MANIFESTATIONS OF CELLULAR INJURY
Cellular Manifestations: Accumulations
Cellular accumulations, also known as infiltrations, occur as a result of not only sublethal injury sustained by cells but also normal (but inefficient) cell function. Common accumulations consist of substances that are normally present, such as fluids and electrolytes, triglycerides (lipids), glycogen, calcium, uric acid, proteins, melanin, and bilirubin. Abnormal accumulations of these substances can occur in the cytoplasm (frequently in the lysosomes) or in the nucleus if (1) the normal, endogenous substance is produced in excess or at an increased rate; (2) an endogenous substance (normal or abnormal) is not effectively catabolized, usually because of lack of a vital lysosomal enzyme; or (3) harmful exogenous materials, such as heavy metals, mineral dusts, or microorganisms, accumulate because of inhalation, ingestion, or infection.
In all storage diseases the cells attempt to digest, or catabolize, the “stored” substances. As a result, excessive amounts of metabolites (products of catabolism) accumulate in the cells and are expelled into the extracellular matrix, where they are taken up by phagocytic cells called macrophages (see Chapter 6). Some of these scavenger cells circulate throughout the body, whereas others remain fixed in certain tissues, such as the liver or spleen. As more and more macrophages and other phagocytes migrate to tissues that are producing excessive metabolites, the affected tissues begin to swell. This is the mechanism that causes enlargement of the liver (hepatomegaly) or the spleen (splenomegaly). Enlargement of one of these organs is a clinical manifestation of many of the storage diseases.
Water
Cellular swelling, the most common degenerative change, is caused by the shift of extracellular water into the cells. In hypoxic injury, movement of fluid and ions into the cell is associated with acute failure of metabolism and loss of ATP production. Normally, the pump that transports sodium ions out of the cell is maintained by the presence of ATP and ATPase, the active-transport enzyme. In metabolic failure caused by hypoxia, reduced ATP and ATPase permit sodium to accumulate in the cell, whereas potassium diffuses outward. The increase of intracellular sodium increases osmotic pressure, which draws more water into the cell (transport mechanisms are described in Chapter 1). The cisternae of the endoplasmic reticulum become distended, rupture, and coalesce to form large vacuoles that isolate the water from the cytoplasm, a process called vacuolation. Progressive vacuolation results in oncosis (which has replaced the old term hydropic degeneration) or vacuolar degeneration (degeneration by water) (Figure 2-24). If cellular swelling affects all cells in an organ, the organ increases in weight and becomes distended and pale.
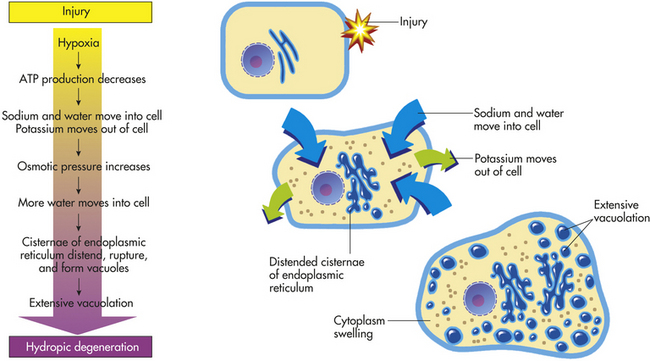
Figure 2-24 The process of oncosis (formerly known as hydropic degeneration). ATP, Adenosine triphosphate.
Cellular swelling is reversible and is considered to be sublethal. It is, in fact, an early manifestation of almost all types of cellular injury, including severe or lethal cell injury. It is also associated with high fever, hypokalemia (abnormally low concentrations of potassium in the blood; see Chapter 3), and certain infections.
Lipids and Carbohydrates
Certain metabolic disorders result in the abnormal intracellular accumulation of carbohydrates and lipids. These substances may accumulate throughout the body but are found primarily in the cells of the spleen, liver, and CNS. Accumulations in cells of the CNS can cause neurologic dysfunction and severe mental retardation. Lipids accumulate in Tay-Sachs, Niemann-Pick, and Gaucher diseases, whereas in the diseases known as mucopolysaccharidoses, carbohydrates are in excess. The mucopolysaccharidoses are progressive disorders that usually involve multiple organs, including the liver, spleen, heart, and blood vessels. The accumulated mucopolysaccharides are found in reticuloendothelial cells, endothelial cells, intimal smooth muscle cells, and fibroblasts throughout the body. These carbohydrate accumulations can cause clouding of the cornea, joint stiffness, and mental retardation.2
Although lipids sometimes accumulate in heart and kidney cells, the most common site of intracellular lipid accumulation, or fatty change, is liver cells. Because hepatic metabolism and secretion of lipids are crucial to proper body function, imbalances and deficiencies in these processes lead to major pathologic changes. Lipid accumulation in liver cells causes an organic condition known as fatty liver, or fatty change (Figure 2-25). As lipids fill the cells, vacuolation pushes the nucleus and other organelles aside. Grossly, the liver looks yellowish and greasy.
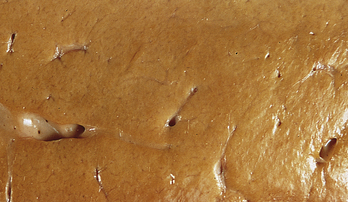
Figure 2-25 Fatty liver. The liver appears yellow. (From Damjanov I, Linder J: Pathology: a color atlas, St Louis, 2000, Mosby.)
Lipid accumulation in liver cells occurs after cellular injury sets one or more of the following mechanisms in motion:
1. Increased movement of free fatty acids into the liver (starvation, for example, increases breakdown of triglycerides in adipose tissue, releasing fatty acids that subsequently enter liver cells)
2. Failure of the metabolic process that converts fatty acids to phospholipids, resulting in the preferential conversion of the fatty acids to triglycerides
3. Increased synthesis of triglycerides from fatty acids (increases in the enzyme, α-glycerophosphatase, which can accelerate triglyceride synthesis)
4. Decreased synthesis of apoproteins (lipid-acceptor proteins)
5. Failure of lipids to bind with apoproteins and form lipoproteins
6. Failure of mechanisms that transport lipoproteins out of the cell
7. Direct damage to the endoplasmic reticulum by free radicals released by alcohol’s toxic effects
Alcohol abuse is one of the most common causes of fatty liver (see Chapter 39). Fatty change caused by alcohol can lead to a form of liver fibrosis called cirrhosis (see What’s New? Cellular Mechanisms of Fibrosis and Reversal). If alcohol intake ceases, the cirrhotic liver can return to a normal size and function. Fatty change from other causes, notably carbon tetrachloride poisoning, is often irreversible.
Glycogen
Intracellular accumulations of glycogen are seen in genetic disorders called glycogen storage diseases and in disorders of glucose and glycogen metabolism. Like water and lipid accumulation, glycogen accumulation results in excessive vacuolation of the cytoplasm. The most common cause of glycogen accumulation is diabetes mellitus, a disorder of glucose metabolism (see Chapter 21).
Proteins
Proteins provide cellular structure and constitute most of the cell’s dry weight. They are synthesized on ribosomes in the cytoplasm from the essential amino acids lysine, threonine, leucine, isoleucine, methionine, tryptophan, valine, phenylalanine, and histidine. Protein accumulation probably damages cells in two ways. First, metabolites, produced when the cell attempts to digest some proteins, are enzymes that when released from lysosomes can damage cellular organelles. Second, excessive amounts of protein in the cytoplasm push against cellular organelles, disrupting organelle function and intracellular communication.
Protein excess accumulates primarily in the epithelial cells of the renal convoluted tubule and in the antibody-forming plasma cells (B lymphocytes) of the immune system. Several types of renal disorders cause excessive excretion of protein molecules in the urine (proteinuria). Normally, little or no protein is present in the urine, and its presence in significant amounts indicates cellular injury and altered cellular function in the glomerular membrane.
Accumulations of protein in B lymphocytes can occur during active synthesis of antibodies during the immune response. The excess aggregates of protein are called Russell bodies. Russell bodies have been identified in multiple myeloma (plasma cell tumor) (see Chapter 27).
Pigments
Pigment accumulations may be normal or abnormal, endogenous (produced within the body) or exogenous (produced outside the body). Endogenous pigments are derived, for example, from amino acids (e.g., tyrosine, tryptophan). They include melanin and the blood proteins—porphyrins, hemoglobin, and hemosiderin (ferritin). Lipid-rich pigments such as lipofuscin (the aging pigment or spots) give a yellow-brown color to cells undergoing slow, regressive, and often atrophic changes. Exogenous pigments include mineral dusts containing silica and iron particles, lead, silver salts, and dyes for tattoos.
Melanin: Melanin accumulates in epithelial cells (keratinocytes) of the skin and retina. It is an extremely important pigment because it protects the skin against long exposure to sunlight and is considered an essential factor in the prevention of skin cancer (see Chapters 12 and 44). Ultraviolet light (e.g., sunlight) stimulates the synthesis of melanin, which probably absorbs ultraviolet rays during subsequent exposure. Melanin also may protect the skin by trapping the injurious free radicals produced by the action of ultraviolet light on skin.
