The Child with Hematologic or Immunologic Dysfunction
HEMATOLOGIC AND IMMUNOLOGIC DYSFUNCTION
IMMUNOLOGIC DEFICIENCY DISORDERS
Human Immunodeficiency Virus Infection and Acquired Immunodeficiency Syndrome
TECHNOLOGIC MANAGEMENT OF HEMATOLOGIC AND IMMUNOLOGIC DISORDERS
On completion of this chapter the reader will be able to:
 Distinguish between the various categories of anemia.
Distinguish between the various categories of anemia.
Describe the prevention of and care of the child with iron deficiency anemia.
Compare sickle cell anemia and β-thalassemia major in relation to pathophysiology and nursing care.
Describe the mechanisms of inheritance and nursing care of the child with hemophilia.
Relate the pathophysiology and clinical manifestations of leukemia.
Demonstrate an understanding of the rationale of therapies for neoplastic disease.
Outline a care plan for the child with neoplastic disease and the family.
Contrast the pathophysiology and management of the immunodeficiency disorders.
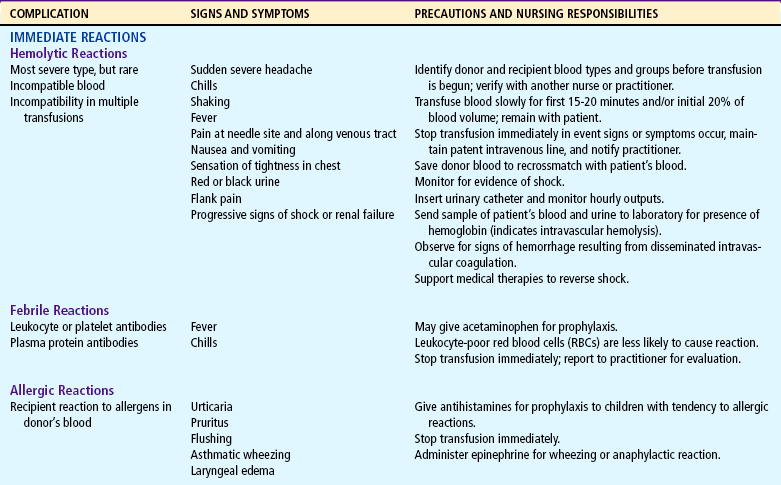
List nursing precautions and responsibilities during blood transfusion.
RELATED TOPICS and ADDITIONAL RESOURCES
 IN TEXT
IN TEXTAdministration of Medication, Ch. 22
Anaphylaxis, Ch. 25
Bone Marrow Aspiration or Biopsy, Ch. 22
Family-Centered Care of the Child During Illness and Hospitalization, Ch. 21
Immunizations, Ch. 10
Impact of the Child’s Chronic Illness or Disability, Ch. 18
Infection Control, Ch. 22
Lumbar Puncture, Ch. 22
Pain Assessment; Pain Management, Ch. 7
Physical Examination, Ch. 6
Preparation for Diagnostic and Therapeutic Procedures, Ch. 22
HEMATOLOGIC AND IMMUNOLOGIC DYSFUNCTION
Several tests can be performed to assess hematologic function, including additional procedures to identify the cause of the dysfunction. The following discussion is limited to a description of the most common and one of the most valuable tests, the complete blood cell count (CBC). Other procedures, such as those related to iron, coagulation, and immune status, are discussed throughout the chapter as appropriate. The nurse should be familiar with the significance of the findings from the CBC (Table 26-1) and aware of normal values for age, which are listed in Appendix C.
TABLE 26-1
Tests Performed as Part of the Complete Blood Cell Count
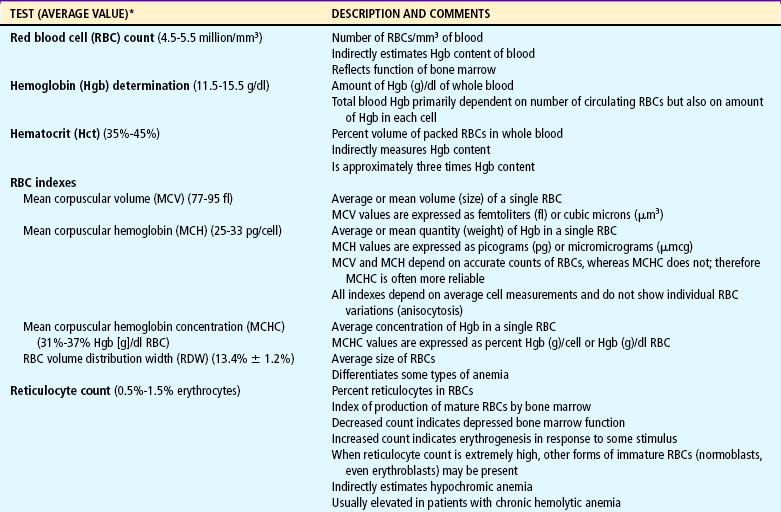

*See Appendix C for normal values according to ages.
As with any disorder, the history and physical examination are essential to identify hematologic dysfunction, and the nurse is often the first person to suspect a problem based on information from these sources. Comments by the parent regarding the child’s lack of energy, food diary of poor sources of iron, frequent infections, and bleeding that is difficult to control offer clues to the more common disorders affecting the blood. A careful physical appraisal, especially of the skin, can reveal findings (e.g., pallor, petechiae, bruising) that may indicate minor or serious hematologic conditions. Nurses need to be aware of the clinical manifestations of blood diseases to assist in recognizing symptoms and establishing a diagnosis.
RED BLOOD CELL DISORDERS
The term anemia describes a condition in which the number of red blood cells (RBCs) or the hemoglobin (Hgb or Hb) concentration is reduced below normal values for age. This diminishes the oxygen-carrying capacity of the blood, causing a reduction in the oxygen available to the tissues. Anemia is the most common hematologic disorder of infancy and childhood and is not a disease itself but an indication or manifestation of an underlying pathologic process.
Classification
Anemias are classified in relation to (1) etiology or physiology, manifested by erythrocyte or Hgb depletion; and (2) morphology, the characteristic changes in RBC size, shape, or color (Box 26-1). Although the morphologic classification is more useful in terms of laboratory evaluation of anemia, the etiologic approach provides direction for planning nursing care. For example, anemia with reduced Hgb concentration may be caused by a dietary depletion of iron, and the principal intervention is replenishing iron stores. The classification of anemias is found in Fig. 26-1.
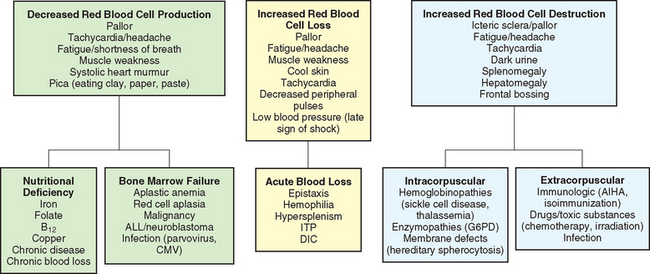
Consequences of Anemia
The basic physiologic defect caused by anemia is a decrease in the oxygen-carrying capacity of blood and consequently a reduction in the amount of oxygen available to the cells. When the anemia has developed slowly, the child usually adapts to the declining Hgb level.
The effects of anemia on the circulatory system can be profound. Because the viscosity of blood depends almost entirely on the concentration of RBCs, the resulting hemodilution of severe anemia decreases peripheral resistance, causing greater quantities of blood to return to the heart. The increased circulation and turbulence within the heart may produce a murmur. Because the cardiac workload is greatly increased, especially during exercise, infection, or emotional stress, cardiac failure may ensue.
Children seem to have a remarkable ability to function well despite low levels of Hgb. Cyanosis (the result of the quantity of deoxygenated Hgb in arterial blood) is typically not evident. Growth retardation, resulting from decreased cellular metabolism and coexisting anorexia, is a common finding in chronic severe anemia and is frequently accompanied by delayed sexual maturation in the older child.
Diagnostic Evaluation
In general, anemia may be suspected based on findings on the history and physical examination, such as lack of energy, easy fatigability, and pallor, but unless the anemia is severe, the first clue to the disorder may be alterations in the CBC, such as decreased RBCs, and decreased Hgb and hematocrit (Hct) levels (see Fig. 26-1). Although anemia is sometimes defined as an Hgb level below 10 or 11 g/dl, this arbitrary cutoff is inappropriate for all children, because Hgb levels normally vary with age (see Table 26-1 and Appendix C).
Other tests specific to a particular type of anemia are employed to determine the underlying cause of anemia. These are discussed in relation to the particular disorder.
Therapeutic Management
The objective of medical management is to reverse the anemia by treating the underlying cause and to make up for any deficiency of blood, blood component, or substance the blood needs for normal functioning. For example, blood or blood cells are replaced after hemorrhage; in nutritional anemias the specific deficiency is replaced.
In patients with severe anemia, supportive medical care may include oxygen therapy, bed rest, and replacement of intravascular volume with intravenous (IV) fluids. The prognosis for anemia depends on the correction of the cause.
Nursing Care Management
The assessment of anemia includes the basic techniques that are applicable to any condition. The age of the infant or child provides some clues regarding the possible etiology of the anemia. For example, iron deficiency anemia occurs more frequently in the toddler between 12 and 36 months of age and during the growth spurt of adolescence.
Racial or ethnic background is significant. For example, the anemias related to abnormal Hgb levels are found in Southeast Asians and persons of African or Mediterranean ancestry. These same groups may be genetically deficient in the enzyme lactase after the period of infancy. Affected individuals are unable to tolerate lactose in the diet, with consequent intestinal irritation and chronic blood loss.
Special emphasis is placed on a careful history to elicit any information that might help identify the cause of the anemia. For example, a statement such as “My child drinks lots of milk” is a frequent finding in toddlers with iron deficiency anemia. An episode of diarrhea may have precipitated temporary lactose intolerance in a young child.
Stool examination for occult (microscopic) blood (Hemoccult test) can identify chronic intestinal bleeding that results from a primary or secondary lactase deficiency. It is also important to understand the significance of various blood tests (see Table 26-1).
Prepare Child and Family for Laboratory Tests.: Usually, several blood tests are ordered, but because they are generally done sequentially rather than at one time, the child is subjected to multiple finger or heel punctures or venipunctures. Laboratory technicians frequently are not aware of the trauma that repeated punctures represent to a child. However, these invasive procedures need not be painful (see Blood Specimens, Chapter 22). For example, the topical application of EMLA (an eutectic mix of lidocaine and prilocaine) or 4% lidocaine (Ela-Max) before needle punctures can eliminate pain (see Pain Management, Chapter 7). Therefore the nurse is responsible for preparing the child and family for the tests by:
Explaining the significance of each test, particularly why the tests are not all done at one time
Encouraging parents or another supportive person to be with the child during the procedure
Allowing the child to play with the equipment on a doll or participate in the actual procedure (e.g., by cleansing the finger with an alcohol swab)
Older children may appreciate the opportunity to observe the blood cells under a microscope or in photographs. This experience is especially important if a serious blood disorder, such as leukemia, is suspected, since it serves as a foundation for explaining the pathophysiology of the disorder.
Bone marrow aspiration is not a routine hematologic test but is essential for definitive diagnosis of the leukemias, lymphomas, and certain anemias.
Decrease Tissue Oxygen Needs.: Because the basic pathologic process in anemia is a decrease in oxygen-carrying capacity, an important nursing responsibility is to assess the child’s energy level and minimize excess demands. The child’s level of tolerance for activities of daily living and play is assessed, and adjustments are made to allow as much self-care as possible without undue exertion. During periods of rest the nurse takes vital signs and observes behavior to establish a baseline of nonexertion energy expenditure. During periods of activity the nurse repeats these measurements and observations to compare them with resting values.
Prevent Complications.: Children who are so severely anemic that they are hospitalized may require oxygen to prevent or reduce tissue hypoxia. Because these children are susceptible to infection, every effort is expended to prevent exposure to infectious agents. All the usual precautions are taken to prevent infection, such as practicing thorough hand washing, selecting an appropriate room in a noninfectious area, restricting visitors or hospital personnel with active infection, and maintaining adequate nutrition. The nurse also observes for signs of infection, particularly temperature elevation and leukocytosis.
IRON DEFICIENCY ANEMIA
Anemia caused by an inadequate supply of dietary iron is the most prevalent nutritional disorder in the United States and the most common mineral disturbance. Children 12 to 36 months of age are at risk for anemia as a result of cow’s milk being a major staple of the child’s diet (Richardson, 2007; Segel, Hirsh, and Feig, 2002). The prevalence of iron deficiency anemia has decreased, probably in part because of families’ participation in the Women, Infants, and Children (WIC) program, which provides iron-fortified formula for the first year of life and routine screening of Hgb levels during early childhood (Bogen, Krause, and Serwint, 2001). Preterm infants are especially at risk because of their reduced fetal iron supply. Adolescents are also at risk because of their rapid growth rate combined with poor eating habits.
Pathophysiology
Iron deficiency anemia can be caused by any number of factors that decrease the supply of iron, impair its absorption, increase the body’s need for iron, or affect the synthesis of Hgb. Although the clinical manifestations and diagnostic evaluation are similar regardless of the cause, the therapeutic and nursing care management depend on the specific reason for the iron deficiency. The following discussion is limited to iron deficiency anemia resulting from inadequate iron in the diet.
During the last trimester of pregnancy, iron is transferred from mother to fetus. Most of the iron is stored in the circulating erythrocytes of the fetus, with the remainder stored in the fetal liver, spleen, and bone marrow. These iron stores are usually adequate for the first 5 to 6 months in a full-term infant but for only 2 to 3 months in preterm infants or multiple births. If dietary iron is not supplied to meet the infant’s growth demands after the fetal iron stores are depleted, iron deficiency anemia results. Physiologic anemia should not be confused with iron deficiency anemia resulting from nutritional causes.
Although most toddlers with iron deficiency anemia are underweight, many infants are overweight because of excessive milk ingestion (known as milk babies). These children become anemic for two reasons: milk, a poor source of iron, is given almost to the exclusion of solid foods, and 50% of iron-deficient infants fed cow’s milk have an increased fecal loss of blood.
Therapeutic Management
After the diagnosis of iron deficiency anemia is made, therapeutic management focuses on increasing the amount of supplemental iron the child receives. This is usually done through dietary counseling and the administration of oral iron supplements.
In formula-fed infants the most convenient and best sources of supplemental iron are iron-fortified commercial formula and iron-fortified infant cereal. Iron-fortified formula provides a relatively constant and predictable amount of iron and is not associated with an increased incidence of gastrointestinal (GI) symptoms, such as colic, diarrhea, or constipation. Infants younger than 12 months of age should not be given fresh cow’s milk because it may increase the risk of GI blood loss occurring from allergy to the milk protein or from GI mucosal damage resulting from a lack of cytochrome iron (heme protein) (Richardson, 2007; Segel, Hirsh, and Feig, 2002). If GI bleeding is suspected, the child’s stool should be guaiac tested on at least four or five occasions to identify any intermittent blood loss.
Dietary addition of iron-rich foods is usually inadequate as the sole treatment of iron deficiency anemia, since the iron is poorly absorbed and thus provides insufficient supplemental quantities of iron. If dietary sources of iron cannot replace body stores, oral iron supplements are prescribed for approximately 3 months. Ferrous iron, more readily absorbed than ferric iron, results in higher Hgb levels. Ascorbic acid (vitamin C) appears to facilitate absorption of iron and may be given as vitamin C–enriched foods and juices with the iron preparation.
If the Hgb level fails to rise after 1 month of oral therapy, it is important to assess for persistent bleeding, iron malabsorption, noncompliance, improper iron administration, or other causes for the anemia. Parenteral (IV or intramuscular [IM]) iron administration is safe and effective, but painful, expensive, and occasionally associated with regional lymphadenopathy or allergic reaction (Andrews, 2003; McKenzie, 2004). Therefore parenteral iron is reserved for children who have iron malabsorption or chronic hemoglobinuria. Transfusions are indicated for the most severe anemia and in cases of serious infection, cardiac dysfunction, or surgical emergency when anesthesia is required. Packed RBCs (2 to 3 ml/kg), not whole blood, are used to minimize the chance of circulatory overload. Supplemental oxygen is administered when tissue hypoxia is severe.
Prognosis.: The prognosis for a child with this condition is very good. However, there is some evidence that if the iron deficiency anemia is severe and longstanding, cognitive, behavioral, and motor impairment may result (Burden, Westerlund, Sivan-Armony, and others, 2007; Andrews, 2003).
Nursing Care Management
An essential nursing responsibility is instructing parents in the administration of iron. Oral iron should be given as prescribed in two divided doses between meals, when the presence of free hydrochloric acid is greatest, since more iron is absorbed in the acidic environment of the upper GI tract. A citrus fruit or juice taken with the medication aids in absorption.
An adequate dosage of oral iron turns the stools a tarry green color. The nurse advises parents of this normally expected change and inquires about its occurrence on follow-up visits. Absence of the greenish black stool may be a clue to poor administration of iron, either in schedule or in dosage. Vomiting or diarrhea can occur with iron therapy. If the parents report these symptoms, the iron can be given with meals and the dosage reduced and then gradually increased until tolerated.
Liquid preparations of iron may temporarily stain the teeth. If possible, the medication should be taken through a straw or given through a syringe or medicine dropper placed toward the back of the mouth. Brushing the teeth after administration of the drug lessens the discoloration.
If parenteral iron preparations are prescribed, iron dextran must be injected deeply into a large muscle mass using the Z-track method. The injection site is not massaged after injection to minimize skin staining and irritation. Because no more than 1 ml should be given in one site, the IV route should be considered to avoid multiple injections. Careful observation is required because of the risk of adverse reactions, such as anaphylaxis, with IV administration. A test dose is recommended before routine use.
Diet.: A primary nursing objective is to prevent nutritional anemia through family education. Because breast milk is a poor iron source after 5 months of lactation, the nurse must reinforce the importance of administering iron supplementation to the exclusively breast-fed infant by 6 months of age (Andrews, 2003; Chandran and Gelfer, 2006; Richardson, 2007). The American Academy of Pediatrics (2005) recommends preterm and low-birth-weight infants or infants with inadequate iron stores at birth receive iron supplements before 6 months of age.
In the formula-fed infant, the nurse discusses with parents the importance of using iron-fortified formula and the introducing solid foods at the appropriate age during the first year of life. Traditionally, cereals are one of the first semisolid foods to be introduced into the infant’s diet at approximately 6 months of age (Chandran and Gelfer, 2006; Glader, 2007). The best solid-food source of iron is commercial iron-fortified cereals. It may be difficult at first to teach the infant to accept foods other than milk. The same principles are applied as those for introducing new foods (see Nutrition, Chapter 10), especially feeding the solid food before the milk. Predominantly milk-fed infants rebel against solid foods, and parents are cautioned about this and the need to be firm in not relinquishing control to the child. It may require intense problem solving on the part of both the family and the nurse to overcome the child’s resistance.
A difficulty encountered in discouraging the parents from feeding milk to the exclusion of other foods is dispelling the popular myth that milk is a “perfect food.” Many parents believe that milk is best for the infant and equate the weight gain with a “healthy child” and “good mothering.” The nurse can also stress that overweight is not synonymous with good health.
Diet education of teenagers is especially difficult, especially because teenage girls are particularly prone to following weight-reduction diets. Emphasizing the effect of anemia on appearance (pallor) and energy level (difficulty maintaining popular activities) may be useful. (See Mineral Imbalances, Chapter 11, and Table 11-2 for sources of iron-rich foods.)
