The Tricarboxylic Acid Cycle
Learning objectives
After reading this chapter you should be able to:
 Outline the sequence of reactions in the tricarboxylic acid (TCA) cycle and explain the purpose of the cycle.
Outline the sequence of reactions in the tricarboxylic acid (TCA) cycle and explain the purpose of the cycle.
Identify the four oxidative enzymes in the TCA cycle and their products.
Identify the two intermediates required in the first step of the TCA cycle and their metabolic sources.
Identify four important metabolic intermediates synthesized from TCA cycle intermediates.
Describe how the TCA cycle is regulated by substrate supply, allosteric effectors, covalent modification, and protein synthesis.
Explain why there is no net synthesis of glucose from acetyl-CoA.
Explain the concept of ‘suicide substrate’ as applied to the TCA cycle.
Introduction
Located in the mitochondrion, the tricarboxylic acid (TCA) cycle, also known as the Krebs or citric acid cycle, is a shared pathway for metabolism of all fuels. It oxidatively strips electrons from acetyl-CoA, which is the common product of catabolism of fat, carbohydrate and proteins, producing the majority of the reduced coenzymes that are used for the generation of adenosine triphosphate (ATP) in the electron transport chain. Although the TCA cycle does not use oxygen in any of its reactions, it requires oxidative metabolism in the mitochondrion for reoxidation of reduced coenzymes. The TCA cycle has two major functions: energy production and biosynthesis (Fig. 14.1).
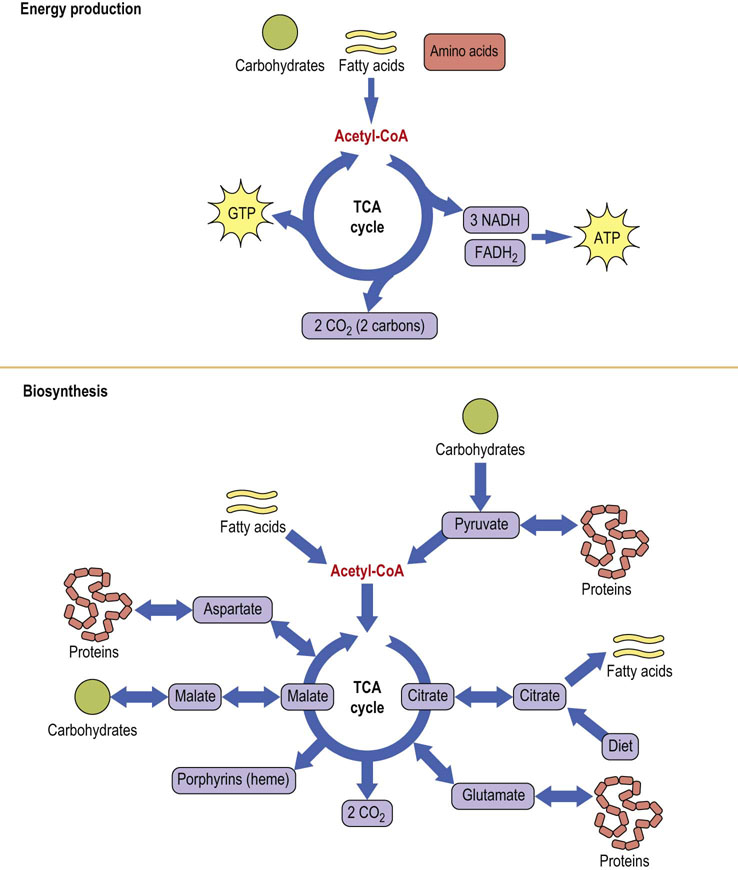
Fig. 14.1 Amphibolic nature of the TCA cycle.
The TCA cycle provides energy and metabolites for cellular metabolism. Because of the catabolic (top) and anabolic (bottom) nature of the TCA cycle, it is described as amphibolic. Acetyl-CoA is the common intermediate between metabolic fuels and the TCA cycle. FADH2, reduced flavin adenine dinucleotide; GDP, guanosine diphosphate; NADH, reduced nicotinamide adenine dinucleotide.
Functions of the tricarboxylic acid cycle
Four oxidative steps provide free energy for ATP synthesis
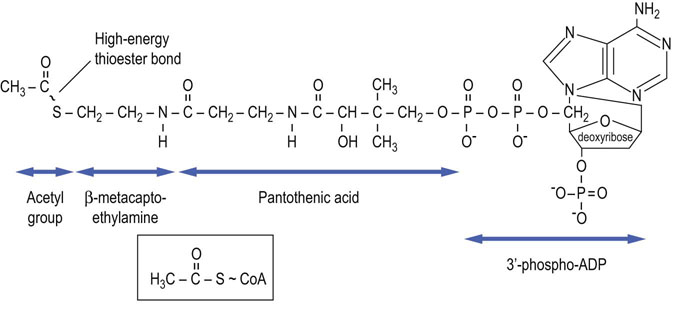
A common end product of carbohydrate, fatty acid and amino acid metabolism, acetyl-CoA (Fig. 14.2) is oxidized in the TCA cycle to produce reduced coenzymes by four redox reactions per turn of the cycle. Three produce reduced nicotinamide adenine dinucleotide (NADH) and another produces reduced flavin adenine dinucleotide (FADH2) (Fig. 9.4). These reduced nucleotides provide energy for ATP synthesis by the electron transport system (see Chapter 9). One high-energy phosphate, guanosine triphosphate (GTP), is also produced in the cycle by substrate-level phosphorylation. Nearly all metabolic carbon dioxide is produced by decarboxylation reactions catalyzed by pyruvate dehydrogenase and TCA cycle enzymes in the mitochondrion.
The TCA cycle provides a common ground for interconversion of fuels and metabolites
In addition to its role in catabolism, the TCA cycle (see Fig. 14.1) participates in the synthesis of glucose from amino acids and lactate during starvation and fasting (gluconeogenesis; Chapter 13). It is also involved in the conversion of carbohydrates to fat following a carbohydrate-rich meal (Chapter 16). It is a source of nonessential amino acids, such as aspartate and glutamate, which are synthesized directly from TCA cycle intermediates. One TCA cycle intermediate, succinyl coenzyme A (succinyl-CoA), serves as a precursor to porphyrins (heme) in all cells, but especially in bone marrow and liver (Chapter 30). Biosynthetic reactions proceeding from the TCA cycle require the input of carbons from intermediates other than acetyl-CoA. Such reactions are known as anaplerotic (building up) reactions.
