Pulmonary Infections
Respiratory tract infections are more frequent than infections of any other organ and account for the largest number of workdays lost in the general population. The vast majority are upper respiratory tract infections caused by viruses (common cold, pharyngitis), but bacterial, viral, mycoplasmal, and fungal infections of the lung (pneumonia) still account for an enormous amount of morbidity and are responsible for one sixth of all deaths in the United States.121 Pneumonia can be very broadly defined as any infection of the lung parenchyma.
Pulmonary defense mechanisms are described in Chapter 8. Pneumonia can result whenever these local defense mechanisms are impaired or the systemic resistance of the host is lowered. Factors that affect resistance in general include chronic diseases, immunological deficiency, treatment with immunosuppressive agents, and leukopenia. The local defense mechanisms of the lung can be interfered with by many factors, such as the following:
Defects in innate immunity (including neutrophil and complement defects) and humoral immunodeficiency typically lead to an increased incidence of infections with pyogenic bacteria. On the other hand, cell-mediated immune defects (congenital and acquired) lead to increased infections with intracellular microbes such as mycobacteria and herpesviruses as well as with microorganisms of very low virulence, such as Pneumocystis jiroveci.
Several other points should be emphasized. First, one type of pneumonia sometimes predisposes to another, especially in debilitated patients. For example, the most common cause of death in viral influenza epidemics is superimposed bacterial pneumonia. Second, although the portal of entry for most pneumonias is the respiratory tract, hematogenous spread from one organ to other organs can occur, and secondary seeding of the lungs may be difficult to distinguish from primary pneumonia. Finally, many patients with chronic diseases acquire terminal pneumonias while hospitalized (nosocomial infection). Bacteria common to the hospital environment may have acquired resistance to antibiotics; opportunities for spread are increased; invasive procedures, such as intubations and injections, are common; and bacteria may contaminate equipment used in respiratory care units.
Pneumonias are classified by the specific etiologic agent, which determines the treatment, or, if no pathogen can be isolated, by the clinical setting in which the infection occurs. The latter considerably narrows the list of suspected pathogens for administering empirical antimicrobial therapy. As Table 15-8 indicates, pneumonia can arise in seven distinct clinical settings (“pneumonia syndromes”), and the implicated pathogens are reasonably specific to each category.
TABLE 15-8 The Pneumonia Syndromes
| COMMUNITY-ACQUIRED ACUTE PNEUMONIA |
| COMMUNITY-ACQUIRED ATYPICAL PNEUMONIA |
| HOSPITAL-ACQUIRED PNEUMONIA |
| ASPIRATION PNEUMONIA |
| Anaerobic oral flora (Bacteroides, Prevotella, Fusobacterium, Peptostreptococcus), admixed with aerobic bacteria (Streptococcus pneumoniae, Staphylococcus aureus, Haemophilus influenzae, and Pseudomonas aeruginosa) |
| CHRONIC PNEUMONIA |
| NECROTIZING PNEUMONIA AND LUNG ABSCESS |
| PNEUMONIA IN THE IMMUNOCOMPROMISED HOST |
SARS, severe acute respiratory syndrome.
COMMUNITY-ACQUIRED ACUTE PNEUMONIAS
Community-acquired pneumonias may be bacterial or viral. Often, the bacterial infection follows an upper respiratory tract viral infection. Bacterial invasion of the lung parenchyma causes the alveoli to be filled with an inflammatory exudate, thus causing consolidation (“solidification”) of the pulmonary tissue. Many variables, such as the specific etiologic agent, the host reaction, and the extent of involvement, determine the precise form of pneumonia. Predisposing conditions include extremes of age, chronic diseases (congestive heart failure, COPD, and diabetes), congenital or acquired immune deficiencies, and decreased or absent splenic function (sickle cell disease or post-splenectomy, which puts the patient at risk for infection with encapsulated bacteria such as pneumococcus).
Streptococcus pneumoniae
Streptococcus pneumoniae, or pneumococcus, is the most common cause of community-acquired acute pneumonia. Examination of Gram-stained sputum is an important step in the diagnosis of acute pneumonia. The presence of numerous neutrophils containing the typical gram-positive, lancet-shaped diplococci supports the diagnosis of pneumococcal pneumonia, but it must be remembered that S. pneumoniae is a part of the endogenous flora in 20% of adults, and therefore false-positive results may be obtained. Isolation of pneumococcifrom blood cultures is more specific but less sensitive (in the early phase of illness, only 20% to 30% of patients have positive blood cultures). Pneumococcal vaccines containing capsular polysaccharides from the common serotypes are used in patients at high risk.
Haemophilus influenzae
Haemophilus influenzae is a pleomorphic, gram-negative organism that is a major cause of life-threatening acute lower respiratory tract infections and meningitis in young children. In adults it is a very common cause of community-acquired acute pneumonia.122 This bacterium is a ubiquitous colonizer of the pharynx, where it exists in two forms: encapsulated (5%) and unencapsulated (95%). Typically, the encapsulated form dominates the unencapsulated forms by secreting an antibiotic called haemocin that kills the unencapsulated H. influenzae.123 Although there are six serotypes of the encapsulated form (types a to f), type b, which has a polyribosephosphate capsule, used to be the most frequent cause of severe invasive disease. With routine use of H. influenzae conjugate vaccines, the incidence of disease caused by the b serotype has declined significantly. By contrast, infections with nonencapsulated forms are increasing. Also called nontypeable forms, they spread along the surface of the upper respiratory tract and produce otitis media (infection of the middle ear), sinusitis, and bronchopneumonia.
