Rejection of Tissue Transplants
Transplant rejection is discussed here because it involves several of the immunological reactions that underlie immune-mediated inflammatory diseases. A major barrier to transplantation is the process of rejection, in which the recipient’s immune system recognizes the graft as being foreign and attacks it.
Mechanisms of Recognition and Rejection of Allografts
Rejection is a complex process in which both cell-mediated immunity and circulating antibodies play a role99; moreover, the contributions of these two mechanisms are often reflected in the histologic features of the rejected organs.
T Cell–Mediated Reactions
The critical role of T cells in transplant rejection has been documented both in humans and in experimental animals. T cell–mediated graft rejection is called cellular rejection, and it involves destruction of graft cells by CD8+ CTLs and delayed hypersensitivity reactions triggered by activated CD4+ helper cells. The major antigenic differences between a donor and recipient that result in rejection of transplants are differences in highly polymorphic HLA alleles. The recipient’s T cells recognize donor antigens from the graft (the allogeneic antigens, or alloantigens) by two pathways, called direct and indirect (Fig. 6-39).100
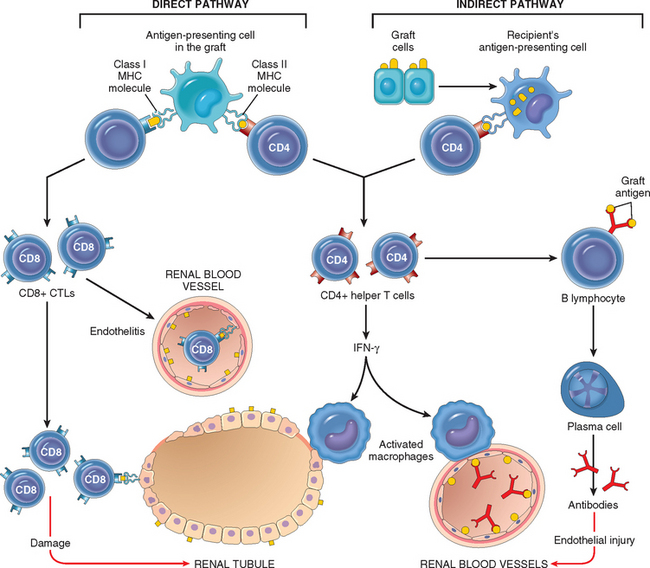
FIGURE 6-39 Recognition and rejection of organ allografts. In the direct pathway, donor class I and class II MHC antigens on antigen-presenting cells in the graft (along with costimulators, not shown) are recognized by host CD8+ cytotoxic T cells and CD4+ helper T cells, respectively. CD4+ cells proliferate and produce cytokines (e.g., IFN-γ), which induce tissue damage by a local delayed hypersensitivity reaction. CD8+ T cells responding to graft antigens differentiate into CTLs that kill graft cells. In the indirect pathway graft antigens are picked up, processed, and displayed by host APCs and activate CD4+ T cells, which damage the graft by a local delayed hypersensitivity reaction and stimulate B lymphocytes to produce antibodies.
Antibody-Mediated Reactions
Although T cells are pivotal in the rejection of organ transplants, antibodies produced against alloantigens in the graft are also important mediators of rejection.101 This process is called humoral rejection, and it can take two forms. Hyperacute rejection occurs when preformed antidonor antibodies are present in the circulation of the recipient. Such antibodies may be present in a recipient who has previously rejected a kidney transplant. Multiparous women who develop anti-HLA antibodies against paternal antigens shed from the fetus may have preformed antibodies to grafts taken from their husbands or children, or even from unrelated individuals who share HLA alleles with the husbands. Prior blood transfusions can also lead to presensitization, because platelets and white blood cells are rich in HLA antigens and donors and recipients are usually not HLA-identical. With the current practice of cross-matching, that is, testing recipient’s serum for antibodies against donor’s cells, hyperacute rejection is no longer a significant clinical problem.
In recipients not previously sensitized to transplantation antigens, exposure to the class I and class II HLA antigens of the donor graft may evoke antibodies. The antibodies formed by the recipient may cause injury by several mechanisms, including complement-dependent cytotoxicity, inflammation, and antibody-dependent cell-mediated cytotoxicity. The initial target of these antibodies in rejection seems to be the graft vasculature. Thus, antibody-dependent acute humoral rejection is usually manifested by a vasculitis, sometimes referred to as rejection vasculitis.
Rejection of Kidney Grafts
Because kidneys were the first solid organs to be transplanted and more kidneys have been transplanted than any other organ, much of our understanding of the clinical and pathologic aspects of solid-organ transplantation is based on studies of renal allografts.
