Musculoskeletal Neoplasms
Neoplasm is defined as a new or abnormal growth of cells and is often used interchangeably with tumor, which means any swelling or mass. Neoplasms are divided into two broad categories: benign and malignant. Benign neoplasms show no tendency to metastasize, are noninvasive, and are usually slow growing. A malignant neoplasm is one that can be invasive or can metastasize (see further discussion, Chapter 9).
Although neoplasms represent a small portion of the spectrum of pathology seen in clinics, their severity and potential for serious consequences necessitate an understanding of their detection and treatment.
The purpose of this chapter is to review the characteristics of primary and secondary musculoskeletal neoplasms. Those that may be encountered by therapists are highlighted. It is hoped that by increasing awareness of the clinical manifestations, earlier detection will be possible.
PRIMARY TUMORS
Description
Primary musculoskeletal tumors are those that have developed from or within tissue in a localized area. Primary musculoskeletal neoplasms can be benign or malignant, soft tissue or bone. A soft tissue tumor may originate from muscle, cartilage, nerve, collagen, adipose, lymph or blood vessel, or skin (see Table 9-1). Common sites in the body and location within the bone vary depending on the type of tumor (Table 26-1; Fig. 26-1).
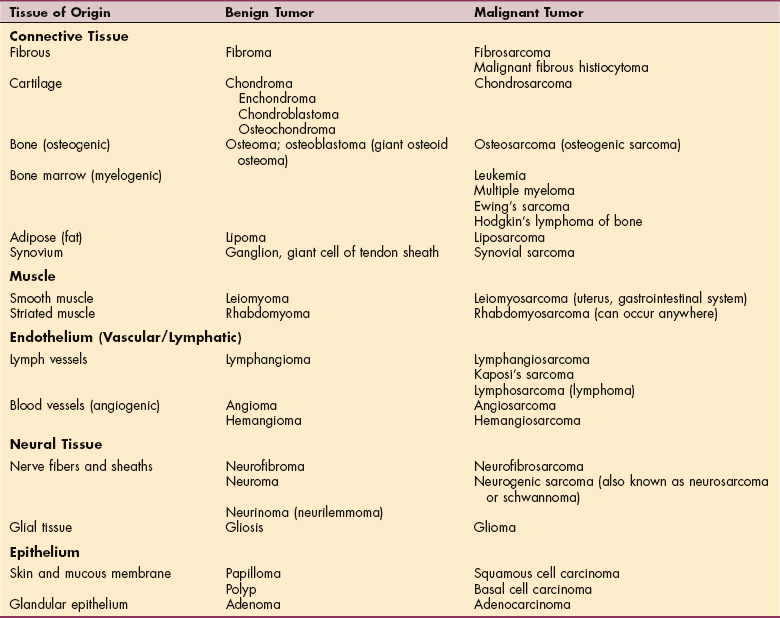
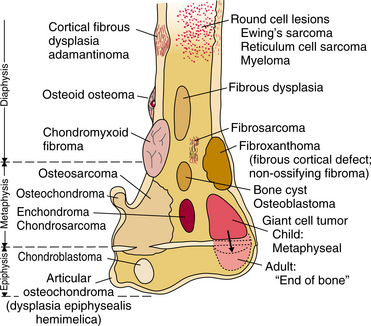
Figure 26-1 Composite diagram illustrating frequent sites of bone tumors. The diagram depicts the end of a long bone that has been divided into the epiphysis, metaphysis, and diaphysis. The epiphysis refers to the articular end of the long bones, which is primarily cartilaginous in the growing child. The metaphysis is the wider part of the shaft of the long bone. The diaphysis refers to the shaft itself. The typical sites of common primary bone tumors are labeled. (From Madewell JE, Ragsdale BD, Sweet DE: Radiologic and pathologic analysis of solitary bone lesions: I. Internal margins, Radiol Clin North Am 19:715, 1981.)
Modern classification of soft tissue tumors recognizes more than 200 benign and approximately 70 malignant (sarcomatous) lesions with a ratio of benign tumors to malignant sarcomas of 100: 1. The focus of this chapter will remain on the most common bone and soft tissue tumors encountered in the physical therapist’s practice.
Other soft or connective tissue tumors (e.g., skin, heart, myeloma, lymphatic, hematologic, neurologic) are discussed elsewhere in this text. An excellent comparison of soft tissue sarcomas in adults and children, including rehabilitation, is available elsewhere.46,59 A review of solid bone cancers occurring during adulthood and the implications for physical therapy is also available.34
Benign Neoplasm
Benign tumors are well differentiated, resemble normal tissue, rarely invade locally, and have low potential for autonomous growth. However, benign does not necessarily mean innocuous. For example, osteoblastomas in the spine may produce serious neurologic problems requiring resection, with additional complications possible from the surgical procedure.
Some benign bone tumors pose difficult evaluation and management decisions and can result in a significant level of impairment. For example, large fibrous defects in weight-bearing bones can cause pathologic fractures. A pathologic fracture refers to bone that has been weakened by local destruction (osteoclastic resorption) from any cause; bone with this type of impairment is more readily fractured than normal bone. This complication is referred to as a pathologic fracture because it occurs through an area of abnormal or pathologic bone.
Although rare, some benign lesions can develop into a malignancy. Benign lesions usually do not cause the constant, severe pain that is commonly associated with progressive malignant disease, but benign tumors can impair blood supply or compress nerve tissue.
Malignant Neoplasm
Malignant primary tumors of bone by definition have the capacity to spread to other sites and often do so aggressively by invading locally and destroying adjacent tissues and by metastasizing to distant sites. Skeletal neoplasms often metastasize to the lungs through the bloodstream.
Fortunately, malignant tumors are not as common as benign lesions; however, this rarity has made it difficult to standardize treatment interventions and management. For this reason, most individuals with malignant primary tumors are referred to regional centers, where valuable experience concerning evaluation and treatment can be gained and then applied to future cases.
Incidence
Primary tumors of the musculoskeletal system are uncommon, although the incidence is difficult to determine because these lesions often escape diagnosis (Table 26-2). Excluding myeloma and skin cancer, as few as 2400 new cases of primary bone tumors and 9200 cases of soft tissue sarcomas are detected annually in the United States with a 3: 1 ratio of men to women affected.54 This does not mean that they are unimportant. A great deal of time is devoted to research, reporting, and educating physicians in the proper management of people with primary tumors. These efforts are indicative of the serious nature of the problem rather than the frequency.
Table 26-2
Relative Frequency of Primary Bone Tumors*
| Benign | |
| Osteochondroma | 35% of benign tumors; 10% of all bone tumors |
| Osteoid osteoma | 10%-12% of benign bone tumors |
| Enchondroma | 10% of benign bone tumors; some report as high as 24% |
| Osteoblastoma | 1%-2% of benign bone tumors |
| Chondroblastoma | <1% of all bone tumors |
| Hemangioma | <1% of all bone tumors |
| Malignant | |
| Metastatic neoplasm | Most common form of bone malignancy; secondary neoplasm of bone |
| Multiple myeloma | Most common primary neoplasm of bone; plasma cell malignancy (bone marrow) |
| Osteosarcoma | 35% of all malignant bone tumors; 15%-20% of primary sarcomas (excluding multiple myeloma) |
| Chondrosarcoma | 25% of malignant bone tumors (excluding multiple myeloma) |
| Ewing’s sarcoma | 16% of malignant bone tumors; second most common in children; fourth overall primary bone tumor for adults and children (after myeloma) |
| Malignant fibrous histiocytoma | 2%-5% of malignant bone tumors (excluding multiple myeloma) |
| Chordoma | 1%-4% of all malignant bone tumors; slow growing but locally aggressive |
| Angiosarcoma | 1.4% of malignant bone tumors (excluding multiple myeloma) |
*Listed by decreasing order of frequency; with the exception of metastatic neoplasm listed, these statistics refer to primary bone tumors.
Data from Dorfman HD, Czerniak B: Bone cancers, Cancer 75(1 suppl):203-210, 1995; Dorfman HD, Czerniak B: Bone tumors, St Louis, 1998, Mosby.
Risk Factors
Little progress has been made in our knowledge of the risk factors involved in the etiopathogenesis of malignant bone tumors. Although bone tumors may have a predilection for certain sites, age groups, and gender, most causes of osteosarcoma are unknown. The main factors implicated are Paget’s disease, Li-Fraumeni syndrome, antineoplastic drugs, ionizing radiation, and hereditary retinoblastoma.36
Exposure to alkylating chemotherapeutic agents such as cyclophosphamide, used in the treatment of acute lymphocytic leukemia, has been associated with subsequent development of osteosarcoma in a small percentage of cases.
Several genetic conditions are related to the development of soft tissue sarcoma (e.g., neurofibromatosis, tuberous sclerosis, basal cell nevus syndrome), but this is only a small number of cases.103 Soft tissue tumors also may be associated with high doses of radiation or exposure to toxic chemicals in the workplace (herbicides, dioxin, preservatives, and so on).
Etiologic Factors and Pathogenesis
The histogenesis of tumors is generally poorly understood, although significant progress has been made toward understanding tumor development as a biologic phenomenon. For a detailed description of the molecular biology of bone formation, apoptosis and its role in bone cancer, and molecular and oncogenetic concepts of bone neoplasia, the reader is referred to Dorfman and Czerniak, 1998.30
Bone Tumors.: To grasp the concepts of tumor formation, one must understand that bone metabolism is a balancing act of bone formation and resorption. The coupling of these two processes usually results in a balance of bone resorption and formation. When metabolic bone disease and neoplastic formations occur, this balance is upset.
Under normal circumstances, bone remodeling involves a fine balance between osteoblast activity, which promotes new bone synthesis, and osteoclasts, which stimulate bone resorption. This balance is disrupted by the presence of malignant cells, resulting in uncoupling of the process of remodeling.
Bone remodeling is a structured process governed by highly specialized cells. Osteoblasts are derived from mesenchymal fibroblast-like cells and are responsible for bone formation. Bone formation is accomplished by synthesizing various collagens, alkaline phosphatase, and other chemicals. A variety of paracrine factors, including tumor necrosis factor α, tumor necrosis factor β, interleukin-1, and prostaglandins are released during the remodeling process and may ultimately contribute to growth of the metastatic cells.53
Cortical bone is most abundant in the outer walls of the shafts of long bones and is quite dense. The haversian canal system, which refers to the concentric rings of lamellae, is found in cortical bone. Cortical bone surrounds the trabecular or cancellous bone, which is the honeycomb-like bone found in the ends of long bones. Trabeculae are aligned with applied stresses in the bone. The metabolic activity is higher in cancellous bone than cortical bone, which accounts for why many disorders that create disturbances in metabolic activity are first noted in cancellous bone.
Bone tumors are considered to be either osteoblastic or osteolytic, although most have characteristics of both processes. The osteoblastic process can be preceded by tumor cells or by normal cells in the host bone reacting to the tumor. Since the host bone continues with the normal process of resorption and bone formation, there will likely be a variety of cell types within the lesion. This makes histologic interpretation difficult.
Neoplastic cells do not themselves destroy bone, but their presence incites local osteoclastic resorption of bone. The cells of certain neoplasms also incite local osteoblastic deposition of normal bone, referred to as reactive bone. The neoplastic cells of the osteogenic group of neoplasms are capable of producing osteoid (young bone that has not undergone calcification) and bone, which are then referred to as tumor bone or neoplastic bone. The radiographic appearance of lesions affecting bone reflects varying proportions of bone resorption (osteolysis) and bone deposition (osteosclerosis)—some of the latter being reactive bone, and some being neoplastic bone.84
Soft Tissue Tumors.: Four types of genetic disorders underlying soft tissue sarcomas have been identified: translocations, gene amplifications, mutations, and complex genetic imbalances. Detection of these molecular changes can guide treatment and may predict response to treatment. Techniques used to detect translocations are very sensitive and in some cases may be used to detect microscopic metastasis.13
Soft tissue sarcomas have a predictable growth pattern, beginning as small masses and often growing in a centripetal pattern. The leading edge of the tumor (reactive zone) contains edema, fibrous tissue, inflammatory cells, and tumor cells. Uncontrolled growth often causes loss of blood supply at the center of the tumor.
Benign soft tissue tumors also have a centripetal growth pattern, but the expansion is more controlled and much slower. Benign lesions tend to be more superficially located compared with malignant lesions, which often grow within tissues under the deep fascia.94
Clinical Manifestations
The clinical features must be well understood to ensure that the diagnostic evaluation proceeds expeditiously. Unfortunately, many tumors are not diagnosed on their initial presentation. This is due to the ambiguous presentation of most tumors in their early stages; rarely does one actually find the case that is described as typical for a given lesion. This may be true regarding benign or malignant tumors, the initial presentation, and the appearance of the lesion.
Pain.: Pain is a hallmark of tumor development, especially with malignant lesions. With bone tumors, intense pain is more likely to occur with rapidly growing lesions caused by pressure or tension on the sensitive periosteum and endosteum. Constant pain that is not dependent on position or activity and is increased with weight-bearing activities is a red flag symptom. The presence of night pain is considered an additional important finding. When the client reports night pain, further questioning is required.
The therapist should ensure that the client is reporting true night pain, which awakens the person from sleep, rather than a pain that makes it difficult to fall asleep. Ask the individual if rolling onto the involved side or painful area awakens him or her. Ascertain whether the pain subsides with movement and change in position, possibly indicating mechanical ischemia or positioning as the cause of the night pain. Determine the effects of eating on pain, as this may be an indicator of gastrointestinal (GI) involvement.
It is also important to remember the common referral patterns for pain. These may give important clues to the origin of symptoms. The varied pain pattern is a result of the nature, site, and rate of growth of the tumor.
Since pain is the overriding symptom in many people who seek treatment, a great deal of information should be obtained concerning the pain. The onset, progression, nature, quality, intensity, and aggravating factors are just some of the factors that may be important in identifying a tumor in the early stages.
Keep in mind that with cancer, pain is not always a measure of disease progression. Some tumors can progress to advanced stages without causing significant pain. Soft tissue tumors can occur in any anatomic region, although most develop in the extremities, usually the legs. These tumors may progress with relatively little pain because the soft tissue allows the growth to occur without putting undue pressure on nerve endings. Any swelling present is often attributed to a minor injury, delaying medical examination.
In fact, clients often report a recent history of trauma, although no scientific evidence directly connects such injury to the inception of soft tissue or bone sarcomas. Instead, such traumatic episodes are thought to call attention to a specific body part or location, thereby increasing the likelihood of detecting an otherwise painless and often innocuous soft tissue mass or bone lesion.103
Fractures.: Pathologic fractures are rare in primary neoplasms, but if the lytic process affects a significant portion of the cortex (over 50%) or occupies 60% of the bone diameter, the risk of fracture increases. A relatively small lytic lesion in the femoral neck that destroys the inferior cortex of the femoral neck also places the client at increased risk. In benign lesions, no other symptoms may warn of the impending fracture.
A history of sudden onset of severe pain may be an indication of a pathologic fracture. Solitary bone cysts, fibrous dysplasia, nonossifying fibroma, and enchondromas may only be detected after presentation with a pathologic fracture. In addition to the tumor itself, other factors such as disuse, treatment (biopsy, radiation), and other health problems (osteoporosis) may increase the risk of pathologic fracture.
Miscellaneous.: Other signs and symptoms often encountered include swelling, fever, and the presence of a mass. Other factors that are useful in screening for serious pathology include unexplained weight loss, failure of rest to provide relief of pain, age, and history of cancer. The history will often give more meaningful information regarding the possibility of skeletal neoplasms than the physical examination.
Swelling.: Swelling surrounding a tumor may not be detectable in a bone tumor, but with soft tissue tumors close to the skin surface, swelling may be one of the first presenting signs. The nature of swelling, including the location, amount, temperature, and tenderness, is somewhat dependent on the vascularity of the lesion.
Mass.: A careful physical examination may reveal a mass or other signs of an inflammatory process. The presence of a mass should raise questions concerning the location, mobility, tenderness, dimensions, and recent changes in any of these factors. As with pain, the size of the mass is not indicative of the severity of the lesion but is one factor to consider. Any change in size, appearance, or other characteristics of a lump, local swelling, or lesion of any kind within the previous 6 weeks to 6 months should be reported to the physician.
Metastases.: Sarcomas spread by hematogenous routes rather than through the lymphatics. The most common site of metastases for individuals with extremity sarcomas is the lung, followed by liver and other bone sites. Anyone diagnosed with soft tissue sarcoma has an approximately 50% chance of local recurrence, since these tumors spread along tissue planes and involve adjacent tissue. Lymph node involvement is uncommon and is often associated with poor prognosis.57
MEDICAL MANAGEMENT
Physical examination, imaging studies (e.g., x-rays, computed tomography [CT], magnetic resonance imaging [MRI]), and biopsy are the primary diagnostic tools.
Physical Examination.
Many tumors cannot be observed or palpated during the physical examination, but if a mass is present its characteristics must be noted. The presence of café au lait spots (associated with neurofibromatosis), skin ulceration, or neurologic findings (e.g., footdrop, calf pain) may be significant.
