Etiology of Cancer: Carcinogenic Agents
Genetic damage lies at the heart of carcinogenesis. What extrinsic agents can inflict such damage? Three classes of carcinogenic agents have been identified: (1) chemicals, (2) radiant energy, and (3) microbial agents. Chemicals and radiant energy are documented causes of cancer in humans, and oncogenic viruses are involved in the pathogenesis of tumors in several animal models and some human tumors. In the following discussion, each class of agent is considered separately; of note, however, several may act in concert or sequentially to produce the multiple genetic abnormalities characteristic of neoplastic cells.
Chemical Carcinogens
More than 200 years ago, the London surgeon Sir Percival Pott correctly attributed scrotal skin cancer in chimney sweeps to chronic exposure to soot. On the basis of this observation, the Danish Chimney Sweeps Guild ruled that its members must bathe daily. No public health measure since that time has achieved so much in the control of a form of cancer. Subsequently, hundreds of chemicals have been shown to be carcinogenic in animals.
Some of the major agents are presented in Table 5–4. A few comments on a handful of these are offered next.
Table 5–4 Major Chemical Carcinogens
| Direct-Acting Carcinogens |
| Alkylating Agents |
| Acylating Agents |
| Procarcinogens That Require Metabolic Activation |
| Polycyclic and Heterocyclic Aromatic Hydrocarbons |
| Aromatic Amines, Amides, Azo Dyes |
| Natural Plant and Microbial Products |
| Others |
Direct-Acting Agents
Direct-acting agents require no metabolic conversion to become carcinogenic. They are in general weak carcinogens but are important because some of them are cancer chemotherapy drugs (e.g., alkylating agents) used in regimens that may cure certain types of cancer (e.g., Hodgkin lymphoma), only to evoke a subsequent, second form of cancer, usually leukemia. This situation is even more tragic when the initial use of such agents has been for non-neoplastic disorders, such as rheumatoid arthritis or Wegener granulomatosis. The associated risk of induced cancer is low, but its existence dictates judicious use of such agents.
Indirect-Acting Agents
The designation indirect-acting refers to chemicals that require metabolic conversion to an ultimate carcinogen. Some of the most potent indirect chemical carcinogens are polycyclic hydrocarbons, present in fossil fuels. For example, benzo[a]pyrene and other carcinogens are formed in the high-temperature combustion of tobacco in cigarette smoking. These products are implicated in the causation of lung cancer in cigarette smokers. Polycyclic hydrocarbons also may be produced from animal fats during the process of broiling meats and are present in smoked meats and fish. The principal active products in many hydrocarbons are epoxides, which form covalent adducts (addition products) with molecules in the cell, principally DNA, but also with RNA and proteins.
The aromatic amines and azo dyes constitute another class of indirect-acting carcinogens. Before its carcinogenicity was recognized, β-naphthylamine was responsible for a 50-fold increased incidence of bladder cancers in heavily exposed workers in the aniline dye and rubber industries. Many other occupational carcinogens are listed in Table 5–2. Because indirect-acting carcinogens require metabolic activation for their conversion to DNA-damaging agents, much interest is focused on the enzymatic pathways that are involved, such as that mediated by the cytochrome P-450–dependent monooxygenases. The genes that encode these enzymes are polymorphic, and enzyme activity varies among different persons. It is widely believed that the susceptibility to chemical carcinogenesis depends at least in part on the specific allelic form of the enzyme inherited. Thus, it may be possible in the future to assess cancer risk in a given patient by genetic analysis of such enzyme polymorphisms.
A few other agents merit brief mention. Aflatoxin B1 is of interest because it is a naturally occurring agent produced by some strains of Aspergillus, a mold that grows on improperly stored grains and nuts. A strong correlation has been found between the dietary level of this food contaminant and the incidence of hepatocellular carcinoma in some parts of Africa and the Far East. Additionally, vinyl chloride, arsenic, nickel, chromium, insecticides, fungicides, and polychlorinated biphenyls are potential carcinogens in the workplace and about the house. Finally, nitrites used as food preservatives have caused concern, since they cause nitrosylation of amines contained in the food. The nitrosamines thus formed are suspected to be carcinogenic.