Melanin is a brown-black pigment derived from the amino acid tyrosine. It is synthesized by epidermal cells called melanocytes and is stored in membrane-bound cytoplasmic vesicles called melanosomes. Melanosomes are particularly abundant in projections of melanocytic cytoplasm, called dendrites, from which they are transmitted to neighboring keratinocytes, where melanin accumulation occurs.10 (Keratinocytes, which constitute 95% of epidermal cells, are discussed with other skin components in Chapter 44.) The dendritic melanocytes form bridges between neighboring keratinocytes and inject melanosomes into the keratinocytes by an unknown mechanism.
Melanin also can accumulate in melanophores (melanin-containing pigment cells), macrophages, or other phagocytic cells in the dermis. Presumably these cells acquire the melanin from nearby melanocytes or from pigment that has been extruded from dying epidermal cells. This is the mechanism that causes freckles.
Although rare, melanin accumulation occurs in the skin of individuals with Addison disease (adrenocortical insufficiency resulting from disorders of the adrenal cortex; see Chapter 21). The increased melaninogenesis (melanin production) seen in Addison disease is caused by the loss of feedback control of adrenocorticotropic hormone (ACTH). Decreased hormonal secretion from the adrenal gland causes increased release of ACTH from the pituitary gland. In Addison disease the increase in melanin occurs presumably because a segment of the ACTH molecule contains the melanin-stimulating hormone (MSH).
An increase in melanin also occurs in the benign form of “pigmented moles” called nevi (Figure 2-26) (see Chapter 44). Malignant melanoma is a cancerous skin tumor that contains melanin and invades normal tissue early and widely and often leads to death.
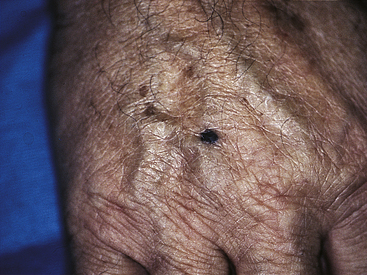
Figure 2-26 Blue nevus, common type. Nevus is dark blue-black, small, and symmetric. (From Damjanov I, Linder J: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
A decrease in melanin production occurs in the inherited disorder of the melanin metabolism called albinism. Albinism is often diffuse, involving all the skin, the eyes, and the hair. Albinism is also related to phenylalanine metabolism. In classic types the person with albinism is unable to convert tyrosine to dopa (3,4-dihydroxyphenylalanine), an intermediary in melanin biosynthesis. Melanin-producing cells are present in normal numbers, but they are unable to make melanin. Individuals with albinism are very sensitive to sunlight and quickly become sunburned. They are also at high risk for skin cancer.
Hemoproteins: Hemoproteins are among the most essential of the normal endogenous pigments. They include hemoglobin and the oxidative enzymes, the cytochromes. Central to an understanding of disorders involving these pigments is knowledge of iron uptake, metabolism, excretion, and storage (see Chapter 25). Hemoprotein accumulations in cells are caused by excessive storage of iron, which is transferred to the cells from the bloodstream. Iron enters the blood from three primary sources: (1) tissue stores, (2) the intestinal mucosa, and (3) macrophages that remove and destroy dead or defective red blood cells. The amount of iron in blood plasma also depends on the metabolism of the major iron-transport protein, transferrin.
Iron is stored in tissue cells in two forms: as ferritin and, when greater levels of iron are present, as hemosiderin. Hemosiderin is a yellow-brown pigment derived from hemoglobin. With pathologic states, excesses of iron cause hemosiderin to accumulate within cells. Accumulation of hemosiderin often occurs in areas of bruising and hemorrhage and in the lungs and spleen after congestion caused by heart failure. With a local hemorrhage, the skin first appears red-blue and then lysis of the escaped red blood cells occur, causing the hemoglobin to be transformed to hemosiderin. The color changes noted in bruising reflect this transformation.
Hemosiderosis is a condition in which excess iron is stored as hemosiderin in the cells of many organs and tissues. This condition is common in individuals who have received repeated blood transfusions or prolonged parenteral administration of iron. Hemosiderosis is also associated with increased absorption of dietary iron, conditions in which iron storage and transport are impaired, and hemolytic anemia. Excessive alcohol ingestion also can lead to hemosiderosis. Normally, absorption of excessive dietary iron is prevented by an iron-absorption process in the intestines. Failure of this process can lead to total-body iron accumulations in the range of 60 to 80 g, compared with normal iron stores of 4.5 to 5 g. Excessive accumulations of iron, such as occur in hemochromatosis (a genetic disorder of iron metabolism and the most severe example of iron overload), are associated with liver and pancreatic cell damage.
Bilirubin is a normal yellow-to-green pigment of bile derived from the porphyrin structure of hemoglobin. Excesses of bilirubin within cells and tissues cause jaundice (icterus), or yellowing of the skin. Jaundice occurs when the bilirubin level exceeds 1.5 to 2 mg/dl of plasma, compared with the normal values of 0.4 to 1 mg/dl. Hyperbilirubinemia occurs with (1) destruction of red blood cells (erythrocytes), such as in hemolytic jaundice; (2) diseases affecting the metabolism and excretion of bilirubin in the liver; and (3) diseases that cause obstruction of the common bile duct, such as gallstones or pancreatic tumors. (For a detailed description of these diseases, see Chapter 39.) Certain drugs, specifically chlorpromazine and other phenothiazine derivatives, estrogenic hormones, and halothane (an anesthetic), can cause the obstruction of normal bile flow through the liver.
Because unconjugated bilirubin is lipid soluble, it can injure the lipid components of the plasma membrane. Albumin, a plasma protein, provides significant protection by binding unconjugated bilirubin in plasma. Unconjugated bilirubin causes two cellular effects: uncoupling of oxidative phosphorylation and a loss of cellular proteins. These two effects could cause structural injury to the various membranes of the cell.
Calcium
Calcium salts accumulate in both injured and dead tissues (Figure 2-27). An important mechanism of cellular calcification is the influx of extracellular calcium in injured mitochondria (see pp. 53 and 80). Another mechanism that causes calcium accumulation in alveoli (gas-exchange airways of the lungs), gastric epithelium, and renal tubules is the excretion of acid at these sites, leading to the local production of hydroxyl ions. Hydroxyl ions result in precipitation of calcium hydroxide (Ca[OH]2) and hydroxyapatite (3Ca3[PO4]2Ca[OH]2), a mixed salt. Damage occurs when calcium salts clump and harden, interfering with normal cellular structure and function.
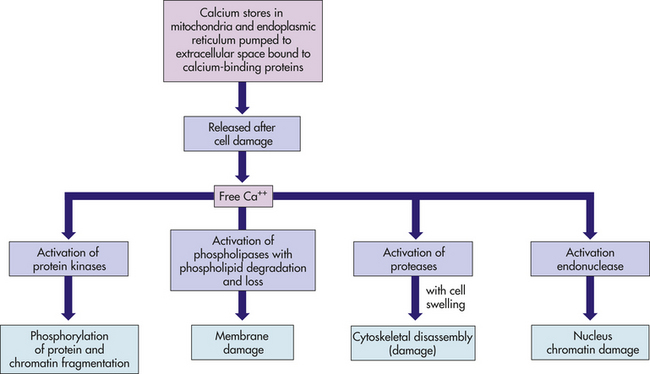
Figure 2-27 Free cytosolic calcium: a destructive agent. Normally calcium is removed from the cytosol by adenosine triphosphate (ATP)–dependent calcium pumps. In normal cells, calcium is bound to buffering proteins, such as calbindin or paralbumin, and is contained in the endoplasmic reticulum and the mitochondria. If there is abnormal permeability of calcium-ion channels, direct damage to membranes, or depletion of ATP (i.e., hypoxic injury), calcium increases in the cytosol. If the free calcium cannot be buffered or pumped out of cells, uncontrolled enzyme activation takes place, causing further damage. Uncontrolled entry of calcium into the cytosol is an important final pathway in many causes of cell death.
Pathologic calcification can be dystrophic or metastatic. Dystrophic calcification is the calcification of dying and dead tissues and occurs in chronic tuberculosis of the lungs and lymph nodes, in arteries with advanced atherosclerosis (narrowing as a result of plaque accumulation), and often in injured heart valves (Figure 2-28). Calcification of the heart valves interferes with opening and closing of the valves, causing heart murmurs (see Chapter 30). Calcification of the coronary arteries predisposes them to severe narrowing and thrombosis, which can lead to myocardial infarction. Another site of dystrophic calcification is the center of tumors. Over time, the center is deprived of oxygen supply, dies, and becomes calcified. The calcium salts appear as gritty, clumped granules that can become hard as stone. When several layers clump together, they resemble grains of sand and are called psammoma bodies.
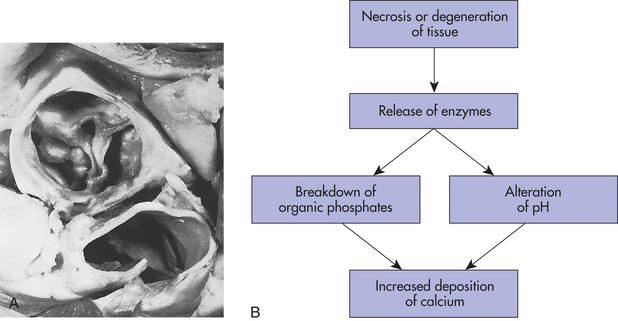
Figure 2-28 Aortic valve calcification. A, This aortic valve was unable to close because of calcification caused by rheumatic heart disease. B, Algorithm showing the dystrophic mechanism of calcification. (A from Damjanov I, Linder J, editors: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
The exact pathogenic mechanisms responsible for dystrophic calcification are unknown. A popular hypothesis is that with progressive deterioration of dead cells, the exposed denatured (changed) proteins preferentially bind with phosphate ions. The phosphate ions then react with calcium ions to form deposits of phosphate carbonate precipitates and, sometimes, crystalline formations of calcium phosphate. Dystrophic calcification develops slowly and is an explicit marker for the site of dead cells.