SICKLE CELL ANEMIA
Sickle cell anemia (SCA) is one of a group of diseases collectively termed hemoglobinopathies, in which normal adult Hgb (Hgb A [HbA]) is partly or completely replaced by abnormal sickle Hgb (HbS). Sickle cell disease (SCD) includes all those hereditary disorders whose clinical, hematologic, and pathologic features are related to the presence of HbS. Even though the term SCD is sometimes used to refer to SCA, this use is incorrect. Other correct terms for SCA are SS and homozygous SCD.
The following are the most common forms of SCD in the United States:
SCA, the homozygous form of the disease (HbSS or SS).
Sickle cell–C disease, a heterozygous variant of SCD, including both HbS and HbC (SC).
Sickle cell–hemoglobin E disease, a variant of SCD in which glutamic acid has been substituted for lysine in the number-26 position of the β-chain (SE).
Sickle thalassemia disease, a combination of sickle cell trait and β-thalassemia trait (Sβthal). β+ refers to the ability to still produce some normal HbA. β° indicates that there is no ability to produce HbA.
Of the SCDs, SCA is the most common form in African-Americans, followed by sickle cell–C disease and sickle thalassemia. Sickle syndromes exist when the HbS is paired with other mutant globins.
SCA is found primarily in 1 in 375 births of African-Americans, 1 in 1200 births of Hispanics, with lower incidence in the other ethnic groups (Driscoll, 2007). The incidence of the disease varies in different geographic locations. Among African-Americans the incidence of sickle cell trait is about 9%. In West Africa the incidence is reported to be as high as 40% among native Africans. The high incidence of sickle cell trait in West Africans is believed by some to be the result of selective protection afforded trait carriers against one type of malaria.
The gene that determines the production of HbS is situated on an autosome and, when present, is always detectable and therefore dominant. Heterozygous persons who have both normal HbA and abnormal HbS are said to have sickle cell trait. Persons who are homozygous have predominantly HbS and have sickle cell anemia. The inheritance pattern is essentially that of an autosomal recessive disorder. Therefore, when both parents have sickle cell trait, there is a 25% chance with each pregnancy of producing an offspring with SCA.
Although the defect is inherited, the sickling phenomenon is usually not apparent until later in infancy because of the presence of fetal Hbg (HbF). As long as the child has predominantly HbF, sickling does not occur because there is less HbS. The newborn with SCA is generally asymptomatic because of the protective effect of HbF (60% to 80% HbF), but this rapidly decreases during the first year, so the child is at risk for sickle cell–related complications (Dover and Platt, 2003; Driscoll, 2007).
Pathophysiology
The clinical features of SCA are primarily the result of (1) obstruction caused by the sickled RBCs and (2) increased RBC destruction (Fig. 26-2). The abnormal adhesion, entanglement, and enmeshing of rigid sickle-shaped cells with one another
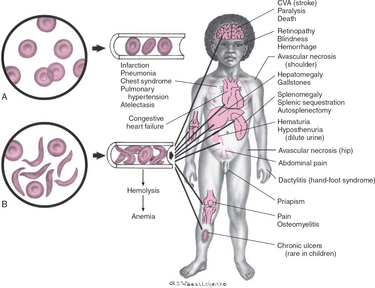
FIG. 26-2 Differences between effects of normal (A) and sickled (B) red blood cells on circulation with related complications. CVA, Cerebrovascular accident.
intermittently block the microcirculation, causing vasoocclusion. The resultant absence of blood flow to adjacent tissues causes local hypoxia, leading to tissue ischemia and infarction (cellular death). Most of the complications seen in SCA can be traced to this process and its impact on various organs of the body. The effect of sickling and infarction on organ structures occurs in the following sequence (see Box 26-2):
BOX 26-2 Clinical Manifestations of Sickle Cell Anemia
Pain in area(s) of involvement
Manifestations related to ischemia of involved areas
Extremities—Painful swelling of hands and feet (sickle cell dactylitis, or hand-foot syndrome), painful joints
Abdomen—Severe pain resembling acute surgical condition
Cerebrum—Stroke, visual disturbances
Chest—Symptoms resembling pneumonia, protracted episodes of pulmonary disease
EFFECTS OF CHRONIC VASOOCCLUSIVE PHENOMENA
Heart—Cardiomegaly, systolic murmurs
Lungs—Altered pulmonary function, susceptibility to infections, pulmonary insufficiency
Kidneys—Inability to concentrate urine, enuresis, progressive renal failure
Liver—Hepatomegaly, cirrhosis, intrahepatic cholestasis
Spleen—Splenomegaly, susceptibility to infection, functional reduction in splenic activity progressing to autosplenectomy
Eyes—Intraocular abnormalities with visual disturbances; sometimes progressive retinal detachment and blindness
Extremities—Avascular necrosis of hip or shoulder; skeletal deformities, especially lordosis and kyphosis; chronic leg ulcers; susceptibility to osteomyelitis
Clinical Manifestations
The clinical manifestations of SCA vary greatly in severity and frequency. The most acute symptoms of the disease occur during periods of exacerbation called crises. There are several types of episodic crises: vasoocclusive, acute splenic sequestration, aplastic, hyperhemolytic, cerebrovascular accident, chest syndrome, and infection. The crises may occur individually or concomitantly with one or more other crises. The episode may be a vasoocclusive crisis (VOC), preferably called a “painful episode,” characterized by distal ischemia and pain; sequestration crisis, a pooling of blood in the liver and spleen with decreased blood volume and shock; aplastic crisis, diminished RBC production resulting in profound anemia; or hyperhemolytic crisis, an accelerated rate of RBC destruction characterized by anemia, jaundice, and reticulocytosis.
Another serious complication is acute chest syndrome (ACS), which is clinically similar to pneumonia. It is the presence of a new pulmonary infiltrate and is associated with chest pain, fever, cough, tachypnea, wheezing, and hypoxia. A cerebrovascular accident (CVA, stroke) is a sudden and severe complication, often with no related illnesses. Sickled cells block the major blood vessels in the brain, resulting in cerebral infarction, which causes variable degrees of neurologic impairment. The current treatment for SCD children who have experienced a stroke is chronic transfusion therapy. Repeat CVAs causing progressively greater brain damage occur in approximately 70% of untreated children who have experienced one stroke (Dover and Platt, 2003).
Diagnostic Evaluation
Newborn screening for SCA is mandatory in most of the United States so that infants can be identified before symptoms occur. At birth the infant has up to 80% of HbF, which does not carry the defect. Because levels of HbS are low at birth, Hgb electrophoresis or other tests that measure Hgb concentrations are indicated. Early diagnosis (before 3 months of age) enables initiation of appropriate interventions to minimize complications. The family is taught to administer prophylactic antibiotics and identify early signs of infection to seek medical therapy as soon as possible.
If SCA is not diagnosed in early infancy, it is likely to manifest symptoms during the toddler and preschool years. SCA is occasionally first diagnosed during a crisis that follows an acute respiratory tract or GI infection. Routine hematologic tests are done to evaluate the anemia. Several specific tests detect the presence of the abnormal Hgb in the heterozygote or the homozygote. For screening purposes the sickleturbidity test (Sickledex) is frequently used because it can be performed on blood from a fingerstick and yields accurate results in 3 minutes. However, if the test is positive, Hgb electrophoresis is necessary to distinguish between those children with the trait and those with the disease. Hemoglobin electrophoresis (“fingerprinting” of the protein) is an accurate, rapid, and specific test for detecting the homozygous and heterozygous forms of the disease, as well as the percentages of the various types of Hgb.
Therapeutic Management
The aims of therapy are (1) to prevent the sickling phenomena, which are responsible for the pathologic sequelae; and (2) to treat the medical emergencies of sickle cell crisis. The successful achievement of the aims depends on prompt nursing interventions, medical therapies, patient and family preventive measures, and use of innovative treatments.
Medical management of a crisis is usually directed toward supportive and symptomatic treatment. The main objectives are to provide (1) rest to minimize energy expenditure and oxygen use; (2) hydration through oral and IV therapy; (3) electrolyte replacement, since hypoxia results in metabolic acidosis, which also promotes sickling; (4) analgesics for the severe pain from vasoocclusion; (5) blood replacement to treat anemia and to reduce the viscosity of the sickled blood; and (6) antibiotics to treat any existing infection (see Ethical Case Study).
ETHICAL CASE STUDY: Sickle Cell Disease
 ETHICAL DECISION MAKING MODEL
ETHICAL DECISION MAKING MODELJoey is a 7-year-old with sickle cell disease. His family belongs to a Jehovah’s Witnesses congregation, and the parents indicate that they are strong in their faith and beliefs. Joey has had several sickle cell pain crises this past year requiring hospitalization. He has never had a blood transfusion, yet the parents have been informed that a blood transfusion is often required to treat the physical problems associated with sickle cell disease. He arrives in the clinic today looking pale, and his hemoglobin level is 4.9 g/dl (baseline level is 8.0 g/dl), with a reticulocyte count of 1.0%. He has had a cough, fever as high as 37.8° C (100° F), and runny nose for the past week. Physical examination reveals a grade II/VI systolic ejection murmur with gallop at the left lower sternal border. His pulse is 120 beats/min, respirations 20 breaths/min, and blood pressure 102/60 mm Hg. Joey states that he feels tired and has no appetite, and he appears listless.
The first priority is to evaluate Joey’s condition. Children with sickle cell disease are anemic, but when their hemoglobin level drops extremely low, the reticulocyte count should increase as the bone marrow tries to produce new red blood cells. When children with sickle cell disease experience aplastic crisis, the bone marrow does not respond to the decreasing hemoglobin level; this situation is exemplified by Joey’s low reticulocyte count. This often occurs after a viral illness, which is indicated in the symptoms experienced by Joey the previous week. The treatment for aplastic crisis in a child with sickle cell disease is blood transfusion, since it can be several weeks before the bone marrow recovers and begins making new red blood cells again. On examination, Joey has clinical symptoms created by the decreasing hemoglobin level.
Treat All Involved with Respect
The physician sits with Joey’s parents to discuss the need for a blood transfusion. The physician explains that without a blood transfusion, Joey’s condition could become severe and he could die. Joey’s mother becomes distraught and states that she cannot give permission to transfuse her son with blood or blood products. The physician listens while the parents discuss their religious beliefs.
The controversy in this case is that Joey’s family are Jehovah’s Witnesses. Individuals of this faith believe that ingesting the blood of any flesh is forbidden and that blood transfusions are equivalent to oral ingestion of blood. Transfusion of whole blood, packed red blood cells, white blood cells, platelets, and plasma (fresh or frozen) is forbidden.
If the parents give consent for a blood transfusion, the family will have to leave the congregation, family, friends, and Jehovah’s Witnesses community. Refusal of transfusion of blood and blood products is a basic component of the Jehovah’s Witnesses faith, and if this precept is broken, the individual loses eternal salvation. Families are often shunned by the Witnesses community, and the child is perceived as an outcast because his future beyond death is affected by the transfusion of blood regardless of its impact on the child’s health.
Joey’s hematology nurse and social worker are asked to spend time with the parents to further explore their concerns regarding the blood transfusion. It becomes evident that the parents cannot give permission for the transfusion. The physician is adamant that the child’s best hope for recovery is to receive a blood transfusion.
For the parents’ sake, it is better to obtain a court order for lifesaving transfusion than to ask their permission. This removes the decision from the parents and prevents the parents and child from being ostracized, since the matter was removed from their control. The nurse’s role is to be an advocate for the family and a resource to other members of the health care team regarding the family’s beliefs.
The eventual outcome if Joey does not receive a blood transfusion is that his hemoglobin level will likely continue to fall as red blood cells are lysed. He will become more ill with ensuing respiratory difficulty, propensity for systemic infections, and eventual cardiac failure and death. If his parents fail to give consent for a blood transfusion, a court order may be obtained and Joey may be given the necessary blood transfusion, which will improve his health status. However, it is possible that Joey will become ill again at some point in the future and will again require a blood transfusion, at which point a court order will again become necessary because the parents will not give consent for such therapy. It is unknown how Joey’s parents and Jehovah’s Witnesses congregation will react to his receiving a blood transfusion.
Administration of pneumococcal and meningococcal vaccines is recommended for these children because of their susceptibility to infection as a result of functional asplenia. In addition to routine immunizations, the child with SCD should receive a yearly influenza vaccination (see Immunizations, Chapter 10). Oral penicillin prophylaxis is also recommended by 2 months of age to reduce the chance of pneumococcal sepsis (see Evidence-Based Practice box) (American Academy of Pediatrics, 2002; National Institutes of Health, National Heart, Lung, and Blood Institute, 2002; Redding-Lallinger and Knoll, 2006).
Short-term oxygen therapy may be helpful if a child has symptoms of respiratory difficulty. Severe hypoxia must be prevented because this causes massive systemic sickling that can be fatal. Although oxygen may prevent more sickling, it usually is not effective in reversing sickling because the oxygen is unable to reach the enmeshed sickled erythrocytes in clogged vessels (Perkins, 2001; Chiocca, 1996). In addition, prolonged administration can depress bone marrow, further aggravating the anemia (Khoury and Grimsley, 1995: Dover and Platt, 2003).
Sickle Cell Anemia and Penicillin Prophylaxis
In children with sickle cell anemia, does prophylaxis with penicillin prevent pneumococcal infection?
CRITICALLY ANALYZE THE EVIDENCE
Administration of oral prophylactic penicillin was compared with the 14-valent pneumococcal vaccine in preventing pneumococcal infection in 242 children between the ages of 6 months and 3 years with homozygous sickle cell disease. In the first 5 years of the trial, there were 11 pneumococcal infections in the pneumococcal vaccine group and higher infection rates in those given the vaccine before 1 year of age. No pneumococcal isolates were found in the group receiving penicillin, although four pneumococcal isolates were found in this group within 1 year of stopping the penicillin prophylaxis at age 3 years. This study supported the use of penicillin prophylaxis to prevent pneumococcal infection in children younger than 3 years of age (John, Ramlal, Jackson, and others, 1984).
In a multicenter, randomized, double-blind, placebo-controlled clinical trial, 105 children received penicillin twice daily; a control group of 110 children received a placebo twice daily. The trial was terminated 8 months early when an 84% reduction in the incidence of pneumococcal infections was observed in the group treated with penicillin compared with the placebo group. There were no deaths in the penicillin group, but three deaths from infection occurred in the placebo group. Researchers stressed the importance of screening children during the neonatal period and prescribing prophylactic penicillin to decrease the morbidity and mortality associated with pneumococcal infection (Gaston, Verter, Woods, and others, 1986).
Zarkowsky, Gallagher, Gill, and others (1986) conducted a retrospective analysis of 178 episodes of bacteremia in children with sickle hemoglobinopathies that occurred during 13,771 patient-years of follow-up (N 5 3451). The predominant pathogen in patients younger than 6 years of age was Streptococcus pneumoniae (66%), and gram-negative organisms were responsible for 50% of the bacteremias in patients 6 years and older. The incidence of pneumococcal bacteremia in children with sickle cell anemia younger than 3 years of age was 6.1 events per 100 patient-years. The results of this study supported prophylactic administration of penicillin for prevention of pneumococcal bacteremia in children younger than 3 years of age.
A cohort study of 315 patients with homozygous sickle cell disease who lived in Jamaica was conducted between June 1973 and December 1981. The patients were divided into three groups to determine whether interventions such as penicillin prophylaxis, parental education in early diagnosis of acute splenic sequestration, and close monitoring in a sickle cell clinic improved survival. A significant decline in deaths from acute splenic sequestration and pneumococcal septicemia and meningitis was found. The research indicated that early detection of sickle cell disease and prophylactic measures could significantly reduce deaths associated with homozygous sickle cell disease (Lee, Thomas, Cupidore, and others, 1995).
In a retrospective longitudinal study conducted from January 1995 through December 1999, 261 children under 4 years of age with sickle cell disease who did not have adequate health insurance were found to have received inadequate refills for antibiotic prophylaxis that placed them at increased risk of developing pneumococcal infection. Study findings showed that an increased number of outpatient visits for preventive care was associated with improved dispensing of prophylactic antibiotic refills (Sox, Cooper, Koepsell, and others, 2003).
Riddington and Owusu-Ofori (2002) conducted a systematic review of randomized controlled trials evaluating the effectiveness of prophylactic antibiotic administration in preventing pneumococcal infection in children with sickle cell disease. The review of published research found that penicillin prophylaxis significantly reduced the risk of pneumococcal infection in children with homozygous sickle cell disease with minimal adverse reactions.
APPLY THE EVIDENCE: NURSING IMPLICATIONS
The evidence demonstrated that penicillin prophylaxis significantly reduces the risk of pneumococcal infection in children with sickle cell anemia. The epidemiologic studies strongly suggest that all children with sickle cell anemia should be started on prophylactic penicillin at 2 months of age. Parents and children with sickle cell anemia should be instructed in the importance of taking the prophylactic penicillin twice daily and seeking medical attention immediately for acute illness, especially if the temperature exceeds 38.3° C (101° F), regardless of the use of prophylaxis.
REFERENCES
Gaston, MH, Verter, JI, Woods, G, et al. Prophylaxis with oral penicillin in children with sickle cell anemia: a randomized trial. N Engl J Med. 1986;314(25):1593–1599.
John, AB, Ramlal, A, Jackson, H, et al. Prevention of pneumococcal infection in children with homozygous sickle cell disease. BMJ. 1984;288(6430):1567–1570.
Lee, A, Thomas, P, Cupidore, L, et al. Improved survival in homozygous sickle cell disease: lessons from cohort study. BMJ. 1995;311(7020):1600–1602.
Riddington, C, Owusu-Ofori, S. Prophylactic antibiotics for preventing pneumococcal infection in children with sickle cell disease. http://www.cochrane.org/reviews/en/ab003427.html, 2002. [retrieved August 19, 2005, from].
Sox, CM, Cooper, WO, Koepsell, TD, et al. Provision of pneumococcal prophylaxis for publicly insured children with sickle cell disease. JAMA. 2003;290(8):1057–1061.
Zarkowsky, HS, Gallagher, D, Gill, FM, et al. Bacteremia in sickle hemoglobinopathies. J Pediatr. 1986;109(4):579–585.
Exchange transfusion, which reduces the number of circulating sickle cells and slows down the vicious circle of hypoxia, thrombosis, tissue ischemia, and injury, has been successful. The procedure is sometimes advocated as a possible preventive technique. A transcranial Doppler (TCD) test identifies the child with SCD who is at high risk for developing a CVA by monitoring the intracranial vascular flow (American Academy of Pediatrics, 2002; Bulas, 2005; Driscoll, 2007). The TCD is performed yearly on children from 2 to 16 years of age. If the TCD is abnormal, the recommended treatment is chronic transfusion therapy (Driscoll, 2007; Segal, Hirsh, and Feig, 2002). However, multiple transfusions carry the risk of transmission of viral infection, hyperviscosity, transfusion reactions, alloimmunization, and hemosiderosis (Redding-Lallinger and Knoll, 2006; Orkin and Nathan, 2003; Driscoll, 2007). After a CVA, blood transfusions are usually given every 3 to 4 weeks to help prevent a repeat stroke. To reduce iron overload from chronic transfusion therapy, chelation therapy may be started (see p. 924).
In children with recurrent life-threatening splenic sequestration, splenectomy may be a lifesaving measure. However, the spleen usually atrophies on its own through progressive fibrotic changes (functional asplenia) by 6 years of age. Prophylactic penicillin postsplenectomy and pneumococcal vaccines have decreased the incidence of pneumococcal sepsis. Packed RBC transfusions are recommended for treatment of splenic sequestration and stroke and preoperatively for most surgical procedures in the child with SCD.