Acetyl-CoA is a common product of many catabolic pathways
The TCA cycle begins with acetyl-CoA (Fig. 14.2), which has three major metabolic precursors. Carbohydrates undergo glycolysis to yield pyruvate (Chapter 12), which can be taken up by mitochondria and oxidatively decarboxylated to acetyl-CoA by the pyruvate dehydrogenase complex. During lipolysis, triacylglycerols are converted to glycerol and free fatty acids, which are taken up by cells and transported into mitochondria, where they undergo oxidation to acetyl-CoA (Chapter 15). Lastly, proteolysis of tissue proteins releases constituent amino acids, many of which are metabolized to acetyl-CoA and TCA cycle intermediates (Chapter 19).
The first version of the TCA cycle, proposed by Krebs in 1937, began with pyruvic acid, not acetyl-CoA. Pyruvic acid was decarboxylated and condensed with oxaloacetic acid through an unknown mechanism to form citric acid. The key intermediate, acetyl-CoA, was not identified until years later. It is tempting to begin the TCA cycle with pyruvic acid, unless it is recognized that fatty acids and many amino acids form acetyl-CoA by pathways that bypass pyruvate. It is for this reason that the TCA cycle is said to begin with acetyl-CoA, not pyruvic acid.
The TCA cycle is located in the mitochondria l matrix
Localization of the TCA cycle in the mitochondrial matrix is important metabolically; this allows identical intermediates to be used for different purposes inside and outside mitochondria. Acetyl-CoA, for example, cannot cross the inner mitochondrial membrane. The main fate of mitochondrial acetyl-CoA is oxidation in the TCA cycle but, in the cytoplasm, it is used for biosynthesis of fatty acids and cholesterol.
Metabolic defects in the TCA cycle are rare
Metabolic defects involving enzymes of the TCA cycle are rare, because functioning of the cycle is absolutely essential to sustain life. Products of energy-producing pathways must be metabolized in the TCA cycle for efficient production of ATP. Any defect in the TCA cycle will limit ATP production, and cells deprived of ATP either die rapidly or are severely impaired functionally. Tissues that use oxygen at rapid rates, such as the central nervous system and muscle, are most susceptible to such defects.
Pyruvate carboxylase
Pyruvate may be directly converted to four different metabolites
Pyruvate is at a crossroads in metabolism. It may be converted in one step to lactate (lactate dehydrogenase), to alanine (alanine aminotransferase, ALT), to oxaloacetate (pyruvate carboxylase), and to acetyl-CoA (pyruvate dehydrogenase complex) (Fig. 14.3). Depending on metabolic circumstances, pyruvate may be routed toward gluconeogenesis (Chapter 13), fatty acid biosynthesis (Chapter 16) or the TCA cycle itself. Pyruvate carboxylase, like most other carboxylases, uses CO2 and the coenzyme biotin (Fig. 14.4), a water-soluble vitamin, and ATP to drive the carboxylation reaction. The enzyme is a tetramer of identical subunits, each of which contains an allosteric site that binds acetyl-CoA, a positive heterotropic modifier. In fact, pyruvate carboxylase has an absolute requirement for acetyl-CoA; the enzyme does not work in its absence. An abundance of mitochondrial acetyl-CoA acts as a signal for the generation of additional oxaloacetate. For example, when lipolysis is stimulated, intramitochondrial acetyl-CoA levels rise, allosterically activating pyruvate carboxylase to produce additional oxaloacetate for gluconeogenesis (Chapter 13).
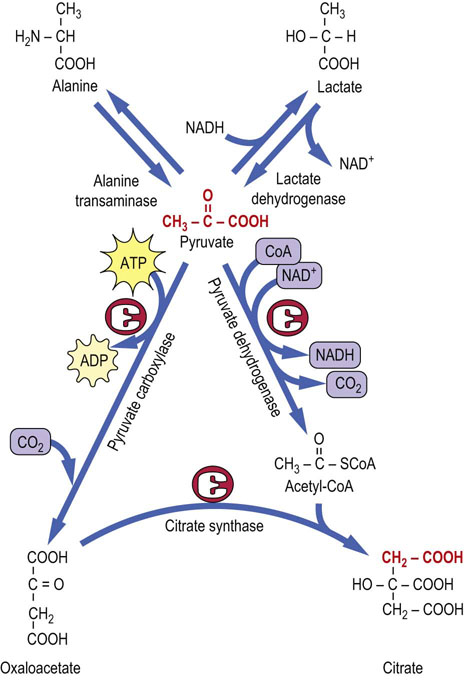
Fig. 14.3 Pyruvate is at the crossroads of metabolism.
Pyruvate is readily formed from lactate or alanine. Acetyl-CoA and oxaloacetate are derived from pyruvate through the catalytic action of pyruvate dehydrogenase and pyruvate carboxylase, respectively. ADP, adenosine diphosphate.
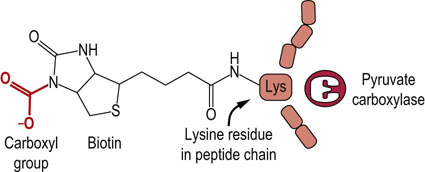
Fig. 14.4 The carboxy-biotin intermediate.
Pyruvate carboxylase catalyzes carboxylation of pyruvate to oxaloacetate. The coenzyme, biotin, is covalently bound to pyruvate carboxylase, and transfers the carbon originating from CO2 to pyruvate (see Chapter 11).