Metastatic calcification consists of mineral deposits that occur in undamaged normal tissues as the result of hypercalcemia (excess of calcium in the blood; see Chapter 3). Conditions that cause hypercalcemia include hyperparathyroidism, toxic levels of vitamin D, hyperthyroidism, idiopathic hypercalcemia of infancy, Addison disease (adrenocortical insufficiency), systemic sarcoidosis, milk-alkali syndrome, and the increased bone demineralization that results from bone tumors, leukemia, and disseminated cancers. Hypercalcemia also can occur in some instances of advanced renal failure with phosphate retention, resulting in hyperparathyroidism.10
Urate
In humans, uric acid (urate) is the major end product of purine catabolism because of the absence of the enzyme urate oxidase. Serum urate concentration is, in general, stable: approximately 5 mg/dl in postpubertal males and 4.1 mg/dl in postpubertal females. Disturbances in maintaining serum urate levels result in hyperuricemia and deposition of sodium urate crystals in the tissues, leading to painful disorders collectively called gout. These disorders include acute arthritis, chronic gouty arthritis, tophus (firm nodular subcutaneous deposits of urate crystals surrounded by fibrosis), and nephritis (inflammation of the nephron).
Chronic hyperuricemia results in the deposition of urate in tissues, cell injury, and inflammation. Because urate crystals are not degraded by lysosomal enzymes, they persist in dead cells.
Systemic Manifestations
Systemic manifestations of cellular injury include a general sense of fatigue and malaise, a loss of well-being, and altered appetite. Fever is frequently present because of biochemicals produced during the inflammatory response (see Chapter 6). Table 2-10 summarizes the most significant systemic manifestations of cellular injury.
Table 2-10
Systemic Manifestations of Cellular Injury
| Manifestation | Cause |
| Fever | Release of endogenous pyrogens (interleukin-1, tumor necrosis factor-α (TNF-α), prostaglandins) from bacteria or macrophages; acute inflammatory response |
| Increased heart rate | Increase in oxidative metabolic processes resulting from fever |
| Increase in leukocytes (leukocytosis) | Increase in total number of white blood cells because of infection; normal is 5000-9000/mm3 (increase is directly related to the severity of the infection) |
| Pain | Various mechanisms, such as release of bradykinins, obstruction, pressure |
| Presence of cellular enzymes in extracellular fluid | Release of enzymes from cells of tissue∗ |
| Lactate dehydrogenase (LDH) (LDH isoenzymes) | Release from red blood cells, liver, kidney, skeletal muscle |
| Creatine kinase (CK) (CK isoenzymes) | Release from skeletal muscle, brain, heart |
| Aspartate aminotransferase (AST; SGOT) | Release from heart, liver, skeletal muscle, kidney, pancreas |
| Alanine aminotransferase (ALT; SGPT) | Release from liver, kidney, heart |
| Alkaline phosphatase (ALP) | Release from liver, bone |
| Amylase | Release from pancreas |
| Aldolase | Release from skeletal muscle, heart |
∗The rapidity of enzyme transfer is a function of the weight of the enzyme and the concentration gradient across the cellular membrane. The specific metabolic and excretory rates of the enzymes determine how long levels of enzymes remain elevated.
CELLULAR DEATH
Two main types of cell death are necrosis and apoptosis. Necrosis, or accidental cell death, occurs after severe and sudden injury. Programmed cell death is often referred to as apoptosis, but other forms have been determined with sophisticated ultrastructural studies (see What’s New? Programmed Cell Death More Than Apoptosis).
Necrosis
Necrosis is the sum of cellular changes after local cell death and the process of cellular lysis and it provokes an inflammatory reaction in surrounding tissue (Figure 2-29). The structural signs that indicate irreversible injury and progression to necrosis are the dense clumping and progressive disruption of genetic material and disruption of the plasma and organelle membranes. In later stages of necrosis, most organelles are disrupted, and karyolysis (nuclear dissolution and lysis of chromatin from the action of hydrolytic enzymes) is under way. In some cells the nucleus shrinks and becomes a small, dense mass of genetic material—a process called nuclear pyknosis. The pyknotic nucleus eventually dissolves (by karyolysis) as a result of the action of hydrolytic lysosomal enzymes on DNA. Karyorrhexis means fragmentation of the nucleus into smaller particles or “nuclear dust.”
WHAT’S NEW?
Programmed Cell Death More Than Apoptosis
With better technologies to study cell death, recent work has revealed at least three distinct forms of cell death. The best studied is apoptosis or type 1 cell death (see p. 84). Type 2 cell death, called autophagic cell death (Greek: auto, oneself; phagy, to eat), refers to any cellular degradative pathway that delivers cytoplasmic products to the lysosome (see Figure 2-29). In autophagic death, membrane sheets, mostly from the endoplasmic reticulum (ER), form cytoplasmic vacuoles that engulf intracellular organelles and cytoplasmic materials. The vesicles fuse with lysosomes, called autolysosomes, in which the contents are digested. This process is thought to be the cell’s major mechanism for degrading organelles and long-lived proteins. Type 3 cell death, the least studied, is characterized by the swelling of intracellular organelles and a non-lysosomal cell death similar to necrosis.
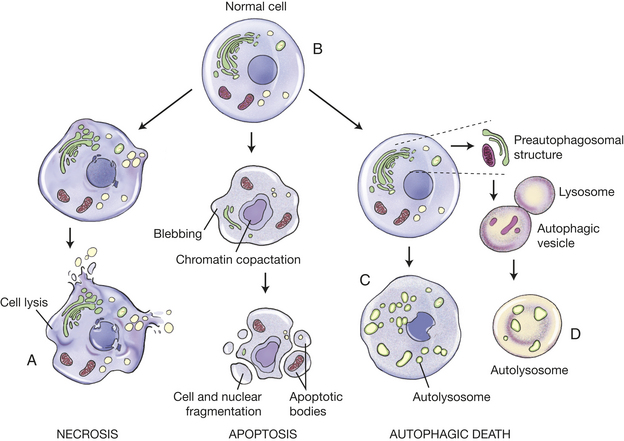
Figure 2-29 Different forms of cell death. A, Schematic illustration of structural changes that characterize cell death by necrosis, apoptosis, and autophagic cell death. Whereas necrotic cell death culminates in cell lysis and provokes immediate inflammation, apoptotic cells are packaged into apoptotic bodies that are then engulfed by adjacent cells without an inflammatory response. Autophagic cell death is characterized by the appearance of cytoplasmic vesicles engulfing bulk cytoplasm and organelles. The contents of the vesicles are digested by the lysosomal system of the same cell after the fusion of the autophagic vesicles with lysosomes. Inset: Formation of autolysosome by fusion of autophagic vesicle and lysosome with the cell undergoing autophagy. B-D, Ultrastructural features of cells undergoing apoptotic and autophagic cell death: (B) a normal cell, (C) an apoptotic cell, and (D) a cell undergoing autophagic cell death are shown. Polyribosomes, mitochondria, and autophagic vacuoles are indicated. Although autophagic vacuoles can be seen in healthy cells and apoptotic cells, they are much more abundant during autophagic cell death. (Redrawn from Bursch et al: J Cell Sci 113:1189-1198, 2000, by permission)
Data from Levine B, Kroemer G: Cell 132(1):27-42, 2008.
Different types of necroses tend to occur in different organs or tissues and sometimes can indicate the mechanism or cause of cellular injury. The four major types of necroses are coagulative, liquefactive, caseous, and fatty. Another type, gangrenous necrosis, is not a distinctive type of cell death but refers to larger areas of tissue death.
Coagulative necrosis, which occurs primarily in the kidneys, heart, and adrenal glands, commonly results from hypoxia caused by severe ischemia or hypoxia caused by chemical injury, especially ingestion of mercuric chloride (Figure 2-30). Coagulation is caused by protein denaturation, which causes the protein albumin to change from a gelatinous, transparent state to a firm, opaque state, similar to that of a cooked egg white. The necrotic tissues appear firm and slightly swollen. Recent evidence indicates that an abnormality in intracellular levels of Ca++ (e.g., increased) may be a critical event in coagulation necrosis.3
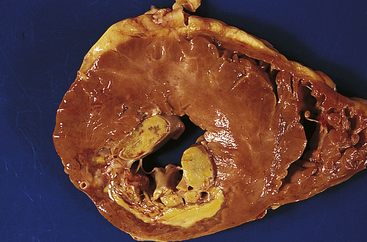
Figure 2-30 Coagulative necrosis of myocardium of posterior wall of left ventricle of heart. A large anemic (white) infarct is readily apparent; note also the necrosis of papillary muscle. (From Damjanov I, Linder J: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
Liquefactive necrosis commonly results from ischemic injury to neurons and glial cells in the brain (Figure 2-31). Dead brain tissue is readily affected by liquefactive necrosis because brain cells are rich in the digestive hydrolytic enzymes and lipids, and the brain contains little connective tissue. As the cells are digested by their own hydrolases, the tissue becomes soft, liquefies, and is walled off from healthy tissue, forming cysts. (Cyst formation is described in Chapter 6.)