Mechanisms of Action of Chemical Carcinogens
Because malignant transformation results from mutations, it should come as no surprise that most chemical carcinogens are mutagenic. Indeed, all direct and ultimate carcinogens contain highly reactive electrophile groups that form chemical adducts with DNA, as well as with proteins and RNA. Although any gene may be the target of chemical carcinogens, the commonly mutated oncogenes and tumor suppressors, such as RAS and TP53, are important targets of chemical carcinogens. Indeed, specific chemical carcinogens, such as aflatoxin B1, produce characteristic mutations in the TP53 gene, such that detection of the “signature mutation” within the TP53 gene establishes aflatoxin as the causative agent. These associations are proving to be useful tools in epidemiologic studies of chemical carcinogenesis.
Carcinogenicity of some chemicals is augmented by subsequent administration of promoters (e.g., phorbol esters, hormones, phenols, certain drugs) that by themselves are nontumorigenic. To be effective, repeated or sustained exposure to the promoter must follow the application of the mutagenic chemical, or initiator. The initiation-promotion sequence of chemical carcinogenesis raises an important question: Since promoters are not mutagenic, how do they contribute to tumorigenesis? Although the effects of tumor promoters are pleiotropic, induction of cell proliferation is a sine qua non of tumor promotion. It seems most likely that while the application of an initiator may cause the mutational activation of an oncogene such as RAS, subsequent application of promoters leads to clonal expansion of initiated (mutated) cells. Forced to proliferate, the initiated clone of cells accumulates additional mutations, developing eventually into a malignant tumor. Indeed, the concept that sustained cell proliferation increases the risk of mutagenesis, and hence promotes neoplastic transformation, also is applicable to human carcinogenesis. For example, endometrial hyperplasia (Chapter 18) and increased regenerative activity that accompanies chronic liver cell injury are associated with the development of cancer in these organs. Were it not for the DNA repair mechanisms discussed earlier, the incidence of chemically induced cancers in all likelihood would be much higher. As mentioned previously, the rare hereditary disorders of DNA repair, including xeroderma pigmentosum, are associated with greatly increased risk of cancers induced by UV light and certain chemicals.
 Summary
Summary
Chemical Carcinogens
• Chemical carcinogens have highly reactive electrophile groups that directly damage DNA, leading to mutations and eventually cancer.
• Direct-acting agents do not require metabolic conversion to become carcinogenic, while indirect-acting agents are not active until converted to an ultimate carcinogen by endogenous metabolic pathways. Hence, polymorphisms of endogenous enzymes such as cytochrome P-450 may influence carcinogenesis.
• After exposure of a cell to a mutagen or an initiator, tumorigenesis can be enhanced by exposure to promoters, which stimulate proliferation of the mutated cells.
• Examples of human carcinogens are direct-acting agents (e.g., alkylating agents used for chemotherapy), indirect-acting agents (e.g., benzopyrene, azo dyes, aflatoxin), and promoters or agents that cause hyperplasia of endometrium or regenerative activity in the liver.
Radiation Carcinogenesis
Radiation, whatever its source (UV rays of sunlight, x-rays, nuclear fission, radionuclides) is an established carcinogen. Unprotected miners of radioactive elements have a 10-fold increased incidence of lung cancers. Follow-up study of survivors of the atomic bombs dropped on Hiroshima and Nagasaki disclosed a markedly increased incidence of leukemia—principally myelogenous leukemias—after an average latent period of about 7 years, as well as increased mortality rates for thyroid, breast, colon, and lung carcinomas. The nuclear power accident at Chernobyl in the former Soviet Union continues to exact its toll in the form of high cancer incidence in the surrounding areas. More recently, it is feared that radiation release from a nuclear power plant in Japan damaged by a massive earthquake and tsunami will result in significantly increased cancer incidence in the surrounding geographic areas.