 Clinical test box Measuring lactate
Clinical test box Measuring lactate
Lactic acid is measured in a clinical setting, because its accumulation can result in rapid death. Lactic acid is produced metabolically by the reversible reduction of pyruvate with NADH by the enzyme lactate dehydrogenase (LDH). Both lactate and pyruvate coexist in metabolic systems, and the ratio of pyruvate : lactate is roughly proportional to the cytosolic ratio of NAD+/NADH. Both lactate and pyruvate contribute to the acidity of biological fluid; however, lactate is usually present at higher concentrations and is more easily measured. Blood lactate may increase in chronic obstructive lung disease and during intense exercise. Its measurement is usually indicated when there is metabolic acidosis, characterized by an elevated anion gap, [Na+] – ([Cl−] + [HCO3−]), indicating the presence of an unknown anion(s) in plasma. Although rare, lactic acidosis can be caused by metabolic defects in energy-producing pathways, such as some of the glycogen storage diseases or in any enzyme in the pathways from pyruvate to the generation of ATP, including the pyruvate dehydrogenase complex, TCA cycle, electron transport system or ATP synthase.
The pyruvate dehydrogenase complex
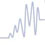
The pyruvate dehydrogenase complex (PDC) serves as a bridge between carbohydrates and the TCA cycle (Fig. 14.5). PDC is one of several α-ketoacid dehydrogenases having analogous reaction mechanisms, including α-ketoglutarate dehydrogenase in the TCA cycle and α-ketoacid dehydrogenases associated with the catabolism of leucine, isoleucine and valine. Its irreversibility explains in part why acetyl-CoA cannot yield a net synthesis of glucose (below). The complex functions as a unit consisting of three principal enzymes:
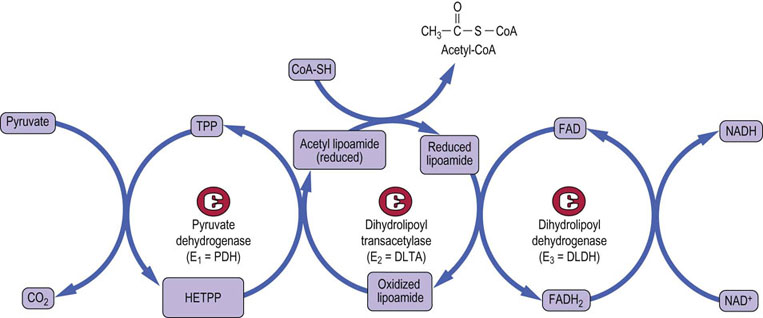
Fig. 14.5 Mechanism of action of the pyruvate dehydrogenase complex.
The three enzyme components of the pyruvate dehydrogenase complex are pyruvate dehydrogenase (E1 = PDH), dihydrolipoyl transacetylase (E2 = DLTA) and dihydrolipoyl dehydrogenase (E3 = DLDH). Pyruvate is first decarboxylated by the thiamine pyrophosphate-containing enzyme (E1), forming CO2 and hydroxyethyl-thiamine pyrophosphate (HETPP). Lipoamide, the prosthetic group on E2, serves as a carrier in the transfer of the 2-carbon unit from HETPP to coenzyme A (CoA). The oxidized, cyclic disulfide form of lipoamide accepts the hydroxyethyl group from HETPP. The lipoamide is reduced and the hydroxyethyl group converted to an acetyl group during this transfer reaction, forming acetyldihydrolipoamide. Following transfer of the acetyl group to CoA, E3 reoxidizes the lipoamide, using FAD, and the FADH2 is in turn oxidized by NAD+, yielding NADH. The net reaction is: Pyr + NAD+ + CoA-SH → acetyl-CoA + NADH + H+ + CO2
Intermediates are tethered to the transacetylase component of the complex during the reaction sequence (Figs 14.5 and 14.6). This optimizes the catalytic efficiency of the enzyme since substrate does not equilibrate into solution.
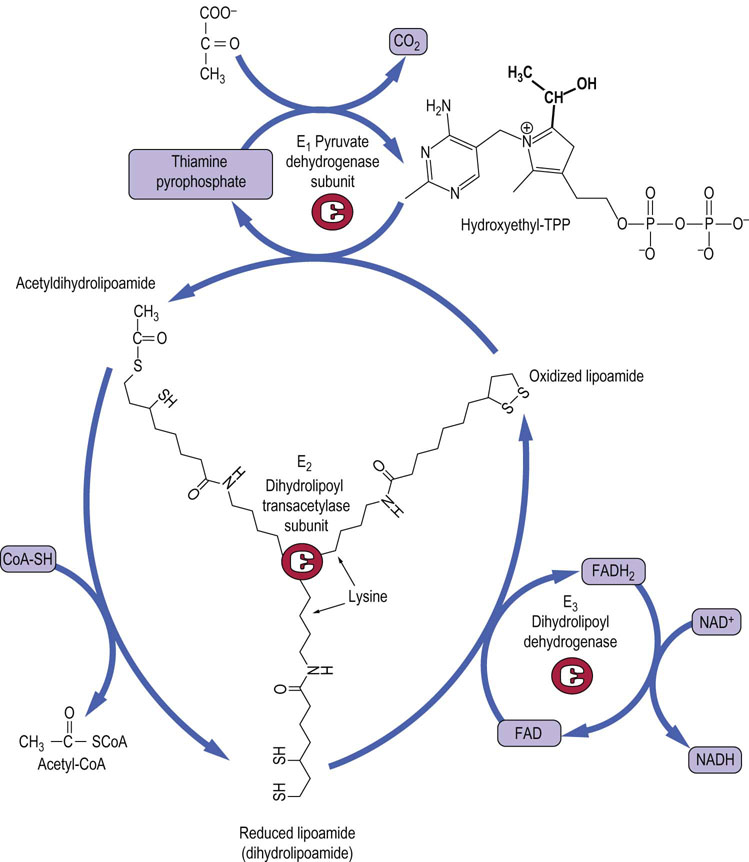
Fig. 14.6 Lipoic acid in the pyruvate dehydrogenase complex.
The coenzyme lipoamide is attached to a lysine residue in the transacetylase subunit of pyruvate dehydrogenase. Lipoamide moves from one active site to another on the transacetylase subunit in a ‘swinging arm’ mechanism. The structures of thiamine pyrophosphate (TPP) and lipoamide are shown.
Two additional enzymes of the complex, pyruvate dehydrogenase kinase and pyruvate dehydrogenase phosphatase, regulate its activity by covalent modification via reversible phosphorylation/dephosphorylation. There are four known isoforms of the kinase, and two of the phosphatase; the relative amounts of each are cell specific.