Pili on the surface of H. influenzae mediate adherence of the organisms to the respiratory epithelium.124 In addition, H. influenzae secretes a factor that disorganizes ciliary beating and a protease that degrades IgA, the major class of antibody secreted into the airways. Survival of H. influenzae in the bloodstream correlates with the presence of the capsule, which, like that of pneumococcus, prevents opsonization by complement and phagocytosis by host cells. Antibodies against the capsule protect the host from H. influenzae infection; hence the capsular polysaccharide b is incorporated in the vaccine against H. influenzae used for children.
H. influenzae pneumonia, which may follow a viral respiratory infection, is a pediatric emergency and has a high mortality rate. Descending laryngotracheobronchitis results in airway obstruction as the smaller bronchi are plugged by dense, fibrin-rich exudate of polymorphonuclear cells, similar to that seen in pneumococcal pneumonias. Pulmonary consolidation is usually lobular and patchy but may be confluent and involve the entire lung lobe. Before a vaccine became widely available, H. influenzae was a common cause of suppurative meningitis in children up to 5 years of age. H. influenzae also causes an acute, purulent conjunctivitis (pink eye) in children and, in predisposed older patients, may cause septicemia, endocarditis, pyelonephritis, cholecystitis, and suppurative arthritis. H. influenzae is the most common bacterial cause of acute exacerbation of COPD.
Moraxella catarrhalis
Moraxella catarrhalis is being increasingly recognized as a cause of bacterial pneumonia, especially in the elderly. It is the second most common bacterial cause of acute exacerbation of COPD. Along with S. pneumoniae and H. influenzae, M. catarrhalis constitutes one of the three most common causes of otitis media in children.
Staphylococcus aureus
Staphylococcus aureus is an important cause of secondary bacterial pneumonia in children and healthy adults following viral respiratory illnesses (e.g., measles in children and influenza in both children and adults). Staphylococcal pneumonia is associated with a high incidence of complications, such as lung abscess and empyema. Intravenous drug abusers are at high risk of developing staphylococcal pneumonia in association with endocarditis. It is also an important cause of hospital-acquired pneumonia, as will be discussed later.
Klebsiella pneumoniae
Klebsiella pneumoniae is the most frequent cause of gram-negative bacterial pneumonia. It commonly afflicts debilitated and malnourished people, particularly chronic alcoholics. Thick and gelatinous sputum is characteristic, because the organism produces an abundant viscid capsular polysaccharide, which the patient may have difficulty expectorating.
Pseudomonas aeruginosa
Although Pseudomonas aeruginosa most commonly causes hospital-acquired infections, it is mentioned here because of its occurrence in cystic fibrosis patients. It is common in patients who are neutropenic and it has a propensity to invade blood vessels with consequent extrapulmonary spread. Pseudomonas septicemia is a very fulminant disease.
Legionella pneumophila
Legionella pneumophila is the agent of Legionnaires’ disease, an eponym for the epidemic and sporadic forms of pneumonia caused by this organism. It also causes Pontiac fever, a related self-limited upper respiratory tract infection. This organism flourishes in artificial aquatic environments, such as water-cooling towers and within the tubing system of domestic (potable) water supplies. The mode of transmission is either inhalation of aerosolized organisms or aspiration of contaminated drinking water. Legionella pneumonia is common in individuals with some predisposing condition such as cardiac, renal, immunological, or hematologic disease. Organ transplant recipients are particularly susceptible. It can be quite severe, frequently requiring hospitalization, and immunosuppressed patients may have fatality rates of up to 50%. Rapid diagnosis is facilitated by demonstration of Legionella antigens in the urine or by a positive fluorescent antibody test on sputum samples; culture remains the gold standard of diagnosis.
Morphology. Bacterial pneumonia has two patterns of anatomic distribution: lobular bronchopneumonia and lobar pneumonia (Fig. 15-31). Patchy consolidation of the lung is the dominant characteristic of bronchopneumonia (Fig. 15-32), while fibrinosuppurative consolidation of a large portion of a lobe or of an entire lobe defines lobar pneumonia (Fig. 15-33). These anatomic but still classic categorizations are often difficult to apply in individual cases because patterns overlap. The patchy involvement may become confluent, producing virtually total lobar consolidation; in contrast, effective antibiotic therapy for any form of pneumonia may limit involvement to a subtotal consolidation. Moreover, the same organisms may produce either pattern depending on patient susceptibility. Most important from the clinical standpoint are identification of the causative agent and determination of the extent of disease.
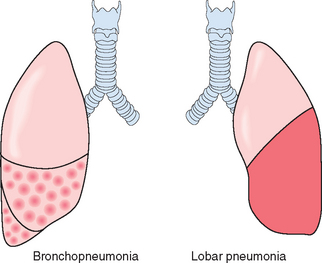


FIGURE 15-33 Lobar pneumonia—gray hepatization, gross photograph. The lower lobe is uniformly consolidated.
