The adverse effects seen with other opioids can be expected with fentanyl patches; however, there are data from observational studies that suggest a lower frequency of constipation (Fine, Portenoy, 2007; Kornick, Santiago-Palma, Moryl, et al., 2003; Malkin, Ackerman, Schein, et al., 2001; Staats, Markowitz, Schein, 2004; Tassinari, Sartori, Tamburini, et al., 2008). Despite the potential for less constipation, a bowel management regimen is recommended during any long-term opioid therapy in patients predisposed to this adverse effect. Nausea and vomiting may also be reduced compared with morphine (Skaer, 2006). Skin reactions such as rash and itching may occur (see Chapter 19 for treatment of opioid-induced adverse effects).
Local heat exposure can increase transdermal drug absorption and plasma concentrations by more than three-fold (Ashburn, Ogden, Zhang, et al., 2003). Patients should be instructed to avoid heating pads, electric blankets, hot tubs, and other sources of sustained heat, as these could increase the rate of absorption and lead to toxicity. This advisory applies to the care of patients receiving transdermal fentanyl in all settings. A case of overdose was reported when an upper body warming blanket was used to treat hypothermia during surgery in a patient who had a transdermal fentanyl patch on her chest for treatment of pre-existing persistent pain (Frolich, Giannotti, Modell, 2001). Patients with fever, which can also increase absorption of transdermal opioids, should be frequently assessed for opioid toxicity as well (Skaer, 2004).
Care must be taken to ensure that all hand-off communication (verbal and written) includes information about the analgesics patients are taking. A case of overdose and life-threatening respiratory depression was reported in a patient with a transdermal fentanyl patch in place for treatment of pre-existing persistent pain without the knowledge of the anesthesiologist who administered epidural opioid analgesia to the patient for postoperative pain control (Alsahaf, Stockwell, 2000). This reinforces the importance of communication regarding medication use across the continuum of care.
Despite changes in aging skin due to decreased water and lipid content, absorption of transdermal medications is not dramatically affected by age. The transdermal route is considered advantageous for older adults because no pills need to be swallowed; patch changes are typically every 2 to 3 days, obviating the need for frequent dosing; and stable plasma concentration reduces the risk of some adverse effects. Older adults are more likely to experience skin irritation, however (Kaestli, Wasilewski-Rasca, Bonnabry, et al., 2008). Warnings regarding improper prescribing of transdermal fentanyl (mentioned previously) should be emphasized when considering analgesic options for older adults.
As with all opioids, caution is recommended with the use of transdermal fentanyl in patients with underlying pulmonary conditions, such as emphysema or chronic obstructive pulmonary disease, as they are predisposed to hypoventilation (Kornick, Santiago-Palma, Moryl, et al., 2003). If respiratory depression occurs, the patient must be monitored closely for at least 24 hours after discontinuation of transdermal fentanyl. If naloxone is necessary, treatment will be needed for a prolonged period and the typical approach involves a naloxone infusion for 12 to 24 hours (see Chapter 19).
Nurses often express concern that transdermal fentanyl will not be effective in cachectic patients because they lack subcutaneous fat. Only one study could be found that addressed this issue. Ten normal-weight and 10 cachectic patients with cancer-related pain were recruited to evaluate whether or not absorption of transdermal fentanyl is affected by cachexia (Heiskanen, Matzke, Haakana, et al., 2009). A transdermal patch with a dose approximately equianalgesic to the patients’ previous opioid dose was applied to each patient’s upper arm and left in place for 3 days. Plasma concentrations at 48 and 72 hours were significantly lower in the cachectic patients. The researchers pointed out that absorption of transdermal fentanyl is governed by skin permeability and local blood flow, not by the amount of subcutaneous adipose tissue a patient has and that other factors, such as xerosis (abnormal dryness of the skin), may have been the cause. Although the researchers called for more studies to confirm their findings, they suggested that transdermal fentanyl may lead to inadequate analgesia in cachectic patients. Transdermal fentanyl may not be the best choice when initiating opioid therapy in a cachectic patient, and pain control must be closely monitored in patients who are already taking transdermal fentanyl and develop cachexia. If analgesia is inadequate, doses should be promptly increased or the patient switched to another opioid formulation.
Parenteral
The parenteral route includes the IM, SC, and IV routes of administration. Following is a discussion of these routes.