Five coenzymes are required for PDC activity: thiamine pyrophosphate, lipoamide (lipoic acid bound in amide linkage to protein), CoA, FAD, and NAD+. Four vitamins are required for their synthesis: thiamine, pantothenic acid, riboflavin and nicotinamide. Deficiencies in any of these vitamins have obvious effects on energy metabolism. For example, increases in cellular concentrations of pyruvate and α-ketoglutarate are found in beriberi because of thiamine deficiency (Chapter 11). In this case, all the proteins are available but the relevant coenzyme is not, and the conversions of pyruvate to acetyl-CoA and α-ketoglutarate to succinyl-CoA are significantly decreased. Symptoms include cardiac and skeletal muscle weakness and neurologic disease. Thiamine deficiency is common in alcoholism, because distilled spirits are devoid of vitamins, and symptoms of beri-beri are often observed.
 Clinical box Pyruvate dehydrogenase complex deficiency
Clinical box Pyruvate dehydrogenase complex deficiency
Most children with this enzyme deficiency present in infancy with delayed development and reduced muscle tone often associated with ataxia and seizures. Some infants have congenital malformations of the brain.
Without mitochondrial oxidation, pyruvate is reduced to lactate. The ATP yield from anaerobic glycolysis is less than a tenth of that produced from complete oxidation of glucose via the tricarboxylic acid cycle. The diagnosis is suggested by elevated lactate, but with a normal lactate/pyruvate ratio, i.e. no evidence of hypoxia. A ketogenic diet and severe restriction of protein (<15%) and carbohydrate (<5%) improve mental development. Such treatment ensures that the cells use acetyl-CoA from fat metabolism. A few children show a reduction in plasma lactate on treatment with large doses of thiamine, but the outlook is generally poor.
Enzymes and reactions of the tricarboxylic acid cycle
The TCA cycle is a sequence of reactions for oxidation of acetyl-CoA to CO2 and reduced nucleotides
The TCA cycle is a sequence of eight enzymatic reactions (Fig. 14.7), beginning with condensation of acetyl-CoA with oxaloacetate (OAA) to form citrate. The oxaloacetate is regenerated on completion of the cycle. Of the four oxidations in the cycle, two involve decarboxylations. Three dehydrogenases produce NADH and one produces FADH2. GTP, a high-energy phosphate, is produced at one step by substrate-level phosphorylation.
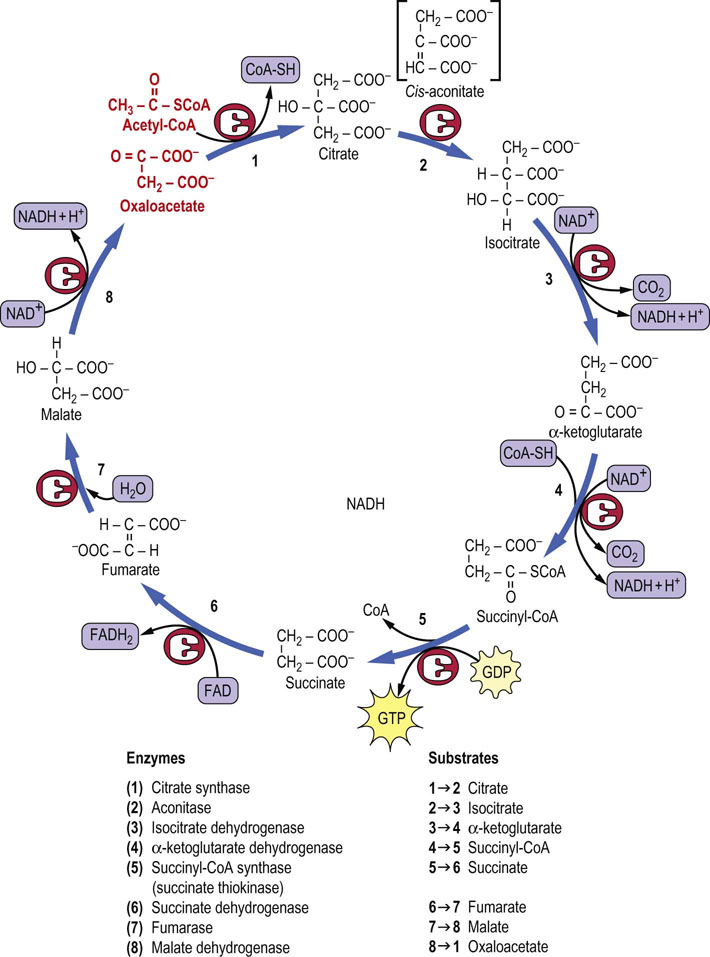
Citrate synthase
Citrate synthase begins the TCA cycle by catalyzing the condensation of acetyl-CoA and oxaloacetate to form citric acid. The reaction is driven by cleavage of the high-energy thioester bond of citroyl-CoA, an intermediate in the reaction. A later TCA cycle enzyme, succinyl-CoA synthetase, utilizes the high-energy thioester bond in succinyl-CoA to produce GTP, a high-energy phosphate (Fig. 14.7).
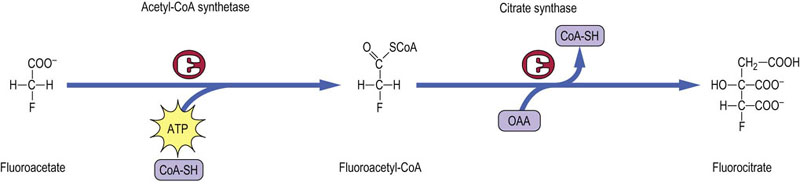
 Advanced concept box Toxicity of fluoroacetate – a suicide substrate
Advanced concept box Toxicity of fluoroacetate – a suicide substrate
Fluoroacetate, originally isolated from plants, is a potent toxin. It is activated as fluoroacetyl-CoA and then condenses with oxaloacetate to form fluorocitrate (Fig. 14.8). Death results from inhibition of the TCA cycle by 2-fluorocitrate, a strong inhibitor of aconitase. Fluoroacetate is an example of a ‘suicide substrate’, a compound that is not toxic per se but is metabolically activated to a toxic product. Thus, the cell is said to commit suicide by converting an apparently harmless substrate to a lethal toxin. Similar processes are involved in the activation of many environmental procarcinogens to carcinogens that induce mutations in DNA.