The most frequent problem for patients with SCA is vasoocclusive pain. The chronic nature of this pain can greatly affect the child’s development. Priapism (continuous or intermittent) is defined as painful erection of the penis. As a vasoocclusive crisis, the priapism event is caused by sickling in sinusoids of the corpora cavernosa and is treated with aspiration from the corpora cavernosa only when conventional approaches fail (Redding-Lallinger and Knoll, 2006). A multidisciplinary approach is best for vasoocclusive pain management that includes pharmacologic treatment, hydration, physical therapy, and complementary treatment (e.g., prayer, spiritual healing, massage, herbs, relaxation, acupuncture, and biofeedback) (Redding-Lallinger and Knoll, 2006; Yoon and Black, 2006). When mild to moderate pain is reported, ibuprofen or acetaminophen (Tylenol) is used initially. If these drugs are not effective alone, codeine can be added. The dosages of both drugs are titrated (adjusted) to a therapeutic level. Opioids such as immediate- and sustained-release morphine, oxycodone, hydromorphone (Dilaudid), and methadone are administered intravenously or orally for severe pain and are given around the clock. Patient-controlled analgesia (PCA) has been used successfully for sickle cell–related pain. PCA reinforces the patient’s role and responsibility in managing the pain and provides flexibility in dealing with pain, which may vary in severity over time (see Pain Management, Chapter 7).
Prognosis.: The prognosis varies, but most patients live into the fifth decade. Most of the time, children are without symptoms and participate in normal activities without restrictions. The greatest risk is usually in children younger than 5 years of age, and the majority of deaths in these children are caused by overwhelming infection. Consequently, SCA is a chronic illness with a potentially terminal outcome. Physical and sexual maturation are delayed in adolescents with SCA. Although adults achieve normal height, weight, and sexual function, the delay may present problems to the adolescent (Dover and Platt, 2003; Redding-Lallinger and Knoll, 2006).
SCD individuals with higher levels of HbF tend to have a milder disease with fewer complications than those with lower levels (Anderson, 2006; Driscoll, 2007). Hydroxyurea is a U.S. Food and Drug Administration–approved medication that increases the production of HbF, reduces endothelial adhesion of sickle cells, and improves the sickle cell hydration (National Institutes of Health, National Heart, Lung, and Blood Institute, 2002). Long-term follow-up of patients taking hydroxyurea alone revealed a 40% reduction in mortality and decreased frequency of vasoocclusive crisis, ACS, hospital admissions, and need for transfusions, thus making SCD crises milder (Anderson, 2006; Steinberg, Barton, Castro, and others, 2003). Pediatric studies has shown that hydroxyurea can be safely used in children (Miller, Zimmerman, Schultz, and others, 2001; Zimmerman, Schultz, Davis, and others, 2004).
Hematopoietic stem cell transplantation (HSCT) offers the only cure for some children, although the mortality rate is approximately 8% and graft failures after transplantation range from 9% to 14% (Dover and Platt, 2003; Driscoll, 2007) (see p. 945).
Nursing Care Management
Educate Family and Child.: Family education begins with an explanation of the disease and its consequences. After this explanation, the most important issues to teach the family are to (1) seek early intervention for problems, such as fever of 38.5° C (101.3° F) or greater; (2) give penicillin as ordered; (3) recognize signs and symptoms of splenic sequestration, as well as respiratory problems that can lead to hypoxia; and (4) treat the child normally. The nurse tells the family that the child is normal but can get sick in ways that other children cannot.
The nurse emphasizes the importance of adequate hydration to prevent sickling and to delay the adhesion-stasis-thrombosis-ischemia cycle in a crisis. It is not sufficient to advise parents to “force fluids” or “encourage drinking.” They need specific instructions on how many daily glasses or bottles of fluid are required. Many foods are also a source of fluid, particularly soups, flavored ice pops, ice cream, sherbet, gelatin, and puddings.
Increased fluids combined with impaired kidney function result in the problem of enuresis. Parents who are unaware of this fact frequently employ the usual measures to discourage bed-wetting, such as limiting fluids at night, and may resort to punishment and shame to force bladder control. Enuresis is treated as a complication of the disease, such as joint pain or some other symptom, to alleviate parental pressure on the child.
 FAMILY FOCUS
FAMILY FOCUSAlthough the pain during a sickle cell crisis is usually severe and opioids are needed, many families fear that their child will become addicted to the narcotic. Unfortunately, misinformed health professionals may foster this unfounded fear, which results in needless suffering. Very few children who receive opioids for severe pain become behaviorally addicted to the drug (American Pain Society, 1999; National Institutes of Health, National Heart, Lung, and Blood Institute, 2002). Families and older children, especially adolescents, need to be reassured that opioids are medically indicated, high doses may be needed, and children rarely become addicted.
Promote Supportive Therapies During Crises.: The success of many of the medical therapies relies heavily on nursing implementation. Management of pain is an especially difficult problem and often involves experimenting with various analgesics, including opioids, and schedules before relief is achieved. Unfortunately, these children tend to be undermedicated, resulting in their “clock watching” and demands for additional doses sooner than might be expected. Often this incorrectly raises suspicions of drug addiction, when in fact the problem is one of improper dosage (see Family Focus box). In choosing and scheduling analgesics, the goal should be prevention of pain.
Any pain program should be combined with psychologic support to help the child deal with the depression, anxiety, and fear that may accompany the disease. This includes regular visits with the child to discuss any concerns during the hospitalization and positive reinforcement of coping skills, such as successful methods of dealing with the pain and compliance with treatment prescriptions. To reduce the negative connotation associated with the term crisis, it is best to say pain episode.
Frequently, heat to the affected area is soothing. Cold compresses are not applied to the area because this enhances sickling and vasoconstriction. Bed rest is usually well tolerated during a crisis, although actual rest depends greatly on pain alleviation and organized schedules of nursing care. Some activity, particularly passive range-of-motion exercises, is beneficial to promote circulation. Usually the best course of action is to let children dictate their activity tolerance.
If blood transfusions or exchange transfusions are given, the nurse has the responsibility of observing for signs of transfusion reaction (see Table 26-5). Because hypervolemia from too-rapid transfusion can increase the workload of the heart, the nurse also is alert to signs of cardiac failure.


In splenic sequestration the size of the spleen is gently measured by abdominal palpation (see Abdomen, Chapter 6). The nurse should be aware of spleen size because increasing splenomegaly is an ominous sign. A decreasing spleen size denotes response to therapy. Vital signs and blood pressure are also closely monitored for impending shock. Anemia is typically not a presenting complication in vasoocclusive crises but is a critical problem in other types of crises. The nurse monitors for evidence of increasing anemia and institutes appropriate nursing interventions (see p. 918). Oxygen is not beneficial in vasoocclusive episodes unless hypoxemia is present (Dover and Platt, 2003; Karayalcin, 2000). It does not reverse sickled RBCs, and if used in the nonhypoxic patient, it will decrease erythropoiesis (Khoury and Grimsley, 1995). Because prolonged use of oxygen can aggravate the anemia, signs of lack of therapeutic benefit, such as restlessness, increased pallor, and continued pain, are reported.
Intake, especially of IV fluids, and output are recorded. The child’s weight should be taken on admission to serve as a baseline for evaluating hydration. Because diuresis can result in electrolyte loss, the nurse also observes for signs of hypokalemia and should be familiar with normal serum electrolyte values to report changes.
Recognize Other Complications.: Nurses also need to be aware of the signs of ACS and CVA, both potentially fatal complications.
Support Family.: Families need the opportunity to discuss their feelings regarding transmitting a potentially fatal, chronic illness to their child. Because of the widely publicized prognosis for children with SCA, many parents express their prevalent fear of the child’s death. Three manifestations of SCD that may appear in the first 2 years of life (dactylitis, severe anemia, leukocytosis) can be predictors of disease severity (Platt, Brambilla, Roose, and others, 1994; Ohls and Christensen, 2007). However, nursing care for the family should be the same as for any family with a child with a life-threatening illness. Particular emphasis is placed on the siblings’ reactions, the stress on the marital relationship, and the childrearing attitudes displayed toward the child (see Chapter 18). Several resources are available to the family with a sickling disorder.*
The nurse advises parents to inform all treating personnel of the child’s condition. The use of medical identification, such as a bracelet, is another way of ensuring awareness of the disease.
If family members have the SCD trait or SCA, genetic counseling is necessary. A primary goal is informing parents who carry the trait, in language they can understand, of the 25% chance with each pregnancy resulting in a child with the disease.
β-THALASSEMIA (COOLEY ANEMIA)
The term thalassemia, which is derived from the Greek word thalassa, meaning “sea,” is applied to a variety of inherited blood disorders characterized by deficiencies in the rate of production of specific globin chains in Hgb. The name appropriately refers to descendants of or those people living near the Mediterranean Sea, who have the highest incidence of the disease, namely Italians, Greeks, and Syrians. Evidence suggests that the high incidence of the disorders among these groups is a result of the selective advantage the trait confers in relation to malaria, as is postulated in SCD. However, the disorder has a wide geographic distribution, probably as a result of genetic migration through intermarriage or possibly as a result of spontaneous mutation.
β-Thalassemia is the most common of the thalassemias and occurs in four forms:
Two heterozygous forms, thalassemia minor, an asymptomatic silent carrier, and thalassemia trait, which produces a mild microcytic anemia
Thalassemia intermedia, which is manifested as splenomegaly and moderate to severe anemia
A homozygous form, thalassemia major (also known as Cooley anemia), which results in a severe anemia that would lead to cardiac failure and death in early childhood without transfusion support
Pathophysiology
Normal postnatal Hgb is composed of two α- and two β-polypeptide chains. In β-thalassemia there is a partial or complete deficiency in the synthesis of the β-chain of the Hgb molecule. Consequently, there is a compensatory increase in the synthesis of α-chains, and γ-chain production remains activated, resulting in defective Hgb formation. This unbalanced polypeptide unit is very unstable; when it disintegrates, it damages RBCs, causing severe anemia.
To compensate for the hemolytic process, an overabundance of erythrocytes is formed unless the bone marrow is suppressed by transfusion therapy. Excess iron from hemolysis of supplemental RBCs in transfusions and from the rapid destruction of defective cells is stored in various organs (hemosiderosis).
Diagnostic Evaluation
The onset of thalassemia major may be insidious and not recognized until the latter half of infancy. The clinical effects of thalassemia major are primarily attributable to (1) defective synthesis of HbA, (2) structurally impaired RBCs, and (3) shortened life span of erythrocytes (Box 26-3).
Hematologic studies reveal the characteristic changes in RBCs (i.e., microcytosis, hypochromia, anisocytosis, poikilocytosis, target cells, and basophilic stippling of various stages). Low Hgb and Hct levels are seen in severe anemia, although they are typically lower than the reduction in RBC count because of the proliferation of immature erythrocytes. Hgb electrophoresis confirms the diagnosis, and radiographs of involved bones reveal characteristic findings.
Therapeutic Management
The objective of supportive therapy is to maintain sufficient Hgb levels to prevent bone marrow expansion and the resulting bony deformities, and to provide sufficient RBCs to support normal growth and normal physical activity. Transfusions are the foundation of medical management. Recent studies have evaluated the benefits of maintaining the child’s Hgb level above 9.5 g/dl, a goal that may require transfusions as often as every 3 to 5 weeks. The advantages of this therapy include (1) improved physical and psychologic well-being because of the ability to participate in normal activities, (2) decreased cardiomegaly and hepatosplenomegaly, (3) fewer bone changes, (4) normal or near-normal growth and development until puberty, and (5) fewer infections.
One of the potential complications of frequent blood transfusions is iron overload. Because the body has no effective means of eliminating the excess iron, the mineral is deposited in body tissues. To minimize the development of hemosiderosis, the new oral iron chelator deferasirox has been shown to be equivalent to deferoxamine (Desferal), a parenteral iron-chelating agent, and more tolerable by patients and families (Morris, Singer, and Walters, 2006; Okpala, 2005).
In some children with severe splenomegaly who demonstrate increased transfusion requirements, a splenectomy may be necessary to decrease the disabling effects of abdominal pressure and to increase the life span of supplemental RBCs. Over time, the spleen may accelerate the rate of RBC destruction and thus increase transfusion requirements. After a splenectomy, children generally require fewer transfusions, although the basic defect in Hgb synthesis remains unaffected. A major postsplenectomy complication is severe and overwhelming infection. Therefore these children continue to receive prophylactic antibiotics with close medical supervision for many years and should receive the pneumococcal and meningococcal vaccines in addition to the regularly scheduled immunizations (see Immunizations, Chapter 10).
Prognosis.: Most children treated with blood transfusion and early chelation therapy survive well into adulthood. The most common cause of death is iron-induced heart disease, multiple organ failure, postsplenectomy sepsis, liver disease, and malignancy (Paley, 2000). HSCT has the best results in the least symptomatic pediatric patients, with an 85% to 90% rate of complication-free survival (Morris, Singer, and Walters, 2006; Orkin and Nathan, 2003; Richardson, 2007).
Nursing Care Management
The objectives of nursing care are to (1) promote compliance with transfusion and chelation therapy, (2) assist the child in coping with the anxiety-provoking treatments and the effects of the illness, (3) foster the child’s and family’s adjustment to a chronic illness, and (4) observe for complications of multiple blood transfusions. Basic to each of these goals is explaining to parents and older children the defect responsible for the disorder, its effect on RBCs, and the potential effects of untreated iron overload (such as diabetes and heart disease). Because the prevalence of this condition is high among families of Mediterranean descent, the nurse also inquires about the family’s previous knowledge about thalassemia. All families with a child with thalassemia should be tested for the trait and referred for genetic counseling.
As with any chronic illness, the family’s needs must be met for optimal adjustment to the stresses imposed by the disorder (see Chapter 18). Sources of information for the family include the Cooley’s Anemia Foundation* and the Northern California Comprehensive Thalassemia Center.† Genetic counseling for the parents and fertile offspring is mandatory, and both prenatal diagnosis using amniocentesis at 20 weeks’ gestation or fetal blood sampling at 10 weeks and screening for thalassemia trait are available.
APLASTIC ANEMIA
Aplastic anemia (AA) refers to a bone marrow failure condition in which the formed elements of the blood are simultaneously depressed. The peripheral blood smear demonstrates pancytopenia or the triad of profound anemia, leukopenia, and thrombocytopenia. Hypoplastic anemia is characterized by a profound depression of RBCs, but normal or slightly decreased white blood cells (WBCs) and platelets.
Etiology
AA can be primary (congenital, or present at birth) or secondary (acquired). The best-known congenital disorder of which AA is an outstanding feature is Fanconi syndrome, a rare hereditary disorder characterized by pancytopenia, hypoplasia of the bone marrow, and patchy brown discoloration of the skin resulting from the deposit of melanin and associated with multiple congenital anomalies of the musculoskeletal and genitourinary systems. The syndrome appears to be inherited as an autosomal recessive trait with varying penetrance; therefore affected siblings may demonstrate several different combinations of defects.
Several etiologic factors contribute to the development of acquired hypoplastic anemia; however, most of the cases are considered idiopathic (Box 26-4). Acquired AA is classified as either severe acquired AA or moderate acquired AA. The following discussion focuses on severe acquired AA, which carries a poorer prognosis and follows a more rapidly fatal course than the primary types.
BOX 26-4 Common Causes of Acquired Aplastic Anemia
Human parvovirus infection, hepatitis, or overwhelming infection
Immune disorders such as eosinophilic fasciitis and hypoimmunoglobulinemia
Drugs such as certain chemotherapeutic agents, anticonvulsants, and antibiotics
Industrial and household chemicals, including benzene and its derivatives, which are found in petroleum products, dyes, paint remover, shellac, and lacquers
Infiltration and replacement of myeloid elements, such as in leukemia or the lymphomas
Idiopathic (In most cases no identifiable precipitating cause can be found.)
Diagnostic Evaluation
The onset of clinical manifestations, which include anemia, leukopenia, and decreased platelet count, is usually insidious. Definitive diagnosis is determined from bone marrow aspiration, which demonstrates the conversion of red bone marrow to yellow, fatty bone marrow. Severe AA is defined as less than 25% bone marrow cellularity with at least two of the following findings: absolute granulocyte count less than 500/mm3, platelet count less than 20,000/mm3, and absolute reticulocyte count less than 40,000/mm3 (Hord, 2007; Shimamura and Guinan, 2003). Moderate AA is defined as more than 25% bone marrow cellularity with the presence of mild or moderate cytopenia (Shimamura and Guinan, 2003; Shende, 2000).
Therapeutic Management
The objectives of treatment are based on the recognition that the underlying disease process is failure of the bone marrow to carry out its hematopoietic functions. Therefore therapy is directed at restoring function to the marrow and involves two main approaches: (1) immunosuppressive therapy to remove the presumed immunologic functions that prolong aplasia or (2) replacement of the bone marrow through transplantation. Bone marrow transplantation is the treatment of choice for severe AA when a suitable donor exists (see p. 945).
Antilymphocyte globulin (ALG) or antithymocyte globulin (ATG) is the principal drug treatment used for AA. The rationale for using ATG is based on the theory that AA may be a result of autoimmunity. ATG and cyclosporine suppress T cell–dependent autoimmune responses but do not cause bone marrow suppression. Cyclosporine is administered orally for several weeks to months. ATG usually is administrated intravenously over 12 to 16 hours for 4 days, after a test dose to check for hypersensitivity. A course may be repeated, depending on the reduction in circulating lymphocytes and the patient’s response. Because of the hypersensitivity response associated with ATG (i.e., fever, chills, myalgias), methylprednisolone is given intravenously to prevent these side effects. Colony-stimulating factor (CSF), and granulocyte-macrophage colony-stimulating factor (GM-CSF) given parenterally, may be used to enhance bone marrow production. Androgens may be used with ATG to stimulate erythropoiesis if the AA is unresponsive to initial therapies.
HSCT should be considered early in the course of the disease if a compatible donor can be found. Transplantation is more successful when performed before multiple transfusions have sensitized the child to leukocyte and human leukocyte antigens (HLA). HSCT is associated with an 85% survival rate in untransfused patients compared with a 70% survival rate in transfused patients (Marsh, 2005; Shende, 2000).
Nursing Care Management
The care of the child with AA is similar to that of the child with leukemia (see p. 930)–specifically, preparing the child and family for the diagnostic and therapeutic procedures, preventing complications from the severe pancytopenia, and emotionally supporting them in the face of a potentially fatal outcome. Information and support are available from the Aplastic Anemia and MDS International Foundation, Inc.*
Because the aspects of nursing care are discussed in the section on leukemia, only the exceptions are presented here. The drug ATG is usually administered by way of a central vein. If not, vigilant care must be directed to the IV infusion to prevent extravasation. Meticulous care of the venous access is essential because of the child’s susceptibility to infection. CSFs are usually given by subcutaneous injection over several days. Chemotherapeutic agents have been reported in the treatment of the relapsed patient with AA after ATG and CSF therapy. Many of the side effects associated with chemotherapy such as nausea and vomiting, alopecia, and mucositis are experienced by children receiving treatment for AA. Specialized care is required for children who have HSCT (see p. 945).
DEFECTS IN HEMOSTASIS
Hemostasis is the process that stops bleeding when a blood vessel is injured. Vascular and plasma clotting factors, as well as platelets, are required. A complex system of clotting, anticlotting, and clot breakdown (fibrinolysis) mechanisms exists in equilibrium to ensure clot formation only in the presence of blood vessel injury and to limit the clotting process to the site of vessel wall injury. Dysfunction in these systems will lead to bleeding or abnormal clotting. Although the coagulation process is complex, clotting depends on three factors: (1) vascular influence, (2) platelet role, and (3) clotting factors.
HEMOPHILIA
The term hemophilia refers to a group of bleeding disorders in which there is a deficiency of one of the factors necessary for coagulation of the blood. Although the symptomatology is similar regardless of which clotting factor is deficient, the identification of specific factor deficiencies allows definitive treatment with replacement agents.
