55 Anticancer drugs
Overview
In this chapter, we deal with cancer and anticancer therapy, emphasising first the pathogenesis of cancer before proceeding to describe the drugs that can be used therapeutically. Finally, we consider the extent to which our new knowledge of cancer biology is leading to new treatments. The use of radioactive isotopes in cancer treatment is beyond the scope of this book.
Introduction
Cancer is a disease characterised by uncontrolled multiplication and spread of abnormal forms of the body’s own cells. It is the second most common cause of death in the developed nations (cardiovascular disease has the dubious distinction of heading that table) and one in three people will be diagnosed with cancer during their lifetime. In the UK, over 365 000 new cases were reported and mortality in 2006 was in excess of 154 000 (Cancer Research UK). Cancer is responsible for approximately one-quarter of all deaths in the UK, with lung and bowel cancer comprising the largest category, closely followed by breast and prostate cancer. Statistics from most other countries in the developed world tell much the same story. At first sight, incidence figures for the past 100 years or so give the impression that the disease is increasing in developed countries, but cancer is largely a disease of later life, and with advances in public health and medical science, many more people now live to an age where they are more liable to contract cancer.
The terms cancer, malignant neoplasm (neoplasm simply means ‘new growth’) and malignant tumour are synonymous. Both benign and malignant tumours manifest uncontrolled proliferation, but the latter are distinguished by their capacity for dedifferentiation, their invasiveness and their ability to metastasise (spread to other parts of the body). In this chapter, we shall be concerned only with the therapy of malignant neoplasia or cancer. The appearance of these abnormal characteristics reflects altered patterns of gene expression in the cancer cells, resulting from inherited or acquired genetic mutations.
There are three main approaches to treating established cancer—surgical excision, irradiation and drug therapy (often called chemotherapy)—and the relative value of each of these approaches depends on the type of tumour and the stage of its development. Chemotherapy may be used on its own or as an adjunct to other forms of therapy.
Compared with that of bacterial diseases, cancer chemotherapy presents a difficult problem. In biochemical terms, microorganisms are both quantitatively and qualitatively different from human cells (see Ch. 49), but cancer cells and normal cells are so similar in most respects that it is more difficult to find general, exploitable, biochemical differences between them. In recent years, the focus of cancer chemotherapy has broadened to include, as well as conventional cytotoxic drugs (which act on all cells, and rely on a small margin of selectivity to be useful as anticancer agents), several drugs that affect either the hormonal regulation of tumour growth, or the defective cell cycle controls that underlie malignancy (see below and Ch. 5). Overall, this has been one of the most fruitful fields of drug development in recent years, in which genomics and biopharmaceuticals have played a major role. The flow of innovation seems set to continue.
The Pathogenesis of Cancer
To understand the action and drawbacks of current anticancer agents and to appreciate the therapeutic hurdles that must be surmounted by putative new drugs, it is important to consider in more detail the pathobiology of this disease.
Cancer cells manifest, to varying degrees, four characteristics that distinguish them from normal cells. These are:
•
uncontrolled
proliferation•
dedifferentiation and loss of function
The Genesis of a Cancer Cell
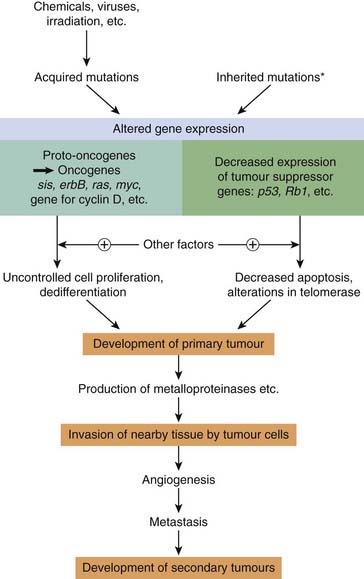
A normal cell turns into a cancer cell because of one or more mutations in its DNA, which can be inherited or acquired, usually through exposure to viruses or carcinogens (e.g. tobacco products, asbestos). A good example is breast cancer; women who inherit a single defective copy of either of the tumour suppressor genes BRCA1 and BRCA2 (see below) have a significantly increased risk of developing breast cancer. However, carcinogenesis is a complex multistage process, usually involving more than one genetic change as well as other, epigenetic factors (hormonal, co-carcinogen and tumour promoter effects, etc.) that do not themselves produce cancer but which increase the likelihood that the genetic mutation(s) will eventually result in cancer.
There are two main categories of genetic change that are important:
1
The activation of
proto-oncogenes to
oncogenes. Proto-oncogenes are genes that normally control cell division, apoptosis and differentiation (see
Ch. 5), but which can be converted to oncogenes that induce malignant change by viral or carcinogen action.
2
The inactivation of
tumour suppressor genes. Normal cells contain genes that have the ability to suppress malignant change—termed tumour suppressor genes (antioncogenes)—and mutations of these genes are involved in many different cancers. The loss of function of tumour suppressor genes can be the critical event in carcinogenesis.
About 30 tumour suppressor genes and 100 dominant oncogenes have been identified. The changes that lead to malignancy are a result of point mutations, gene amplification or chromosomal translocation, often caused by viruses or chemical carcinogens.
The Special Characteristics of Cancer Cells
Uncontrolled Proliferation
Many healthy cells, in the bone marrow and the epithelium of the gastrointestinal tract for example, have the property of continuous rapid division, and it is not generally true that cancer cells proliferate faster than normal cells. Some cancer cells multiply slowly (e.g. those in plasma cell tumours) and some much more rapidly (e.g. the cells of Burkitt’s lymphoma). The significant issue is that cancer cells have escaped from the mechanisms that normally regulate cell division and tissue growth. It is this, rather than their rate of proliferation, that distinguishes them from normal cells.