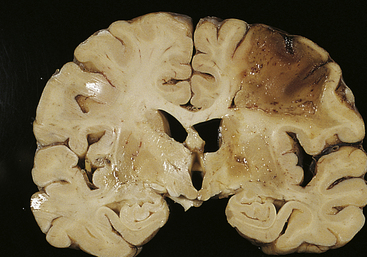
Figure 2-31 Liquefactive necrosis. Liquefactive necrosis of the brain developed at a large cerebral infarct caused by ischemia. (From Damjanov I, Linder J: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
Liquefactive necrosis can also result from bacterial infection, particularly by staphylococci, streptococci, and Escherichia coli. In this case the hydrolases are released from the lysosomes of neutrophils, which are phagocytes attracted to the infected area to kill the bacteria. Liquefaction of bacterial cells and neighboring tissue cells by neutrophilic hydrolases results in the accumulation of pus.
Caseous necrosis, which commonly results from tuberculous pulmonary infection, particularly by Mycobacterium tuberculosis, is a combination of coagulative and liquefactive necrosis (Figure 2-32). The dead cells disintegrate, but the debris is not digested completely by hydrolases. Tissues appear soft and granular and resemble clumped cheese, which gives this type of necrosis its name. A granulomatous inflammatory wall encloses areas of caseous necrosis.
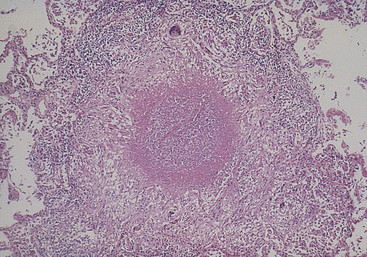
Figure 2-32 Granuloma with central caseous necrosis typical of pulmonary tuberculosis. (From Damjanov I, Linder J: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
Fat necrosis, which occurs in the breast, pancreas, and other abdominal structures, is cellular dissolution caused by powerful enzymes called lipases (Figure 2-33). Lipases break down triglycerides, releasing free fatty acids, which then combine with calcium, magnesium, and sodium ions, creating soaps (a process known as saponification). The necrotic tissue appears opaque and chalk white.
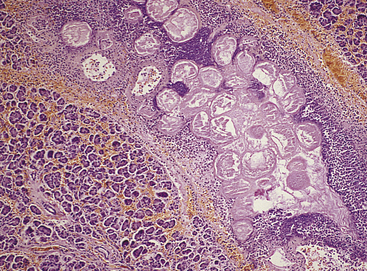
Figure 2-33 Fat necrosis of pancreas. Interlobular adipocytes are necrotic; these are surrounded by acute inflammatory cells. (From Damjanov I, Linder J: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
Gangrenous necrosis, a term commonly used in surgical clinical practice, refers to death of tissue and results from severe hypoxic injury, commonly occurring because of arteriosclerosis, or blockage, of major arteries, especially in the lower leg. With hypoxia and subsequent bacterial invasion, the tissues can undergo necrosis. Dry gangrene is usually the result of coagulative necrosis. The skin becomes very dry and shrinks, resulting in wrinkles, and its color changes to dark brown or black (Figure 2-34). Wet gangrene develops when neutrophils invade the site, causing liquefactive necrosis. This usually occurs in internal organs, causing the site to become cold, swollen, and black. A foul odor is present, produced by pus, and if systemic symptoms become severe, death can ensue.
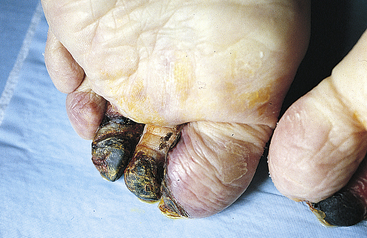
Figure 2-34 Gangrene of toes. Dry gangrene. (From Damjanov I: Pathology for the health-related professions, ed 2, Philadelphia, 2000, Saunders.)
Gas gangrene, a special type of gangrene, is caused by infection of injured tissue by one of many species of Clostridium. These anaerobic bacteria produce hydrolytic enzymes and toxins that destroy connective tissue and cellular membranes and cause bubbles of gas to form in muscle cells. Gas gangrene can be fatal if enzymes lyse the membranes of red blood cells, destroying their oxygen-carrying capacity. Death is the result of shock. The condition is treated with antitoxins and supplemental oxygen delivered in a hyperbaric (pressurized) chamber.
Apoptosis
Apoptosis (Greek for “dropping off”) is an important distinct type of cell death84 that differs from necrosis in several respects (Figure 2-35). Apoptosis is an active process of cellular self-destruction, called programmed cell death, that is implicated in both normal and pathologic tissue changes.85 Newer sophisticated studies have revealed that programmed cell death includes other forms than just apoptosis (see What’s new? Programmed Cell Death More than Apoptosis). Cells need to die, otherwise endless proliferation would lead to gigantic bodies. Every day an average adult may create 10 billion new cells and kill off the same number. Apoptosis is responsible for local deletion of cells during normal embryonic development, bone cells dying during turnover, lymphocytes dying during receptor repertoire selection, infected cells, and so on. It has been shown to play a major role in endocrine-dependent tissues that are undergoing atrophic change. Apoptosis can occur spontaneously in malignant tumors and in normal, rapidly proliferating cells treated with cancer chemotherapeutic agents and ionizing radiation.86 Defects in apoptosis can cause cancer.87 Its significance in aging is unknown; however, apoptosis is required to maintain a balance between cell proliferation and cell death.87
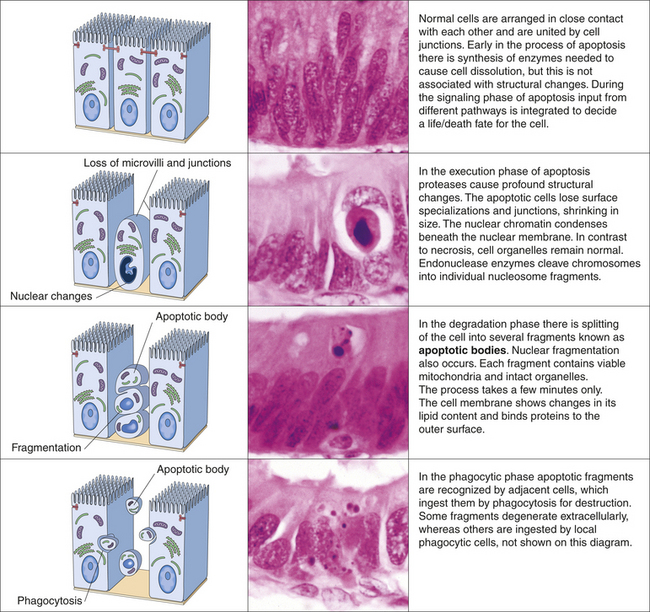
Figure 2-35 Apoptosis. Apoptosis of cells is a programmed and energy-dependent process designed specifically to switch cells off and eliminate them. This controlled pattern of cell death, termed programmed cell death, is very different from that which occurs as a direct result of acute alterations, for example, trauma. (From Stevens A, Lowe J: Pathology, ed 2, London, 2000, Mosby.)
Necrosis and apoptosis affect tissues differently. Unlike necrosis, apoptosis affects scattered, single cells. Apoptosis is nuclear, and cytoplasmic shrinkage of a cell (i.e., unlike necrosis, in which cells swell and lyse) is followed by fragmentation into membrane-bound fragments and subsequent phagocytosis by neighboring, healthy cells.10 Apoptosis depends on a tightly regulated cellular program for its initiation and execution.86 Programmed cell death involves enzymes that cut up other proteins (proteases) that are, themselves, activated by proteolytic activity in response to signals that induce apoptosis.88 These proteases are called caspases, a family of aspartic acid–specific proteases. The activated suicide caspases cleave, and thereby activate, other members of the family resulting in an amplifying “suicide” cascade. The activated caspases then cleave other key proteins in the cell, killing it quickly and neatly.88–90 Two different pathways converge on caspase activation called the mitochondrial pathway and the death receptor pathway (Figure 2-36). Cells that die by apoptosis release chemical factors that recruit phagocytes that quickly engulf the remains of the dead cell, thus reducing chances of inflammation. With necrosis, cell death is not neat because cells that die as a result of acute injury swell, burst, and spill their contents all over their neighbors, likely causing a damaging inflammatory response.88 Molecular helpers in apoptosis are present in different subcellular compartments, including the plasma membrane, cytosol, mitochondria, and nucleus. The progression of apoptosis depends on the interplay among these compartments and the exchange of specific signaling molecules.86
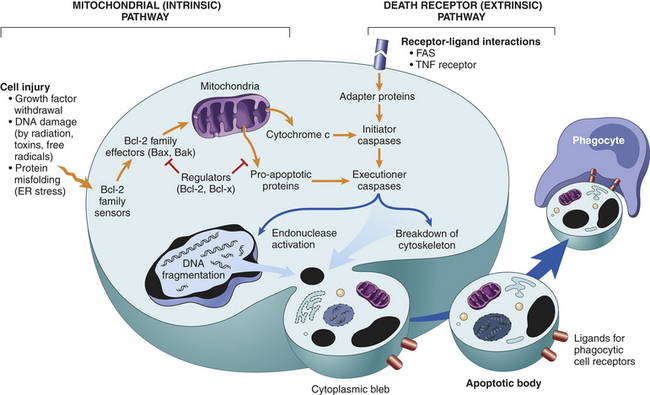
Figure 2-36 Mechanisms of apoptosis. The two pathways of apoptosis differ in their induction and regulation, and both culminate in the activation of “executioner” caspases. The induction of apoptosis is dependent on the balance between pro- and anti-apoptotic signals and intracellular proteins. The figure shows the pathways that induce apoptotic cell death, and the anti-apoptotic proteins that inhibit mitochondrial leakiness and cytochrome c–dependent caspase activation and thus function as regulators of mitochondrial apoptosis. (From from Kumar V, Abbas A, Fausto N: Robbins and Cotran pathologic basis of disease, ed 8, Philadelphia, 2007, Saunders.)