Therapeutic irradiation of the head and neck can give rise to papillary thyroid cancers years later. The oncogenic properties of ionizing radiation are related to its mutagenic effects; it causes chromosome breakage, translocations, and, less frequently, point mutations. Biologically, double-stranded DNA breaks seem to be the most important form of DNA damage caused by radiation.
The oncogenic effect of UV rays merits special mention because it highlights the importance of DNA repair in carcinogenesis. Natural UV radiation derived from the sun can cause skin cancers (melanomas, squamous cell carcinomas, and basal cell carcinomas). At greatest risk are fair-skinned people who live in locales such as Australia and New Zealand that receive a great deal of sunlight. Nonmelanoma skin cancers are associated with total cumulative exposure to UV radiation, whereas melanomas are associated with intense intermittent exposure—as occurs with sunbathing. UV light has several biologic effects on cells. Of particular relevance to carcinogenesis is the ability to damage DNA by forming pyrimidine dimers. This type of DNA damage is repaired by the nucleotide excision repair pathway. With extensive exposure to UV light, the repair systems may be overwhelmed, and skin cancer results. As mentioned earlier, patients with the inherited disease xeroderma pigmentosum have a defect in the nucleotide excision repair pathway. As expected, there is a greatly increased predisposition to skin cancers in this disorder.
Summary
Radiation Carcinogenesis
• Ionizing radiation causes chromosome breakage, translocations, and, less frequently, point mutations, leading to genetic damage and carcinogenesis.
• UV rays induce the formation of pyrimidine dimers within DNA, leading to mutations. Therefore, UV rays can give rise to squamous cell carcinomas and melanomas of the skin.
Viral and Microbial Oncogenesis
Many DNA and RNA viruses have proved to be oncogenic in animals as disparate as frogs and primates. Despite intense scrutiny, however, only a few viruses have been linked with human cancer. The following discussion focuses on human oncogenic viruses. Also discussed is the emerging role of the bacterium H. pylori in gastric cancer.
Oncogenic RNA Viruses
The study of oncogenic retroviruses in animals has provided spectacular insights into the genetic basis of cancer. However, only one retrovirus, the human T cell lymphotropic virus-1 (HTLV-1), has been demonstrated to cause cancer in humans. HTLV-1 is associated with a form of T cell leukemia/lymphoma that is endemic in certain parts of Japan and the Caribbean basin but is found sporadically elsewhere, including the United States. Similar to the human immunodeficiency virus (HIV), HTLV-1 has tropism for CD4+ T cells, and this subset of T cells is the major target for neoplastic transformation. Human infection requires transmission of infected T cells through sexual intercourse, blood products, or breastfeeding. Leukemia develops only in about 3% to 5% of infected persons after a long latent period of 20 to 50 years.
There is little doubt that HTLV-1 infection of T lymphocytes is necessary for leukemogenesis, but the molecular mechanisms of transformation are not clear. The HTLV-1 genome does not contain a viral oncogene, and in contrast with certain animal retroviruses, no consistent integration site next to a cellular oncogene has been discovered. Indeed, the long latency period between initial infection and development of disease suggests a multistep process, during which many oncogenic mutations are accumulated.