Since synovial sarcoma, rhabdomyosarcoma, and epithelioid sarcoma can metastasize via the lymphatics, examination of the lymph nodes is essential.94 A tumor overlying bone and muscle can be evaluated by contracting the muscle and checking for movement or change in consistency of the tumor.
Radiographic Examination.
Radiographs also help differentiate between bone and soft tissue involvement. Plain radiographs are a mainstay in the detection and evaluation of many skeletal tumors. In many cases, skeletal tumors are found incidentally on routine radiographs for associated injuries. The radiograph provides unique information concerning skeletal tumors. MRI has emerged as the most useful imaging tool for evaluating soft tissue tumors, although biopsy is essential for a definitive histologic diagnosis.
The location of the tumor will give many clues to the type of lesion (see Fig. 26-1). Some tumors develop exclusively in the epiphysis, whereas others develop in the diaphysis of long bones. Bone tumors tend to predominate in those ends of long bones that undergo the greatest growth and remodeling and hence have the greatest number of cells and amount of cell activity (shoulder and knee regions).
When small tumors, presumably detected early, are analyzed, preferential sites of tumor origin become apparent within each bone, as shown in Fig. 26-1. This suggests a relationship between the type of tumor and the anatomic site affected. In general, a tumor of a given cell type arises in the field in which the homologous normal cells are most active. These regional variations suggest that the composition of the tumor is affected or may be determined by the metabolic field in which it arises.
The effect that the tumor has on bone is described as destructive or lytic if the normal bone pattern is disrupted. Approximately 50% of the bone must be destroyed before the lesion can be detected. This may be evident by an irregular, erosive border surrounding the lesion; loss of trabeculae; or disruption of the cortex.
The response of surrounding bone to the tumor is another important feature to note on plain radiographs. Sclerotic borders give an indication of the growth characteristics of the tumor. A well-defined border with definite sclerotic margins is seen with a slow-growing lesion.
A tumor with a permeated or moth-eaten appearance (i.e., an area with multiple holes with irregular edges randomly distributed) with an expansive cortical shell indicates an aggressive malignant lesion (Fig. 26-2). Codman’s triangle, a triangular-shaped area of reactive bone, is formed when the neoplasm has eroded the cortex, ele- vating the periosteum and producing reactive bone in the angle where it is still attached (Fig. 26-3).
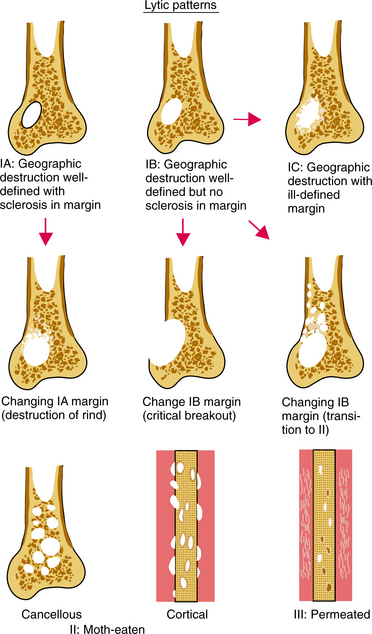
Figure 26-2 Schematic diagram of patterns of bone destruction (types IA, IB, IC, II, and III) and their margins. Arrows indicate the most common transitions or combinations of these margins. Transitions imply increased activity and a greater probability of malignancy. (From Madewell JE, Ragsdale BD, Sweet DE: Radiologic and pathologic analysis of solitary bone lesions: I. Internal margins, Radiol Clin North Am 19:715, 1981.)
Figure 26-3 Schematic diagram of periosteal reactions. The arrows indicate that the continuous reactions may be interrupted. (From Ragsdale BD, Madewell JE, Sweet DE: Radiologic and pathologic analysis of solitary bone lesions: II. Periosteal reactions, Radiol Clin North Am 19:749, 1981.)
The tumor’s location, its effect on bone, and the local bone response to the lesion are just some of the radiographic features to be noted and will help in planning the rest of the evaluation.
Imaging.
Radionuclide bone scan (scintigraphy), CT, MRI, angiography, and ultrasonography all have a place in the evaluation of bone lesions. Bone scans help locate skip metastases and the presence of bone metastases as well as metastatic bone lesions, and they assess tumor activity by the amount of radioisotope uptake in and around the tumor. Greater uptake indicates a more aggressive and malignant tumor.
CT scans are the most sensitive technique in detecting pulmonary metastases and also provide detailed information about the interaction between the tumor and various components of the bone (e.g., bone cortex, cancellous bone, reactive bone).
MRI is valuable in determining the extent of the marrow involvement and soft tissue masses outside the bone. The surgical team uses the information provided by an MRI to help visualize the involvement of the tumor and to plan limb salvage techniques.
Angiography plays an important role when limb-sparing surgery is being considered by providing information regarding the neovascularity of the tumor and mapping the vascular anatomy. Ultrasonography is a noninvasive imaging method that can be used to determine the size and consistency of a soft tissue mass. It may be used to establish intraarterial access for subsequent chemotherapy.56
Biopsy.
A biopsy is the definitive diagnostic procedure in both bone and soft tissue tumors and is usually performed after physical examination and imaging. This procedure can take many forms. The decision to do an open or incisional, core needle or fine-needle biopsy, or excisional biopsy is based on the location and type of tumor.
Laboratory Tests.
Various laboratory studies are used to detect, diagnose, and differentiate musculoskeletal neoplasms. Laboratory tests that may be of value include the complete blood count (CBC), urinalysis, erythrocyte sedimentation rate (ESR) (elevated in Ewing’s sarcoma), serum calcium (elevated in metastatic bone disease), phosphorus (decreased with “brown tumors” associated with hyperthyroidism), alkaline phosphatase (elevated in osteosarcoma and Paget’s disease), and serum protein electrophoresis (abnormal in metastatic bone disease).
Serum levels of alkaline phosphatase and calcium are often elevated with metastatic disease. Elevated alkaline phosphatase and lactic dehydrogenase (LDH) also occur with osteosarcoma (see Table 40-5).
STAGING AND GRADING.
The purpose of much of the extensive workup once a tumor is identified is to determine the grade and stage of the tumor. Grading determines the histologic characteristics, such as the extent of anaplasia or differentiation of the cells from grade I, indicating cells that are very differentiated, to grade IV, those that are undifferentiated.
Staging of a tumor is concerned with the extent of its growth, both local and distant. The tumor-node-metastasis (TNM) staging system (see Box 9-2) reflects the degree of local extension at the primary tumor site, involvement of local nodes, and presence of metastasis. This classification group is strongly correlated with survival.
No universally accepted staging system for musculoskeletal neoplasms exists because of the low incidence of such tumors, their heterogeneous nature and unpredictable behavior, and disagreement as to the relative importance of prognostic factors.76 The surgical staging system of Enneking is used for soft tissue and bone tumors and includes prognostic variables such as the histologic grade of the tumor, location of the tumor, and presence or absence of metastases (Table 26-3).33 The American Joint Committee on Cancer (AJCC) also provides staging for soft tissue sarcomas.3 Staging helps in planning and standardizing the intervention strategy for these rare lesions.
Table 26-3
Enneking Staging System for Bone and Soft Tissue Tumors
| Stage | Grade | Site |
| Stage 0 | G0 (benign neoplasm) | |
| Stage IA* | G1 (low grade; locally inactive or latent tumor with low probability of metastases) | T1 (tumor is contained within the bone and involves only one compartment; i.e., single compartment = individual bone with its medullary cavity) |
| Stage IB | G1 (low grade; active, slow growth) | T1 (tumor extends into soft tissue) |
| Stage IIA | G2 (high grade; aggressive tumor with high metastatic potential) | T1 (tumor is contained within the bone) |
| Stage IIB | G2 (high grade; aggressive) | T2 (tumor extends beyond cortex into adjacent soft tissue, joint, epidural space, or other bone) |
| Stage III | Any grade | Metastases present |
For staging according to the American Joint Committee on Cancer (AJCC), see National Comprehensive Cancer Network (NCCN) Practice guidelines in oncology: soft tissue sarcoma, vol 2, 2007. Available at www.nccn.org [page 28]. Accessed May 30, 2008.
*The suffixes A and B in this system indicate A, intracompartmental or B, extracompartmental lesions.
From Dorfman HD, Czerniak B: Bone tumors, St Louis, 1998, Mosby.
Grading sarcomas has been one of the most important contributions pathologists have made to the treatment of sarcomas. There is not one single, individual grading scheme that works well for all sarcomas. Outcomes do not always correspond to grades, and some tumors are “ungradable.” Diagnosis and grading are increasingly based on tissue obtained by core needle biopsy, which presents its own challenges.29
TREATMENT.
Once a tumor has been identified and staged, decisions about management and intervention can be considered. Treatment ranges from observation in the case of some benign bone tumors to surgical intervention. Principles of treatment are similar for some of the malignant bone tumors such as Ewing’s sarcoma and osteosarcoma. Chemotherapy or surgery alone cures few people. Multimodal measures are needed for a long-term successful response.4
Complete tumor resection is the best surgical strategy and is attempted whenever possible. A marginal excision removes the tumor at its border, resulting in some of the tumor remaining. A wide excision (sometimes referred to as an en bloc incision) removes some of the normal surrounding tissue, leaving none of the tumor. Soft tissue sarcomas often require wide excision to reduce the recurrence rate. Radical resection may be required in which the entire involved bone and all the tissue compartments adjacent to the tumor are removed.
The spine, sacrum, pelvis, ankle, hand, mediastinum, and chest wall are just a few examples of bone cancer locations that make surgery difficult. When local excision has positive margins (not all the cancer was removed), local control may be increased with radiation and chemotherapy regimens. Immunotherapy and biotherapy are additional treatment methods used to prevent cancer recurrence.4
Limb salvage or limb-sparing procedures have largely replaced amputation as the principal method to eradicate primary sarcomas. The three phases to any limb-sparing procedure are (1) resection of the tumor, (2) reconstruction of the skeletal area involved, and (3) soft tissue and muscle transfer to complete the reconstruction.
Obtaining a wide surgical margin while preserving limb viability and function remains the challenge to the medical team, requiring close coordination of surgical, medical, and oncologic staff. Often, soft tissue reconstruction is necessary to provide wound coverage after tumor removal.
The use of radiation is recommended for some tumors such as Ewing’s sarcoma and myeloma, but many malignant tumors are not affected by radiation. For some soft tissue tumors, adjunctive radiation is used in an attempt to limit the degree of surgical excision needed. In general, radiation is not recommended for benign conditions. Irradiation creates a suboptimal tissue bed susceptible to wound breakdown, seroma, and hematoma formation and infection, which may complicate the success of soft tissue reconstruction.103
Because hematogenous spread occurs early in musculoskeletal tumors, chemotherapy is also used to help eradicate malignant tumors. For example, combination chemotherapy has resulted in increased survival rates in clients with Ewing’s sarcoma and rhabdomyosarcoma as well. When chemotherapy is combined with other modalities such as surgery and radiation, less toxic doses can be used.
Future improvements in treatment may come about as a clearer understanding of cellular and molecular pathways of pathogenesis is elucidated. The development of less toxic, more specific therapies remains an important challenge. Newer strategies under investigation include stem cell transplantation, gene therapy, biotherapy such as biologic response modifiers, antibody targeting of immunotoxins to tumor cells, and vaccines designed to elicit T-cell immunity with specificity for tumor peptides.
As discussed in Chapter 9, modern clinical oncology is moving toward tailored therapy according to genetic profiling. Treatment can be stratified with different intensities prescribed based on the genetic characteristics of the individual cancer. Individuals with a poor prognosis may do better with aggressive therapies such as stem cell transplantation for an improved cure rate. Gene silencing techniques may make it possible for the development of specific drugs that will target malignant cells without causing damage to normal tissue.7
PROGNOSIS.
The prognosis is based in part on the type of tumor and whether it is benign or malignant. Survival is influenced by the grade of malignancy, tumor stage, and achieved surgical margins. A high grade and evidence of metastasis are associated with a poor prognosis for all neoplasms of bone or soft tissue regardless of the staging system that is used.76 Tumor extension into both anterior and posterior columns of a vertebra is correlated with a poor outcome. Incomplete resections are more likely to result in tumor recurrence with subsequent surgeries and increased risk for complications and poor outcome.106
Slow-growing tumors should be followed for prolonged periods, to determine the natural history and to identify the ultimate prognosis. Prognosis can vary from 3-to 5-year survival rates for clients with sarcomas and myeloma, to tumors that are asymptomatic. Successfully treated individuals may develop severe late effects, including second cancers (e.g., radiation-induced sarcomas or treatment-related leukemia), particularly after high-dose therapy with an alkylating agent, and chemotherapy-induced cardiomyopathy.6
RECURRENCE.
People with recurrent disease generally have a poor prognosis but need to undergo a complete reevaluation of the extent of the disease to determine this more specifically. The prognosis depends on the type of therapy given previously, duration of remission, and extent of metastases. Recurrence or progression of tumor during initial therapy is generally incurable.6 The lung is the most common initial site of distant metastases for the majority of soft tissue and bone sarcomas. Other sites may include distant osseous sites, bone marrow, and lymph nodes.
PRIMARY BENIGN BONE TUMORS
Overview and Incidence
Bone islands are oval, usually small, sclerotic lesions of bone. They are one of the most common benign bone lesions. Bone islands have been observed in all bones and may present as solitary or multiple lesions. The lesion is well defined and made up of cortical bone with a well-developed haversian canal system. The borders blend in with the surrounding bone. The presence of spicules of cortical bone extending from the margins to the surrounding trabeculae is characteristic. A prevalence of 14% has been reported for spinal bone islands.81
MEDICAL MANAGEMENT
When the bone islands are small (less than 1 cm), diagnosis with plain radiographs is adequate. They are usually oblong and align themselves with the axis of the bone. A bone scan is usually normal, confirming the absence of malignancy. The emphasis is not on intervention but on the judicious use of diagnostic tools. Biopsies should be avoided, as they are usually unnecessary. Although some bone islands can enlarge, they do not transform into malignant lesions.
Osteoid Osteoma
Overview, Incidence, and Etiologic Factors
Osteoid osteoma is a rare benign vascular osteoblastic lesion. It is often found in the cortex of long bones such as the femur and tibia but may occur in almost any bone except the skull. The tumors occur near the end of the diaphysis (Fig. 26-4). Osteoid osteoma accounts for about 10% to 12% of benign bone tumors. Most of these lesions are found in men under the age of 25. The cause of osteoid osteoma remains unknown.
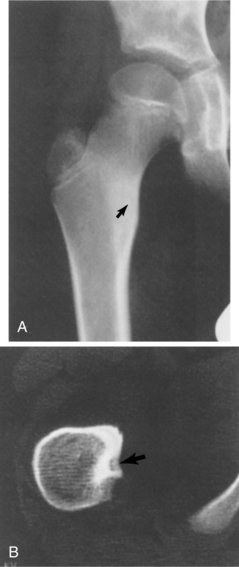
Figure 26-4 Osteoid osteoma. A, Bony sclerosis with cortical thickening is seen in this person with pain in the proximal femur. A faint lucency (arrow) can be seen in the area of sclerosis, which is the nidus of an osteoid osteoma. B, A computed tomographic (CT) scan through the nidus shows it to lie just dorsal to the lesser trochanter (arrow). This is a characteristic appearance of an osteoid osteoma with CT. (From Helms C: Fundamentals of skeletal radiology: benign cystic lesions, Philadelphia, 1989, WB Saunders.)
Pathogenesis
Pathologic study shows areas of immature bone surrounded by prominent osteoblasts and osteoclasts. The lesion is vascular, but no cartilage is present. Osteoid osteoma is probably a “reactive” bone-forming lesion rather than a true neoplasm, consisting of a small, round nidus (nest) of osteoid tissue surrounded by reactive bone sclerosis.
The zone of sclerosis is not an integral part of the tumor and represents a secondary reversible change that gradually disappears after the removal of the nidus. Osteoid osteomas are not progressive and rarely grow larger than 1 cm in diameter. They are uncalcified and therefore radiolucent.
Clinical Manifestations
Gradually increasing and persistent local pain in the area of the tumor, described as a dull ache, is the primary complaint. The pain is often worse at night and is characteristically relieved by aspirin and other nonsteroidal antiinflammatory drugs (NSAIDs). Pain relief may be due to the inhibitory effect on prostaglandins produced by osteoid osteomas. Systemic symptoms are uncommon.
When the lesion is located near a joint, synovial effusion may develop and interfere with joint function, with local muscle atrophy developing.84 A significant leg length discrepancy can occur, caused by the increased growth rate of affected bone in young individuals with open growth plates.
