DISORDERS OF JOINTS
The American College of Rheumatology (ACR) recognizes 13 groups of joint disease—arthropathies. Most of these disorders can be placed into two major categories: noninflammatory joint disease and inflammatory joint disease. With recent improvements in detection methods, conditions such as osteoarthritis that were previously classified as noninflammatory have now had inflammatory pathways identified.
Osteoarthritis
Osteoarthritis (OA) has been commonly classified as noninflammatory joint disease. However, new information has made it clear that specific markers of inflammation are present in OA.51–53 The discovery of inflammation in the joints also has emerged as an important feature of OA. The use of MRI and arthroscopy has made it clear that osteoarthritic changes are not defined by changes noted on x-ray films alone. Consequently OA has been reclassified as inflammatory joint disease (Figure 42-21).
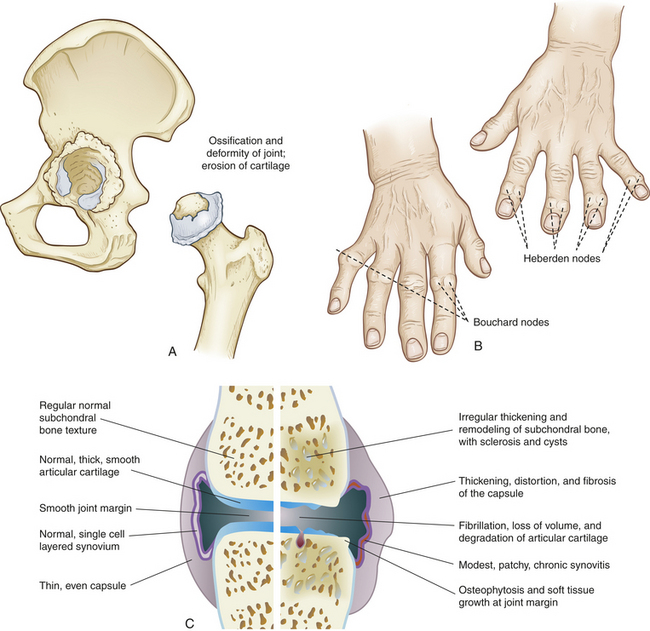
Figure 42-21 Osteoarthritis (OA) A, Cartilage and degeneration of the hip joint resulting from osteoarthritis. B, Heberden nodes and Bouchard nodes. C, Characteristics of OA. Normal versus osteoarthritic synovial joint.
Medical clinicians are somewhat divided about use of the terms degenerative joint disease and osteoarthritis. The term osteoarthritis has been used in most recent communications and appears to be the more accepted term in the European literature.
Osteoarthritis (OA) is a common age-related disorder of synovial joints. It is characterized by local areas of loss and damage of articular cartilage, new bone formation of joint margins (osteophytosis), subchondral bone changes, variable degrees of mild synovitis and thickening of the joint capsule (see Figure 42-21). Pathology centers on load-bearing areas. Advancing disease reveals narrowing of the joint space because of cartilage loss, bone spurs (osteophytes), and sometimes changes in the subchondral bone. OA can arise in any synovial joint but is commonly found in the hands, knees, hips, and spine. It is less common in people younger than 40 years but rises in incidence with age. OA is a multifactorial disease involving environmental-lifestyle factors and genetics.
OA is generally distributed throughout the peripheral and central joints of the body and affects adult men more than women until after age 55. Although the exact causes of OA are unclear, they involve low-grade inflammation, calcification of articular cartilage, genetic alterations, and metabolic disorders.54
With aging, the quality and quantity of the proteoglycans in cartilage decrease in direct proportion to the severity of OA. Evidence also suggests that OA involves a complex interaction of cytokines, growth factors, matrix metalloproteinases, and enzymes25 (see Pathophysiology).
OA can be caused by any condition that damages cartilage directly; subjects the joint surfaces or underlying bone to chronic, excessive, or abnormal forces; or causes instability in the joint. Specific risk factors include the following:
1. Trauma, particularly sprains, strains, joint dislocations, and fractures
2. Long-term mechanical stress associated with athletics, ballet dancing, or repetitive physical tasks worsened by obesity
3. The presence of inflammation in joint structures, during which inflammatory cells release enzymes capable of digesting cartilage cells
4. Joint instability caused by damage to supporting structures, such as the joint capsule, ligaments, or tendons
5. Neurologic disorders (e.g., diabetic neuropathy, Charcot neuropathic joint) in which pain and proprioceptive reflexes are diminished or lost, increasing the tendency for abnormal movement, positioning, or weightbearing
6. Congenital or acquired skeletal deformities
7. Hematologic or endocrine disorders, such as hemophilia, which causes chronic bleeding into the joints, or hyperparathyroidism, which causes bone to lose calcium
8. Drugs (e.g., colchicine, indomethacin, steroids) that stimulate the activity of collagen-digesting enzymes in the synovial membrane
All of these factors alter articular cartilage in some way and accelerate the rate of cartilage loss.
PATHOPHYSIOLOGY The primary defect in OA is loss of articular cartilage. Early in the disease, the articular cartilage loses its glistening appearance, becoming yellow-gray or brownish gray. As the disease progresses, surface areas of the articular cartilage flake off and deeper layers develop longitudinal fissures (fibrillation). The cartilage becomes thin and may be absent over some areas, leaving the underlying bone (subchondral bone) unprotected. Consequently, the unprotected subchondral bone becomes sclerotic (dense and hard). Cysts sometimes develop within the subchondral bone and communicate with the longitudinal fissures in the cartilage. Pressure builds in the cysts until the cystic contents are forced into the synovial cavity, breaking through the articular cartilage on the way. As the articular cartilage erodes, cartilage-coated osteophytes may grow outward from the underlying bone and alter the bone contours and joint anatomy. These spur-like bony projections enlarge until small pieces, called joint mice, break off into the synovial cavity. If osteophyte fragments irritate the synovial membrane, synovitis and joint effusion result. The joint capsule also becomes thickened and at times adheres to the deformed underlying bone, which may contribute to the limitation of movement (see Figure 42-21).
Articular cartilage is probably lost through enzymatic breakdown of the cartilage matrix—the proteoglycans, glycosaminoglycans, and collagen. First, the enzymes break down the macromolecules of proteoglycans, glycosaminoglycans, and collagen into large, diffusible fragments. Then the fragments are taken up by the cartilage cells (chondrocytes) and digested by the cell’s own lysosomal enzymes. (Processes of cellular uptake and lysosomal digestion are described in Chapter 1.) The loss of proteoglycans from articular cartilage is a hallmark of the osteoarthritic process.
Enzymatic destruction of articular cartilage begins in the matrix, with destruction of proteoglycans and collagen fibers. Enzymes, particularly stromelysin and acid metalloproteinase, affect proteoglycans by interfering with assembly of the proteoglycan subunit or the proteoglycan aggregate (see Chapter 41); these enzymes are markedly elevated in OA. Changes in the conformation of proteoglycans disrupt the pumping action that regulates movement of water and synovial fluid into and out of the cartilage. Without the regulatory action of the proteoglycan pump, cartilage imbibes too much fluid and becomes less able to withstand the stresses of weightbearing. Also with aging, the proteoglycan content is decreased and water content in cartilage can be increased by as much as 8%, affecting the strength of the cartilage. Persons with OA, even those with fairly extensive cartilage destruction, have elevated levels of proteoglycans or fragments of proteoglycans in their synovial fluid, perhaps indicative of a more pronounced reparative phase. Other studies indicate that inflammatory cytokines, such as IL-1 (see Chapter 7 for discussion of cytokines), play a major role in cartilage degradation in part through induction of nitric oxide synthase (iNOS) and nitric oxide (NO) increased generation. Cytokines release and activate proteolytic and collagenolytic enzymes, causing an imbalance of cell responses to growth factor activity.
Some of the enzymes that degrade collagen (e.g., chemokines and metalloproteinases) are produced by the synovium.53 With comparisons to atherosclerotic lesions, cartilage calcification also appears to involve apoptosis. Chondrocyte apoptosis is increased in OA cartilage and is directly correlated with hydroxyapatite crystal deposition.55 NO stimulates apoptosis in chondrocytes. The resultant cartilage destruction initiates the IL-1β and TNF-α pathways of inflammation.53,56 Genetic deficiencies of inhibitors of calcification also may contribute to “run away” calcification (Figure 42-22). Collagen breakdown destroys the fibrils that give articular cartilage its tensile strength and exposes the chondrocytes to mechanical stress and enzyme attack. Thus a cycle of destruction begins that involves all the components of articular cartilage—proteoglycans, collagen fibers, and chondrocytes.
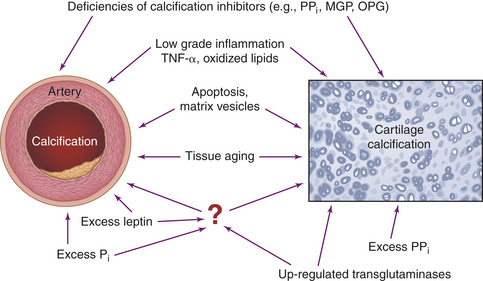
Figure 42-22 Mechanisms driving artery and cartilage calcification. All specified factors have been demonstrated to promote calcification in either arteries and/or cartilage (see text). MGP, (matrix GLA-protein): OPG, osteoprotegerin; PPi, inorganic pyrophosphate. (From Rutsch F, Terkeltaub R: Joint Bone Spine 72:110-118, 2005.)
CLINICAL MANIFESTATIONS Clinical manifestations of OA typically appear during the fifth or sixth decade of life, although asymptomatic articular surface changes are common after age 40. Pain and stiffness in one or more joints, usually weightbearing or load-bearing joints, are the first symptoms of the disease. Use-related joint pain relieved by rest is a key feature. Examination usually shows general involvement of peripheral and central joints. Peripheral joints most often involved are in the hands, wrists, knees, and feet. Central joints most often affected are in the lower cervical spine, lumbosacral spine, shoulders, and hips.
Joint structures are capable of generating a limited number of signs and symptoms. The primary signs and symptoms of joint disease are pain, stiffness, enlargement or swelling, tenderness, limited range of motion, muscle wasting, partial dislocation, and deformity.
Pain and stiffness are the predominant symptoms of OA. They are usually aggravated by weightbearing or use of the joint and relieved by resting the joint. Nocturnal pain is usually not relieved by rest and may be accompanied by paresthesias (numbness, tingling, prickling). Sometimes pain is referred to another part of the body. For example, osteoarthritis of the lumbosacral spine may mimic sciatica, causing severe pain in the back of the thigh along the course of the sciatic nerve. OA in the lower cervical spine may cause brachial neuralgia (pain in the arm) aggravated by movement of the neck. Osteoarthritic conditions in the hip cause pain that may be referred to the lower thigh and knee area.