The genome of HTLV-1 contains, in addition to the usual retroviral genes, a unique region called pX. This region contains several genes, including one called TAX. The TAX protein has been shown to be necessary and sufficient for cellular transformation. By interacting with several transcription factors, such as NF-κB, the TAX protein can transactivate the expression of genes that encode cytokines, cytokine receptors, and costimulatory molecules. This inappropriate gene expression leads to autocrine signaling loops and increased activation of promitogenic signaling cascades. Furthermore, TAX can drive progression through the cell cycle by directly binding to and activating cyclins. In addition, TAX can repress the function of several tumor suppressor genes that control the cell cycle, including CDKN2A/p16 and TP53. From these and other observations, the following scenario is emerging (Fig. 5–31): The TAX gene turns on several cytokine genes and their receptors (e.g., the interleukins IL-2 and IL-2R and IL-15 and IL-15R), setting up an autocrine system that drives T cell proliferation. Of these cytokines, IL-15 seems to be more important, but much remains to be defined. Additionally, a parallel paracrine pathway is activated by increased production of granulocyte-macrophage colony-stimulating factor, which stimulates neighboring macrophages to produce other T cell mitogens. Initially, the T cell proliferation is polyclonal, because the virus infects many cells, but because of TAX-based inactivation of tumor suppressor genes such as TP53, the proliferating T cells are at increased risk for secondary transforming events (mutations), which lead ultimately to the outgrowth of a monoclonal neoplastic T cell population.
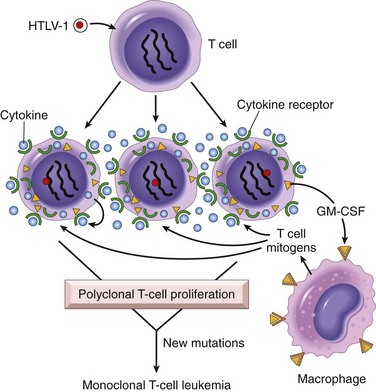
Figure 5–31 Pathogenesis of human T cell lymphotropic virus (HTLV-1)–induced T cell leukemia/lymphoma. HTLV-1 infects many T cells and initially causes polyclonal proliferation by autocrine and paracrine pathways triggered by the TAX gene. Simultaneously, TAX neutralizes growth inhibitory signals by affecting TP53 and CDKN2A/p16 genes. Ultimately, a monoclonal T cell leukemia/lymphoma results when one proliferating T cell suffers additional mutations.
Summary
Oncogenic RNA Viruses
• HTLV-1 causes a T cell leukemia that is endemic in Japan and the Caribbean.
• The HTLV-1 genome encodes a viral TAX protein, which turns on genes for cytokines and their receptors in infected T cells. This sets up autocrine and paracrine signaling loops that stimulate T cell proliferation. Although this proliferation initially is polyclonal, the proliferating T cells are at increased risk for secondary mutations that lead to the outgrowth of a monoclonal leukemia.
Oncogenic DNA Viruses
As with RNA viruses, several oncogenic DNA viruses that cause tumors in animals have been identified. Four DNA viruses—HPV, Epstein-Barr virus (EBV), Kaposi sarcoma herpesvirus (KSHV, also called human herpesvirus-8 [HHV-8]), and hepatitis B virus (HBV)—are of special interest because they are strongly associated with human cancer. KSHV and Kaposi sarcoma are discussed in Chapter 4. The others are presented here.
Human Papillomavirus
Scores of genetically distinct types of HPV have been identified. Some types (e.g., 1, 2, 4, and 7) cause benign squamous papillomas (warts) in humans (Chapters 18 and 21). Genital warts have low malignant potential and are also associated with low-risk HPVs, predominantly HPV-6 and HPV-11. By contrast, high-risk HPVs (e.g., types 16 and 18) cause several cancers, particularly squamous cell carcinoma of the cervix and anogenital region. In addition, at least 20% of oropharyngeal cancers, particularly those arising in the tonsils, are associated with HPV.
The oncogenic potential of HPV can be related to products of two early viral genes, E6 and E7. Together, they interact with a variety of growth-regulating proteins encoded by proto-oncogenes and tumor suppressor genes. The E7 protein binds to the retinoblastoma protein and releases the E2F transcription factors that normally are sequestered by Rb, promoting progression through the cell cycle. Of interest, E7 protein from high-risk HPV types has a higher affinity for Rb than does E7 from low-risk HPV types. E7 also inactivates the CDKIs CDKN1A/p21 and CDNK1B/p27. The E6 protein has complementary effects. It binds to and mediates the degradation of p53. By analogy with E7, E6 from high-risk HPV types has a higher affinity for p53 than does E6 from low-risk HPV types. Also of interest, in benign warts the HPV genome is maintained in a nonintegrated episomal form, while in cancers the HPV genome is randomly integrated into the host genome. Integration interrupts the viral DNA, resulting in overexpression of the oncoproteins E6 and E7. Furthermore, cells in which the viral genome has integrated show significantly more genomic instability.