Aging and Altered Cellular and Tissue Biology
Aging is usually defined as a normal physiologic process that is universal and inevitable. The basic mechanisms of aging depend on presumably irreversible and universal processes at the cellular and molecular level. To understand aging requires the separation of irreversible processes from potentially reversible mechanisms (i.e., those that result from disease or age-related debilities)—a very difficult task!
Aging traditionally has not been considered a disease because it is “normal”; disease is usually considered “abnormal.” Conceptually, this distinction seems clear until the concept of “injury” or “damage” is introduced; disease has been defined by some pathologists as the result of injury. Aging has been defined as the time-dependent loss of structure and function that proceeds very slowly and in such small increments that it appears to be the result of the accumulation of small, imperceptible injuries—a gradual result of wear and tear.
Injuries may result from unavoidable and universal microinsults caused by continual bombardment by ultraviolet light, countless mechanical insults, and reactions to metabolites.91,92 In this context the distinction between aging and disease is unclear because the aging process increases the vulnerability to disease. For example, some degree of atrophy of the brain is considered normal in old age until it proceeds far enough to cause clinically significant disability and is then called disease. Likewise, most humans have atherosclerosis, and the plaques progress with age, but at what point in this progression is it considered abnormal? These conceptual distinctions have given rise to two general categories of theories of aging. The first category proposes that aging is the result of the accumulation of random injuries and events. The second category proposes that aging is the result of a genetically controlled developmental program, or built-in self-destructive processes. A classic experiment done by Hayflick84 demonstrated that fibroblasts are limited to a finite number of generations (40 to 60 doublings). Hayflick himself, however, reports this theory of aging is now losing support. “The weight of evidence indicates that genes do not drive the aging process. …”93 Rather it is the molecular health (fidelity) —structure and function—that underlies aging. Increasing molecular disorder, or entropy, causes aging. Molecular disorder is due to random targeted events (i.e., stochastic) that affect cellular renewal and repair. The loss of molecular order ultimately exceeds repair and turnover capacity and thus increases vulnerability to pathology or age-associated disease.93 (No matter what conceptual distinction is used as a basis, it seems clear that even in the absence of disease, the individual’s frailty increases with age, and death inevitably results! Or as Dick Cavett laments, “Don’t worry about aging, it won’t go on forever.”
Normal Life Span
The maximal life span of humans is between 80 and 100 years and does not vary significantly among populations. However, in primitive societies, few individuals reach the maximal life span; most die in infancy and the early years.94 In societies with improved sanitation, housing, nutrition, and healthcare, many individuals attain the maximal life span. Although the maximal life span has not changed significantly over time, life expectancy has increased—but not for all Americans. In each successive age group from 65 years and older, women outnumber men; thus women have a greater life expectancy than men. Increases in life expectancy have resulted in a larger older adult population and, for some, inherent problems of disability, disease, and socioeconomic hardship.
Life Expectancy Differences Across America
Life expectancy is the average number of years of life remaining at a given age. Although the slow but steady rise in life expectancy has occurred generally in the United States, disparities exist among various counties. A surprising government-sponsored study by Harvard researchers found life expectancy actually declined in a number of counties (e.g., smallest unit of analysis) from 1983 to 1999, particularly for women (see What’s New? Decline in Life Expectancy in Some U.S. Counties).
Theories and Mechanisms of Aging
Relatively little “indisputable” knowledge exists on the subject of aging. Gaining support, however, is that aging is the result of cellular damage and molecular disorder. Table 2-11 presents the historical development of aging research. Numerous theories exist about the causes of aging. Many of these theories overlap, interact, and are similar. Some of them have focused on a single mechanism—the so-called magic bullet approach to arrest aging. It is doubtful that a single theory will explain all the mechanisms of aging. Emerging from active investigation are some common mechanisms. These mechanisms are included in the What’s New? The Emerging Focus on the Biology of Aging.
Table 2-11
| Theory | Year | Proponent |
| Waste product theory | 1923 | Carrell and Ebeling |
| Wear-and-tear theory | 1924 | Pearl |
| Rate of living theorya | 1928 | Pearl |
| Neuroendocrine theory (including DHEA and melatonin) | 1947 | Korenchevsky and Jones |
| Free-radical theory | 1955 | Harman |
| Collagen theoryb | 1957 | Verzar |
| Metabolic theorya | 1957; 1961 | Carlson et al; Johnson et al |
| Somatic mutation theory | 1959 | Sziliard |
| Error-catastrophe theory | 1963; 1970 | Orgel |
| Cross-linking theoryb | 1968 | Bjorksten |
| Programmed senescence theory | 1969 | Hayflick |
| Immunologic theory | 1969 | Walform |
| Mitochondrial theory | 1972 | Harman |
| Evolution theory | 1977 | Kirkwood |
NOTE: Theories with the same superscript may represent the same theory.
Data from Schneider EL: Theories of aging: a perspective. In Warner HR et al, editors: Modern biological theories of aging, New York, 1987, Raven; Madison HE: Theories of aging. In Lueckenotte A: Gerontologic nursing, ed 2, St Louis, 2000, Mosby; Hayflick L: How and why we age, New York, 1996, Ballantine Books.
Degenerative Extracellular Changes
Extracellular factors that affect the aging process include the binding of collagen; the increase in free radicals’ effects on cells; the structural alterations of fascia, tendons, ligaments, bones, and joints; and peripheral vascular disease, particularly arteriosclerosis (see Chapter 30).
Aging affects the extracellular matrix with increased cross-linking (e.g., aging collagen becomes more insoluble, chemically stable, but rigid, resulting in a decrease of cell permeability), decreased synthesis, and increased degradation of collagen. These changes, together with the disappearance of elastin and changes in proteoglycans and plasma proteins, cause disorders of the ground substance that result in dehydration and wrinkling of the skin (see Chapter 44). Other age-related defects in the extracellular matrix include skeletal muscle alterations (e.g., atrophy, decreased tone, loss of contractility), cataracts, diverticula, hernias, and rupture of intervertebral disks.
Free radicals of oxygen that result from oxidative cellular metabolism, called oxidative stress (e.g., respiratory chain, phagocytosis, prostaglandin synthesis), are known to damage tissues during the aging process (Figure 2-37). The oxygen radicals produced include superoxide radical, hydroxyl radical, and hydrogen peroxide (see p. 54). These oxygen products are extremely reactive and can damage nucleic acids, destroy polysaccharides, oxidize proteins, peroxidize unsaturated fatty acids, and kill and lyse cells. Oxidant effects on target cells can give rise to malignant transformation, presumably through DNA damage. That progressive and cumulative damage from oxygen radicals may lead to harmful alterations in cellular function is consistent with those alterations of aging. This hypothesis is founded on the wear-and-tear theory of aging, which states that damages accumulate with time, decreasing the organism’s ability to maintain a steady state. Because these oxygen-reactive species not only can permanently damage cells but also may lead to cell death, there is new support for their role in the aging process.
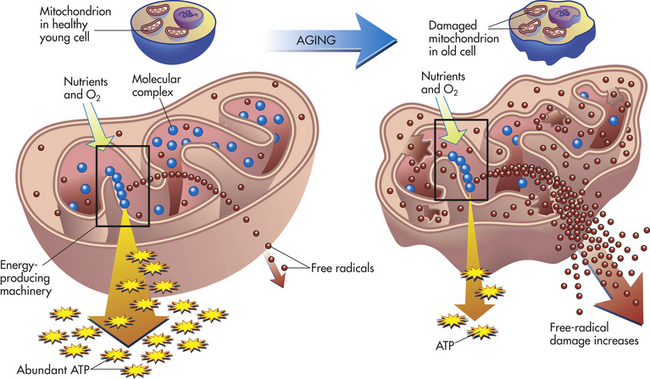
Of much interest is the relationship between aging and the disappearance or alteration of extracellular substances important for vessel integrity. With aging, lipid, calcium, and plasma proteins are deposited in vessel walls. These depositions cause serious basement membrane thickening and alterations in smooth muscle functioning, resulting in arteriosclerosis. Arteriosclerosis is a progressive disease that causes serious problems in the aged individual, including stroke, myocardial infarction, renal disease, and peripheral vascular disease.
