Cardiomyopathies
The term cardiomyopathy (literally, heart muscle disease) is used to describe heart disease resulting from an abnormality in the myocardium.89,90 Diseases of the myocardium usually produce abnormalities in cardiac wall thickness and chamber size, and mechanical and/or electrical dysfunction, and are associated with significant morbidity and mortality. Although chronic myocardial dysfunction secondary to ischemia, valvular abnormalities, or hypertension can cause ventricular dysfunction (see previous sections of this chapter), these conditions are not considered to be cardiomyopathies.
Clinical manifestations associated with the cardiomyopathies are either confined to the heart or can be a part of a generalized systemic disorder; in either situation, cardiac dysfunction is a key problem. Primary cardiomyopathies are diseases predominantly confined to the heart muscle, whereas secondary cardiomyopathies have myocardial involvement as a component of a systemic or multiorgan disorder. In most cases the mechanism by which the noncardiac problem affects the heart is well understood. In other disorders such as diabetes, the pathogenesis of the cardiac dysfunction is less obvious.91 A major advance in our understanding of cardiomyopathies is the increasing appreciation that many cases have underlying genetic causes,91,92 which we will discuss as we review the major categories of cardiomyopathy.
Cardiomyopathies of diverse etiology may have a similar morphologic appearance, and a clinician encountering a person with myocardial disease is usually unaware of the underlying cause. Hence the clinical approach is largely determined by which one of three clinical, functional, and pathologic patterns is present (Fig. 12-30 and Table 12-10): (1) dilated cardiomyopathy (DCM), (2) hypertrophic cardiomyopathy (HCM), or (3) restrictive cardiomyopathy. Another rare form of cardiomyopathy, left ventricular noncompaction, is characterized by a distinctive “spongy” appearance of the left ventricular myocardium. This congenital disorder is frequently associated with heart failure or arrhythmias and other clinical symptomatology; it may be diagnosed in children or adults as either an isolated finding or associated with other congenital heart anomalies, such as complex cyanotic congenital heart disease.93 Gene mutations affecting myocardial ion channel function have also been included in recent classifications of primary cardiomyopathy (this group of disorders has been discussed in the context of sudden cardiac death). For the purposes of this discussion, arrhythmogenic right ventricular cardiomyopathy (arrhythmogenic right ventricular dysplasia), discussed below, is considered a variant of DCM.

FIGURE 12-30 Representations of the three distinctive forms of myocardial disease. Ao, aorta; LA, left atrium; LV, left ventricle.

Within the hemodynamic patterns of myocardial dysfunction, there is a spectrum of clinical severity, and overlap of clinical features often occurs between groups. Moreover, each of these patterns can be either idiopathic or due to one of numerous specific identifiable causes (Table 12-11).
TABLE 12-11 Conditions Associated with Heart Muscle Diseases
| CARDIAC INFECTIONS |
| TOXINS |
| METABOLIC |
| NEUROMUSCULAR DISEASE |
| STORAGE DISORDERS AND OTHER DEPOSITIONS |
| INFILTRATIVE |
| IMMUNOLOGICAL |
Endomyocardial biopsies are used in the diagnosis and management of individuals with myocardial disease and in cardiac transplant recipients.94,95 Endomyocardial biopsy involves inserting a device (called a bioptome) transvenously into the right side of the heart and using its jaws to snip a small piece of septal myocardium, which is then analyzed by a pathologist.
DILATED CARDIOMYOPATHY
The term dilated cardiomyopathy (DCM) is applied to a form of cardiomyopathy characterized by progressive cardiac dilation and contractile (systolic) dysfunction, usually with concomitant hypertrophy. It is sometimes called congestive cardiomyopathy.
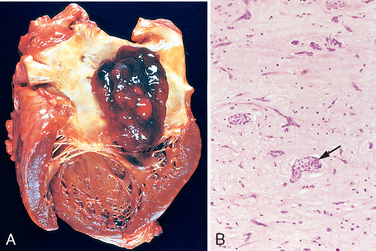
Morphology. In DCM the heart is usually enlarged, heavy (often weighing two to three times normal), and flabby, due to dilation of all chambers (Fig. 12-31). Mural thrombi are common and may be a source of thromboemboli. There are no primary valvular alterations, and mitral (or tricuspid) regurgitation, when present, results from left (or right) ventricular chamber dilation (functional regurgitation). Either the coronary arteries are free of significant narrowing or the obstructions present are insufficient to explain the degree of cardiac dysfunction.