Morphology. On the basis of the morphology and the underlying mechanism, rejection reactions are classified as hyperacute, acute, and chronic. The morphologic changes in these patterns are described below as they relate to renal transplants. Similar changes may occur in any other vascularized organ transplant and are discussed in relevant chapters.
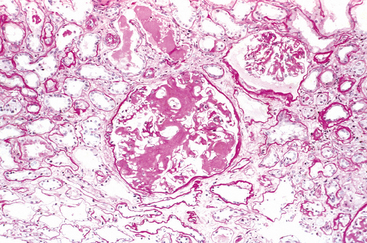
Hyperacute Rejection. This form of rejection occurs within minutes or hours after transplantation. A hyperacutely rejecting kidney rapidly becomes cyanotic, mottled, and flaccid, and may excrete a mere few drops of bloody urine. Immunoglobulin and complement are deposited in the vessel wall, causing endothelial injury and fibrin-platelet thrombi (Fig. 6-40A). Neutrophils rapidly accumulate within arterioles, glomeruli, and peritubular capillaries. As these changes become diffuse and intense, the glomeruli undergo thrombotic occlusion of the capillaries, and fibrinoid necrosis occurs in arterial walls. The kidney cortex then undergoes outright necrosis (infarction), and such nonfunctioning kidneys have to be removed.
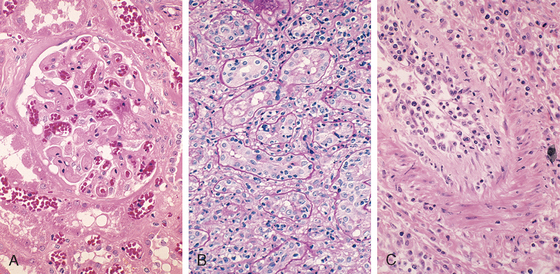
FIGURE 6-40 Morphology of hyperacute and acute graft rejection. A, Hyperacute rejection of a kidney allograft showing endothelial damage, platelet and thrombin thrombi, and early neutrophil infiltration in a glomerulus. B, Acute cellular rejection of a kidney allograft with inflammatory cells in the interstitium and between epithelial cells of the tubules. C, Acute humoral rejection of a kidney allograft (rejection vasculitis) with inflammatory cells and proliferating smooth muscle cells in the intima.
(Courtesy of Dr. Helmut Rennke, Department of Pathology, Brigham and Women’s Hospital and Harvard Medical School, Boston, MA.)
Acute Rejection. This may occur within days of transplantation in the untreated recipient or may appear suddenly months or even years later, after immunosuppression has been used and terminated. In any one patient, cellular or humoral immune mechanisms may predominate. Histologically, humoral rejection is associated with vasculitis, whereas cellular rejection is marked by an interstitial mononuclear cell infiltrate.
Acute cellular rejection is most commonly seen within the initial months after transplantation and is heralded by clinical and biochemical signs of renal failure (Chapter 20). Histologically, there may be extensive interstitial mononuclear cell infiltration and edema as well as mild interstitial hemorrhage (Fig. 6-40B). As might be expected, immunohistochemical staining reveals both CD4+ and CD8+ T lymphocytes, which express markers of activated T cells, such as the α chain of the IL-2 receptor. Glomerular and peritubular capillaries contain large numbers of mononuclear cells that may also invade the tubules, causing focal tubular necrosis. In addition to causing tubular damage, CD8+ T cells may injure vascular endothelial cells, causing a so-called endothelitis. The affected vessels have swollen endothelial cells, and at places the lymphocytes can be seen between the endothelium and the vessel wall. The recognition of cellular rejection is important because, in the absence of an accompanying humoral rejection, patients respond well to immunosuppressive therapy. Cyclosporine, a widely used immunosuppressive drug, is also nephrotoxic, and hence the histologic changes resulting from cyclosporine may be superimposed.
Acute humoral rejection (rejection vasculitis) is mediated by antidonor antibodies, and hence it is manifested mainly by damage to the blood vessels. This may take the form of necrotizing vasculitis with endothelial cell necrosis, neutrophilic infiltration, deposition of immunoglobulins, complement, and fibrin, and thrombosis. Such lesions are associated with extensive necrosis of the renal parenchyma. In many cases, the vasculitis is less acute and is characterized by marked thickening of the intima with proliferating fibroblasts, myocytes, and foamy macrophages (Fig. 6-40C). The resultant narrowing of the arterioles may cause infarction or renal cortical atrophy. The proliferative vascular lesions mimic arteriosclerotic thickening and are believed to be caused by cytokines that cause proliferation of vascular smooth muscle cells. Deposition of the complement breakdown product C4d in allografts is a strong indicator of humoral rejection, because C4d is produced during activation of the complement system by the antibody-dependent classical pathway.101,102 The importance of making this diagnosis is that it provides a rationale for treating affected patients with B cell–depleting agents.