Cellular Aging
Cellular changes characteristic of aging include atrophy, decreased function, and loss of cells, possibly caused by apoptosis. Loss of cellular function from any of these causes initiates the compensatory mechanisms of hypertrophy and hyperplasia of remaining cells, which can lead to metaplasia, dysplasia, and neoplasia. All these changes can alter receptor placement and function, nutrient pathways, secretion of cellular products, and neuroendocrine control mechanisms. In the aged cell, DNA, RNA, cellular proteins, and membranes are most susceptible to injurious stimuli. DNA is particularly vulnerable to such injuries as breaks, deletions, and additions. Although DNA can repair itself with time, the aged cell’s capacity for DNA repair is decreased. Lack of DNA repair increases the cell’s susceptibility to mutations that may be lethal or may promote the development of neoplasia (see What’s New? The Emerging Focus on the Biology of Aging).
Mitochondria are the organelles responsible for the generation of most of the energy used by eukaryotic cells. Mitochondrial DNA (mtDNA) encodes some of the proteins of the electron transfer chain, the system necessary for the conversion of adenosine diphosphate (ADP) to ATP. Mutations in mtDNA can deprive the cell of ATP, and mutations are correlated with the aging process. The most common age-related mtDNA mutation in humans is a large rearrangement called the 4977 deletion, or common deletion, and is found in humans more than 40 years old. It is a deletion that removes all or part of 7 of the 13 protein-encoding mtDNA genes and 5 of the 22 tRNA genes. Individual cells containing this deletion have a condition known as heteroplasmy. Heteroplasmy levels rise with aging and are tissue dependent.95–97
The production of ROS under physiologic conditions is associated with activity of the respiratory chain in aerobic ATP production. Therefore, increased mitochondrial activity per se can be an “oxidative stress” to cells. The production of ROS is markedly increased in many pathologic conditions in which the respiratory chain is impaired. Because mtDNA, which is essential for normal oxidative phosphorylation, is located in proximity to the ROS-generating respiratory chain, it is more oxidatively damaged than is nuclear DNA. Cumulative damage of mtDNA is implicated in the aging process as well as in the progression of such common diseases as diabetes, cancer, and heart failure.
Tissue and Systemic Aging
It is probably safe to say that every physiologic process can be shown to function less efficiently with increasing age. The most characteristic tissue change with age is a progressive stiffness or rigidity that affects many systems, including the arterial, pulmonary, and musculoskeletal systems. A consequence of blood vessel and organ stiffness is a progressive increase in peripheral resistance to blood flow. The movement of intracellular and extracellular substances also usually decreases with age as does the diffusion capacity of the lung. Blood flow through organs decreases; for example, renal plasma flow decreases.
Changes in the endocrine and immune systems include thymus atrophy. Although this occurs at puberty, causing a decreased immune response to T-dependent antigens (foreign proteins), increased autoantibodies and immune complexes (antibodies bound to antigen) and an overall decrease in the immunologic tolerance for the host’s own cells further diminish the effectiveness of the immune system later in life. The reproductive system loses ova in women, and spermatogenesis in men is decreased. Responsiveness to hormones decreases in the breast and endometrium.
The stomach experiences decreases in the rate of emptying and secretion of hormones and hydrochloric acid. Muscular atrophy diminishes mobility by decreasing motor tone and contractility. Sarcopenia, the loss of muscle mass and strength, can occur into old age. The skin of the aged individual is affected by atrophy and wrinkling of the epidermis and alterations in underlying dermis, fat, and muscle.
Total body changes include a decrease in height; a reduction in circumference of the neck, thighs, and arms; widening of the pelvis; and lengthening of the nose and ears. Several of these changes are the result of tissue atrophy and decreased bone mass caused by osteoporosis and osteoarthritis. Body composition changes with age.98 With middle age there is an increase in body weight (men gain until 50 years of age and women until 70 years) and fat mass followed by a decrease in stature, weight, fat-free mass (FFM) (FFM includes all minerals, proteins, and water plus all other constituents except lipids), and body cell mass at older ages. As fat increases, total body water decreases. Increased body fat and centralized fat distribution (abdominal) are associated with non–insulin-dependent diabetes and heart disease. Total body potassium also decreases because of decreased cellular mass. An increased sodium/potassium ratio suggests that the decreased cellular mass is accompanied by an increased extracellular compartment.
Although some of these alterations are probably inherent in aging, others represent consequences of aging. Advanced age increases susceptibility to disease, and death occurs after an injury or insult because of diminished cellular, tissue, and organic function. To determine that an individual “died of old age” would be a monumental if not impossible task.
Frailty
Frailty is imprecisely defined as a wasting syndrome of aging, leaving a person vulnerable to falls, functional decline, disease, and death.99 The syndrome is complex, involving decreased protein synthesis, sarcopenia, neuroendocrine and muscular decline, and immune dysfunction. The clinical condition of frailty includes decreased lean body mass (sarcopenia), osteopenia, cognitive impairment, and anemia.100 The most evident feature is sarcopenia. The altered skeletal muscle mass is associated with fatigue, weakness, imbalance, altered gait, and speed.101 Recent studies raise the possibility of dysregulated inflammatory processes and ROS as being involved or central to patterns of aging and frailty.100 Several physiologic gender differences may explain differing levels of frailty: (1) higher baseline levels of muscle mass for men may be protective against frailty, (2) testosterone and growth hormone can provide advantages in muscle mass maintenance, (3) cortisol is more dysregulated in older women than older men, (4) alterations in immune function and immune responsiveness to sex steroids make men more vulnerable to sepsis and infection and women more vulnerable to chronic inflammatory conditions and muscle mass loss, and (5) lower levels of activity and caloric intake may influence greater susceptibility to frailty in women.102
SOMATIC DEATH
Somatic death is death of the entire person. Unlike the changes that follow cellular death in a live body, postmortem change is diffuse and does not involve components of the inflammatory response. Within minutes of death, manifestations of postmortem change appear, eliminating any difficulty in determining that death has occurred. The most notable manifestations are complete cessation of respiration and circulation. The surface of the skin usually becomes pale and yellowish; however, the lifelike color of the cheeks and lips may persist after death from causes such as carbon monoxide poisoning, drowning, and chloroform poisoning.103
Body temperature falls gradually immediately after death and then more rapidly (approximately 1.0° to 1.5° F/hr) until, after 24 hours, body temperature equals that of the environment.104 After death caused by certain infective diseases, body temperature may continue to rise for a short time. Postmortem reduction of body temperature is called algor mortis.
Blood pressure within the retinal vessels decreases, causing muscle tension to decrease and the pupils to become dilated. The face, nose, and chin begin to look “sharp” or “peaked” as blood and fluids drain away.103 Gravity causes blood to settle in the most dependent, or lowest, tissues, which develop a purple discoloration called livor mortis. Incisions at this time usually fail to cause bleeding. The skin loses its elasticity and transparency.
Within 6 hours after death, acidic compounds accumulate within the muscles because of the breakdown of carbohydrate and depletion of ATP. This interferes with ATP-dependent detachment of myosin from actin (contractile proteins), and muscle stiffening, or rigor mortis, sets in. The smaller muscles are usually affected first, particularly the muscles of the jaw. Within 12 to 14 hours, rigor mortis usually affects the entire body.
Signs of putrefaction—state of decay with foul-smelling odor—are generally obvious about 24 to 48 hours after death. Rigor mortis gradually diminishes, and the body becomes flaccid in 12 to 14 hours. Putrefactive changes vary depending on the temperature of the environment. The most visible is greenish discoloration of the skin, particularly on the abdomen. The discoloration is thought to be related to the diffusion of hemolyzed blood into the tissues and the production of sulfhemoglobin.105 Slippage or loosening of the skin from underlying tissues occurs at the same time. After this, swelling or bloating of the body and liquefactive changes occur, sometimes causing opening of the body cavities. At a microscopic level, putrefactive changes are associated with the release of enzymes and lytic dissolution called postmortem autolysis.
