Emile L. Boulpaep
In the preceding chapters, we examined cardiovascular regulation at several different levels. Powerful systemic mechanisms operate over both the short term and the long term to control mean arterial pressure and cardiac output. Operating independently of these are local mechanisms of control that regulate blood flow at the microcirculatory level. In addition, individual organs have their own unique tools for managing specific circulatory requirements. In this chapter, we put it all together and learn how the cardiovascular system integrates the complex systemic, local, and individualized regulatory mechanisms in response to the demands of everyday life.
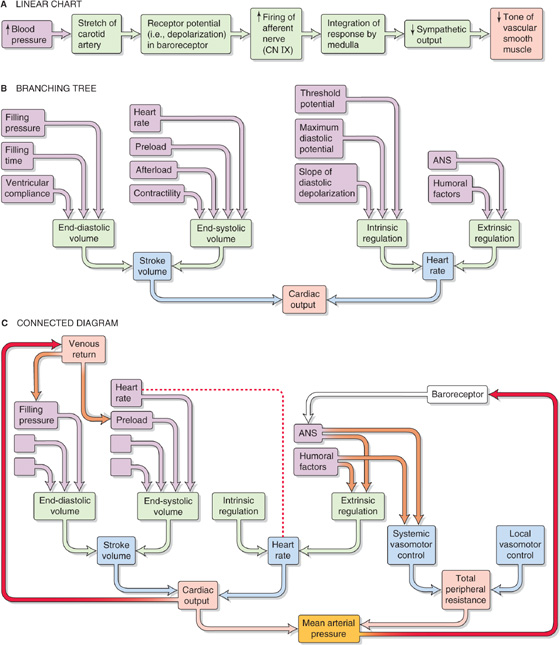
In the previous chapters, we often presented physiological responses in a linear sequence or on a linear chart. For instance, we might represent the carotid-baroreceptor feedback loop (see Chapter 23) as a linear sequence of events (Fig. 25-1A). However, cardiovascular parameters and associated physiological responses are often related by multiple factors, requiring a more complex diagram called a branching tree (or an algorithmic tree). For example, in our discussion of the control of cardiac output (see Chapter 23), we started with the knowledge that cardiac output depends on two parameters—stroke volume and heart rate. Therefore, with our very first step, we come to a fork in the road—an example of a branching tree (Fig. 25-1B). At the next level, we encounter a pair of forks because stroke volume and heart rate both depend on two parameters. Finally, at the third level, we see that each of the determinants of stroke volume and heart rate depends on multiple factors (i.e., multiple forks).
Figure 25-1 Patterns of cardiovascular control. A, The baroreceptor reflex is depicted as if increased blood pressure affected a single stretch receptor, which ultimately would influence a single effector (i.e., vascular smooth muscle). B, Cardiac output depends on multiple parameters, which in turn depend on multiple parameters, and so on. However, we ignore potential interactions among parameters. C, A branching tree represents the control of mean arterial pressure. The left limb repeats B. Superimposed on this simple tree are three more complex interactions: (1) feedback loops (red arrows), (2) two occurrences of the same parameter (connected by the red dashed line), and (3) examples of parameters exerting effects on two different branches of the tree (brown arrows). ANS, autonomic nervous system.
The control of some cardiovascular parameters is even more complex, requiring that we graft branches from smaller trees. For example, we know from Ohm’s law that mean arterial pressure depends on both cardiac output (and all the elements in its branching tree in Fig. 25-1B) and total peripheral resistance, which requires a branching tree of its own (Fig. 25-1C). Moreover, sometimes an element in one part of the “forest” interacts with another element that is far away. A physiological system with such complex interactions is best represented by a connected diagram, which may include feedback loops (Fig. 25-1C, red arrows), parameters that appear more than once in the tree (connected by a red dashed line), or factors that modulate parameters in two different branches of the tree (connected by brown arrows). Although not shown in Figure 25-1C, several feedback loops may impinge on a single element, and some loops are more dominant than others. The complex interactions among parameters make it difficult to distinguish factors of overriding importance from those of lesser weight. Moreover, when one disturbs a single parameter in a complex physiological system, the initial state of other parameters determines the end-state of the system. In previous chapters, we have chosen situations that artificially isolate one portion of the cardiovascular system (e.g., heart, microcirculation) to explain in a simple way the homeostatic control mechanisms that govern that subsystem. However, conditions isolating subsystems rarely apply to a real person.
How can we evaluate which parameters are crucial? As an example, consider one subsystem, the heart. Let us assume that we can rigorously analyze all determinants of cardiac function—such as Starling’s law, force-velocity relationships, effect of heart rate on contractility, and so forth. These analyses take the form of mathematical expressions, which we can combine by systems analysis to create a model that describes the behavior of the entire heart—at least theoretically. How can we test whether our model is reasonable? We can compare the physiological response of the heart in vivo with the response predicted by a computer simulation of the model. Using this approach, we may be able to establish whether we have used the correct feedback loops, whether we have assigned proper values to various elements, and whether we have assigned the proper weight to each interaction. In this way, we can use any agreement between the experimental data and the performance of the model as evidence—but not proof—that the concepts contained in the model are reasonable.
In performing a systems analysis of the entire cardiovascular system, we must consider the interrelationships among the various “subsystems” summarized in the central yellow block of Figure 25-2. Not surprisingly, we cannot fully understand how a particular disturbance affects the overall circulation unless we consider all subsystems in an integrated fashion. For instance, consider the effects of administering norepinephrine, which has a high affinity for α1 adrenoceptors, less for β1 adrenoreceptors, and far less for β2 adrenoceptors. These receptors are present, in varying degrees, in both the blood vessels and the heart. A branching tree would predict the following. Because α1 adrenoceptors (high affinity) are present in most vascular beds, we expect widespread vasoconstriction. Because β2 adrenoceptors (low affinity) are present in only a few vascular beds, we predict little vasodilation. Because β1 adrenoceptors (intermediate affinity) are present in pacemaker and myocardial cells of the heart, we would anticipate an increase in both heart rate and contractility and therefore an increase in cardiac output.
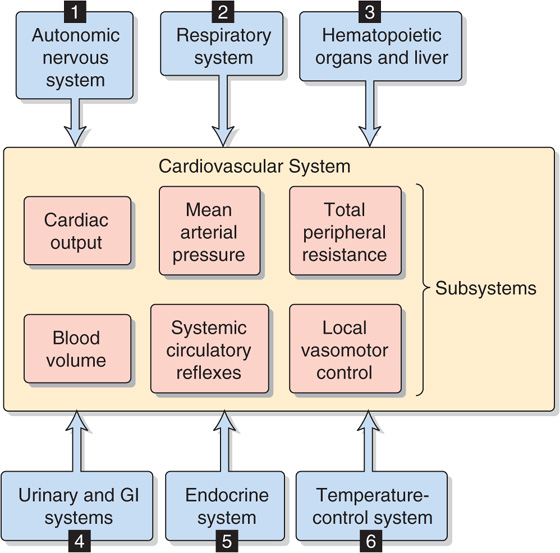
Figure 25-2 Interaction among cardiovascular subsystems and noncirculatory systems. GI, gastrointestinal.
Although our analysis predicts that the heart rate should increase, in most cases, the dominant effect of intravenous norepinephrine injection is to slow down the heart. The explanation, which relies on a connected diagram, is that increased peripheral resistance (caused by stimulation of α1 receptors) and increased cardiac output (caused by stimulation of β1 receptors) combine to cause a substantial rise in mean arterial pressure. The baroreceptor reflex (see red arrow on right in Fig. 25-1C) then intervenes to instruct the heart to slow down (see Chapter 23). However, bradycardia may not occur if many vascular beds were dilated before the administration of norepinephrine; in this case, the rise in blood pressure would be modest. Bradycardia might also not occur if the baroreceptor reflex were less sensitive, as would be the case in a chronically hypertensive patient (see Chapter 23 for the box on this topic). Thus, the effect of intravenous administration of norepinephrine on heart rate depends on the preexisting state of various subsystems.