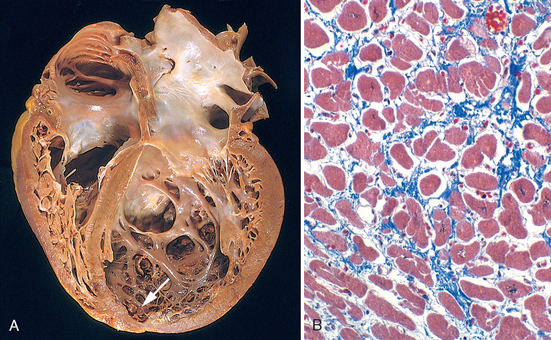
FIGURE 12-31 Dilated cardiomyo-pathy. A, Four-chamber dilatation and hypertrophy are evident. There is granular mural thrombus (arrow) at the apex of the left ventricle (on the right in this apical four-chamber view). The coronary arteries were patent. B, Histologic section demonstrating variable myocyte hypertrophy and interstitial fibrosis (collagen is highlighted as blue in this Masson trichrome stain).
The histologic abnormalities in DCM are nonspecific and usually do not point to a specific etiologic agent. Moreover, the severity of morphologic changes may not reflect either the degree of dysfunction or the patient’s prognosis. Most muscle cells are hypertrophied with enlarged nuclei, but some are attenuated, stretched, and irregular. Interstitial and endocardial fibrosis of variable degree is present, and small subendocardial scars may replace individual cells or groups of cells, probably reflecting healing of previous ischemic necrosis of myocytes caused by hypertrophy-induced imbalance between perfusion and demand.
Pathogenesis.
Although many individuals with DCM have a familial (genetic) form, DCM can also result from various acquired myocardial insults or interactions of genetics and the environment that ultimately may yield a similar clinicopathologic pattern.96 These include (1) myocarditis (an inflammatory disorder that precedes the development of cardiomyopathy in at least some cases, and is sometimes caused by viral infections), (2) toxicities (including adverse effects of chemotherapeutic agents and chronic alcoholism, a history of which can be elicited in 10% to 20% of patients), and (3) childbirth. Each of these subgroups are summarized in Figure 12-32 and described below.
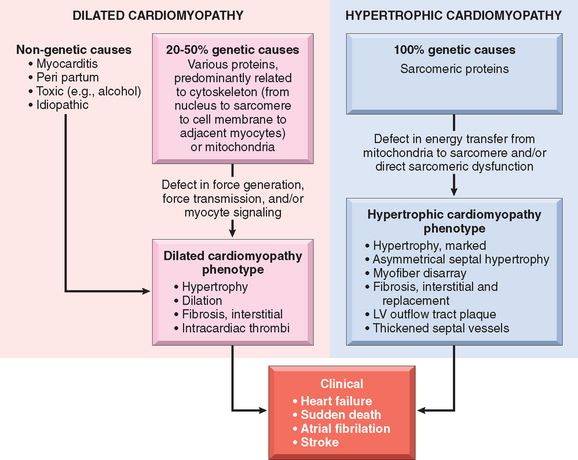
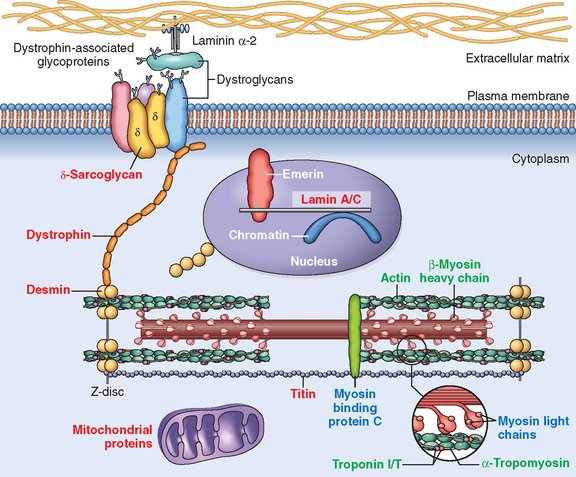
FIGURE 12-32 Causes and consequences of dilated and hypertrophic cardiomyopathy. Some dilated cardiomyopathies and virtually all hypertrophic cardiomyopathies are genetic in origin. The genetic causes of dilated cardiomyopathy involve mutations in any of a wide variety of proteins, predominantly of the cytoskeleton, but also the sarcomere, mitochondria, and nuclear envelope. In contrast, the mutated genes that cause hypertrophic cardiomyopathy encode proteins of the sarcomere. Although these two forms of cardiomyopathy differ greatly in subcellular basis and morphologic phenotypes, they share a common set of clinical complications. LV, left ventricle.