To summarize, infection with high-risk HPV types simulates the loss of tumor suppressor genes, activates cyclins, inhibits apoptosis, and combats cellular senescence. Thus, it is evident that many of the hallmarks of cancer discussed earlier are driven by HPV proteins. However, infection with HPV itself is not sufficient for carcinogenesis. For example, when human keratinocytes are transfected with DNA from HPV-16, -18, or -31 in vitro, they are immortalized, but they do not form tumors in experimental animals. Cotransfection with a mutated RAS gene results in full malignant transformation. These data strongly suggest that HPV, in all likelihood, acts in concert with other environmental factors (Chapter 18). However, the primacy of HPV infection in the causation of cervical cancer is attested to by the near-complete protection from this cancer by anti-HPV vaccines.
Epstein-Barr Virus
EBV was the first virus linked to a human tumor, Burkitt lymphoma. Over the last 40 years, however, EBV has been discovered with the cells of a surprisingly diverse list of tumors, including B cell lymphomas in patients with defective T cell immunity (e.g., those infected with HIV), a subset of Hodgkin lymphoma, nasopharyngeal carcinoma, a subset of T cell lymphomas, gastric carcinomas, NK cell lymphomas, and even, in rare instances, sarcomas, mainly in the immunosuppressed.
Burkitt lymphoma is endemic in certain parts of Africa and is sporadic elsewhere. In endemic areas, tumor cells in virtually all affected patients carry the EBV genome. The molecular basis for B cell proliferations induced by EBV is complex. EBV uses the complement receptor CD21 to attach to and infect B cells. In vitro, such infection leads to polyclonal B cell proliferation and generation of B lymphoblastoid cell lines. One of the EBV-encoded genes, called LMP1 (latent membrane protein 1) acts as an oncogene, and its expression in transgenic mice induces B cell lymphomas. LMP1 promotes B cell proliferation by activating signaling pathways, such as NF-κB and JAK/STAT, which mimic B cell activation by the B cell surface molecule CD40. Concurrently, LMP1 prevents apoptosis by activating BCL2. Thus, the virus “borrows” a normal B cell activation pathway to promote its own replication by expanding the pool of cells susceptible to infection. Another EBV-encoded protein, EBNA2, transactivates several host genes, including cyclin D and the src family of proto-oncogenes. In addition, the EBV genome contains a viral cytokine, vIL-10, that was pirated from the host genome. This viral cytokine can prevent macrophages and monocytes from activating T cells and killing virally infected cells.
In immunologically normal persons, EBV-driven polyclonal B cell proliferation is readily controlled, and the affected patient either remains asymptomatic or experiences a self-limited episode of infectious mononucleosis (Chapter 11). Evasion of the immune system seems to be a key step in EBV-related oncogenesis. In regions of the world in which Burkitt lymphoma is endemic, concomitant (endemic) malaria (or other infections) impairs immune competence, allowing sustained B cell proliferation. Of interest, although LMP1 is the primary transforming oncogene in the EBV genome, it is not expressed in EBV-associated Burkitt lymphoma, presumably because it also is one of the major viral antigens recognized by the immune system. Infected cells expressing viral antigens such as LMP-1 are kept in check by the immune system. Lymphoma cells may emerge only when translocations activate the MYC oncogene, a consistent feature of this tumor. MYC may substitute for LMP1 signaling, allowing the tumor cells to downregulate LMP1 and evade the immune system. Of note, in nonendemic areas, 80% of tumors are negative for EBV, but virtually all tumors possess MYC translocations. This observation suggests that although non-African Burkitt lymphomas are triggered by mechanisms other than EBV, these cancers develop by similar pathways.