Aconitase
Aconitase is an iron-sulfur protein (Chapter 9) that isomerizes citrate to isocitrate through the enzyme-bound intermediate cis-aconitate. The two-step reaction is reversible and involves dehydration followed by hydration. Although citrate is a symmetric molecule, aconitase works specifically on the oxaloacetate end of citrate, not the end derived from acetyl-CoA (Fig. 14.9). Such stereochemical specificity occurs because of the geometry of the active site of aconitase (Figs 14.10 and 14.11). A cytosolic protein with aconitase activity, known as IRE-BP (iron-response element binding protein), functions in the regulation of iron storage.
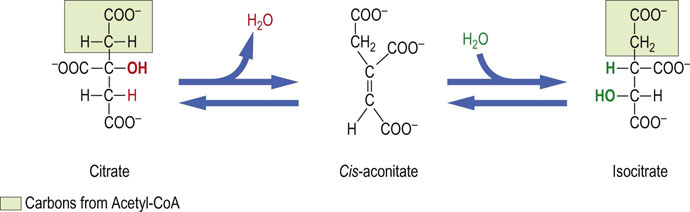
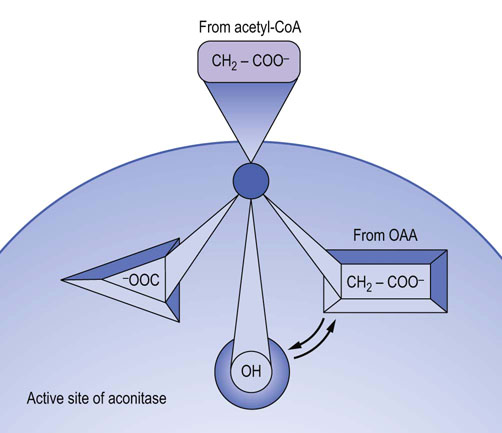
Fig. 14.10 Stereochemistry of the aconitase reaction.
Aconitase converts achiral citrate to a specific chiral form of isocitrate. Binding of the adjacent C-3 hydroxyl (OH) and carboxylate (COO–) groups of citrate on the enzyme surface places the carboxymethyl (—CH2—COO−) group, derived from the oxaloacetate end of the molecule, in touch with the third binding locus in the active site of aconitase. This assures the transfer of the OH group to the CH2 group derived from oxaloacetate, indicated by arrows, rather than that derived from the acetyl group. OAA, oxaloacetate.
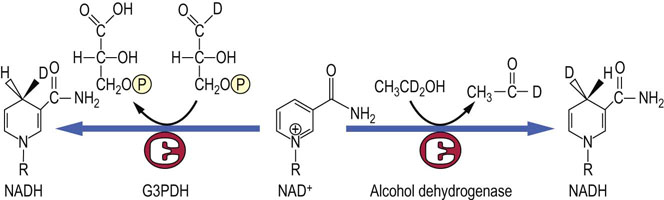
Fig. 14.11 Stereochemistry of the reduction of NAD+ by dehydrogenases.
Alcohol dehydrogenase places the hydrogen ion on the front face of the nicotinamide ring, while glyceraldehyde-3-phosphate dehydrogenase (G3PDH) places the hydrogen on the back face of the ring. The two positions can be discriminated using deuterated (D) substrates.
Advanced concept box Stereospecificity of enzymes
Aconitase catalyzes isomerization at the oxaloacetate end of the citrate molecule. However, citrate has no asymmetric centers; it is achiral. How does aconitase know ‘which end is up’? The answer lies in the nature of citrate binding to the active site of aconitase, a process known as three-point attachment. As shown in Figure 14.10, because of the geometry of the active site of aconitase, there is only one way for citrate to bind. This ‘three-point binding’ places the oxaloacetate carbons in the proper orientation for the isomerization reaction, while the carbons derived from acetyl-CoA are excluded from the active site.
Although citrate is a symmetric or achiral molecule, it is termed ‘prochiral’ because it is converted to a chiral molecule, isocitrate. Similar types of three-point binding processes are involved in transaminase reactions that produce exclusively L-amino acids from ketoacids. The reduction of the nicotinamide ring by NAD(H)-dependent dehydrogenases is also stereospecific. Some dehydrogenases place the added hydrogen exclusively on the front face of the nicotinamide ring (viewed with the amide group to the right), while others add hydrogen only to the back face (see Fig. 14.11 and Chapter 6).
Isocitrate dehydrogenase and α-ketoglutarate dehydrogenase
Isocitrate dehydrogenase and the α-ketoglutarate dehydrogenase complex catalyze two sequential oxidative decarboxylation reactions in which NAD+ is reduced to NADH, and CO2 is released. The first of these enzymes, isocitrate dehydrogenase, catalyzes the conversion of isocitrate to α-ketoglutarate. It is an important regulatory enzyme that is inhibited under energy-rich conditions by high levels of NADH and ATP, and is activated when NAD+ and ADP are produced by metabolism. Inhibition of this enzyme following a carbohydrate meal causes intramitochondrial accumulation of citrate, which is then exported to the cytosol for lipogenesis (Chapter 16). Citrate is also an allosteric effector, inhibiting phosphofructokinase-1 (Chapter 13) and activating acetyl-CoA carboxylase (Chapter 16).
The second dehydrogenase, the α-ketoglutarate dehydrogenase complex, catalyzes the oxidative decarboxylation of α-ketoglutarate to NADH, CO2 and succinyl-CoA, a high-energy thioester compound. Like the pyruvate dehydrogenase complex, this enzyme complex contains three subunits having the same designations as pyruvate dehydrogenase (E1, E2 and E3). E3 is identical in the two complexes and is encoded by the same gene. The reaction mechanisms and the cofactors thiamine pyrophosphate, lipoate, CoA, FAD and NAD+ are the same. Both enzymes begin with an α-keto acid, pyruvate or α-ketoglutarate, and both form the CoA esters, acetyl-CoA or succinyl-CoA, respectively.