Clinical Features.
DCM may occur at any age, including in childhood, but it most commonly affects individuals between the ages of 20 and 50. It presents with slowly progressive signs and symptoms of CHF such as shortness of breath, easy fatigability, and poor exertional capacity. In the end stage, patients often have ejection fractions of less than 25% (normal, ∼50% to 65%). Fifty percent of patients die within 2 years, and only 25% survive longer than 5 years, but some severely affected patients may unexpectedly improve on therapy. Secondary mitral regurgitation and abnormal cardiac rhythms are common. Death is usually attributable to progressive cardiac failure or arrhythmia and can occur suddenly. Embolism from dislodgment of an intracardiac thrombus can occur. Cardiac transplantation is frequently done, and long-term ventricular assist may be beneficial in some patients. Interestingly, mechanical cardiac assist may induce lasting regression of cardiac dysfunction in some patients.104
Arrhythmogenic Right Ventricular Cardiomyopathy (Arrhythmogenic Right Ventricular Dysplasia)
Arrhythmogenic right ventricular cardiomyopathy (ARVC), or arrhythmogenic right ventricular dysplasia, is an inherited disease of the cardiac muscle that causes right ventricular failure and various rhythm disturbances, particularly ventricular tachycardia or fibrillation that can lead to sudden death, primarily in young people.105 Left-sided involvement with left-sided heart failure may also occur. Morphologically, the right ventricular wall is severely thinned because of loss of myocytes, with extensive fatty infiltration and fibrosis (Fig. 12-34). The condition appears to have autosomal-dominant inheritance and variable penetrance. The disease seems to be related to defective cell adhesion proteins in the desmosomes that link adjacent cardiac myocytes. Naxos syndrome is a disorder characterized by arrhythmogenic right ventricular cardiomyopathy and hyperkeratosis of plantar palmar skin surfaces that is associated with mutations in the gene encoding plakoglobin.106
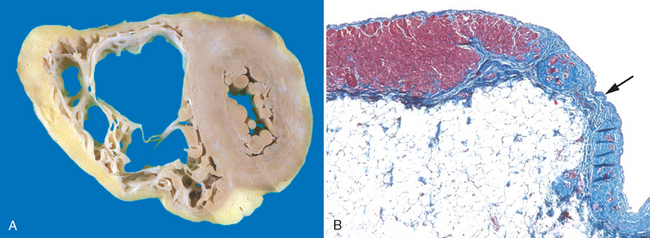
FIGURE 12-34 Arrhythmogenic right ventricular cardiomyopathy. A, Gross photograph, showing dilation of the right ventricle and near-transmural replacement of the right ventricular free-wall by fat and fibrosis. The left ventricle has a virtually normal configuration. B, Histologic section of the right ventricular free wall, demonstrating replacement of myocardium (red) by fibrosis (blue, arrow) and fat (Masson trichrome stain).
HYPERTROPHIC CARDIOMYOPATHY
Hypertrophic cardiomyopathy (HCM) is characterized by myocardial hypertrophy, poorly compliant left ventricular myocardium leading to abnormal diastolic filling, and in about one third of cases, intermittent ventricular outflow obstruction. It is the leading cause of left ventricular hypertrophy unexplained by other clinical or pathologic causes.107 As discussed below, HCM is caused by mutations in genes encoding sarcomeric proteins. Since the incidence of unexplained cardiac hypertrophy is approximately 1 in 500, HCM may be the most common cardiovascular disorder caused by single gene mutations. The heart is thick-walled, heavy, and hypercontracting, in striking contrast to the flabby, hypocontracting heart of DCM. HCM causes primarily diastolic dysfunction; systolic function is usually preserved. The two most common diseases that must be distinguished clinically from HCM are deposition diseases of the heart (e.g., amyloidosis, Fabry’s disease) and hypertensive heart disease coupled with age-related subaortic septal hypertrophy (see “Hypertensive Heart Disease”). Occasionally, valvular or congenital subvalvular aortic stenosis can also mimic HCM.