Though they occur rarely in the spine, if present, they are found in the lower thoracic or lumbar spine located in the posterior vertebral arch. The tumor can lead to joint pain and dysfunction, often delaying the diagnosis by masquerading as a more common problem such as an overuse syndrome.105
Spine involvement may result in an unexplained backache or painful type of scoliosis with unilateral spasticity of spinal muscles. Some people with vertebral lesions may have clinical symptoms suggestive of a neurologic disorder, lumbar disc disease, or both.30 In the case of spine involvement, neurologic deficits can be caused by extradural compression.105
MEDICAL MANAGEMENT
Radiographs can be diagnostic for osteoid osteoma, although these are often normal early in the course. Later, a small (less than 1 cm) translucency or nidus forms, surrounded by sclerotic bone. When the tumor is not easily identified on radiographs (e.g., vertebral nidus), further testing is required, such as a bone scan (scintigraphy), which will show a focal uptake of the radiotracer. Plain films may not be adequate when the tumor is intraarticular; in such cases, CT or MRI can be used to accurately locate the nidus.
TREATMENT AND PROGNOSIS.
In those tumors that are symptomatic, surgical excision of the nidus may be indicated. Since the tumor is small, excision is usually sufficient, although bone grafting may be needed depending on the size and location of the tumor. Recurrence is rare, and a full recovery is common. Osteoid osteomas have no potential for malignant transformation.105 Differences in the expected rate of recovery may occur depending on the location of the tumor and the extent of excision required.
Osteoblastoma
Osteoblastoma is another reactive but benign bone lesion similar to osteoid osteoma, only larger, with a tendency to expand. Some aggressive forms of osteoblastoma have been recognized. Unlike osteoid osteoma, osteoblastomas are often found in the spine, sacrum, and flat bones. Osteoblastomas involve the spine in approximately 35% of affected individuals, with the cervical spine affected in up to 39% of those people.26
Those found in the long bones are usually in the diaphysis, although as with most tumors, they can be seen elsewhere (Fig. 26-5). The histologic makeup of osteoblastoma is very similar to that of an osteoid osteoma. In fact, sometimes it is size alone that differentiates the two, with osteoblastoma being the larger. The lesions are osteolytic and have a sclerotic border.
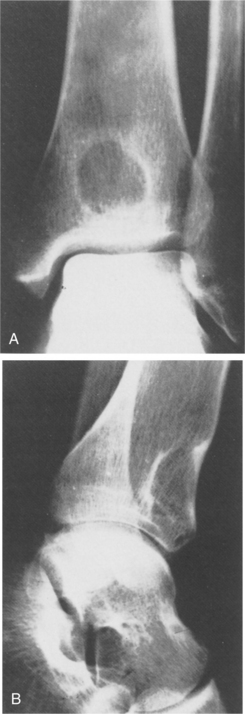
Figure 26-5 Genuine (conventional) osteoblastoma of the tibia in a 24-year-old woman. Anteroposterior (A) and lateral (B) radiographs show a round radiolucent lesion with slightly sclerotic borders at the lower and anterior aspect of the tibia. (From Gitelis S, Schajowicz F: Osteoid osteoma and osteoblastoma, Orthop Clin 20:320, 1989.)
An aggressive osteoblastoma represents a borderline lesion between benign osteoblastoma and osteosarcoma. It is very rare and not discussed further in this text.
Incidence
Osteoblastoma occurs most often in men less than 30 years old, but cases have been reported in children as young as 2 years old and adults in their seventies.105 Osteoblastoma is a rare osteoblastic tumor that makes up only 1% to 2% of all benign bone tumors.
Clinical Manifestations
When the tumor is located in the spine, the pedicles are often affected. Pain is the common presentation; it is not relieved with aspirin as occurs with osteoid osteoma. In general, the pain of osteoblastoma is not as severe as with osteoid osteoma, especially at night. Tenderness over the lesion is expected. With a spinal location, a functional scoliosis may be observed. In some cases a neurologic deficit may be present, which can mimic other, more common causes of nerve compression. Metastases and even death have been reported with the aggressive variant, which can behave in a fashion similar to that of osteosarcoma.
MEDICAL MANAGEMENT
Osteoblastoma is seen on plain radiographs, but when it is located in the spine, other imaging techniques are also useful. The lesion can have variations in its appearance. Often it looks like a large osteoid osteoma with a well-defined radiolucency in the central portion and a thin, sclerotic border. It also can be similar to an aneurysmal bone cyst that is expansile, lytic, and has a soap bubble appearance (see Fig. 26-3). CT and MRI are valuable in localizing the tumor and determining the extent of tissue involved. An aggressive lesion can expand beyond the cortex and involve soft tissue.
TREATMENT.
In the long bones, curettage (scraping to remove the contents of the bone cavity) is often adequate. A wider excision is sometimes recommended because of the unpredictable nature of osteoblastoma and high recurrence rate (up to 15%). Recurrence is often attributed to incomplete resection.
Extramarginal excisions can result in the need to perform reconstructive procedures using autografts or allografts and internal fixation when the tumor is located in the diaphysis of long bones. If the joint is affected, implants may be needed. In the spine, removal of the tumor may lead to instability, which may require fusion and internal fixation.
In the cervical spine, their presence so close to neurovascular structures (e.g., vital blood vessels and the spinal cord) makes treatment of this problem very complex. Embolization (either partial or complete) may be done first before surgery. Embolization is a nonsurgical, minimally invasive procedure using metal sponges or other devices to purposefully block blood flow. Surgery to remove the tumor is then done within 24 hours of the embolization. When necessary, bone defect filling and instrumented fusion may be done.26
PROGNOSIS.
Ninety percent to 95% of osteoblastomas are cured by the initial treatment,35 but even with careful removal of the tumors, they recur in about 10% of affected individuals.87 There is a risk of malignant transformation into an osteosarcoma, which can sometimes be determined early. Appropriate intervention with adjunctive chemotherapy or radiation is the current standard of care. Embolization before marginal resection may reduce the rate of recurrence.26
PRIMARY MALIGNANT BONE TUMORS
Primary malignant bone tumors are relatively rare, representing about 6% to 7% of all pediatric neoplasms. Osteosarcomas are the most frequent type, followed by Ewing’s sarcoma. Osteosarcomas make up over half of all malignant bone tumors; Ewing’s sarcomas account for one-third of all primary malignant bone tumors (Table 26-4).36
Table 26-4
Malignant Bone Tumors*

*In order of descending frequency.
Adapted from Damjanov I: Pathology for the health professions, ed 3, Philadelphia, 2006, Saunders.
Osteosarcoma
Osteosarcoma, also known as osteogenic sarcoma, is an extremely malignant tumor with destructive lesions and abundant sclerosis, both from the tumor itself and from reactive bone formation. A characteristic of osteosarcoma is the production of osteoid by malignant, neoplastic cells. This is seen on photomicrographs and is one of the features used to help differentiate this tumor. Resected specimens usually show that the cortex has been broken by the destructive tumor. Although various types of osteosarcoma exist, including parosteal, periosteal, telangiectatic, and small cell, only the most common, conventional intramedullary osteosarcoma, is discussed here.
Incidence
Osteosarcoma is the second most frequent malignant condition of bone, accounting for 15% to 20% of all primary bone tumors; only myeloma is seen more often. Osteosarcoma occurs most often in male children, adolescents, and young adults under the age of 30, with a peak frequency during the adolescent growth spurt and another smaller peak in people older than 50 years.30
Osteosarcoma can develop in many bones but is more common in long bones, the site of the most active epiphyseal growth. The distal femur (knee) is the most common site, followed by the proximal tibia and proximal fibula (50% are located in the knee region), proximal humerus, pelvis, and occasionally the mandible, vertebrae, or scapula.
Etiologic and Risk Factors
Osteosarcomas can be primary or secondary. Certain genetic or acquired conditions increase the risk of osteosarcoma (e.g., retinoblastoma, Paget’s disease of bone, enchondromatosis, ionizing radiation). Alterations of multiple chromosomes and their extra copies have been demonstrated but only in distinct clinical subsets of osteosarcoma. Secondary osteosarcomas are those that develop from other lesions such as Paget’s disease, chronic osteomyelitis, osteoblastoma, or giant cell tumor.
Pathogenesis
The mechanisms involved in the development of osteosarcomas are still obscure. Osteosarcoma originates from primitive (poorly differentiated) cells from the osteoblasts of the mesenchyme. This suggests that early osteoprogenitor cells with the ability for chondroblastic differentiation are affected in the development of osteosarcoma. Whether a protective mechanism in the process of bone development is turned off (suppressed) or differentiation activity is altered remains unknown.
Osteosarcoma grows rapidly and is locally destructive. It may be osteosclerotic (producing considerable neoplastic or tumor bone), or it may arise from more primitive cells and remain predominantly osteolytic, eroding the cortex of the metaphyseal region and resulting in pathologic fracture. As it continues to grow beyond the confines of the bone, the tumor lifts the periosteum, resulting in the formation of reactive bone in the angle between elevated periosteum and bone called Codman’s triangle (see Fig. 26-3).
Clinical Manifestations
Osteosarcoma seems to appear in bones undergoing an active growth phase and appears at the epiphyseal plate of rapidly growing bone in adolescents. The long bones such as the distal femur, proximal humerus, and proximal tibia have a relatively more active growth period than other bones, which makes them more vulnerable (Fig. 26-6).
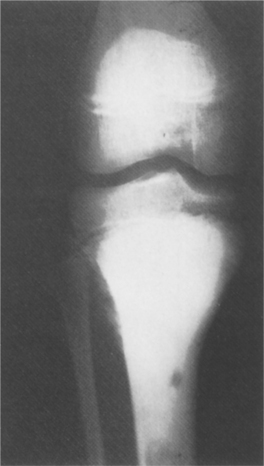
Figure 26-6 Osteosarcoma. An extremely sclerotic lesion in the proximal tibia of a child is noted, which is characteristic of an osteogenic sarcoma. (From Helms C: Fundamentals of skeletal radiology: benign cystic lesions, Philadelphia, 1989, WB Saunders.)
Pain that has continued for several weeks to months is the presenting complaint. The tumor is often located in the metaphysis but does not cross the physis. Even so, joint pain and tenderness can be present as the lesion penetrates the cortex and invades the joint capsule, also spreading to other nearby structures (e.g., tendons, fat, muscles).
Since osteosarcoma can be a rapidly destructive tumor, the pain increases, and swelling may develop in just a few weeks, accompanied by some limitation of motion. Systemic symptoms are rare, although occasional fever may occur. This aggressive neoplasm is very vascular, and the overlying skin is usually warm. Metastases appear in the lungs early in 90% of cases and occur in 20% to 25% of cases at the time of presentation.6
MEDICAL MANAGEMENT
Diagnosis is often delayed, especially when swelling is minimal, as is often the case in early stages.46 X-rays should be done with any complaint of bone pain, especially around the knee. Plain radiographs often reflect dramatic changes and obvious tumor formation, but important findings can also be subtle.
CT scans and especially MRI are used to evaluate the extent of disease. In Fig. 26-7 plain films of a pelvis demonstrate minimal changes that could easily be dismissed as insignificant. The CT scan, however, reveals a large osteosarcoma involving the ilium. More commonly, radiographs show a rapidly growing lesion with poorly defined margins, and a permeated or moth-eaten appearance in the lytic area.
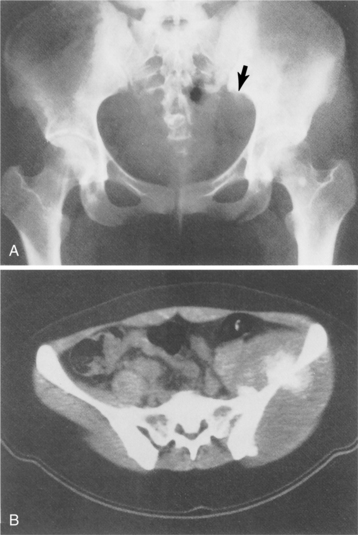
Figure 26-7 Osteosarcoma. A, A subtle sclerotic lesion is seen in the left ilium adjacent to the sacroiliac joint that was initially diagnosed as osteitis condensans ilii, a benign entity. Because of persistent pain, the person returned for a follow-up visit, and a small amount of cortical destruction on the pelvic brim was noted (arrow). B, A computed tomographic scan was performed, which showed a large tissue mass and new bone tumor around the ilium, which is characteristic of an osteogenic sarcoma. (From Helms C: Fundamentals of skeletal radiology: benign cystic lesions, Philadelphia, 1989, WB Saunders.)
A biopsy is performed to determine the histologic makeup of the lesion. Serum alkaline phosphatase level is often elevated, but this is not diagnostic.
TREATMENT.
Because osteosarcoma is relatively resistant to radiation therapy, complete surgical removal of the primary tumor and any metastases is essential to cure.6 The current surgical thinking is to use limb-sparing techniques (segmental resection and replacement with bone graft or implant) whenever possible (Fig. 26-8).
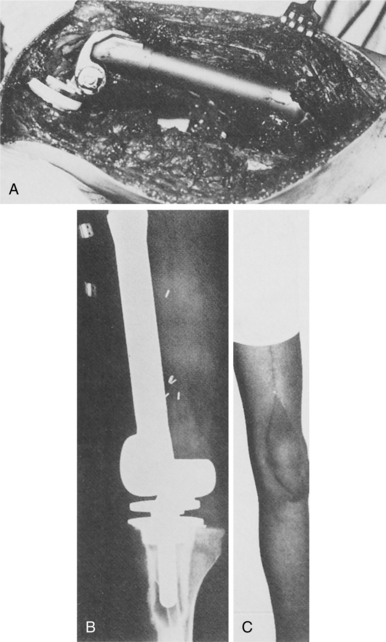
Figure 26-8 Osteosarcoma of the distal femur in a 17-year-old boy. A, Intraoperative photograph following resection of the distal femur. B, Postoperative radiograph of custom-made, rotating-hinge prosthesis. C, Follow-up clinical photograph (3 years after surgery). Soft tissue coverage of the prosthesis by latissimus dorsi myocutaneous free flap with acceptable cosmesis. (From Klein M, Kenan S, Lenis M: Osteosarcoma: clinical and pathologic considerations, Orthop Clin 20:343, 1989.)
The use of a noninvasive expandable prosthesis for skeletally immature children and adolescents following limb salvage for malignant tumors in the leg has been reported. The Repiphysis prosthesis for pediatric osteosarcoma is an expandable metal rod that replaces the bone and does not require repeated procedures to lengthen as the child’s other leg grows. Painless electromagnetic rays are used to expand the rod slowly without compromise to the surrounding skin and muscle.43
Another creative procedure called rotationplasty removes the cancerous portion of the bone below the knee then uses the remaining bottom segment of the leg and ankle joint as a new knee. The surgeon removes the affected bone, rotates the lower portion of the leg 180 degrees so the foot faces the opposite direction, and reattaches it to the upper femoral area. Nerves, muscles, and blood supply are preserved. The posterior-facing ankle now functions as a weight-bearing knee joint in a specially fitted prosthesis (Fig. 26-9). Although the outcome is visually unusual, such a procedure improves gait and knee function and prevents amputation.100
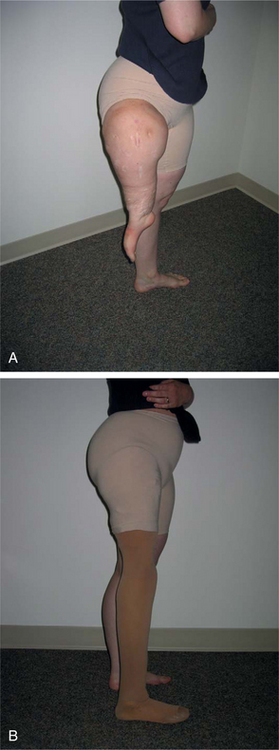
Figure 26-9 Rotationplasty for osteosarcoma. The primary reason for rotationplasty is to enhance the person’s mobility as a prosthesis user. Placing the ankle joint in the position of the knee creates a functional, natural knee, and the toes provide important sensory feedback to the brain. A, Rotationplasty removes the cancerous portion of the femur (proximal to the midshaft of the femur), then rotates the lower portion of the leg 180 degrees so the foot faces the opposite direction. The proximal tibia is fused to the distal femur; the remaining bottom segment of the leg and ankle joint function as a new knee. B, Standing on the prosthesis with the cover on it. (Courtesy Kevin Carroll, Hanger Prosthetics and Orthotics, Orlando, FL.)
When the child takes off the prosthesis, the cosmesis of seeing a foot turned backward may not be acceptable. In such cases, children and families may still prefer endoprosthetic reconstruction or even amputation. Younger children (less than 10 years old) seem better able to adapt psychologically and physically to the rotationplasty.64
The tibia turn-up is another important procedure that is an option in cases of osteosarcoma (Fig. 26-10). The leg is amputated above the knee, and the tibia bone from the lower leg is inverted, or turned up, making it possible for the ankle end of the tibia to be fused to the bottom of the femur. The muscles are then sutured back onto the tibia.19,20
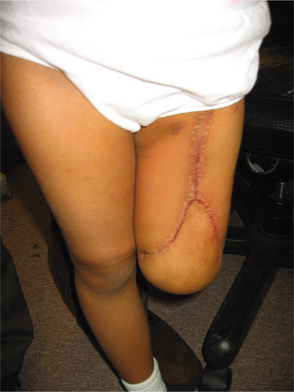
Figure 26-10 Tibia turn-up procedure. Sarcoma just below lesser trochanter in a 7-year old girl. There were three surgical options for this client: (1) transtrochanteric amputation (major loss of limb), (2) tibia turn-up procedure (shown here), or (3) rotationplasty (see Fig. 26-9). The tibia turn-up procedure was chosen for cosmetic reasons with excellent functional outcomes with the use of a prosthesis. The tibia turn-up procedure avoids high-level transfemoral amputation and provides an outcome similar to that of a knee disarticulation amputation. (Courtesy Kevin Carroll, Hanger Prosthetics and Orthotics, Orlando, FL.)