The actual mechanisms of joint pain are complex and poorly understood, but several explanations are possible. The pain could be caused by articular distention and stretching of the fibrous joint capsule, which has an abundant nerve supply. In addition, inflammation of the joint capsule causes fibrous shrinking, so that movement of the joint in any direction causes painful stretching. Pain also can arise from the subchondral or periarticular bone (Figure 42-23).
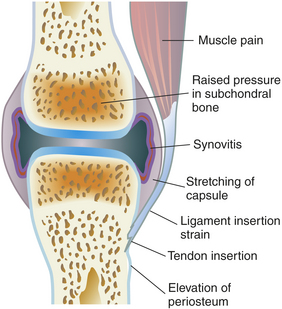
The origin of joint stiffness is unknown. Joint stiffness is generally defined as difficulty in initiating joint movement, immobility, or a loss of range of motion. The stiffness usually occurs as joint movement begins and dissipates within 30 minutes. Enlargement and bulging of joint contour, commonly described as swelling, may be caused by bone enlargement or the proliferation of osteophytes around the margins of the joint. Swelling also occurs if inflammatory exudate or blood enters the joint cavity, thereby increasing the volume of synovial fluid. This condition, termed joint effusion, is caused by (1) the presence of osteophyte fragments in the synovial cavity, (2) drainage of cysts from diseased subchondral bone, or (3) acute trauma to joint structures, resulting in hemorrhage and inflammatory exudation into the synovial cavity.
Range of motion is limited to some degree, depending on the extent of cartilage degeneration. Frequently, joint motion is accompanied by sounds of crepitus, creaking, or grating. Hypermobility and subluxation of joints occur in OA secondary to a neurologic disorder.
As OA of the lower extremity progresses, the person may begin to limp noticeably. Having a limp is distressing because it affects the person’s independence and ability to do activities of daily living. The affected joint is also more symptomatic after use, such as at the end of the day.
EVALUATION AND TREATMENT Evaluation consists of clinical assessment and radiologic studies, CT scan, arthroscopy, and MRI. Treatment is based on severity of symptoms. Conservative treatment includes rest of the involved joint until inflammation, if present, subsides; range-of-motion exercise to prevent joint capsule contraction; use of a cane, crutches, or walker to decrease weightbearing; weight loss if obesity is present; and analgesic and anti-inflammatory drug therapy to reduce swelling and pain. Glucosamine, a nutraceutical, has shown some success in reducing the pain and progression of OA. Other alternative therapies, including magnetic bracelets and acupuncture, seem to improve symptoms in some people. Intra-articular injection of high-molecular-weight viscosupplements, particularly hyaluronic acid, also has been successful in decreasing knee pain with OA.57 Surgery is used to improve joint movement, correct deformity or malalignment, or create a new joint with artificial implants. There are nearly 250,000 total hip replacements yearly in the United States, most of which are related to OA.
Classic Inflammatory Joint Disease
Inflammatory joint disease commonly is called arthritis. Inflammatory joint disease is characterized by inflammatory damage or destruction in the synovial membrane or articular cartilage and by systemic signs of inflammation (fever, leukocytosis, malaise, anorexia, hyperfibrinogenemia). See discussion on OA (considered part of inflammatory joint disease).
Inflammatory joint disease can be infectious or noninfectious. In infectious inflammatory joint disease, inflammation is caused by invasion of the joint by bacteria, mycoplasmas, viruses, fungi, or protozoa. These agents can invade the joint through a traumatic wound, surgical incision, or contaminated needle, or they can be delivered by the bloodstream from sites of infection elsewhere in the body, typically bones, heart valves, or blood vessels. In noninfectious inflammatory joint disease, which is the most common form, inflammation is caused by immune reactions or the deposition of crystals of monosodium urate in and around the joint. Rheumatoid arthritis and ankylosing spondylitis are noninfectious inflammatory diseases caused by disturbances in immune reactions58; gouty arthritis is a noninfectious inflammatory disease caused by crystal deposition.
Rheumatoid Arthritis
Rheumatoid arthritis (RA) is a systemic inflammatory autoimmune disease associated with swelling and pain in multiple joints. (Autoimmune disease is described in Chapter 8.) The first joint tissue to be affected is the synovial membrane, which lines the joint cavity (see Chapter 41 and Figure 42-9). Multiple immunoregulatory cytokines (such as interleukins, B-cells, and matrix metalloproteinases) contribute to joint damage. Eventually, inflammation may spread to the articular cartilage, fibrous joint capsule, and surrounding ligaments and tendons, causing pain, joint deformity, and loss of function (Figure 42-24). The joints most commonly affected are in the fingers, feet, wrists, elbows, ankles, and knees, but the shoulders, hips, and cervical spine also may be involved, as well as the tissues of the lungs, heart, kidneys, and skin.
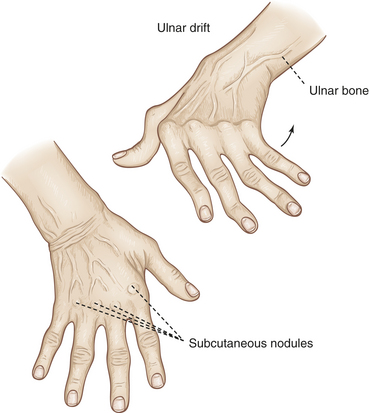
Figure 42-24 Rheumatoid arthritis of the hand. Note swelling from chronic synovitis of metacarpophalangeal joints, marked ulnar drift, subcutaneous nodules, and subluxation of metacarpophalangeal joints with extension of proximal interphalangeal joints and flexion of distal joints. Note also deformed position of thumb. Hand has wasted appearance. (From Mourad L: Orthopedic disorders, St Louis, 1991, Mosby.)
RA affects 1% to 2% of adults and, like most autoimmune diseases, develops most often in women, with a female/male ratio of 3:1. The frequency of RA increases from the third decade on, affecting 5% or more of the population ages 70 years and older. Besides inflammation of the joints, RA can cause fever, malaise, rash, lymph node or spleen enlargement, and Raynaud phenomenon (transient lack of circulation to the fingertips and toes).
Despite intensive research, the cause of RA remains obscure. Proposals of the initiating event include an infectious agent or other environmental exposure but genetic, hormonal, and reproductive factors may contribute to developing RA. The initiating event unleashes an immune response that results in inflammation of the lining of the joint—the synovial membrane. RA probably occurs in a genetically susceptible person because of an aberrant immune response to an unidentified antigen. A key genetic element has been localized to the human leukocyte antigen-death receptor 4 (HLA-DR4), HLA-DRB1 (death receptor beta), and HLA-DP genes of the major histocompatibility complex. Infectious microorganisms that may play a role in the cause of RA include bacteria, mycoplasmas, and viruses (especially Epstein-Barr virus) (Table 42-7). With long-term or intensive exposure to the antigen, normal antibodies (immunoglobulins [Ig]) become autoantibodies—antibodies that attack host tissues (self-antigens). Because they are usually present in individuals with rheumatoid arthritis, the transformed antibodies are termed rheumatoid factors (RFs). The RFs usually consist of two classes of immunoglobulin antibodies (antibodies for IgM and IgG) but occasionally involve antibodies for IgA. Their main antigenic targets are portions of the immunoglobulin molecules. RFs bind with their target self-antigens in blood and synovial membrane, forming immune complexes (antigen-antibody complexes) (see Chapter 7).
RA has a higher incidence in women, with evidence of hormonal involvement because disease symptoms lessen during pregnancy and exacerbate in the postpartal period. Evidence for endocrine involvement in RA tissues and cells includes (1) presence of androgen and estrogen receptors, (2) high concentrations of biologically active steroids, (3) key enzymes of steroid metabolism, and (4) significant changes of estrogen to androgen ratio. These data strongly suggest that individual immune cells, including synovial macrophages, may behave as steroid-sensitive cells. Most studies on the influence of exogenous hormones and risk of RA have focused on oral contraceptive pills, with inconsistent findings. Fewer studies have been done with hormone replacement; however, interest has emerged on the role of estrogen and autoimmunity. RA also has seasonal variations, being worse in winter months.
PATHOPHYSIOLOGY The pathogenesis of rheumatoid arthritis is summarized in Figure 42-25.
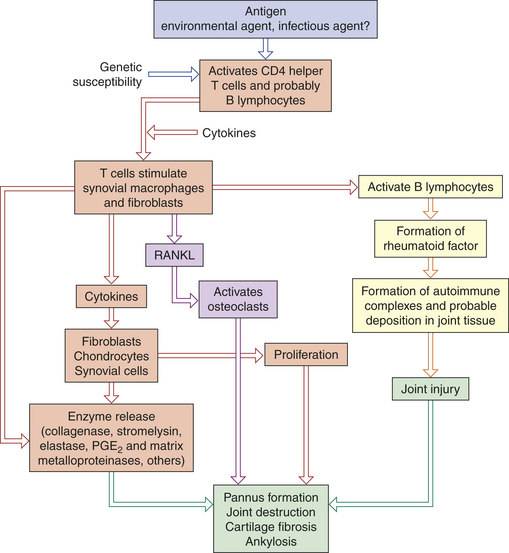
Figure 42-25 Emerging model of pathogenesis of rheumatoid arthritis. Rheumatoid arthritis is an autoimmune disease of a genetically susceptible host triggered by an unknown antigenic agent. Chronic autoimmune reaction with activation of CD4+ helper T cells and possibly other lymphocytes and the local release of inflammatory cytokines and mediators eventually destroys the joint. T cells stimulate cells in the joint to produce cytokines that are key mediators of synovial damage. Apparently immune complex deposition also plays a role. Tumor necrosis factor (TNF) and interleukin-1 (IL-1), as well as some other cytokines, stimulate synovial cells to proliferate and produce other mediators of inflammation, such as prostaglandins (PGE2) matrix metalloproteinases, and enzymes that all contribute to destruction of cartilage. Activated T cells and synovial fibroblasts also produce receptor activator of nuclear factor Kβ ligand (RANKL), which activates the osteoclasts and promotes bone destruction. Pannus is a mass of synovium and synovial stroma with inflammatory cells, granulation tissue, and fibroblasts that grows over the articular surface and causes its destruction.
Cartilage damage in RA is the result of several processes:
1. CD4 T helper cells, and other cells in the synovial fluid become activated, promoting cytokine release and activating B lymphocytes
2. Recruitment and retention of inflammatory cells in the joint sublining region
3. Vicious cycle of altered cytokine and signal transduction pathways
4. Possible immune complex deposition and resultant inflammatory molecule release
5. RANKL release and osteoclast activation
6. Angiogenesis, or growth of new blood vessels in the synovium
Several types of leukocytes are attracted out of the circulation and into the synovial membrane. The phagocytes of inflammation (neutrophils and macrophages) ingest the immune complexes and, in the process of doing so, release powerful enzymes that degrade synovial tissue and articular cartilage (Figure 42-26). The immune system’s B and T lymphocytes are also activated. The B lymphocytes are stimulated to produce more RFs, and the T lymphocytes eventually cause release of enzymes that amplify and perpetuate the inflammatory response.59 Destruction of the extracellular matrix possibly leads to significant disability in individuals with RA.60 Cartilage destruction is mediated by processes from the synovium and cellular invasion into the matrix. These processes may be facilitated by oxidative stress and alterations in deoxyribonucleic acid (DNA) repair mechanisms, causing mutation in key genes.60 In addition RANKL is expressed by various cells in the synovium and induces osteoclast maturation and activation, thus producing increased bone resorption (see p. 1580).