Intramuscular (IM)
Although commonly used, the IM route of administration is not recommended for pain management and should be abandoned (APS, 2003; Miaskowski, Cleary, Burney, et al., 2005). The IM route has numerous disadvantages and essentially no advantages. Disadvantages include painful administration, unreliable absorption with a 30- to 60-minute lag time to peak effect, and a rapid drop in action compared with oral administration (APS, 2003). Chronic IM administration can result in sterile abscess and fibrosis of muscle and soft tissue. Other complications include nerve damage, hematoma, bleeding, cellulitis, tissue necrosis, and gangrene (Prettyman, 2005). The IM route is a particularly poor choice for older adults, who have decreased muscle mass, and children, who will endure severe pain rather than accept an IM injection (APS, 2003; Prettyman, 2005).
Early research demonstrated that the effects of IM meperidine vary considerably in different individuals and within the same individual (Austin, Stapleton, Mather, 1980b). IM administration of meperidine produced as much as a five-fold difference between individuals in the time to reach peak concentration. Any one patient given meperidine at different times of the day demonstrated a two-fold difference in time to peak concentration. At the dose given, pain control was poor during the first 4-hour dosing interval, and satisfactory pain control did not occur until the third or fourth dose. Pain control after an injection usually was not achieved until 45 minutes had passed, lasted only 75 to 90 minutes, and increased steadily to severe levels by the fourth hour after injection. Presumably, these results apply to any opioid given by the IM route.
In addition to being ineffective, the IM route of administration is dangerous, especially for opioid-naïve patients. Unreliable absorption makes it difficult to predict peak times of the opioid administered. As noted previously, typical IM meperidine doses may not result in satisfactory pain control until the third or fourth dose. If doses are administered at relatively short intervals, it is possible to overshoot the effective dose, a situation that is made more likely to occur by the delay in peak effect after IM injection. If injections are given in the setting of changing circulatory status (e.g., before or after a period of hypotension), the possibility that IM absorption can change further complicates decisions about the dosing interval and the monitoring required to reduce the risk of overshooting.
The unpredictability of the IM route warrants much closer monitoring of patients receiving opioids by this route than is customarily practiced. Careful nurse monitoring of respiratory status and sedation is critical to help ensure that clinically significant respiratory depression does not go undetected (see Chapter 19 for more on sedation and respiratory depression). Many institutions have established monitoring protocols for patients receiving IM opioids that are similar to those developed for IV PCA and epidural analgesia. For example, for postoperative patients, respiratory status and sedation level are assessed every hour for the first 12 hours, every 2 hours for the next 12 hours if stable, and then every 4 hours if stable after initiation of opioid therapy (Pasero, 2009b).
Reducing the Discomfort of IM Injections
Although not recommended, IM injections continue to be used. Clinicians may lack the skill and knowledge required for delivering analgesics by other routes, such as the IV or epidural route, or the drug may be available only in the IM form, such as with immunizations. When the IM route cannot be avoided, pain and tissue trauma of injections may be reduced by improving the technique used to administer them. Topical local anesthetics should be applied whenever possible, particularly in patients who require repeated IM injections and when the solution being injected is viscous (see Chapter 28 for detailed discussion of topical local anesthetic creams). The site of the IM injection can influence the onset of analgesia, the analgesic plasma level, and injection discomfort. The ventrogluteal site is the primary site of IM injection, but the vastus lateralis is preferred because it contains no major nerves or blood vessels. Injection into the deltoid muscle, which is well-perfused, will provide a faster and higher plasma level than an injection into a less well-perfused muscle like the gluteal muscle (Buxton, 2006); however, because of the small size of the deltoid muscle and the proximity of major nerves and blood vessels, this site is appropriate only for small volumes of medications. Box 14-7 provides suggestions for reducing the discomfort of IM injections, and Figures 14-4 and 14-5 provides illustrations of the various injection sites.
Guidelines
Box 14-7
Reducing the Discomfort of IM Injections
1. Match syringe size as closely as possible to volume injected.
2. Use a filter needle to draw up medication from vial or ampule.
3. If a prefilled syringe is used to draw up more medication, instill the complete dose into another syringe.
4. Change to a new dry, sterile needle before injecting.
5. If medication has dripped from the needle of a prefilled unit-dose syringe, wipe it clean with a dry, sterile pad before injecting.
6. Assess the amount of subcutaneous tissue at the site to determine needle length required to penetrate the muscle. Generally, use a 1.5-inch needle for adults and a 1-inch needle for children and infants as young as 4 months old and possibly as young as 2 months old.