Tibia turn-up is an alternative that people may consider when the appearance of a rotationplasty seems too extreme. Tibia turn-up is also an option when cancer occurs in the thigh that might otherwise require a high-level above-knee amputation (Fig. 26-11). By having the tibia fused to the femur, these individuals now have a long residual limb that will be easier to fit with a prosthesis, providing them with increased function. Although these individuals will wear an above-knee prosthesis with a mechanical knee, their comfort and mobility will usually exceed that of above-knee prosthesis users with a short residual limb.19
Figure 26-11 Rotationplasty or tibia turn-up can be a good alternative to high-level above-knee amputations such as this. (Courtesy Kevin Carroll, Hanger Prosthetics and Orthotic, Orlando, FL.)
Rotationplasty and tibia turn-up techniques both make allowances for the natural process of growth that extends into young adulthood. Before surgery, x-rays and other tests are performed to determine how much growth will occur in the sound leg. Growth plates at the hip account for 30% of growth in the femur, while plates at the knee contribute the remaining 70%. In the lower leg, plates at the ankle account for 40% of growth in the tibia and fibula, while those at the knee contribute the remaining 60%. Therefore, if the growth plates on either side of the knee are completely removed during amputation, the surgeon may choose to make the residual limb a little longer to compensate. Oftentimes, however, a growth plate can be salvaged, enabling the femur to grow naturally. If, in the future, the amputated side begins to grow more than desired, the surgeon can stop the growth by suturing the growth plate.19
Many factors such as age, remaining growth, expected functional outcome, and prognosis are considered in making the best treatment choices for osteosarcoma. Chemotherapy often precedes surgery. Chemotherapy is evaluated by its effect on the client and tumor. Chemotherapy may also help lessen the chance of skip lesions, or multiple foci of tumor that can cause recurrence of the tumor after surgery. New discoveries about the molecular genetics of osteosarcoma eventually may lead to effective gene therapy for osteosarcoma.
PROGNOSIS.
Until the 1970s, surgery for osteosarcoma consisted of amputation or disarticulation. The 5-year survival rate at that time was about 20%, with frequent pulmonary micrometastasis.25 Today the use of adjunctive (preoperative) chemotherapy with surgery results in 5-year cure rates of 70% to 80%. The majority of affected individuals (more than 90%) have limb-sparing surgery.101 Surgery alone will probably allow pulmonary metastasis to occur. Individuals who develop lung metastases have a 20% to 30% 5-year survival rate.
Even with chemotherapy, the outcome is dependent on the stage at diagnosis and the ability of the surgeon to achieve a tumor-free margin. Local recurrence is a poor prognostic sign. Local recurrence of craniofacial lesions after treatment is 50% for mandibular tumors and even higher for maxillary and skull lesions (80% and 75%, respectively); metastases occur in about one third of craniofacial osteosarcomas.30
In older people, osteosarcoma may develop as a complication of Paget’s disease, in which case the prognosis is extremely grave.84
26-5 SPECIAL IMPLICATIONS FOR THE THERAPIST
See previous discussion and Special Implications for the Therapist: Primary Tumors earlier in the chapter.
Malignant neoplasms usually necessitate aggressive intervention, and therefore rehabilitation is more intensive, prolonged, and individualized. Extensive surgery, such as limb-sparing techniques, has provided therapists with an opportunity to assist these clients in maximizing their function (Fig. 26-12). When musculoskeletal structures are involved, it is important to be aware of reduced tensile strength of malignant tissue as compared with uninvolved bone tissue.
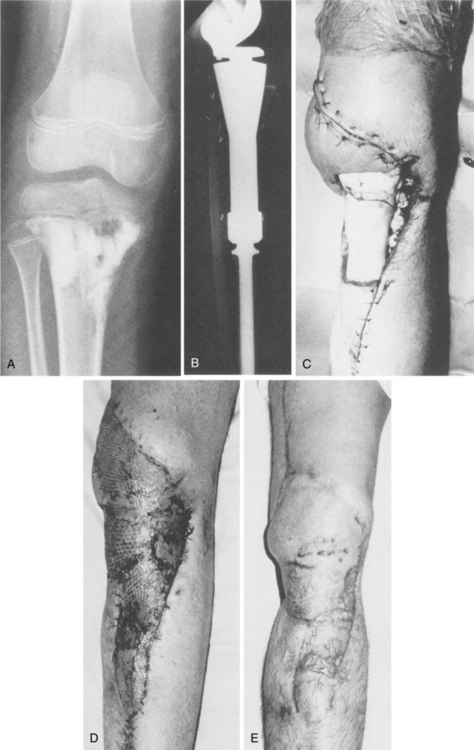
Figure 26-12 Use of a free muscle transfer to salvage an infected massive prosthesis. A, Preoperative radiograph of a 9-year-old boy with an osteosarcoma. B, After radical resection, an expandable prosthesis was inserted. C, When infection occurred, with subsequent breakdown of the wound, the prosthesis was removed, and the area was widely débrided. A spacer of antibiotic-impregnated methacrylate was inserted. D, Infection was controlled, and the knee was reconstructed with another prosthesis and a free latissimus transfer. E, A satisfactory result was obtained, sparing the leg. (From Hausman M: Microvascular applications in limb-sparing tumor surgery, Orthop Clin 20:434, 1989.)
Preoperative Assessment19
People who have been diagnosed with cancer and are faced with the impending amputation of a leg find themselves in a state of shock and grief. Parents of children who are born with a lower limb difference experience similar emotions. Under these circumstances, it is difficult to talk openly with a surgeon about amputation and to meet with a prosthetist to discuss future prosthetic needs. The fact that the majority of these clients are children, teenagers, and young adults only increases the level of anxiety. Yet beneath the surface of these painful conversations are seeds of hope: amputation can save a person’s life, preoperative consultations can help people make better decisions, and children who are fitted early with a prosthesis can lead very active lives.19
Knowing what the options are before surgery can enable individuals and their families to make the best choice for each specific situation. Not all surgeons are aware of limb-sparing procedures, some of which involve bone replacement with human or laboratory-grown bone, or prosthetic implants. Some surgeons may make what they consider the most “conservative” recommendation: a standard above-knee amputation at a point significantly higher than the site of the cancer. To be as informed as possible, the best course of action is to consult with one or more orthopedic oncologists at a comprehensive cancer center. Parents of children with congenital lower limb differences should seek the advice of a pediatric orthopedic physician. Treatment of these conditions requires highly specialized physicians and medical facilities.19
The therapist can be instrumental in discussing the Van Nes rotationplasty and tibia turn-up, two surgical procedures that may increase client mobility as prosthesis users. At first glance, both procedures appear somewhat extreme and are difficult for people to visualize. However, the long-term positive results experienced by most people are impressive. Ideally, rotationplasty gives the affected individual a level of function that may be equivalent to that of a below-knee prosthesis user, even though he or she has experienced an above-knee amputation. The goal of tibia turn-up is to provide the person who faces a high-level, above-knee amputation with a longer, stronger residual limb onto which the prosthetic socket can lock.19
Since these tumors are treated at regional medical centers, the initial phases of rehabilitation may be implemented by therapists with a great deal of experience working with clients with malignant neoplasms and those who have undergone various reconstructive surgical procedures.
When the client returns home a local therapist may be called on to continue the rehabilitation program. Communication with the therapist at the regional medical center to confirm initial management plan, progression, and prognosis is recommended.
The use of a new tool, Functional Mobility Assessment (FMA), has been examined in clients with lower extremity sarcoma. FMA requires the individual to physically perform functional mobility tasks and provides a reliable and valid measure of objective functional outcome and may help therapists guide children and adolescents in returning to daily activities.64
As might be expected, rehabilitation following limbsparing surgery or rotationplasty focuses on retraining muscles and increasing weight bearing and balance, ROM, and strength.
Chondrosarcoma
Chondrosarcoma is usually a relatively slow-growing malignant neoplasm that arises either spontaneously in previously normal bone or as the result of malignant change in a preexisting nonmalignant lesion, such as an osteochondroma or an enchondroma. The pelvic and shoulder girdles are common sites of tumor and related pain, as are the proximal and distal femur, proximal humerus, and ribs.
Chondrosarcoma is the second most common solid malignant tumor of bone in adults (after osteosarcoma, third after myeloma). These tumors can be primary or secondary. Primary chondrosarcomas are more common, but their origin is idiopathic. Secondary tumors are those that arise from previously benign cartilaginous tumors or from a preexisting condition such as Paget’s disease.
Men in their forties to sixties are those most likely to be affected by primary chondrosarcoma.
Pathogenesis
In general, chondrosarcomas develop from cells committed to cartilaginous differentiation. The neoplastic cartilaginous cells produce cartilage rather than the osteoid seen with osteosarcoma. Alterations of programmed cell death (apoptosis) may play a significant role in the pathogenesis of low-to intermediate-grade chondrosarcomas, whereas high-grade lesions most likely develop by means of a multistep mechanism involving multiple transforming genes and tumor suppressor genes.30
Chondrosarcoma is classified by location of the lesion: central, peripheral, or juxtacortical. With central chondrosarcoma, the neoplastic tissue is compressed inside the bone, and areas of necrosis, cystic change, and hemorrhage are common. Peripheral chondrosarcoma arises outside the bone and then invades the bone. The juxtacortical chondrosarcoma is thought to be periosteal (affecting the periosteum) or parosteal (affecting the outer surface of the periosteum) in origin. Chondrosarcomas can be graded based on their microscopic appearance. The presence of a chondroid matrix, extent of necrosis, and type of cells are some of the grading standards used.
Clinical Manifestations
Pain is the most common presenting complaint, although this is a slow-growing tumor, so in some cases the tumor can exist for years without symptoms. The lesion can range from a slow-growing lesion to an aggressive malignancy capable of metastasizing to other organs. The metastatic potential of chondrosarcoma is less than for osteosarcoma. The majority of chondrosarcomas are grade I or II, which rarely metastasize. When metastasis occurs, it is via the hematogenous route to the lungs, others bones, or organs.
MEDICAL MANAGEMENT
On radiograph the tumor often shows an expansile lesion in the diaphysis of long bones with cortical thickening and destruction of the medullary bone (Fig. 26-13). The appearance is somewhat variable depending on the rate of growth and the host bone response. Biopsy is important not only for accurate diagnosis but also for guiding treatment. Chondrosarcoma can develop on the surface of bone or present as multicentric, involving several bones.
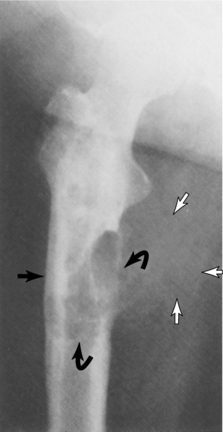
Figure 26-13 Characteristic radiographic features of chondrosarcoma include thickening of the cortex (closed arrow); destruction of the medullary and cortical bone (curved arrows); and soft tissue mass (open arrows). Note the characteristic punctate calcifications in the proximal part of the tumor. (From Greenspan A: Tumors of cartilage origin, Orthop Clin 20:359, 1989.)
TREATMENT.
Treatment of chondrosarcoma is surgical, with complete tumor removal. Wide resections or limb-sparing procedures are often required, and internal fixation after tumor removal to prevent fracture may be recommended. As with osteosarcoma, radiation therapy is ineffective. Due to the slow-growing nature of this malignancy, chemotherapy is limited in its effectiveness.
PROGNOSIS.
The prognosis is dependent on the aggressiveness and stage of the lesion. For example, a grade I lesion is unlikely to metastasize, and if it is completely resected, a good prognosis follows with 80% chance of cure. A grade III lesion is much more likely to metastasize. Undifferentiated lesions found in the pelvis or any bone where complete resection is difficult have a poorer prognosis. Secondary chondrosarcomas are usually of a low-grade malignancy and have a good prognosis with adequate intervention.
Ewing’s Sarcoma
Ewing’s sarcoma is a malignant nonosteogenic primary tumor that can arise in bone or soft tissue.44 It is the second most common primary malignant bone tumor of children, adolescents, and young adults and the fourth most common overall, although it only accounts for approximately 3% of all pediatric malignancies.51 Most tumors of this type (80%) occur in young people under the age of 20; approximately 225 new cases are diagnosed each year in the United States.9 Ewing’s sarcoma has been reported in children as young as 5 months, but occurs rarely in the black population.
Although this type of bone tumor was noted as early as 1866, it was not until 1921 that James Ewing described his experience with the lesion. The pelvis and lower extremity are the most common sites. Unlike with many tumors, no predilection for a certain part of the bone is evident.
Risk Factors, Etiologic Factors, and Pathogenesis
Based on different levels of scientific evidence, the main risk factors related to Ewing’s sarcoma include Caucasian race, parental occupation (exposure to pesticides, herbicides, fertilizers), and parental smoking.36
Cytogenetic studies show that 95% of these tumors are derived from a specific genetic translocation between chromosomes 11 and 22, although the molecular oncogenesis remains unknown. The formation of the EWS-FLI1 fusion protein from the chromosomal translocation contributes to the pathogenesis of Ewing’s sarcoma by modulating the expression of target genes.
Ewing’s sarcoma is composed of islands of small, uniformly round cells of neural origin characterized by strong membrane expression of CD99.9,27 It is the least differentiated tumor in a group of neuroectodermally derived lesions in bone and soft tissue. These morphologic features are characteristic enough to serve as useful diagnostic markers.58
The tumor is soft, sometimes viscous, with hemorrhagic necrosis caused by the rapid tumor growth outpacing its blood supply. The cortical bone is affected through the haversian canals. The medullary cavity is affected, and infiltration of the bone marrow can progress extensively without radiographic evidence of bone destruction. When the tumor perforates the cortex of the bone shaft and elevates the periosteum, the consequent reactive bone formation causes layered calcification referred to as an “onion-skin” appearance seen radiographically (Fig. 26-14).
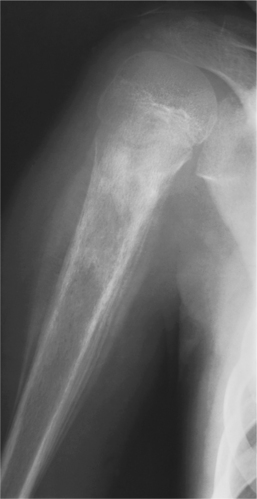
Figure 26-14 Ewing’s sarcoma of the humerus. Bone destruction is seen in the proximal metadiaphysis. The cortex is infiltrated and a multilaminar periosteal reaction with an onion-skin appearance is present medially; Codman’s triangles are present on the lateral aspect. (From Grainger RG, Allison D: Grainger and Allison’s diagnostic radiology: a textbook of medical imaging, ed 4, Philadelphia, 2001, Churchill Livingstone.)
Clinical Manifestations
As with other malignant bone tumors, local bone pain is the most common presenting symptom after an injury (e.g., sports-related injury), a factor that sometimes delays diagnosis. Ewing’s sarcoma presents most often in the long (tubular) bones (e.g., femur, tibia, fibula, humerus) and the pelvis. Less often, the ribs, scapula, vertebrae, feet, and craniofacial bones are involved.
Swelling occurs in approximately 70% of all cases, and both pain and swelling are usually progressive. The pain may be intermittent, which also delays diagnosis. There may be a palpable or observable mass. Pathologic fractures occur at the site of the tumor in long bones but only in 5% to 10% of cases. In young children, flulike symptoms, including a low-grade fever, may be present, which may lead to the mistaken diagnosis of osteomyelitis.45
Ewing’s sarcoma frequently metastasizes to other bones, especially late in the course of the disease. When the cervical or lumbar spine is involved, neurologic deficit may lead to a mistaken diagnosis of disc disease.45
MEDICAL MANAGEMENT
Anyone suspected of having Ewing’s sarcoma is staged for both local and metastatic disease. Radiographs show an obvious lytic process with a moth-eaten appearance involving a diffuse area of bone (Fig. 26-15). As mentioned, an onion-skin formation may be seen, which is due to layers of reactive bone (see Fig. 26-14). On radiographs the appearance may not differentiate this lesion from osteomyelitis or osteosarcoma.