At this point, the net carbon yield of the TCA cycle is zero, i.e. two carbons were introduced as acetyl-CoA and two carbons were liberated as CO2. Note, however, that because of the asymmetry of the aconitase reaction, neither of the CO2 molecules produced in this first round trip through the TCA cycle originates from the carbons of the acetyl-CoA, because they are derived from the oxaloacetate end of the citrate molecule. Both of the carbons that originated from acetyl-CoA remain in TCA cycle intermediates, and may appear in compounds produced in biosynthetic reactions branching from the TCA cycle, including glucose, aspartic acid and heme. However, because of the loss of two CO2 molecules at this point, there is no net synthesis of these metabolites from acetyl-CoA.
Animals cannot perform net synthesis of glucose from acetyl-CoA. This is an especially important concept in the understanding of starvation, diabetes and ketogenesis, because large amounts of acetyl-CoA are generated from fatty acids, but it does not yield a net synthesis of glucose. ‘Net’ synthesis is invoked, because labeled carbons from acetyl-CoA eventually appear in glucose, making it appear that glucose is synthesized from acetyl-CoA. However, the investment of the two carbons of acetyl-CoA is dissipated by the two decarboxylation reactions in the TCA cycle. Net synthesis also means that in order for continued operation of the TCA cycle for gluconeogenesis or biosynthesis of metabolites, anaplerotic reactions must supply carbons to the cycle in forms other than acetyl-CoA (see below).
Clinical box Deficiencies in pyruvate metabolism in the TCA cycle
A 7-month-old-child showed progressive neurologic deterioration characterized by loss of coordination and muscle tone. He was unable to keep his head upright and had great difficulty moving his limbs, which were limp. He also suffered from unrelenting acidosis. Administration of thiamine had no effect. Measurements showed that he had elevated blood levels of lactate, α-ketoglutarate and branched-chain amino acids. The child died a week later. Liver, brain, kidney, skeletal muscle, and heart were examined postmortem, and all gluconeogenic enzymes were shown to have normal activities, but both pyruvate dehydrogenase and α-ketoglutarate dehydrogenase were deficient. The defective component was shown to be dihydrolipoyl dehydrogenase (E3), which is a single gene component required by all of the α-ketoacid dehydrogenases.
This is an example of one of the many variants of Leigh's disease, which is a group of disorders that are all characterized by lactic acidosis. Lactic acid accumulates under anaerobic conditions or because of any enzyme defect in the pathway from pyruvate to the synthesis of ATP. In this case, there are defects in both the pyruvate dehydrogenase and α-ketoglutarate complexes, as well as other α-keto acid dehydrogenase complexes required for the catabolism of branched-chain amino acids. The failure of aerobic metabolism leads to increases in blood levels of lactate, α-ketoglutarate and branched-chain amino acids. Tissues dependent on aerobic metabolism, such as brain and muscle, are most severely affected, so that the clinical picture includes impaired motor function, neurologic disorders and mental retardation. These diseases are rare, but deficiencies in pyruvate carboxylase and all the components of the pyruvate dehydrogenase complex (PDH) have been described, including the associated kinase and phosphatase enzymes (Fig. 14.12).
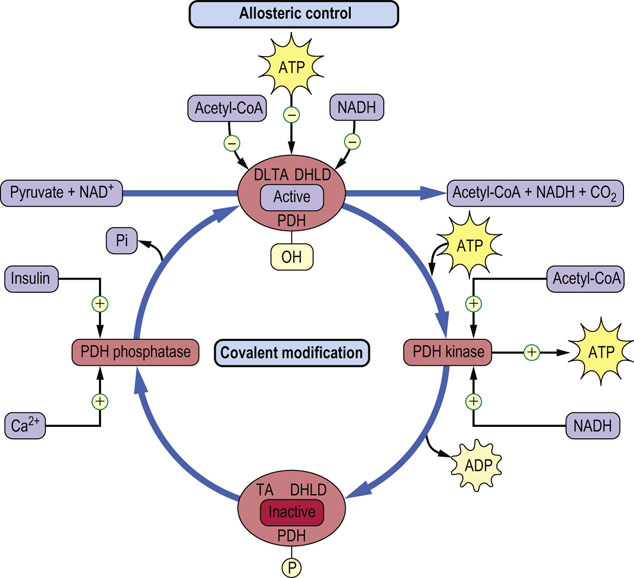
Fig. 14.12 Regulation of the pyruvate dehydrogenase complex.
The pyruvate dehydrogenase complex regulates the flux of pyruvate into the TCA cycle. NAD(H), ATP and acetyl-CoA exert both allosteric and covalent control of enzyme activity. PDH, pyruvate dehydrogenase; TA, dihydrolipoyl transacetylase; DHLD, dihydrolipoamide dehydrogenase subunit.
Succinyl-CoA synthetase
Succinyl-CoA synthetase (succinate thiokinase) catalyzes the conversion of energy-rich succinyl-CoA to succinate and free CoA. The free energy of the thioester bond in succinyl-CoA is conserved by formation of GTP from GDP and inorganic phosphate (Pi). Because a high-energy thioester serves as the driving force for the synthesis of GTP, this is a substrate-level phosphorylation reaction, like the reactions catalyzed by phosphoglycerate kinase and pyruvate kinase in glycolysis (Chapter 12). GTP is used by enzymes such as phosphoenol-pyruvate carboxykinase (PEPCK) in gluconeogenesis (Chapter 13), and in several steps in protein synthesis (Chapter 34) and cell signaling (Chapter 13) but is also readily equilibrated with ATP by the enzyme nucleoside diphosphate kinase:
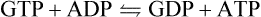
The next three reactions in the TCA cycle illustrate a common theme in metabolism for introducing a carbonyl group into a molecule:
This same sequence occurs in the form of enzyme-bound intermediates during the oxidation of fatty acids (Chapter 15).
Succinate dehydrogenase
Succinate dehydrogenase is a flavoprotein containing the prosthetic group FAD. As described in Chapter 9, this enzyme is embedded in the inner mitochondrial membrane where it is a part of complex II (succinate-Q reductase). The reaction involves oxidation of succinate to the trans-dicarboxylic acid fumarate, with reduction of FAD to FADH2.
Fumarase
Fumarase stereospecifically adds water across the trans double bond of fumarate to form the α-hydroxy acid, L-malate.