Morphology. The essential feature of HCM is massive myocardial hypertrophy, usually without ventricular dilation (Fig. 12-35). The classic pattern is disproportionate thickening of the ventricular septum as compared with the free wall of the left ventricle (with a ratio greater than 1 : 3), frequently termed asymmetric septal hypertrophy. In about 10% of cases, however, the hypertrophy is symmetrical throughout the heart. On cross-section, the ventricular cavity loses its usual round-to-ovoid shape and may be compressed into a “banana-like” configuration by bulging of the ventricular septum into the lumen (Fig. 12-35A). Although marked hypertrophy can involve the entire septum, it is usually most prominent in the subaortic region. Often present are endocardial thickening or mural plaque formation in the left ventricular outflow tract and thickening of the anterior mitral leaflet. Both findings are a result of contact of the anterior mitral leaflet with the septum during ventricular systole, and they correlate with echocardiographically demonstrated functional left ventricular outflow tract obstruction during midsystole.
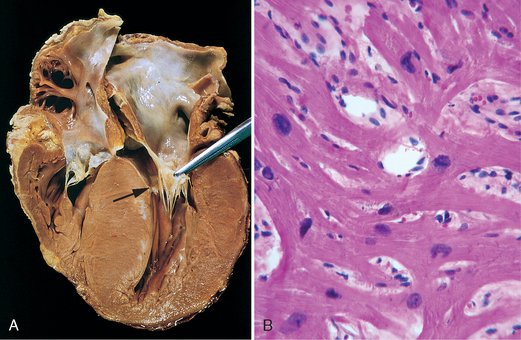
FIGURE 12-35 Hypertrophic cardiomyopathy with asymmetric septal hypertrophy. A, The septal muscle bulges into the left ventricular outflow tract, and the left atrium is enlarged. The anterior mitral leaflet has been moved away from the septum to reveal a fibrous endocardial plaque (arrow) (see text). B, Histologic appearance demonstrating disarray, extreme hypertrophy, and branching of myocytes as well as the characteristic interstitial fibrosis (collagen is blue in this Masson trichrome stain).
The most important histologic features of the myocardium in HCM are (1) extensive myocyte hypertrophy to a degree unusual in other conditions, with transverse myocyte diameters frequently greater than 40 μm (normal, ∼15 μm); (2) haphazard disarray of bundles of myocytes, individual myocytes, and contractile elements in sarcomeres within cells (termed myofiber disarray); and (3) interstitial and replacement fibrosis (Fig. 12-35B).
Pathogenesis.
HCM is caused by mutations in any one of several genes that encode sarcomeric proteins.108 In most cases the pattern of transmission is autosomal dominant with variable penetrance. Remaining cases seem to be sporadic. More than 400 different mutations have been found in nine different genes in HCM, most being missense mutations. Mutations causing HCM are found most commonly in the gene encoding β-myosin heavy chain (β-MHC), with the genes for cardiac TnT, α-tropomyosin, and myosin-binding protein C (MYBP-C) being the next most frequently mutated. Mutations in β-MHC, MYBP-C, and TnT account for 70% to 80% of all cases of HCM. Different affected families may have distinct mutations involving the same protein. For example, approximately 50 different mutations of β-MHC are known to cause HCM. The prognosis of HCM varies widely and correlates strongly with specific mutations. Although it is clear that these genetic defects are critical to the etiology of HCM, the sequence of events leading from mutations to disease is still poorly understood. To make matters even more complicated, changes in certain genes, depicted in Figure 12-33, can give rise to both HCM and DCM.
Clinical Features.
The basic physiologic abnormality in HCM is reduced stroke volume due to impaired diastolic filling, which results from the reduced chamber size and compliance of the massively hypertrophied left ventricle. In addition, approximately 25% of patients with HCM have dynamic obstruction to the left ventricular outflow. The limitation of cardiac output and a secondary increase in pulmonary venous pressure cause exertional dyspnea. Auscultation discloses a harsh systolic ejection murmur, caused by ventricular outflow obstruction as the anterior mitral leaflet moves toward the ventricular septum during systole. Because of the massive hypertrophy, high left ventricular chamber pressure, and frequently abnormal intramural arteries, focal myocardial ischemia commonly results, even in the absence of concomitant coronary artery disease; thus, anginal pain is frequent. The major clinical problems in HCM are atrial fibrillation, mural thrombus formation leading to embolization and possible stroke, intractable cardiac failure, ventricular arrhythmias, and, not infrequently, sudden death, especially in some affected families. Indeed, HCM is one of the most common causes of sudden, otherwise unexplained death in young athletes.109
The natural history of HCM is highly variable. Most patients can be helped by treatment with drugs that decrease heart rate and contractility, such as β-adrenergic blockers. Some benefit may also be gained from reduction of the mass of the septum, which relieves the outflow tract obstruction. This can be achieved either by surgical excision of muscle or carefully controlled septal infarction, which is induced by infusion of alcohol through a catheter.