In about 80% of all cases of hemophilia, the inheritance pattern is demonstrated as X-linked recessive. The two most common forms of the disorder are factor VIII deficiency (hemophilia A, or classic hemophilia) and factor IX deficiency (hemophilia B, or Christmas disease). Von Willebrand disease (vWD) is another hereditary bleeding disorder characterized by a deficiency, abnormality, or absence of the protein called von Willebrand factor (vWF) and a deficiency of factor VIII. Unlike hemophilia, vWD affects both males and females. The following discussion is primarily concerned with factor VIII deficiency, which accounts for 80% to 85% of all hemophilia cases.
Pathophysiology
The basic defect of hemophilia A is a deficiency of factor VIII (antihemophilic factor [AHF]). AHF is produced by the liver and is necessary for the formation of thromboplastin in phase I of blood coagulation. The less AHF found in the blood, the more severe the disease. Individuals with hemophilia have two of the three factors required for coagulation: vascular influence and platelets. Therefore they may bleed for longer periods, but not at a faster rate.
Bleeding into subcutaneous and intramuscular tissue is common. Hemarthrosis, which is bleeding into a joint space, is the most frequent type of internal bleeding. Bony changes and crippling deformities occur after repeated bleeding episodes over several years. Signs of hemarthrosis are swelling, warmth, redness, pain, and loss of movement. Bleeding in the neck, mouth, or thorax is serious because the airway can become obstructed. Intracranial hemorrhage can have fatal consequences and is one of the major causes of death. Hemorrhage anywhere along the GI tract can lead to anemia, and bleeding into the retroperitoneal cavity is especially hazardous because of the large space for blood to accumulate. Hematomas in the spinal cord can cause paralysis.
Diagnostic Evaluation
Overt, prolonged hemorrhage is readily apparent; bleeding into tissues is less apparent (Box 26-5). The diagnosis is usually made from a history of bleeding episodes, evidence of X-linked inheritance (only one third of the cases are new mutations), and laboratory findings. The tests specific for hemophilia plasma depend on specific factors for a reaction to occur, such as the partial thromboplastin time (PTT). Specific determination of factor deficiencies requires assay procedures normally performed in specialized laboratories. Carrier detection is possible in classic hemophilia using deoxyribonucleic acid (DNA) testing and is an important consideration in families in which female offspring may have inherited the trait.
BOX 26-5 Clinical Manifestations of Hemophilia
Prolonged bleeding anywhere from or in the body
Hemorrhage from any trauma–Loss of deciduous teeth, circumcision, cuts, epistaxis, injections
Excessive bruising, even from a slight injury, such as a fall
Subcutaneous and intramuscular hemorrhages
Hemarthrosis (bleeding into the joint cavities), especially the knees, ankles, and elbows
Therapeutic Management
The primary therapy for hemophilia is replacement of the missing clotting factor. The products available are factor VIII concentrate from pooled plasma or a genetically engineered recombinant, to be reconstituted with sterile water immediately before use, and DDAVP (1-deamino-8-d-arginine vasopressin), a synthetic form of vasopressin that increases plasma factor VIII and vWF levels and is the treatment of choice in mild hemophilia and vWD if the child shows an appropriate response. DDAVP is not effective in the treatment of severe hemophilia A, severe vWD, or any form of hemophilia B. Vigorous therapy is instituted to prevent chronic crippling effects from joint bleeding.
Other drugs may be included in the therapy plan, depending on the source of the hemorrhage. Corticosteroids are given for hematuria, acute hemarthrosis, and chronic synovitis. Nonsteroidal antiinflammatory drugs (NSAIDs), such as ibuprofen, are effective in relieving pain caused by synovitis; however, they must be used with caution because they inhibit platelet function (Curry, 2004; National Hemophilia Foundation, 2006). Oral administration or local application of 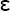 -aminocaproic acid (Amicar) prevents clot destruction; however, its use is limited to mouth or trauma surgery, and a dose of factor concentrate must be given first.
-aminocaproic acid (Amicar) prevents clot destruction; however, its use is limited to mouth or trauma surgery, and a dose of factor concentrate must be given first.
A regular program of exercise and physical therapy is an important aspect of management. Physical activity within reasonable limits strengthens muscles around joints and may decrease the number of spontaneous bleeding episodes.
Treatment without delay results in more rapid recovery and a decreased likelihood of complications; therefore most children are treated at home. The family is taught the technique of venipuncture and to administer the AHF to children older than 2 to 3 years of age. The child learns the procedure for self-administration at 8 to 12 years of age. Home treatment is highly successful, and the rewards, in addition to the immediacy, are less disruption of family life, fewer school or work days missed, and enhancement of the child’s self-esteem and independence.
Primary prophylaxis in hemophilia patients has proved to be effective in preventing bleeding complications by administrating periodic factor replacement. Primary prophylaxis involves the infusion of factor VIII concentrate on a regular basis before the onset of joint damage. Secondary prophylaxis involves the infusion of factor VIII concentrate on a regular basis after the child experiences his or her first joint bleed. The infusions are given three times a week. Aggressive factor replacement may be a cost-effective alternative to primary prophylaxis. This involves the infusion of a high dose of factor VIII concentrate when a joint bleed occurs, followed by 2 days of more standard doses of factor VIII concentrate with consideration of additional treatment every other day for one week (Montgomery, Gill, and Scott, 2003).
Prognosis.: Although there is no cure for hemophilia, its symptoms can be controlled and its potentially crippling deformities greatly reduced or even avoided. Today many children with hemophilia function with minimal or no joint damage. They are normal children with an average life expectancy in every respect but one: they have a tendency to bleed, which is a significant inconvenience but not necessarily a life-threatening event.
Gene therapy may prove to be a treatment option in the future. This therapy involves introducing a working copy of the factor VIII gene into a patient who has a flawed copy of the gene. Problems exist with appropriate selection of the vector, identification of the cell for gene expression, and control of side effects (Montgomery, Gill, and Scott, 2003).
Nursing Care Management
The earlier a bleeding episode is recognized, the more effectively it can be treated. Signs that indicate internal bleeding are especially important to recognize. Children are aware of internal bleeding and are reliable in telling the examiner where an internal bleed is. In addition to the manifestations described (see Box 26-5), the nurse maintains a high level of suspicion when a child with hemophilia demonstrates signs such as headache, slurred speech, loss of consciousness (from cerebral bleeding), and black tarry stools (from GI bleeding).
Prevent Bleeding.: The goal of prevention of bleeding episodes is directed toward decreasing the risk of injury. Prevention of bleeding episodes is geared mostly toward appropriate exercises to strengthen muscles and joints and to allow age-appropriate activity. During infancy and toddlerhood the normal acquisition of motor skills creates innumerable opportunities for falls, bruises, and minor wounds. Restraining the child from mastering motor development can foster more serious long-term problems than allowing the behavior. However, the environment should be made as safe as possible, with close supervision during playtime to minimize incidental injuries.
For older children the family usually needs assistance in preparing for school. A nurse who knows the family can be instrumental in discussing the situation with the school nurse and in jointly planning an appropriate activity schedule. Because almost all persons with hemophilia are boys, the physical limitations in regard to active sports may be a difficult adjustment, and activity restrictions must be tempered with sensitivity to the child’s emotional and physical needs. Use of protective equipment, such as padding and helmets, is particularly important, and noncontact sports, especially swimming, walking, jogging, tennis, golf, fishing, and bowling, are encouraged (National Hemophilia Foundation, 2006).
To prevent oral bleeding, some readjustment in terms of dental hygiene may be needed to minimize trauma to the gums, such as use of a water irrigating device, softening the toothbrush in warm water before brushing, or using a spongetipped disposable toothbrush. A regular toothbrush should be soft bristled and small.
Because any trauma can lead to a bleeding episode, all persons caring for these children must be aware of their disorder. These children should wear medical identification, and older children should be encouraged to recognize situations in which disclosing their condition is important, such as during dental extraction or injections. Health personnel need to take special precautions to prevent the use of procedures that may cause bleeding, such as IM injections. The subcutaneous route is substituted for IM injections whenever possible. Venipunctures for blood samples are usually preferred for these children. There is usually less bleeding after the venipuncture than after finger or heel punctures. Neither aspirin nor any aspirin-containing compound should be used. Acetaminophen is a suitable aspirin substitute, especially for controlling pain at home.
Recognize and Control Bleeding.: As noted, the earlier a bleeding episode is recognized, the more effectively it can be treated. Factor replacement therapy should be instituted according to established medical protocol, and supportive measures may be implemented, such as RICE, which stands for Rest, Ice, Compression, and Elevation. When parents and older children are taught such measures beforehand, they can be prepared to initiate immediate treatment. Plastic bags of ice or cold packs should be kept in the freezer for such emergencies. However, such measures do not take the place of factor replacement.
Prevent Crippling Effects of Bleeding.: As a result of repeated episodes of hemarthrosis, incompletely absorbed blood in the joints, and limitation of motion, bone and muscle changes occur that result in flexion contractures and joint fixation. During bleeding episodes the joint is elevated and immobilized. Active range-of-motion exercises are usually instituted after the acute episode. This allows the child to control the degree of exercise and discomfort. If an exercise program is instituted in the home, a physical therapist or public health nurse may need to supervise compliance with the regimen. Rarely, orthopedic intervention, such as casting, application of traction, or aspiration of blood, may be necessary to preserve joint function. Diet is also an important consideration, since excessive body weight can increase the strain on affected joints, especially the knees, and predispose the child to hemarthrosis. Consequently, calories need to be supplied in accordance with energy requirements.
Support Family and Prepare for Home Care.: Genetic counseling is essential as soon as possible after diagnosis. Unlike many other disorders in which both parents carry the trait, the feeling of responsibility for this condition usually rests with the mother. Without an opportunity to discuss her feelings, the marital relationship can suffer. Technology is now available to identify carriers in approximately 80% of cases and may reduce the anxiety regarding childbearing in women who may be at risk of carrying the defective gene, such as sisters or maternal aunts of an affected male. The discovery of factor concentrates has greatly changed the outlook for these children. Bleeding can be minimized, and the child can live a much more normal, unrestricted life. Children are taught to take responsibility for their disease at an early age. They learn their limitations, other preventive measures, and self-administration of the prophylactic AHF.
The needs of families who have children with hemophilia are best met through a comprehensive team approach of physicians (pediatrician, hematologist, orthopedist), nurse practitioner, nurse, social worker, and physical therapist. Parent-group discussions are beneficial in meeting those needs often best met by similarly affected families. For example, with the improved prognosis for these children, parents and adolescents with hemophilia face vocational and financial problems, in addition to concern over future childbearing. After children reach 21 years of age, many insurance companies will no longer carry them. This can be disastrous in terms of the cost of treatment. The National Hemophilia Foundation* and the Canadian Hemophilia Society† provide numerous services and publications for both health providers and families. Financial support is particularly important. A person with severe hemophilia may require factor replacement therapy and other medical treatments that cost in excess of $70,000 to $90,000 a year.
Children who have become infected with HIV through transfusions and factor replacement products are faced with the consequences of this dreaded disease. Consequently, they need the support of health professionals, especially in the areas of safe sexual practices to avoid disease transmission and public education regarding acquired immunodeficiency syndrome (AIDS) and ways to deal with public reactions to persons who have AIDS (see p. 939).
IDIOPATHIC THROMBOCYTOPENIC PURPURA
Idiopathic thrombocytopenic purpura (ITP) is an acquired hemorrhagic disorder characterized by (1) thrombocytopenia, excessive destruction of platelets; (2) purpura, a discoloration caused by petechiae beneath the skin; and (3) normal bone marrow with normal or increased number of immature platelets (megakaryocytes) and eosinophils. Although the cause is unknown, it is believed to be an autoimmune response to disease-related antigens. It is the most frequently occurring thrombocytopenia of childhood. The greatest frequency of occurrence is between 2 and 10 years of age.
The disease occurs in one of two forms: an acute, self-limiting course or a chronic condition (greater than 6 months’ duration). The acute form is most often seen after upper respiratory tract infections; after the childhood diseases measles, rubella, mumps, and chickenpox; or after infection with parvovirus B19.
Diagnostic Evaluation
The diagnosis is suspected on the basis of clinical manifestations (Box 26-6). In ITP the platelet count is reduced to below 20,000/mm3; therefore tests that depend on platelet function, such as the tourniquet test, bleeding time, and clot retraction, are abnormal. Although there is no definitive test on which to establish a diagnosis of ITP, several are usually performed to rule out other disorders in which thrombocytopenia is a manifestation, such as systemic lupus erythematosus, lymphoma, or leukemia.
Therapeutic Management
Management of ITP is primarily supportive, since the course of the disease is self-limited in the majority of cases. Activity is restricted at the onset while the platelet count is low and while active bleeding or progression of lesions is occurring. Treatment for acute presentation is symptomatic and has included prednisone, IV immune globulin (IVIG), and anti-D antibody. These are not curative therapies. Anti-D antibody is a relatively new therapy for ITP. Infusion of anti-D antibody causes a transient hemolytic anemia in the patient. Along with the clearance of antibody-coated RBCs, there is prolonged survival of platelets resulting from the blockade of the Fc receptors of the reticuloendothelial cells. The platelet count does not increase until 48 hours after an infusion of anti-D antibody; therefore it is not appropriate therapy for patients who are actively bleeding. The benefits of choosing anti-D antibody therapy over prednisone or IVIG is that anti-D antibody can be given in one dose over 5 to 10 minutes and is significantly less expensive than IVIG. Historically, patients who are treated with prednisone may first undergo a bone marrow examination to rule out leukemia. Therefore the use of anti-D antibody alleviates the need for a bone marrow examination. Patients must meet certain criteria before the administration of anti-D antibody (Box 26-7). Premedication with acetaminophen 5 to 10 minutes before infusion is recommended.
BOX 26-7 Criteria for Anti-D Antibody Therapy
Age between 1 and 19 years; Rh(D)-positive blood type
Normal white blood count and hemoglobin level for age; platelet count of 20,000/mm3
No history of reaction to plasma products
No known immunoglobulin A deficiency
Absence of Evans syndrome (characterized by the combination of idiopathic thrombocytopenic purpura and autoimmune hemolytic anemia)
No suspicion of lupus erythematosus or other collagen-vascular disorder
Splenectomy is reserved for those patients in whom ITP has persisted for 1 year or longer. It is the only treatment associated with long-term remission for 60% to 90% of children. Splenectomy removes the risk of hemorrhage but increases the risk of septicemia (Buchanan, 2005; Scott and Montgomery, 2007). Before splenectomy is considered, waiting until the child is older than 5 years of age is generally recommended because of the increased risk of bacterial infection. Pneumococcal and meningococcal vaccines are recommended before splenectomy (see Immunizations, Chapter 10). The child also receives penicillin prophylaxis after splenectomy. The length of prophylactic therapy is controversial, but in general, a minimum of 3 years is recommended.
Nursing Care Management
Nursing care is largely supportive and should include teaching regarding possible side effects of therapy and limitation in activities while the child’s platelet count is 50,000 to 100,000/mm3. Children with ITP should not participate in any contact sports, bike riding, skateboarding, in-line skating, gymnastics, climbing, or running. Parents are encouraged to engage their children in quiet activities and to prevent any injuries to the child’s head. The harmful effects of using aspirin and NSAIDs to control pain are critical for these children; therefore salicylate substitutes (such as acetaminophen) are always used. As in any condition with an uncertain outcome, the family needs emotional support.
DISSEMINATED INTRAVASCULAR COAGULATION
Disseminated intravascular coagulation (DIC), also known as consumption coagulopathy, is characterized by diffuse fibrin deposition in the microvasculature, consumption of coagulation factors, and endogenous generation of thrombin and plasmin. DIC is a secondary disorder of coagulation that occurs as a complication of a number of pathologic processes, such as hypoxia, acidosis, shock, and endothelial damage. It can result from many severe systemic diseases, such as congenital heart disease, necrotizing enterocolitis, gram-negative bacterial sepsis, rickettsial infections, and some severe viral infections.
Pathophysiology
DIC occurs when the first stage of the coagulation process is abnormally stimulated. Although no well-defined sequence of events occurs, two distinct phases can be identified. First, when the clotting mechanism is triggered in the circulation, thrombin is generated in greater amounts than can be neutralized by the body. Consequently, there is rapid conversion of fibrinogen to fibrin, with aggregation and destruction of platelets. If local and widespread fibrin deposition in blood vessels takes place, obstruction and eventual necrosis of tissues occur. Second, the fibrinolytic mechanism is activated, causing extensive destruction of clotting factors. With a deficiency of clotting factors, the child is vulnerable to uncontrollable hemorrhage into vital organs. An additional complication is damage and hemolysis of RBCs (Fig. 26-3).
Diagnostic Evaluation
DIC is suspected when the patient has an increased tendency to bleed (Box 26-8). Hematologic findings include prolonged prothrombin time (PT), PTT, and thrombin time (TT). There is a profoundly depressed platelet count, fragmented RBCs, and depleted fibrinogen.
Therapeutic Management
Treatment of DIC is directed toward control of the underlying or initiating cause, which in most instances stops the coagulation problem spontaneously. Platelets and fresh-frozen plasma may be needed to replace lost plasma components, especially in the child whose underlying disease remains uncontrolled. The extremely ill newborn infant may require exchange transfusion with fresh blood. The IV administration of heparin to inhibit thrombin formation is most often restricted to patients who have not responded to treatment of the underlying disease or replacement of coagulation factors and platelets.
Nursing Care Management
The goals of nursing care are to be aware of the possibility of DIC in the severely ill child and to recognize signs that might indicate its presence. The skills needed to monitor IV infusion and blood transfusions and to administer heparin are the same as for any child receiving these therapies (see p. 943). (See Chapter 18 for care of the child with a life-threatening illness.)
EPISTAXIS (NOSEBLEEDING)
Isolated and transient episodes of epistaxis, or nosebleeding, are common in childhood. The nose, especially the septum, is a highly vascular structure, and bleeding usually results from direct trauma, including blows to the nose, foreign bodies, and nose picking; or from mucosal inflammation associated with allergic rhinitis and upper respiratory tract infections. The bleeding ordinarily stops spontaneously or with minimal pressure and requires no medical evaluation or therapy.
Recurrent epistaxis and severe bleeding may indicate an underlying disease, particularly vascular abnormalities, leukemia, thrombocytopenia, and clotting factor deficiency diseases (e.g., hemophilia, vWD). Nosebleeds are sometimes associated with administration of aspirin, even in normal amounts. Persistent episodes of epistaxis require medical evaluation.
Nursing Care Management
In the event of a nosebleed, an essential intervention is to remain calm. Otherwise, the child will become more agitated, the blood pressure will increase, and the child will not cooperate. Although in most instances a nosebleed is not serious, it can be upsetting to family members as well. They need reassurance that the loss of blood is not serious and that the bleeding usually stops within 10 to 15 minutes.
To control the bleeding, the child is instructed to sit up and lean forward (not to lie down) to avoid aspiration of blood. Most of the nosebleeding originates in the anterior part of the nasal septum and can be controlled by applying pressure to the soft lower portion of the nose with the thumb and forefinger (see Emergency Treatment box). During this time the child breathes through the mouth.
In the event that hemorrhage continues, the child should be evaluated by a practitioner, who may pack the nose with epinephrine-soaked gauze. After a nosebleed, petroleum or water-soluble jelly can be inserted into each nostril to prevent crusting of old blood and to lessen the likelihood of the child’s picking at the nose and restarting the hemorrhage. If a child has numerous nosebleeds, factors believed to increase the likelihood of bleeds are eliminated, such as discouraging nose picking or altering the household humidity by placing a cool-mist humidifier in the child’s room. Repeated bleeding episodes lasting longer than 30 minutes may be an indication to refer the child for evaluation for the possibility of a bleeding disorder.
NEOPLASTIC DISORDERS
Neoplastic disorders are the leading cause of death from disease in children past infancy, and almost half of all childhood cancers involve the blood or blood-forming organs. Leukemias and lymphomas are discussed here. Malignant solid tumors of childhood are discussed elsewhere in relation to the tissues or organs involved.