In lobar pneumonia, four stages of the inflammatory response have classically been described: congestion, red hepatization, gray hepatization, and resolution. Current effective antibiotic therapy frequently slows or halts the progression. In the first stage of congestion the lung is heavy, boggy, and red. It is characterized by vascular engorgement, intra-alveolar fluid with few neutrophils, and often the presence of numerous bacteria. The stage of red hepatization that follows is characterized by massive confluent exudation with neutrophils, red cells, and fibrin filling the alveolar spaces (Fig. 15-34A). On gross examination, the lobe now appears distinctly red, firm, and airless, with a liver-like consistency, hence the term hepatization. The stage of gray hepatization follows with progressive disintegration ofred cells and the persistence of a fibrinosuppurative exudate (Fig. 15-34B), giving the gross appearance of a grayish brown, dry surface. In the final stage of resolution the consolidated exudate within the alveolar spaces undergoes progressive enzymatic digestion to produce granular, semifluid debris that is resorbed, ingested by macrophages, expectorated, or organized by fibroblasts growing into it (Fig. 15-34C). Pleural fibrinous reaction to the underlying inflammation, often present in the early stages if the consolidation extends to the surface (pleuritis), may similarly resolve. More often it undergoes organization, leaving fibrous thickening or permanent adhesions.
Foci of bronchopneumonia are consolidated areas of acute suppurative inflammation. The consolidation may be patchy through one lobe but is more often multilobar and frequently bilateral and basal because of the tendency of secretions to gravitate into the lower lobes. Well-developed lesions are slightly elevated, dry, granular, gray-red to yellow, and poorly delimited at their margins (see Fig. 15-32). Histologically, the reaction usually elicits a suppurative, neutrophil-rich exudate that fills the bronchi, bronchioles, and adjacent alveolar spaces (Fig. 15-34A).
Complications of pneumonia include (1) tissue destruction and necrosis, causing abscess formation (particularly common with type 3 pneumococci or Klebsiella infections); (2) spread of infection to the pleural cavity, causing the intrapleural fibrinosuppurative reaction known as empyema; and (3) bacteremic dissemination to the heart valves, pericardium, brain, kidneys, spleen, or joints, causing metastatic abscesses, endocarditis, meningitis, or suppurative arthritis.
Clinical Course.
The major symptoms of community-acquired acute pneumonia are abrupt onset of high fever, shaking chills, and cough productive of mucopurulent sputum; occasional patients may have hemoptysis. When fibrinosuppurative pleuritis is present it is accompanied by pleuritic pain and pleural friction rub. The whole lobe is radiopaque in lobar pneumonia, whereas there are focal opacities in bronchopneumonia.
The clinical picture is markedly modified by the administration of antibiotics. Treated patients may be relatively afebrile with few clinical signs 48 to 72 hours after the initiation of antibiotics. The identification of the organism and the determination of its antibiotic sensitivity are the keystones to appropriate therapy. Fewer than 10% of patients with pneumonia severe enough to merit hospitalization now succumb, and in most such instances death may be attributed either to a complication, such as empyema, meningitis, endocarditis, or pericarditis, or to some predisposing influence, such as debility or chronic alcoholism.
COMMUNITY-ACQUIRED ATYPICAL (VIRAL AND MYCOPLASMAL) PNEUMONIAS
The term primary atypical pneumonia was initially applied to an acute febrile respiratory disease characterized by patchy inflammatory changes in the lungs, largely confined to the alveolar septa and pulmonary interstitium. The term atypical denotes the moderate amount of sputum, no physical findings of consolidation, only moderate elevation of white cell count, and lack of alveolar exudate. The pneumonitis is caused by a variety of organisms, the most common being Mycoplasma pneumoniae. Mycoplasma infections are particularly common among children and young adults. They occur sporadically or as local epidemics in closed communities (schools, military camps, and prisons). Other etiologic agents are viruses, including influenza virus types A and B, the respiratory syncytial viruses, human metapneumovirus, adenovirus, rhinoviruses, rubeola, and varicella viruses; Chlamydia pneumoniae; and Coxiella burnetii (Q fever).124,125 In some cases the cause cannot be determined. Any one of these agents can cause merely an upper respiratory tract infection, recognized as the common cold, or a more severe lower respiratory tract infection. Factors that favor such extension of the infection include extremes of age, malnutrition, alcoholism, and underlying debilitating illnesses.
The common pathogenetic mechanism is attachment of the organisms to the upper respiratory tract epithelium followed by necrosis of the cells and an inflammatory response. When the process extends to the alveoli there is usually interstitial inflammation, but there may also be some outpouring of fluid into alveolar spaces, so that on chest x-ray the changes may mimic bacterial pneumonia. Damage to and denudation of the respiratory epithelium inhibit mucociliary clearance and predispose to secondary bacterial infections.
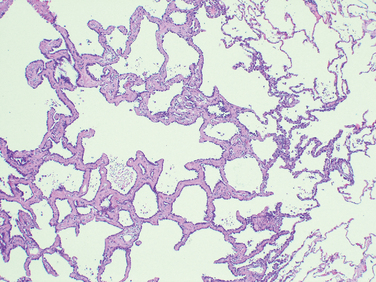
Morphology. All causal agents produce essentially similar morphologic patterns. The lung involvement may be quite patchy or may involve whole lobes bilaterally or unilaterally. The affected areas are red-blue and congested. The pleura is smooth, and pleuritis or pleural effusions are infrequent.