Abrasion 64
Algor mortis 90
Anoxia 52
Apoptosis 84
Asphyxial injury 68
Atrophy 47
Atypical hyperplasia 49
Autophagic vacuole 47
Avulsion 64
Bilirubin 79
Blast injury 72
Blow back 66
Blunt force injury 62
Callus 49
Carbon monoxide (CO) 59
Carboxyhemoglobin 59
Caseous necrosis 82
Caspase 85
Cellular accumulation (infiltration) 76
Cellular swelling 76
Chemical asphyxiant 68
Choking asphyxiation 68
Chopping wound 66
Coagulative necrosis 82
Compensatory hyperplasia 49
Contact range entrance wound 66
Contusion 62
Cyanide 68
Decompression sickness (caisson disease) 72
Drowning 68
Dry gangrene 83
Dysplasia (atypical hyperplasia) 49
Dystrophic calcification 79
Entrance wound 66
Epidural hematoma 63
Ethanol 59
Exit wound 67
Fat necrosis 83
Fat-free mass (FFM) 90
Fatty change 76
Fetal alcohol syndrome (FAS) 61
Frailty 90
Free radical 54
Gangrenous necrosis 83
Gas gangrene 84
Hanging strangulation 68
Heat cramp 72
Heat exhaustion 72
Heat stroke 72
Hematoma 62
Hemoprotein 79
Hemosiderin 79
Hemosiderosis 79
Hepatocyte growth factor (HGF) 49
Hormonal hyperplasia 49
Hydrogen sulfide (sewer gas) 68
Hyperglycemia 69
Hyperlipidemia 69
Hyperplasia 48
Hyperthermic injury 72
Hypertrophy 47
Hypolipidemia 69
Hypothermic injury 71
Hypoxia 52
Incised wound 65
Indeterminate-range (distant) entrance wound 67
Intermediate-range entrance wound 66
Ionizing radiation 73
Irreversible injury 51
Ischemia 52
Karyolysis 81
Karyorrhexis 81
Laceration 64
Lead 56
Life expectancy 86
Ligature strangulation 68
Lipid-acceptor protein (apoprotein) 55
Lipid peroxidation 54
Lipofuscin 47
Liquefactive necrosis 82
Livor mortis 90
Manual strangulation 68
Maximal life span 86
Mechanoporation 75
Melanin 78
Metaplasia 50
Metastatic calcification 79
Muzzle imprint 66
Necrosis 81
Noise 75
Oncosis (vacuolar) degeneration 76
Oxidative stress 54
Pathologic atrophy 47
Pathologic hyperplasia 49
Physiologic atrophy 47
Postmortem autolysis 90
Postmortem change 90
Psammoma body 79
Puncture wound 66
Pyknosis 81
Reperfusion (reoxygenation) injury 54
Reversible injury 51
Rigor mortis 90
Sarcopenia 90
Shored exit wound 67
Somatic death 90
Stab wound 65
Stippling 66
Strangulation 68
Subdural hematoma 62
Suffocation 68
Tattooing 66
Ubiquitin-proteasome pathway 47
Up-regulation of proteasome 47
Urate 81
Vacuolation 53
Wet gangrene 83
REFERENCES
1. Dahlmann, B. Role of proteasomes in disease. BMC Biochem. 2007;8(Suppl):S3.
2. Damjanov, I., Linder, J. Anderson’s pathology, ed 10. St Louis: Mosby; 1996.
3. Bicknell, K.A., Coxon, C.H., Brooks, G. Can the cardiomyocyte cell cycle be reprogrammed? J Mol Cell Cardiol. 2007;42(4):706–721.
4. Bottaro, D.P., et al. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science. 1991;251(4995):802–804.
5. Alberts, B., et al. Molecular biology of the cell, ed 5. New York: Garland, 2008.
6. Degnim, A.C., et al. Stratification of breast cancer risk in women with atypia: a Mayo cohort study. J Clin Oncol. 2007;25(19):2672–2677.
7. Reis-Filho, J.S., Lakhani, S.R. The diagnosis and management of pre-invasive breast disease: genetic alterations in pre-invasive lesions. Breast Cancer Res. 2004;5:313–319.
8. Kumar, V., Abbas, A., Fausto, N. Robbins and Cotran pathologic basis of disease, ed 7. Philadelphia: Saunders; 2005.
9. Valko, M., et al. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39(1):44–84.
10. Li, C., Jackson, R.M. Reactive species mechanisms of cellular hypoxia—reoxygenation injury. Am J Physiol Cell Physiol. 2002;282:C227–C241.
11. Murphy, E., Steenbergen, C. Mechanisms underlying acute protection from cardiac ischemia—reperfusion injury. Physiol Rev. 2008;88(2):581–609.
12. Burwell, L.S., Brookes, P.S. Mitochondria as a target for the cardioprotective effects of nitric oxide in ischemia-reperfusion injury. Antioxid Redox Signal. 2008;10(3):579–599.
13. Papaharalambus, C.A. Basic mechanisms of oxidative stress and reactive oxygen species in cardiovascular injury. Trends Cardiovasc Med. 2007;17(2):48–54.
14. Gunnell, D., Murray, V., Hawton, K. Use of paracetamol (acetaminophen) for suicide and nonfatal poisoning: worldwide patterns of use and misuse. Suicide Life Threat Behav. 2000;30:313–326.
15. Lai, M.W., et al. Annual report of the American Association of Poison Control Centers’ national poisoning and exposure database. Clin Toxicol (Phila). 2005;44(6-7):803–932. [2006].
16. Heard, K.J. Acetlycysteine for acetaminophen poisoning. N Engl J Med. 2008;359(3):285–292.
17. Murray, K.F., et al. Drug-associated hepatotoxicity and acute liver failure. J Pediatr Gastroenterol Nutr. 2008;47(5):395–405.
18. Markel, H. Getting the lead out: the Rhode Island Lead Pain Trials and Their Impact on Children’s Health. JAMA. 2007;297(24):2773–2775.
19. Roberts, J.W., et al. Reducing dust, lead, dust mites, bacteria, and fungi in carpets by vacuuming. Arch Environ Contam Toxicol. 1999;36(4):477–484.
20. Schanne, F.A., Long, G.J., Rosen, J.F. Lead induced rise in intracellular free calcium is mediated through activation of protein kinase C in osteoblastic bone cells. Biochem Biophys Acta. 1997;1360(3):247–254.
21. Meo, S.A., Al-Khlawi, T. Health hazards of welding fumes. Saudi Med J. 2003;24(11):1176–1182.
22. Holbrook, J. Cigarette smoking. In: Rom W.H., ed. Environmental and occupational medicine. Boston: Little, Brown, 1993.
23. Cassel C.K., et al, eds. Geriatric medicine, ed 2, New York: Springer-Verlag, 1990.
24. Lieber, C.S. Microsomal ethanol-oxidizing system (MEOS): the first 30 years (1968-1998)—a review. Alcohol Clin Exp Res. 1999;23(6):991–1007. [Review].
25. Agarwal, D.P. Genetic polymorphisms of alcohol metabolizing enzymes. Pathol Biol (Paris). 2001;49(9):703–709. [Review].
26. Booyse, F.M., et al. Mechanisms by which alcohol and wine polyphenols affect coronary heart disease risk. Ann Epidemiol. 2007;17(5 Suppl):S24–S31.
27. Sesso, H.D. Alcohol and cardiovascular health: recent findings. Am J Cardiovasc Drugs. 2001;1(3):167–172.
28. O’Keefe, J.H., Bybee, K.A., Lavie, C.J. Alcohol and cardiovascular health: the razor-sharp double-edged sword. Am J Coll Cardiol. 2007;50(11):1009–1014.
29. May, J.J. Occupational hearing loss. Am J Ind Med. 2000;37(1):112–120.
30. Punjabi, N.M. The epidemiology of adult obstructive sleep apnea. Proc Am Thorac Soc. 2008;5(2):136–143.
31. Guilleminault, A., Abad, V.C. Obstructive sleep apnea syndromes. Med Clin North Am. 2004;88(3):611–630.
32. Lieber, C.S. Metabolism of alcohol. Clin Liver Dis. 2005;9(1):1–35.
33. Lieber, C.S. Alcoholic liver disease: new insights in pathogenesis lead to new treatments. J Hepatol. 2000;32(1 Suppl):113–128.
34. Clark, C.M., et al. Structural and functional brain integrity of fetal alcohol syndrome in nonretarded cases. Pediatrics. 2000;105(5):1096.
35. Saito, M., et al. Ethanol alters lipid profiles and phosphorylation status of AMP-activated protein kinase in the neonatal mouse brain. J Neurochem. 2007;103(3):1208–1218.
36. Stratton K., Howe C., Battaglia F., eds. Fetal alcohol syndrome: diagnosis, epidemiology, prevention and treatment. Washington, DC: National Academy Press, 1996.
37. Mattson, S.N., Riley, E.P. Brain anomalies in fetal alcohol syndrome. In: Abel E.A., ed. Fetal alcohol syndrome: from mechanism to prevention. Boca Raton, FL: CRC Press, 1996.
38. Cortese, B.M., et al. Magnetic resonance and spectroscopic imaging in prenatal alcohol-exposed children: preliminary findings in the caudate nucleus. Neurotoxicol Teratol. 2006;28(5):597–606.
39. Boonstra, J., et al. The epidermal growth factor. Cell Biol Int. 1995;19(5):413–430.
40. Henderson, G.I., et al. Ethanol, oxidative stress, reactive aldehydes, and the fetus. Front Biosci. 1999;15(4):D541.
41. Lieber, C.S. CYP2EI: from ASH to NASH. Hepatol Res. 2004;28(1):1–11.
42. Donohue, T.M., Jr., et al. Role of the proteasome in ethanol-induced liver pathology. Alcohol Clin Exp Res. 2007;31(9):1446–1459.
43. Clarkson, T.W., Magos, L., Myers, G.I. The toxicology of mercury—current exposures and clinical manifestations. N Engl J Med. 2003;349(18):1731–1737.
44. Ahlquist, M., et al. Serum mercury concentrations in relation to survival, symptoms, and disease: results from the prospective population study of women in Gothenburg, Sweden. Acta Odontol Scand. 1999;57:168–174.
45. Bjorkman, L., Pedersen, N.L., Lichtenstein, P. Physical and mental health related to dental amalgam fillings in Swedish twins. Community Dent Oral Epidemiol. 1996;24(4):260–267.
46. Saxe, S.R., et al. Dental amalgam and cognitive function in older women: findings from the Nun Study. J Am Dent Assoc. 1995;126(11):1495–1501.