As discussed above, HCM is a disease caused by mutations in proteins of the sarcomere, and DCM is mostly associated with abnormalities of cytoskeletal proteins (Fig. 12-33). DCM seems to be a disease of abnormal force generation, force transmission, or myocyte signaling. In the past, HCM has been considered a disorder of sarcomeres that impairs cardiac function and induces a compensatory hypertrophic response. However, recent evidence suggests that HCM may stem from a defect in energy transfer from its source of generation (mitochondria) to its site of use (sarcomeres), leading to subcellular energy deficiency. Despite these etiologic differences, there are some common mechanistic and clinicopathologic threads between HCM and the genetic and acquired forms of DCM, as summarized in Figure 12-32.
RESTRICTIVE CARDIOMYOPATHY
Restrictive cardiomyopathy is a disorder characterized by a primary decrease in ventricular compliance, resulting in impaired ventricular filling during diastole. Because the contractile (systolic) function of the left ventricle is usually unaffected, the functional abnormality can be confused with that of constrictive pericarditis or HCM.110 Restrictive cardiomyopathy may be idiopathic or associated with distinct diseases or processes that affect the myocardium, principally radiation fibrosis, amyloidosis, sarcoidosis, metastatic tumors, or the deposition of metabolites that accumulate due to inborn errors of metabolism.
Morphology. The ventricles are of approximately normal size or slightly enlarged, the cavities are not dilated, and the myocardium is firm and noncompliant. Biatrial dilation is commonly observed. Microscopically, there may be only patchy or diffuse interstitial fibrosis, which can vary from minimal to extensive. However, endomyocardial biopsy will often reveal a specific etiology. An important specific subgroup is amyloidosis (described later).
Several other restrictive conditions require brief mention. Endomyocardial fibrosis is principally a disease of children and young adults in Africa and other tropical areas, characterized by fibrosis of the ventricular endocardium and subendocardium that extends from the apex upward, often involving the tricuspid and mitral valves. The fibrous tissue markedly diminishes the volume and compliance of affected chambers and so induces a restrictive functional defect. Ventricular mural thrombi sometimes develop, and indeed there is a suggestion that the fibrous tissue results from the organization of mural thrombi. The etiology is unknown.
Loeffler endomyocarditis also results in endomyocardial fibrosis, typically with large mural thrombi. It is similar in morphology to the tropical disease, but in addition to the cardiac changes, there is often a peripheral eosinophilia (i.e., elevated blood eosinophils) and eosinophilic infiltrates in other organs. The release of toxic products of eosinophils, especially major basic protein, is postulated to initiate endomyocardial necrosis, followed by scarring of the necrotic area, layering of the endocardium by thrombus, and finally organization of the thrombus. It is now recognized that some patients with Loeffler endomyocarditis have a myeloproliferative disorder associated with chromosomal rearrangements involving either the PDGFRα or PDGFRβ genes (Chapter 13). These rearrangements produce fusion genes that encode constitutively active PDGFR tyrosine kinases. Treatment of such patients with the tyrosine kinase inhibitor imatinib has resulted in hematologic remissions associated with reversal of the endomyocarditis, which otherwise is often rapidly fatal.
Endocardial fibroelastosis is an uncommon heart disease of obscure etiology characterized by focal or diffuse fibroelastic thickening usually involving the mural left ventricular endocardium. It is most common in the first 2 years of life, and is accompanied by aortic valve obstruction or other congenital cardiac anomalies in about one third of cases. Diffuse involvement may be responsible for rapid and progressive cardiac decompensation and death.
MYOCARDITIS
Under the designation myocarditis are a diverse group of pathologic entities in which infectious microorganisms and/or an inflammatory process cause myocardial injury.111 These disorders must be distinguished from conditions, such as ischemic heart disease, in which injuries caused by other mechanisms lead to inflammation secondarily.
Etiology and Pathogenesis.
In the United States, viral infections are the most common cause of myocarditis. Coxsackieviruses A and B and other enteroviruses probably account for most of the cases. Other less common etiologic agents include cytomegalovirus, HIV, and a host of other agents (listed in Table 12-12). The responsible virus can sometimes be identified by serologic studies or, more recently, identification of viral nucleic acids (DNA or RNA) in myocardial biopsies. Whether viruses cause the myocardial injury directly or initiate a destructive immune response is unclear.
TABLE 12-12 Major Causes of Myocarditis
| INFECTIONS |
| IMMUNE-MEDIATED REACTIONS |
| UNKNOWN |
HIV, human immunodeficiency virus.