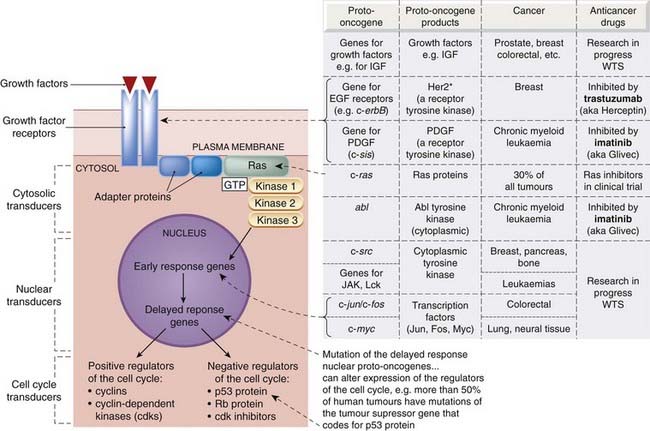
What are the changes that lead to the uncontrolled proliferation of tumour cells? Inactivation of tumour suppressor genes or transformation of proto-oncogenes into oncogenes can confer autonomy of growth on a cell and thus result in uncontrolled proliferation by producing changes in several cellular systems (see Fig. 55.1), including:
•
growth factors, their receptors and signalling pathways
•
the
cell cycle transducers, for example cyclins, cyclin-dependent kinases (cdks) or the cdk inhibitors
•
the
apoptotic machinery that normally disposes of abnormal cells
•
local blood vessels, resulting from tumour-directed angiogenesis.
Potentially all the genes coding for the above components could be regarded as oncogenes or tumour suppressor genes (see Fig. 55.2), although not all are equally prone to malignant transformation. It should be understood that malignant transformation of several components is needed for the development of cancer.
Resistance to apoptosis
Apoptosis is programmed cell death (Ch.5), and genetic mutations in the antiapoptotic genes are usually a prerequisite for cancer; indeed, resistance to apoptosis is a hallmark of the disease. It can be brought about by inactivation of proapoptotic factors or by activation of antiapoptotic factors.
Telomerase expression
Telomeres are specialised structures that cap the ends of chromosomes—like the small metal tubes on the end of shoelaces—protecting them from degradation, rearrangement and fusion with other chromosomes. Furthermore, DNA polymerase cannot easily duplicate the last few nucleotides at the ends of DNA, and telomeres prevent loss of the ‘end’ genes. With each round of cell division, a portion of the telomere is eroded, so that eventually it becomes non-functional. At this point, DNA replication ceases and the cell becomes senescent.
Rapidly dividing cells, such as stem cells and those of the bone marrow, the germline and the epithelium of the gastrointestinal tract, express telomerase, an enzyme that maintains and stabilises telomeres. While it is absent from most fully differentiated somatic cells, about 95% of late-stage malignant tumours do express the enzyme, and it is this that may confer ‘immortality’ on cancer cells.
The control of tumour-related blood vessels
The factors described above lead to the uncontrolled proliferation of individual cancer cells, but other factors, particularly blood supply, determine the actual growth of a solid tumour. Tumours 1–2 mm in diameter can obtain nutrients by diffusion, but any further expansion requires angiogenesis, the development of new blood vessels. Angiogenesis occurs in response to growth factors produced by the growing tumour (see Griffioen & Molema, 2000).
Dedifferentiation and Loss of Function
The multiplication of normal cells in a tissue begins with division of the undifferentiated stem cells giving rise to daughter cells. These daughter cells eventually differentiate to become the mature cells of the relevant tissue, ready to perform their programmed functions. For example, when fibroblasts mature, they secrete and organise extracellular matrix; mature muscle cells are capable of contraction. One of the main characteristics of cancer cells is that they dedifferentiate to varying degrees. In general, poorly differentiated cancers multiply faster and carry a worse prognosis than well-differentiated cancers.
Invasiveness
Normal cells are not generally found outside their ‘designated’ tissue of origin. This is because, during differentiation and tissue or organ growth, normal cells develop certain spatial relationships with respect to each other. These relationships are maintained by various tissue-specific survival factors that prevent apoptosis (see Ch. 5). In this way, any cells that escape accidentally lose these survival signals and die.
Consequently, although the cells of the normal mucosal epithelium of the rectum proliferate continuously as the lining is shed, they remain as a lining epithelium. A cancer of the rectal mucosa, by comparison, invades other tissues forming the rectum and often the tissues of other pelvic organs. Cancer cells have not only lost, through mutation, the restraints that act on normal cells, but they also secrete enzymes (e.g. metalloproteinases; see Ch. 5) that break down the extracellular matrix, enabling them to move around.
Metastasis
Metastases are secondary tumours (‘secondaries’) formed by cells that have been released from the initial or primary tumour and which have reached other sites through blood vessels or lymphatics, by transportation on other cells or as a result of being shed into body cavities. Metastases are the principal cause of mortality and morbidity in most cancers and constitute a major problem for cancer therapy.