In trying to understand the integrated response of the cardiovascular system to an insult, we must include in our analysis not only all the subsystems of the cardiovascular system but also the pertinent control systems outside the circulation (see blue boxes in Fig. 25-2):
1. Autonomic nervous system. Part of the autonomic nervous system (ANS) is intimately involved in cardiovascular control (e.g., high-pressure baroreceptor response). On the other hand, a generalized activation of the ANS, such as occurs with the fight-or-flight response (see Chapter 14), also affects the circulation.
2. Respiratory system. We have already seen that ventilatory activity converts the intrinsic bradycardia response to tachycardia during stimulation of the peripheral chemoreceptors (see Chapter 23). In addition, the action of the respiratory muscles during inspiration causes intrathoracic pressure to become more negative, thereby increasing venous return. A third example is that the evaporative loss of water during breathing reduces total body water and, ultimately, blood volume.
3. Hematopoietic organs and liver. These systems control blood composition in terms of cell constituents and plasma proteins. The hematocrit and large proteins (e.g., fibrinogen) are major determinants of blood viscosity (see Chapter 17) and therefore of blood flow. Because the plasma proteins also determine colloid osmotic pressure, they are a major component of the Starling forces, which determine the distribution of extracellular fluid between the interstitium and the blood plasma. (See Chapter 20.)
4. Gastrointestinal and urinary systems. Because the gastrointestinal tract and kidneys are the principal organs determining input and output of electrolytes and water, they are mainly responsible for controlling the volume and electrolyte composition of extracellular fluid. Extracellular fluid volume plays a central role in the long-term control of blood pressure (see Chapter 40).
5. Endocrine system. Part of the endocrine system is intimately involved in cardiovascular control (e.g., epinephrine release by the adrenal medulla). Other hormones influence the cardiovascular system either because they are vasoactive agents (see Chapter 23) or because they regulate fluid volume and electrolyte composition by acting on the kidney and gastrointestinal system.
6. Temperature control system. The cardiovascular system is a major effector organ for thermoregulation, carrying blood from the body core to the skin, where heat loss then occurs (see Chapter 59). In part, this heat loss occurs as sweat glands secrete fluid that then evaporates. However, the loss of extracellular fluid volume reduces the effective circulating volume (see Chapter 23).
Inclusion of control elements outside the circulation (Fig. 25-2) in our connected diagram of the cardiovascular system (Fig. 25-1C) would expand the computer model to include hundreds of independent and dependent variables. Rather than trying to grasp such an exhaustive model, we work here through the integrated cardiovascular responses to four important circulatory “stresses”: (1) orthostasis (i.e., standing up), (2) emotional stress, (3) exercise, and (4) hemorrhage.
About two thirds of the total blood volume resides in the systemic veins (see Chapter 19). When a recumbent subject assumes an upright position, the blood shifts from the central blood volume reservoirs and other veins to large veins in the dependent limbs. In discussing Figure 17-8B, we assumed that the cardiovascular system somehow made the adjustments necessary to keep right atrial pressure (RAP) constant at about +2 mm Hg. Indeed, unless compensatory mechanisms intervene, blood redistribution will lower not only arterial blood pressure but also venous return and thus cardiac output.
To illustrate the effect of blood redistribution on venous return, we will represent the entire circulatory system by a horizontal, distensible cylinder 180 cm in length (the height of our person) and 3 cm in radius (Fig. 25-3A). This cylinder holds ~5000 mL of blood (the normal blood volume). We know that immediately after a cardiac arrest, the entire vascular system will have a mean systemic filling pressure (MSFP) of ~7 mm Hg (see Chapter 23). The MSFP is the pressure in the circulation that would remain in the absence of any pumping or any gravity effects. Thus, if a subject is recumbent and has no heartbeat, and if the cardiovascular system is filled with a normal blood volume of 5000 mL, the overall compliance of the system will produce a uniform pressure of ~7 mm Hg. If we were to transfuse an additional 100 mL of blood into our cylinder, the MSFP would rise by ~1 mm Hg. We can conclude that the compliance, expressed as a normalized distensibility (see Chapter 19), is


Figure 25-3 Model of the orthostatic redistribution of blood. A, A horizontal tube (3-cm radius, 180 cm long) contains the entire blood volume (5 L). With no blood flow, pressure inside the tube is uniform and corresponds to a mean systemic filling pressure of 7 mm Hg. B, With the cylinder upright (orthostasis), pressure gradually increases toward the bottom, causing increasingly greater distention of this compliant tube. Because blood volume has shifted to the bottom, the upper level of the blood column is 30 cm below the level of the heart, preventing venous return. C, Reducing compliance of the tube by half also causes distention to fall by half. With the reduced shift of blood volume, the upper level of the blood column now just reaches the heart.
Thus, for every 2% increase in blood volume, the MSFP of the cylinder increases by 1 mm Hg.
What will happen to our cylinder if we now turn it upright? This position is called orthostasis (from the Greek orthos [upright] + histanai [to stand]). Because we have a vertical column of blood 180 cm tall, we must now consider gravity (Fig. 25-3B). The highest pressures will be at the bottom of the cylinder. (Figure 25-3B shows that orthostasis causes venous pressure at the ankle to rise from 5 to 100 mm Hg.) Therefore, our cylinder will distend maximally at the bottom, and this distention represents a shift in blood volume. The bottom of the cylinder (corresponding to the “dependent areas” of a person) gains volume, whereas the top (i.e., corresponding to the cranial portion of a person) loses blood volume. In fact, there would be no blood at all at the top of the cylinder.
By just how much would the column of blood fall in our upright, 180-cm-tall cylinder? If the overall vascular volume distensibility were 0.02/(mm Hg), then the actual height of the blood column inside the cylinder would be only about 100 cm. If the heart were 50 cm below the top of the cylinder, then the top of the column would now be ~30 cm below the level of the heart. Therefore, there would be no blood to return to the heart. Moreover, the RAP would be negative (−30 cm H2O, or about −22 mm Hg). Because the heart cannot create a vacuum this large at its input by “sucking” blood—in fact, the heart must be filled by a positive RAP—cardiac output would fall to zero. (See Note: Calculation of Distension in an Upright Cylinder)
If our model predicts that orthostasis should cause RAP to fall to −22 mm Hg, how is it that the body manages to maintain RAP at about +2 mm Hg in the upright position? The answer is that pooling of blood in the dependent vessels is much less pronounced during orthostasis than would be predicted by Figure 25-3B, where ~2.2 L disappeared from the top of the cylinder. The actual amount of pooling in both legs of a real person is only ~500 mL. Four major factors help reduce pooling and maintain RAP. (See Note: Hypothetical Volume of Pooled Blood during Orthostasis)
Nonuniform Initial Distribution of Blood In our cylinder example, the blood was initially distributed evenly throughout the length of the cylinder. In a recumbent human, however, most of the blood in large veins is located in the central blood volume (see Chapter 19), that is, the vessels near the heart. If a large fraction of the blood had started off in the head, the orthostatic shift of blood would have been more dramatic, as in Figure 25-3B. The majority of the 500 mL of blood that pools in the legs during orthostasis comes from the intrathoracic vascular compartments. What is the sequence of events by which blood volume redistributes during orthostasis? As one stands, the output from the heart for a number of beats exceeds the venous return into the thoracic pool. This excess blood ends up filling the vessels in the dependent regions of the body. The result is a net transfer of blood—by way of the heart—from the intrathoracic vascular compartments to the dependent vessels.