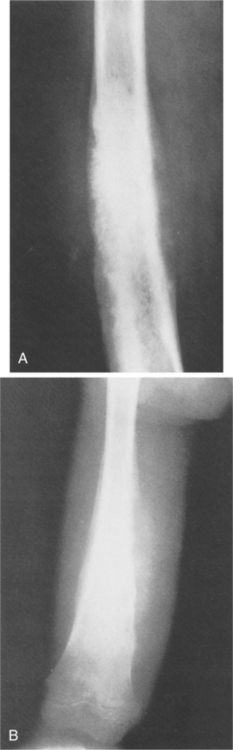
Figure 26-15 Ewing’s sarcoma. A, A mixed lytic-sclerotic lesion in the femur of a child with periostitis that is amorphous and sunburst that is characteristic of Ewing’s sarcoma. B, This is a predominantly sclerotic process with large amounts of sunburst periostitis in the diaphysis of a femur that, on biopsy, was found to be Ewing’s sarcoma. (From Helms C: Fundamentals of skeletal radiology: benign cystic lesions, Philadelphia, 1989, WB Saunders.)
An elevated ESR may be noted but is not diagnostic. CT, MRI, and bone scans can help diagnose and define the extent of the tumor. MRI is more sensitive than CT scan in assessing soft tissue involvement and bone marrow spread. The MRI or CT scan is repeated after several cycles of chemotherapy to better assess the response to chemotherapy and help plan further treatment of the local site with radiation or surgery.
Metastatic disease is evaluated at the time of presentation with chest x-ray or chest CT scan looking for pulmonary metastases. Bone scan to detect bone metastases, bone marrow aspirate at a site far from the local tumor site, and tumor biopsy are used to assess the spread of the disease and help with staging and treatment planning. Researchers are investigating the use of real-time polymerase chain reaction (PCR) to provide accurate quantitative estimates of circulating tumor burden in this disease.67
TREATMENT.
Significant progress has been made in the management of Ewing’s sarcoma in the past 25 years. Cure requires intensive therapy to control both local and distant disease. Multimodal treatment can include chemotherapy, radiotherapy, immunotherapy or biotherapy, embolization, and surgery.5,9 Local tumors are very responsive to high-dose radiation. In some cases, radiation is associated with the additional morbidities of second malignancy and a significant adverse impact on both cardiac and pulmonary function.89
Effective combination chemotherapy has been developed to eradicate distant metastases. Selective surgery in the treatment of primary Ewing’s sarcoma can result in amputation, but the development of limb-sparing techniques has reduced amputations considerably. There is no ideal method of reconstruction in limb salvage surgery. The choice of method is individualized based on many factors, including age; location and extent of the tumor; preferences of the client or, in the case of a child, the family; the availability of surgical facilities and expertise; and cost of the procedure.98
Targeted therapies using drugs against the insulin-like growth factor receptor I (IGF-IR or CD99) are under clinical investigation. CD99 is a cell surface transmembrane protein that is highly expressed in Ewing’s sarcoma. Neutralizing IGR-IR functions has been shown in animal studies to significantly affect tumor cells by causing massive apoptosis of Ewing’s sarcoma cells, thus reducing their malignant potential.87 Studies incorporating intensive therapy followed by stem cell infusion show no clear benefit.9,32
PROGNOSIS.
Although Ewing’s sarcoma is extremely malignant with a high frequency of both metastatic spread and local recurrence, the prognosis for clients with this tumor is improving steadily. Just a few decades ago only about 5% to 10% of clients with Ewing’s sarcoma lived longer than 5 years after detection. The 5-year survival rate is now in excess of 70% if metastasis has not occurred at the time of diagnosis and treatment.51
People with Ewing’s sarcoma of distal sites such as the bones of the hands and feet have a much better prognosis than people with lesions in central sites such as the pelvis and sacrum. Tumors larger than 8 to 10 cm have a significantly poorer outcome than smaller tumors.97
Long-term survival is determined by the presence or absence of metastasis and the site and extent of the local tumor58; only about 25% of individuals with metastatic disease at the time of diagnosis survive 5 years.9
Many individuals without metastasis are remaining continuously disease free at 5 and 10 years. As many as 35% of clients will have metastatic disease at the time of diagnosis, usually to the lung. More than four metastatic nodules is a poor prognostic indicator, whereas good response to chemotherapy (e.g., decrease in the size of the tumor mass, greater than 95% tumor kill) is a favorable prognostic sign.78,102
There is much debate about the role of age at diagnosis. Some studies show older age to be associated with poorer outcome; others show no association between age and survival. It may be that younger children with small, well-defined, distal lesions have the best prognosis.88 With the increase in long-term survival rates following improved treatment intervention, the problems of late local recurrence, late functional impairment secondary to complications of radiation therapy, and radiation-induced sarcomas are on the rise.97
Chordoma
Chordomas are usually slow-growing but locally aggressive malignant neoplasms. Chordomas account for 1% to 4% of all malignant bone tumors, primarily affecting older adults.11,106
Chordomas do not have a capsule and tend to infiltrate into neighboring soft tissues. Metastases can occur to the liver, lungs, lymph nodes, peritoneum, skin, heart, brain, and distant regions of the spine but often remain asymptomatic and are discovered only on postmortem examination. Metastases occur most often when there is local recurrence of the primary tumor.65
Clinical Manifestations
Most chordomas arise in the midline of the body, involving the clivus (central skull base) in half the cases. One third of all chordomas occur in the sacrum, with the remaining found in the cervical and lumbar spine. The high cervical region, especially C2, is affected most often.106 Clival chordomas are frequently midline lesions whose posterior growth may breach the dura and invaginate the brainstem.
Clinical manifestations based on the biologic behavior of chordoma appears to differ from person to person. The most common presenting symptom is pain; generally, symptoms depend on the location of the tumor. For example, clival chondromas may cause headaches, visual disturbances, dysphagia, muscle weakness, and even hemiparesis.50
Night pain or pain at rest that is not relieved by analgesics is a red flag finding. Other symptoms can include bowel and/or bladder dysfunction, gait disturbances, and motor impairment.
MEDICAL MANAGEMENT
The mainstay of treatment for chordoma is aggressive surgical resection. Complete resection of the tumor is not always possible, especially when it is located in the high cervical region. Adjuvant therapy (radiation and/or chemotherapy) may be administered before and/or after surgery. Recurrence is seen, often requiring subsequent treatment. Metastases require resection and chemotherapy unless metastases are too extensive for systemic treatment.65
PROGNOSIS.
Although chordoma is a relatively slow-growing tumor, it has a high incidence of local recurrence and poor long-term prognosis.104,106 Cancer recurrence often necessitates repeat surgical procedures with risk for complications.
Metastases are becoming more common as people with chordomas live longer as a result of more aggressive surgical and adjuvant treatments. Researchers hope to identify markers that will help predict which tumors will behave aggressively in order to direct treatment toward early diagnosis and intervention for people with aggressive tumors.65
Giant Cell Tumor
Giant cell tumor of bone is a distinct, locally aggressive neoplasm that accounts for approximately 5% of all primary bone tumors. Although classically considered benign, these tumors are now considered a low-grade (malignant) sarcoma because of their high rate of recurrence and potential for malignant transformation.28
The tumor most frequently involves the epiphyseal ends of long tubular bones in skeletally mature adults between the ages of 20 and 55 years of age, with a peak age incidence in the third decade of life. Giant cell tumor occurs more often in Chinese people (up to 20% of the population are affected) compared to Caucasians in Western countries.
Sixty percent occur around the knee; 10% to 12% involve the distal radius. The bones of the hand and wrist are rarely affected.52 Although the sacrum can be affected, it is extremely rare in the vertebrae.
Etiology and Pathogenesis
The etiology of giant cell tumor is unknown. The tumor cells have been reported to produce chemoattractants that can attract osteoclasts and osteoclast precursors.90
Pathologic examination of the neoplasm reveals a tumor that is soft, friable (easily breaks apart), fleshy, and red-brown with yellow areas. The tumor usually extends to but not into the articular cartilage. Destruction of the bone cortex with expansion into soft tissue can occur (Fig. 26-16). Hemorrhage, cyst formation, and necrosis can be seen on gross pathology. Hemorrhage and necrosis (often accompanied by pathologic fracture) occur often in the weight-bearing bones. The tumors can be locally invasive (into bone and soft tissue) with extensive bone destruction and cortical expansion.
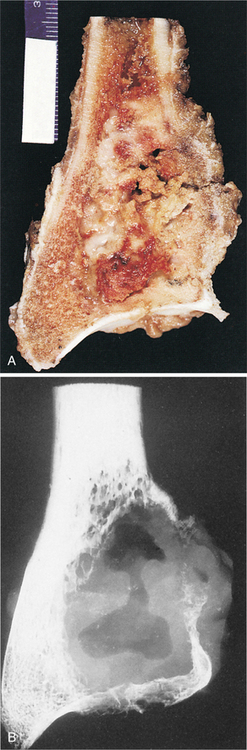
Figure 26-16 Giant cell tumor. Gross morphologic features of giant cell tumor. A, Bisected distal end of radius with well-demarcated tumor mass expanding bone contour. Tumor tissue is red-brown with yellow septations. B, Radiographic presentation of tumor showing focal destruction of cortex. (From Dorfman HD, Czerniak B: Bone tumors, St Louis, 1998, Mosby.)
Clinical Manifestations
Pain on weight bearing with pathologic fracture may be the presenting clinical feature when tumors occur in the weight-bearing bones. Sacral tumors may present with localized pain in the low back radiating to one or both of the legs. Abdominal discomfort and bowel and bladder symptoms may be present.80
Pulmonary metastases referred to as benign pulmonary implants occur in 1% of cases. The nodules grow slowly and can be excised effectively. They are usually asymptomatic and discovered when a routine chest x-ray is taken. Multiple lung lesions or progressive spread can result in death.
MEDICAL MANAGEMENT
Diagnosis is by x-ray and confirmed by histologic assessment with findings typical of this particular type of tumor observed. Treatment is with surgical excision; bone grafting to fill the cavity and further reconstruction may be needed.95
Recurrence is usually confined to the bone and does not extend to the soft tissue. Recurrence after excision occurs in up to one third of clients. Recurrence usually occurs within 3 years of the index removal but can occur many years (even decades) later. Although it is rare, giant cell tumors can transform to a malignant form.42
Radiation is not a mainstay of treatment but may be utilized in cases of surgically inaccessible or incompletely resectable lesions.66 The use of adjuvant treatment such as chemotherapy and radiotherapy is debated. Studies using radiofrequency ablation, bone substitutes, and liquid nitrogen and phenol as alternative therapies are underway.41
MULTIPLE MYELOMA
Multiple myeloma is a hematopoietic neoplasm involving bone marrow. It is a primary bone cancer with plasma cell proliferation and is one of a group of disorders called plasma cell dyscrasias (see Chapter 14 for a detailed description of this condition).
Skeletal involvement is most common in the spine, pelvis, and skull, because bone marrow is found in high concentrations in these structures. Deep bone pain is often present clinically, and radiographs may demonstrate osteopenia and punched-out areas of bone with sclerotic borders (in flat bones) (Fig. 26-17).
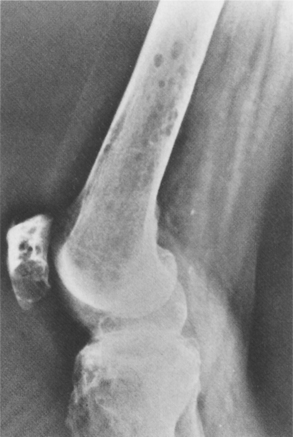
Figure 26-17 Multiple myeloma. Small lucencies in the distal femur, proximal tibia, and patella. (From Ghelman B: Radiology of bone tumors, Orthop Clin 20:307, 1989.)
The prognosis is generally poor, with most people dying from the disease within 1 to 3 years after the diagnosis is made.
PRIMARY SOFT TISSUE TUMORS
Common benign soft tissue tumors include lipoma, ganglia, popliteal cyst (Baker cyst), nerve sheath tumor (neurofibroma and schwannoma), and desmoid tumors.
Lipoma is the most common soft tissue tumor, generally occurring during middle age and late adulthood and comprised of mature fat cells. These tumors are usually superficially located in the subcutaneous tissue and remain asymptomatic. Occasionally a lipoma of the breast will grow large enough to cause tenderness and block lymphatic drainage, requiring removal. Even without surgical excision, lipomas are unlikely to ever undergo malignant transformation, but recurrence is possible if the lesion, including microscopic cells, is not completely removed.
Ganglia arise from a joint capsule or tendon sheath, usually on the dorsal aspect of the wrist but sometimes on the volar aspect of the wrist or on the lower extremity. Pain or tenderness may or may not be present; pressure on a nerve can cause focal neurologic symptoms.
Popliteal cyst, more commonly referred to as a Baker cyst, is a subtype of ganglion that often communicates with a joint space. A Baker cyst is most often palpated behind the knee in older adults with osteoarthritis. Rupture of the cyst or hemorrhage from the joint into the cyst causes episodes of severe pain. Swelling distal to the lesion (calf and foot) may also occur.
Nerve sheath tumor is a tumor of the nerve sheath arising in a peripheral nerve and growing concentrically from the center of the nerve. Neurofibromas infiltrate the nerve and splay apart the individual nerve fibers. They are usually superficially located, painless, and benign but can sometimes degenerate into cancer.
Neurofibromas can occur as a single lesion or in greater numbers as part of a collection of symptoms in association with von Recklinghausen’s disease (neurofibromatosis) and schwannomas. Neurofibromas contain cells and features of Schwann cells but also contain fibroblasts and perineural cells. Both neurofibromas and schwannomas are benign, grow slowly, and can be cured surgically.1,60
Schwannomas and neurofibromas arise from the coverings of peripheral and cranial nerves. Schwannomas arise from Schwann cells as the name suggests. Schwannoma is a rare tumor of the sheath or lining around the peripheral nerves. It starts in the Schwann cells, which is how it gets its name. Schwann cells help form the cover around the nerves called the myelin sheath. Schwannomas can be benign or malignant. The malignant type is called neurosarcoma or neurogenic sarcoma.
In the benign form, growth is slow and painless. The tumor stays on the outside of the nerve. The benign form does not spread to other areas and is not likely to cause death. But if it grows large enough to put pressure on the nerve, then pain, numbness, and even paralysis can occur.
Schwannomas can arise from Schwann cells covering the vestibular portion of cranial nerve VIII, causing benign acoustic neurinomas. Although the tumors grow slowly and are considered benign, they can compress the eighth cranial nerve, resulting in hearing loss and tinnitus. Vestibular function is lost but slowly enough that the body compensates; for this reason vertigo is uncommon with acoustic neuromas. Large tumors can compress the cerebellum and brainstem, resulting in ataxia and hydrocephalus. The affected individual may also experience facial paralysis if the trigeminal nerve is compressed.60
Schwannomas can also occur as intradural extramedullary tumors, most often in individuals with neurofibromatosis. Multiple schwannomas in this population group are common. The tumors often extend through the intervertebral foramen into the abdomen or thoracic cavity. Compression causes local or radicular pain and may progress to include symptoms of spinal cord compression (e.g., motor weakness, sensory disturbances, autonomic changes). As with all benign schwannomas, surgical resection is curative.
Malignant Soft Tissue Tumors
Soft tissue sarcomas are a heterogenous group of rare tumors that arise predominantly from the embryonic mesoderm and present most often as an asymptomatic mass. They can occur anywhere in the body, but most originate in the extremities (59%), trunk (19%), retroperitoneum (15%), or head and neck (9%).23
Sarcomas account for 1% of all newly diagnosed adult cancers. In 2007, there were 9220 cases of soft tissue tumors, including heart tumors.54 The incidence is much higher in children, constituting 15% of annual pediatric malignancies.
Types of Soft Tissue Sarcomas.: Currently there are more than 50 histologic types of soft tissue sarcoma that have been identified. The most common are malignant fibrous histiocytoma, leiomyosarcoma, liposarcoma, synovial sarcoma, and malignant peripheral nerve sheath tumors. Rhabdomyosarcoma is the most common soft tissue sarcoma of childhood (Table 26-5).23
Table 26-5
Soft Tissue Sarcoma*
*Listed in approximate descending order of prevalence. Most soft tissue sarcomas are rare; some (as labeled) are extremely rare.
Malignant schwannoma, also known as neurosarcoma or neurogenic sarcoma, is a rare nerve sheath tumor of the peripheral nerves arising from Schwann cells or within existing neurofibromas. They can occur anywhere in the body but are often located on the flexor surface of the extremities. They are usually slow growing and painless, often present for years.94 When pressure is placed on the involved nerve, then pain, paresthesia, and paralysis may occur.
Rhabdomyosarcomas constitute more than half of all soft tissue sarcomas in children under 15 years of age. Occurrence in adults is possible but relatively rare.74 Eighty percent of the affected individuals are Caucasian. Boys are affected slightly more than girls; approximately 250 children in the United States are diagnosed each year with rhabdomyosarcoma.6,73
Rhabdomyosarcoma is a malignancy of striated muscle but can occur sporadically at any site in the body (e.g., bladder, prostate, head and neck, limbs, testes, muscle) and is of unknown cause. Symptoms are site dependent, but the tumor presents as a painless mass in the soft tissues. About one third of all people with rhabdomyosarcoma have readily resectable tumors, half do not, and in about half of all cases, regional lymphatic spread at diagnosis is evident, with a much less favorable prognosis.72 Diagnosis is often delayed as lesions are frequently attributed to sports-related trauma.