As discussed above, dislodgment or aberrant migration of normal cells would lead to programmed cell death as a result of withdrawal of the necessary antiapoptotic factors. Cancer cells that metastasise have undergone a series of genetic changes that alter their responses to the regulatory factors that control the cellular architecture of normal tissues, enabling them to establish themselves ‘extraterritorially’. Tumour-induced growth of new blood vessels locally (see above) favours metastasis.
Secondary tumours occur more frequently in some tissues than in others. For example, metastases of mammary cancers are often found in lung, bone and brain. The reason for this is that breast cancer cells express chemokine receptors such as CXR4 (see Ch. 17) on their surfaces, and chemokines that recognise these receptors are expressed at high level in these tissues but not in others (e.g. kidney), facilitating the selective accumulation of cells at these sites.
General Principles of Cytotoxic Anticancer Drugs
In experiments with rapidly growing transplantable leukaemias in mice, it has been found that a given therapeutic dose of a cytotoxic drug1 destroys a constant fraction of the malignant cells. Thus a dose that kills 99.99% of cells, if used to treat a tumour with 1011 cells, will still leave 10 million (107) viable malignant cells. As the same principle holds for fast-growing tumours in humans, schedules for chemotherapy are aimed at producing as near a total cell kill as possible because, in contrast to the situation that occurs in microorganisms, little reliance can be placed on the host’s immunological defence mechanisms against the remaining cancer cells.
One of the major difficulties in treating cancer is that tumour growth is usually far advanced before cancer is diagnosed. Let us suppose that a tumour arises from a single cell and that the growth is exponential, as it may well be during the initial stages. ‘Doubling’ times vary, being, for example, approximately 24 h with Burkitt’s lymphoma, 2 weeks in the case of some leukaemias, and 3 months with mammary cancers. Approximately 30 doublings would be required to produce a cell mass with a diameter of 2 cm, containing 109 cells. Such a tumour is within the limits of diagnostic procedures, although it could easily go unnoticed. A further 10 doublings would produce 1012 cells, a tumour mass that is likely to be lethal, and which would measure about 20 cm in diameter if it were one solid mass.
However, continuous exponential growth of this sort does not usually occur. In the case of most solid tumours (for example of lung, stomach, uterus and so on), as opposed to leukaemias (tumours of white blood cells), the growth rate falls as the neoplasm grows. This is partly because the tumour outgrows its blood supply, and partly because not all the cells proliferate continuously. The cells of a solid tumour can be considered as belonging to three compartments:
1
Compartment A consists of dividing cells, possibly being continuously in cell cycle.
2
Compartment B consists of resting cells (G
0 phase) which, although not dividing, are potentially able to do so.
3
Compartment C consists of cells that are no longer able to divide but which contribute to the tumour volume.
Essentially, only cells in compartment A, which may form as little as 5% of some solid tumours, are susceptible to the main current cytotoxic drugs, as is explained below. The cells in compartment C do not constitute a problem, but it is the existence of compartment B that makes cancer chemotherapy difficult, because these cells are not very sensitive to cytotoxic drugs and are liable to re-enter compartment A following chemotherapy.
Most current anticancer drugs, particularly cytotoxic agents, affect only one characteristic aspect of cancer cell biology—cell division—but have no specific inhibitory effect on invasiveness, the loss of differentiation or the tendency to metastasise. In many cases, the antiproliferative action results from an action during S phase of the cell cycle, and the resultant damage to DNA initiates apoptosis (see above). Furthermore, because their main target is cell division, they will affect all rapidly dividing normal tissues, and thus they are likely to produce, to a greater or lesser extent, the following general toxic effects:
•
bone marrow toxicity (myelosuppression) with decreased leukocyte production and thus decreased resistance to infection
•
loss of hair (alopecia)
•
damage to
gastrointestinal epithelium (including oral mucous membranes)
•
depression of growth in children
They can also, in certain circumstances, be themselves carcinogenic. Rapid cell destruction also entails extensive purine catabolism, and urates may precipitate in the renal tubules and cause kidney damage. Finally, in addition to specific toxic effects associated with individual drugs, virtually all cytotoxic drugs produce severe nausea and vomiting, which has been called the ‘inbuilt deterrent’ to patient compliance in completing a course of treatment with these agents.
Cancer pathogenesis and cancer chemotherapy: general principles 
•
Cancer arises as a result of a series of genetic and epigenetic changes, the main genetic lesions being:
–
inactivation of tumour suppressor genes
–
the activation of oncogenes (mutation of the normal genes controlling cell division and other processes).
•
Cancer cells have four characteristics that distinguish them from normal cells:
–
uncontrolled proliferation
–
loss of function because of lack of capacity to differentiate
–
the ability to metastasise.
•
Cancer cells have uncontrolled proliferation often because of changes in:
–
growth factors and/or their receptors
–
intracellular signalling pathways, particularly those controlling the cell cycle and apoptosis
•
This may be supported by tumour-related angiogenesis.
•
Most anticancer drugs are antiproliferative—most damage DNA and thereby initiate apoptosis. They also affect rapidly dividing normal cells and are thus likely to depress bone marrow, impair healing and depress growth. Most cause nausea, vomiting, sterility, hair loss and teratogenicity.