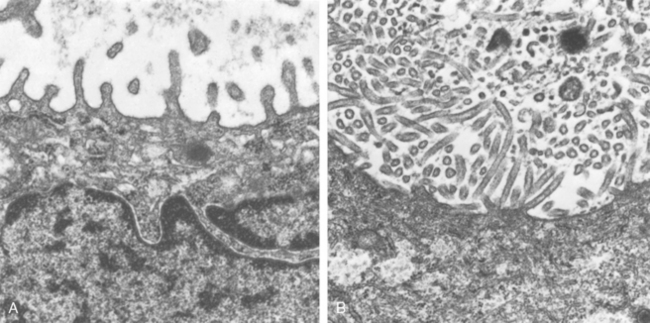
The histologic pattern depends on the severity of the disease. Predominant is the interstitial nature of the inflammatory reaction, virtually localized within the walls of the alveoli. The alveolar septa are widened and edematous and usually have a mononuclear inflammatory infiltrate of lymphocytes, macrophages, and occasionally plasma cells. In acute cases neutrophils may also be present. The alveoli may be free from exudate, but in many patients there is intra-alveolar proteinaceous material and a cellular exudate. When complicated by ARDS, characteristically pink hyaline membranes lining the alveolar walls are present (see Fig. 15-3). Eradication of the infection is followed by reconstitution of the normal architecture of the lung.
Superimposed bacterial infection modifies the histologic picture by causing ulcerative bronchitis, bronchiolitis, and bacterial pneumonia. Some viruses, such as herpes simplex, varicella, and adenovirus, may be associated with necrosis of bronchial and alveolar epithelium and acute inflammation. Characteristic viral cytopathic changes are described in Chapter 8.
Clinical Course.
The clinical course is extremely varied. Many cases masquerade as severe upper respiratory tract infections or as chest colds. Even individuals with welldeveloped atypical pneumonia have few localizing symptoms. Cough may be absent, and the major manifestations may consist only of fever, headache, muscle aches, and pains in the legs. The edema and exudation are both strategically located to cause mismatching of ventilation and blood flow and thus evoke symptoms out of proportion to the scanty physical findings.
The ordinary sporadic form of the disease is usually mild with a low mortality rate, below 1%. Interstitial pneumonia, however, may assume epidemic proportions with intensified severity and greater mortality, as documented in the devastating influenzal pandemics of 1915 and 1918 and the many smaller epidemics since then. Secondary bacterial infection by staphylococci or streptococci is common in such circumstances.
Influenza Infections
The genome of influenza virus is composed of eight helices of single-stranded RNA, each encoding a single gene and each bound by a nucleoprotein that determines the type of influenza virus (A, B, or C). The spherical surface of influenza virus is a lipid bilayer (envelope) containing the viral hemagglutinin and neuraminidase, which determine the subtype of the virus (H1 to H3; N1 or N2). Host antibodies to the hemagglutinin and neuraminidase prevent and ameliorate, respectively, future infection with the influenza virus. Two mechanisms account for the clearance of primary influenza virus infection: cytotoxic T cells kill virus-infected cells, and an intracellular anti-influenza protein (called Mx1) is induced in macrophages by the cytokines IFN-α and IFN-β.126
Influenza viruses of type A infect humans, pigs, horses, and birds and are the major cause of pandemic and epidemic influenza infections. A single subtype of influenza virus A predominates throughout the world at a given time.127 Epidemics of influenza occur through mutations of the hemagglutinin and neuraminidase that allow the virus to escape most host antibodies (antigenic drift). Pandemics, which are longer and more widespread than epidemics, may occur when both the hemagglutinin and the neuraminidase are replaced through recombination of RNA segments with those of animal viruses, making all individuals susceptible to the new influenza virus (antigenic shift). Polymerase chain reaction (PCR) analysis of influenza virus from the lungs of a soldier who died in the 1918 influenza pandemic that killed between 20 million and 40 million people worldwide identified a swine influenza virus belonging to the same family of influenza viruses causing illness today.128 Current antiviral drugs are effective against recombinant influenza viruses bearing the 1918 hemagglutinin, neuraminidase, and matrix genes.129 Influenza virus types B and C, which do not show antigenic drift or shift, infect mostly children, who develop antibodies that prevent reinfection. Rarely, influenza virus may cause interstitial myocarditis or, after aspirin therapy, Reye syndrome (Chapter 18).
Avian influenza refers to strains of influenza which primarily infect birds. One such strain with the antigenic type H5N1 is of great concern because infection is frequently lethal in humans (approximately 60%) and since 2003 the virus is spreading throughout the world in wild and domestic birds. As of the fall of 2008, a total of 387 H5N1 influenza virus infections in humans have been reported to the WHO. Nearly all cases of H5N1 influenza in humans have been acquired by close contact with domestic birds. The severity of the disease results from the ability of the virus to cause widespread infection in the human body, instead of infection being limited to the lung. The tissue tropism of H5N1 influenza is increased due to the unusual structure of its hemagglutinin protein. Cleavage of viral hemagglutinin by host proteases is required for the influenza virus to enter host cells. The hemagglutinin protein of H5N1 influenza virus, and other highly pathogenic influenza viruses, is unusual in that it can be cleaved by ubiquitous proteases in the human, while the hemagglutinin of less virulent influenza virus stains can only be cleaved by proteases found in limited organs, including the lung. Fortunately, the transmission of the current H5N1 virus is inefficient. Most patients with H5NI infection present with pneumonia. However, if antigenic recombination occurs between H5N1 influenza and a strain of influenza which is highly infectious for humans, sustained human-to-human transmission could give rise to a pandemic, similar to the Spanish pandemic of 1918. This concern has spurred efforts to develop a vaccine.130
Morphology. Viral upper respiratory infections are marked by mucosal hyperemia and swelling with a predominantly lymphomonocytic and plasmacytic infiltration of the submucosa accompanied by overproduction of mucus secretions. The swollen mucosa and viscous exudate may plug the nasal channels, sinuses or the Eustachian tubes, and lead to suppurative secondary bacterial infection. Virus-induced tonsillitis with enlargement of the lymphoid tissue within the Waldeyer ring is frequent in children, although lymphoid hyperplasia is not usually associated with suppuration or abscess formation, such as is encountered with streptococci or staphylococci.