Other sites of metastases include the lungs, bone, and bone marrow. Tumors are aggressive and must be excised whenever possible. If tumors are too large to remove surgically, preoperative chemotherapy is used first to shrink the tumor. This allows for the possibility of complete resection and possibly a lower dose of radiation to achieve local control. Reduced exposure to radiation may decrease the late effects of radiation.6
Over the last 30 years, the prognosis for children with rhabdomyosarcoma has improved dramatically with the use of multiagent chemotherapy, aggressive surgery for local disease, and more precise delivery of radiation therapy. Prognosis depends on the type of gross residual tumor (histology), location of the tumor, and the presence and number of metastases at the time of diagnosis. Age and completeness of resection are additional prognostic factors.83
Current 5-year survival in rhabdomyosarcoma reaches 60% to 70% in nonmetastatic cases and remains below 20% in metastatic situations.32 Children with relapsed, recurrent, or metastatic sarcomas represent a complex challenge for the pediatric oncologist.24,47
Other common soft tissue sarcomas include malignant fibrous histiocytoma, liposarcoma, synovial sarcoma, epithelioid sarcoma, and clear cell sarcoma. Malignant fibrous histiocytoma is now recognized as the most common (although still occurring rarely) soft tissue sarcoma in adults, primarily affecting men 50 to 70 years old. Malignant fibrous histiocytoma occurs as a deep-seated mass that typically enlarges to 5 cm or more by the time of diagnosis and is usually located on the leg, especially the thigh.
Liposarcoma is a soft tissue malignancy with a peak incidence between ages 40 and 60 years. These are slow-growing lesions that can achieve a large size (10 to 15 cm), usually located in the thigh but occasionally retroperitoneally, causing pain and weight loss. Synovial sarcoma occurs most often in a young adult as a slow-growing mass of the extremities, often located near the knee. These lesions are painful and tender to palpation and often present similarly to a Baker cyst or ganglion. Reclassification of this sarcoma will eventually reflect the fact that the synovium is not involved in this type of sarcoma.
Epithelioid sarcoma, a small, firm, slow-growing mass, typically occurs in young adults on the extensor surface of an extremity but can also occur on the shoulder. These can develop deep enough to be undetectable on physical examination. Epithelioid sarcoma can look like a rheumatoid nodule, ganglion, or draining abscess and is often confused with a benign lesion.
Clear cell sarcoma arises deep to the dermis, has a uniform growth pattern, and is often located on tendons or aponeuroses. In rare cases this type of tumor can also originate in the spinal nerve roots with dissemination to the vertebral bodies, resulting in cauda equina. In approximately 20% of individuals the tumor has a dark appearance resulting from production of melanin, and it is often confused with benign soft tissue tumors.94
Etiology and Risk Factors
Soft tissue sarcomas do not seem to develop from malignant changes of benign soft tissue tumors. Specific inherited genetic alterations are associated with an increased risk of soft tissue sarcomas. Distinct chromosomal translocations that code for oncoproteins are associated with certain histiologic subtypes of soft tissue sarcomas. Oncogenes identified in the development of soft tissue sarcomas include MDM2, N-myc, c-erbB2, and members of the ras family.23 Ras proteins regulate cell proliferation, survival, and differentiation and are activated by mutations in many cancers.
Risk factors for soft tissue sarcomas include radiation therapy for cancer of the breast, cervix, testes, or lymphatic system with a mean latency period of approximately 10 years. Other risk factors include occupational exposure to chemicals, including herbicides and wood preservatives. Chronic lymphedema following axillary dissection is an additional risk factor for the development of lymphangiosarcoma.23
Pathogenesis
All sarcomas share a mesodermal cellular origin, but research has not been able to completely identify the pathogenesis involved. Sarcomas probably do not originate from normal tissue but arise from aberrant differentiated and proliferative malignant mesenchymal cell formations. There are some genetic origins that have been specifically identified for individual sarcoma types. Many sarcoma-linked oncogenes appear to be triggered by viruses; sequencing of these viruses may eventually allow for the development of specific antibodies against oncogenic activation.96
Clinical Manifestations
Soft tissue sarcomas present most often as painless, asymptomatic masses. They can grow quite large before being observed but do not usually produce pain when compressing surrounding structures. Metastasis occurs primarily hematogenously, with lymph node dissemination in rare cases.
MEDICAL MANAGEMENT
Diagnostic imaging, fine-needle aspiration, biopsy, and clinical studies are the mainstay of diagnosis. X-rays are used to look for lung metastases; CT scans and contrast-enhanced techniques provide details of high-grade lesions and large tumors, and assess the extent of tumor burden and proximity to vital structures. MRI is the preferred imaging modality for sarcomas of the extremities.48
Staging of soft tissue sarcomas follows the AJCC method of staging based on anatomic location (depth), grade, size of the tumor, and presence of distant or nodal metastases (nodal status). Metastases occur to the lungs first, but also to the bone, brain, and liver. Intracompartmental or extracompartmental extension of extremity sarcomas is important for surgical decision making and planning.71
TREATMENT.
Treatment depends on the type of tumor, stage, and location. For example, a multidisciplinary approach is taken for people with soft tissue sarcomas of the extremities. Surgical excision with clear margins combined with radiation yields good local control, but metastasis and death remain significant problems, especially for those individuals who have sarcomas at sites other than the extremities.
Systemic therapy (i.e., cytotoxic chemotherapy) is effective only for certain histologic subtypes; the adverse toxic side effects in individuals who do not respond to chemotherapy negate the routine use of this form of treatment. Many studies with randomized controlled trials have now shown that chemotherapy does not improve disease-free and overall survival in people with soft tissue sarcomas.39,79,92
Likewise, there are few supportive data to show that the use of preoperative chemotherapy can improve survival rates. Studies are underway to combine systemic chemotherapy with radiosensitizers and concurrent external beam radiation in hopes of treating microscopic disease, thus producing favorable local as well as systemic results.
There has been a gradual change in the local treatment of soft tissue sarcomas from amputation to a more conservative, limb-sparing, function-preserving approach combined with radiation.79 Amputation may be required for high-grade extremity sarcomas in about 5% of people whose tumor cannot be removed while still preserving function using limb-sparing techniques.12,23 See previous discussion in Primary Tumors in this chapter.
PROGNOSIS.
The overall 5-year survival rate for soft tissue sarcomas of all stages remains about 50% to 60%.82 Death from recurrence and metastatic complications occurs within 2 to 3 years of the initial diagnosis in 80% of cases. Despite improvements in local control rates, individuals with high-risk soft tissue sarcomas have poor long-term results.
Advanced, metastatic sarcomas are always incurable; management is palliative. Factors associated with a poorer prognosis include age older than 60, tumors larger than 5 cm, and high-grade histology.71 Individuals with leiomyosarcomas, clear cell sarcomas, and malignant fibrous histiocytomas may have a poorer survival rate compared with those individuals who have fibrosarcomas, liposarcomas, and neurofibrosarcomas.63
Cartilaginous Tumors
Many tumors of cartilaginous origin can occur. Three of the more common tumors of a cartilaginous origin are the enchondroma, osteochondroma, and chondrosarcoma. Cartilage tumors involving some parts of the skeleton (e.g., small bones of the hands and feet) are almost always benign, whereas cartilaginous lesions of the ribs, sternum, and flat bones such as the pelvis and scapula are more likely to be aggressive.30
Determining the aggressiveness of cartilaginous tumors is especially difficult, and even the histologic differentiation is troublesome. Sometimes the presence of pain or the development of pain in a previously diagnosed benign cartilaginous tumor such as an enchondroma is all that raises suspicion of a malignant process or transformation.
Benign Cartilaginous Tumors
Overview and Incidence.: Enchondroma is a common, benign tumor arising from residual islands of cartilage in the metaphysis of bones (Fig. 26-18). The tubular bones of the hands and feet (phalanges, metacarpals, metatarsals) are common sites, although the long tubular bones (femur, humerus) can be affected. They are rarely seen in sites most commonly affected by chondrosarcoma (trunk bones). Enchondromas account for approximately 10% of benign skeletal tumors. They are seen in people between the ages of 20 and 40 but can occur at any age in both men and women.
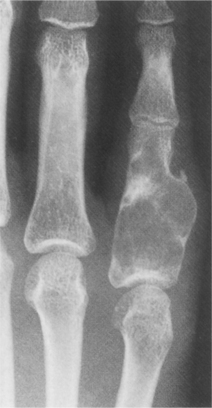
Pathogenesis and Clinical Manifestations.: Cartilaginous tumors are lesions in which cartilage is produced, rather than osteoid as in the osteosarcomas. These lesions are classified as chondromas. Enchondral ossification is the process by which most bones in the skeleton are formed—that is, bone is slowly absorbed from the inner cortex while at the same time, periosteal reactive bone is deposited on the outer surface. A cartilaginous model exists as a precursor to mature bone. A tumor may then develop from cartilage islands displaced from the growth plate during development. This is thought to occur perhaps secondary to trauma or to an abnormality in the growth plate.
Histologically, enchondroma consists of hyaline cartilage appearing as lobules rimmed with a narrow band of reactive bone. These may be difficult to differentiate from a slow-growing chondrosarcoma, and in a small percentage of cases, a single enchondroma (usually in a large, long bone) does undergo malignant change to become a chondrosarcoma.84
Enchondromas may be asymptomatic. In some cases, some swelling may occur. When present in the hands, pain may be a symptom of pathologic stress fracture.
MEDICAL MANAGEMENT
In those cases where no symptoms are present, plain radiographs or bone scans performed for other reasons reveal the tumor as an incidental finding. Once detected, differentiating the lesion from a chondrosarcoma is crucial. The radiograph and clinical history, not the histologic makeup, are the most informative. Radiographs of enchondromas do not show cortical destruction. Pain without evidence of a fracture is also suspicious of malignancy rather than enchondroma.
TREATMENT AND PROGNOSIS.
Curettage is a common form of treatment, with or without bone grafting, depending on the size and location of the lesion. Clients with enchondromas in the hand may develop stress fractures, which often respond to splinting. Recurrence of enchondromas after curettage is less than 5%, and malignant transformation occurs in 2% of all cases (usually adults).84
Overview and Incidence.: Osteochondroma is the most common primary benign neoplasm of bone, accounting for 90% of all benign bone tumors.40 A continuous osseous outgrowth of bone with a cartilaginous cap is characteristic (Fig. 26-19). The outgrowth arises from the metaphysis of long bones and extends away from the nearest epiphysis. The metaphyses of long bones, especially the distal femur, proximal humerus, and proximal tibia, are common sites. The flat bones of the ilium and scapula can also be involved.40
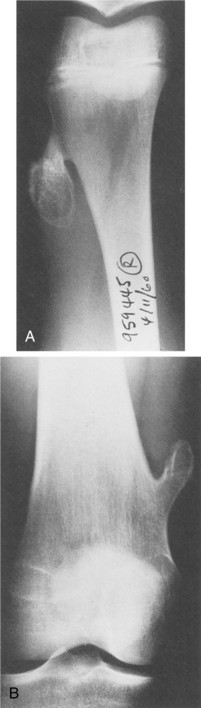
Figure 26-19 Osteochondroma. Two radiographs (A and B) showing mature osteochondroma: stalked lesion pointing toward the diaphysis and away from the growth plate. (From Bogumill G, Schwamm H: Orthopaedic pathology, Philadelphia, 1984, WB Saunders.)
The incidence of osteochondroma is unknown. Some reports indicate that men are affected more often, but this may be due to the fact that it is often an incidental finding, and men may be more likely to have a radiograph taken during the second decade of life when the lesion is usually seen.
Pathogenesis.: Osteochondromas appear to result from aberrant epiphyseal development. They are an extension of normal bone capped by cartilage that forms a prominent “tumor” (lump, swelling), sometimes referred to as osteocartilaginous exostosis. The younger the individual, the larger is the cartilage cap, because during the growing years, an osteochondroma has its own epiphyseal plate from which it grows.84
The lesion will usually cease growing when the individual reaches skeletal maturity. The central portion of the lesion is normal medullary bone. The lesion may begin as a displaced fragment of epiphyseal cartilage that penetrates a cortical defect and continues to grow.
Clinical Manifestations.: In some people, a hard mass will be detected, sometimes present for many years. When the tumor is palpable it may, owing to the cartilaginous cap, feel much larger than is apparent on radiographs.
Osteochondromas are not painful lesions in themselves, but they may interfere with the function of surrounding soft tissues such as tendons, nerves, or bursae. Blood vessels can also be compromised by the tumors (Fig. 26-20), and if tumors are sufficiently large, they may even limit joint motion.
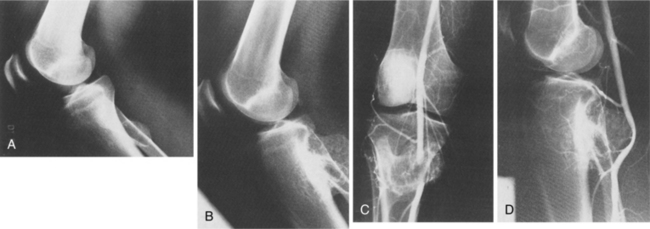
Figure 26-20 Osteochondroma of the proximal fibula in a young man. A, Lateral radiograph of the right knee obtained when the patient was 17 years old demonstrates an exophytic lesion arising from the proximal fibula. B, Lateral radiograph obtained 8 years later shows considerable interim growth of the osteochondroma, although a smooth outline is maintained. C, Anteroposterior and D, lateral angiograms demonstrate displacement and marked narrowing of the distal popliteal artery by the tumor. (From Guidici M, Moser R, Kransdorf M: Cartilaginous bone tumors, Radiol Clin North Am 31:247, 1993.)
Synovial osteochondromatosis can occur secondary to benign proliferation of the synovium and presents as multiple loose bodies within a joint.
MEDICAL MANAGEMENT
Plain radiographs may show a slender stalk of bone directed away from the nearest growth plate. This is referred to as a pedunculated osteochondroma. A sessile osteochondroma has a broad base of attachment (Fig. 26-21). In both types the most important feature to note is the continuity of the cortex between the host bone and the tumor.
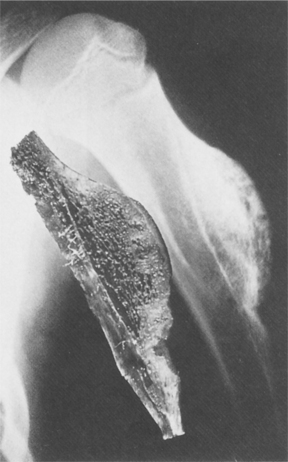
Figure 26-21 Osteochondroma. Radiograph and gross specimen of the sessile osteochondroma. Note the cartilaginous component causing the radiographic defect in the distal portion. Note also incorporation of hematopoietic tissue into the base of the osteochondroma. (From Bogumill G, Schwamm H: Orthopaedic pathology, Philadelphia, 1984, WB Saunders.)
CT and MRI are not commonly used in the diagnostic workup of benign lesions, but if atypical clinical mani- festations or recent changes in the appearance of the lesion on plain radiographs are evident, MRI may be indicated. For example, MRI can demonstrate the continuity of the marrow between the tumor and the host bone, thereby ruling out a periosteal osteosarcoma.
TREATMENT AND PROGNOSIS.
Since osteochondromas usually cease their growth at skeletal maturity, no intervention is needed unless they are symptomatic or interfering with normal limb function. Removal of the lesion is sometimes required when symptoms such as vascular compromise, chronic bursitis, or pain develop secondarily. Rarely, an osteochondroma can transform into a chondrosarcoma. Those symptomatic lesions that are removed have a very low recurrence rate.
Fibrous Lesions
Fibrous (fibroosseous) lesions (also referred to as fibrous dysplasia) within bone are a common osseous anomaly of mesenchymal tissue. They are usually solitary lesions found in the femur, skull, humerus, and tibia. Adolescents and young adults are affected. These lesions vary from small, fibrous cortical defects to larger fibrous dysplasias. Although most are benign, fibrosarcoma has many of the features of osteosarcoma. Many children have defects in the metaphysis, but most resolve spontaneously. Those that persist are seen in young adult men. The distal femur and tibia are common sites.
Pathogenesis and Clinical Manifestations
The hallmark of this disease is the inability of bone-forming tissue to produce mature bone. The process is arrested at the level of woven bone; even if a large amount of osteoid tissue is produced, it cannot or does not mature to lamellar bone. The pathogenesis is unknown, but it appears the underlying molecular mechanism involves the fundamental cell differentiation process.30
Although the defect occurs in the metaphysis, during normal bone growth the defect may be displaced into the diaphysis. Microscopic examination reveals disorganized, haphazard deposits characteristic of woven bone, sometimes accompanied by local hemorrhage and serous fluid accumulation. Growth of lesions is often stabilized during puberty.