Anticancer Drugs
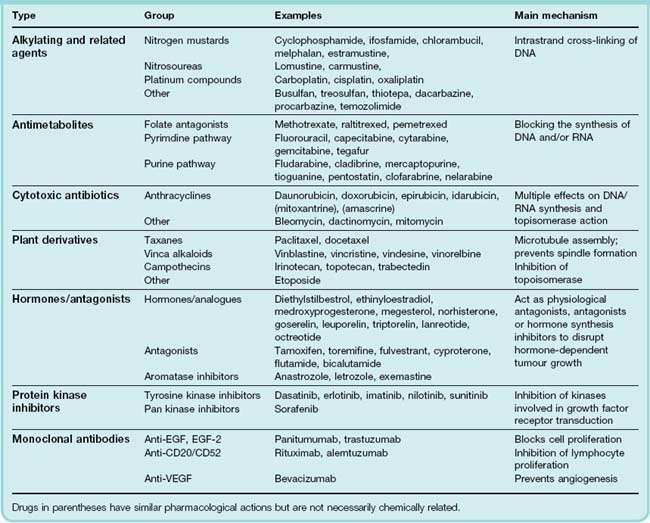
The main anticancer drugs can be divided into the following general categories:
•
Cytotoxic drugs. The mechanism of action of these drugs is discussed more fully below and summarised in
Table 55.1; they include:
–
alkylating agents and related compounds, which act by forming covalent bonds with DNA and thus impeding replication
–
antimetabolites, which block or subvert one or more of the metabolic pathways involved in DNA synthesis
–
cytotoxic antibiotics, i.e. substances of microbial origin that prevent mammalian cell division
–
plant derivatives (vinca alkaloids, taxanes, campothecins): most of these specifically affect microtubule function and hence the formation of the mitotic spindle.
•
Hormones, of which the most important are steroids (e.g. glucocorticoids, oestrogens and androgens) as well as drugs that suppress hormone secretion or antagonise hormone action.
•
Monoclonal antibodies: these are generally only of use in particular types of cancer.
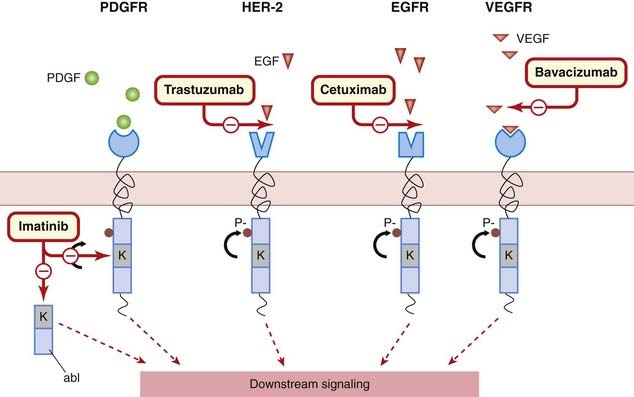
•
Protein kinase inhibitors: these drugs inhibit protein (usually tyrosine) kinases that transduce growth signals in rapidly dividing cells. They have a rather restricted use.
•
Miscellaneous agents that do not easily fit into the above categories.
The clinical use of anticancer drugs is the province of the specialist, who selects treatment regimens appropriate to the patient with the objective of curing, prolonging life or providing palliative therapy.2 There are over 80 drugs available in the UK, which are often used in combination. Here we discuss mechanisms of action and the main unwanted effects of commonly used anticancer agents. A recent textbook (Airley, 2009) provides detailed information.
Alkylating Agents and Related Compounds
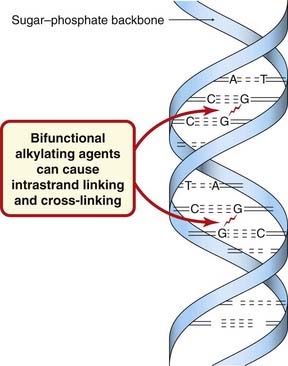
Alkylating agents and related compounds contain chemical groups that can form covalent bonds with particular nucleophilic substances in the cell. With alkylating agents themselves, the main step is the formation of a carbonium ion—a carbon atom with only six electrons in its outer shell. Such ions are highly reactive and react instantaneously with an electron donor such as an amine, hydroxyl or sulfhydryl group. Most of the cytotoxic anticancer alkylating agents are bifunctional, i.e. they have two alkylating groups (Fig. 55.3).
The nitrogen at position 7 (N7) of guanine, being strongly nucleophilic, is probably the main molecular target for alkylation in DNA (Fig. 55.3), although N1 and N3 of adenine and N3 of cytosine may also be affected. A bifunctional agent, by reacting with two groups, can cause intra- or interchain cross-linking (Fig. 55.3). This interferes not only with transcription, but also with replication, which is probably the critical effect of anticancer alkylating agents. Other effects of alkylation at guanine N7 are excision of the guanine base with main chain scission, or pairing of the alkylated guanine with thymine instead of cytosine, and eventual substitution of the GC pair by an AT pair. Their main impact is seen during replication (S phase), when some zones of the DNA are unpaired and more susceptible to alkylation. This results in a block at G2 (see Fig. 55.3) and subsequent apoptotic cell death.
All alkylating agents depress bone marrow function and cause gastrointestinal disturbances. With prolonged use, two further unwanted effects occur: depression of gametogenesis (particularly in men), leading to sterility, and an increased risk of acute non-lymphocytic leukaemia and other malignancies.
Alkylating agents are among the most commonly employed of all anticancer drugs. A large number are available for use in cancer chemotherapy (some dozen are approved in the UK at the time of writing). Only a few commonly used ones will be dealt with here.