Nonuniform Distensibility of the Vessels In Figure 25-3B, we assumed a relative distensibility of 0.02/(mm Hg). If we had instead used a value of 0.01/(mm Hg)—that is, if the vessels were less distensible—standing would cause a less dramatic shift of blood to the dependent vessels, ~1.4 L (Fig. 25-3C) instead of ~2.2 L (Fig. 25-3B). As a result, the height of the blood column would fall from 180 to only 130 cm (Fig. 25-3C) in the upright position, rather than to 100 cm (Fig. 25-3B). Assuming a lower distensibility for the leg veins is reasonable because small vessels are far stiffer than larger ones, such as the aorta and vena cava. With the lower relative distensibility of 0.01/(mm Hg), the column of blood would just reach the heart. Indeed, when a subject stands quietly, the zero effective pressure level—the height in the body where vascular pressure equals atmospheric pressure—is about at the level of the right atrium. Obviously, if the circulatory system reduces its distensibility even further through the regulated contraction of vascular smooth muscle (discussed later), the height of the column of blood will increase and improve venous return. (See Note: Calculation of Distension in an Upright Cylinder)
Muscle Pumps An important compensation for blood pooling during orthostasis comes from skeletal muscle contraction. When a person stands, the muscles of the legs and abdomen tighten. The presence of valves in the veins, as well as intermittent muscular movement, contributes to the flow of blood upward along the veins (see Figs. 22-7C and 24-6). Vessels of the abdominal region remain nearly unaffected by orthostasis because the abdominal viscera are contained in a water-filled jacket that is maintained by the tone of the abdominal muscles.
Autonomic Reflexes Because of decreased venous return, cardiac output tends to fall by ~20% soon after one assumes an erect position. However, the fall in cardiac output would be even greater in the absence of autonomic reflexes. The decreased venous return leads to a fall in RAP, which in turn leads to a decrease in stroke volume and thus arterial pressure. High-pressure baroreceptors (see Chapter 23) sense this decrease in arterial pressure, which leads to an increased sympathetic output that raises vascular tone throughout the body and increases heart rate and contractility. Together, the constriction of arterioles (which raises total peripheral resistance) and the increased heart rate restore the systemic mean arterial pressure, despite a small decrease in stroke volume. In the dependent regions of the body, the sympathetic response also increases the tone of the veins, thereby decreasing their diameter and their capacity (compare B and C in Fig. 25-3). (See Note: Baroreceptor Responses in Orthostasis)
In summary, of the four factors that contribute to the stability of RAP during orthostasis, two are anatomical (i.e., nonuniformities of initial blood volume distribution and distensibility) and two are physiological (i.e., muscle pumps and autonomic reflexes). The two physiological mechanisms are both important. Indeed, after a lumbar sympathectomy, patients tend to faint when standing. However, after some months, they are able to compensate, perhaps by using the muscle pumps more effectively or by enhancing the sympathetic response of the heart.
The extent of the orthostatic response—how much the heart rate or peripheral vascular resistance increases under the control of the ANS—depends on a variety of factors (Table 25-1), which involve nearly the entire cardiovascular system. Because these factors may differ from person to person or within any one individual according to the circumstances, the orthostatic response is highly variable. We now discuss two examples of this variability.
Table 25-1 Factors Influencing the Degree of Orthostatic Response*
Total blood volume |
Distribution of blood volume |
Size of vessels in dependent regions of the body |
Vascular distensibility |
Mean systemic filling pressure (pressure in the absence of cardiac output) |
Level at which zero effective pressure is normally located in a particular individual |
Degree of tilt |
Skeletal muscle tone; strength and rate of intermittent contraction of skeletal muscles |
Vascular sufficiency |
Abdominal muscle tone |
Temperature |
Response of low-pressure receptors |
Response of high-pressure baroreceptors |
Activity of the sympathetic system |
Initial heart rate |
Initial myocardial contractility |
Sensitivity of vascular smooth muscle to sympathetic stimulation |
*That is, by how much does standing up increase heart rate and peripheral vascular resistance?
Postural Hypotension In very sensitive subjects lying on a tilt table, a sudden orthostatic tilt can cause such a large fall in arterial pressure that the individual becomes dizzy or even faints. Fainting is caused by a transient fall in arterial pressure that causes cerebral perfusion to become inadequate.
Temperature Effects In a cool environment, where the arterioles in the lower extremities are constricted, the initial dip in arterial pressure can be small, despite the decrease in stroke volume. The explanation is that the high arteriolar resistance delays the transfer of blood from the thoracic pool to the legs. As a result, the sympathetic response to the small drop in mean arterial pressure may already be in effect before further pooling can occur. In a warm environment, where the arterioles in the skin are more dilated, orthostasis leads to faster transfer of blood from the thoracic pool to the legs so that—before the sympathetic response can develop—the initial decreases in stroke volume, mean arterial pressure, and pulse pressure can be large. Thus, soldiers standing quietly at attention in hot weather are more likely to faint than are soldiers marching in a cold environment because of differences in muscle pump activity and vasoconstriction. (See Note: Effects of Temperature on Venous Pooling)
Emotional responses vary greatly among people. A severe emotional reaction can resemble the fight-or-flight response in animals (see Chapter 14). This defense reaction causes a generalized increase in skeletal muscle tone and increased sensory attention.
Fight-or-flight behavior is an extreme example of an integrated acute stress response that originates entirely within the central nervous system (CNS), without involvement of peripheral sensors or reflexes. The response is due to the activation of sensory centers in the cortex (Fig. 25-4), which activate a part of the limbic system called the amygdalae. The amygdalae in turn activate the locus coeruleus (see Chapter 13), which is in the pons, as well as hypothalamic nuclei. Noradrenergic neurons in the locus coeruleus project to nearly every part of the CNS (see Fig. 13-7), including the hypothalamic paraventricular nucleus (PVN), which produces both an endocrine and an ANS response. The endocrine response of the PVN involves (1) release of arginine vasopressin (AVP) by magnocellular neurons in the PVN (see Fig. 40-8), thereby reducing urine output (see Chapter 38); and (2) release of corticotropin-releasing hormone (see Chapter 50) by parvocellular neurons in the PVN, activating the hypothalamic-pituitary-adrenal axis and thereby releasing cortisol, which is important for the metabolic response to stress. The ANS response of the PVN involves projections to (1) autonomic nuclei in the brainstem (dorsal motor nucleus of the vagus, rostral ventrolateral medulla, and NTS) that are part of the medullary cardiovascular center (see Chapter 23) and (2) direct projections to the spinal intermediolateral column (Fig. 25-4).
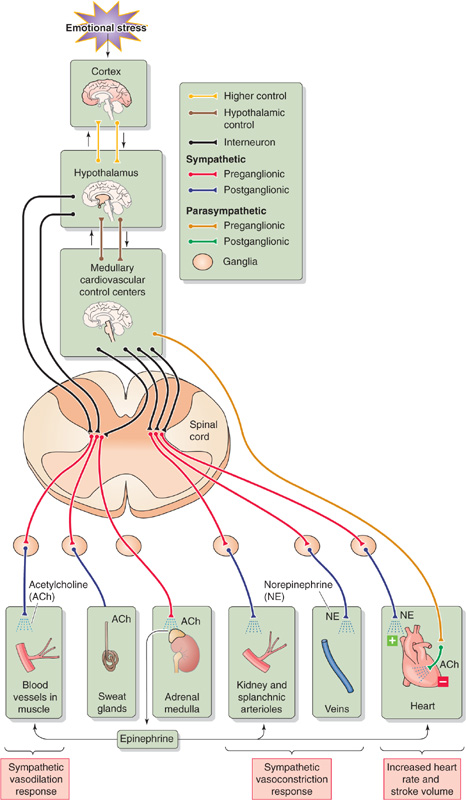
Figure 25-4 Fight-or-flight response.
The overall fight-or-flight response involves the following:
1. Skeletal muscle blood flow. In animals—but perhaps not in humans—activation of postganglionic sympathetic cholinergic neurons (see Chapter 14) directly causes a rapid increase in blood flow to skeletal muscle (see Chapter 23). In humans as well as in other mammals, flow also increases secondarily more slowly and less dramatically because the adrenal medulla releases epinephrine, which acts on β2 adrenoceptors on skeletal muscle blood vessels. The result is dilation and an increase in blood flow. In a full-blown fight-or-flight reaction, muscle exercise generates metabolites that further increase skeletal muscle blood flow (see Chapter 24). Of course, humans may not exercise skeletal muscle in responding to internal stress (e.g., anxiety or panic).