Most fibrous defects are asymptomatic. Some individuals experience mild to moderate pain with swelling or deformity of the affected site. The more extensive the disease, the earlier the onset of symptoms. Pathologic fractures may be the initial symptom and occur where large lesions exist. Depending on the specific form of dysplasia, affected bones include ribs; craniofacial bones; long, tubular bones; and pelvis.
There may be associated extraskeletal symptoms such as hyperpigmentation of the skin (café au lait spots corresponding to the site of musculoskeletal involvement) or endocrine dysfunction (e.g., early menarche in females, acromegaly, hyperthyroidism, hyperparathyroidism, Cushing’s syndrome).
MEDICAL MANAGEMENT
Plain radiographs are usually diagnostic. Fibrous defects are usually observed as an incidental finding on radiographs. The lesion has an irregular shape with a thin sclerotic border (Fig. 26-22). Even though this is a benign lesion, treatment sometimes requires surgery. When the dysplasia thins the cortex of a weight-bearing bone or occupies more than one half of the diameter of the bone, the risk of pathologic fracture increases. Benign fibrous defects generally have a good prognosis.
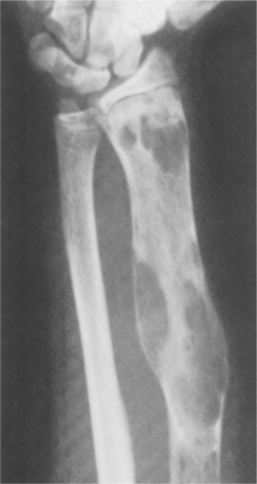
Figure 26-22 Fibrous dysplasia. A predominantly lytic lesion with some sclerosis and expansion is seen in the distal half of the radius in a child. A long lesion in a long bone typifies fibrous dysplasia. Although parts of this lesion indeed have a ground-glass appearance, most of it does not. Expansion and bone deformity like this are commonly seen in fibrous dysplasia. (From Helms C: Fundamentals of skeletal radiology: benign cystic lesions, Philadelphia, 1989, WB Saunders.)
Implications for the therapist are similar to those with other benign bone tumors.
METASTATIC TUMORS
Overview
Cancer commonly metastasizes to bone; skeletal involvement represents the third most common site of metastatic spread (after lung and liver).91 Secondary or metastatic neoplasms refer to those lesions that originate in other organs of the body.
All malignant tumors have the capability to spread to bone; the skeleton is the third most common site of metastatic carcinoma, exceeded only by lung and liver. Malignant tumors that have metastasized to the bone are the most common neoplasm of the bone.
Although all of the factors that affect the timing and location of metastasis are not known, some patterns do exist. Cancer metastases (both carcinomas and sarcomas) to bone are a common clinical problem, because the cancers that cause them are prevalent and often metastasize.15 Primary cancers responsible for 75% of all bone metastases include prostate, breast, lung, kidney, and thyroid.
Common sites for breast cancer to metastasize include the pelvis, ribs, vertebrae, and proximal femur. Lung cancer can metastasizes to the bone early in the disease, remaining asymptomatic until widespread dissemination has taken place; therefore, treatment is often not successful. Neoplasms in the kidney metastasize to the vertebrae, pelvis, and proximal femur in about 40% of the cases. The prostate is the most common source of skeletal metastases in men.
Early detection is important for successful intervention. Thera pists should be aware of this possible cause of lumbar spine and hip pain, especially in men over age 50. Cancer of the thyroid is uncommon but does metastasize to bone. Women are affected by bone metastases from the thyroid three times more often than men. Therapists should remember that the development of metastasis may be delayed and may even occur after removal of a cancerous thyroid. For discussions of specific primary cancers, see the relevant chapters.
Incidence and Etiology
Metastatic bone neoplasms are much more common than primary bone lesions; about half of all individuals with cancer (except skin cancer) will develop bone metastases at some point. Incidence increases to 80% of individuals with advanced cancer. The incidence of bone metastasis is expected to increase with the prolonged survival associated with improved antineoplastic therapies now available. The spine is the site most commonly affected, with more than 50% of the metastases involving the spine93—usually the thoracic or lumbar spine, much less often the cervical spine, and rarely the atlantoaxial region.2
In the spine, the size of the vertebral body may influence the distribution of metastases. The larger lumbar vertebral bodies are more commonly affected than the smaller thoracic or cervical vertebrae. Neurologic compromise is more likely to occur when metastatic lesions affect the thoracic spine because of the smaller ratio between the diameter of the spinal canal and the spinal cord within the thoracic spine.99
Risk Factors
Risk factors are those related to the primary cancer. For some cancers the risk factors are well documented, and efforts to educate individuals on health risks should be stressed. Adequate exercise, proper diet and nutrition, and avoidance of tobacco use are the primary preventive measures. It is likely that the increase in incidence of spinal (and other) metastases can be attributed to the improving survival of clients with cancer.99
Pathogenesis
The pathophysiology of metastasis is not completely understood, but new information on the biology of tumor metastases derived from advanced techniques in molecular pathology is contributing new insight daily (see the section on Invasion and Metastases in Chapter 9). The development of metastatic disease, regardless of the eventual target organ, usually follows a common pathway.
Cancer can spread through the bloodstream, lymphatic system, or by direct extension into adjacent tissue. Hematogenous spread of the cancer is most common, and therefore skeletal metastases are found in areas of bones with a good blood supply. These include the vertebrae, ribs, skull, and proximal femur and humerus.
The skeletal vasculature represents a significant proportion of the body’s total vasculature. At the same time, the vertebral plexus of veins has no valves, so that the retrograde venous pressure is often increased in the abdominal and chest regions. This enables the retrograde blood flow to bypass the caval system, reaching the bones of the vertebral column instead via the extradural Batson venous plexus.8,30 Batson’s plexus may be the route by which breast cancer cells metastasize directly to the thoracic spine.99
The unique vasculature of the spine contributes to the high rate of spinal involvement in metastatic disease. The vertebral venous system is a valveless channel that extends from the sacrum to the skull. Venous connections to this system exist from the breasts, lungs, thyroid gland, kidneys, and prostate gland. Cells from the primary tumor mass enter the circulation by traversing either the walls of small blood vessels in normal tissue or those of vessels induced by the tumor itself.69 Once having gained access to the vertebral vein system, tumor cells can travel to distant organ sites. There are also direct connections to the vertebrae, ribs, pelvis, skull, and the shoulder and pelvic girdles.34
From a biologic point of view, it is very unlikely that the abundance of the vascular network within the bone is the only factor that predisposes to metastasis, because metastases rarely develop in other tissues that have an equally rich vascular supply. It is proposed that the biologic conditions of bone tissue must be important factors in promoting the growth of tumor cells that reach the marrow through the venous and arterial blood network.30
The development of skeletal metastasis involves a series of events that begins when a tumor cell separates from the primary site, enters the blood system, and then extravasates from the blood vessel to the secondary site.99 Adhesion molecules control separation and clustering of cancerous cells. The presence or absence of certain molecules controls the ability of cells to metastasize. Various types of adhesion molecules have been implicated. Cadherins, integrins, and selectins each have distinct properties that can regulate the propensity for a primary lesion to metastasize to a specific organ.
Metastasis to bone often results in osteolysis, because cancer cells secrete a number of paracrine factors that stimulate osteoclast function. The cancer tries to destroy the bone (lytic process), and in response, the bone attempts to grow new bone (blastic process) to surround the cancer. If the cancer overwhelms the bone, it becomes weak and fractures easily. Bone metastases may be lytic (most common), blastic, or mixed. Lesions originating from the breast, lung, kidney, and thyroid are usually lytic. Blastic metastases are commonly associated with advanced carcinomas of the prostate and sometimes the breast.70
Clinical Manifestations
Although as many as 50% of people with breast or prostate metastasis have no bone pain, pain remains the most common presenting symptom, often characterized as sharp, severe, worse at night, transient or intermittent in the early course but eventually constant in more advanced cancer, and mechanical (see the section on Cancer Pain in Chapter 9).
Bone pain of a mechanical nature associated with skeletal metastases occurs as a result of significant bone destruction, joint instability, mechanical insufficiency, and fracture. It is often incapacitating and persistent despite local and systemic therapies. Long bone or vertebral fractures with or without spinal cord compression may be the first indication of advanced disease. Spinal cord compression, the most serious complication of bone metastasis, occurs secondary to increased pressure on the spinal cord or as a result of vertebral collapse. Classic signs and symptoms of cord compression include pain, numbness, and/or paralysis.34
Pain may also arise from a biologic origin for a number of reasons. It may occur as a result of rapid growth of the tumor stretching the periosteum. Increased blood flow or angiogenesis (sometimes giving a throbbing or pulsatile sensation) and the release of cytokines at the site of the metastases gives rise to bone pain. And neuropeptides elaborated by or acting on bone-associated nerves in the endosteum can result in bone pain.31,68 Because the skeleton provides both form and support, growing tumors that deform the cortical bone contribute to activity-associated pain. This type of pain is often intermittent and related to weight bearing and movement.68
Bone often functions as a metastatic conduit for peripheral nerves, as bone metastases travel hematogenously from distal body parts to the central nervous system. Therefore, bone tumor growth and invasion into surrounding tissues can result in neuritic pain syndromes, plexopathies, and spinal cord compression.
These pain syndromes contribute to increasing loss of mobility and bed rest, the effects of which are increasing generalized weakness, risk of thromboembolism, hypercalcemia, atelectasis, and pneumonia. The latter occur particularly in anyone with painful rib metastases. Mechanical failure or pathologic bone fracture may occur as a result of prolonged immobilization (osteoporosis). As with primary tumors, pathologic fractures can occur directly from the tumor itself or from the secondary effects of intervention.68
Metabolic changes can also occur as a result of the disease or the treatment, increasing the risk of fracture. In people with multiple metastases, the resultant hypercalcemia may cause anorexia, nausea, vomiting, general weakness, and depression. Unexplained weight loss is typically a late sign of metastatic disease.
Left untreated, hypercalcemia may lead to diffuse osteoporosis, renal insufficiency, and dehydration (see Chapter 5). These symptoms may be relieved (and possibly prevented)75 by the use of bisphosphonates (e.g., intravenous pamidronate [Aredia] and clodronate; oral ibandronate and clodronate), small molecules that inhibit osteoclast-induced bone resorption. The reactive bone formation stimulated by these lesions accounts for the elevation of serum alkaline phosphatase.84
MEDICAL MANAGEMENT
A history of malignancy raises the suspicion of recurrent disease or a metastatic lesion. The evaluation of an individual with a previous history of cancer or a current malignancy and bone pain begins with a physical examination and basic radiographic studies. Spinal metastasis may be evident by the loss of the pedicle as seen in the anteroposterior view of a standard spinal radiograph.
Another early manifestation is the pathologic fracture of the lesser trochanter. Since much of the bone matrix must be destroyed before the lytic process is noted on radiographs, plain films are not sensitive and are not useful in early detection but are more important in staging and treatment planning.
Whole-body bone scans are much more sensitive for early detection of skeletal metastasis but are not useful in predicting fractures. Approximately one third of those with skeletal metastatic disease have positive bone scan findings yet negative radiographic results. Scans are also used to determine the extent of dissemination.
CT and MRI also have roles in delineating various types of metastasis and assessing the size and extent of the lesion. More advanced technology using single-photon emission tomography (SPET) allows for better determination of anatomical location of the areas of radioisotope uptake.85,99 Biopsy is sometimes necessary to confirm a diagnosis when the primary source is not known. CT-guided biopsy is used to assess spinal lesions; diagnostic accuracy is greater for lytic lesions (93%) compared to sclerotic lesions (76%).62
Other diagnostic tests may include serum chemistries, urinalysis, serum protein electrophoresis, and prostate-specific antigen determination (for men). Biochemical markers of bone turnover such as N-telopeptide and pyridinium cross-links (pyridinoline and deoxypyridinoline) may provide information on bone dynamics that reflect diseases activity in bone.
Several studies have shown bone markers to be elevated in people with documented evidence of metastatic bone disease. Increased levels are also observed in some people without clinical evidence of bone metastases, when compared with normal subjects. Rises in such markers may be the first indication of bone involvement and possibly a useful early diagnostic sign of progression.
Preliminary data suggest that bone marker level correlates with the extent of metastatic disease and the number of skeletal sites involved. Markers of bone turnover may be helpful in identifying those individuals likely to respond to bisphosphonate treatment and as a means of monitoring the effectiveness of bisphosphonate therapy in the management of bone metastases.61
TREATMENT.
Therapeutic interventions may depend, in part, on the extent of involvement. The person with localized disease may be offered potentially curative therapy, whereas the individual with extensive skeletal and visceral involvement may only benefit from palliative treatment.53
Treatment of bone metastasis is problematic, costly, and primarily palliative. Prolonging survival is not always possible, so improving function with pain relief, local control of disease, and bone stability is often the primary goal. This is becoming more important as treatment for primary cancers improves. Individuals may die from the primary tumor or from the metastasis (e.g., breast cancer). When survival rates and longevity increase, the likelihood of skeletal metastasis increases.
Intervention for skeletal neoplasms requires a multidisciplinary approach to optimize therapy options and coordinate their sequencing. Intervention modalities may include endocrine therapy (for breast and prostate cancer), chemotherapy, biotherapy (immunotherapy), use of bone-seeking radioisotopes (a therapy that has analgesic and antitumor effects), and bisphosphonates to suppress bone resorption. These are often combined with other localized interventions such as surgery and site-directed radiation therapy.
Surgery is rarely curative but can be an effective therapy to decompress neural tissue for resolution of symptoms and/or restoration of function, reduce anxiety, improve mobility and function, facilitate nursing care, preempt fracture (i.e., repair bony lesions before they fracture), and control local tumor when nonsurgical therapies fail.70,103
Pathologic fractures that occur in the femur and humerus often require surgical stabilization. Intramedullary fixation with interlocking devices to limit motion at the fracture site is indicated in many instances. The desire to restore normal anatomy must be weighed against the reality that the individual may have a terminal disease. An estimated life expectancy of at least 6 months is desirable before extensive joint reconstructive procedures are carried out.49
Where the risk of fracture is great, as when more than 50% of the cortex is destroyed, prophylactic nailing of the femur may be indicated (Fig. 26-23). See the discussion of fractures and fracture treatment, including newly developing fracture treatment procedures, in Chapter 27.
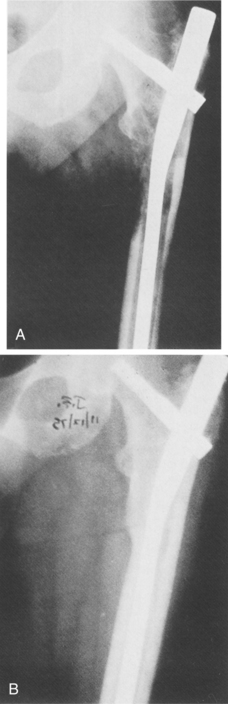
Figure 26-23 A, Prophylactic fixation in a 63-year-old woman with an impending fracture secondary to breast metastasis treated by Zickel nailing. B, Complete healing of this subtrochanteric lesion 5 months after radiation and chemotherapy. (From Habermann E, Lopez R: Metastatic disease of bone and treatment of pathologic fracture, Orthop Clin 20:475, 1989.)
Spinal metastases can cause severe pain, instability, and spinal cord compression with neurologic compromise. Pathologic fractures of the spine can be immobilized in an appropriate spinal brace, but a progressive neurologic deficit is an indication for surgical intervention. Surgery can take the form of decompression, posterior stabilization, excision, and reconstruction or prosthetic replacement. Vertebroplasty or kyphoplasty may be considered for the person with a vertebral compression fracture and minimal bone deformity.37
PROGNOSIS.
Although management of the skeletal metastasis may be successful in terms of restoring stability to a pathologic fracture, the prognosis for the primary cancer is still guarded. Only rarely is the skeletal metastasis actually the cause of death. Skeletal morbidity includes bone pain, hypercalcemia, pathologic fracture, spinal cord or nerve root compression, and immobility, all of which can impact mortality rates.22
The median survival for people with tumors that have metastasized to the bone is determined by the type of tumor (e.g., prostate: 29 months; breast: 23 months; kidney: 12 months; lung: less than 4 months). The overall median survival after detection of bone metastases is approximately 19 months; this significant amount of time allows for interventions that can dramatically improve a person’s quality of life and functional independence.31
Favorable prognostic factors include indolent nature of the primary lesion (e.g., prostate cancer); well-differentiated tumor on histologic examination; a long recurrence-free survival (greater than 3 years); sclerotic lesion on radiograph as opposed to a lytic lesion, especially after treatment; a single bone lesion; a single system involved with metastatic disease; low tumor markers; no vital organ involvement; and general good condition of the individual.