Malate dehydrogenase
Malate dehydrogenase catalyzes the oxidation of L-malate to oxaloacetate, producing NADH, completing one round trip through the TCA cycle. The oxaloacetate may then react with acetyl-CoA, continuing the cycle of reactions.
Advanced concept box The malonate block
The malate dehydrogenase reaction played an important role in the elucidation of the cyclic nature of the TCA cycle. Addition of tricarboxylic acids (citrate, aconitate) and α-ketoglutarate was known to catalyze pyruvate metabolism – we now know that this is the result of formation of catalytic amounts of oxaloacetate from these intermediates. In 1937, Krebs found that malonate, the 3-carbon dicarboxylic acid homologue of succinate and competitive inhibitor of succinate dehydrogenase, blocked metabolism of pyruvate by minced muscle preparations. He also showed that malonate inhibition of pyruvate metabolism led to accumulation not only of succinate but also of citrate and α-ketoglutarate, suggesting that succinate was a product of pyruvate metabolism and that the tricarboxylic acids might be intermediates in this process. Interestingly, fumarate and oxaloacetate also stimulated pyruvate oxidation and led to accumulation of citrate and succinate during malonate block, suggesting that the 3- and 4-carbon acids might combine to form the tricarboxylic acids. The experiments with fumarate indicated that there were two paths between fumarate and succinate, one involving reversal of the succinate dehydrogenase reaction, which was inhibited during malonate block, and the other involving conversion of fumarate to succinate through a series of organic acids. These observations, combined with Krebs' experience a few years earlier in characterization of the urea cycle (Chapter 19), led to his description of the TCA cycle.
Energy yield from the tricarboxylic acid cycle
During the course of the TCA cycle, each mole of acetyl-CoA generates sufficient reduced nucleotide coenzymes for synthesis of ~9 moles ATP by oxidative phosphorylation.
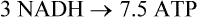
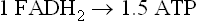
Together with the GTP synthesized by substrate-level phosphorylation in the succinyl-CoA synthetase (succinate thiokinase) reaction, a total of ~10 ATP equivalents is available per mole of acetyl-CoA. Thus, complete metabolism of a mole of glucose through glycolysis, the pyruvate dehydrogenase complex and the TCA cycle yields ≈30–32 moles ATP (Table 14.1). (The actual ATP yield depends on the route of transport of redox equivalents to the mitochondrion, i.e. about 5 moles of ATP by the malate aspartate shuttle and about 3 moles of ATP by the glycerol phosphate shuttle (Chapter 9).) In contrast, only 2 moles of ATP (net) are recovered by anaerobic glycolysis in which glucose is converted to lactate (Chapter 12).
Table 14.1
ATP yield from glucose during oxidative metabolism
| Reaction | Mechanism | Moles ATP/mole Glc |
| Hexokinase | Phosphorylation | –1 |
| Phosphofructokinase | Phosphorylation | –1 |
| G3PDH | NADH, oxidative phosphorylation | +5 (+3)* |
| Phosphoglycerate kinase | Substrate-level phosphorylation | +2 |
| Pyruvate kinase | Substrate-level phosphorylation | +2 |
| Pyruvate dehydrogenase | NADH, oxidative phosphorylation | +5 |
| Isocitrate dehydrogenase | NADH, oxidative phosphorylation | +5 |
| α-ketoglutarate dehydrogenase | NADH, oxidative phosphorylation | +5 |
| Succinyl-CoA synthetase | Substrate-level phosphorylation (GTP) | +2 |
| Succinate dehydrogenase | FADH2, oxidative phosphorylation | +3 |
| Malate dehydrogenase | NADH, oxidative phosphorylation | +5 |
| TOTAL | 32 (30)* |
The yields of ATP shown are approximate, because they are measured experimentally with live, isolated mitochondria and there is some variability. Recent work suggests that the actual yields of ATP from NADH and FADH2 are about 2.5 and 1.5, respectively, yielding approximately 30–32 moles of ATP per mole of glucose. The oxidation of glucose in a bomb calorimeter yields 2870 kJ/mol (686 cal/mol), while the synthesis of ATP requires 31 kJ/mol (7.3 kcal/mol). Aerobic metabolism of glucose is therefore about 40% efficient (2870 kJ/mol glucose/31 kJ/mole ATP = 93 theoretical moles of ATP/mol glucose; 36/93 = 39%).
*Electrons from cytosolic NADH can result in the synthesis of about 5 moles of ATP per mole of glucose via the malate–aspartate shuttle, but only about 3 via the glycerol-3-phosphate shuttle per mole of glucose (Chapter 9).
Anaplerotic (‘building up’) reactions
As shown in Figure 14.1, many TCA cycle intermediates participate in biosynthetic processes, which deplete TCA cycle intermediates. For example, the synthesis of 1 mole of heme requires 8 moles of succinyl-CoA. The TCA cycle would cease to function if the intermediates were not replenished, because acetyl-CoA cannot yield a net synthesis of oxaloacetate. Anaplerotic (building up) reactions provide the TCA cycle with intermediates other than acetyl-CoA to maintain activity of the cycle. Pyruvate carboxylase is a prime example of an enzyme that catalyzes an anaplerotic reaction. It converts pyruvate to oxaloacetate, which is required for initiation of the cycle. Malic enzyme in the cytoplasm also converts pyruvate to malate, which can enter the mitochondrion as a substrate for the TCA cycle. Aspartate is also a precursor of oxaloacetate by a transamination reaction, and α-ketoglutarate can be produced through an aminotransferase reaction from glutamate, as well as by the glutamate dehydrogenase reaction. Several other ‘glucogenic’ amino acids (Chapter 19) may also serve as sources of pyruvate or TCA cycle intermediates, guaranteeing that the cycle never stalls because of a lack of intermediates.
Regulation of the tricarboxylic acid cycle
Pyruvate dehydrogenase and isocitrate dehydrogenase regulate TCA cycle activity
There are several levels of control of the TCA cycle. In general, the overall activity of the cycle depends on the availability of NAD+ for the dehydrogenase reactions. This, in turn, is linked to the rate of NADH consumption by the electron transport system, which ultimately depends on the rate of ATP utilization and production of ADP by metabolism (see Table 14.1). Thus, as ATP is used for metabolic work, ADP is produced, then NADH is consumed by the electron transport system for ATP production, and NAD+ is produced. The TCA cycle is activated, fuels are consumed, and more NADH is produced so that more ATP may be made. The mitochondrial level of NAD+ provides a link between work (ATP utilization) and fuel consumption (Chapter 9).