2. Cutaneous blood flow. The sympathetic response causes little change in blood flow to skin unless it stimulates sweating. The neural pathway involves sympathetic cholinergic neurons (see Chapter 14), which release acetylcholine and perhaps vasodilatory neurotransmitters (e.g., calcitonin gene–related peptide, vasoactive intestinal peptide). The acetylcholine causes the secretion of sweat and possibly also the local formation of kinins (see Chapter 23). These kinins increase capillary permeability and presumably also dilate arterioles but constrict venules (i.e., increasing the midcapillary pressure). The result would be an increased filtration of fluid from the skin capillaries into the interstitium, causing dermal swelling.
3. Adrenal medulla. Preganglionic sympathetic neurons stimulate the chromaffin cells to release epinephrine, which causes vasodilation in muscle (through β2 adrenoceptors) and vasoconstriction in the kidney and splanchnic beds (through α1 adrenoceptors).
4. Renal and splanchnic blood flow. In virtually all vascular beds, increased sympathetic output causes vasoconstriction and thereby decreases blood flow. The systemic release of epinephrine also vasoconstricts these vascular beds rich in α1 adrenoceptors.
5. Veins. Most veins constrict in response to sympathetic output.
6. The heart. Increased sympathetic output and decreased vagal output cause a rise in heart rate and contractility, so that cardiac output increases.
7. Blood volume. High plasma levels of AVP reduce urine output and maintain blood volume.
8. Mean arterial pressure. Depending on the balance of vasodilation and vasoconstriction, the overall result of vascular resistance changes may be either a decrease or an increase in total peripheral resistance. Nevertheless, because cardiac output increases, the net result of an increased cardiac output and a resistance change is an increase in arterial pressure.
About one fifth of humans experience one or more episodes of fainting during adolescence. This type of fainting is known as vasodepressor syncope or vasovagal syncope (VVS). About 40% of syncope cases seen in outpatient settings are vasovagal in nature. VVS can occur in response to a sudden emotional stress, phlebotomy, the sight of blood, or acute pain. Fainting usually starts when the individual is standing or sitting, rarely when the individual is recumbent. The loss of consciousness is due to a transient fall in perfusion pressure to the brain. The “playing dead” reaction in animals is the equivalent of VVS in humans.
VVS originates with activation of specific areas in the cerebral cortex. Indeed, stimulation of areas in the anterior cingulate gyrus can trigger a faint. Although the exact trigger is not known, VVS has been attributed to activation of the Bezold-Jarisch reflex. This reflex—originally described as the cardiorespiratory response to the intravenous injection of veratrum alkaloids—causes bradycardia, hypotension, and apnea. In experimental animals, stimulation of arterial baroreceptors or ventricular baroreceptors (see Chapter 23) by any of a host of chemicals—veratrum alkaloids, nicotine, capsaicin, histamine, serotonin, snake and insect venoms—can also trigger the Bezold-Jarisch reflex. In patients, coronary injection of contrast material or of thrombolytic agents can cause VVS, presumably by stimulating ventricular receptors. It is possible that these chemical stimuli activate the same stretch-sensitive TRPC channels of arterial baroreceptors (see Chapter 23) that are usually activated by high blood pressure. In humans, triggers clearly distinct from those known to initiate a Bezold-Jarisch reflex can also elicit VVS. Whatever the actual trigger, vagal afferents carry signals to higher CNS centers, which act through autonomic nuclei in the medulla to cause a massive stimulation of the parasympathetic system and abolition of sympathetic tone.
VVS involves changes in several parameters (Fig. 25-5):
1. Total peripheral resistance. A massive vasodilation results from the removal of sympathetic tone from the resistance vessels of the skeletal muscle, splanchnic, renal, and cerebral circulations. The resulting fall in blood pressure fails to activate a normal baroreceptor response.
2. Cardiac output. Intense vagal output to the heart causes bradycardia and decreased stroke volume, resulting in a marked decrease in cardiac output. Because atropine, a muscarinic receptor blocker, does not reliably prevent syncope, decreased sympathetic tone to the heart may also play a role in causing bradycardia.
3. Arterial pressure. The combination of a sudden decrease in both total peripheral resistance and cardiac output causes a profound fall in mean arterial pressure.
4. Cerebral blood flow. The fall in mean arterial pressure causes global cerebral ischemia. If the decreased cerebral blood flow persists for only a few seconds, the result is dizziness or faintness. If it lasts for ~10 seconds, the subject loses consciousness. The stress underlying the common faint also may provoke hyperventilation, which lowers arterial PCO2 (see Chapter 31). The resulting constriction of cerebral blood vessels (see Chapter 24) further impairs cerebral blood flow, which increases the likelihood of a faint.
5. Other manifestations of altered ANS activity. Pallor of the skin and sweating (beads of perspiration) are signs that often appear before the loss of consciousness. Intense vagal stimulation of the gastrointestinal tract may cause epigastric pain that is interpreted as nausea. Mydriasis (pupillary dilation) as well as visual blurring can also result from parasympathetic stimulation. (See Note: Ocular Symptoms and Signs Associated with Fainting)
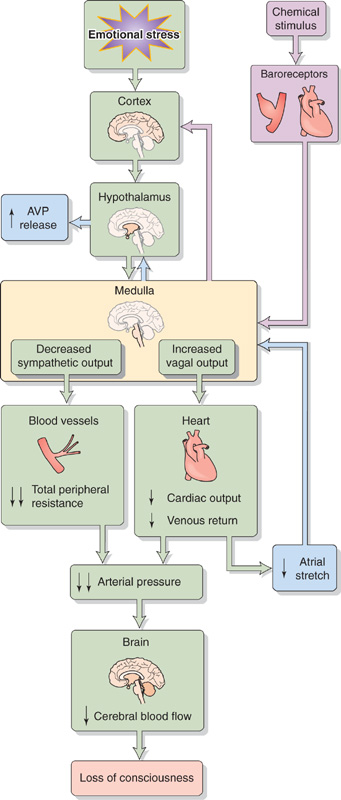
Figure 25-5 Vasovagal syncope. AVP, arginine vasopressin.
Fainting is more likely to occur in a warm room, after a volume loss (e.g., dehydration or hemorrhage), or after standing up or other maneuvers that tend to lower mean arterial pressure. You might think that these stresses would trigger baroreceptor responses that increase cardiac output and vascular resistance, thereby making fainting less likely. However, the same integrated pattern of brain activity that orchestrates VVS also appears to suppress the expected baroreceptor reflexes that would otherwise counteract the syncope. (See Note: Suppression of the Classical Baroreceptor Response in Vasovagal Syncope)
After regaining consciousness, the patient often notices oliguria (reduced urine output), caused by high plasma levels of AVP (also known as antidiuretic hormone; see Chapter 38). Elevated levels of AVP can result in part from the reduced atrial stretch that occurs during periods of decreased venous return (see Chapter 23). The pallor and nausea that persist after fainting may also result from the high levels of circulating AVP.
Adaptation to exercise probably places the greatest demands on circulatory function. The main feature of the cardiovascular response to exercise is an increased cardiac output, up to 4 or 5 times the resting cardiac output. The increase in cardiac output during exercise is the result of increased heart rate (~3 times control) more than of increased stroke volume (~1.5 times control). The cardiovascular response to exercise has both early and late components and originates from higher centers in the CNS (early), from mechanical and chemical changes triggered by contracting skeletal muscle (delayed), and from various reflexes (delayed).
Contracting skeletal muscle produces cardiovascular changes that mimic many of those that occur during exercise. In fact, early physiologists believed that these skeletal muscle effects triggered all of the cardiovascular changes associated with exercise. We now know that these skeletal muscle effects are important only for the delayed cardiovascular responses in exercise. Muscle contraction directly affects the cardiovascular system in two ways (Fig. 25-6)—a mechanical response that increases venous return and a chemical response that dilates blood vessels in active muscle.