Leukemias
Leukemia, cancer of the blood-forming tissues, is the most common form of childhood cancer. The annual incidence is 3 to 4 cases per 100,000 Caucasian children younger than 15 years of age (Margolin, Steuber, and Poplack, 2006; Pearce and Sills, 2005). It is more common in males and Caucasians, with the peak onset between 2 and 5 years of age (Pearce and Sills, 2005; Margolin, Steuber, and Poplack, 2006). It is one of the forms of cancer that has demonstrated dramatic improvements in survival rates. Current long-term disease-free survival for children with acute lymphoid leukemia approaches 80% (Pui, Relling, and Downing, 2004; Pearce and Sills, 2005), whereas acute nonlymphoid leukemia has a nearly 50% survival rate (Pearce and Sills, 2005). (See also Prognosis, p. 931.)
Classification
Leukemia is a broad term given to a group of malignant diseases of the bone marrow and lymphatic system. Research has revealed that it is a complex disease of varying heterogeneity. Consequently, classification has become increasingly complex, sophisticated, and essential, since identification of the subtype of leukemia has therapeutic and prognostic implications. The following is a brief overview of the major classification systems currently being used.
Morphology.: Two forms are generally recognized in children: acute lymphoid leukemia (ALL) and acute nonlymphoid (myelogenous) leukemia (ANLL or AML). Synonyms for ALL include lymphatic, lymphocytic, lymphoblastic, and lymphoblastoid leukemia. Usually the terms stem cell or blast cell leukemia also refer to the lymphoid type. Synonyms for the AML type include granulocytic, myelocytic, monocytic, myelogenous, monoblastic, and monomyeloblastic.
Cytochemical markers—Several chemical stains (e.g., terminal deoxynucleotidyl transferase [TdT]) aid in differentiation between ALL and ANLL.
Chromosome studies—Chromosome analysis has become an important tool in the diagnosis of ALL. For example, children with trisomy 21 have 20 times the risk of other children for developing ALL. Children with more than 50 chromosomes on the leukemic cells (hyperdiploid) have the best prognosis (Margolin, Steuber, and Poplack, 2006). Translocations of chromosomes also found on the leukemic cells can denote good prognosis, as in the trisomies 4 and 10, or a poor prognosis, as in the t(9:22) or Philadelphia chromosome.
Cell-surface immunologic markers—Cell-surface antigens have permitted differentiation of ALL into three broad classes: non-T, non-B ALL; B-cell ALL; and T-cell ALL. Children with non-T, non-B ALL have the best prognosis, especially if they have the common ALL antigen, known as CALLA positive (or CD 10+), on their cell surfaces (Margolin, Steuber, and Poplack, 2006).
Pathophysiology
Leukemia is an unrestricted proliferation of immature WBCs in the blood-forming tissues of the body. Although not a “tumor” as such, the leukemic cells demonstrate the same neoplastic properties as solid cancers. Therefore the resulting pathologic condition and clinical manifestations are caused by infiltration and replacement of any tissue of the body with nonfunctional leukemic cells. Highly vascular organs, such as the spleen and liver, are the most severely affected.
To understand the pathophysiology of the leukemic process, it is important to clarify two common misconceptions. First, although leukemia is an overproduction of WBCs, most often in the acute form the leukocyte count is low (thus the term leukemia). Second, these immature cells do not deliberately attack and destroy the normal blood cells or vascular tissues. Cellular destruction takes place by infiltration and subsequent competition for metabolic elements (Table 26-2).
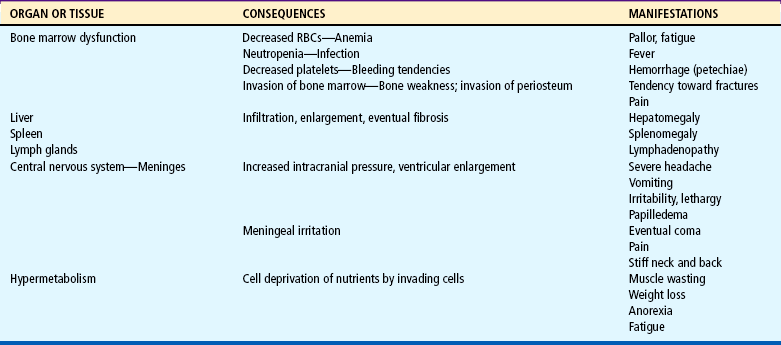
In all types of leukemia the proliferating cells depress the production of formed elements of the blood in bone marrow by competing for and depriving the normal cells of the essential nutrients for metabolism. The most frequent presenting signs and symptoms of leukemia are a result of infiltration of the bone marrow. The three main consequences are (1) anemia from decreased RBCs, (2) infection from neutropenia, and (3) bleeding from decreased platelet production. The invasion of the bone marrow with leukemic cells gradually causes a weakening of the bone and a tendency toward fractures. As leukemic cells invade the periosteum, increasing pressure causes severe pain.
The spleen, liver, and lymph glands demonstrate marked infiltration, enlargement, and eventually fibrosis. Hepatosplenomegaly is typically more common than lymphadenopathy. The next most important site of involvement is the CNS secondary to leukemic infiltration, which may cause increased intracranial pressure (see Box 28-1).
Leukemic cells may also invade the testes, kidneys, prostate, ovaries, GI tract, and lungs. With long-term survivors becoming more common, such sites of leukemia invasion, especially the testes, are becoming more important clinically.
Diagnostic Evaluation
Leukemia is usually suspected based on the history, physical manifestations (see Table 26-2), and a peripheral blood smear that contains immature forms of leukocytes, frequently combined with low blood counts. Definitive diagnosis is based on flow cytometry of the cells obtained in the bone marrow aspiration or biopsy. Flow cytometry identifies the specific type of blast cell. Typically, the bone marrow is hypercellular, with primarily blast cells. After the diagnosis is confirmed, a lumbar puncture is performed to determine whether there is any CNS involvement. A few children will have CNS involvement at diagnosis, although most are asymptomatic.
Therapeutic Management
Treatment of leukemia involves the use of chemotherapeutic agents, with or without cranial irradiation, in four phases: (1) induction therapy, which achieves a complete remission or less than 5% leukemic cells in the bone marrow; (2) CNS prophylactic therapy, which prevents leukemic cells from invading the CNS; (3) intensification therapy (consolidation), which eradicates residual leukemia cells, followed by delayed intensification, which prevents emergence of resistant leukemic clones; and (4) maintenance therapy, which serves to maintain the remission phase. Although the combination of drugs and radiation may vary according to institutions, the prognostic or risk characteristics of the patient, and the type of leukemia being treated.
Hematopoietic Stem Cell Transplantation.: HSCT has been used successfully for treating children who have ALL and AML. HSCT is not recommended for children with ALL during the first remission because of the excellent results possible with chemotherapy. Because of the poorer prognosis in children with AML, HSCT may be considered during the first remission when a suitable donor is available (Bollard, Krance, and Heslop, 2006).
HSCT may be not only from antigen-matched related donors, but also from matched unrelated donors or mismatched donors. Peripheral blood stem cell transplants are capable of differentiating into specialized cells of the hematologic system and can be obtained from related or unrelated donors or from umbilical cord blood. Regardless of the type of transplant, it is accompanied by significant morbidity and mortality, including graft-vs-host disease (GVHD), overwhelming infection, or severe organ damage.
Prognosis
The most important prognostic factors for determining long-term survival for children with ALL (in addition to treatment) are (1) the initial WBC count, (2) the child’s age at the time of diagnosis, (3) the type of cell involved, (4) the sex of the child, and (5) karyotype analysis. Children with a normal or low WBC count and who have non-T, non-B ALL and are CALLA positive have a much better prognosis than those with a high count or other cell types. Children diagnosed between 2 and 9 years of age have consistently demonstrated a better outlook than those diagnosed before 2 or after 10 years of age, and girls appear to have a more favorable prognosis than boys. Children with a DNA index greater than 1.16 (hyperdiploid) and translocation of chromosomes 4 and 10 have a better prognosis (Margolin, Steuber, and Poplack, 2006).
Late Effects of Treatment
Although vigorous treatment of childhood cancers has resulted in dramatically improved survival rates, increasing concern surrounds late effects–adverse changes related to treatment modalities–and recurrence of the disease process. Almost no organ is exempt, and almost every antineoplastic agent, especially irradiation, is responsible for some adverse effect.
The most devastating late effect is development of a second malignancy. Children who received cranial irradiation at age 5 years or younger are most susceptible to developing brain tumors (Silverman and Sallan, 2003; Bhatia, 2004). Treatment with an anthracycline is associated with cardiomyopathy; cranial irradiation and intrathecal chemotherapy are associated with cognitive and neuropsychologic deficits, which are just a few of the long-term sequelae. Consequently, close monitoring for late effects is essential, especially with the advent of additional clinical trials.
Nursing Care Management
Nursing care of the child with leukemia is directly related to the therapeutic regimen. General psychologic interventions during each phase of therapy are discussed in Chapter 18. The Nursing Care Plan for the Child with Cancer appears below.
Prepare Child and Family for Diagnostic and Therapeutic Procedures.: From the time before diagnosis to cessation of therapy, children must undergo several tests; the most traumatic are bone marrow aspiration, bone marrow biopsy, and lumbar punctures. Multiple finger sticks and venipunctures for blood analysis and drug infusion are common occurrences. Therefore the child needs an explanation of each procedure and what can be expected. In addition, effective pharmacologic measures, including conscious and unconscious sedation, and nonpharmacologic strategies are used to reduce discomfort associated with these painful procedures.
Relieve Pain.: The effective use of analgesia is especially important when the malignant process is uncontrolled and causes acute pain. Dosages of opioids (narcotics) are adjusted, or titrated, to the child’s needs and administered around the clock for optimal pain control. Nonpharmacologic strategies should be implemented as needed but are not substitutes for pharmacologic management. The reader is encouraged to review the principles of pain assessment and management presented in Chapter 7 and Preparation for Diagnostic and Therapeutic Procedures, Chapter 22, when caring for a child with leukemia.
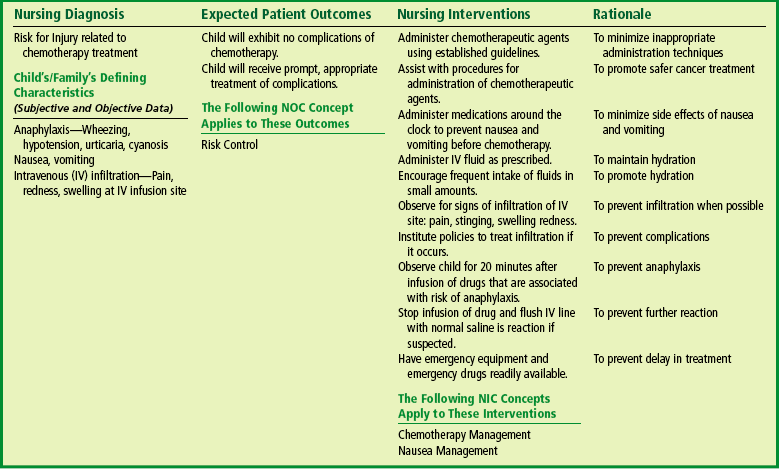
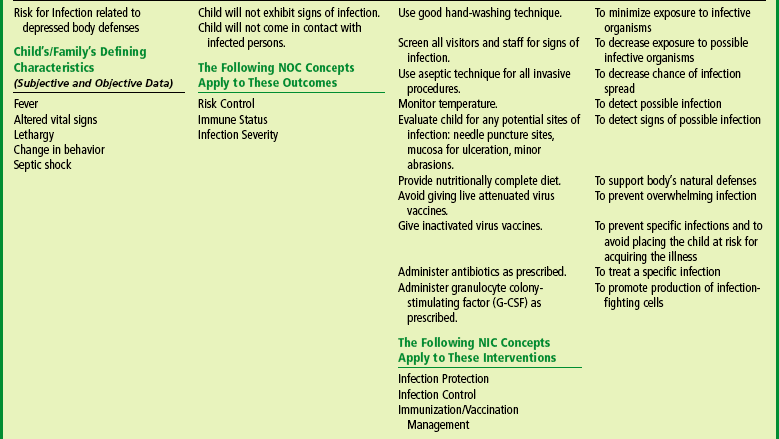





Prevent Complications of Myelosuppression.: The leukemic process and most of the chemotherapeutic agents cause myelosuppression. The reduced numbers of blood cells result in secondary problems of infection, bleeding tendencies, and anemia. Supportive care involves both medical and nursing management. Because these are so closely linked, they are discussed together.
Infection: A frequent complication of treatment for childhood cancer is overwhelming infection secondary to neutropenia. The child is most susceptible to overwhelming infection during three phases of the disease: (1) at the time of diagnosis and relapse when the leukemic process has replaced normal leukocytes; (2) during immunosuppressive therapy; and (3) after prolonged antibiotic therapy, which predisposes the child to the growth of resistant organisms. However, the use of granulocyte colony-stimulating factor (GCSF) has reduced the incidence and duration of infection in children receiving treatment for cancer.
The first defense against infection is prevention. When the child is hospitalized, the nurse employs all measures to control transfer of infection. These typically include the use of a private room, restriction of all visitors and health personnel with active infection, and strict hand-washing technique with an antiseptic solution. In some research centers, special germ-free environments are available during complete myelosuppression from intensive chemotherapy or for bone marrow transplant.
The child is evaluated for potential sites of infection (e.g., mucosal ulceration; skin abrasion; skin tear, such as a hangnail) and observed for any elevation in temperature. To identify the source of infection, chest radiographs and blood, stool, urine, and nasopharyngeal cultures are taken. IV antibiotics are administered, and if this therapy is prolonged, a venous access device, such as a peripherally inserted central catheter or intermittent infusion device (saline lock or PRN adaptor), is used to maintain IV access.
Prevention of infection continues to be a priority after discharge from the hospital. Ordinarily, the child is allowed to return to school when the WBC count is at a satisfactory level, usually an absolute neutrophil count (ANC) greater than 500/mm3 (see Nursing Care Guidelines box). At all times, family members are encouraged to practice good hand washing to prevent introducing pathogens into the home. The child may need to be isolated from school contacts in the event of an outbreak of a childhood disease, especially chickenpox.
Nutrition is another important component of infection prevention. An adequate protein-caloric intake provides the child with better host defenses against infection and increased tolerance to chemotherapy and irradiation. However, providing optimal nutrition during periods of anorexia and vomiting from chemotherapy is a tremendous challenge (see Feeding the Sick Child, Chapter 22).
Hemorrhage: Before the use of transfused platelets, hemorrhage was a leading cause of death in patients with leukemia. Now most bleeding episodes can be prevented or controlled with the administration of platelet concentrates or platelet-rich plasma.
Because infection increases the tendency toward hemorrhage, and because bleeding sites become more easily infected, skin punctures are avoided whenever possible. When finger sticks, venipunctures, IM injections, and bone marrow aspirations are performed, aseptic technique must be employed, along with continued observation for bleeding. Meticulous mouth care is essential, since gingival bleeding with resultant mucositis is a frequent problem. Because the rectal area is prone to ulceration from various drugs, feces and urine are removed immediately, and the perianal area is washed. Using rectal temperatures is avoided to prevent trauma. Children are advised to avoid activities that might cause injury or bleeding, such as riding bicycles or skateboards, climbing trees or playground equipment, and playing contact sports.
Platelet transfusions are generally reserved for active bleeding episodes that do not respond to local treatment and that may occur during induction or relapse therapy. Epistaxis and gingival bleeding are the most common. The nurse teaches parents and older children measures to control nosebleeding (see pp. 929–930). Pressure at the site without disturbing clot formation is the general rule.
During bleeding episodes the parents and child need much emotional support. Often parents will request a platelet transfusion, unaware of the need for trying local measures first. The nurse can be instrumental in allaying anxiety by acknowledging the feelings of the child and family and explaining the reason for delaying a platelet transfusion until absolutely necessary.
Anemia: Initially, anemia may be profound from complete replacement of the bone marrow by leukemic cells. During induction therapy, blood transfusions may be necessary. The usual precautions in caring for the child with anemia are instituted (see p. 914).
Use Precautions in Administering and Handling Chemotherapeutic Agents.: In addition to the nurse’s many responsibilities in regard to the child and family, nurses must also use safeguards to protect themselves. Handling chemotherapeutic agents may present risks to handlers and to their offspring, although the exact degree of risk is not known. Many chemotherapeutic agents are vesicants (sclerosing agents) that can cause severe cellular damage if even minute amounts of the drug infiltrate surrounding tissue. Only nurses experienced with chemotherapeutic agents should administer vesicants. Guidelines are available* and must be followed exactly to prevent tissue damage to patients. Interventions for extravasation vary, but each nurse should be aware of the institution’s policies and implement them at once.
In addition to extravasation, a potentially fatal complication is anaphylaxis, especially from l-asparaginase, teniposide (VM-26), etoposide (VP-16), bleomycin, and cisplatin. Nursing responsibilities include prevention of, recognition of, and preparation for serious reactions. Prevention begins with a careful history for known allergies.
Most children with cancer have a venous access device, which facilitates administration of IV drugs. During treatment and remission, many drugs are taken orally at home. Compliance with the medication schedule is essential, and nurses play an important role in educating the family about the drugs and encouraging adherence to the plan.
Manage Problems of Drug Toxicity.: Chemotherapy presents several nursing challenges. The complexity of the treatment protocols is often overwhelming to families. In addition, each therapy is associated with a number of predictable side effects. Nurses must be aware of these side effects and use judgment in recognizing reactions, as well as toxicities.†
Nausea and Vomiting: The nausea and vomiting that occur shortly after administration of several of the drugs and from cranial or abdominal radiation can be profound. The serotonin-receptor antagonists (e.g., ondansetron, granisetron) are effective in the control of nausea and vomiting occurring after emetogenic chemotherapy and radiotherapy. When combined with dexamethasone, these agents are the treatment of choice in the prevention of delayed emesis (Berde, Billett, and Collins, 2006).
The most beneficial regimen for antiemetic control has been the administration of the antiemetic before the chemotherapy begins. The goal is to prevent the child from ever experiencing nausea or vomiting, thus preventing development of anticipatory symptoms (the conditioned response of developing nausea and vomiting before receiving the drug).
Anorexia: Loss of appetite is a direct consequence of the chemotherapy or irradiation. It is a major problem for parents because it is the one area they feel responsible for, particularly when so many other facets of care are outside their control. There are no universally successful techniques for encouraging a sick child to eat. However, the guidelines in Chapter 22 can be helpful during the anorexic period and can prevent additional problems during the remission.
Some children still do not eat despite these approaches. When loss of appetite and weight persist, the nurse should investigate the family situation to determine whether any factors (e.g., conditioned aversion to food, environmental stress related to eating, controlling behavior, anger) might be contributing to the problem. Nasogastric tube feedings or total parenteral nutrition may be implemented for children with significant nutritional problems.
Mucosal Ulceration: One of the most distressing side effects of several drugs is GI mucosal cell damage, which can produce ulcers anywhere along the alimentary tract. Oral ulcers greatly compound anorexia because eating is extremely uncomfortable, but the following interventions may be helpful: (1) provide a bland, moist, soft diet appropriate for the child’s age and preferences; (2) use a soft sponge toothbrush (Toothettes) or cotton-tipped applicator; (3) provide frequent mouthwashes with normal saline (using a solution of 1 teaspoon of table salt and 1 pint of water) or sodium bicarbonate mouth rinses (using a solution of 1 teaspoon of baking soda in 1 quart of water); and (4) use local anesthetics (e.g., Chloraseptic lozenges) or nonprescription preparations without alcohol (e.g., hydrocortisone dental paste [Orabase], antiseptic mouth rinse [UlcerEase], diphenhydramine [Benadryl] and aluminum and magnesium hydroxide [Maalox] solution). Although local anesthetics are effective in temporarily relieving the pain, many children dislike the taste and numb feeling they produce.