7. Use the ventrogluteal site (Figure 14-4, A) as the primary site of injection for patients of any age. As an alternative, the vastus lateralis site (Figure 14-4, B) is preferred over the dorsogluteal site (Figure 14-4, C) because it contains no major nerves or vessels. The deltoid site (Figure 14-4, D) is associated with less site pain and fewer local adverse effects from vaccines compared with the vastus lateralis site.
8. Position patient correctly:
• Side-lying: Flex knee of the side on which injection will be given, then pivot leg forward from the hip 20 degrees.
• Supine: Flex knee of the side on which injection will be given.
• Prone: Point toes in to internally rotate the femur.
• Standing and using dorsogluteal site: Lean against a counter, put weight on side opposite injection site, and point toes on injection side toward opposite foot.
• Prone or on side and using dorsogluteal site: Point toes inward to internally rotate femur and relax muscle.
9. Allow skin antiseptic to dry completely.
10. Use the Z-track method (Figure 14-5, A): Pull skin down or to one side, insert needle quickly and smoothly at a 90-degree angle, inject slowly (10 sec/mL), wait 10 sec before smoothly withdrawing the needle, and apply pressure at the site.
Figure 14-5 A, Z-track method for administering IM injections. The Z-track method involves pulling the skin down or to one side, inserting the needle quickly and smoothly at a 90-degree angle, injecting slowly (10 sec/mL), and waiting 10 seconds before smoothly withdrawing the needle and applying pressure at the site. The Z-track left after injection prevents the deposit of medication through sensitive tissue. B, Pinch-grasp technique for administering IM injections. The pinch-grasp technique involves grasping the muscle, pulling it about ½ to 1 inch toward the person administering the injection, and applying a pinching pressure hard enough to cause mild discomfort. The injection is given at a 90-degree angle. A, From Potter, P. A., & Perry, A. G. (1997). Fundamentals of nursing, ed 4, St Louis, Mosby. B, From Pasero, C., & McCaffery, M. (2011). Pain assessment and pharmacologic management, p. 399, St. Louis, Mosby. May be duplicated for use in clinical practice.
11. Limit volume of injection; if the muscle is large and healthy, up to 4 mL may be injected. If injections are to be given on an ongoing basis, consider concentrating the medication to reduce the volume.
From Pasero, C., & McCaffery, M. (2011). Pain assessment and pharmacologic management, p. 397, St. Louis, Mosby. Data from Barnhill, B. J., Holbert, M. D., Jackson, N. M., et al. (1996). Using pressure to decrease the pain of intramuscular injections. J Pain Symptom Manage, 12(1), 52-58; Beecroft, P. C., & Kongelbeck, S. R. (1994). How safe are intramuscular injections? AACN Clin Issues, 5(2), 207-215; Beyea, S. C., & Nicoll, L. H. (1996). Back to basics: Administering IM injections the right way, Am J Nurs, 96(1), 34-35; Feldman, H. R. (1987). Practice may make perfect but research makes a difference. Nursing, 87(7), 82; Meissner, J. E. (1987). Using the vastus lateralis for injections. Nursing, 87(7), 82; Rettig, F. M., & Southby, J. (1982). Using different body positions to reduce discomfort from dorsogluteal injection. Nurs Res, 31, 219. Pasero C, McCaffery M. May be duplicated for use in clinical practice.