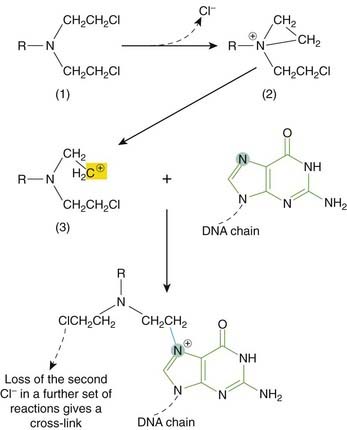
Nitrogen mustards
Nitrogen mustards are related to the ‘mustard gas’ used during the First World War; their basic formula (R-N-bis-(2-chloroethyl)) is shown in Figure 55.4. In the body, each 2-chloroethyl side-chain undergoes an intramolecular cyclisation with the release of a Cl−. The highly reactive ethylene immonium derivative so formed can interact with DNA (see Figs 55.3 and 55.4) and other molecules.
Cyclophosphamide is probably the most commonly used alkylating agent. It is inactive until metabolised in the liver by the P450 mixed function oxidases (see Ch. 9). It has a pronounced effect on lymphocytes and can also be used as an immunosuppressant (see Ch. 26). It is usually given orally or by intravenous injection but may also be given intramuscularly. Important toxic effects are nausea and vomiting, bone marrow depression and haemorrhagic cystitis. This last effect (which also occurs with the related drug ifosfamide) is caused by the metabolite acrolein and can be ameliorated by increasing fluid intake and administering compounds that are sulfhydryl donors, such as N-acetylcysteine or mesna (sodium-2-mercaptoethane sulfonate). These agents interact specifically with acrolein, forming a non-toxic compound. See also Chapters 9 and 57. Other nitrogen mustards used include melphalan and chlorambucil.
Estramustine is a combination of chlormethine (mustine) with an oestrogen. It has both cytotoxic and hormonal action, and is generally used for the treatment of prostate cancer.
Nitrosoureas
Examples include lomustine and carmustine. As they are lipid soluble and cross the blood–brain barrier, they may be used against tumours of the brain and meninges. However, most nitrosoureas have a severe cumulative depressive effect on the bone marrow that starts 3–6 weeks after initiation of treatment.
Other alkylating agents
Busulfan has a selective effect on the bone marrow, depressing the formation of granulocytes and platelets in low dosage and of red cells in higher dosage. It has little or no effect on lymphoid tissue or the gastrointestinal tract. It is used in chronic granulocytic leukaemia.
Dacarbazine, a prodrug, is activated in the liver, and the resulting compound is subsequently cleaved in the target cell to release an alkylating derivative. Unwanted effects include myelotoxicity and severe nausea and vomiting. Temozolomide is a related compound with a restricted usage (malignant glioma).
Procarbazine inhibits DNA and RNA synthesis and interferes with mitosis at interphase. Its effects may be mediated by the production of active metabolites. It is given orally, and its main use is in Hodgkin’s disease. It causes disulfiram-like actions with alcohol (see Ch. 56), exacerbates the effects of central nervous system depressants and, because it is a weak monoamine oxidase inhibitor, can produce hypertension if given with certain sympathomimetic agents (see Ch. 46). It causes the usual unwanted effects, and can be leukaemogenic, carcinogenic and teratogenic. Allergic skin reactions may necessitate cessation of treatment.
Other alkylating agents in clinical use include thiotepa and treosulfan.
Platinum compounds
Cisplatin is a water-soluble planar coordination complex containing a central platinum atom surrounded by two chlorine atoms and two ammonia groups. Its action is analogous to that of the alkylating agents. When it enters the cell, Cl− dissociates, leaving a reactive complex that reacts with water and then interacts with DNA. It causes intrastrand cross-linking, probably between N7 and O6 of adjacent guanine molecules, which results in local denaturation of DNA.
Cisplatin has revolutionised the treatment of solid tumours of the testes and ovary. Therapeutically, it is given by slow intravenous injection or infusion. It is seriously nephrotoxic, and strict regimens of hydration and diuresis must be instituted. It has low myelotoxicity but causes very severe nausea and vomiting. The 5-HT3 receptor antagonists (e.g. ondansetron; see Chs 15, 29 and 38) are very effective in preventing this and have transformed cisplatin-based chemotherapy. Tinnitus and hearing loss in the high-frequency range may occur, as may peripheral neuropathies, hyperuricaemia and anaphylactic reactions.
Carboplatin is a derivative of cisplatin. Because it causes less nephrotoxicity, neurotoxicity, ototoxicity, nausea and vomiting than cisplatin (although it is more myelotoxic), it is sometimes given on an outpatient basis. Oxaliplatin is another platinum-containing compound with a restricted application.
Anticancer drugs Alkylating agents and related compounds
•
Alkylating agents have groups that form covalent bonds with cell substituents; a carbonium ion is the reactive intermediate. Most have two alkylating groups and can cross-link two nucleophilic sites such as the N7 of guanine in DNA. Cross-linking can cause defective replication through pairing of alkylguanine and thymine, leading to substitution of AT for GC, or it can cause excision of guanine and chain breakage.
•
Their principal effect occurs during DNA synthesis and the resulting damage triggers apoptosis.
•
Unwanted effects include myelosuppression, sterility and risk of non-lymphocytic leukaemia.
•
The main alkylating agents are:
–
nitrogen mustards, for example
cyclophosphamide, which is activated to give aldophosphamide, then converted to phosphoramide mustard (the cytotoxic molecule) and acrolein (which causes bladder damage that can be ameliorated by mesna). Cyclophosphamide myelosuppression affects particularly the lymphocytes
–
nitrosoureas, for example
lomustine, may act on non-dividing cells, can cross the blood–brain barrier and cause delayed, cumulative myelotoxicity.