In laryngotracheobronchitis and bronchiolitis there is vocal cord swelling and abundant mucus exudation. Impairment of bronchociliary function invites bacterial superinfection with more marked suppuration. Plugging of small airways may give rise to focal lung atelectasis. In the more severe bronchiolar involvement widespread plugging of secondary and terminal airways by cell debris, fibrin, and inflammatory exudate may, when prolonged, cause organization and fibrosis, resulting in obliterative bronchiolitis and permanent lung damage.
Human Metapneumovirus (MPV)
Human MPV, a paramyxovirus discovered in 2001, is found worldwide and is associated with upper and lower respiratory tract infections, most commonly in young children, elderly subjects, and immunocompromised patients. Human MPV can cause severe infections such as bronchiolitis and pneumonia and is responsible for 5% to 10% of hospitalizations and 12% to 20% of outpatient visits of children suffering from acute respiratory tract infections. Such infections are clinically indistinguishable from those caused by human respiratory syncytial virus. The first human MPV infection occurs during early childhood, but reinfections are common throughout life, especially in older subjects. Molecular methods such as reverse transcriptase–PCR are the preferred diagnostic modality because of fastidious growth in cell culture. No commercial treatments are yet available for human MPV, although ribavirin has shown activity both in vitro and in animal models. Live attenuated vaccines produced by genetically altered virus have also shown good efficacy in animals.131
Severe Acute Respiratory Syndrome (SARS)
SARS first appeared in November 2002 in the Guangdong Province of China and subsequently spread to Hong Kong, Taiwan, Singapore, Vietnam, and Toronto, where large outbreaks also occurred.132 The ease of travel between continents clearly contributed to this pandemic. Between Fall 2002 and Spring 2003, there were more than 8000 cases of SARS, including 774 deaths. The worldwide epidemic was halted, perhaps in part because of public health measures, and the last cases of SARS were laboratory-associated infections reported in April 2004.125
After an incubation period of 2 to 10 days, SARS begins with a dry cough, malaise, myalgias, fever, and chills. A third of patients improve and resolve the infection, but the rest progress to severe respiratory disease with shortness of breath, tachypnea, and pleurisy, and nearly 10% of patients die from the illness, for which there is no specific treatment.
The cause of SARS is a previously undiscovered coronavirus. Nearly a third of upper respiratory infections are caused by coronaviruses, but the SARS virus differs from previously known coronaviruses in that it infects the lower respiratory tract and spreads throughout the body. The SARS virus seems to have been first transmitted to humans through contact with wild masked palm civets that are eaten in China. Subsequent cases were spread person-to-person, mainly through infected respiratory secretions, although some cases may have been contracted from stool.
SARS can be diagnosed either by detection of the virus by PCR or by detection of antibodies to the virus. Levels of the virus are low initially and peak 10 days after onset of illness, so testing of different specimens (respiratory secretions, blood, and stool) collected on several days may be needed to detect the virus. Detection of antibodies specific for the SARS virus is a very sensitive and specific test; however, patients may not have a measurable antibody response for up to 28 days after infection.
The pathophysiology of SARS is not understood, nor is it known why the virus moved from animals to humans. Most human SARS coronaviruses have a 29-nucleotide deletion in the RNA when compared to the virus found in wild animals, which may enhance its transmission or pathogenicity. In patients who have died of SARS the lungs show diffuse alveolar damage and multinucleated giant cells. Coronaviruses can be seen within pneumocytes by electron microscopy.
HOSPITAL-ACQUIRED PNEUMONIA
Hospital-acquired pneumonias are defined as pulmonary infections acquired in the course of a hospital stay. They are common in patients with severe underlying disease, immunosuppression, prolonged antibiotic therapy, or invasive access devices such as intravascular catheters. Patients on mechanical ventilation are at particularly high risk. Superimposed on an underlying disease (that caused hospitalization), hospital-acquired infections are serious and often lifethreatening complications. Gram-negative rods (Enterobacteriaceae and Pseudomonas species) and S. aureus are the most common isolates; unlike community-acquired pneumonias, S. pneumoniae is not a major pathogen.
ASPIRATION PNEUMONIA
Aspiration pneumonia occurs in markedly debilitated patients or those who aspirate gastric contents either while unconscious (e.g., after a stroke) or during repeated vomiting. These patients have abnormal gag and swallowing reflexes that predispose to aspiration. The resultant pneumonia is partly chemical because of the extremely irritating effects of the gastric acid, and partly bacterial (from the oral flora). Typically, more than one organism is recovered on culture, aerobes being more common than anaerobes. This type of pneumonia is often necrotizing, pursues a fulminant clinical course, and is a frequent cause of death. In those who survive, lung abscess is a common complication.