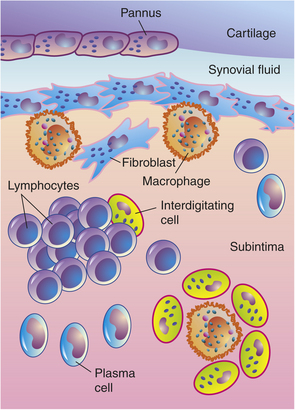
Figure 42-26 Synovitis. Inflamed synovium showing typical arrangements of macrophages and fibroblastic cells.
Inflammatory and immune processes have several damaging effects on the synovial membrane. Along with the swelling caused by leukocyte infiltration, the synovial membrane undergoes hyperplastic thickening as its cells proliferate and enlarge abnormally. As synovial inflammation progresses to involve its blood vessels, small venules become occluded by the hypertrophied endothelial cells, fibrin, platelets, and inflammatory cells, which decrease vascular flow to the synovial tissue. Compromised circulation, coupled with increased metabolic needs because of hypertrophy and hyperplasia, causes hypoxia and metabolic acidosis. Acidosis stimulates the release of hydrolytic enzymes from synovial cells into the surrounding tissue, initiating erosion of the articular cartilage and inflammation in the supporting ligaments and tendons.
Inflammation causes hemorrhage, coagulation, and fibrin deposition on the synovial membrane, in the intracellular matrix, and in the synovial fluid. Over denuded areas of the synovial membrane, fibrin develops into granulation tissue called pannus. Pannus is composed of several different cells; however, the two types primarily responsible for joint damage are osteoclasts and synovial fibroblasts.61 (Granulation tissue is the tissue produced earliest in the process of healing; see Chapter 6.) Synovial fibroblasts also produce additional proinflammatory cells, cytokines, matrix metalloproteinases, and other cells involved in cartilage damage (Figure 42-27, B).
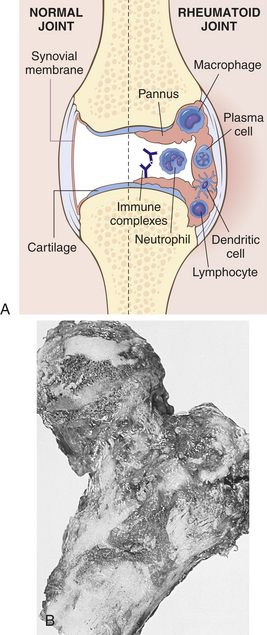
Figure 42-27 Rheumatoid arthritis A, Schematic view of the joint lesion. B, Advanced rheumatoid arthritis involving femur. There is prominent proliferation of synovium and almost complete destruction of overlying articular cartilage. (A modified from Feldmann M: Nat Rev Immunol 2:364, 2002; B from Rosai J: Ackerman’s surgical pathology, ed 8, St Louis, 1996, Mosby.)
CLINICAL MANIFESTATIONS The onset of RA is usually insidious, although as many as 15% of cases have an acute onset. RA begins with general systemic manifestations of inflammation, including fever, fatigue, weakness, anorexia, weight loss, and generalized aching and stiffness. Local manifestations also appear gradually over weeks or months. Typically the joints become painful, tender, and stiff. Pain early in the disease is caused by pressure from swelling; later it is caused by sclerosis of subchondral bone and new bone formation. Stiffness usually lasts for about 1 hour after arising in the morning and is thought to be related to synovitis. Initially most commonly involved are the metacarpophalangeal (MCP) joints, proximal interphalangeal (PIP) joints, and wrists, with later involvement of larger weight-bearing joints.
Joint swelling, which is widespread and symmetric, is caused by increasing amounts of inflammatory exudate (leukocytes, plasma, plasma proteins) in the synovial membrane, hyperplasia of inflamed tissues, and formation of new bone. On palpation, the swollen joint feels warm and the synovial membrane feels “boggy.” The skin over the joint may have a ruddy, cyanotic hue and may look thin and shiny.
An inflamed joint may lose some of its mobility. Even mild synovitis can lead to loss of range of motion, which becomes evident after inflammation subsides. Extension becomes limited and is eventually lost if flexion contractures form. Loss of range of motion can progress to permanent deformities of the fingers, toes, and limbs, including ulnar deviation of the hands, boutonnière and swan-neck deformities of the finger joints, plantar subluxation of the metatarsal heads of the foot, and hallux valgus (angulation of the great toe toward the other toes). Flexion contractures of the knees and hips are also common.
Joint deformities cause the physical limitations experienced by persons with RA. Loss of joint motion is quickly followed by secondary atrophy of the surrounding muscles. With secondary muscle atrophy the joint becomes unstable, which further aggravates joint pathology.
Two complications of chronic RA are caused by an excessive amount of inflammatory exudate in the synovial cavity. One complication is the formation of cysts in the articular cartilage or subchondral bone. Occasionally these cysts communicate with the skin surface (usually the sole of the foot) and can drain through passages called fistulae. The second complication is rupture of a cyst or of the synovial joint itself, usually caused by strenuous physical activity that places excessive pressure on the joint. Rupture releases inflammatory exudate into adjacent tissues, thereby spreading inflammation.
Extrasynovial rheumatoid nodules, or swellings, are observed in areas of pressure or trauma in 20% of individuals with RA. Each nodule is an aggregate of inflammatory cells surrounding a central core of fibrinoid and cellular debris. T lymphocytes are the predominant leukocytes in the nodule; B lymphocytes, plasma cells, and phagocytes are found around the periphery. Nodules are found most often in subcutaneous tissue over the extensor surfaces of elbows and fingers. Less common sites are the scalp, back, feet, hands, buttocks, and knees.
Rheumatoid nodules also may invade the skin, cardiac valves, pericardium, pleura, lung parenchyma, and spleen. These nodules are identical to those encountered in some individuals with rheumatic fever and are characterized by central tissue necrosis surrounded by proliferating connective tissue. Also noted are large numbers of lymphocytes and occasional plasma cells. Acute glaucoma may result, with nodules forming on the sclera. Pulmonary involvement may result in diffuse pleuritis or multiple intraparenchymal nodules. Together, the occurrence of pulmonary nodules and pneumoconiosis (chronic inflammation of the lungs from inhalation of dust) creates Caplan syndrome. Diffuse pulmonary fibrosis may occur because of immunologically mediated immune complex deposition.
Rheumatoid nodules within the heart may cause valvular deformities, particularly of the aortic valve leaflets. Pericardial effusion or other pericardial problems occur in almost 50% of RA patients. Lymphadenopathy of the nodes close to the affected joints may develop. Rheumatoid nodules within the spleen result in splenomegaly. Involvement of blood vessels results in an acute necrotizing vasculitis, characteristic of that noted in other immunologic/inflammatory states. Thromboses of such involved vessels may give rise to myocardial infarctions, cerebrovascular occlusions, mesenteric infarction, kidney damage, and vascular insufficiency in the hands and fingers (Raynaud phenomenon). Vascular changes are noted primarily in individuals receiving steroid therapy; thus there is some concern that the therapy may play a role in initiating these lesions. Changes in skeletal muscle are often noted in the form of nonspecific atrophy secondary to joint dysfunction.
EVALUATION AND TREATMENT Evaluation of RA is by physical examination, roentgenography of the joint, and serologic tests for RF and circulating immune complexes. The ACR lists the following diagnostic criteria for RA:
1. Morning stiffness for longer than 1 hour
2. Arthritis of three or more joint areas
5. Rheumatoid nodules over extensor surfaces or bony prominences
The presence of four or more of the criteria is diagnostic of RA. Criteria 1 through 4 with joint signs or symptoms must be present for 6 weeks.
Treatment is conservative or surgical. Conservative treatment includes rest of the inflamed joint and whole-body rest for several hours daily, use of hot and cold packs, physical therapy, antineoplastic medications, a diet high in calories and vitamins, corticosteroids, anti-inflammatory drugs, immunosuppressants, and disease-modifying antirheumatic drugs (DMARDs) taken orally or by injection. Biologic agents that target cytokines have demonstrated significant improvement in outcomes for both adult and juvenile RA.62 Surgical synovectomy may be done early in the disease to decrease inflammatory effusion and remove pannus. Surgery is used to correct deformity or mechanical deficiency in intermediate or late stages of the disease and includes arthrodesis, arthroplasty, or total joint replacement. Interestingly, total fasting induces a substantial reduction in joint pain, swelling, morning stiffness, and other symptoms in individuals with RA.
Ankylosing Spondylitis
Ankylosing spondylitis (AS) (spondyloarthritis) is a chronic, inflammatory joint disease characterized by stiffening and fusion (ankylosis) of the spine and sacroiliac joints. Although the etiology of AS is unknown, it is associated with HLA-B27. Although inflammation is the primary pathologic process in both RA and AS, the two diseases possibly differ in the primary site of inflammation and the end result. In RA the primary site of inflammation is the synovial membrane, resulting in the destruction and instability of synovial joints. In AS, the primary pathologic site has classically been proposed as the enthesis (the point at which ligaments, tendons, and the joint capsule are inserted into bone) and the end results are fibrosis, ossification, and fusion of the joint, primarily the sacroiliac joints and the vertebral column.63 Recent data from MRI studies, however, show that synovitis and bone marrow inflammation, rather than enthesis, explain the alteration of AS in the sacroiliac joints.64,65
The prevalence of AS in the United States is approximately 0.5% to 1% among whites, 3% to 4% among blacks, and 18% to 50% in various nations of American Indians. Worldwide, the disease appears to be most prevalent in whites. The prevalence of AS in males is at least 10 times greater than previously considered. It affects men three times as often as women. In women, AS may affect the peripheral joints of the appendicular skeleton rather than the axial skeleton, progress less rapidly, and cause less dramatic spinal changes. Many individuals with AS remain undiagnosed.
Primary AS usually develops in late adolescence or young adulthood, with peak incidence at about 20 years of age. Secondary AS affects older age groups and is often associated with other inflammatory diseases (e.g., psoriatic arthropathy, inflammatory bowel disease, Reiter syndrome).