•
Platinum compounds (e.g.
cisplatin) cause intrastrand linking in DNA. Cisplatin has low myelotoxicity but causes severe nausea and vomiting, and can be nephrotoxic. It has revolutionised the treatment of germ cell tumours.
Resistance to Anticancer Drugs
The resistance that neoplastic cells manifest to cytotoxic drugs is said to be primary (present when the drug is first given) or acquired (developing during treatment with the drug). Acquired resistance may result from either adaptation of the tumour cells or mutation, with the emergence of cells that are less susceptible or resistant to the drug and consequently have a selective advantage over the sensitive cells. The following are examples of various mechanisms of resistance. See Mimeault et al. (2008) for an up-to-date appraisal of this issue.
•
Decreased accumulation of cytotoxic drugs in cells as a result of the increased expression of cell surface, energy-dependent drug transport proteins. These are responsible for multidrug resistance to many structurally dissimilar anticancer drugs (e.g. doxorubicin, vinblastine and dactinomycin; see
Gottesman et al., 2002). An important member of this group is
P-glycoprotein (P-gp/MDR1; see
Ch. 8). The physiological role of P-glycoprotein is thought to be the protection of cells against environmental toxins. It functions as a hydrophobic ‘vacuum cleaner’, picking up foreign chemicals, such as drugs, as they enter the cell membrane and expelling them. Non-cytotoxic agents that reverse multidrug resistance are being investigated as potential adjuncts to treatment.
•
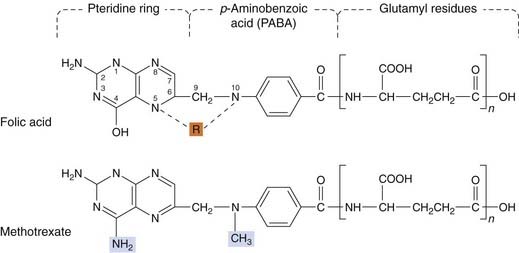
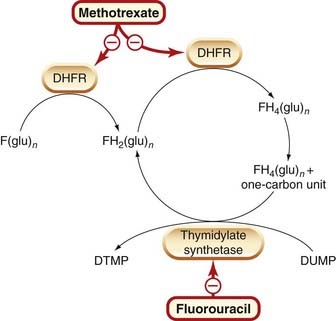
A decrease in the amount of drug taken up by the cell (e.g. in the case of methotrexate).
•
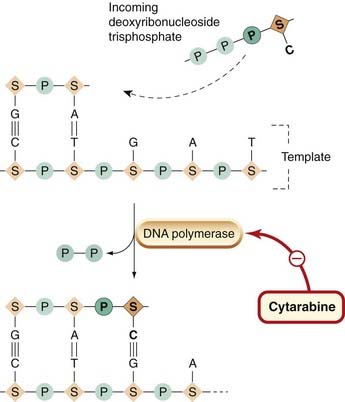
Insufficient activation of the drug. Some drugs require metabolic activation to manifest their antitumour activity. If this fails, they may no longer be effective. Examples include conversion of fluorouracil to FDUMP, phosphorylation of cytarabine and conversion of mercaptopurine to a fraudulent nucleotide.
•
Increase in inactivation (e.g. cytarabine and mercaptopurine).
•
Increased concentration of target enzyme (methotrexate).
•
Decreased requirement for substrate (crisantaspase).
•
Increased utilisation of alternative metabolic pathways (antimetabolites).
•
Rapid repair of drug-induced lesions (alkylating agents).
•
Altered activity of target, for example modified topoisomerase II (doxorubicin).
•
Mutations in various genes, giving rise to resistant target molecules. For example, the p53 gene and overexpression of the
Bcl-2 gene family (several cytotoxic drugs).
Future Developments
As the reader will have judged by now, our current approach to cancer chemotherapy embraces an eclectic mixture of drugs and techniques, all designed to target selectively cancer cells. Real therapeutic progress has been achieved, although ‘cancer’ as a disease (actually many different diseases with a similar outcome) has not been defeated and remains a massive challenge for future generations of researchers. In this therapeutic area, probably more than in any other, the debate about the risk–benefit of treatment and the patient quality of life issues has taken centre stage and remains a major area of concern. These sensitive issues have been explored by Duric & Stockler (2001) and Klastersky & Paesmans (2001).
The quest for less toxic forms of therapy is, of course, central to anticancer initiatives, and many new drugs or novel combination regimens are in clinical trial or at earlier stages of development (see, for example, Kurtz et al., 2003). What follows is a selection of new and different approaches to the treatment of cancer that may bear fruit over the next decade.
Angiogenesis and metalloproteinase inhibitors
Tumour cells produce metalloproteinases and angiogenic factors that facilitate tumour growth, invasion of normal tissue and metastases. Targeting the mechanisms involved could provide us with drugs that block metastases. Several existing drugs already target this process (e.g. bevacizumab) and it is likely that this area will see further development (see Griffioen & Molema, 2000; Thijssen et al., 2007).
Cyclo-oxygenase inhibitors
There is strong epidemiological and experimental evidence suggesting that chronic use of cyclo-oxygenase (COX) inhibitors (see Ch. 26) protects against cancer of the gastrointestinal tract and possibly other sites as well. The COX-2 isoform is overexpressed in about 85% of cancers, and prostanoids originating from this source may activate signalling pathways that enable cells to escape from apoptotic death. The COX-2 inhibitor celecoxib reduces mammary and gastrointestinal cancer incidence in animal models and causes regression of existing tumours. It is in trial in humans as an inhibitor of a familial type of colon tumour. Overall, COX-2 is now considered to be a potentially important target for anticancer drug development although, ironically, some argue that the mechanism of action is unrelated to COX inhibition. The literature is daunting and often controversial; see Karamouzis & Papavassiliou (2004) for recent comment.