LUNG ABSCESS
The term “pulmonary abscess” describes a local suppurative process within the lung, characterized by necrosis of lung tissue. Oropharyngeal surgical procedures, sinobronchial infections, dental sepsis, and bronchiectasis play important roles in their development.
Etiology and Pathogenesis.
Although under appropriate circumstances any pathogen can produce an abscess, the commonly isolated organisms include aerobic and anaerobic streptococci, S. aureus, and a host of gram-negative organisms. Mixed infections often occur because of the important causal role played by inhalation of foreign material.133 Anaerobic organisms normally found in the oral cavity, including members of the Bacteroides, Fusobacterium, and Peptococcus species, are the exclusive isolates in about 60% of cases. The causative organisms are introduced by the following mechanisms:
When all these causes are excluded, there are still cases in which no reasonable basis for the abscess formation can be identified. These are referred to as primary cryptogenic lung abscesses.
Morphology. Abscesses vary in diameter from lesions of a few millimeters to large cavities of 5 to 6 cm. They may affect any part of the lung and may be single or multiple. Pulmonary abscesses due to aspiration are more common on the right (because of the more vertical right main bronchus) and are most often single. Abscesses that develop in the course of pneumonia or bronchiectasis are usually multiple, basal, and diffusely scattered. Septic emboli and pyemic abscesses are multiple and may affect any region of the lungs.
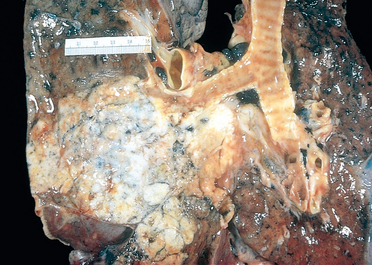
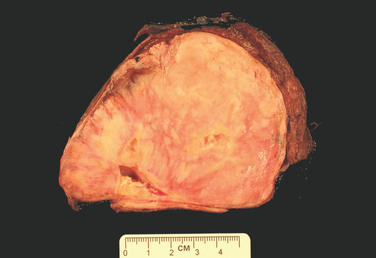
The abscess cavity might be filled with suppurative debris. If there is communication with an air passage, the contained exudate may be partially drained to create an air-containing cavity. Superimposed saprophytic infections are prone to flourishing within the already necrotic debris of the abscess cavity. Continued infection leads to large, fetid, green-black, multilocular cavities with poor demarcation of their margins, designated gangrene of the lung. The cardinal histologic change in all abscesses is suppurative destruction of the lung parenchyma within the central area of cavitation (Fig. 15-35). In chronic cases considerable fibroblastic proliferation produces a fibrous wall.
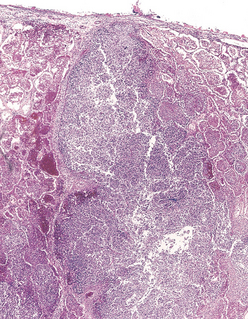
Clinical Course.
The manifestations of pulmonary abscesses are much like those of bronchiectasis and are characterized principally by cough, fever, and copious amounts of foul-smelling purulent or sanguineous sputum. Fever, chest pain, and weight loss are common. Clubbing of the fingers and toes may appear within a few weeks after the onset of an abscess. Diagnosis of this condition can be only suspectedfrom the clinical findings and must be confirmed radiologically. Whenever an abscess is discovered in older individuals, it is important to rule out an underlying carcinoma, because this is present in 10% to 15% of cases.
The course of abscesses is variable. With antimicrobial therapy, most resolve leaving behind a scar. Complications include extension of the infection into the pleural cavity, hemorrhage, the development of brain abscesses or meningitis from septic emboli, and (rarely) secondary amyloidosis (type AA).
CHRONIC PNEUMONIA
Chronic pneumonia is most often a localized lesion in the immunocompetent patient, with or without regional lymph node involvement. Typically, the inflammatory reaction is granulomatous, and is caused by bacteria (e.g., M. tuberculosis) or fungi (e.g., Histoplasma capsulatum). Tuberculosis of the lung and other organs was described in Chapter 8. Here we will discuss chronic pneumonias caused by fungi.
Histoplasmosis, blastomycosis, and coccidioidomycosis are discussed together because (1) they are granulomatous diseases of the lungs that may resemble tuberculosis, (2) they are caused by fungi that are thermally dimorphic in that they grow as hyphae that produce spores at environmental temperatures but grow as yeasts (spherules or ellipses) at body temperature within the lungs, and (3) each fungus is geographic in that it causes disease primarily among immunocompetent individuals living along the Ohio and Mississippi rivers and in the Caribbean (Histoplasma), in the central and southeastern United States (Blastomyces), and in the Southwest and Far West of the United States and in Mexico (Coccidioides).
Histoplasmosis
Histoplasma capsulatum infection is acquired by inhalation of dust particles from soil contaminated with bird or bat droppings that contain small spores (microconidia), the infectious form of the fungus. Like M. tuberculosis, H. capsulatum is an intracellular parasite of macrophages. The clinical presentations and morphologic lesions of histoplasmosis also strikingly resemble those of tuberculosis, including (1) a self-limited and often latent primary pulmonary involvement, which may result in coin lesions on chest radiography; (2) chronic, progressive, secondary lung disease, which is localized to the lung apices and causes cough, fever, and night sweats; (3) localized lesions in extrapulmonary sites, including mediastinum, adrenals, liver, or meninges; and (4) a widely disseminated disease in immunocompromised patients.