The cause of AS is unknown, but the disease is strongly associated with the presence of histocompatibility antigen HLA-B27 on the chromosomes of affected individuals, suggesting a genetic predisposition to the disease. Not all HLA-B27 subtypes, however, are associated with AS. Klebsiella, chlamydia-delivered peptides, or other “triggers” may perpetuate the inflammatory response.66
PATHOPHYSIOLOGY AS has a strong association with HLA-B27. Several hypotheses have been proposed to explain this association including the arthritogenic peptide theory that proposes that certain B-27 alleles bind certain arthritogenic peptides because of their specific anchoring proteins. Cartilage antigens are proposed as the targets for the immune response and the presentation of such antigens to CD8+ T cells. In the early phases of AS, T cells and macrophages invade and cause erosion of the cartilage at different sites. Based on these observations, it has recently been proposed that the cartilage is the primary target for the immune response.67 Aggrecan, a proteoglycan, forms a major part of the extracellular matrix of cartilage and helps maintain its stability. A specific CD4+ T cell response to proteins derived from aggrecan has been found in animals and humans. Although these T cells have been found in AS, their role as a causative agent in AS remains unclear and necessitates future study.
AS involves inflammation of fibrocartilage in cartilaginous joints, primarily the vertebrae. The fibrous tissue of the joint capsule, the cartilage that surrounds intervertebral disks, the entheses, and periosteum are infiltrated by inflammatory cells. As inflammatory cells (chiefly macrophages) and lymphocytes infiltrate and erode bone and fibrocartilage in joint structures, repair begins. Repair of cartilaginous structures begins with the proliferation of fibroblasts. Fibroblasts synthesize and secrete collagen. The collagen becomes organized into fibrous scar tissue that eventually undergoes calcification and ossification. With time, all the cartilaginous structures of the joint are replaced by ossified scar tissue, causing the joint to fuse, or lose flexibility.
Repair of eroded bone begins with osteoblast activation and proliferation. Osteoblasts lay down new bone (callus), which is remodeled and replaced by compact, lamellar bone. Bone repair changes the contour of the bone’s surface because the new bone grows outward to form a new enthesis with the end of the eroded ligament. The new enthesis, which forms on top of the old one, is called a syndesmophyte. As calcification of the spinal ligaments progresses, the vertebral bodies lose their concave anterior contour and appear square. On radiographs the spine assumes the classic “bamboo spine” appearance of AS.
CLINICAL MANIFESTATIONS The most common signs and symptoms of early AS are low back pain and stiffness. Typically the individual with primary disease develops low back pain during the early 20s. The pain is at first insidious but progressively becomes persistent. It is often worse after prolonged rest and is alleviated by physical activity. Early morning stiffness usually accompanies the low back pain, and the individual typically has difficulty sitting up or twisting the spine. Forward flexion, rotation, and lateral flexion of the spine are restricted and painful. Early pain and resultant loss of motion are caused by the underlying inflammation and reflex muscle spasm rather than by soft tissue or bony fusion.
As the disease progresses, the normal convex curve of the lower spine (lumbar lordosis) diminishes and concavity of the upper spine (kyphosis) increases. The individual becomes increasingly stooped. The thoracic spine becomes rounded, the head and neck are held forward on the shoulders, and the hips are flexed (Figure 42-28).
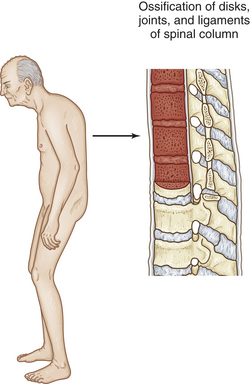
Figure 42-28 Ankylosing spondylitis. Characteristic posture and primary pathologic sites of inflammation and resulting damage.
Inflammation in the tendon insertions of the many costosternal and costovertebral muscles can cause pleuritic chest pain and restricted chest movement. The pain is usually worse on inspiration. Movement in the diaphragm is normal and full. Pressure on the anterior chest wall over the sternum, ribs, and costal cartilages may cause tenderness. Tenderness over the pelvic brim may cause discomfort at night and interfere with sleep because turning onto the iliac crests causes pain. Tenderness over the ischial tuberosities may make sitting on hard seats unbearable. Tenderness in the heels may contribute to a limp or the cautious placement of the feet during walking.
Along with low back pain, many individuals have peripheral joint involvement, uveitis, fibrotic changes in the lungs, cardiomegaly, aortic incompetence, amyloidosis, and Achilles tendinitis. Symptoms may include fatigue, weight loss, low-grade fever, hypochromic anemia, and an increased erythrocyte sedimentation rate.68
EVALUATION AND TREATMENT Diagnosis of AS is made from the history and physical examination, roentgenograms, MRI, and serum analysis for the presence of the histocompatibility antigen HLA-B27. Erythrocyte sedimentation rate is elevated throughout the disease (normal is 0 to 9 mm/hr in males, 0 to 2 mm/hr in females). Alkaline phosphatase levels often are elevated. Treatment of individuals with AS consists of physical therapy to maintain skeletal mobility and prevent the natural progression of contractures. Prevention of deformity and maintenance of mobility require a continuous program of physical therapy. Exercises are performed several times each day to maintain chest expansion, full extension of the spine, and complete range of motion in the proximal joints. The long-term morbidity of AS has been previously underestimated.
Nonsteroidal anti-inflammatory drugs (NSAIDs) often provide relief of symptoms within 48 hours. Analgesic medications are prescribed to suppress some of the pain and stiffness and to facilitate exercise. The medications do not prevent disease progression, but they do provide relief from symptoms (see What’s New? Cox II Inhibitors and Side Effects). DMARDs such as gold, methotrexate, and sulfasalazine have little or no effect in AS. Three TNF inhibitors (etanercept, infliximab, and adalimumab) have been approved for severe AS.
Antibiotics may improve treatment outcomes in AS but results are conflicting.69,70 Surgical procedures, such as osteotomy, total hip replacement, cervical spinal fusion, and radiation therapy are sometimes used to provide relief for individuals with end-stage disease or intolerable deformity. Persons should stop smoking to lessen pulmonary problems.
Gout
Gout is a syndrome caused by an inflammatory response to uric acid production or excretion resulting in high levels of uric acid in the blood (hyperuricemia) and in other body fluids, including synovial fluid. Although hyperuricemia is essential for the development of gout, it is not the only factor. Other factors include age (rare before 30 years), genetic predisposition (X-linked alteration of enzyme hypoxanthine-guanine phosphoribosyltransferase [HGPRT]), excessive alcohol consumption, obesity, certain drugs (especially thiazides), and lead toxicity. When the uric acid reaches a certain concentration in fluids, it crystallizes, forming insoluble precipitates that are deposited in connective tissues throughout the body. Crystallization in synovial fluid causes acute, painful inflammation of the joint, a condition known as gouty arthritis. With time, crystal deposition in subcutaneous tissues causes the formation of small, white nodules, or tophi, that are visible through the skin. Crystal aggregates deposited in the kidneys can form urate renal stones and lead to renal failure.
In classic gouty arthritis, monosodium urate crystals form and cause joint inflammation. Pseudogout is caused by the formation of calcium pyrophosphate dihydrate (CPPD) crystals. The effect of either crystal is the same—the onset of a cytokine-mediated acute inflammatory response (see Chapter 6).
Gout is rare in children and premenopausal women and is uncommon in males younger than 30 years. The peak age of onset in males is between 40 and 50 years, whereas it is somewhat later in females. The risk of developing gouty arthritis is similar in males and females for a particular urate concentration. The plasma urate concentration is an important determinant of the risk of developing gout (Table 42-8).
Table 42-8
Mean Urate Concentrations by Age and Gender
| Characteristic | Mean Urate Levels |
| Prepuberty | 3.5 mg/dl |
| Males (at puberty) | Steep rise to 5.2 mg/dl |
| Females (puberty to premenopause) | Slow rise to ≅4 mg/dl |
| Females (after menopause) | 4.7 mg/dl |
| Hyperuricemia | |
| Men | 7 mg/dl |
| Women | 6 mg/dl |
Uric acid is a byproduct of protein metabolism that normally assists with removal of nitrogen waste from the body.71 When ionized, uric acid can form salts with various cations, but 98% of extracellular uric acid is in the form of monosodium urate (uric acid salt). At any time the proportion of uric acid or urate is pH dependent, so the ratio of these two forms varies considerably in urine (Figure 42-29).
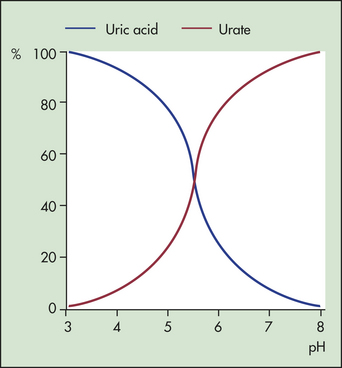
Figure 42-29 Effect of pH on uric acid and urate equilibrium. At pH 5.7, equal amounts of uric acid and urate are present in the solution. (Redrawn from Klippel JH, Dieppe PA, editors: Rheumatology, ed 2, London, 1998, Mosby-Wolfe.)
The solubility of urate and uric acid is critical to the development of crystals. Urate is more soluble in plasma, synovial fluid, and urine than in aqueous solutions. The solubility of uric acid in urine rises dramatically as the pH increases to more than 4. There is little change, however, in the solubility of urate within the normal pH range that exists in the plasma, synovial fluid, and other tissues. Decreasing temperatures cause both urate and uric acid solubility to fall. The pathways of production of uric acid are shown in Figure 42-30.
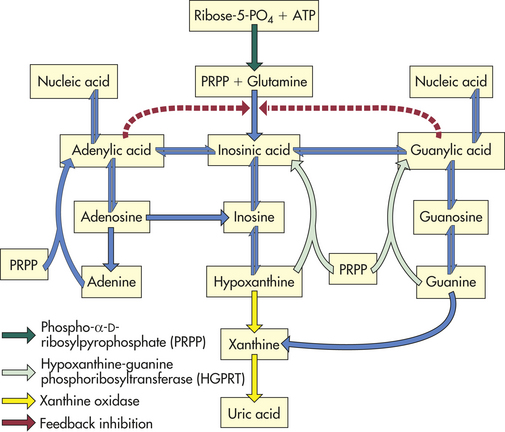
Figure 42-30 Production of uric acid. The major pathways involved in purine nucleotide synthesis. (Redrawn from Klippel JH, Dieppe PA, editors: Rheumatology, ed 2, London, 1998, Mosby-Wolfe.)
PATHOPHYSIOLOGY The pathophysiology of gout is closely linked to purine metabolism (or cellular metabolism of purines) and kidney function. At the cellular level, purines are synthesized to purine nucleotides, which are used in the synthesis of nucleic acids, adenosine triphosphate, cyclic adenosine monophosphate (cAMP), and cyclic guanosine monophosphate (GMP). Uric acid is a breakdown product of purine nucleotides (uric acid synthesis and elimination are illustrated in Figure 42-31). Some individuals with gout have an accelerated rate of purine synthesis accompanied by an overproduction of uric acid. Other individuals break down purine nucleotides at an accelerated rate that also results in an overproduction of uric acid. Production of uric acid can be the result of an increased turnover of nucleic acids, which is associated with an increased turnover of cells at other body sites. The increased turnover of nucleic acids leads to increased levels of uric acid with a compensatory increase in purine synthesis. A deficiency of the enzyme HGPRT (see earlier) can lead to an increased production of uric acid. A complete absence of HGPRT is uncommon but can occur in the X-linked Lesch-Nyhan syndrome, with males at risk for hyperuricemia, neurologic alterations, and sometimes gouty arthritis.72 The majority of individuals with gout, however, have an unknown metabolic defect, which is referred to as primary gout. When the etiology is known, it is referred to as secondary gout.