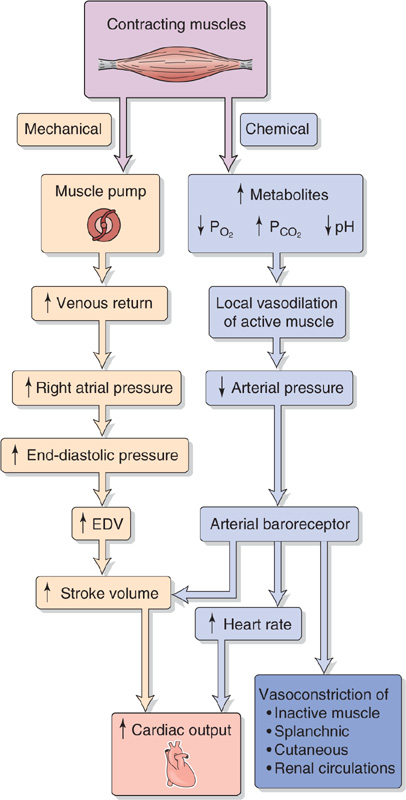
Figure 25-6 An early model of how exercise affects cardiovascular function. EDV, end-diastolic volume.
Mechanical Response: Increased Venous Return The pumping action of contracting skeletal muscle improves venous return (see Chapters 22 and 24). As a result, RAP, ventricular end-diastolic pressure, and end-diastolic volume should all increase. According to the Starling mechanism (see Chapter 22), the result should be an increase in stroke volume.
Chemical Response: Local Vasodilation in Active Muscle Enhanced skeletal muscle metabolism produces multiple changes in the chemistry of the interstitial fluid. The PO2 and pH fall, whereas other metabolites (CO2, lactic acid, K+, and adenosine) accumulate. Moreover, the accumulation of metabolites causes interstitial osmolarity to increase. After a small delay that follows the onset of muscle contraction, the developing chemical changes cause the arterioles to dilate (see Chapters 20 and 24), which may lead to an initial fall in arterial pressure. However, this fall is transient because of the intervening baroreceptor response (see Chapter 23), which increases heart rate and stroke volume, both of which enhance cardiac output. At the same time, the baroreceptor reflex vasoconstricts inactive muscle regions as well as the splanchnic, renal, and cutaneous circulations.
Cardiovascular physiologists believed for a long time that the mechanical and chemical limbs in Figure 25-6, both of which originate in active muscle, are responsible for the increased cardiac output during exercise. However, not only is Figure 25-6 incomplete, some of its predictions are incorrect or do not occur in the predicted order. As far as the mechanical effect on venous return is concerned, the model predicts a rise in ventricular end-diastolic pressure and thus end-diastolic volume. As far as the chemical effect is concerned, the model predicts that it should take time for chemical changes in active skeletal muscle to produce local vasodilation. Therefore, there ought to be a time lag between the initiation of exercise and the fall in mean arterial pressure that triggers the baroreceptor response (e.g., increased heart rate).
In the 1950s, Rushmer tested these predictions on trained unanesthetized dogs. Recording the mechanical limb of Figure 25-6, he found that at the onset of exercise, left ventricular end-diastolic pressure does not rise and that left ventricular end-diastolic volume diminishes rather than increases. These findings cast doubt on the primary role of the Starling mechanism in raising stroke volume during exercise. Recording the chemical limb of Figure 25-6, Rushmer found no transient fall in mean arterial pressure, thus casting doubt on the importance of the baroreceptor reflex. Furthermore, he saw no delay between the onset of exercise and the increase of heart rate, thus calling into question the idea that the chemically induced vasodilation is at the root of the tachycardia.
The explanation for the discrepancies between the predicted and actual findings is the presence of a central command that rapidly activates the sympathetic division of the ANS.
During exercise, a central command controls the parallel activation of both the motor cortex (see Chapter 16) and cardiovascular centers. The central command involves such brain areas as the medial prefrontal cortex (involved in the mental state of thinking and planning exercise) as well as the insula and anterior cingulate gyrus, which are cortical parts of the limbic system (see Chapter 14). Indeed, the medial prefrontal cortex receives multiple limbic inputs. Moreover, both cortical centers modulate stress-related sympathetic outflow, including the sympathetic outflow related to exercise. Rushmer and his colleagues explored various sites in the diencephalon (see Chapter 10) of dogs to determine if stimulation of any of them might mimic the integrated sympathetic response to exercise. In the 1950s, Rushmer found that stimulation of the H2 fields of Forel in the ventral thalamus or neurons in the periventricular gray matter of the hypothalamus reproduced all the details of the cardiac response to exercise, even though the muscles of the dog were completely quiescent. The central command centers project to the lateral hypothalamus, rostral ventrolateral medulla, and NTS to make autonomic adaptations appropriate for exercise (Fig. 25-7): (See Note: Central Commands for Exercise Originating Outside the Medulla)
1. Increased cardiac output. Increased sympathetic output to the heart causes early tachycardia and increased contractility, resulting in a rapid upsurge of cardiac output.
2. Vasoconstriction. Sympathetic output from the medulla causes vasoconstriction in inactive muscle regions as well as in the renal, splanchnic, and cutaneous circulations. The net effect is to make more blood available for diversion to the contracting muscles. Except during maximal exercise, the increase in splanchnic and renal resistance does not result in a fall in local blood flow to the abdominal viscera and kidneys. Rather, because the arterial pressure increases along with the renal and splanchnic vascular resistance, the absolute blood flow remains close to resting levels in these tissues, even as the flow to the skeletal muscle increases markedly. However, fractional blood flow (i.e., local blood flow normalized to cardiac output) does fall in these regions. In the early phase of exercise, skin blood flow also decreases. However, cutaneous blood flow eventually rises, reflecting the attempt of the temperature regulatory system to prevent body temperature from rising too much (see Chapter 59).
3. Early vasodilation in active muscle. In dogs—although not in humans or other primates—at the initiation of exercise, central command stimulates hypothalamic neurons whose axons bypass the medullary cardiovascular centers and synapse on preganglionic sympathetic neurons in the spinal cord (see Chapter 23). The postganglionic neurons synapse on cholinergic sympathetic vasodilator fibers that innervate the vascular smooth muscle of skeletal muscle and trigger early peripheral vasodilation in active skeletal muscle. As discussed in the next section, the delayed local “chemical” response later reinforces this vasodilation.
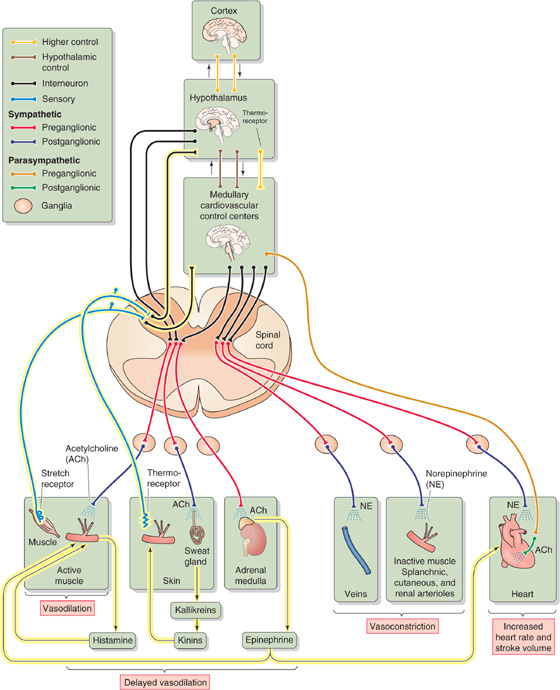
Figure 25-7 Integrated cardiovascular response to exercise. The hypothalamus orchestrates an early response, which includes vasodilation of active muscle (the mechanism of which is controversial in humans), vasoconstriction of certain inactive tissues, and increased cardiac output. In addition to those responses shown in Figure 25-6, the delayed responses (highlighted in yellow) include release of histamine, kallikreins, and epinephrine, leading to delayed vasodilation. Delayed local chemical responses from contracting muscles sustain early cardiovascular responses. Cutaneous thermoreceptors trigger delayed vasodilation in the skin.
The increased alertness that accompanies the anticipation of exercise can elicit all the components of the early response to exercise. The early response resembles the fight-or-flight reaction. In humans, the anticipatory cardiovascular adjustments prepare the body for the increased metabolism of the exercising skeletal muscle. For sprinters at the start of a 100-m dash, the anticipatory response prepares them to deliver an increased cardiac output and simultaneously to divert the increased blood flow away from tissues that do not require increased blood flow.