Unfavorable prognostic factors include the following31:
• Aggressive nature of the primary lesion (e.g., lung cancer)
• Poorly differentiated tumor on histology
• Short recurrence-free survival (less than 1 year)
The risk of pathologic fracture is greater in osteolytic lesions of the long bones. A direct relationship exists between the degree of cortical destruction and the risk of pathologic fracture. When cortical destruction is less than 25% to 35%, the risk for fracture is low. Destruction greater than 50% correlates with a much higher risk for pathologic fracture. The presence of pain with weight-bearing activities indicates compromised structural integrity and therefore also places the individual at greater risk of fracture.34,38
References
1. Abeloff, MD, Wolff, AC, Wood, WC, et al. Cancer of the breast. In Abeloff MD, Armitage JO, Lichter AS, et al, eds.: Clinical oncology, ed 3, Philadelphia: Elsevier, 2004.
2. Abu, WA, Provencher, M. Primary bone and metastatic tumors of the cervical spine. Spine. 1998;23(24):2767–2777.
3. American Joint Committee on Cancer (AJCC). Cancer staging manual, ed 6. New York: Springer, 2002.
4. Anderson, P. Improving outcomes in difficult bone cancers using multimodality therapy. Curr Oncol Rep. 2006;8(6):415–422.
5. Anderson, PM, Pearson, M. Novel therapeutic approaches in pediatric and young adult sarcomas. Curr Oncol Rep. 2006;8(4):310–315.
6. Arndt, CAS, Crist, WM. Common musculoskeletal tumors of childhood and adolescence. N Engl J Med. 1999;341(5):342–352.
7. Avigad, S, Yaniv, I. Novel approaches for the management of patients with Ewing sarcoma. Future Oncol. 2006;2(5):659–665.
8. Batson, OV. The role of the vertebral veins in metastatic processes. Ann Intern Med. 1942;16:38–45.
9. Bernstein, M. Ewing’s sarcoma family of tumors: current management. Oncologist. 2006;11(5):503–519.
10. Boone, L. Hemipelvectomy surgery for the excision of synovial sarcomas of the pelvic bones. Rehabil Oncol. 2000;18(2):8–9, 16.
11. Boriani, S, Chevalley, F, Weinstein, JN, et al. Chordoma of the spine above the sacrum: treatment and outcome. Spine. 1996;21:1569–1577.
12. Brennan, MF. The role of multimodality therapy in soft-tissue sarcoma. Ann Surg. 1991;214:328–336.
13. Bui Nguyen Binh, M, Collin, F, Coindre, JM. Soft tissue sarcomas: update on molecular data. Cancer Radiother. 2006;10(1-2):15–21.
14. Bunting, RW. Rehabilitation of cancer patients with skeletal metastases. Clin Orthop Relat Res. 1995;312:197–200.
15. Bunting, RW. Rehabilitation of the patient with bone metastases. Rehabil Oncol. 2000;18(1):24–25.
16. Bunting, RW, et al. Functional outcome of pathologic fracture secondary to malignant disease in a rehabilitation hospital. Cancer. 1992;69:98–102.
17. Bunting, RW, et al. Pathologic fracture risk in rehabilitation of patients with bony metastases. Clin Orthop. 1985;192:222–227.
18. Bunting, RW, Shea, B. Bone metastasis and rehabilitation. Cancer. 2001;92(4 suppl):1020–1028.
19. Carroll, K, Know your options: an explanation of the rotationplasty and tibia turn-up procedures. In Motion. 2005;15(2). [on-line journal]. Available on-line at http://www.amputee-coalition.org/inmotion/mar_apr_05/rotationplasty.html Accessed February 28, 2007.
20. Carroll, K, Radical innovation in above-knee amputation. Orthop Tech Rev 2006. Available on-line at http://www.orthopedictechreview.com/issues/articles/2006-03_06.asp. Accessed March 7, 2007.
21. Coleman, M. Oncology lecture. Baltimore: University of Maryland, School of Medicine, Department of Physical Therapy, 1998.
22. Coleman, RE. Skeletal complications of malignancy. Cancer. 1997;80(8 suppl):1588–1594.
23. Cormier, JN. Soft tissue sarcomas. CA Cancer J Clin. 2004;54(2):94–109.
24. Czauderna, P. Role of surgery in the management of soft tissue tumors. Future Oncol. 2006;2(5):667–673.
25. Dahlin, DC, Coventry, MB. Osteosarcoma: a study of 600 cases. J Bone Joint Surg Am. 1967;49:101–110.
26. Denaro, V. Surgical management of cervical spine osteoblastomas. Clin Orthop Relat Res. 2006;455:190–195.
27. Devaney, K, et al. MIC2 detection in tumors of bone and adjacent soft tissues. Clin Orthop. 1995;310:176–187.
28. DeVita, VT. ed 7. Cancer: principles and practice of oncology. Philadelphia: Lippincott Williams & Wilkins; 2004;2.
29. Deyrup, AT, Weiss, SW. Grading of soft tissue sarcomas. Histopathology. 2006;48(1):42–50.
30. Dorfman, HD, Czerniak, B. Bone tumors. St Louis: Mosby, 1998.
31. Egan, MP. Surgical and physical therapy considerations for patients with bone metastases. Rehabil Oncol. 2001;19(2):25–26.
32. Ek, ET, Choong, PF. The role of high-dose therapy and autologous stem cell transplantation for pediatric bone and soft tissue sarcomas. Expert Rev Anticancer Ther. 2006;62:225–237.
33. Enneking, WF. A system of staging musculoskeletal neoplasms. Clin Orthop. 1986;153:106–120.
34. Esser, C, Wruble, ER. A literature review of solid bone cancers occurring during adulthood and the implications for physical therapy. Acute Perspect. 2001;10(1, 2):30–35.
35. Fechner, R, Mills, S. Tumors of the bones and joints. Washington, DC: Armed Forces Institute of Pathology, 1992.
36. Ferris, I, Tortajada, J. Risk factors for pediatric malignant bone tumors. Ann Pediatr. 2005;63(6):537–547.
37. Fourney, DR. Percutaneous vertebroplasty and kyphoplasty for painful vertebral body fractures in cancer patients. J Neurosurg. 2003;98(1 suppl):21–30.
38. Frassica, FJ, et al. Metastatic bone disease: evaluation, clinicopathologic features, biopsy, fracture risk, nonsurgical treatment and supportive management. Instruct Course Lect. 2000;49:453–459.
39. Frustaci, S. Adjuvant chemotherapy for adult soft tissue sarcomas of the extremities and girdles: results of the Italian randomized cooperative trial. J Clin Oncol. 2001;19:1238–1247.
40. Gentles, C. Diagnosis: soft-tissue chondroma. Orthopedics. 2007;30(3):241–243.
41. Gibbs, CP. Beyond bone grafting: techniques in the surgical management of benign bone tumors. Instr Course Lect. 2005;54:497–503.
42. Gitelis, S. Recurrence of a giant-cell tumor with malignant transformation to a fibrosarcoma twenty-five years after primary treatment. J Bone Joint Surg. 1989;71A:757–761.
43. Gitelis, S. The use of closed expandable prosthesis for pediatric sarcomas. Chir Organi Mov. 2003;88(4):327–333.
44. Grier, HE. The Ewing family of tumors: Ewing’s sarcoma and primitive neuroectodermal tumors. Pediatr Clin North Am. 1997;44:991–1004.
45. Grubb, MR, et al. Primary Ewing’s sarcoma of the spine. Spine. 1994;19:309–313.
46. Gudas, SA. Rehabilitation of pediatric and adult sarcomas. Rehabil Oncol. 2000;18(3):10–13.
47. Gupta, AA, Pappo, AS. New drugs for the treatment of metastatic or refractory soft tissue sarcomas in children. Future Oncol. 2006;2(5):675–685.
48. Hanna, SL. MR imaging of malignant soft-tissue tumors. Magn Reson Imaging Clin N Am. 1995;3:629–650.
49. Harrington, K. Orthopaedic management of extremity and pelvic lesions. Clin Orthop. 1995;312:136–147.
50. Harsh, G, Pedicled rhinotomy for clival chordomas invaginating the brainstem. Neurosurg Focus. 2001;10(3):E8. Available on-line at http://www.medscape.com/viewarticle/405691. Accessed November 13, 2006.
51. Hendershot, E. Treatment approaches for metastatic Ewing’s sarcoma. J Pediatr Oncol Nurs. 2005;22(6):339–352.
52. James, SL, Davis, AM. Giant-cell tumours of bone of the hand and wrist: a review of imaging findings and differential diagnoses. Eur Radiol. 2005;15(9):1855–1866.
53. Janjan, N. Bone metastases: approach to management. Semin Oncol. 2001;28(4 suppl 11):28–34.
54. Jemal, A. Cancer statistics, 2007. CA Cancer J Clin. 2007;57(1):43–66.
55. Kawamura, H, et al. Restoring normal gait after limb salvage procedures in malignant bone tumors of the knee. Scand J Rehab Med. 1999;31:77–81.
56. Kelley, CH, Thomas, R. Osteogenic sarcoma. In: Miaskowski C, Buchsel P, eds. Oncology nursing: assessment and clinical care. St Louis: Mosby, 1999.
57. Kelley, CH, Thomas, R. Soft tissue sarcomas. In: Miaskowski C, Buchsel P, eds. Oncology nursing: assessment and clinical care. St Louis: Mosby, 1999.
58. Khoury, JD. Ewing sarcoma family of tumors. Adv Anat Pathol. 2005;12(4):212–220.
59. Lawrence, W. Soft tissue sarcomas in adults and children: a comparison. CA Cancer J Clin. 1994;44(4):197–199.
60. Lenhard, RE, Osteen, RT, Gansler, T. Clinical oncology. Atlanta: American Cancer Society, 2001.
61. Lipton, A, et al. Use of markers of bone turnover for monitoring bone metastases and the response to therapy. Semin Oncol. 2001;28(4 suppl 11):54–59.
62. Lis, E. Percutaneous CT-guided biopsy of osseous lesion of the spine in patients with known or suspected malignancy. Am J Neuroradiol. 2004;25:1583–1588.
63. Mankin, HJ, Hornicek, FJ. Diagnosis, classification, and management of soft tissue sarcomas. Cancer Control. 2005;12(1):5–21.
64. Marchese, VG. Assessing functional mobility in survivors of lower-extremity sarcoma: reliability and validity of a new assessment tool. Pediatr Blood Cancer. 2007;49(2):183–189.
65. McPherson, CM, Suki, D, McCutcheon, IE, et al. Metastatic disease from spinal chordoma: a 10-year experience. J Neurosurg Spine. 2006;5:277–280.
66. Mendenhall, WM. Giant cell tumor of bone. Am J Clin Oncol. 2006;29(1):96–99.
67. Merino, ME, et al. Immunomagnetic purging of Ewing’s sarcoma from blood and bone marrow: quantitation by real-time polymerase chain reaction. J Clin Oncol. 2001;19(16):3649–3659.
68. Mertens, WC, et al. Systemic bone-seeking radionuclides for palliation of painful osseous metastases: current concepts. CA Cancer J Clin. 1998;48(6):361–374.
69. Mundy, GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Natl Rev Cancer. 2002;2:584–593.
70. Murphy-Fertak, LA, Yasko, AW. Bone tumors. In: Boyer KL, et al, eds. Primary care oncology. Philadelphia: WB Saunders, 1999.
71. National Cancer Institute (NCI) Treatment statement for health professionals: adult soft tissue sarcoma [updates posted 2007]. Available on-line at http://www.meb.uni-bonn.de/cancer.gov/CDR0000062820 Accessed May 17, 2007
72. Neville, H, et al. Preoperative staging, prognostic factors and outcome in extremity rhabdomyosarcoma: a preliminary report from the Intergroup Rhabdomyosarcoma Study IV (1991-1997). J Pediatr Surg. 2000;35(2):317–321.
73. Pappo, AS, Shapiro, DN, Crist, WM. Rhabdomyosarcoma: biology and treatment. Pediatr Clin North Am. 1997;44:953–972.
74. Parham, DM, Ellison, DA. Rhabdomyosarcomas in adults and children: an update. Arch Pathol Lab Med. 2006;130(10):1454–1465.
75. Paterson, AH. Adjuvant bisphosphonate therapy: the future. Semin Oncol. 2001;28(4 suppl 11):81–85.
76. Peabody, TD, Gibbs, CP, Simon, MA. Evaluation and staging of musculoskeletal neoplasms. J Bone Joint Surg. 1998;80A(8):1204–1218.
77. Pfalzer, L. Oncology: examination, diagnosis, and treatment: medical and surgical considerations. In: Myers RS, ed. Saunders manual of physical therapy practice. Philadelphia: WB Saunders, 1995.
78. Picci, P, et al. Prognostic significance of histopathologic response to chemotherapy in nonmetastatic Ewing’s sarcoma of the extremities. J Clin Oncol. 1993;11:1763–1769.
79. Pisters, PW. Evidence-based recommendations for local therapy for soft tissue sarcomas. J Clin Oncol. 2007;25(8):1003–1008.
80. Randall, RL. Giant cell tumor of the sacrum. Neurosurg Focus. 2003;15(2):E13.
81. Resnick, D, Hemcek, AA, Haghighi, P. Spinal enostoses (bone islands). Radiology. 1983;147:373.
82. Ries, LAG SEER cancer statistics review, 1975-2003. Bethesda, MD: National Cancer Institute; 2006. Available on-line at http://seer.cancer.gov/csr/1975_2003/ Accessed May 15, 2007.
83. Rodeberg, D, Paidas, C. Childhood rhabdomyosarcoma. Semin Pediatr Surg. 2006;15(1):57–62.
84. Salter, RB. Textbook of disorders and injuries of the musculoskeletal system, ed 3. Baltimore: Williams & Wilkins, 1999.
85. Savelli, G, et al. The role of bone SPET study in diagnosis of single vertebral metastases. Anticancer Res. 2000;20(2B):1115–1120.
86. Schneider, M, Sabo, D, Gerner, HJ, et al. Destructive osteoblastoma of the cervical spine with complete neurologic recovery. Spinal Cord. 2002;40(5):248–252.
87. Scotlandi, K. Targeted therapies in Ewing’s sarcoma. Adv Exp Med Biol. 2006;587:13–22.
88. Scurr, M, Judson, I. How to treat the Ewing’s family of sarcomas in adult patients. Oncologist. 2006;11(1):65–72.
89. Shamberger, RC, Grier, HE. Ewing’s sarcoma/primitive neuroectodermal tumor of the chest wall. Semin Pediatr Surg. 2001;10(3):153–160.
90. Skubitz, KM, Manivel, JC. Giant cell tumor of the uterus: case report and response to chemotherapy. BMC Cancer. 2007;7:46.
91. Smukler, A, Govindan, R. Management of bone metastasis. Contemp Oncol. 2002;13(1):1–10.
92. Tierney, JF. Adjuvant chemotherapy for localized resectable soft-tissue sarcoma of adults: meta-analysis of individual data, Sarcoma Meta-analysis Collaboration. Lancet. 1997;350:1647–1654.
93. Toma, S. Metastatic bone tumors. Clin Orthop. 1993;295:246–251.
94. Tortolani, PJ. Soft-tissue tumors: distinguishing the benign from the malignant. J Musculoskelet Med. 1999;16(8):469–476.
95. Turcotte, RE. Giant cell tumor of bone. Orthop Clin North Am. 2006;37(1):L35–L51.
96. Tuveson, D, Fletcher, J. Signal transduction pathways in sarcoma as targets for therapeutic intervention. Curr Opin Oncol. 2001;13:249–255.
97. Vlasak, R, Sim, FH. Ewing’s sarcoma. Pediatr Orthoped Oncol. 1996;27(3):591–603.
98. Wafa, H, Grimer, RJ. Surgical options and outcomes in bone sarcoma. Expert Rev Anticancer Ther. 2006;6(2):239–248.
99. White, AP. Metastatic disease of the spine. J Am Acad Orthop Surg. 2006;14(11):587–598.
100. Wick, JM, Alexander, KM. Rotationplasty-a unique surgical procedure with a functional outcome. AORN J. 2006;84(2):190–214.
101. Wilkins, RM. Reconstruction options for pediatric bone tumors about the knee. J Knee Surg. 2005;18(4):305–309.
102. Wunder, JS et al Chemotherapy-induced tumor necrosis as a prognostic indicator in surgically treated Ewing’s sarcoma. Presentation at Joint Meeting of EMSOS-AMSTS, Florence, Italy, May 1995
103. Yasko, AW, et al. Sarcomas of soft tissue and bone. In: Lenhard RE, Osteen RT, Gansler T, eds. Clinical oncology. Atlanta: American Cancer Society, 2001.
104. Yonemoto, T, Tatezaki, S, Takenouchi, T, et al. The surgical management of sacrococcygeal chordoma. Cancer. 1999;85:878–883.
105. Zileli, M. Osteoid osteomas and osteoblastomas of the spine. Neurosurg Focus. 2003;15(5):E5.
106. Zileli, M. Primary tumors of the cervical spine: a retrospective review of 35 surgically managed cases. Spine J. 2007;7(2):165–173.