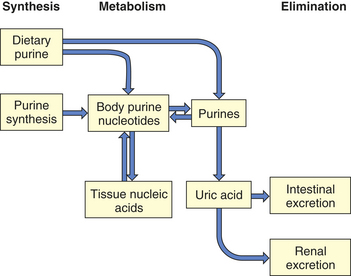
Figure 42-31 Uric acid synthesis and elimination. Uric acid is derived from ingested purines or synthesized from ingested foods, as well as being recycled following cell breakdown. Uric acid is then eliminated through the kidneys and gastrointestinal tract. (Redrawn from Klippel JH, Dieppe PA, editors: Rheumatology, ed 2, London, 1998, Mosby-Wolfe.)
Most uric acid is eliminated from the body through the kidneys. Urate is filtered at the glomerulus and undergoes reabsorption and excretion within the renal tubules. In primary gout, urate excretion by the kidneys is sluggish. The sluggish excretion may be the result of a decrease in glomerular filtration of urate or an acceleration in urate reabsorption. In addition, monosodium urate crystals are deposited in renal interstitial tissues, causing impaired urine flow. (Kidney function is described in Chapter 35.)
The exact process by which crystals of monosodium urate are deposited in joints and induce gouty arthritis is unknown. However, several mechanisms may be involved, including the following:
1. Monosodium urate precipitates at the periphery of the body, where lower body temperatures may reduce the solubility of monosodium urate
2. Decreased albumin or glycosaminoglycan levels, which cause decreased urate solubility
3. Changes in ion concentration and decreases of pH that enhance urate deposition
The monosodium urate crystals may form in the synovial fluid or in the synovial membrane, cartilage, or other connective tissues in joints and elsewhere, such as in the heart, earlobes, and kidneys. Evidence suggests that an acute attack of gout is the result of the formation of crystals rather than the releasing of the crystals from connective tissues into the synovial fluid.
Monosodium urate crystals can stimulate and perpetuate the inflammatory response (Figure 42-32). The presence of the crystals triggers the acute inflammatory response. Initiation of the complement system activates cytokines and produces other substances, called chemoattractants, that draw neutrophils out of the circulation to begin phagocytizing (ingesting) the crystals.
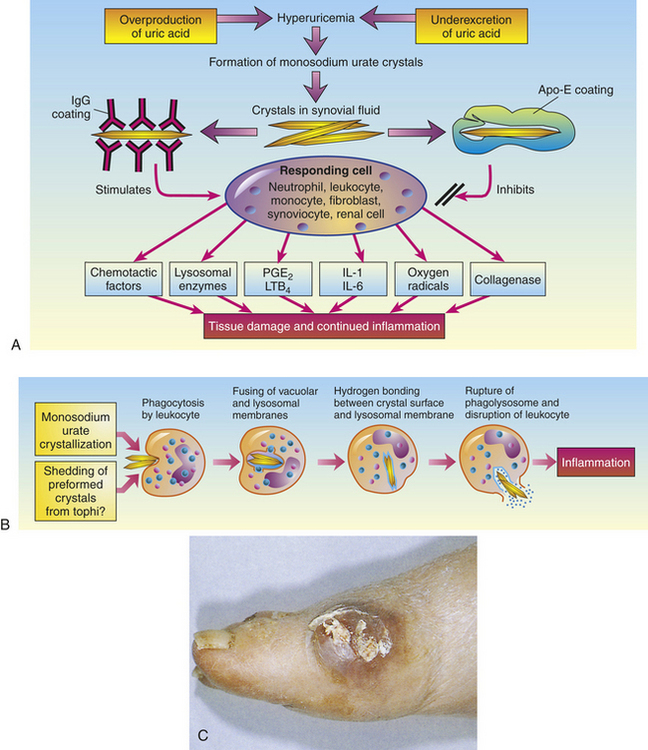
Figure 42-32 Pathogenesis of acute gouty arthritis A, Depending on the urate crystal coating, a variety of cells may be stimulated to produce a wide range of inflammatory mediators. B, Sequence of events in the production of inflammation response to urate crystals. C, Gouty tophus on right foot. Apo-E, Apolipoprotein E; IgG, immunoglobulin G; IL, interleukin; LTB4, leukotriene B4; PGE, prostaglandin E. (C from Dieppe P et al: Arthritis and rheumatism in practice, London, 1991, Gower.)
Crystals coated with IgG are thought to react with crystallizable fragment (Fc) receptors on the surface of the responding cell (see Figure 42-32), thereby promoting phagocytosis with the formation of a phagolysosome. When the phagolysosomal enzymes strip the IgG from the surface of the crystal, the hydrogen bands on the surface of the crystal can induce membrane breakdown of the phagolysosome and cause rupture of the cell within. Recent evidence indicates that apolipoprotein-E coating of urate crystals will inhibit phagocytosis and the cellular response (see Figure 42-32, A).
A variety of inflammatory mediators are released during the crystal/cell response, including chemotactic factors, lysosomal enzymes, eicosanoids, prostaglandin E (PGE2), IL-1 and IL-6, reactive oxygen species, and collagenase (see Figure 42-32, B). Some of these mediators stimulate the influx of neutrophils, monocytes, and lymphocytes. (Acute inflammation and phagocytosis are described in Chapter 6.)
Within the joint fluid, urate crystals react particularly with neutrophils and monocytes. Tissue damage begins to occur, principally when the neutrophils release the contents of their phagolysosomes. These contents also perpetuate inflammation. At an early phase of an acute gouty attack, synovial microtophi have been demonstrated. As the process continues, numerous microtophi may be present on the synovial membrane (see Figure 42-32, C).
CLINICAL MANIFESTATIONS Gout is manifested by (1) an increase in serum urate concentration (hyperuricemia), (2) recurrent attacks of monoarticular arthritis (inflammation of a single joint), (3) deposits of monosodium urate monohydrate (tophi) in and around the joints, (4) renal disease involving glomerular, tubular, and interstitial tissues and blood vessels, and (5) the formation of renal stones. These manifestations appear in three clinical stages:
1. Asymptomatic hyperuricemia: the serum urate level is elevated but arthritic symptoms, tophi, and renal stones are not present; may persist throughout life.
2. Acute gouty arthritis: attacks develop with increased serum urate concentrations; tends to occur with sudden or sustained increases of hyperuricemia but also can be triggered by trauma, drugs, and alcohol.
3. Tophaceous gout: the third and chronic stage of disease; can begin as early as 3 years or as late as 40 years after the initial attack of gouty arthritis. Progressive inability to excrete uric acid expands the urate pool until urate crystal deposits (tophi) appear in cartilage, synovial membranes, tendons, and soft tissue.
Trauma is the most common aggravating factor. The great toe is subject to chronic strain in walking, and subsequently an acute gout attack may follow long walks. Trauma associated with occupations, such as truck driving, also may precipitate an attack.
Attacks of gouty arthritis occur abruptly, usually in a peripheral joint. The primary symptom is severe pain. Approximately 50% of the initial attacks occur in the metatarsophalangeal joint of the great toe. The other 50% involve the heel, ankle, instep of the foot, knee, wrist, or elbow. The pain is usually noticed at night. Within a few hours the affected joint becomes hot, red, and extremely tender and may be slightly swollen. Lymphangitis and systemic signs of inflammation (leukocytosis, fever, elevated sedimentation rate) occasionally are present. Untreated, mild attacks usually subside in several hours but may persist for 1 or 2 days. Severe attacks may persist for several days or weeks. After recovery, the symptoms resolve completely. Intervals between acute attacks of gouty arthritis are called intercritical periods. Some individuals never have a second attack; others experience subsequent attacks within days to as long as 5 to 10 years after the first.
The helix of the ear is the most common site of tophi, which are the characteristic diagnostic lesions of chronic gout. Each tophus consists of a deposit of urate crystals, surrounded by a granuloma made up of mononuclear phagocytes (macrophages) that have developed into epithelial and giant cells. (Granuloma formation is described in and illustrated in Chapter 6.)
Tophaceous deposits produce irregular swellings of the fingers, hands, knees, and feet. Tophi commonly form lumps along the ulnar surface of the forearm, the tibial surface of the leg, the Achilles tendon, and the olecranon bursa. Tophi may produce marked limitation of joint movement and eventually cause grotesque deformities of the hands and feet. Although the tophi themselves are painless, they often cause progressive stiffness and persistent aching of the affected joint. Tophi in the upper extremities may cause nerve compressions such as carpal tunnel syndrome. Tophi in the lower extremities may cause tarsal tunnel syndrome. They also may erode and drain through the skin.
Renal stones are 1000 times more prevalent in individuals with primary gout than in the general population. The stones can be the size of a grain of sand or a piece of gravel, or they can accumulate in massive deposits called staghorn calculi. They range from pale yellow to brown to reddish black, depending on their composition. Some stones consist of pure monosodium urate; others are calcium oxalate or calcium phosphate. Renal stones can form in the collecting tubules, pelvis, or ureters, causing obstruction, dilation, and atrophy of the more proximal tubules and leading eventually to acute renal failure. Stones deposited directly in renal interstitial tissue initiate an inflammatory reaction that leads to chronic renal disease and progressive renal failure.
TREATMENT The aims of gout treatment are to terminate the acute gouty attack as promptly as possible, prevent recurring attacks, prevent or reverse complications associated with urate deposits in the joints and kidneys, and prevent formation of kidney stones. Acute gouty arthritis is treated with antiinflammatory drugs. The drugs of choice are NSAIDs and xanthine oxidase inhibitors, such as allopurinol and febuxostat. Colchicine is used in individuals unable to tolerate NSAIDs. Hydrocortisone may be injected into the joint to relieve pain. Drugs that block IL-1 have shown promise.73 Ice also may relieve some of the inflammation of the joint. Weightbearing on the involved joint is avoided until the acute attack subsides. The individual is put on a low-purine diet, with high fluid intake to increase urinary output. Antihyperuricemic drugs are given to reduce serum urate concentrations.
DISORDERS OF SKELETAL MUSCLE
The common symptoms of disorders of skeletal muscle are muscle weakness and fatigue. In many cases, neural, traumatic, and psychogenic causes provide an adequate explanation for the failure to generate force (weakness) or sustain force (fatigue) seen in myopathies. The pathophysiologic mechanisms in some of the metabolic and inflammatory muscle diseases have been explored, but the cause of many of the myopathies remains obscure. The complex interaction between muscles and nerves affects muscular function as well. Only inherited and acquired disorders of skeletal muscles are discussed here.