In addition to the events orchestrated by the command center, the integrated cardiovascular response to exercise includes the following.
1. Exercise pressor reflex. A neural drive called the exercise pressure reflex originates within the exercising muscle itself. Contraction activates stretch receptors that sense muscle tension and may also activate chemoreceptors that sense metabolites. Signals from these receptors travel through small thinly myelinated (Aδ or group III in Table 12-1) and unmyelinated (C or group IV) sensory fibers from skeletal muscle to the spinal cord and then on to the medullary cardiovascular control centers. This sensory input reinforces the central input to the cardiovascular control center and thus sustains the sympathetic outflow.
2. Arterial baroreflexes. Elevated mean arterial pressure resulting from high cardiac output and vasoconstriction outside active muscle would normally slow the heart. However, during exercise, central command resets the sensitivity of the arterial baroreflex so that the heart slows only at much higher arterial pressures. Conversely, if massive vasodilation in exercising skeletal muscle would reduce total peripheral resistance, the baroreceptor reflex maintains mean arterial pressure.
3. Vasodilation triggered by metabolites in skeletal muscle. Metabolites released locally (Fig. 25-7) dilate the resistance vessels and recruit capillaries that had received no blood flow at rest (see Chapter 24). As a result of this decrease in resistance, in concert with the increase in cardiac output, blood flow to active skeletal muscle can be as much as 20 times that to resting skeletal muscle. This vasodilator effect of metabolites thus more than overcomes any vasoconstrictive tendency produced by norepinephrine.
4. Increased venous return. The central command discussed in the preceding section explains the early increase in cardiac output during exercise. The mechanical and the chemical limbs described in Figure 25-6 further sustain the high cardiac output. Mechanically, the muscle pump increases venous return, and stroke volume rises by the Starling mechanism. Chemical mediators cause a vasodilation of active muscle beds that results in the rapid mobilization of blood from the central blood volume to exercising muscle.
5. Histamine release. Cells near the arterioles may release their intracellular stores of histamine, a potent vasodilator (see Table 20-7). Although these histamine-containing cells are quiescent when sympathetic nerves release norepinephrine, they release histamine when sympathetic tone wanes. Because of relaxation of the arterioles and precapillary sphincters, the pressure in the muscle capillaries rises, leading to increased extravasation of fluid and enhanced lymph flow.
6. Epinephrine release. During severe exercise, preganglionic sympathetic fibers to the adrenal medulla stimulate epinephrine release. The systemic effects of circulating epinephrine on cardiac β1 adrenoceptors enhance the neural effects on the heart, thus increasing cardiac output. Circulating epinephrine also acts on vascular β2 adrenoceptors, augmenting vasodilation mainly in skeletal muscle and heart.
7. Regulation of body core temperature. As exercise continues, increased metabolism causes body core temperature to rise, activating temperature-sensitive cells in the hypothalamus. This activation has two effects, both of which promote heat loss through the skin as part of a temperature regulatory response. (See Chapter 59.) First, the hypothalamus signals the medulla to inhibit its sympathetic vasoconstrictor outflow to the skin, thereby increasing cutaneous blood flow. Recall that vasoconstriction is part of the early response to exercise (see earlier). Second, the hypothalamus activates sympathetic cholinergic fibers to sweat glands, causing an increase in sweat production as well as an indirect cutaneous vasodilation that may involve kinin formation. In addition, these neurons may co-release neurotransmitters that directly dilate cutaneous vessels (see Chapter 24).
If a person rapidly loses more than 10% or 20% of total blood volume from a large vein, the inadequate intravascular volume causes sequential decreases in central blood volume, venous return, ventricular filling, stroke volume, cardiac output, and thus mean arterial pressure. However, if the blood loss comes from a large peripheral artery, the mean arterial pressure in central arteries does not fall until cardiac output falls secondary to decreased venous return. Of course, if the blood loss occurs from a blown aortic aneurysm, mean arterial pressure falls immediately.
Large hemorrhages, in which one loses 30% or more of total blood volume, produce hypovolemic shock. Shock is a state of peripheral circulatory failure that is characterized by inadequate perfusion of the peripheral tissues. During shock, the systolic arterial pressure is usually below 90 mm Hg, and the mean arterial pressure is below 70 mm Hg. For reasons that will become clear, by the time one records a significant fall in mean arterial pressure, other signs of shock are evident. The first signs may be narrowing of the pulse pressure and a sensation of faintness when sitting or standing. The subject in hypovolemic shock has cold and moist (i.e., “clammy”) skin as well as a rapid and weak pulse. Moreover, urine output drops to less than 25 mL/hr, even though fluid intake had been normal.
After its abrupt initial fall, arterial pressure tends to return to normal (Fig. 25-8, red curve), although blood pressure falls irreversibly (blue dashed curve) in some cases. Under favorable circumstances, the body restores blood pressure toward normal values by mobilizing two lines of defense. First, circulatory control mechanisms act on the heart and blood vessels to restore cardiac output and to increase peripheral resistance. Second, mechanisms of capillary exchange and fluid conservation restore the intravascular volume.
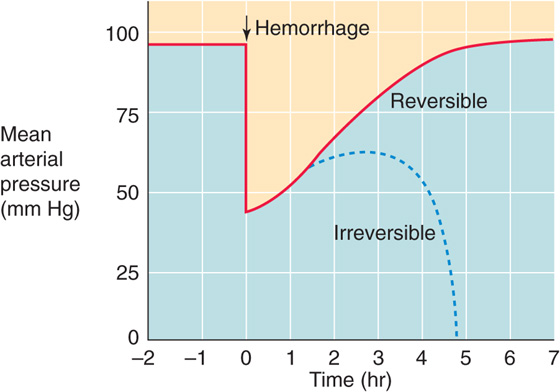
Figure 25-8 Changes in blood pressure with hemorrhage. At time zero, the investigator removes enough blood from the experimental animal to lower mean arterial pressure to 45 mm Hg.
Several cardiovascular reflexes cooperate to compensate for the fall in mean arterial pressure. These reflexes originate from four major groups of receptors (numbered 1 to 4 in Fig. 25-9).
1. High-pressure baroreceptors. The fall in arterial pressure leads to a decrease in the firing rate of afferents from the carotid and aortic baroreceptors (see Chapter 23). The resulting enhanced sympathetic output and diminished vagal output increase heart rate and cardiac contractility and also produce venoconstriction and selective arteriolar constriction. These responses cooperate to re-establish the arterial pressure.
2. Low-pressure baroreceptors. Reduced blood volume directly decreases effective circulating volume, which in turn lessens the activity of low-pressure stretch receptors (see Chapter 23). The resulting increased sympathetic outflow causes vasoconstriction in a number of vascular beds, particularly the kidney, reducing glomerular filtration rate and urine output. In response to decreased stretch, low-pressure receptors at various sites in the circulation ultimately have divergent effects on heart rate. The atrial stretch receptors also instruct the hypothalamus to enhance release of AVP, which reduces renal water excretion (see Chapter 38). During shock, the vasoconstrictor effects of AVP appear to be important for maintaining peripheral vascular resistance. Reduced atrial stretch also lowers the level of circulating atrial natriuretic peptide (see Chapter 23), thereby reducing salt and water loss by the kidneys (see Chapter 40).
3. Peripheral chemoreceptors. As blood pressure drops, perfusion of the carotid and aortic bodies declines, causing local hypoxia near the glomus cells and an increase in the firing rate of the chemoreceptor afferents, a response enhanced by increased sympathetic tone to the peripheral chemoreceptor vessels (see Chapter 32). Increased chemoreceptor discharge leads to increased firing of the sympathetic vasoconstrictor fibers and ventilatory changes that indirectly increase heart rate (see Chapter 23).
4. Central chemoreceptors. Severe hypotension results in brain ischemia, which leads to a fall in the PO2 of brain extracellular fluid as well as a rise in PCO2 and a fall in pH. The acidosis has a profound effect on the central chemoreceptors in the medulla, leading to a sympathetic output several-fold more powerful than that caused by baroreceptor reflexes (see Chapter 23).