In patients with deficient T cell function, including those with HIV and organ transplant recipients, EBV-infected B cells undergo polyclonal expansion, producing lymphoblastoid-like cells. In contrast with Burkitt lymphoma, the B lymphoblasts in immunosuppressed patients do express viral antigens, such as LMP-1, that are recognized by T cells. These potentially lethal proliferations can be subdued if T cell immunity can be restored, as may be achieved by withdrawal of immunosuppressive drugs in transplant recipients.
Nasopharyngeal carcinoma is endemic in southern China and some other locales, and the EBV genome is found in all tumors. LMP-1 is expressed in the carcinoma cells and, as in B cells, activates the NF-κB pathway. Furthermore, LMP1 induces the expression of pro-angiogenic factors such as VEGF, FGF-2, MMP-9, and COX-2, which may contribute to oncogenesis. How EBV enters epithelial cells is unclear, as these cells fail to express the CD21 protein that serves as the EBV receptor in B cells.
Summary
Oncogenic DNA Viruses
• HPV is associated with benign warts, as well as cervical cancer.
• The oncogenicity of HPV is related to the expression of two viral oncoproteins, E6 and E7; they bind to Rb and p53, respectively, neutralizing their function.
• E6 and E7 from high-risk strains of HPV (which give rise to cancers) have higher affinity for their targets than do E6 and E7 from low-risk strains of HPV (which give rise to benign warts).
• EBV is implicated in the pathogenesis of Burkitt lymphomas, lymphomas in immunosuppressed patients (HIV infection or organ transplant recipients), some forms of Hodgkin lymphoma, uncommon T cell and NK cell tumors, nasopharyngeal carcinoma, a subset of gastric carcinoma, and rarely sarcomas.
• Certain EBV gene products contribute to oncogenesis by stimulating a normal B cell proliferation pathway. Concomitant compromise of immune competence allows sustained B cell proliferation, leading eventually to development of lymphoma, with occurrence of additional mutations such as t(8;14) leading to activation of the MYC gene.
Hepatitis B and Hepatitis C Viruses
The epidemiologic evidence linking chronic HBV and hepatitis C virus (HCV) infection with hepatocellular carcinoma is strong (Chapter 15). It is estimated that 70% to 85% of hepatocellular carcinomas worldwide are due to infection with HBV or HCV. However, the mode of action of these viruses in tumorigenesis is not fully elucidated. The HBV and HCV genomes do not encode any viral oncoproteins, and although the HBV DNA is integrated within the human genome, there is no consistent pattern of integration in liver cells. Indeed, the oncogenic effects of HBV and HCV are multifactorial, but the dominant effect seems to be immunologically mediated chronic inflammation with hepatocyte death leading to regeneration and genomic damage. Although the immune system generally is thought to be protective, recent work has demonstrated that in the setting of unresolved chronic inflammation, as occurs in viral hepatitis or chronic gastritis caused by H. pylori (see further on), the immune response may become maladaptive, promoting tumorigenesis.
As with any cause of hepatocellular injury, chronic viral infection leads to the compensatory proliferation of hepatocytes. This regenerative process is aided and abetted by a plethora of growth factors, cytokines, chemokines, and other bioactive substances produced by activated immune cells that promote cell survival, tissue remodeling, and angiogenesis. The activated immune cells also produce other mediators, such as reactive oxygen species, that are genotoxic and mutagenic. A key molecular step seems to be activation of the nuclear factor-κB (NF-κB) pathway in hepatocytes caused by mediators derived from the activated immune cells. Activation of the NF-κB pathway within hepatocytes blocks apoptosis, allowing the dividing hepatocytes to incur genotoxic stress and to accumulate mutations. Although this seems to be the dominant mechanism in the pathogenesis of virus-induced hepatocellular carcinoma, both HBV and HCV also contain proteins within their genomes that may more directly promote the development of cancer. The HBV genome contains a gene known as HBx, and hepatocellular cancers develop in mice transgenic for this gene. HBx can directly or indirectly activate a variety of transcription factors and several signal transduction pathways. In addition, viral integration can cause secondary rearrangements of chromosomes, including multiple deletions that may harbor unknown tumor suppressor genes.