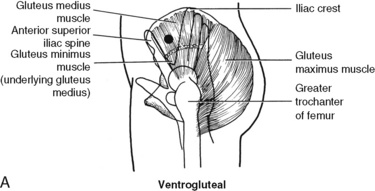
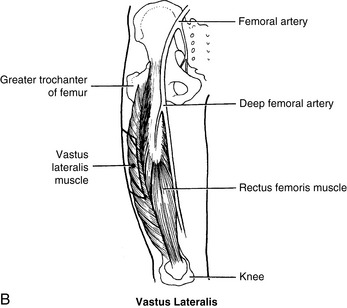
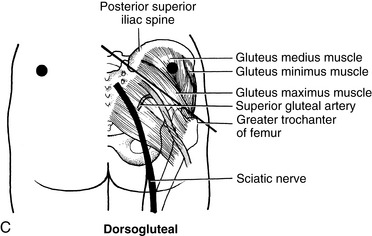
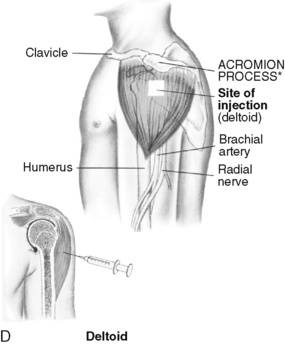
Figure 14-4 A, Ventrogluteal site for IM injections. To locate the ventrogluteal site, palpate the greater trochanter of the femur with the heel of the hand. The index and middle fingers are spread to form a V from the anterior superior iliac spine to just below the iliac crest. The triangle formed between the index finger, the middle fingers, and the crest of the ilium is the injection site. B, Vastus lateralis site for IM injections. The vastus lateralis site is the preferred site in patients of all ages. The belly of the muscle is one-third the distance between the greater trochanter and the knee. In adults, the site for injection is from one hand’s breadth below the greater trochanter to one hand’s breadth above the knee. The site is located on the medial outer aspect in the center third portion of the thigh in children. C, Dorsogluteal site for IM injections. To locate the dorsogluteal site for injection, draw an imaginary line from the posterior iliac spine to the greater trochanter of the femur. Because this line is lateral and parallel to the sciatic nerve, a site selected laterally and superiorly will be away from the nerve and the superior gluteal artery. D, Deltoid site for IM injections. To locate the densest area of the deltoid muscle and to avoid the radial nerve and deep brachial artery, locate the site 1 to 2 inches below the acromion process. A-C, From Pagliaro, A. M., & Pagliaro, L. A. (1986). Pharmacologic aspects of nursing, St Louis, Mosby. D, From Wong, D. L. (1997). Whaley and Wong’s essentials of pediatric nursing, ed 5, St Louis, Mosby.
The Z-track method for administering IM injections has been found to be less painful than the traditional injection technique (Beyea, Nicoll, 1996; Keen, 1986). The traditional IM technique allows leakage of the solution from the site, which can cause significant pain and irritation. The Z-track method involves pulling the skin down or to one side before the injection, then injecting slowly, removing the needle smoothly, and then allowing the skin to return to its original position (Figure 14-5, A). This prevents painful leakage by sealing the injected solution into the musculature.
A pinch-grasp technique has been shown to reduce the discomfort of IM injections into the deltoid muscle. This technique involves grasping the muscle, pulling it about ½ to 1 inch toward the person administering the injection, and applying a pinching pressure hard enough to cause mild discomfort. Resting the wrist about 3 inches from the site of injection, the injection is given at a 90-degree angle (Locsin, 1985) (Figure 14-5, B). Before pinching the skin, warn the patient and explain the purpose.
Manual pressure over the injection site may help reduce the pain associated with dorsogluteal IM injections of immune globulin in adults. In one study, investigators applied pressure over the injection site with the noninjecting thumb until resistance was felt, then maintained that pressure for 10 seconds in patients in group 1 (Barnhill, Holbert, Jackson, et al., 1996). They applied no manual pressure over the injection sites of patients in group 2. For both groups, the skin at the injection site was held taut between the noninjecting thumb and index finger, and the immune globulin was injected over a period of 5 to 10 seconds. Patients in group 1 reported significantly less injection pain on injection compared with patients in group 2.
The use of cold in addition to manual pressure to reduce the pain of IM injection also has been studied (Hillman, Jarman, 1986). Ice and “slight” pressure applied to the IM injection site for 15 to 20 seconds before injection was found to reduce pain at deeper penetrations than without ice and pressure. Application of cold alone can be an effective pain reliever in some patients. A 5-minute ice massage to the site contralateral to that used for a bone marrow aspiration reduced pain during the aspiration procedure (Hudziak, 1983). It seems likely that ice massage would be effective in reducing the pain of injection as well.
Even gentle slapping of the area before an IM injection can reduce its discomfort (Goodfriend, 1987). Obviously, before the slap, the patient should be warned and the reason explained.
Subcutaneous (SC)
The SC route can be used for continuous infusion (CI) of opioids, particularly in patients with persistent pain who are unable to take oral medications and who do not have central venous access (Mikkelsen, Butler, Huerta, et al., 2000; Stevens, Ghazi, 2000; Vascello, McQuillan, 2006). The SC route also can be used for repeated injections, and along with brief IV infusion or IV bolus, is preferred when this type of drug administration is required. Although it is a second-line route, it is a well accepted approach for medication delivery at end of life (Anderson, Shreve, 2004). The SC route obviates the need for normal GI function (Anderson, Shreve, 2004), and a trial of this route may be considered when patients experience dose-limiting adverse effects with oral opioids (Bourdeanu, Loseth, Funk, 2005), require a large number of tablets or patches for pain control, or require parenteral opioids because of bowel obstruction but have limited venous access (Miaskowski, Cleary, Burney, et al., 2005). Other candidates include patients with confusion or other mental status changes that make the oral route contraindicated because of the risk of aspiration (Miaskowski, Cleary, Burney, et al., 2005). The SC route is rarely used for acute pain management because onset is slow.