Antisense oligonucleotides
Genetic approaches are seen by many experts as the hope for the future. Antisense oligonucleotides (see Ch. 59) are synthetic sequences of single-stranded DNA complementary to specific coding regions of mRNA, which can inhibit gene expression. An antisense drug, augmerosen, downregulates the antiapoptotic factor Bcl-2. In an early clinical trial, it sensitised malignant melanoma to standard anticancer drugs.
Gene therapy
The introduction of engineered genes, antisense oligonucleotides or siRNA by gene therapy (see Ch. 59) offers, in principle, enormous advantages over conventional approaches in terms of selective toxicity to cancer cells. There are many technical problems yet to be solved with the delivery of the genes, (e.g. p53 or growth factor antisense DNA) into the target cells. There have been clinical trials, some of which showed modest success (see, for example, Wolf & Dwayne Jenkins, 2002, on ovarian cancer trials), but progress has been disappointingly slow.
Reversal of multidrug resistance
Several non-cytotoxic drugs (e.g. verapamil) that inhibit P-glycoprotein can reverse multidrug resistance. Other drugs with this action are being investigated. In addition, the use of antibodies, immunotoxins, antisense oligonucleotides (see above) or liposome-encapsulated agents may be useful in the elimination of cells with multidrug resistance (reviewed by Gottesman & Pastan, 1993).
Telomerase is known to be important in maintaining cancer cell viability. Several strategies for controlling its activity have been reviewed by Keith et al. (2004).
References and Further Reading
General textbook
Airley R. Anticancer drugs. Chichester: Wiley-Blackwell; 2009. (Recent textbook covering all aspects from basic pharmacology to clinical use)
Mechanisms of carcinogenesis
Buys C.H.C.M. Telomeres, telomerase and cancer. N. Engl. J. Med.. 2000;342:1282-1283. (Clear, concise coverage)
Chambers A.F., Groom A.C., MacDonald I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer. 2, 2002. 563-557. (Review; stresses the importance of metastases in most cancer deaths, discusses the mechanisms involved in metastasis and raises the possibility of targeting these in anticancer drug development)
Griffioen A., Molema G. Angiogenesis: potentials for pharmacologic intervention in the treatment of cancer, cardiovascular diseases and chronic inflammation. Pharmacol. Rev.. 2000;52:237-268. (Comprehensive review covering virtually all aspects of angiogenesis and the potential methods of modifying it to produce an antineoplastic effect)
Haber D.A., Fearon E.R. The promise of cancer genetics. Lancet. 1998;351:1-8. (Excellent coverage; detailed tables of mutations in proto-oncogenes and tumour suppressor genes in human cancers)
Mimeault M., Hauke R., Batra S.K. Recent advances on the molecular mechanisms involved in the drug resistance of cancer cells and novel targeting therapies. Clin. Pharmacol. Ther.. 2008;83:673-691. (Comprehensive review covering all aspects of this field)
Talapatra S., Thompson C.B. Growth factor signalling in cell survival: implications for cancer treatment. J. Pharmacol. Exp. Ther.. 2001;298:873-878. (Succinct overview of death receptor-induced apoptosis, the role of growth factors in preventing it and potential drugs that could be used to promote cell death)
Weinberg R.A. How cancer arises. Sci. Am.. 1996;Sept:42-48. (Simple, clear overview, listing main oncogenes, tumour suppressor genes and the cell cycle; excellent diagrams)
Zörnig M., Hueber A.-O., Baum W., Evan G. Apoptosis regulators and their role in tumorigenesis. Biochim. Biophys. Acta. 2001;1551:F1-F37. (Extensive review describing the genes and mechanisms involved in apoptosis, and summarising the evidence that impaired apoptosis is a prerequisite for cancer development)
Anticancer therapy
Gottesman M.M., Fojo T., Bates S.E. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat. Rev. Cancer. 2002;2:48-56. (Outlines cellular mechanisms of resistance; describes ATP-dependent transporters, emphasising those in human cancer; considers resistance reversal strategies)
Houghton A.N., Scheinberg D. Monoclonal antibody therapies—a ‘constant’ threat to cancer. Nat. Med.. 2000;6:373-374. (Lucid article; very useful diagram)
Krause D.S., Van Etten R. Tyrosine kinases as targets for cancer therapy. N. Engl. J. Med.. 2005;353:172-187. (Excellent review on tyrosine kinases as targets; good diagrams and tables as well as a highly readable style)
Kurtz J.-E., Emmanuel A., Natarajan-Ame S., et al. Oral chemotherapy in colorectal cancer treatment: review of the literature. Eur. J. Int. Med.. 2003;14:18-25. (Discusses potential new leads in colorectal cancer; good tables summarising recent advances and clinical trials)
Norman K.L., Farassati F., Lee P.W.K. Oncolytic viruses and cancer therapy. Cytokine Growth Factor Rev.. 2001;12:271-282. (Describes mechanisms of action and efficacy of three oncolytic viruses in clinical trial)
Overall C.M., López-Otin C. Strategies for MMO inhibition in cancer: innovations for the post-trial era. Nat. Rev. Cancer. 2002;2:6577-7672. (Review of matrix metalloproteinases and their role in tumour metastasis; also discusses various approaches that could be used to target metalloproteinases, thus producing new anticancer drugs)