Other preparations that may be used to prevent or treat mucositis include chlorhexidine gluconate (Peridex) because of its dual effectiveness against candidal and bacterial infections, antifungal troches (lozenges) or mouthwash, and lip balm (e.g., Aquaphor) to keep the lips moist. Agents that should not be used include lemon glycerin swabs (irritate eroded tissue and can decay teeth), hydrogen peroxide (delays healing by breaking down protein), and milk of magnesia (dries mucosa).
Stomatitis may cause such difficulty with eating that the child may require hospitalization for hydration, parenteral nutrition, and pain control (often with IV morphine). The child will usually choose the foods that are best tolerated, and the nurse should encourage parents to relax any eating pressures. Because the stomatitis is a temporary condition, the child can resume good food habits after the ulcers heal. Dental hygiene can become a serious problem for children with orthodontic appliances. Sometimes it may be necessary to remove the braces to allow chemotherapy to continue.
Rectal ulcers are managed by meticulous toilet hygiene, warm sitz baths after each bowel movement, and use of an occlusive ointment or dressing applied to the ulcerated area to promote epithelialization. Stool softeners are necessary to prevent further discomfort. Parents are advised to record bowel movements, since the child may voluntarily avoid defecation to prevent discomfort. Rectal thermometers and suppositories are contraindicated because insertion may further traumatize the area.
Neuropathy: Vincristine and, to a lesser extent, vinblastine can cause various neurotoxic effects. Nursing interventions for management of these effects include (1) administering stool softeners or laxatives for severe constipation caused by decreased bowel innervation; (2) maintaining good body alignment and, if patient is on bed rest, using a footboard or high-top shoes to minimize or prevent footdrop; (3) carrying out safety measures during ambulation because of weakness and numbing of the extremities, which may cause difficulty in walking or fine hand movement; and (4) providing a soft or liquid diet for severe jaw pain.
Hemorrhagic Cystitis: Sterile hemorrhagic cystitis, a side effect of chemical irritation to the bladder from cyclophosphamide, can be decreased and often prevented by (1) promoting a liberal fluid intake (at least one and a half times the recommended daily fluid requirement); (2) frequent voiding immediately after feeling the urge, before bed, and after arising; (3) administering the drug early in the day to allow for sufficient oral intake and voiding; and (4) administering mesna (an agent that provides protection to the bladder) as ordered. If oral home administration is prescribed, the family needs specific instructions regarding exactly how much fluid the child must have.
Alopecia: Hair loss is a common side effect of several chemotherapeutic drugs and cranial irradiation, although not all children lose their hair during drug therapy. It is better to warn children and parents of this side effect than to allow them to think that it is only a remote possibility. A soft cotton cap is the most comfortable head wear for children. Polyester increases perspiration and causes itching. Other options include scarves, hats, or a wig.
The nurse should also inform the family that hair regrows in 3 to 6 months and may be of a different color and texture. Frequently the hair is darker, thicker, and curlier than before. If the child chooses not to wear a wig, attention to some type of head covering, especially in cold climates and during exposure to sun, and scalp hygiene are important. The scalp should be washed like any other body part.
Moon Face: Short-term steroid therapy produces no acute toxicities and produces two beneficial reactions: increased appetite and a sense of well-being. However, it does produce alterations in appearance, which, although not clinically significant, can be extremely distressing to older children. One of these is moon face, in which the child’s face becomes rounded and puffy. It is helpful to reassure the child that after cessation of the drug, the facial shape will return to normal. Unlike hair loss, little can be done to camouflage this obvious change. If the child resumes activity early in the course of treatment, the change may be less noticeable to peers than after a long absence.
Mood Changes: Shortly after beginning steroid therapy, children experience a number of mood changes that range from feelings of well-being and euphoria to depression and irritability. If parents are unaware of these drug-induced changes, they may become unduly concerned. The nurse should warn them of the reactions and encourage them to discuss the behavioral changes with each other and the child.
Provide Emotional Support.: An important aspect of continued emotional support involves the prognosis. Although leukemia is no longer invariably fatal, it must be remembered that survival statistics are only average estimates and apply to those children treated with the latest protocols since diagnosis. For the low-risk child the chances may be better, but for the high-risk child they may be significantly poorer. Of those who do survive after discontinuing therapy, some will relapse. Therefore, at present, only the passage of time is positive confirmation of the child’s being ultimately “cured” of the disease. Remission, even in excess of 5 years, cannot be equated with a cure. With increasing concern regarding late effects of treatment, continued surveillance of the child’s health status is needed. The nurse who is working with family members must individualize information regarding the “numbers” and the potential risks. An understanding of each member’s emotional needs, as well as competent care of physical ones, is essential to the positive, growth-promoting support of the family. Comprehensive emotional support for the family of the child with a potentially fatal illness is discussed in Chapter 18.
Lymphomas
Pediatric lymphomas are the third most common group of malignancies in children and adolescents. The lymphomas, a group of neoplastic diseases that arise from the lymphoid and hematopoietic systems, are divided into Hodgkin disease and non-Hodgkin lymphoma (NHL). These diseases are further subdivided according to tissue type and extent of disease. NHL is more prevalent in children younger than 14 years of age, whereas Hodgkin disease is prevalent in adolescence and the young adult period, with a striking increase between ages 15 and 19 years.
HODGKIN DISEASE
Hodgkin disease is a neoplastic disease that originates in the lymphoid system and primarily involves the lymph nodes. It predictably metastasizes to nonnodal or extralymphatic sites, especially the spleen, liver, bone marrow, and lungs, although no tissue is exempt from involvement (Fig. 26-4). It is classified according to four histologic types: (1) lymphocytic predominance, (2) nodular sclerosis, (3) mixed cellularity, and (4) lymphocytic depletion. Accurate staging of the extent of disease is the basis for treatment protocols and expected prognoses.
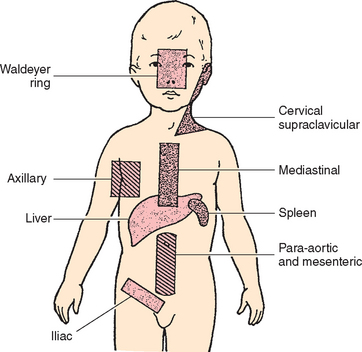
The Ann Arbor staging system assigns a stage based on the number of sites of lymph node involvement, presence of extranodal disease, and history of any symptoms. Patients are classified as A if asymptomatic and as B if they have the following symptoms: temperature of 38° C (100.4° F) or higher for 3 consecutive days, drenching night sweats, or unexplained loss of body weight (10% or more) over the preceding 6 months (Hudson, Onciu, and Donaldson, 2006).
Asymptomatic enlarged cervical or supraclavicular lymphadenopathy is the most common presentation of Hodgkin disease. Other systemic symptoms may be manifested, including fever, weight loss, night sweats, cough, abdominal discomfort, anorexia, nausea, and pruritus. Because multiple organs may be involved, diagnosis is based on several tests and the extent of metastatic disease. Tests include a CBC, erythrocyte sedimentation rate, serum copper, ferritin level, fibrinogen, immunoglobulins, uric acid level, liver function tests, T-cell function studies, and urinalysis. Radiographic tests include computed tomography (CT) scans of the neck, chest, abdomen, and pelvis; a gallium scan (identifies metastatic or recurrent disease); a chest x-ray film; and, if clinically indicated, a bone scan to identify metastatic disease.
Although used rarely, lymphangiography may be performed. This is visualization of the lymphatic circulation of the lower extremities, groin, ileopelvic and abdominal-aortic regions, and thoracic duct by way of a radiopaque medium injected in the feet or hands.
A lymph node biopsy is essential to establish histologic diagnosis and staging. The presence of Reed-Sternberg cells is characteristic of Hodgkin disease. These large cells, which are multilobed and nucleated with abundant cytoplasm and a typically halolike clear zone around the nucleolus, are often described as having an “owl’s eyes” appearance (Hudson, Onciu, and Donaldson, 2006). A bone marrow aspiration or biopsy is usually performed. With the advent of CT and gallium scans to identify metastatic disease and multiagent chemotherapy to eradicate metastatic disease, a laparotomy without splenectomy is avoided except in a few selected cases.
Therapeutic Management
The primary modalities of therapy are radiation and chemotherapy. Each may be used alone or in combination based on the clinical staging. Radiation may involve only the involved field (IF), an extended field (EF) (involved areas plus adjacent nodes), or total nodal irradiation (TNI), depending on the extent of involvement.
An effective combination of chemotherapy widely used is MOPP (mechlorethamine, vincristine [Oncovin], procarbazine, prednisone) or ABVD (adriamycin, bleomycin, vinblastine, dacarbazine). However, this therapy combination has caused severe late effects, especially secondary malignancies. Other drug combinations such as COPP (cyclophosphamide, vincristine, prednisone, procarbazine) as a substitute for MOPP have minimized late effects.
Follow-up care of children no longer receiving therapy is essential to identify relapse and secondary cancers. In children with splenectomy resulting from laparotomy or splenic irradiation, prophylactic antibiotics are administered for an indefinite period. Also, immunizations against pneumococci and meningococci are recommended before the splenectomy.
Nursing Care Management
Nursing care involves the same objectives as for patients with other types of cancer, specifically: (1) preparation for diagnostic and operative procedures, (2) explanation of treatment side effects, and (3) child and family support (see Chapter 18). Because this is most often a disease of adolescents and young adults, the nurse must have an appreciation of their psychologic needs and reactions during the diagnostic and treatment phases (see Nursing Care Plan, pp. 932–934).
The most common side effect of irradiation is fatigue. This is particularly difficult for active, outgoing school-age children and adolescents, since it prevents them from keeping up with their peers. Sometimes adolescents will push themselves to the point of physical exhaustion rather than admit and succumb to the decreased activity tolerance. The nurse cautions parents to observe for behavior such as extreme fatigue at the end of the day, falling asleep at the dinner table, inability to concentrate on homework, or an increased susceptibility to infection. A regular bedtime and scheduled rest periods are important for these children, especially during chemotherapy, when myelosuppression increases the risk of infection and debilitation. Before discharge the nurse should discuss a feasible school schedule with the parents and child.
An area of concern for adolescents is the high risk of sterility from irradiation and chemotherapy. Both drugs, particularly procarbazine and alkylating agents, and irradiation to the gonads can lead to infertility. Adolescents should be informed of these side effects early in the course of the diagnosis and treatment. Sperm banking is now offered at many cancer centers before the initiation of treatment in adolescent boys. Sexual function is not altered, although the appearance of secondary sexual characteristics and menstruation may be delayed in the pubescent child. Delayed sexual maturation may be an extremely sensitive and stressful issue for children (see Chapter 17).
NON-HODGKIN LYMPHOMA
NHL occurs more frequently in children than Hodgkin disease. NHL is diagnosed in approximately 750 to 800 children each year in the United States (Link and Weinstein, 2006). Histologic classification of childhood NHL is strikingly different from that of Hodgkin disease, as demonstrated in the following statements:
The disease is usually diffuse rather than nodular.
The cell type is either undifferentiated or poorly differentiated.
Dissemination occurs early, more often, and rapidly.
Mediastinal involvement and invasion of meninges are common.
NHL exhibits a variety of morphologic, cytochemical, and immunologic features, not unlike the diversity seen in leukemia. Classification is based on the histologic pattern: (1) lymphoblastic, (2) Burkitt or non-Burkitt, or (3) large cell. Immunologically these cells are also classified as T cells; B cells; or non-T, non-B cells (lacking immunologic properties). The clinical staging system used in Hodgkin disease is of little value in NHL, although it has been modified and other systems have been developed.
Diagnostic Evaluation
Because the clinical presentation of most children with NHL is widespread disseminated disease, thorough pathologic staging is unnecessary. Clinical manifestations depend on the anatomic site and extent of involvement. These manifestations include many of those seen in Hodgkin disease and leukemia, as well as organ symptoms related to pressure from enlargement of adjacent lymph nodes, such as intestinal or airway obstruction, cranial nerve palsies, and spinal paralysis.
Recommendations for staging include a surgical biopsy of an enlarged node, histopathologic confirmation of disease with cytochemical and immunologic evaluation, bone marrow examination, radiographic studies (especially tomograms of the lungs and GI organs), and lumbar puncture.
Therapeutic Management
The treatment protocols for NHL include aggressive use of irradiation and chemotherapy. Similar to leukemic therapy, the protocols include induction, consolidation, and maintenance phases, some with intrathecal chemotherapy. Several antineoplastic agents used in the treatment of NHL include vincristine, prednisone, l-asparaginase, methotrexate, 6-mercaptopurine, cytarabine, cyclophosphamide, anthracyclines, and teniposide or etoposide (Link and Donaldson, 2003; Link and Weinstein, 2006).
Nursing Care Management
Nursing care of the child with NHL is similar to that required for children with leukemia. Many of the same drugs are employed, although the schedules differ. Because of the intense chemotherapy, nursing care is primarily directed toward managing the side effects of these agents and providing supportive care to the child and family (see Nursing Care Plan, pp. 932–934).
IMMUNOLOGIC DEFICIENCY DISORDERS
A number of disorders can cause profound, often life-threatening alterations within the body’s immune system. The most serious are those conditions that completely depress immunity, such as severe combined immunodeficiency disease (SCID). However, the one disorder that generates the most anxiety, within both the family and the community at large, is HIV infection/AIDS.
Several classifications of immune dysfunction exist. AIDS, SCID, and Wiskott-Aldrich syndrome (WAS) are syndromes wherein the body is unable to mount an immune response. The immune response can also be misdirected. In autoimmune disorders, antibodies, macrophages, and lymphocytes attack healthy cells.
HUMAN IMMUNODEFICIENCY VIRUS INFECTION AND ACQUIRED IMMUNODEFICIENCY SYNDROME
Since the first cases of AIDS were identified in the early 1980s, HIV infection has generated intense medical investigation. Research has led to early diagnosis of and improved medical treatments for HIV infection, changing this disease from a rapidly fatal one to a chronic, but terminal, disease of childhood.
Epidemiology
The first AIDS cases in the pediatric population in the United States were identified in children born to HIV-infected mothers and in children who received blood products. More than 90% of these children acquired the disease perinatally from their mothers. Smaller numbers of children were infected through the transfusion of contaminated blood or blood products before 1985 or were infected through sexual abuse. In contrast, sexual activity and IV drug use are major sources of HIV infection in adolescents.
The estimated number of children with perinatally acquired AIDS peaked in 1992; subsequent years have seen significant declines. This trend is a result of implementation of recommended HIV counseling and voluntary testing practices and the use of highly active antiretroviral therapy (HAART) to prevent perinatal transmission. HAART, typically a combination of two nucleoside analog reverse transcriptase inhibitors and a protease inhibitor, is the current standard in the United States for the treatment of HIV-infected pregnant women, and it has significantly reduced the transmission of HIV (Cibulka, 2006; Perinatal HIV Guidelines Working Group, 2007). Routine HIV screening and voluntary testing for pregnant women are recommended (Centers for Disease Control and Prevention, 2006), and guidelines for the use of antiretroviral drugs in HIV-infected pregnant women to reduce perinatal transmission are available (Perinatal HIV Guidelines Working Group, 2007).
Etiology
HIV is a retrovirus that is transmitted by lymphocytes and monocytes. It is found in the blood, semen, vaginal secretions, and breast milk. It has an incubation period of months to years (Ezekowitz and Stockman, 2003). There are different strains of HIV. HIV-2 is prevalent in Africa, whereas HIV-1 is the dominant strain in the United States and elsewhere. Horizontal transmission of HIV occurs through intimate sexual contact or parenteral exposure to blood or body fluids containing visible blood. Perinatal (vertical) transmission occurs when an HIV-infected pregnant woman passes the infection to her infant. There is no evidence that casual contact between infected and uninfected individuals can spread the virus.
Pathophysiology
The HIV virus primarily infects a specific subset of T lymphocytes, the CD4 + T cells. The virus takes over the machinery of the CD4 + lymphocyte, using it to replicate itself, rendering the CD4 + cell dysfunctional. The CD4 + lymphocyte count gradually decreases over time, leading to progressive immunodeficiency. The count eventually reaches a critical level below which there is substantial risk of opportunistic illnesses, followed by death.
Clinical Manifestations
Common clinical manifestations of HIV infection in children are varied (Box 26-9). The diagnosis of AIDS is associated with certain illnesses or conditions. The most common AIDS-defining conditions observed among American children are listed in Box 26-10. Other problems in these children may include short stature, malnutrition, and cardiomyopathy. CNS abnormalities resulting from HIV infection may include neuropsychologic deficits; developmental disabilities; and deficits in motor skills, communication, and behavioral functioning.
Diagnostic Evaluation
For children 18 months of age and older, the HIV enzyme-linked immunosorbent assay (ELISA) and Western blot immunoassay are performed to determine HIV infection. In infants born to HIV-infected mothers, these assays will be positive because of the presence of maternal antibodies derived transplacentally. Maternal antibodies may persist in the infant up to 18 months of age. Therefore other diagnostic tests are employed, most commonly the HIV polymerase chain reaction (PCR) for detection of proviral DNA. With this technique, more than 95% of infected infants can be diagnosed by 1 to 4 months of age (Ezekowitz and Stockman, 2003; Goldschmidt and Fogler, 2006).
The Centers for Disease Control and Prevention (1994) has developed a classification system to describe the spectrum of HIV disease in children (Table 26-3). The system indicates the severity of clinical signs and symptoms and the degree of immunosuppression. Mild signs and symptoms include lymphadenopathy, parotitis, hepatosplenomegaly, and recurrent or persistent sinusitis or otitis media. Moderate signs and symptoms include lymphoid interstitial pneumonitis (LIP) and a variety of organ-specific dysfunctions or infections. Severe signs and symptoms include AIDS-defining illnesses with the exception of LIP. Children with LIP have a better prognosis than those with other AIDS-defining illnesses. In children whose HIV infection is not yet confirmed, the letter E (vertically exposed) is placed in front of the classification. The immune categories are based on CD4 + lymphocyte counts and percentages. Age adjustment of these numbers is necessary because normal counts, which are relatively high in infants, decline steadily until 6 years of age, when they reach adult norms (Table 26-4).
TABLE 26-3
Pediatric HIV Infection Classification*

*Children whose human immunodeficiency virus infection status is not confirmed are classified by using the above table with the letter E (for perinatally exposed) placed before the appropriate classification code (e.g., EN2).
†Both category C and lymphoid interstitial pneumonitis in category B are reportable to state and local health departments as acquired immunodeficiency syndrome.
From Centers for Disease Control and Prevention: 1994 Revised classification system for human immunodeficiency virus infection in children less than 13 years of age, MMWR Recomm Rep 43(RR-12):1-10, 1994.
TABLE 26-4
Immunologic Categories Based on Age-Specific CD4 + T-Lymphocyte Counts and Percent of Total Lymphocytes

From Centers for Disease Control and Prevention: 1994 Revised classification system for human immunodeficiency virus infection in children less than 13 years of age, MMWR Recomm Rep 43(RR-12):1-10, 1994.
Therapeutic Management
The goals of therapy for HIV infection include slowing the growth of the virus, preventing and treating opportunistic infections, and providing nutritional support and symptomatic treatment. Antiretroviral drugs work at various stages of the HIV life cycle to prevent reproduction of functional new virus particles. Although not a cure, these drugs can suppress viral replication, preventing further deterioration of the immune system, and thus delay disease progression. Classes of antiretroviral agents include nucleoside reverse transcriptase inhibitors (e.g., zidovudine, didanosine, stavudine, lamivudine, abacavir), nonnucleoside reverse transcriptase inhibitors (e.g., nevirapine, delavirdine, efavirenz), nucleotide reverse transcriptase inhibitors (e.g., adefovir), protease inhibitors (e.g., indinavir, saquinavir, ritonavir, nelfinavir, amprenavir), and adjunctive antiretrovirals (e.g., hydroxyurea). Combinations of these drugs are used to forestall the emergence of drug resistance. Antiretroviral therapy regimens and guidelines are continually evolving. Therapy is lifelong, making adherence difficult. Laboratory markers (CD4 + lymphocyte count, viral load) assist in monitoring both disease progression and response to therapy.