The pathogenesis of histoplasmosis is incompletely understood. It is known that macrophages are the major target of infection. H. capsulatum may be internalized into macrophages after opsonization with antibody. Histoplasma yeasts can multiply within the phagosome, and lyse the host cells. Histoplasma infections are controlled by helper T cells that recognize fungal cell wall antigens and heat-shock proteins and subsequently secrete IFN-γ, which activates macrophages to kill intracellular yeasts. In addition, Histoplasma induces macrophages to secrete TNF, which recruits and stimulates other macrophages to kill Histoplasma. Lacking cellular immunity, patients with acquired immunodeficiency syndrome are susceptible to disseminated infection with Histoplasma, which is an opportunistic pathogen in this disease.
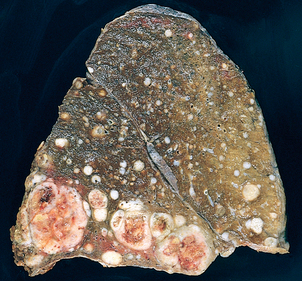
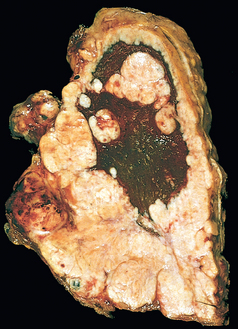
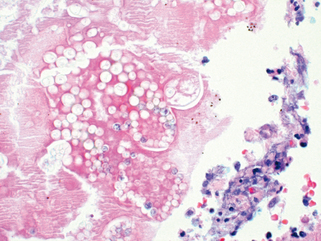
Morphology. In the lungs of otherwise healthy adults, Histoplasma infections produce epithelioid cell granulomas, which usually undergo caseation necrosis and coalesce to produce large areas of consolidation but may also liquefy to form cavities (seen in patients with COPD). With spontaneous or drug control of the infection, these lesions undergo fibrosis and concentric calcification (tree-bark appearance) (Fig. 15-36A). Histologic differentiation from tuberculosis, sarcoidosis, and coccidioidomycosis requires identification of the 3- to 5-μm thin-walled yeast forms that may persist in tissues for years.
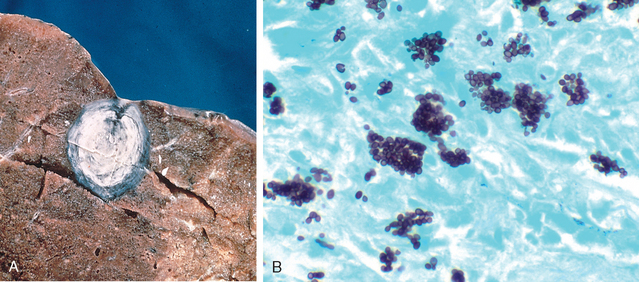
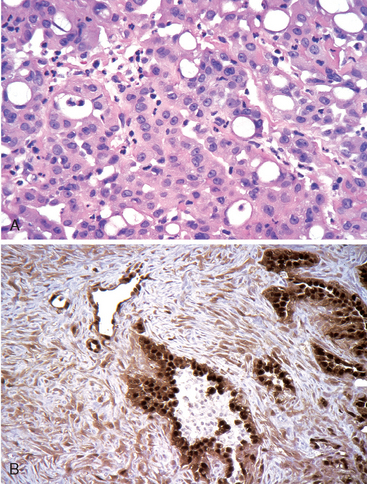
FIGURE 15-36 Histoplasmosis. A, Laminated Histoplasma granuloma of the lung. B, Histoplasma capsulatum yeast forms fill phagocytes in the lung of a patient with disseminated histoplasmosis (silver stain).
In fulminant disseminated histoplasmosis, which occurs in immunosuppressed individuals, epithelioid cell granulomas are not formed; instead, there are focal accumulations of mononuclear phagocytes filled with fungal yeasts throughout the tissues and organs of the body (Fig. 15-36B).
The diagnosis of histoplasmosis is established by culture or identification of the fungus in tissue lesions. In addition, serologic tests for antibodies and antigen are also available. Antigen detection in body fluids is most useful in the early stages, because antibodies are formed 2 to 6 weeks after infection.134
Blastomycosis
Blastomyces dermatitidis is a soil-inhabiting, dimorphic fungus that is remarkably difficult to isolate. It is a cause of disease in people living in or visiting the central and southeastern United States; infection also occurs in Canada, Mexico, the Middle East, Africa, and India. There are three clinical forms: pulmonary blastomycosis, disseminated blastomycosis, and a rare primary cutaneous form that results from direct inoculation of organisms into the skin. Pulmonary blastomycosis most often presents as an abrupt illness with productive cough, headache, chest pain, weight loss, fever, abdominal pain, night sweats, chills, and anorexia. Chest radiographs reveal lobar consolidation, multilobar infiltrates, perihilar infiltrates, multiple nodules, or miliary infiltrates. The upper lobes are most frequently involved. The process may resolve spontaneously, persist, or progress to a chronic lesion.