The most common opioid analgesics administered by SCCI are hydromorphone and morphine (Fine, Portenoy, 2007; Mikkelsen, Butler, Huerta, et al., 2000). Oxymorphone, fentanyl, and levorphanol also have been administered subcutaneously. A 6-day randomized, cross-over design study found SC fentanyl to be as effective with similar adverse effects and better bowel function compared with SC morphine (Hunt, Fazekas, Thorne, et al., 1999). The researchers suggested a conversion ratio of morphine 10 mg to fentanyl 150 mcg when given subcutaneously. Administration of methadone is more likely to produce irritation by the SC route and is not preferred for this reason (Fine, Portenoy, 2007); however, some suggest it can be managed by this route with low opioid concentrations, frequent site rotation, and co-administration of dexamethasone (Lynch, 2005). Others recommend flushing of the access site with normal saline to minimize irritation so that sites can be maintained for prolonged periods without the need for dose limitation or medications added to prevent irritation (Hum, Fainsinger, Bielech, 2007) See Chapter 15, for research on the stability and compatibility of many of the analgesics administered by SCCI.
Absorption and distribution vary depending on the placement of the needle and the patient’s adipose tissue; however, cachexia is not a contraindication to SC analgesia. For conversion from oral morphine to SC morphine, a relative equivalency ratio of 3:1 is used, the same ratio used to calculate doses for the IV route (Coyle, Mauskop, Maggard, et al., 1986; Mikkelsen, Butler, Huerta, et al., 2000; Moulin, Johnson, Murray-Parsons, 1992). Although not contraindicated in older patients, this population may require a lower initial SC dose than younger adults (Aubrun, Bunge, Langeron, et al., 2003). An early study showed that patients prefer SC continuous infusion over IV infusion, citing greater and easier mobility and better pain control among the reasons (Bruera, Brennels, Michaud, et al., 1987).
SCCI administration is disliked by some patients, especially children. Although the technique of administration is relatively simple to master (see the paragraphs that follow), it does require a greater expertise than the oral route. Sterile technique is used to establish a site of infusion, and the patient and caregivers must become familiar and skilled in the use of needles, syringes, an infusion pump, and other equipment. The SC injection site must be changed at least weekly, and the potential for local tissue injury at the infusion site must be monitored.
High-concentration opioid formulations are used for SCCI infusion because infusion volumes are limited in most situations. Infusion pumps with the capability of delivering in tenths of a milliliter are necessary to accommodate the high-concentration/low-volume infusions. Most patients can absorb 2 or 3 mL/h, and some can absorb as much as 5 mL/h (Anderson, Shreve, 2004; Coyle, 1996). When necessary, some patients can even tolerate an infusion up to 8 mL/h for a few hours. Breakthrough doses may be provided as SC clinician-administered or PCA boluses (50% of hourly infusion administered as often as every 15 minutes) (Anderson, Shreve, 2004). Like other infusions, the need for bolus doses for breakthrough pain, if this is permitted, can be used to adjust the hourly dose (Anderson, Shreve, 2004). In patients requiring high infusion volumes, two sites can be used to deliver the required amount. For example, one site can be used for the infusion and one for breakthrough boluses. A four-way stopcock can be used to branch an infusion from one infusion pump to two sites (Gordon, 2008). As noted, most patients are able to tolerate the same site for seven days, and patients have been maintained on SC infusions for more than a year (Anderson, Shreve, 2004).
The volume of infusion during SCCI can be greatly increased by the addition of hyaluronidase to the infusate. Hyaluronidase typically is used for hypodermoclysis, or subcutaneous hydration. Although some authors recommend against this approach for the enhanced SC delivery of drugs (Anderson, Shreve, 2004), there is a favorable clinical experience, and the technique should be considered when an opioid is being delivered by SCCI and the required dose cannot be administered within the small volume of infusion. Hyaluronidase at a conventional dose of 150 units in 250 mL or 500 mL can routinely permit infusion rates greater than 100 mL/h (and significantly higher when needed) (see Figure 14-6 and Box 14-8 for guidelines on SC administration).