Pneumocystis carinii pneumonia (PCP) is the most common opportunistic infection of children infected with HIV. It occurs most frequently between 3 and 6 months of age. All infants born to HIV-infected women should receive prophylaxis during the first year of life, according to guidelines from the Centers for Disease Control and Prevention (1995) and the American Academy of Pediatrics (2000a). Trimethoprimsulfamethoxazole (TMP-SMZ) is the agent of choice. If adverse effects are experienced with TMP-SMZ, dapsone or pentamidine can be used.
Prophylaxis is often employed for other opportunistic infections, such as disseminated Mycobacterium avium-intracellulare complex (MAC), candidiasis, or herpes simplex. IVIG has been helpful in preventing recurrent or serious bacterial infections in some HIV-infected children.
Immunization against common childhood illnesses is recommended for all children exposed to and infected with HIV (American Academy of Pediatrics, 2000b). Varicella (chickenpox) vaccine and measles-mumps-rubella (MMR) vaccine can be administered if there is no evidence of severe immunocompromise. Because antibody production to vaccines may be poor or decrease over time, immediate prophylaxis after exposure to several vaccine-preventable diseases (e.g., measles, varicella) is warranted. It should be recognized that children receiving IV gamma globulin prophylaxis may not respond to the MMR vaccine (Centers for Disease Control and Prevention, 2003).
HIV infection often leads to marked failure to thrive and multiple nutritional deficiencies. Nutritional management may be difficult because of recurrent illness, diarrhea, and other physical problems. Intensive nutritional interventions should be instituted when the child’s growth begins to slow or weight begins to decrease.
Prognosis.: Early recognition and improved medical care have changed HIV disease from a rapidly fatal illness to a chronic disease. After the introduction of combination antiretroviral therapy, the numbers of new AIDS cases and deaths declined substantially. Between 1995 and 1998, the annual number of AIDS cases declined by 38% and deaths declined by 63% (Centers for Disease Control and Prevention, 2003; 2007). The annual number of AIDS cases has remained stable in children less than 13 years of age since 1998 (Klause and Johnson, 2007).
Nursing Care Management
Education concerning transmission and control of infectious diseases, including HIV infection, is essential for children with HIV infection and anyone involved in their care. The basic tenets of standard precautions should be presented in an age-appropriate manner, with careful consideration of the educational levels of the individuals (see Infection Control, Chapter 22). Safety issues, including appropriate storage of special medications and equipment (e.g., needles and syringes), are emphasized.
Unfortunately, relatives, friends, and others in the general public may be fearful of contracting HIV infection, and criticism and ostracism of the child and family may occur. In an effort to protect the child, the family may limit the child’s activities outside the home. Although certain precautions are justified in limiting exposure to sources of infections, they must be tempered with concern for the child’s normal developmental needs. Both the family and the community need ongoing education about HIV to dispel many of the myths that have been perpetuated by uninformed persons.*
Prevention is a key component of HIV education. Educating adolescents about HIV is essential in preventing HIV infection in this age-group. Education should include the routes of transmission, the hazards of IV and other recreational drug use, and the value of sexual abstinence and safe sex practices. Such education should be a part of anticipatory guidance provided to all adolescent patients. Nurses can also encourage adolescents at risk to undergo HIV counseling and testing. In addition to identifying infected teenagers and getting them into care, such counseling affords adolescents an opportunity to learn about, and possibly change, their risky behaviors.
The multiple complications associated with HIV disease are potentially painful (Ezekowitz and Stockman, 2003; Sullivan and Woda, 2003). Aggressive pain management is essential for these children to have an acceptable quality of life. Their pain may be due to infections (e.g., otitis media, dental abscess), encephalopathy (e.g., spasticity), adverse effects of medications (e.g., peripheral neuropathy), or an unknown source (e.g., deep musculoskeletal pain). Sources of pain are related not only to disease processes, but also to various treatments these children often undergo, including venipunctures, lumbar punctures, biopsies, and endoscopies. Ongoing assessment of pain is crucial and is most easily accomplished in older children who are able to communicate. Nonverbal and developmentally delayed children are more difficult to assess. The nurse should be alert for signs of pain such as emotional detachment, lack of interactive play, irritability, and depression. Effective pain management depends on the appropriate use of pharmacologic agents, including EMLA cream, acetaminophen, NSAIDs, muscle relaxants, and opioids. Tolerance to opioids may indicate increased dosing; monitored use ensures safety. Nonpharmacologic interventions (e.g., guided imagery, hypnosis, relaxation, and distraction techniques) are useful adjuncts.
 FAMILY FOCUS
FAMILY FOCUSCaregivers and the Infant with Human Immunodeficiency Virus Infection
Unlike other fatal pediatric diseases, human immunodeficiency virus (HIV) infection is associated with special family alterations. The infant infected in utero faces multiple physical and parental problems. Because the mother is infected, she may be ill or dying and therefore unable to care for the child. If possible, grandparents or other relatives may assume care. Foster care is often difficult to arrange because of the nature of the disease, especially in relation to the social stigma and the child’s multiple medical needs. When these children are hospitalized, the importance of consistent caregivers, especially primary nurses, who attend to the youngsters’ physical, developmental, and emotional needs, cannot be overemphasized. However, primary nurses may face the risk of overinvolvement and must be aware of the boundaries of a therapeutic relationship.
Common psychosocial concerns include disclosing the diagnosis to the child, making custody plans when the parent is infected, and anticipating the loss of a family member. Other stressors may include financial difficulties, HIV-associated stigma, efforts to keep the diagnosis secret, other infected family members, and the multiple losses associated with HIV. Most mothers of these children are single mothers who are also HIV infected. As primary caretakers, they often attend to the needs of their child first, neglecting their own health in the process (see Family Focus box). The nurse can encourage the mother to receive regular health care. Family members are often involved in the care of the child, particularly if the mother has symptomatic illness. After the mother’s death, a grandparent or other relative typically assumes responsibility for care of the child. The nurse can provide support and encouragement for the new surrogate parent, particularly during the transition phase. If no family member is available, the child may be placed in a foster or group home. Nursing is an integral part of the multidisciplinary team necessary for the successful management of the complex medical and social problems of these families.
The Child with Human Immunodeficiency Virus Infection
DIAGNOSIS (PROBLEM IDENTIFICATION)
After a thorough assessment, several nursing diagnoses are evident:
Risk for Infection related to impaired body defenses, presence of infective organisms
Altered Nutrition: Less Than Body Requirements related to recurrent illness, diarrheal losses, loss of appetite, oral candidiasis
Impaired Social Interaction related to physical limitations, hospitalizations, social stigma of infection with human immunodeficiency virus (HIV)
Chronic Pain related to disease process
Interrupted Family Processes related to having a child with a dreaded and life-threatening disease
Anticipatory Grieving related to having a child with a potentially fatal illness
Interrupted Family Processes related to a situational crisis
The effectiveness of nursing interventions for the child with HIV infection and his or her family is determined by continual assessment and evaluation of care based on the following guidelines:
Observe child and family for their ability to cope with the situation and comply with care.
Observe for and document child’s emotional and physical needs, especially assessment of adherence to medications.
Evaluate for signs of infection and disease progression.
Educate child and family on importance of disease prevention.
Interview and observe child and family for evidence of their understanding of the condition.
Children with HIV infection attend daycare centers and schools. It is well established that the risk of HIV transmission in these settings is minimal. These institutions are required to follow Centers for Disease Control and Prevention and Occupational Safety and Health Administration (OSHA) guidelines for infection control measures. Standard precautions describing proper management of blood and body fluids should also be followed. It is recommended that school personnel receive current HIV information and include it in the health education curriculum for kindergarten through twelfth grade (American Academy of Pediatrics, 2000a, 1999). School nurses play a vital role in educating the school staff, students, and parents. They are also invaluable in monitoring the needs of known affected children.
Confidentiality is a major issue in daycare or school attendance. Parents and legal guardians have the right to decide whether they inform these agencies of their child’s HIV diagnosis. Unfortunately, myths about HIV infection continue to exist, and the family often wishes to avoid any potential criticism or ostracism of the child.
SEVERE COMBINED IMMUNODEFICIENCY DISEASE
SCID is a defect characterized by absence of both humoral and cell-mediated immunity. The terms Swiss-type lymphopenic agammaglobulinemia (an autosomal recessive form of the disease) and X-linked lymphopenic agammaglobulinemia have been used to describe this disorder, which, as the names imply, can follow either mode of inheritance.
Susceptibility to infection occurs early in life, most often in the first month of life. The child suffers from chronic infection, fails to completely recover from an infection, is frequently reinfected, and is infected with unusual agents. Failure to thrive is a consequence of the persistent illnesses.
Diagnosis is usually based on a history of recurrent, severe infections from early infancy; a familial history of the disorder; and specific laboratory findings, which include lymphopenia, lack of lymphocyte response to antigens, and absence of plasma cells in the bone marrow. Documentation of immunoglobulin deficiency is difficult during infancy because of the normally delayed response of infants in producing their own immunoglobulins and material transfer of immunoglobulin G (IgG).
Therapeutic Management
The definitive treatment for SCID is HSCT from a histocompatible donor, a haplo-identical donor (usually a parent), or a matched unrelated donor. IVIG infusions and PCP prophylaxis are used to augment the humoral immunity until the transplant is performed. Several investigators are attempting gene therapy with some success, but there is a potential complication of insertional mutagenesis (Buckley, 2007).
Nursing Care Management
Nursing care focuses on preventing infection and supporting the child and family. The care is consistent with that needed for HSCT for any condition (see p. 945). Because the prognosis for SCID is very poor if a compatible bone marrow donor is not available, nursing care is directed at supporting the family in caring for a child with a life-threatening illness (see Chapter 18). Genetic counseling is essential because of the modes of transmission in either form of the disorder.
WISKOTT-ALDRICH SYNDROME
WAS is an X-linked recessive disorder characterized by a triad of abnormalities: (1) thrombocytopenia, (2) eczema, and (3) immunodeficiency of selective functions of B lymphocytes and T lymphocytes. A defective gene has been identified and designated the WAS protein (Bonilla and Geha, 2003; Fleisher, 2006). At birth the presenting symptoms may be bloody diarrhea as a result of thrombocytopenia. As the child grows older, recurrent infection and eczema become more severe, and the bleeding becomes less frequent.
Eczema is typical of the allergic type and easily becomes superinfected. Chronic infection with herpes simplex is a frequent problem and may lead to chronic keratitis of the eye with loss of vision. Chronic pulmonary disease, sinusitis, and otitis media result from repeated infections. In those children who survive the bleeding episodes and overwhelming infections, malignancy presents an additional risk to survival. Medical treatment involves:
Counteracting the bleeding tendencies with platelet transfusions
Using IV gamma globulin to provide passive immunity
Administering prophylactic antibiotics to prevent and control infection.
The only curative therapy is HSCT from a matched donor (Buckley, 2007).
Nursing Care Management
Because of the poor prognosis for these children, the main nursing consideration is supporting the family in the care of a fatally ill child (see Chapter 18). Physical care is directed at controlling the problems imposed by the disorder. The measures used to control bleeding are similar to those for hemophilia and vWD (see previous discussions). Another major goal is prevention or control of infection. Because eczema is a troublesome problem, nursing measures specific to this condition are especially important (see Chapter 30). The genetic implications of this X-linked recessive disorder differ little from those of any other X-linked disorder.
TECHNOLOGIC MANAGEMENT OF HEMATOLOGIC AND IMMUNOLOGIC DISORDERS
Technologic advances in blood banking and transfusion medicine enable the administration of only the blood component needed by the child, such as packed RBCs in anemia or platelets for bleeding disorders. However, regardless of the blood component infused, all transfusions have some risks. Therefore nurses need to be aware of the possible complications and the appropriate interventions. Table 26-5 summarizes the major hazards of transfusions, the signs and symptoms typically associated with each, and nursing responsibilities. General guidelines that apply to all transfusions include:
Take vital signs, including blood pressure, before administering blood to establish baseline data for intratransfusion and posttransfusion comparison, then every 15 minutes for 1 hour while blood is infusing and on completion of transfusion.
Check the identification of the recipient with the donor’s blood group and type, regardless of the blood product being used.
Administer the first 50 ml of blood or 20% of the volume (whichever is smaller) slowly and stay with the child.
Administer with normal saline on a piggyback setup or have normal saline available.
Administer blood through an appropriate filter to eliminate particles in the blood and prevent the precipitation of formed elements; gently shake the container frequently.
Use blood within 30 minutes of its arrival from the blood bank; if it is not used, return to the blood bank–do not store in the regular unit refrigerator.
Infuse a unit of blood (or the specified amount) within 4 hours. If the infusion will exceed this time, the blood should be divided into appropriately sized quantities by the blood bank, and the unused portion refrigerated under controlled conditions.
If a reaction of any type is suspected, take vital signs, stop the transfusion, maintain a patent IV line with normal saline and new tubing, notify the practitioner, and do not restart the transfusion until the child’s condition has been medically evaluated.
Although hemolytic reactions are rare, ABO incompatibility remains the most common cause of death from blood transfusion, and human error is usually responsible (administration of the wrong type to the patient or mislabeling of the blood product) (Norville and Bryant, 2002; Bell, 2007). Hemolysis can also cause the release of large quantities of phospholipids, which are capable of stimulating DIC (see p. 929). Acute kidney shutdown and eventual renal failure are a result of renal vasoconstriction from antigen-antibody complexes derived from the RBC surface.
Blood is usually administered to children by infusion pump; therefore the usual precautions and management related to pumps apply. When the blood is started with a standard transfusion set, the filter chamber is filled to allow the total filter to be used. The drip chamber is partially filled with blood to permit counting of the drops. In adjusting the flow rate, it is important to remember that blood administration sets do not use microdrops (60 drops/ml) but regular drops (usually 10 or 15 drops/ml).
HEMATOPOIETIC STEM CELL TRANSPLANTATION
HSCT is used to establish healthy hematopoiesis in both malignant and nonmalignant disease. Candidates for transplantation are children who have disorders that are unlikely to be cured by other means. Most HSCT patients undergo intensive ablative therapy using high-dose combination chemotherapy with or without total body irradiation (Bollard, Krance, and Heslop, 2006). After the immune system is suppressed to prevent rejection of the transplanted marrow, the stem cells harvested from the bone marrow, peripheral blood, or the umbilical vein of the placenta are given to the patient by IV transfusion. The newly transfused stem cells will begin to repopulate the ablative bone marrow. In essence, a new blood-forming organ will be accepted by the recipient.
The selection process for a suitable donor and the potential complications in transplantation are related to the HLA system complex. Some of the major HLA antigens are A, B, C, D, and DR. There is a wide diversity for each of these HLA loci. There are more than 20 different HLA-A antigens that can be inherited and more than 40 different HLA-B antigens.
The genes are inherited as a single unit or haplotype. A child inherits one unit from each parent; thus a child and each parent have one identical and one nonidentical haplotype. Because the possible haplotype combinations among siblings follow the laws of mendelian genetics, there is a one-in-four chance that two siblings have two identical haplotypes and are perfectly matched at the HLA loci.
The importance of HLA matching is to prevent the serious complication known as GVHD. Because the child’s immune system is essentially rendered nonfunctional, there is little difficulty with bone marrow rejection by the recipient. However, the donor’s marrow may contain antigens not matched to the recipient’s antigens, which begin attacking body cells. The more closely the HLA systems match, the less likely GVHD is to develop. However, it can occur even with a perfect HLA match, since there are as yet unidentified and thus unmatched histocompatibility antigens (Bollard, Krance, and Heslop, 2006).
Different types of HSCT are now performed in children with cancer. Allogeneic HSCT involves matching a histocompatible donor with the recipient. However, allogeneic HSCT is limited by the presence of a suitable marrow donor.
Because of the limited numbers of patients having HLA-identical siblings, other types of allogeneic transplants have evolved. Umbilical cord blood stem cell transplantation is an established, rich source of hematopoietic stem cells for use in children with cancer. Because stem cells can be found with high frequency in the circulation of newborns, cord blood transplantation has become an alternative for some children. The benefit of using umbilical cord blood is the blood’s relative immunodeficiency at birth, allowing for partially matched unrelated cord blood transplants to be successful, with a lower risk of GVHD-related problems (Bollard, Krance, and Heslop, 2006; Ryan, Kristovich, Haugen, and others, 2002).
Autologous HSCTs use the patient’s own marrow that was collected from disease-free tissue, frozen, and sometimes treated to remove malignant cells. Children with solid tumors such as neuroblastoma, Hodgkin disease, NHL, rhabdomyosarcoma, Ewing sarcoma, and Wilms tumor have been treated with autologous HSCTs.
Peripheral stem cell transplants (PSCTs) are also used in children with cancer. PSCT, a type of autologous transplant, differs in the way stem cells are collected from the patient. CSF is first given to stimulate the production of many stem cells (Ryan, Kristovich, Haugen, and others, 2002). After the WBC count is high enough, the stem cells are collected by an apheresis machine. This machine filters out peripheral stem cells from whole blood, returning the remainder of the blood cells and plasma to the child. Stem cells have been collected in very small children without problems (Lipton, 2003). The peripheral stem cells are then frozen until the patient is ready for the PSCT.
Nursing Care Management
The care of children undergoing HSCT is similar to that of any child receiving chemotherapy and radiotherapy. The hospitalization is typically 3 to 6 weeks in an isolated environment, during which time the child is subjected to numerous procedures and side effects of therapy. Throughout this long ordeal the family is concerned with successful engraftment and fear of fatal complications (see Family Focus box). Consequently, nurses involved with the child and family need to provide sensitive care and maintain a supportive attitude during the many crises that may arise. If the procedure is not successful, the families need care consistent with that required by the family of any child with a life-threatening disorder (see Chapter 18).
APHERESIS
Apheresis is the removal of blood from an individual, separation of the blood into its components, retention of one or more of these components, and reinfusion of the remainder of the blood into the individual. Apheresis is most often used to remove large quantities of platelets from healthy adult donors. These transfusion products have greatly prolonged the survival of patients with hematologic and oncologic diseases.
The Decision for a Hematopoietic Stem Cell Transplant
A family’s decision for a child to undergo hematopoietic stem cell transplantation (HSCT) may be fraught with challenges. Often the child is facing certain death from the malignancy. The preparation of the child for the transplant also places the patient at great medical risk.
Once the preparatory regimen is begun and the child’s immune system is destroyed, there is no turning back. Unlike kidney transplantation, HSCT does not have a “rescue” procedure, such as dialysis, for supportive therapy. If the donor is a sibling, the issue of his or her marrow “saving” the brother or sister can be a concern, especially if the transplant fails. Parents often must leave the home to stay at the transplant center and encounter additional stressors such as arranging child care, taking a leave from work, and managing finances. The patient faces the greatest stress–fear of HSCT failure or life-threatening complications.
This technique is used to remove peripheral blood stem cells (PBSCs) from children before they receive HSCT or high-dose chemotherapy or radiotherapy, which is severely toxic to the bone marrow. These PBSCs can then be used to restore the child’s bone marrow. Apheresis is also used as a therapeutic modality. The blood component that is diseased or toxic is separated from the blood, and the remainder is returned to the individual. Therapeutic apheresis is considered part of standard therapy for many diseases. Plasma is selectively removed from individuals with hyperviscosity, life-threatening complications of myasthenia gravis, Guillain-Barré syndrome, thrombotic thrombocytopenic purpura, and certain drug overdoses. WBCs are removed from individuals with high-WBC-count leukemia.