Reed J.C. Apoptosis-based therapies. Nat. Rev. Drug Discov.. 2002;1:111-121. (Excellent coverage, useful tables, good diagrams)
Savage D.G., Antman K.H. Imatinib mesylate—a new oral targeted therapy. N. Engl. J. Med.. 2002;346:683-693. (Review with detailed coverage of this drug for chronic myelogenous leukaemia; very good diagrams)
White C.A., Weaver R.L., Grillo-López. Antibody-targeted immunotherapy for treatment of malignancy. Annu. Rev. Med.. 2001;52:125-145. (Clear, comprehensive review; includes tables of monoclonals and radiolabelled monoclonals in clinical trial)
New directions and miscellaneous
Adjei A.A. Blocking oncogenic Ras signaling for cancer therapy. J. Natl. Cancer Inst.. 2001;93:1062-1074. (Gives details of Ras processing, activation, mutations, cytoplamsic targets and physiological role, and outlines therapeutic implications)
Anderson W.F. Gene therapy scores against cancer. Nat. Med.. 2000;6:862-863. (Short crisp article)
Armstrong A.C., Eaton D., Ewing J.C. Cellular immunotherapy for cancer. Br. Med. J.. 2001;323:1289-1293. (Brief discussion of rationale and possible future exploitation of tumour cell and dendritic cell vaccines and T cell therapy)
Carter P. Improving the efficacy of antibody-based cancer therapies. Nat. Rev. Cancer. 2001;1:118-128. (Review considering the possible future use of monoclonal antibodies to treat cancer; lists antibodies in advanced clinical trials)
Duric V., Stockler M. Patients’ preferences for adjuvant chemotherapy in early breast cancer. Lancet Oncol.. 2001;2:691-697. (The title is self-explanatory; deals with patients’ assessment of quality of life issues)
English J.M., Cobb M.H. Pharmacological inhibitors of MAPK pathways. Trends Pharmacol. Sci.. 2002;23:40-45. (Lists mitogen-activated protein kinases and discusses small-molecule inhibitors under investigation)
Favoni R.E., de Cupis A. The role of polypeptide growth factors in human carcinomas: new targets for a novel pharmacological approach. Pharmacol. Rev.. 2000;52:179-206. (Thorough review that describes 14 growth factor families, their signalling pathways and their possible role in cancer; it also deals with drug action on signalling pathways)
Gottesman M.M., Pastan I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu. Rev. Biochem.. 1993;62:385-427.
Karamouzis M.V., Papavassiliou A.G. COX-2 inhibition in cancer therapeutics: a field of controversy or a magic bullet? Expert Opin. Investig. Drugs. 2004;13:359-372. (Good review of the field of COX inhibitors in cancer therapy)
Keith W.N., Bilsland A., Hardie M., Evans T.R. Drug insight: cancer cell immortality—telomerase as a target for novel cancer gene therapies. Nat. Clin. Pract. Oncol.. 2004;1:88-96.
Klastersky J., Paesmans M. Response to chemotherapy, quality of life benefits and survival in advanced non-small lung cancer: review of literature results. Lung Cancer. 2001;34:S95-S101. (Another paper that addresses quality of life issues surrounding chemotherapy)
Smith I.E. New drugs for breast cancer. Lancet. 2002;360:790-792. (Succinct coverage)
Thijssen V.L., van Beijnum J.R., Mayo K.H., Griffioen A.W. Identification of novel drug targets for angiostatic cancer therapy; it takes two to tango. Curr. Pharm. Des.. 2007;13:3576-3583.
Wolf J.K., Dwayne Jenkins A. Gene therapy for ovarian cancer (review). Int. J. Oncol.. 2002;21:461-468. (Very readable review of ovarian cancer and basic concepts in gene therapy, coupled with a round-up of data on compounds in clinical trial)
Useful Web resources
http://www.cancer.org/ (The US equivalent of the Web site below. The best sections for you are those marked Health Information Seekers and Professionals)
http://www.cancerresearchuk.org (The Web site of Cancer Research UK, the largest cancer charity in the UK. Contains valuable data on the epidemiology and treatment of cancer, including links to clinical trials. An excellent resource)
 Crisantaspase is a preparation of the enzyme asparaginase, given intramuscularly or intravenously. It converts asparagine to aspartic acid and ammonia, and is active against tumour cells, such as those of acute lymphoblastic leukaemia, that have lost the capacity to synthesise asparagine and therefore require an exogenous source. As most normal body cells are able to synthesise asparagine, the drug has a fairly selective action and has very little suppressive effect on the bone marrow, the mucosa of the gastrointestinal tract or hair follicles. It may cause nausea and vomiting, central nervous system depression, anaphylactic reactions and liver damage.
Crisantaspase is a preparation of the enzyme asparaginase, given intramuscularly or intravenously. It converts asparagine to aspartic acid and ammonia, and is active against tumour cells, such as those of acute lymphoblastic leukaemia, that have lost the capacity to synthesise asparagine and therefore require an exogenous source. As most normal body cells are able to synthesise asparagine, the drug has a fairly selective action and has very little suppressive effect on the bone marrow, the mucosa of the gastrointestinal tract or hair follicles. It may cause nausea and vomiting, central nervous system depression, anaphylactic reactions and liver damage.