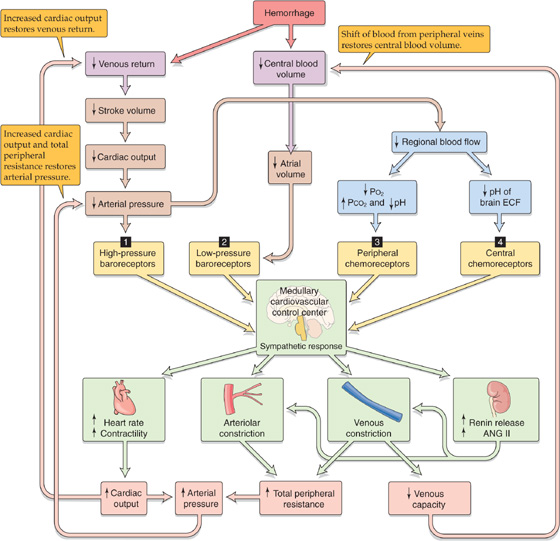
Figure 25-9 Integrated response to hemorrhage. Blood loss triggers four kinds of receptors (numbered 1 to 4) to produce an integrated response orchestrated by the medulla. ANG, angiotensin; ECF, extracellular fluid.
These four reflex pathways have in common the activation of a massive sympathetic response that results in the release of norepinephrine from postganglionic sympathetic neurons. In addition, the sympathetic response triggers the adrenal medulla to release epinephrine and norepinephrine roughly in proportion to the severity of the hemorrhage. Lowering of the mean arterial pressure to 40 mm Hg causes circulating levels of epinephrine to rise 50-fold, and norepinephrine, 10-fold. The consequences of the four combined reflex actions are the following responses (Fig. 25-9).
Tachycardia and Increased Contractility Increased sympathetic activity increases heart rate roughly in proportion to the volume of shed blood. Thus, the degree of tachycardia is an index of the severity of the hemorrhage. Increased sympathetic tone increases myocardial contractility but can increase stroke volume only after venous return also improves.
Arteriolar Constriction Sympathetic constriction of the resistance vessels is most pronounced in the blood vessels of the extremities, skin, skeletal muscle, and abdominal viscera. Although both precapillary and postcapillary resistance vessels constrict, the precapillary response initially dominates. As a result, capillary pressure falls precipitously, leading to the transcapillary refill discussed in the next section. Renal blood flow falls rapidly after hemorrhage as a result of the fall in blood pressure but recovers after a few minutes because of autoregulation (see Chapter 34). The responses of both high- and low-pressure stretch receptors lead to enhanced sympathetic vasoconstrictor traffic to the kidney. Although renal blood flow has a high threshold for this sympathetic traffic, the sympathetic vasoconstriction eventually overrides renal autoregulation if arterial pressure remains low or continues to fall. In hypovolemic shock, renal blood flow falls to a proportionately greater extent than does cardiac output, which explains why severe hemorrhage often results in acute renal failure. Blood flow in the medulla of the kidney is less compromised than in the cortex, leading to “medullary washout” of the hypertonic interstitial fluid in the renal medulla (see Chapter 38) and an inability to produce a concentrated urine. Both coronary blood flow and cerebral blood flow initially fall after hemorrhage, but autoregulation can largely restore blood flow to normal.
Venous Constriction The fall in blood volume with hemorrhage occurs primarily in the large capacitance vessels, especially those that contain the central blood volume. These vessels are very sensitive to sympathetic stimulation (which causes constriction) and less so to local metabolites (which causes dilation). The sympathetic venous constriction decreases both the capacity and the compliance of the large veins, thereby tending to restore central venous pressure. In addition, sympathetic venous constriction increases postcapillary resistance, which is important for transcapillary refill (see later).
Circulating Vasoactive Agonists As already discussed, sympathetic stimulation of the adrenal medulla causes circulating epinephrine levels to rise. In addition, sympathetic stimulation of the granular cells in the juxtaglomerular apparatus of the kidney leads to an increased release of renin and, ultimately, increased plasma levels of angiotensin II (ANG II; see Chapter 23). In hemorrhagic shock, ANG II rises to concentrations that are vasoconstrictive. Activation of the sympathetic system also triggers sympathetic cholinergic stimulation of the sweat glands (see Chapter 24), causing the patient’s extremities to become clammy.
With moderate blood losses (10% to 20%), these four responses can increase total peripheral resistance sufficiently to keep arterial pressure at about normal levels. However, cardiac output remains depressed.
The reflexes discussed in the preceding section compensate for the principal consequences of blood loss—decreased blood pressure and reduced cardiac output. The responses discussed here compensate for the primary disturbance, the loss of blood volume.
Transcapillary Refill The movement of fluid from the interstitium to the blood plasma is the major defense against reduced blood volume. Starling forces (see Chapter 20) are critically important during hemorrhage and hypovolemic shock. Immediately after hemorrhage, a phase of hemodilution develops, as was first observed during World War I, when medics noted that injured soldiers arrived at the first-aid station with diluted blood (i.e., low hematocrit). Within an hour, interstitial fluid replaces ~75% of the shed blood volume. Studies performed around World War II showed that the dilution of hemoglobin is more pronounced than the dilution of plasma proteins after hemorrhage. Therefore, not only do fluid and electrolytes move from the interstitium to the blood, but proteins also enter the vascular compartment.
Transcapillary refill involves two steps. The first is fluid movement from the interstitium to the vasculature. Capillary hydrostatic pressure (Pc) (Fig. 25-10A) depends on arteriolar and venular pressures as well as on the relation of the precapillary to the postcapillary resistance (see Chapter 19). Immediately after the hemorrhage, the upstream arteriolar pressure and the downstream venular pressure both fall, causing Pc to fall (Fig. 25-10B). The Starling forces thus produce a large net movement of fluid and small electrolytes from the interstitium into the capillaries. As compensation occurs, total peripheral resistance increases, in part restoring arteriolar pressure (Fig. 25-10C). However, because precapillary resistance increases more than does postcapillary resistance, Pc remains relatively low, sustaining the net movement of fluid into the capillaries. Normally, little protein would enter with the fluid because nonfenestrated capillary walls reflect proteins very effectively (see Chapter 20). The entry of protein-free fluid into the capillary gradually modifies the three other Starling forces. First, the interstitial fluid volume decreases, lowering interstitial hydrostatic pressure (Pif). Second, the plasma proteins become diluted, so that capillary colloid osmotic pressure (πc) falls. Finally, the removal of a protein-free solution from the interstitium raises colloid osmotic pressure in the bulk interstitium (πif) and in the subglycocalyx fluid (πsg; see Chapter 20). The result of these dissipating Starling forces is that transcapillary refill gradually wanes and eventually ceases.
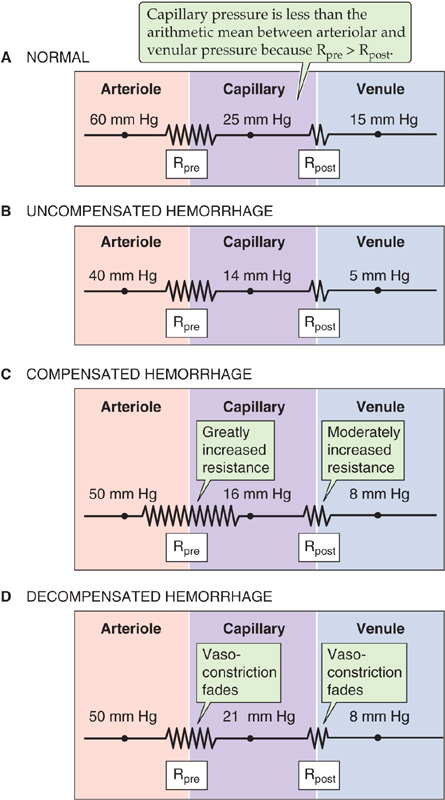
Figure 25-10 Effect of hemorrhage on capillary hydrostatic pressure. A, In this figure (which is similar to Fig. 19-4A), Rpre and Rpost are the precapillary and postcapillary resistances, respectively. Here, the ratio Rpost/Rpre = 0.35. B, The fall in capillary hydrostatic pressure reverses the Starling forces, causing net movement of fluid from the interstitium to the capillary lumen. C, Sympathetic stimulation increases total peripheral resistance (Rpost/Rpre = 0.25 in this example). D, Capillary pressure rises (Rpost/Rpre = 0.45 in this example).