Chronic Rejection. In recent years acute rejection has been significantly controlled by immunosuppressive therapy, and chronic rejection has emerged as an important cause of graft failure.103 Patients with chronic rejection present clinically with a progressive renal failure manifested by a rise in serum creatinine over a period of 4 to 6 months. Chronic rejection is dominated by vascular changes, interstitial fibrosis, and tubular atrophy with loss of renal parenchyma (Fig. 6-41). The vascular changes consist of dense, obliterative intimal fibrosis, principally in the cortical arteries. These vascular lesions result in renal ischemia, manifested by glomerular loss, interstitial fibrosis and tubular atrophy, and shrinkage of the renal parenchyma. The glomeruli may show scarring, with duplication of basement membranes; this appearance is sometimes called chronic transplant glomerulopathy. Chronically rejecting kidneys usually have interstitial mononuclear cell infiltrates of plasma cells and numerous eosinophils.
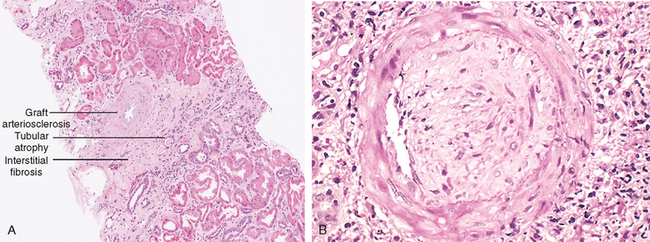
FIGURE 6-41 Chronic rejection of a kidney allograft. A, Changes in the kidney in chronic rejection. B, Graft arteriosclerosis. The vascular lumen is replaced by an accumulation of smooth muscle cells and connective tissue in the vessel intima.
(Courtesy of Dr. Helmut Rennke, Department of Pathology, Brigham and Women’s Hospital and Harvard Medical School, Boston, MA.)
Methods of Increasing Graft Survival
The value of HLA matching between donor and recipient varies in different solid-organ transplants. In kidney transplants, there is substantial benefit if all the polymorphic HLA alleles are matched (both inherited alleles of HLA-A, -B and DR). However, HLA matching is usually not even done for transplants of liver, heart, and lungs, because other considerations, such as anatomic compatibility, severity of the underlying illness, and the need to minimize the time of organ storage, override the potential benefits of HLA matching.
Except in the case of identical twins, who obviously express the same histocompatibility antigens, immunosuppressive therapy is a practical necessity in all other donor-recipient combinations.104 The mainstay of immunosuppression is the drug cyclosporine. Cyclosporine works by blocking activation of a transcription factor called nuclear factor of activated T cells (NFAT), which is required for transcription of cytokine genes, in particular, the gene that encodes IL-2. Additional drugs that are used to treat rejection include azathioprine (which inhibits leukocyte development from bone marrow precursors), steroids (which block inflammation), rapamycin and mycophenolate mofetil (both of which inhibit lymphocyte proliferation), and monoclonal anti–T-cell antibodies (e.g., monoclonal anti-CD3 and antibodies against the IL-2 receptor α chain [CD25], which opsonize and eliminate the cells and may also block T-cell activation). Another, more recent, strategy for reducing antigraft immune responses is to prevent host T cells from receiving costimulatory signals from dendritic cells during the initial phase of sensitization. This can be accomplished by interrupting the interaction between the B7 molecules on the dendritic cells of the graft donor with the CD28 receptors on host T cells, for example, by administration of proteins that bind to B7 costimulators.
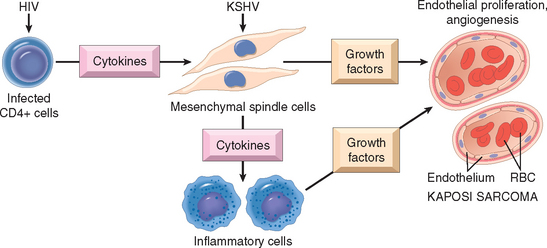
Although immunosuppression prolongs graft survival, it carries its own risks. The price paid in the form of increased susceptibility to opportunistic infections is not small. These patients are also at increased risk for developing EBV-induced lymphomas, human papillomavirus–induced squamous cell carcinomas, and Kaposi sarcoma (Chapter 11), all probably the result of reactivation of latent viral infections because of diminished host defenses. To circumvent the untoward effects of immunosuppression, much effort is being devoted to induce donor-specific tolerance in graft recipients.105 For instance, giving donor cells to graft recipients may prevent reactions to the graft, perhaps because the donor inoculum contains cells, such as immature dendritic cells, that induce tolerance to the donor alloantigens. This approach may result in long-term mixed chimerism, in which the recipient lives with the injected donor cells. Other strategies being tested include injecting regulatory T cells at the time of transplantation, and promoting the death of alloreactive T cells in the recipient.