Nonviral agents are also important causes of infectious myocarditis, particularly the protozoa Trypanosoma cruzi, the agent of Chagas disease.112 Chagas disease is endemic in some regions of South America; myocardial involvement is present in most infected individuals. About 10% of patients die during an acute attack; others develop a chronic immune-mediated myocarditis that may progress to cardiac insufficiency 10 to 20 years later. Trichinosis is the most common helminthic disease associated with myocarditis. Parasitic diseases, including toxoplasmosis, and bacterial infections, including Lyme disease and diphtheria, can also cause myocarditis. In the case of diphtheritic myocarditis, toxins released by Corynebacterium diphtheriae seem to be responsible for the myocardial injury. Myocarditis occurs in approximately 5% of patients with Lyme disease, a systemic illness caused by the bacterial spirochete Borrelia burgdorferi (Chapter 8). Lyme myocarditis manifests primarily as a self-limited conduction system disorder that frequently requires a temporary pacemaker. Myocarditis occurs in many patients with AIDS. Two types have been identified: (1) inflammation and myocyte damage without a clear etiologic agent and (2) myocarditis caused by HIV directly or by an opportunistic pathogen.
There are also noninfectious causes of myocarditis. Myocarditis can be caused by hypersensitivity reactions (hypersensitivity myocarditis), often to drugs such as antibiotics, diuretics, or antihypertensive agents. Myocarditis can also be associated with systemic diseases of immune origin, such as rheumatic fever, systemic lupus erythematosus, and polymyositis. Cardiac sarcoidosis and rejection of a transplanted heart (see Figure 12-40) are also considered forms of myocarditis.

FIGURE 12-40 Complications of heart transplantation. A, Cardiac allograft rejection typified by lymphocytic infiltrate, with associated damage to cardiac myocytes. B, Graft coronary arteriosclerosis, demonstrating severe diffuse concentric intimal thickening producing critical stenosis. The internal elastic lamina (arrow) and media are intact (Movat pentachrome stain, elastin black).
(B, Reproduced by permission from Salomon RN et al.: Human coronary transplantation-associated arteriosclerosis. Evidence for chronic immune reaction to activated graft endothelial cells. Am J Pathol 138:791, 1991.)
Morphology. During the active phase of myocarditis the heart may appear normal or dilated; some hypertrophy may be present depending on disease duration. In advanced stages the ventricular myocardium is flabby and often mottled by either pale foci or minute hemorrhagic lesions. Mural thrombi may be present in any chamber.
During active disease, myocarditis is most frequently characterized by an interstitial inflammatory infiltrate associated with focal myocyte necrosis (Fig. 12-36). A diffuse, mononuclear, predominantly lymphocytic infiltrate is most common (Fig. 12-36A). Although endomyocardial biopsies are diagnostic in some cases, they can be spuriously negative because inflammatory involvement of the myocardium may be focal or patchy. If the patient survives the acute phase of myocarditis, the inflammatory lesions either resolve, leaving no residual changes, or heal by progressive fibrosis.
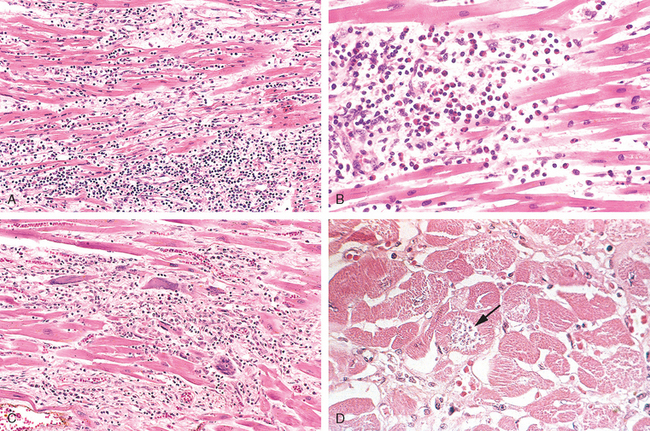
FIGURE 12-36 Myocarditis. A, Lymphocytic myocarditis, associated with myocyte injury. B, Hypersensitivity myocarditis, characterized by interstitial inflammatory infiltrate composed largely of eosinophils and mononuclear inflammatory cells, predominantly localized to perivascular and expanded interstitial spaces. C, Giant-cell myocarditis, with mononuclear inflammatory infiltrate containing lymphocytes and macrophages, extensive loss of muscle, and multinucleated giant cells. D, The myocarditis of Chagas disease. A myofiber distended with trypanosomes (arrow) is present along with inflammation and necrosis of individual myofibers.