47. Lauterbach, M., et al. Neurological outcomes in children with and without amalgam-related mercury exposure: seven years of longitudinal observations in a randomized trial. J Am Dent Assoc. 2008;139(2):138–145.
48. Powell, L.W., Kerr, J.F.R. Pathology of the liver in hemochromatosis. Pathobiol Annu. 1975;5:317–337.
49. Available at www.ewg.org/mercury1. Accessed 2008.
50. Thompson, W.W., et al. Early thimerosal exposure and neuropsychological outcomes at 7 to 10 years. N Engl J Med. 2007;357(13):1281–1292.
51. Pichichero, M.E., et al. Mercury concentrations and metabolism in infants receiving vaccines containing thimerosal: a descriptive study. Lancet. 2002;360(9347):1737–1741.
52. Smith, J.C., Farris, F.F. Methyl mercury pharmacokinetics in man: a reevaluation. Toxicol Appl Pharmacol. 137(2), 1996. [254].
53. Baumgartner, R.N., et al. Age-related changes in sex hormones affect the sex difference in serum leptin independently of changes in body fat. Metabolism. 1999;48(3):378–384.
54. Fraker, P.J., Lill-Elghanian, D.A. The many roles of apoptosis in immunity as modified by aging and nutritional status. J Nutr Health Aging. 2004;8(1):56–63.
55. Stanton, B., Galbraith, J. Drug trafficking among African-American early adolescents: prevalence, consequences, and associated behaviors and beliefs. Pediatrics. 1994;93(6, Pt 2):1039–1043.
56. Centers for Disease Control and Prevention. Injury statistics website. Washington, DC: Centers for Disease Control and Prevention; 2002.
57. Kopec, D., et al. The state-of-the-art in the reduction of medical errors. Stud Health Technol Inform. 2006;121:126–137.
58. Holick, M.F. Vitamin D: importance in the prevention of cancers, type 1 diabetes, and osteoporosis. Am J Clin Nutr. 2004;79(3):362–372.
59. Camara, A.K., et al. Hypothermia augments reactive oxygen species detected in the guinea pig isolated perfused heart. Am J Physiol Heart Circ Physiol. 2004;286(4):H1289–H1299.
60. Rauen, U., de Groot, H. Mammalian cell injury induced by hypothermia—the emerging role for reactive oxygen species. Biol Chem. 2002;383(3-4):477–488.
61. Bartels-Stringer, M., et al. Preserved vascular reactivity of rat renal arteries after cold storage. Cryobiology. 2004;48(1):95–98.
62. Osorio, R.A., et al. Reactive oxygen species in pregnant rats: effects of exercise and thermal stress. Comp Biochem Physiol C Toxicol Pharmacol. 2003;135(1):89–95.
63. Rauen, U., et al. Hypothermia: injury/cold-induced apoptosis—evidence of an increase in chelatable iron causing oxidative injury in spite of low O−2/H2O2 formation. FASEB J. 2000;14(13):1953–1964.
64. Roy, S.B., et al. Haemodynamic studies in high altitude pulmonary oedema. Br Heart J. 1969;31(1):52–58.
65. U.S. Department of Health and Human Services: Report on carcinogens, ed 11, 2004. Available at ntp.nichs.nih.gov/ntp/roc/toc11.html.
66. Cologne, J.B., et al. Effects of radiation on incidence of primary liver cancer among atomic bomb survivors. Radiat Res. 1999;152(4):364–373.
67. Gilbert, E.S., et al. Liver cancers in Mayak workers. Radiat Res. 2000;154(3):246–252.
68. Yap, J., et al. Sarcoma as a second malignancy after treatment for breast cancer. Int J Radiat Oncol Biol Phys. 2002;52(5):1231–1237.
69. Preston, D.L., Shimizu, Y., Pierce, D.A. Studies of mortality of atomic bomb survivors. Report 13: solid cancer and noncancer disease mortality. Radiat Res. 2003;160:381–407.
70. Hoel, D.G. Ionizing radiation and cardiovascular disease. Ann N Y Acad Sci. 2006;1076:309–317.
71. U.S. Food and Drug Administration: What are the radiation risks from CT?, 2002. Available at www.fda.gov/edrh/et/risks.html
72. Siu, T.L., Morley, J.W., Coroneo, M.T. Toxicology of the retina: advances in understanding the defence mechanisms and pathogenesis of drug- and light-induced retinopathy. Clin Experiment Ophthalmol. 2008;36(2):176–185.
73. Available at www.ccoks.ca/oshansevers/ergonomics/lighting-flicker.html. Accessed 2008.
74. Wilkins, A.J., Wilkinson, P. A tint to reduce eye-strain from fluorescent lighting? Preliminary observations. Ophthalmic Physiol Opt. 1991;11(2):172–175.
75. Rubin, G.S., et al. A prospective, population-based study of the role of visual impairment in motor vehicle crashes among older drivers: the SEE study. Invest Ophthalmol Vis Sci. 2007;48(4):1483–1491.
76. Mainster, M.A., Timberlake, G.T. Why HID headlights bother older drivers. Br J Opthalmol. 2003;87(1):113–117.
77. De Flora, S., D’Agostini, F. Halogen lamp carcinogenicity. Nature. 1992;356:569.
78. Bloom, E., et al. Halogen lamp phototoxicity. Dermatology. 1996;193(3):207–211.
79. Barbee, K.A. Mechanical cell injury. Ann N Y Acad Sci. 2005;1066:67–84.
80. Van Epps, J.S., Vorp, D.A. Mechanopathobiology of atherogenesis: a review. J Surg Res. 2007;142(1):202–217.
81. Keyserling, W.M., Armstrong, T.J. Ergonomics. In Last J.M., Wallace R.B., eds.: Maxey-Roseneau-Last: public health and preventive medicine, ed 13, Norwalk, Conn: Appleton & Lange, 1992.
82. NIDCD Statistics about hearing disorders, ear infections, and deafness. Washington, DC: National Institutes of Health; 2005. Available at www.nidcd.nih.gov/health/statistics/nearing.asp.
83. Halpern, N.A., et al. Hearing loss in critical care: an unappreciated phenomenon. Crit Care Med. 1999;27(1):211–219.
84. Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965;37:614–636.
85. Kerr, J.F.R., Searle, J. Apoptosis: its nature and kinetic role. In: Meyn R.E., Withers H.R., eds. Radiation biology in cancer research. New York: Raven, 1980.
86. Wyllie, A.H., Kerr, J.F.R., Currie, A.R. Cell death: the significance of apoptosis. Int Rev Cytol. 1980;68:251–306.
87. Basu, A. Involvement of protein kinase C-delta in DNA damage-induced apoptosis. J Cell Mol Med. 2003;7(4):341–350.
88. Alberts, B., et al. Essential cell biology: an introduction to the molecular biology of the cell, ed 2. New York: Garland, 2004.
89. Amour, A., et al. General considerations for proteolytic cascades. Biochem Soc Trans. 2004;32(Pt 1):15–16.
90. Logue, S.E., Martin, S.J. Caspase activation cascades in apoptosis. Biochem Soc Trans. 2008;36(Pt 1):1–19.
91. Johnson H.A., Johnson H.A., eds. Is aging physiological or pathological? In Johnson HA ed: Relations between normal aging and disease. New York: Raven, 1985.
92. Vijg, J. Somatic mutations and aging: a re-evaluation. Mutat Res. 2000;447(1):117–135.
93. Hayflick, L. Biological aging is no longer an unsolved problem. Ann N Y Acad Sci. 2007;1100:1–13. [Review].
94. Poehlman, E.T., et al. Physiological predictors of increasing total and central adiposity in aging men and women. Arch Intern Med. 1995;155(22):2443–2448.
95. Butow, R.A., Avadhani, N.G. Mitochondrial signaling: the retrograde response. Mol Cell. 2004;14(1):1–15.
96. Maassen, J.A., et al. Mitochondrial diabetes: molecular mechanisms and clinical presentation. Diabetes. 2004;52(Suppl1):S103–S109.
97. Samuels, D.C. Mitochondrial DNA repeats constrain the life span of mammals. Trends Genet. 2004;20(5):226–229.
98. Baumgartner, R.N., et al. Cross-sectional age differences in body composition in persons 60+ years of age. J Gerontol A Biol Sci Med Sci. 1995;50(6):M307–M316.
99. Muhlberg, W., Sieber, C. Sarcopenia and frailty in geriatric patients: implications for training and prevention. Z Gerontol Geriatr. 2004;37(1):2–8.
100. Ershler, W.B. A gripping reality: oxidative stress, inflammation, and the pathway to frailty. J Appl Physiol. 2007;103:3–5.
101. Greenlund, L.J., Nair, K.S. Sarcopenia—consequences, mechanisms, and potential therapies. Mech Ageing Dev. 2003;124:287–299.
102. Gillick, M. Pinning down frailty. J Gerontol A Biol Sci Med Sci. 2001;56(3):M134–M135.
103. Shennan, T. Postmortems and morbid anatomy, ed 3. Baltimore: William Wood; 1935.
104. Minckler, J., Anstall, H.B., Minckler, T.M. Pathobiology: an introduction. St Louis: Mosby; 1971.
105. Richter, C., et al. Oxidants in mitochondria: from physiology to diseases. Biochim Biophys Acta. 1995;1271(1):67–74.