Secondary Muscular Dysfunction
Muscular symptoms arise from a variety of causes unrelated to the muscle itself. Secondary muscular phenomena (contracture, stress-related muscle tension, immobility) are common disorders that influence muscular function.
Contractures
Contractures can be pathologic or physiologic. A physiologic muscle contracture occurs in the absence of a muscle action potential in the sarcolemma. Muscle shortening is explained on the basis of failure of the calcium pump in the presence of ATP. A physiologic contracture is seen in McArdle disease (muscle myophosphorylase deficiency) and malignant hyperthermia. The contracture is usually temporary if the underlying pathology is reversed.
A pathologic contracture is a permanent muscle shortening caused by muscle spasm or weakness. Heel cord (Achilles tendon) contractures are examples of pathologic contractures. They are associated with plentiful ATP and occur in spite of a normal action potential. The most common form of contracture is seen in such conditions as muscular dystrophy (see Chapter 43) and central nervous system (CNS) injury. Contractures also may develop secondary to scar tissue contraction in the flexor tissues of a joint, for example, contracture of burned tissues in the antecubital area of the forearm leading to a flexion contracture.
Stress-Induced Muscle Tension
Abnormally increased muscle tension has been associated with chronic anxiety, as well as a variety of stress-related muscular symptoms, including neck stiffness, back pain, and headache.74,75 Abnormalities in the CNS, reticular activating system, and autonomic nervous system (ANS) have been implicated. For example, as an individual progressively relaxes, the amplitude of the knee-jerk reflex diminishes. Conversely, individuals with absent reflexes increase tension by such maneuvers as teeth clenching or hand grip. The underlying pathophysiology may be related to the fact that as a muscle contracts, the muscle spindle is activated. This gamma-feedback system produces a series of impulses that are transmitted to the brain by the sensitive 1A afferent fibers. Unconscious tension is thought to increase the activity of the reticular activating system as well. This influences increasing firing of the efferent loop of the gamma fibers and produces further muscle contraction and increases muscle tension. ANS function that regulates increased blood flow to the muscle during sympathetic activity may be related to increased muscle contraction tension.
Various forms of treatment have been used to reduce the muscle tension associated with stress. Progressive relaxation training, yoga, meditation, and biofeedback are examples of stress reduction therapies. Biofeedback uses an integrated electromyogram (EMG) to make recordings from the skin surface. The goal is to teach the individual to control tension that has been functioning maladaptively. It is particularly useful in individuals who have a connection between skeletal muscle tension and pain.
Progressive relaxation training emphasizes the individual’s ability to perceive the difference between tension and relaxation. This technique involves sequential tensing and a relaxing environment. The individual is taught to practice this routine daily, often with the use of audiotaped instructions. By teaching the individual to recognize excessive contraction of skeletal muscle, one hopes to enhance the ability to relax specific muscle groups to relieve tension and thus reduce CNS as well as ANS arousal.
Fibromyalgia
Fibromyalgia is a chronic musculoskeletal syndrome characterized by widespread joint and muscle pain, fatigue, and tender points. Increased sensitivity to touch (i.e., tender points), the absence of systemic or localized inflammation, and fatigue and sleep disturbances are common. Because the symptoms are vague and the etiology unknown, fibromyalgia has been primarily diagnosed by exclusion. More recently, however, fibromyalgia has become a diagnosis based on specific criteria. A common misdiagnosis has been chronic fatigue syndrome but there is overlap of symptoms between these two conditions.76 From 80% to 90% of individuals affected are women, and the peak age is 30 to 50 years. Although the incidence is unknown, the prevalence is reported to be 2% to almost 6% and increases with age. The ACR classification criteria include diffuse soft tissue pain of at least 3 months’ duration and pain on palpation of at least 11 of 18 tender points (Figure 42-33). It is more common than RA, but its cause is still unknown.
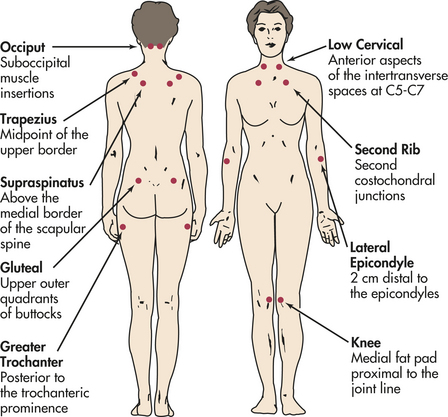
Figure 42-33 Location of specific tender points for diagnostic classification of fibromyalgia. (Redrawn from Freundlich B, Leventhal L: The fibromyalgia syndrome. In Schumacher HR Jr, Klippel JH, Koopman WJ, editors: Primer on the rheumatic diseases, ed 11, Atlanta, 1997, Arthritis Foundation. Copyright 1997. Reprinted with permission of the Arthritis Foundation.)
The etiology of fibromyalgia has been debated for more than a century. It is unlikely that it is caused by a single factor. The most common precipitating factors include the following:
Certain rheumatic diseases, such as RA or systemic lupus erythematosus, may coexist if not initially manifest with fibromyalgia. In addition, fibromyalgia may overlap with myofascial pain syndromes77 (Table 42-9).
Table 42-9
Comparison of Fibromyalgia and Myofascial Pain Syndromes
| Variable | Fibromyalgia | Myofascial Pain |
| Location | Generalized | Regional |
| Examination | Tender points | Trigger points |
| Response to local therapy | Not sustained | Curative |
| Gender | Female/male ratio: 10:1 | Equal or unknown |
| Systemic features | Characteristic | Unknown |
PATHOPHYSIOLOGY Fibromyalgia as a chronic pain syndrome is defined by subjective symptoms and not unique pathophysiologic characteristics. Individuals with fibromyalgia have lowered mechanical and thermal pain thresholds, high pain ratings for provoking stimuli, and altered temporal summation of pain stimuli.77,78 These data provide some evidence of altered pain processing. Aggregation of fibromyalgia within families and other coexisting conditions such as irritable bowel syndrome, chronic fatigue, and mood disorders suggest a major role for neuroendocrine and stress-response alterations (see Chapter 10). Altered circadian activity of several neuroendocrine axes and ANS dysfunction have been reported.79 Corticotropin-releasing hormone (CRH) locus ceruleus–norepinephrine (LC/NE), their peripheral effectors as well as the hypothalamic-pituitary-adrenal (HPA) axis are the main components of the stress system. Impaired functioning of the HPA axis and LC/NE system may be associated with fibromyalgia.
CLINICAL MANIFESTATIONS The prominent symptom of fibromyalgia is diffuse, chronic pain. The locations of nine pairs of tender points for diagnostic classification of fibromyalgia are shown in Figure 42-33. The pain often begins in one location, especially the neck and shoulders, but then becomes more generalized. People describe the pain as burning or gnawing. Fatigue is profound. The effect on everyday life is considerable.80 Some investigators have found that the majority of women experienced pain and fatigue for more than 90% of their time awake.81 Fatigue is most notable when arising from sleep and during the midafternoon. Headaches, symptoms of irritable bowel syndrome, and excess sensitivity to cold (Raynaud-like) are reported in 50% of individuals.
Almost 25% of individuals seek psychologic support for depression. Anxiety, particularly in regard to their diagnosis and future, is almost universal. Again, the only reliable finding on examination is the presence of multiple tender points.
EVALUATION AND TREATMENT Because the manifestations of chronic, generalized pain and fatigue are present in many musculoskeletal (e.g., rheumatic) disorders, these disorders should be considered in the diagnosis of fibromyalgia (Tables 42-10 and 42-11).
Table 42-10
Differential Diagnosis of Fibromyalgia
| Differential Diagnosis | Helpful Differential Features |
| Rheumatoid arthritis∗ | Synovitis, serologic tests, elevated erythrocyte sedimentation rate (ESR) |
| Systemic lupus | Dermatitis, serositis (renal, central erythematosus∗ nervous system, etc.) |
| Polymyalgia rheumatica∗ | Elevated ESR, older adults, response to corticosteroids |
| Myositis | Increased muscle enzymes, weakness more than pain |
| Hypothyroidism∗ | Abnormal thyroid function tests |
| Neuropathies | Clinical and electrophysiologic evidence of neuropathy |
∗Fibromyalgia may also more commonly coexist with these conditions.
Data from Klippel JH, Dieppe PA, editors: Rheumatology, ed 2, London, 1998, Mosby-Wolfe.
Table 42-11
Concomitant Conditions with Fibromyalgia
| Concomitant Condition | Relationship to Fibromyalgia |
| Depression | Present in 25%-60% of fibromyalgia cases |
| Irritable bowel | Present in 50%-80% of fibromyalgia cases |
| Migraine | Present in 50% of fibromyalgia cases |
| Chronic fatigue syndrome (CFS) | 70% of CFS cases meet criteria for fibromyalgia |
| Myofascial pain | May be a localized form of fibromyalgia |
Data from Klippel JH, Dieppe PA, editors: Rheumatology, ed 2, London, 1998, Mosby-Wolfe.
No one regimen of medication has proved successful for fibromyalgia. Medications that improve sleep may be helpful as well as Vitamin D supplementation. Anti-inflammatories have been used despite the fact there is no evidence of tissue inflammation, but these medications have not been effective. Certain CNS-active medications, most notably pregabalin, were significantly better than placebo in controlled trials.82 Treatment consists of a combination of patient education, medication, exercise, and cognitive therapy. Box 42-3 illustrates some of these modalities.
Chronic Fatigue Syndrome
Chronic fatigue syndrome (CFS) is a debilitating and complex disorder characterized by profound fatigue lasting six months or longer that is not improved by bed rest and may worsen with physical and mental activity. CFS is thought to be the result of alterations of multiple ecologically and biologically interrelated homeostatic mechanisms.83 CFS may affect more than one million people in the U.S., but fewer than 20% of sufferers have been diagnosed.84 Women are affected four times as often as men and rarely in children and is most common in the 40s and 50s.
PATHOPHYSIOLOGY There are numerous hypotheses regarding the cause of CFS and research has implicated a number of contributory factors. To date, no specific etiologic factor has been identified. The Centers for Disease Control (CDC) recently instituted a public health research program for a 5-year strategic plan to explore neurologic, psychiatric, and biologic connections to CFS.
Functional MRI (fMRI) has shown lower blood perfusion to the brain stem in CFS individuals.85 Other researchers have shown apparent central nervous system abnormalities, immunologic dysregulation, and higher-than-normal pro-inflammatory cytokines.86, 87 Certain points in skeletal muscle may be affected by oxidative stress reactions (see Chapter 2), accounting for the muscle pain and fatigue associated with CFS.88
CLINICAL MANIFESTATIONS Unrestful sleep is a hallmark of CFS. Defining symptoms include debilitating fatigue made worse by physical or mental exercise (postexertional fatigue), muscle pain, noninflammatory joint pain, headaches, flu-like symptoms, and memory or concentration problems. Other common symptoms include bloating, morning joint stiffness, chest or jaw pain, chills and night sweats, visual disturbances, sore throat and tender axillary or cervical lymph nodes. In order to diagnose CFS, these symptoms need to be present for at least 6 months. Symptoms and their consequences can be severe.