Although not a DNA virus, HCV also is strongly linked to the pathogenesis of liver cancer. The molecular mechanisms used by HCV are less well defined than those for HBV. In addition to chronic liver cell injury and compensatory regeneration, components of the HCV genome, such as the HCV core protein, may have a direct effect on tumorigenesis, possibly by activating a variety of growth-promoting signal transduction pathways.
Summary
Hepatitis B and Hepatitis C Viruses
• Between 70% and 85% of hepatocellular carcinomas worldwide are due to infection with HBV or HCV.
• The oncogenic effects of HBV and HCV are multifactorial, but the dominant effect seems to be immunologically mediated chronic inflammation, with hepatocellular injury, stimulation of hepatocyte proliferation, and production of reactive oxygen species that can damage DNA.
• The HBx protein of HBV and the HCV core protein can activate a variety of signal transduction pathways that also may contribute to carcinogenesis.
Helicobacter pylori
First incriminated as a cause of peptic ulcers, H. pylori now has acquired the dubious distinction of being the first bacterium classified as a carcinogen. Indeed, H. pylori infection is implicated in the genesis of both gastric adenocarcinomas and gastric lymphomas.
The scenario for the development of gastric adenocarcinoma is similar to that for HBV- and HCV-induced liver cancer. It involves increased epithelial cell proliferation on a background of chronic inflammation. As in viral hepatitis, the inflammatory milieu contains numerous genotoxic agents, such as reactive oxygen species. The sequence of histopathologic changes consists of initial development of chronic inflammation/gastritis, followed by gastric atrophy, intestinal metaplasia of the lining cells, dysplasia, and cancer. This sequence takes decades to complete and occurs in only 3% of infected patients. Like those of HBV and HCV, the H. pylori genome also contains genes directly implicated in oncogenesis. Strains associated with gastric adenocarcinoma have been shown to contain a “pathogenicity island” that contains cytotoxin-associated A gene (CagA). Although H. pylori is noninvasive, CagA is injected into gastric epithelial cells, where it has a variety of effects, including the initiation of a signaling cascade that mimics unregulated growth factor stimulation.
As mentioned previously, H. pylori is associated with an increased risk for the development of gastric lymphomas as well. The gastric lymphomas are of B cell origin, and because the transformed B cells grow in a pattern resembling that of normal mucosa-associated lymphoid tissue (MALT), they also have been referred to as MALT lymphomas (Chapter 11). Their molecular pathogenesis is incompletely understood but seems to involve strain-specific H. pylori factors, as well as host genetic factors, such as polymorphisms in the promoters of inflammatory cytokines such as IL-1β and tumor necrosis factor (TNF). It is thought that H. pylori infection leads to the activation of H. pylori–reactive T cells, which in turn cause polyclonal B cell proliferation. In time, a monoclonal B cell tumor emerges in the proliferating B cells, perhaps as a result of accumulation of mutations in growth regulatory genes. In keeping with this model, early in the course of disease, eradication of H. pylori “cures” the lymphoma by removing antigenic stimulus for T cells.
Summary
Helicobacter pylori
• H. pylori infection has been implicated in both gastric adenocarcinoma and MALT lymphoma.
• The mechanism of H. pylori–induced gastric cancers is multifactorial, including immunologically mediated chronic inflammation, stimulation of gastric cell proliferation, and production of reactive oxygen species that damage DNA. H. pylori pathogenicity genes, such as CagA, also may contribute by stimulating growth factor pathways.
• It is thought that H. pylori infection leads to polyclonal B cell proliferations and that eventually a monoclonal B cell tumor (MALT lymphoma) emerges as a result of accumulation of mutations.