The second step in transcapillary refill is the appearance of plasma proteins in the blood. These proteins probably enter the blood across fenestrae of the mesenteric and hepatic capillaries, two regions in which the interstitium has a very high interstitial colloid osmotic pressure. In addition, hemorrhage rapidly stimulates albumin synthesis by the liver. Plasmapheresis, the artificial removal of plasma proteins from the blood, has the same effect on albumin synthesis, suggesting that a reduction in the concentration of plasma proteins per se stimulates the liver to make albumin.
Water from the intracellular compartments ultimately replaces the lost interstitial fluid. What the driving force is for water to leave the cells and to enter the interstitium is somewhat controversial. The blood osmolality often rises after hemorrhage, presumably reflecting interstitial hyperosmolality. The additional interstitial osmoles come from ischemic tissues that release the products of proteolysis, glycolysis, and lipolysis. Therefore, interstitial hyperosmolality may provide the osmotic drive for the movement of water from the intracellular to the interstitial compartment.
Renal Conservation of Salt and Water Arterial hypotension and lowered renal blood flow reduce the glomerular filtration rate (see Chapter 34) and therefore diminish the urinary excretion of salt and water. In addition to the direct hemodynamic effects, the reduced effective circulating volume promotes the renal retention of Na+ by four mechanisms, which are discussed more fully in Chapter 40. First, reduced effective circulating volume activates the reninangiotensin cascade, increasing aldosterone release and thus enhancing salt and water reabsorption by the distal nephron. Second, increased sympathetic nerve activity promotes Na+ retention by altering renal hemodynamics, enhancing renin release and stimulating Na+ reabsorption by renal tubule cells. Third, the release of AVP reduces water excretion. Finally, reduced effective circulating volume inhibits release of atrial natriuretic peptide and thus promotes renal Na+ retention. Therefore, the overall response of the kidney to blood loss is to reduce the excretion of water and salt, thereby contributing to the conservation of extracellular fluid. However, the renal response only conserves fluid; by itself, it does not add any water to the extracellular fluid. (See Chapter 40.)
Thirst The blood hyperosmolality caused by hemorrhage (discussed two paragraphs before) stimulates thirst osmoreceptors. A far more potent stimulus for thirst—as well as salt appetite—is the reduced effective circulating volume and blood pressure caused by severe hemorrhage. (See Chapter 40.) These urges, if fulfilled, actually provide some of the raw materials for replacement of the blood lost in hemorrhage.
In some cases, hemorrhagic shock can be irreversible. After an initial fall in arterial pressure and perhaps some recovery, arterial pressure and the perfusion of peripheral tissues may inexorably deteriorate (Fig. 25-8, blue dashed curve). Moreover, in these cases, the fall in arterial pressure does not reverse even if the physician intervenes at this time and replaces the volume of blood lost as the result of hemorrhage.
The best experimental model for irreversible hemorrhagic shock is prolonged hypotension. Typically, the researcher acutely removes blood from an experimental animal—thereby reducing blood pressure to some low target value—and then clamps the mean arterial pressure at this low target by either removing or infusing blood as the normal physiological responses evolve. Studies of this kind reveal that hemorrhagic shock can become irreversible as a result of the failure of multiple response components: (1) the vasoconstrictor response, (2) the capillary refill response, (3) the cardiac response, and (4) the CNS response.
Failure of the Vasoconstrictor Response With prolonged hemorrhagic hypotension, the total peripheral resistance—which first increases in response to sympathetic stimulation—tends to return to prehemorrhage levels. This failure to maintain vasoconstriction has several origins. First, desensitization of the vascular adrenoceptors or depletion of neurotransmitters in the nerve terminals close to the blood vessels may cause “sympathetic escape.” Second, the ischemic tissues release metabolites and other vasodilator compounds that act on local blood vessels, thereby counteracting the vasoconstricting stimuli. In the late phases of irreversible shock, humans may become completely unresponsive to a range of vasoconstrictor drugs. Third, plasma AVP levels may have fallen substantially from the peak value during the early phase of hemorrhage—perhaps reflecting a decreased ability of the low-pressure baroreceptor reflex to trigger hypothalamic neurons to release AVP or a depletion of AVP stores. Under these conditions, restoration of AVP levels to their initial peak can markedly increase blood pressure.
Failure of the Capillary Refill Some blood vessels are able to sustain the initial increase in resistance better than others are. Over time, precapillary sphincters fail first, followed by the precapillary resistance vessels (i.e., arterioles), the postcapillary resistance vessels, and the capacitance vessels. Figure 25-10D shows an example in which, after prolonged hypotension, the precapillary constrictor response has fully faded, whereas the postcapillary response is partially maintained. Because the ratio Rpost/Rpre has risen, the midcapillary pressure (see Chapter 19) increases from 16 mm Hg (Fig. 25-10C) to 21 mm Hg (Fig. 25-10D). You will recall that early during hemorrhage, the net Starling forces reverse, favoring the movement of fluid from the interstitium to the blood. The gradual increase of Rpost/Rpre (Fig. 25-10D) reverses this reversal, so that fluid once again leaves the capillary, even though the blood volume has not yet been restored. Thus, after the initial large influx of water and electrolytes into the capillary lumen, not only does transcapillary refill decline, but a net loss occurs. This phenomenon contributes to the hemoconcentration that occasionally occurs in prolonged hemorrhagic states.
Failure of the Heart Several factors may contribute to the weakening of the heart. Acidosis reduces [Ca2+]i in the myocardium and thus reduces contractility. In severe cases, subendocardial hemorrhage and necrosis of the heart muscle render the myocardium nonfunctional. Various organs may also release cardiotoxic shock factors, which exert a negative inotropic effect on the heart (see Chapter 22). Ultimately, hypovolemic shock converts to cardiogenic shock.
Central Nervous System Depression Moderate ischemia, by its effects on the central chemoreceptors, stimulates the cardiovascular control centers in the brain. However, prolonged cerebral ischemia depresses neural activity throughout the brain, thereby weakening the sympathetic output and in turn causing a decay in both vascular and cardiac responses. A progressive fall in the circulating levels of catecholamines of adrenal origin further worsens the outcome.
Books and Reviews
Butler PJ, Jones DR: Physiology of diving of birds and mammals. Physiol Rev 1997; 77:837-899.
Gutierrez G, Reines HD, Wulf-Gutierrez ME: Clinical review: Hemorrhagic shock. Crit Care 2004; 8:373-380.
Kunze DL: Role of baroreceptor resetting in cardiovascular regulation: Acute resetting. Fed Proc 1995; 44:2408-2411.
Persson PB: Modulation of cardiovascular control mechanisms and their interaction. Physiol Rev 1996; 76:193-244.
Williamson JW, Fadel PJ, Mitchell JH: New insights into central cardiovascular control during exercise in humans: A central command update. Exp Physiol 2006; 91:51-58.
Journal Articles
Gulli G, Cooper VL, Claydon VE, Hainsworth R: Prolonged latency in the baroreflex mediated vascular resistance response in subjects with postural related syncope. Clin Auton Res 2005; 15:207-212.
Guyton AC, Coleman TG, Cowley AW Jr, et al: A systems analysis approach to understanding long-range arterial blood pressure control and hypertension. Circ Res 1974; 35:159-176.
Jacobsen TN, Morgan BJ, Scherrer U, et al: Relative contributions of cardiopulmonary and sinoaortic baroreflexes in causing sympathetic stimulation in the human skeletal muscle circulation during orthostatic stress. Circ Res 1993; 73:367-378.
Mellander S: Comparative studies on the adrenergic neurohormonal control of resistance and capacitance blood vessels in the cat. Acta Physiol Scand Suppl 1960; 50:1-86.
Smith OA Jr, Rushmer RF, Lasher EP: Similarity of cardiovascular responses to exercise and to diencephalic stimulation. Am J Physiol 1960; 198:1139-1142.