Hypersensitivity myocarditis has interstitial infiltrates, principally perivascular, composed of lymphocytes, macrophages, and a high proportion of eosinophils (Fig. 12-36B). A morphologically distinctive form of myocarditis of uncertain cause, called giant-cell myocarditis, is characterized by a widespread inflammatory cellular infiltrate containing multinucleate giant cells interspersed with lymphocytes, eosinophils, plasma cells, and macrophages. Focal to frequently extensive necrosis is present (see Fig. 12-36C). This variant carries a poor prognosis.
The myocarditis of Chagas disease is rendered distinctive by parasitization of scattered myofibers by trypanosomes accompanied by an inflammatory infiltrate of neutrophils, lymphocytes, macrophages, and occasional eosinophils (see Fig. 12-36D).
Clinical Features.
The clinical spectrum of myocarditis is broad. At one end the disease is entirely asymptomatic, and such patients recover completely without sequelae; at the other extreme is the precipitous onset of heart failure or arrhythmias, occasionally with sudden death. Between these extremes are the many levels of involvement associated with symptoms such as fatigue, dyspnea, palpitations, precordial discomfort, and fever. The clinical features of myocarditis can mimic those of acute MI. Occasionally, patients develop dilated cardiomyopathy as a late complication of myocarditis.
OTHER CAUSES OF MYOCARDIAL DISEASE
Cardiotoxic Drugs.
Cardiac complications of cancer therapy are an important clinical problem.113 Cardiotoxicity can be associated with conventional chemotherapeutic agents, targeted drugs such as tyrosine kinase inhibitors, and certain forms of immunotherapy.114,115 The anthracyclines doxorubicin and daunorubicin are the chemotherapeutic agents that are most often associated with toxic myocardial injury, which often leads to a dilated cardiomyopathy and heart failure. Anthracycline toxicity is dose-dependent (cardiotoxicity becomes progressively more frequent above a total dose of 500 mg/m2) and is attributed primarily to peroxidation of lipids in myocyte membranes.
Many other pharmaceuticals and other agents, such as lithium, phenothiazines, chloroquine, and cocaine, have been implicated in myocardial injury and sometimes sudden death. Common findings in hearts injured by many of these chemicals and drugs (including diphtheria exotoxin and doxorubicin) are myofiber swelling, cytoplasmic vacuolization, and fatty change. With discontinuance of the toxic agent these changes may resolve completely, leaving no apparent sequelae. Sometimes, however, more extensive damage produces myocyte necrosis, which can evolve to a dilated cardiomyopathy.
Catecholamines.
Foci of myocardial necrosis with contraction bands, often associated with a sparse mononuclear inflammatory infiltrate consisting mostly of macrophages, can occur in individuals with a pheochromocytoma, a tumor that elaborates catecholamines (Chapter 24). This is considered to be a manifestation of the general problem of “catecholamine effect,” also seen in association with intense autonomic stimulation (secondary to intracranial lesions or stress), or the exogenous administration of large doses of vasopressor agents such as dopamine.116 Sudden, intense emotional or physical stress can also induce acute left ventricular dysfunction due to myocardial stunning, a phenomenon known as Takotsubo cardiomyopathy.117 Cocaine also causes similar catecholamine-mediated damage. The mechanism of catecholamine cardiotoxicity is uncertain, but it seems to relate either to direct toxicity of catecholamines to cardiac myocytes via calcium overload or to focal vasoconstriction in the coronary arterial macro- or microcirculation in the face of an increased heart rate. The mononuclear cell infiltrate is probably a secondary reaction to the foci of myocyte cell death. Similar changes may be encountered in individuals who have recovered from hypotensive episodes or have been resuscitated from a cardiac arrest; in such cases the damage is a result of ischemia-reperfusion (see earlier) with subsequent inflammation.
Amyloidosis.