Transplantation of Other Solid Organs
In addition to the kidney, a variety of organs, such as the liver (Chapter 18), heart (Chapter 12), lungs, and pancreas, are also transplanted. The rejection reaction against liver transplants is not as vigorous as might be expected from the degree of HLA disparity. The molecular basis of this “privilege” is not understood.
Transplantation of Hematopoietic Cells
Use of hematopoietic stem cell transplants for hematologic malignancies, certain nonhematologic cancers, aplastic anemias, thalassemias, and certain immunodeficiency states is increasing. Transplantation of genetically engineered hematopoietic stem cells may also be useful for somatic cell gene therapy, and is being evaluated in some immunodeficiencies. Hematopoietic stem cells are usually obtained from the bone marrow but may also be harvested from peripheral blood after they are mobilized from the bone marrow by administration of hematopoietic growth factors. In most of the conditions in which bone marrow transplantation is indicated, the recipient is irradiated to destroy the immune system (and sometimes, cancer cells) and to create a graft bed. Several features distinguish bone marrow transplants from solid-organ transplants. Two problems that are unique to bone marrow transplantation are graft-versus-host (GVH) disease and immunodeficiency.
GVH disease occurs in any situation in which immunologically competent cells or their precursors are transplanted into immunologically crippled recipients, and the transferred cells recognize alloantigens in the host.106 It is seen most commonly in the setting of bone marrow transplantation but, rarely, may occur following transplantation of solid organs rich in lymphoid cells (e.g., the liver) or transfusion of unirradiated blood. When immune-compromised recipients receive normal bone marrow cells from allogeneic donors, the immunocompetent T cells present in the donor marrow recognize the recipient’s HLA antigens as foreign and react against them. To try to minimize GVH disease, bone marrow transplants are done between donor and recipient that are HLA-matched using sensitive DNA sequencing methods for molecular typing of HLA alleles.
Acute GVH disease occurs within days to weeks after allogeneic bone marrow transplantation. Although any organ may be affected, the major clinical manifestations result from involvement of the immune system and epithelia of the skin, liver, and intestines. Involvement of skin in GVH disease is manifested by a generalized rash that may lead to desquamation in severe cases. Destruction of small bile ducts gives rise to jaundice, and mucosal ulceration of the gut results in bloody diarrhea. Although tissue injury may be severe, the affected tissues are usually not heavily infiltrated by lymphocytes. It is believed that in addition to direct cytotoxicity by CD8+ T cells, considerable damage is inflicted by cytokines released by the sensitized donor T cells.
Chronic GVH disease may follow the acute syndrome or may occur insidiously. These patients have extensive cutaneous injury, with destruction of skin appendages and fibrosis of the dermis. The changes may resemble systemic sclerosis (discussed earlier). Chronic liver disease manifested by cholestatic jaundice is also frequent. Damage to the gastrointestinal tract may cause esophageal strictures. The immune system is devastated, with involution of the thymus and depletion of lymphocytes in the lymph nodes. Not surprisingly, the patients experience recurrent and life-threatening infections. Some patients develop manifestations of autoimmunity, postulated to result from the grafted CD4+ helper T cells reacting with host B cells and stimulating these cells, some of which may be capable of producing autoantibodies.
Because GVH disease is mediated by T lymphocytes contained in the donor bone marrow, depletion of donor T cells before transfusion virtually eliminates the disease. This protocol, however, has proved to be a mixed blessing: GVH disease is ameliorated, but the incidence of graft failures and EBV-related B-cell lymphoma and the recurrence of disease in leukemic patients increase. It seems that the multifaceted T cells not only mediate GVH disease but also are required for engraftment of the transplanted marrow stem cells, suppression of EBV-infected B-cell clones, and control of leukemic cells. The latter, called graft-versus-leukemia effect, can be quite dramatic. Deliberate induction of graft-versus-leukemia effect by infusion of allogeneic T cells is being used in the treatment of chronic myelogenous leukemia when patients relapse after bone marrow transplantation.
Immunodeficiency is a frequent complication of bone marrow transplantation. The immunodeficiency may be a result of prior treatment, myeloablative preparation for the graft, a delay in repopulation of the recipient’s immune system, and attack on the host’s immune cells by grafted lymphocytes. Affected individuals are profoundly immunosuppressed and are easy prey to infections. Although many different types of organisms may infect patients, infection with cytomegalovirus is particularly important. This usually results from activation of previously silent infection. Cytomegalovirus-induced pneumonitis can be a fatal complication.