Figure 14-6 Subcutaneous infusion needle placement. This image shows sites for SC infusion needle placement, which may be attached to an ambulatory infusion pump. X marks sites that interfere with mobility. Other sites to consider include upper arms and thighs. Sites should be rotated. As appears in Pasero, C., & McCaffery, M. Pain assessment and pharmacologic management, p. 401, St. Louis, Mosby. Pasero C, McCaffery M. May be duplicated for use in clinical practice.
Successful SCCI administration in the home depends on the ability of the family and community health care system to manage the technology in the home (Anderson, Shreve, 2004). The cost and insurance coverage must be considered (Fine, Portenoy, 2007). Because an infusion device and supplies are necessary, SCCI administration usually is more expensive than oral administration. Programmable portable pumps and disposable pumps are available; disposable infusion devices tend to be more expensive than the programmable infusion pumps (Moulin, Johnson, Murray-Parsons, 1992). Important points to address when establishing a plan for parenteral opioid analgesic infusion in the home include determining who will assume primary responsibility for the technologic aspects of the infusion, educating the patient and family regarding the infusion and pain and adverse effect assessment, and ensuring appropriate monitoring and follow-up (Coyle, Cherny, Portenoy, 1995) (see Box 14-8).
Intravenous (IV)
The IV route is most efficient when an immediate analgesic effect is required, such as for acute, severe escalating pain. It allows for rapid titration. The IV route is most commonly used for short courses of therapy in a hospitalized setting, where patients can be closely monitored because many who benefit from IV analgesia are opioid-naïve, such as surgical patients. The IV route may also be used temporarily for patients with cancer pain or noncancer pain who require rapid titration (Bourdeanu, Loseth, Funk, 2005). It is an alternative for patients who are unable to take oral opioid analgesics (Hanks, Cherny, Fallon, 2004; Miaskowski, Cleary, Burney, et al., 2005). In the terminally ill patient, bowel obstruction and dose-limiting adverse effects with other systemic opioids are common reasons for long-term IV infusion. Central lines are placed whenever possible for long-term IV infusion.
When patients are switched from oral dosing to parenteral administration, they typically report greater effectiveness with the IV route, but this should not be misconstrued. Opioids are not more effective when given by the IV route than by other routes; they are simply more bioavailable (100%) because first-pass hepatic metabolism is avoided (Stevens, Ghazi, 2000). A patient may very well feel less pain and fewer adverse effects, but this only reflects relatively higher plasma concentrations of the drug. The IV route may avoid certain adverse effects, such as nausea, caused when the drug is taken orally.
Methods of IV administration include bolus, continuous infusion (CI, basal rate), and PCA. A steady state is better maintained with a continuous infusion compared with the bolus method; however, continuous opioid infusions must be used with caution in opioid-naïve patients (Pasero, McCaffery, 2004) (see Chapter 17).
Duration of analgesia after an IV bolus is determined by the kinetics of the drug and the dose administered; the higher the dose, usually the longer the duration. Compared to equianalgesic doses by other routes, IV administration produces the highest peak concentration. Peak concentration is associated with toxicity (e.g., sedation, nausea), and it is therefore possible that adverse effects with repeated IV boluses are more severe than repeated boluses by other routes (Hanks, Cherny, Fallon, 2004). To decrease the peak effect and lower the level of toxicity, IV boluses may be administered more slowly (e.g., 10 mg of morphine over a 15-minute period) or smaller doses may be administered more often (e.g., 5 mg of morphine every 60 to 90 minutes).
There are drawbacks to the use of the IV route. IV administration depends on venous availability and ability to maintain patency. Sterile technique is required to prevent systemic infection. The IV route is generally more expensive and requires more expertise to use than the oral route (Vascello, McQuillan, 2006). If continuous IV infusion or IV PCA is used, equipment and tubings are required. Patients accustomed to the independence of oral analgesics may find adjusting to the IV route difficult.
Conclusion
This chapter presented the many routes of administration for opioid analgesics. The oral route is the most common for all types of pain. The oral and transdermal routes are first-line choices for long-term opioid therapy. The IV route is used when rapid analgesia is required. There are many second-line routes of administration, and it is often necessary to switch routes of administration during care, particularly in patients with advanced illness. For all types of pain, opioids should be administered by the least invasive and safest route capable of producing satisfactory analgesia.