Nursing Care Management
Difficult venous access and small blood volume can limit the ability to use this therapy in the infant and young child. Education of the family and child includes the purposes of the therapy and the technology.
Specially trained individuals perform the apheresis procedure. Attention focuses on rate of removal, blood component separation, and reinfusion of blood into the child. Vital signs are monitored, and the child is continuously observed for any adverse reactions secondary to the circulatory volume changes and the anticoagulant used.
When apheresis components are infused, nursing measures differ depending on whether the product is autologous (blood component from the child) or allogeneic (blood component from another individual). Autologous components are the child’s own blood; therefore a major precaution is proper identification to ensure the correct component. The rate of infusion should be adjusted to the child’s tolerance. If the product is allogeneic, all precautions for blood transfusions apply.
Anemia is defined as reduction of RBCs or Hgb concentration to levels below normal for age; disorders are classified either by etiology and physiology or by morphology.
The nurse’s role in treatment of anemia is to assist in establishing a diagnosis, prepare the child for laboratory tests, administer prescribed medications, decrease tissue oxygen needs, implement safety precautions, and observe for complications.
The main nursing goal in prevention of nutritional anemia is parent education regarding correct feeding practices.
Sickle cell anemia is a hereditary hemoglobinopathy caused by normal adult Hgb (HbA) being partly or completely replaced by sickle Hgb (HbS).
Nursing care of the child with sickle cell anemia focuses on teaching the family how to prevent and recognize sickle cell problems; managing pain during crises; and helping the child and parents adjust to a lifelong, chronic disease.
Nursing care of the child with β-thalassemia includes observing for complications of multiple blood transfusions, assisting the child in coping with the effects of illness, and fostering parent-child adjustment to long-term illness.
Causes of acquired AA include irradiation, drugs, industrial and household chemicals, infections, and infiltration and replacement of myeloid elements; however, the majority cases are idiopathic.
Clotting depends on three processes: vascular spasm, platelet aggregation, and coagulation and clot formation.
Nursing care of the child with hemophilia involves preventing bleeding by decreasing the risk of injury, recognizing and managing bleeding with factor replacement, preventing the crippling effects of joint degeneration, and preparing and supporting the child and family for home care.
Goals in the care of the child with leukemia are to prepare the family for diagnostic and therapeutic procedures, prevent complications of myelosuppression, manage problems of irradiation and drug toxicity, and provide continued emotional support.
The lymphomas include Hodgkin lymphoma and NHL and are disorders involving the lymphoid system.
Immunodeficiency disorders render the affected individual unable to fight infectious organisms.
HIV infection is primarily acquired in infants during pregnancy or birth from an infected mother and in adolescents from engaging in high-risk behaviors.
Blood transfusions supply needed blood components.
HSCT replaces the diseased or malfunctioning bone marrow with viable blood stem cells.
Apheresis is the selective removal of a blood component. It can be used to supply cellular elements needed for therapy (i.e., platelets or stem cells) or to remove diseased components.
REFERENCES
American Academy of Pediatrics, Committee on Infectious Diseases, Pickering L, eds. Red book: 2006 report of the Committee on Infectious Diseases, ed 27, Elk Grove Village, Ill: The Academy, 2006.
American Academy of Pediatrics. Breastfeeding and the use of human milk. Pediatrics. 2005;115(2):496–506.
American Academy of Pediatrics, Section on Hematology/Oncology, Committee on Genetics. Health supervision for children with sickle cell disease. Pediatrics. 2002;109(3):526–536.
American Academy of Pediatrics, Committee on Pediatric AIDS. Identification and care of HIV-exposed and HIV-infected infants, children, and adolescents in foster care. Pediatrics. 2000;106(1):149–153.
American Academy of Pediatrics, Committee on Pediatric AIDS. Technical report: perinatal human immunodeficiency virus testing and prevention of transmission. Pediatrics. 2000;106(6):1–12.
American Academy of Pediatrics, Committee on Pediatric AIDS, Committee on Infectious Diseases. Issues related to human immuno-deficiency virus transmission in schools, child care, medical settings, the home, and community. Pediatrics. 1999;104(2):318–324.
American Pain Society. Guidelines for the management of acute and chronic pain in sickle-cell disease. Glenview, Ill: The Society, 1999.
Anderson, N. Hydroxyurea therapy: improving the lives of patients with sickle cell disease. Pediatr Nurs. 2006;32(6):541–543.
Andrews, NC. Disorders of iron metabolism and sideroblastic anemia. In Nathan D, Orkin SH, Ginsburg D, et al, eds.: Nathan and Oski’s hematology of infancy and childhood, ed 6, Philadelphia: Saunders, 2003.
Bell, MD. Red blood cell transfusions. Pediatr Rev. 2007;28(8):299–304.
Berde, CB, Billett, AM, Collins, JJ. Symptom management in supportive care. In Pizzo PA, Poplack DG, eds.: Principles and practice of pediatric oncology, ed 5, Philadelphia: Lippincott, 2006.
Bhatia, S. Epidemiology. In: Wallace WHB, Green DM, eds. Late effects of childhood cancer. London: Arnold, 2004.
Bogen, DL, Krause, JP, Serwint, JR. Outcome of children identified as anemic by routine screening in an inner-city clinic. Arch Pediatr Adolesc Med. 2001;155:366–371.
Bollard, CM, Krance, RA, Heslop, HE. Hematopoietic stem cell transplantation in pediatric oncology. In Pizzo PA, Poplack DG, eds.: Principles and practice of pediatric oncology, ed 5, Philadelphia: Lippincott, 2006.
Bonilla, FA, Geha, RS. Primary immunodeficiency diseases. In Nathan D, Orkin SH, Ginsburg D, et al, eds.: Nathan and Oski’s hematology of infancy and childhood, ed 6, Philadelphia: Saunders, 2003.
Buchanan, GR. Thrombocytopenia during childhood: what the pediatrician need to know. Pediatr Rev. 2005;26(11):401–409.
Buckley, RH. Evaluation of the immune system. In Behrman RE, Kliegman RM, Jenson HTS, et al, eds.: Nelson textbook of pediatrics, ed 18, Philadelphia: Saunders, 2007.
Bulas, D. Screening children for sickle cell vasculopathy: guidelines for transcranial Doppler evaluation. Pediatr Radiol. 2005;35:235–241.
Burden, MJ, Westerlund, AJ, Sivan-Armony, R, et al. An event-related potential study of attention and recognition memory in infants with iron-deficiency anemia. Pediatrics. 2007;120(2):336–345.
Carley, A. Anemia: when is it iron deficiency? Pediatr Nurs. 2003;29(2):127–133.
Centers for Disease Control and Prevention revised edition HIV/AIDS surveillance report, 2005. Atlanta: The Centers; 2007;Vol 17:1–54. http://www.cdc.gov/hiv/topics/surveillance/resources/reports [available online at].
Centers for Disease Control and Prevention. Revised recommendations for HIV testing of adults, adolescents and pregnant women in health-care settings. MMWR. 2006;55(RR-14):1–13.
Centers for Disease Control and Prevention. Advancing HIV prevention: new strategies for a changing epidemic—United States. MMWR. 2003;52(15):329–332.
Centers for Disease Control and Prevention. 1995 revised guidelines for prophylaxis against Pneumocystis carinii pneumonia for children infected with or perinatally exposed to human immunodeficiency virus. MMWR. 1995;44(RR-4):1–11.
Centers for Disease Control and Prevention. 1994 revised classified system for human immunodeficiency virus infection in children less than 13 years of age. MMWR. 1994;43(RR-12):1–10.
Chandran, L, Gelfer, P. Breastfeeding: the essential principles. Pediatr Rev. 2006;27(11):409–417.
Chiocca, EM. Sickle cell crisis: severe pain and potential tissue necrosis are the major concerns. Am J Nurs. 1996;96(9):49.
Cho, S, Cheng, AC, Cheng, MCK. Oral care for children with leukemia. Hong Kong Med J. 2000;6(2):203–208.
Cibulka, NJ. Mother-to-child transmission of HIV in the United States. AJN. 2006;106(7):56–63.
Curry, H. Bleeding disorder basics. Pediatr Nurs. 2004;30(5):402–405.
Dover, GJ, Platt, OS. Sickle cell disease. In Nathan D, Orkin SH, Ginsburg D, et al, eds.: Nathan and Oski’s hematology of infancy and childhood, ed 6, Philadelphia: Saunders, 2003.
Driscoll, MC. Sickle cell disease. Pediatr Rev. 2007;28(7):259–267.
Ezekowitz, RAB, Stockman, JA, III. Hematologic manifestations of systemic diseases. In Nathan D, Orkin SH, Ginsburg D, et al, eds.: Nathan and Oski’s hematology of infancy and childhood, ed 6, Philadelphia: Saunders, 2003.
Fleisher, TA. Primary immune deficiencies: windows into the immune system. Pediatr Rev. 2006;27(10):363–373.
Glader, B. Anemias of inadequate production. In Behrman RE, Kliegman RM, Jenson HTS, et al, eds.: Nelson textbook of pediatrics, ed 18, Philadelphia: Saunders, 2007.
Goldschmidt, RH, Fogler, JA. Opportunities to prevent HIV transmission to newborns. Pediatrics. 2006;117(1):208–209.
Hord, JD. The acquired pancytopenia. In Behrman RE, Kliegman RM, Jenson HTS, et al, eds.: Nelson textbook of pediatrics, ed 18, Philadelphia: Saunders, 2007.
Hudson, MM, Onciu, M, Donaldson, SS. Hodgkin’s disease. In Pizzo PA, Poplack DG, eds.: Principles and practice of pediatric oncology, ed 4, Philadelphia: Lippincott, 2006.
Karayalcin, G. Hemolytic anemia (sickle cell anemia). In Lanzkowsky P, ed.: Manual of pediatric hematology and oncology, ed 3, San Diego: Academic Press, 2000.
Khoury, H, Grimsley, E. Oxygen inhalation in nonhypoxic sickle cell patients during vaso-occlusive crisis. Blood. 1995;86(10):3998.
Klause, BD, Johnson, M. Paradigm shift: new testing guidelines for HIV. Adv Nurse Pract. 2007;15(3):59–93.
Link, MP, Donaldson, SS. The lymphomas and lymphadenopathy. In Nathan D, Orkin SH, Ginsburg D, et al, eds.: Nathan and Oski’s hematology of infancy and childhood, ed 6, Philadelphia: Saunders, 2003.
Link, MP, Weinstein, HJ. Malignant non-Hodgkin lymphomas in children. In Pizzo PA, Poplack DG, eds.: Principles and practice of pediatric oncology, ed 5, Philadelphia: Lippincott, 2006.
Lipton, JM. Peripheral blood as a stem cell source for hematopoietic cell transplantation in children: is the effort in vein? Pediatr Transplant. 2003;7(Suppl 3):65–70.
Margolin, JF, Steuber, CP, Poplack, DG. Acute lymphoblastic leukemia. In Pizzo PA, Poplack DG, eds.: Principles and practice of pediatric oncology, ed 4, Philadelphia: Lippincott, 2006.
Marsh, JCW. Management of acquired aplastic anemia. Blood Rev. 2005;19:143–151.
McKenzie, SB. Anemias of disordered iron metabolism and heme synthesis. In: McKenzie SB, ed. Clinical laboratory hematology. Upper Saddle River, NJ: Pearson Prentice Hall, 2004.
Miller, M, Zimmerman, S, Schultz, W, et al. Hydroxyurea therapy for pediatric patients with hemoglobin SC disease. J Pediatr Hematol Oncol. 2001;23(5):306–308.
Montgomery, RR, Gill, JC, Scott, JP. Hemophilia and von Willebrand disease. In Nathan D, Orkin SH, Ginsburg D, et al, eds.: Nathan and Oski’s hematology of infancy and childhood, ed 6, Philadelphia: Saunders, 2003.
Morris, CR, Singer, ST, Walters, MC. Clinical hemoglobinopathies: iron, lungs and new blood. Curr Opin Hematol. 2006;13:407–418.
National Hemophilia Foundation, Bleeding Disorders Information Center. Newly diagnosed: parents FAQ 2006. http://www.hemophilia.org/bdi/bdi_newly7c.htm. [retrieved March 25, 2008, from].
National Institutes of Health, National Heart, Lung, and Blood Institute, Division of Blood Disease and Resources. The management of sickle cell disease. Bethesda, Md: NHLBI Health Information Network, 2002. [NIH Pub No 02-2117].
Norville, R, Bryant, R. Blood component deficiencies. In Baggott CR, Kelly KP, Fochtman D, et al, eds.: APON nursing care of children and adolescents with cancer, ed 3, Philadelphia: Saunders, 2002.
Ohls, R, Christensen, RD. Hemoglobin disorders. In Behrman RE, Kliegman RM, Jenson HTS, et al, eds.: Nelson textbook of pediatrics, ed 18, Philadelphia: Saunders, 2007.
Okpala, I. New therapies for sickle cell disease. Hematol Oncol Clin North Am. 2005;19:975–987.
Orkin, SH, Nathan, DG. The thalassemias. In Nathan D, Orkin SH, Ginsburg D, et al, eds.: Nathan and Oski’s hematology of infancy and childhood, ed 6, Philadelphia: Saunders, 2003.
Paley, C. Hemolytic anemia (thalassemias). In Lanzkowsky P, ed.: Manual of pediatric hematology and oncology, ed 3, San Diego: Academic Press, 2000.
Pearce, JM, Sills, RH. Childhood leukemia. Pediatr Rev. 2005;26(3):96–104.
Perinatal HIV Guidelines Working Group, Public Health Service Task Force. Recommendations for use of antiretroviral drugs in pregnant HIV-infected women for maternal health and interventions to reduce perinatal HIV transmission in the United States. http://www.aidsinfo.nih.gov/ContentFiles/PerinatalGL.pdf, 2007. [retrieved March 24, 2008, from].
Perkins, S. Disorders of hematopoiesis. In Collins RD, Swerdlow SH, eds.: Pediatric hematopathology, ed 1, Philadelphia: Churchill Livingstone, 2001.
Platt, OS, Brambilla, DJ, Roose, WF, et al. Mortality in sickle cell disease: life expectancy and risk factors for early death. N Engl J Med. 1994;330:1639–1644.
Pui, CH, Relling, MV, Downing, JR. Acute lymphoblastic leukemia. N Engl J Med. 2004;350:1535–1548.
Redding-Lallinger, R, Knoll, C. Sickle cell disease—pathophysiology and treatment. Curr Probl Pediatr Adolesc Health Care. 2006;36(10):346–376.
Richardson, M. Microcytic anemia. Pediatr Rev. 2007;28(1):5–13.
Ryan, LG, Kristovich, KM, Haugen, MS, et al. Hematopoietic stem cell transplantation. In Baggott CR, Kelly KP, Fochtman D, et al, eds.: Nursing care of children and adolescents with cancer, ed 3, Philadelphia: Saunders, 2002.
Scott, JP, Montgomery, RR. Platelet and blood vessel disorder. In Behrman RE, Kliegman RM, Jenson HTS, et al, eds.: Nelson textbook of pediatrics, ed 18, Philadelphia: Saunders, 2007.
Segel, GB, Hirsch, MG, Feig, SA. Managing anemia in a pediatric office practice, part I. Pediatr Rev. 2002;23(3):75–83.
Shende, A. Bone marrow failure. In Lanzkowsky P, ed.: Manual of pediatric hematology and oncology, ed 3, San Diego: Academic Press, 2000.
Shimamura, A, Guinan, EC. Acquired aplastic anemia. In Nathan D, Orkin SH, Ginsburg D, et al, eds.: Nathan and Oski’s hematology of infancy and childhood, ed 6, Philadelphia: Saunders, 2003.
Silverman, LB, Sallan, SE. Acute lymphoblastic leukemia. In Nathan D, Orkin SH, Ginsburg D, et al, eds.: Nathan and Oski’s hematology of infancy and childhood, ed 6, Philadelphia: Saunders, 2003.
Steinberg, MH, Barton, F, Castro, O, et al. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment. JAMA. 2003;289(13):1645–1651.
Sullivan, JL, Woda, BA. Lymphohistiocytic disorders. In Nathan D, Orkin SH, Ginsburg D, et al, eds.: Nathan and Oski’s hematology of infancy and childhood, ed 6, Philadelphia: Saunders, 2003.
Walsh, TJ, Roilides, E, Groll, AH. Infectious complications in pediatric cancer patients. In Pizzo PA, Poplack DG, eds.: Principles and practice of pediatric oncology, ed 4, Philadelphia: Lippincott, 2006.
Yoon, SL, Black, S. Comprehensive, integrative management of pain for patients with sickle-cell disease. J Altern Complement Med. 2006;12(10):995–1001.
Zimmerman, S, Schultz, W, Davis, J, et al. Sustained long-term hematologic efficacy of hydroxyurea at maximum tolerated dose in children with sickle cell disease. Blood. 2004;103(6):2039–2045.
*Sickle Cell Disease Association of America, Inc., 231 E. Baltimore St., Suite 800, Baltimore, MD 21202; (410) 528-1555, (800) 421-8453; fax: (410) 528-1495; e-mail: scdaa@sicklecelldisease.org; http://www.sicklecelldisease.org; Sickle Cell Information Center, PO Box 109, Grady Memorial Hospital, 80 Jesse Hill Jr Drive SE, Atlanta, GA 30303; (404) 616-3572; fax: (404) 616-5998; e-mail: aplatt@emory.edu; http://www.scinfo.org; National Heart, Lung, and Blood Institute, PO Box 30105, Bethesda, MD 20824-0105; (301) 592-8573, fax: (240) 629-3246; http://www.nhlbi.nih.gov. Sickle Cell Disease in Newborns and Infants: A Guide for Parents, Pub No AHCPR 93-0564. Available from the AHCPR Publications Clearinghouse, PO Box 8547, Silver Spring, MD 20907-8547; (800) 358-9295; http://www.ahcpr.gov. Guideline for the Management of Acute and Chronic Pain in Sickle-Cell Disease is available from the American Pain Society, 4700 W. Lake Ave., Glenview, IL 60025-1485; (847) 375-4715; fax: (877) 734-8758; e-mail: info@ampainsoc.org; http://www.ampainsoc.org.
*330 Seventh Ave., No. 900, New York, NY 10001; (800) 522-7222; fax: (212) 279-5999; http://www.cooleysanemia.org.
†747 52nd St., Oakland, CA 94609; (510) 428-3885, ext. 4398; http://www.thalassemia.com.
*PO Box 310, Churchton, MD 20733 USA; (800) 747-2820, (410) 867-0242; fax: (410) 867-0240; e-mail: help@aamds.org; http://www.aamds.org.
*116 W. 32nd St., 11th Floor, New York, NY 10001; (800) 42-HANDI, (212) 328-3700; fax: (212) 328-3777; e-mail: handi@hemophilia.org; http://www.hemophilia.org.
†625 President Kennedy Ave., Suite 505, Montreal, Quebec H3A 1K2; (800) 668-2686, (514) 848-0503; fax: (514) 848-9661; e-mail: chs@hemophilia.ca; http://www.hemophilia.ca.
*Cancer Chemotherapy Guidelines can be obtained from the Oncology Nursing Society, 125 Enterprise Drive, Pittsburgh, PA 15275; (866) 257-4ONS, (412) 859-6100; fax: (877) 369-5497; e-mail: customer.service@ons.org; http://www.ons.org.
†Detailed chemotherapeutic agents are outlined in Wilson D, Hockenberry MJ: Wong’s clinical manual of pediatric nursing, ed 7, St Louis, 2008, Mosby, pp 301-307.
*Additional information is available from the AIDS Hotline: (800) 342-2437 (342-AIDS).