Amyloidosis is a prototypical myocardial disorder caused by deposition of an abnormal substance in the heart. Amyloidosis (Chapter 6) is caused by insoluble extracellular fibrillar deposits of protein fragments that are prone to forming β-pleated sheets.118 Cardiac amyloidosis may appear along with systemic amyloidosis or be restricted to the heart, particularly in the aged (senile cardiac amyloidosis).119 In senile cardiac amyloidosis, the amyloid deposits generally occur in the ventricles and atria. Senile cardiac amyloidosis occurs in older individuals and is caused by the deposition of transthyretin, a normal serum protein synthesized in the liver that is responsible for transporting thyroxine and retinol-binding protein. Senile cardiac amyloidosis120 has a far better prognosis than systemic amyloidosis. Mutant forms of transthyretin can accelerate cardiac (and associated systemic) amyloidosis. For example, the risk of isolated cardiac amyloidosis is four times greater in African Americans than in Caucasians after 60 years of age because 4% of African Americans have a transthyretin mutation leading to the substitution of isoleucine for valine at position 122. This substitution produces an amyloidogenic form of transthyretin (responsible for autosomal-dominant familial transthyretin amyloidosis). Isolated atrial amyloidosis can also occur secondary to deposition of atrial natriuretic peptide, but its clinical significance is uncertain.
Cardiac amyloidosis most frequently produces a restrictive cardiomyopathy, but it can also be asymptomatic or manifest as dilation, arrhythmias, or symptoms mimicking those of ischemic or valvular disease. These varied presentations depend on the predominant location of the deposits, which can be found in the interstitium, conduction system, vasculature, or valves.
Morphology. The heart varies in consistency from normal to firm and rubbery. Usually the chambers are of normal size, but in some cases they are dilated and have thickened walls. Numerous small, semitranslucent nodules resembling drips of wax may be seen at the atrial endocardial surface, particularly on the left. Eosinophilic deposits of amyloid may be found in the interstitium, conduction tissue, valves, endocardium, pericardium, and small intramural coronary arteries; they can be distinguished from other hyaline deposits by special stains such as Congo red, which produces classic apple-green birefringence when viewed under polarized light (Fig. 12-37). Amyloid deposits often form rings around cardiac myocytes and capillaries. Intramural arteries and arterioles may have sufficient amyloid in their walls to compress and occlude their lumens, inducing myocardial ischemia (“small-vessel disease”).
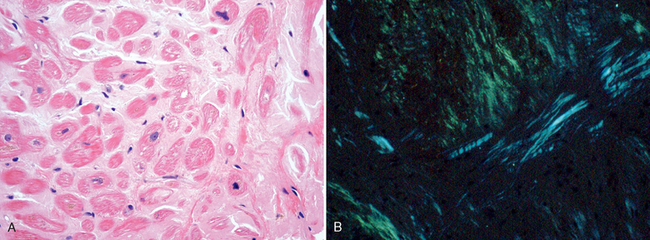
Iron Overload.
Iron overload can result from either hereditary hemochromatosis (Chapter 18) or multiple blood transfusions (hemosiderosis). The heart in each is usually dilated. Iron deposition is more prominent in ventricles than atria and in the myocardium than in the conduction system. It is thought that iron causes systolic dysfunction by interfering with metal-dependent enzyme systems or by inducing oxygen free-radical injury.
Morphology. Grossly, the myocardium of the iron-overloaded heart is rust-brown in color but is usually otherwise indistinguishable from that of idiopathic dilated cardiomyopathy. Microscopically, there is marked accumulation of hemosiderin within cardiac myocytes, particularly in the perinuclear region, demonstrable with a Prussian blue stain. This is associated with varying degrees of cellular degeneration and fibrosis. Ultrastructurally, the cardiac myocytes contain abundant perinuclear siderosomes (ironcontaining lysosomes).
Hyperthyroidism and Hypothyroidism.
Cardiac manifestations are among the earliest, most consistent features of hyperthyroidism and hypothyroidism, and they reflect direct and indirect effects of thyroid hormones on the cells of the heart.121 In hyperthyroidism (Chapter 24), tachycardia, palpitations, and cardiomegaly are common; supraventricular arrhythmias occasionally appear. Cardiac failure is uncommon; when it occurs in this setting, it is usually superimposed on other cardiac diseases. In hypothyroidism (Chapter 24), cardiac output is decreased, due to reductions in stroke volume and heart rate. Increased peripheral vascular resistance and decreased blood volume result in narrowing of the pulse pressure, prolongation of circulation time, and decreased flow to peripheral tissues.
Morphology. In hyperthyroidism the gross and histologic features are those of nonspecific hypertrophy and can also include ischemic foci. In well-advanced hypothyroidism (myxedema) the heart is flabby, enlarged, and dilated. Histologic features of hypothyroidism include myofiber swelling with loss of striations and basophilic degeneration, accompanied by interstitial mucopolysaccharide-rich edema fluid. A similar fluid sometimes accumulates within the pericardial sac. The term myxedema heart has been applied to these changes.