Morphology. In the normal host the lung lesions of blastomycosis are suppurative granulomas. Macrophages have a limited ability to ingest and kill B. dermatitidis, and the persistence of the yeast cells leads to continued recruitment of neutrophils. In tissue, B. dermatitidis is a round, 5- to 15-μm yeast cell that divides by broad-based budding. It has a thick, double -contoured cell wall and multiple nuclei (Fig. 15-37). Involvement of the skin and larynx is associated with marked epithelial hyperplasia, which may be mistaken for squamous cell carcinoma.
Coccidioidomycosis
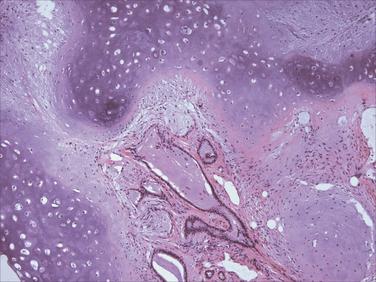
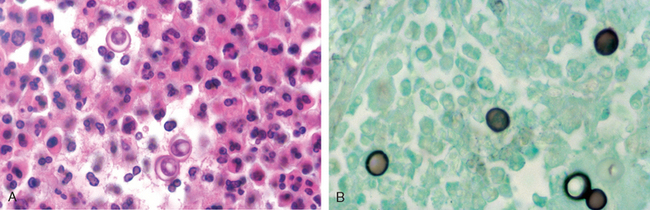
Almost everyone who inhales the spores of Coccidioides immitis becomes infected and develops a delayed-type hypersensitivity to the fungus, so more than 80% of people in endemic areas of the southwestern and western United States have a positive skin test reaction. One reason for the high rate of infectivity by C. immitis is that infective arthroconidia, when ingested by alveolar macrophages, block fusion of the phagosome and lysosome and so resist intracellular killing. As is the case with Histoplasma, most primary infections with C. immitis are asymptomatic, but 10% of people have lung lesions, fever, cough, and pleuritic pains, accompanied by erythema nodosum or erythema multiforme (the San Joaquin Valley fever complex). Less than 1% of people develop disseminated C. immitis infection, which frequently involves the skin and meninges.
Morphology. The primary and secondary lung lesions of C. immitis are similar to the granulomatous lesions of Histoplasma. Within macrophages or giant cells, C. immitis is present as thick-walled, nonbudding spherules 20 to 60 μm in diameter, often filled with small endospores. A pyogenic reaction is superimposed when the spherules rupture to release the endospores (Fig. 15-38). Rare progressive C. immitis disease involves the lungs, meninges, skin, bones, adrenals, lymph nodes, spleen, or liver. At all these sites, the inflammatory response may be purely granulomatous, pyogenic, or mixed. Purulent lesions dominate in patients with diminished resistance and with widespread dissemination.
PNEUMONIA IN THE IMMUNOCOMPROMISED HOST
The appearance of a pulmonary infiltrate, with or without signs of infection (e.g., fever), is one of the most common and serious complications in patients whose immune defenses are suppressed by disease, immunosuppressive therapy for organ transplants, chemotherapy for tumors, or irradiation.135 A wide variety of so-called opportunistic infectious agents, many of which rarely cause infection in normal hosts, can cause these pneumonias, and often more than one agent is involved. Mortality from these opportunistic infections is high. Table 15-9 lists some of the opportunistic agents according to their prevalence and whether they cause local or diffuse pulmonary infiltrates. The differential diagnosis of such infiltrates includes drug reactions and involvement of the lung by tumor. The specific infections are discussed in Chapter 8. Of these, the ones that commonly involve the lung can be classified according to the etiologic agent: (1) bacteria (P. aeruginosa, Mycobacterium species, L. pneumophila, and Listeria monocytogenes), (2) viruses (cytomegalovirus [CMV] and herpesvirus), and (3) fungi (P. jiroveci, Candida species, Aspergillus species, the Phycomycetes, and Cryptococcus neoformans).
TABLE 15-9 Causes of Pulmonary Infiltrates in Immunocompromised Hosts
| Diffuse Infiltrate | Focal Infiltrate |
|---|---|
| COMMON | |
| Cytomegalovirus | Gram-negative rods |
| Pneumocystis jiroveci | Staphylococcus aureus |
| Drug reaction | Aspergillus |
| Candida | |
| Malignancy | |
| UNCOMMON | |
| Bacteria | Cryptococcus |
| Aspergillus | Mucor |
| Cryptococcus | Pneumocystis jiroveci |
| Malignancy | Legionella pneumophila |
PULMONARY DISEASE IN HUMAN IMMUNODEFICIENCY VIRUS INFECTION
Pulmonary disease continues to be the leading cause of morbidity and mortality in HIV-infected individuals. Although the use of potent antiretroviral agents and effective chemoprophylaxis has markedly altered the incidence and outcome of pulmonary disease in HIV-infected persons, the plethora of infectious agents and other pulmonary lesions make diagnosis and treatment a distinct challenge. Some of the individual microbial agents afflicting HIV-infected individuals have already been discussed; this section will focus only on the general principles of HIV-associated pulmonary disease.
Finally, it is useful to remember that pulmonary disease in HIV-infected persons may result from more than one cause, and even common pathogens may present with atypical manifestations. Therefore, the diagnostic work-up of these patients may be more extensive (and expensive) than would be necessary in an immunocompetent individual.