There are several regulatory enzymes that affect the activity of the TCA cycle. The activity of the pyruvate dehydrogenase complex, and therefore the supply of acetyl-CoA from glucose, lactate and alanine, is regulated by allosteric and covalent modifications (see Fig. 14.12). The products of the pyruvate dehydrogenase reaction, NADH and acetyl-CoA, as well as ATP, act as negative allosteric effectors of the enzyme complex. In addition, the pyruvate dehydrogenase complex has associated kinase and phosphatase enzymes that modulate the degree of phosphorylation of regulatory serine residues in the complex. NADH, acetyl-CoA and ATP activate the kinase, which phosphorylates and inactivates the enzyme complex. In contrast, when these three compounds are low in concentration, the enzyme complex is activated allosterically and by dephosphorylation by the phosphatase. This is an important regulatory process during fasting and starvation, when gluconeogenesis is essential to maintain blood glucose concentration. Active fat metabolism during fasting leads to increased NADH and acetyl-CoA in the mitochondrion, which leads to inhibition of pyruvate dehydrogenase and blocks the utilization of carbohydrate for energy metabolism in the liver. Under this condition, pyruvate, from such intermediates as lactate and alanine, is directed toward gluconeogenesis. Conversely, insulin stimulates pyruvate dehydrogenase by activating the phosphatase in response to dietary carbohydrates. This directs carbohydrate-derived carbons into fatty acids (lipogenesis) via citrate synthase (Chapter 16). Ca2+ also affects PDC phosphatase activity, in response to the increase in intracellular Ca2+ during muscle contraction (Chapter 20).
Oxaloacetate is required for entry of acetyl-CoA into the TCA cycle but, at times, the availability of oxaloacetate appears to regulate the activity of the cycle. This occurs especially during fasting when levels of ATP and NADH, derived from fat metabolism, are increased in the mitochondrion. The increase in NADH shifts the malate : oxaloacetate equilibrium toward malate, directing TCA cycle intermediates toward malate, which is exported to the cytosol for gluconeogenesis (Chapter 13). Meanwhile, acetyl-CoA derived from fat metabolism is directed toward synthesis of ketone bodies because of the lack of oxaloacetate, regenerating CoA-SH and leading to the increase in ketone bodies in plasma during fasting (Chapter 15).
Isocitrate dehydrogenase is a major regulatory enzyme within the TCA cycle. It is subject to allosteric inhibition by ATP and NADH and stimulation by ADP and NAD+. During consumption of a high carbohydrate diet under resting conditions, the demand for ATP is diminished and the level of carbohydrate-derived intermediates increases. Under these circumstances, increased insulin levels stimulate the pyruvate dehydrogenase complex, and the accumulation of ATP and NADH inhibits isocitrate dehydrogenase, causing a mitochondrial accumulation of citrate. The citrate is then exported to the cytosol for synthesis of fatty acids, which are exported from the liver for storage in adipose tissue as triglycerides. With an increase in energy demand, e.g. during muscle contraction, NAD+ and ADP accumulate, and they stimulate isocitrate dehydrogenase.
Induction and repression, as well as proteolysis of enzyme proteins, such as pyruvate carboxylase and those in the pyruvate dehydrogenase complex and the TCA cycle, also play an important regulatory role. In fact, all of the TCA cycle and associated enzymes are synthesized in the cytoplasm and transported through a complex series of steps into the mitochondrion. Regulation can occur at the level of translation, transcription and intracellular transport. Diet, for example, is known to control expression of four pyruvate dehydrogenase kinases; one of them is induced in response to a high-fat diet and is repressed in response to a high-carbohydrate diet. Unfortunately, the regulation of the TCA cycle at genetic and transport levels is not as well understood, although it is clearly important for understanding the pathogenesis of a wide range of contemporary health problems, such as diabetes and obesity. Three TCA cycle enzymes, succinate dehydrogenase, fumarate hydratase and isocitrate dehydrogenase, are described as tumor suppressors because genetic defects in these enzymes are associated with human cancers (see Cardaci and Ciriolo and Yang et al. in Further Reading).
Summary
The TCA cycle is the central, common pathway by which fuels are oxidized, and it also participates in major biosynthetic pathways.
In its oxidative role, major products of the TCA cycle are GTP and the reduced coenzymes NADH and FADH2, which furnish large amounts of free energy for the synthesis of ATP by oxidative phosphorylation.
In its biosynthetic role, the TCA cycle provides essential intermediates for the synthesis of glucose, fatty acids, amino acids and heme, as well as the ATP required for their biosynthesis.
The activity of the TCA cycle is tightly regulated by substrate supply, by allosteric effectors and control of gene expression so that fuel consumption is coordinated with energy production.
Cardaci, S, Ciriolo, MR. TCA cycle defects and cancer: When metabolism tunes redox state. Int J Cell Biol. 2012; 2012:161837.
Marin-Valencia, I, Roe, CR, Pascual, JM. Pyruvate carboxylase deficiency: mechanisms, mimics and anaplerosis. Mol Genet Metab. 2010; 101:9–17.
Patel, KP, O'Brien, TW, Subramony, SH, et al. The spectrum of pyruvate dehydrogenase complex deficiency: clinical, biochemical and genetic features in 371 patients. Mol Genet Metab. 2012; 106:385–394.
Qi, F, Pradhan, RK, Dash, RK, et al. Detailed kinetics and regulation of mammalian 2-oxoglutarate dehydrogenase. BMC Biochem. 2011; 12:53.
Sudheesh, NP, Ajith, TA, Janardhanan, KK, et al. Palladium alpha-lipoic acid complex formulation enhances activities of Krebs cycle dehydrogenases and respiratory complexes I–IV in the heart of aged rats. Food Chem Toxicol. 2009; 47:2124–2128.
Yang, M, Soga, T, Pollard, PJ, et al. The emerging role of fumarate as an oncometabolite. Front Oncol. 2012; 2:85.