EVALUATION AND TREATMENT Diagnosis of CFS is often delayed or missed because there is no biologic marker or specific laboratory test for CFS, many CFS symptoms are shared with other illnesses, individuals with CFS do not necessarily look sick, and symptoms typically have a variable course. The CDC recommends considering a diagnosis of CFS if the following two criteria are met89:
1. Unexplained, persistent fatigue that is not due to to ongoing exertion, is not substantially relieved by rest, is of new onset (not lifelong) and results in a significant reduction of previous levels of activity.
2. Four or more of the following symptoms are present for six months or more:
Treatment of CFS is primarily based on the person’s individual needs and involves consideration of psychosocial factors as well as symptomatic and supportive care. Acknowledging the validity of the person’s symptoms is important. Collaborative decision-making between patient and healthcare provider with regard to treatment, professional counseling, medication, diet and activity helps CFS sufferers deal with the limitations imposed by the disease. Alternative therapies such as acupuncture, massage, and therapeutic touch can relieve patient anxiety.
Disuse Atrophy
The term disuse atrophy describes the pathologic reduction in normal size of muscle fibers after prolonged inactivity from bed rest, trauma (casting), or local nerve damage. The effects of muscular deconditioning associated with lack of physical activity may be apparent in a matter of days. Oxidative stress from lack of muscle activity causes decreased protein synthesis and increased proteolysis, leading to muscle atrophy.90 The normal individual on bed rest loses muscle strength from baseline levels at a rate of 3% per day. Bed rest also is associated with cardiovascular, skeletal, and other organ system changes.
Certain genes and transcription factors play a role in muscle atrophy. Measures to prevent atrophy include frequent forceful isometric muscle contractions and passive lengthening exercises. If reuse is not restored within 1 year, regeneration of muscle fibers becomes impaired.
Muscle Membrane Abnormalities
Two defects of the muscle membrane (plasma membrane of the muscle fiber) have been linked to clinical syndromes: the hyperexcitable membrane seen in the myotonic disorders and the intermittently unresponsive membrane seen in periodic paralyses. Although these are infrequent disorders, research into the pathologic processes has led to an improved understanding of the cell membrane.
Myotonia
Myotonia is a delayed relaxation after such voluntary muscle contractions as grip, eye closure, or muscle percussion. The distinctive “dive-bomber” noise, audible on needle EMG, is caused by the prolonged depolarization of the muscle membrane. Because the depolarization is not terminated by neuromuscular-blocking agents, such as curare, the abnormality has been localized at the muscle membrane; the basic defect is due to ion channel dysfunction. (These structures are described in Chapter 1.)
Myotonia can be reproduced by removing extracellular chloride, thus reducing chloride conductance across the plasma membrane. The delicate balance in which sodium diffuses into the intracellular fluid, potassium diffuses out of the intracellular fluid, and chloride is in flux is thus interrupted. Because the normal diffusion processes (described in Chapter 3) stabilize the membrane, the shift in chloride ions is thought to increase membrane excitability. The chloride abnormality may explain the resting membrane hyperexcitability, but it does not explain the delayed relaxation present in myotonia and has not been detected in human myotonia.
Myotonia is seen in several disorders: myotonia congenita, paramyotonia congenita, myotonic muscular dystrophy, and some forms of periodic paralysis. Most are inherited disorders and are mild in symptomatology, with the exception of myotonic muscular dystrophy (see p. 1633). Myotonia is treated by drugs that reduce muscle fiber excitability, such as procaine, procainamide, phenytoin, and quinine preparations. Recent treatments include acetazolamide and dichlorphenamide; both are carbonic anhydrase inhibitors.
Periodic Paralysis
During an attack of periodic paralysis the muscle membrane is unresponsive to neural stimuli and the resting membrane potential is reduced from −90 to −45 mV. Periodic paralysis can be either hyper- or hypokalemic. The disorder is often inherited in an autosomal dominant pattern, although it can be seen in hyperthyroidism.
The paralysis, which leaves the individual flaccid and weak, does not affect the respiratory muscles. Many individuals exhibit myotonia on examination. In most cases the weakness is accompanied by a change in serum potassium, although in some individuals the change may be negligible. Cardiac dysrhythmias have been present during attacks. Although the biochemical defect remains unknown, changes in the muscle membrane and sarcoplasmic reticulum have been described.
Hypokalemic periodic paralysis is triggered by high-carbohydrate meals, prolonged bed rest, or emotional stress. (The effect of potassium on the resting membrane potential is discussed in Chapter 3.) Glucose and insulin infusions and oral potassium loading are used as provocative tests; oral and intravenous potassium can relieve acute attacks. Treatment includes potassium-sparing diuretics and a high-salt diet. Acetazolamide, dichlorphenamide, and a low-salt diet are useful for long-term therapy. Hyperkalemic periodic paralysis is caused by a genetic mutation. Attacks are usually less severe than with the hypokalemic form. Treatment includes small carbohydrate-rich meals, light exercise, and intravenous calcium gluconate.
Metabolic Muscle Diseases
Disorders in muscle metabolism can be caused by endocrine abnormalities or diseases of energy metabolism, such as glycogen storage disease, enzyme deficiencies, and abnormalities in lipid metabolism and mitochondrial function.
Endocrine Disorders
Often the systemic effects of hormonal imbalance overshadow the individual’s muscular symptoms. For example, individuals with thyrotoxicosis may have signs of proximal weakness, paresis of the extraocular muscles (exophthalmic ophthalmoplegia), and rarely, hypokalemic periodic paralysis. Hypothyroidism is often associated with a decrease in muscle mass and strength, with weak, flabby skeletal muscles and sluggish movements.
Thyroid hormone is believed to regulate muscle protein synthesis and electrolyte balance. Changes in muscle protein synthesis and electrolyte balance may therefore explain the changes in muscle mass and contractility seen in endocrine disorders. The muscle symptoms subside with appropriate treatment of the primary hormonal disorder.
Diseases of Energy Metabolism
Muscle relies on carbohydrates, such as glycogen and lipids (free fatty acids), for energy. When stored glycogen or lipids cannot be used because of a lack of the enzyme necessary to convert energy for contraction, the individual experiences cramps, fatigue, and exercise intolerance. Disorders of muscle metabolism can be self-limiting, such as is seen in McArdle disease and some lipid disorders, or cause widespread irreparable muscle destruction, as in acid maltase deficiency.
McArdle Disease: McArdle disease, or glycogen myophosphorylase deficiency, was the first myopathy in which a single enzyme defect was identified (Figure 42-34). Individuals with McArdle disease lack muscle phosphorylase, which is responsible for the breakdown of glycogen in muscle. Normally after the body uses the short-term ATP and phosphocreatine stores, intramuscular lactic acid accumulates as glycogen is used (see Chapter 41). The individual with McArdle disease is not able to break down glycogen or produce lactic acid.
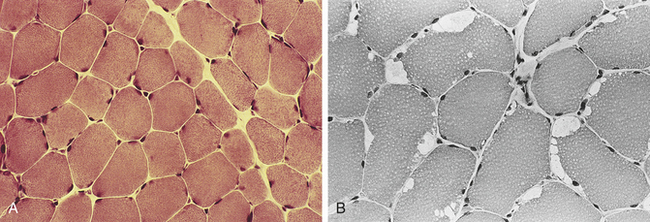
Figure 42-34 McArdle disease. A, Normal muscle fibers. B, Muscle fibers of McArdle disease. Note the enlarged (white) peripheral vacuoles. (From Damjanov I, Linder J, editors: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
The altered energy production manifests itself in exercise intolerance, fatigue, and painful muscle cramps. When exercise is carried to an extreme, painful muscle contracture and myoglobinuria develop. Some individuals describe a “second wind” phenomenon, in which exercise tolerance increases if they slow their pace once the initial sensation of fatigue commences. This may be caused by the use of free fatty acids as a secondary source of energy. As the disease progresses, some individuals have pronounced muscle weakness and wasting. Other organs are not involved because the absence of phosphorylase is limited to muscle. Generally, individuals with McArdle disease learn to adapt their daily routine to avoid muscle symptoms. Usually the diagnosis of McArdle disease is made by the histochemical evaluation of myophosphorylase activity in frozen sections. There is no staining of myofibrils in affected individuals.
Acid Maltase Deficiency: Acid maltase deficiency is an uncommon glycogen storage disease associated with an accumulation of glycogen in the lysosomes of muscle cells and the cells of other tissues. The usual pathways of glycogen degradation are preserved. The absence of the enzyme acid maltase is responsible for the abnormality in glycogen metabolism, although the exact mechanism is unknown. It is an autosomal recessive disorder, with the gene located on the long arm of chromosome 17.
The infantile form, Pompe disease, is an autosomal recessive disease that causes lysosomal glycogen accumulation. The adult form tends to be less dramatic than the infantile type. The infantile form is recognized shortly after birth by hypotonia, dysreflexia, and an enlarged heart, tongue, and liver. Hypertrophy of these tissues is thought to be the result of glycogen deposition. Children die of cardiac or respiratory failure within 1 year of diagnosis. Recombinant human acid alpha-glucosidase administration has been shown to decrease mortality.91,92 The adult variety becomes evident subacutely. The muscular symptoms resemble those of muscular dystrophy or polymyositis. A distinguishing feature in adults may be severe respiratory muscle weakness.
Myoadenylate Deaminase Deficiency: An enzyme deficiency that produces changes in skeletal muscle and is associated with exercise intolerance is myoadenylate deaminase deficiency (MDD). Because these individuals lack myoadenylate deaminase, they have a poor capacity for sustained energy production. Myoadenylate deaminase is the catalytic enzyme that forms phosphocreatine and ATP during exercise through a metabolic pathway that binds the purine and phosphate molecules that constitute ATP. Persons with MDD differ from those with McArdle disease in that during the ischemic exercise test, lactate production is normal when ATP and phosphocreatine are synthesized. The enzyme defect has been reported to be common, but in practice it may rarely be recognized as a cause of exercise intolerance.
Lipid Deficiencies: Disorders of lipid metabolism are uncommon but account for severe changes in muscle metabolism. The lipid content of muscle cells consists of the free fatty acids, which are oxidized in the mitochondria. These acids require carnitine and the enzyme carnitine palmityl transferase (CPT) to transport metabolic byproducts and energy to the myofibrils. Individuals with CPT deficiency have mild muscular symptoms but can experience bouts of renal failure caused by myoglobinuria. Individuals with a deficiency of carnitine alone have progressive muscle weakness and can experience sudden exacerbations.
Measuring the CPT and carnitine content in muscle aids in the diagnosis. Cells in the muscle biopsy show vacuoles and lipid deposits. Treatments with riboflavin, medium-chain triglycerides, oral carnitine, and prednisone have been suggested.
Inflammatory Muscle Diseases: Myositis
Viral, Bacterial, and Parasitic Myositis
Viral, bacterial, and parasitic infections of varying severity are known to produce inflammatory changes in skeletal muscle, a group of conditions collectively described by the term myositis. In tuberculosis and sarcoidosis, chronic inflammatory changes and granulomas are found in muscle, as well as in other affected tissues. In trichinellosis, Trichinella larvae reside in infected pork and, after ingestion, migrate to the intestinal mucosa and from there to the lymphatics. Symptoms include severe pain, rash, and muscle stiffness. Treatment includes administration of corticosteroids and antiparasitic agents, such as mebendazole or albendazole. Unfortunately, once trichinella larvae are established they may reside for years in the muscles. Toxoplasmosis, a common parasitic infection, is also associated with a generalized polymyositis that responds rapidly to therapy.
In the tropics, more prevalent disorders include bacterial infections with S. aureus and parasites such as cysticercus, the larva of the tapeworm Taenia solium. Viral infections can be associated with an acute myositis. Muscle pain, tenderness, signs of inflammation, and CK elevation are common manifestations of viral myositis. The self-limiting symptoms of muscle aches and pains during a bout of influenza may actually be a subacute form of viral myopathy.
Polymyositis and Dermatomyositis
Polymyositis (generalized muscle inflammation) and dermatomyositis (polymyositis accompanied by skin lesions) are the most common inflammatory muscle diseases requiring long-term care. Prevalence rates may be about 6 per 1 million persons.
PATHOPHYSIOLOGY Polymyositis and dermatomyositis are characterized by inflammation of connective tissue and muscle fibers that presumably causes the extensive necrosis and destruction of muscle fibers. The agent that causes the muscle inflammation has not been identified, but recent findings strongly suggest an autoimmune connection93, 94(Figure 42-35). Innate and adaptive immune responses are activated in these myopathies. There appears to be a genetic component in symptom manifestation.95 This family of diseases is now designated as autoimmune because of the presence of autoantibodies in the serum of many individuals. Studies have shown that the inflammatory cells that surround the perimysial and perivascular sites are selectively enriched in B cells and helper T cells in those with dermatomyositis.96 There is less vascular involvement in polymyositis, and most of the inflammatory cells, including B cells, T cells, and macrophages, surround the muscle fibers and fascicles.
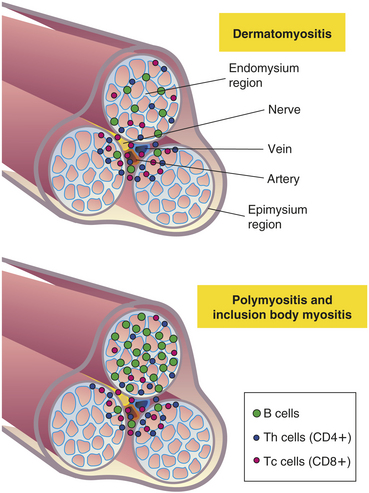
Figure 42-35 Distribution of CD4+ and CD8+ lymphocytes in different clinical forms of myositis. Dermatomyositis shows perivascular and CD4+ T cells (helper T-cells). Polymyositis shows mostly CD8+ T cells (cytotoxic T-cells).
CLINICAL MANIFESTATIONS The acute symptoms include many of those seen in any inflammatory process: malaise, fever, muscle swelling, pain and tenderness, lethargy, and listlessness. In adults, weakness of the shoulder and pelvic girdle muscles is a primary manifestation of polymyositis. Both illnesses are usually associated with a symmetric proximal muscle weakness and initially can be confused with other myopathies. A thorough evaluation is required to exclude other disorders. Clinical features common in both polymyositis and dermatomyositis are dysphagia, reduced esophageal motility, vasculitis, Raynaud phenomenon, cardiomyopathy, and interstitial pulmonary fibrosis. Some patients have other coexisting collagen vascular disorders, such as RA, systemic lupus erythematosus (SLE), and progressive systemic sclerosis (formerly called scleroderma).
The presence of skin rash, calcinosis, and eyelid edema most often suggests dermatomyositis (Figure 42-36). The skin rash is purple (heliotrope) and involves the eyelids, face, chest, and extensor surfaces of the extremities. Dermatomyositis is slightly more common in children and older adults, with an onset before age 15 or after age 50. The adult with dermatomyositis occasionally has underlying malignancies. Calcinosis, with calcium deposition in the subcutaneous tissue, can be a severe long-term complication of dermatomyositis.
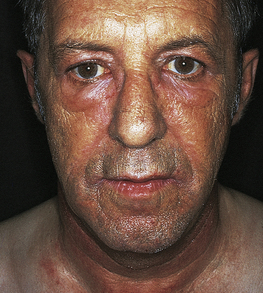
Figure 42-36 Dermatomyositis. Heliotrope (violaceous) discoloration around the eyes and periorbital edema. (From Habif TP: Clinical dermatology, ed 3, St Louis, 1996, Mosby.)
EVALUATION AND TREATMENT The muscle biopsy is striking in dermatomyositis, with most individuals showing inflammatory cells grouped around blood vessels and atrophy of cells in muscle fascicles. This change, perifascicular atrophy, is absent in polymyositis. Creatine kinase (CK) and sedimentation rate are often extremely elevated in both disorders. EMG abnormalities include signs of muscle irritability and myopathic changes—usually large numbers of low-amplitude action potentials of brief duration. The EMG also shows a typical “myopathic” pattern, with short, low-amplitude polyphasic potentials, as well as signs of marked muscle irritability. Muscle biopsy is indispensable for determining a diagnosis of polymyositis or dermatomyositis as opposed to other myotonic diseases. MRI reveals inflammation and edema of the muscles.
Treatment primarily includes immunosuppressive drugs, although they are not always successful if uniformly applied. Most clinicians choose corticosteroids initially, usually prednisone on a daily or alternating-day schedule, tapering the dosage as the symptoms subside. Successful treatment with azathioprine, methotrexate, and cyclophosphamide also has been reported. Creatine supplements and physical therapy may improve muscle strength.97
Myopathy
Myopathy is the term applied to a primary muscle disorder. Many pathologic processes affect muscles and cause loss of functional muscle cells. Myopathies affect muscle strength, tone, and bulk. Primary muscle disease is invariably associated with weakness—usually marked weakness. The distribution of the weakness in myopathy is usually symmetric and proximal, although occasionally the weakness is predominantly distal, such as in myotonic dystrophy. The weakness is associated with mild fatigue. Tone is decreased, as are the tendon reflexes. Atrophy may be present. Some myopathies are associated with muscle hypertrophy as in cretinism and the familial progressive muscular dystrophies of childhood, in which hypertrophied muscles are rubbery and weak. Fasciculations are not present with myopathy because no denervation is present. No sensory changes are found. (Specific neurologic-associated myopathies are discussed in Chapter 16.)
Toxic Myopathies
A number of agents, including corticosteroids, chloroquine, alcohol, phenytoin, azathioprine, organophosphates, and reverse transcriptase inhibitors, have been shown to cause toxic myopathy.98 The incidence of acute alcoholic myopathy has been estimated at up to 20% of individuals admitted with acute alcoholic withdrawal.
The pathologic abnormalities include necrosis of individual muscle fibers, particularly type II fibers; whole segments can be found in the same stage of degeneration. The mechanisms by which alcohol affects the muscle include disturbances of energy cell turnover, gene dysregulation, and initiation of apoptosis.99
Acute alcoholic myopathy can range from benign cramps and pain resolving in a matter of hours to severe weakness and markedly increased CK associated with myoglobinuria and renal failure. Individuals are prone to repeated attacks following recovery. The only treatment is abstinence from alcohol and improved nutrition. The individual with chronic alcoholic myopathy often has a coexisting peripheral neuropathy that complicates the diagnosis.
Rhabdomyolysis and myoglobinuria are often caused by sedatives and narcotics, particularly street heroin, clofibrate (a hypolipidemic agent), and the antifibrinolytic aminocaproic acid. Drugs that induce hypokalemia, such as amphotericin B, licorice, and azathioprine, also have been reported to cause myalgia and myopathy.
Repeated intramuscular injections have been associated also with changes in muscle fibers. Local necrosis of muscle fiber and elevated CK have been reported after intramuscular injections of certain cephalosporins, lidocaine, diazepam, and digoxin; these effects were not produced with injections of saline. When drugs are injected over long periods, a chronic focal myopathy develops. Proliferation of connective tissue in both the muscle fiber and overlying skin and subcutaneous tissue has been reported. Over time, segments of the muscles, particularly the deltoid and quadriceps, are converted into fibrotic bands. Pathophysiologic mechanisms for these changes include repeated needle trauma and infection, along with the nonphysiologic acidity and alkalinity of the injected material.
Muscle Tumors
Rhabdomyoma is an extremely rare benign tumor of muscle that generally occurs in the tongue, neck muscles, larynx, uvula, nasal cavity, axilla, vulva, and heart. These tumors are usually treated by surgical excision and do not recur.
Rhabdomyosarcoma
The malignant tumor of striated muscle is called rhabdomyosarcoma. These tumors are highly malignant, with rapid metastasis. They are located in the muscle tissue of the head, neck, and genitourinary tract in 75% of cases. The remainder are in the trunk and extremities.
Three types of rhabdomyosarcoma are differentiated on pathologic section: pleomorphic, embryonal, and alveolar. The pleomorphic, or spindle cell, type is considered to be one of the most highly malignant tumors of the extremities seen in adulthood. Embryonal tumors are most commonly seen in childhood and appear on biopsy to be shaped like a tadpole or tennis racquet. Alveolar-type tumors appear lattice-like and look like lung tissue alveoli.
The diagnosis of rhabdomyosarcoma is made by history, physical examination, serologic testing, CT, and MRI, and is confirmed by incisional biopsy and examination of the specimen by a pathologist. On electron microscopy the tissue demonstrates myofilaments and Z-band material; CT scan helps define the tissue borders. Staging is based on pathologic grade of the tumor and is helpful in determining prognosis and treatment.
Treatment consists of a combination of surgical excision, radiation therapy, and systemic chemotherapy. Overall survival of childhood rhabdomyosarcoma has improved over the past decades but adult survival remains poor (see Chapter 43).
Other Tumors
Metastatic deposits of tumors in muscles are rare in spite of the extensive vascular supply of skeletal muscles. It is suggested that local pH or metabolic changes prevent metastatic involvement from other tumors. When adjacent carcinomas do cause muscle damage, it is usually related to the compression of tissue and resultant muscle atrophy.
Acid maltase deficiency 1610
Acute gouty arthritis 1605
Age-related bone loss 1582
Aggrecan 1600
Ankylosing spondylitis (AS) (spondyloarthritis) 1600
Arthropathy 1592
Asymptomatic hyperuricemia 1605
Avulsion 1573
Biofeedback 1606
Bowing fracture 1569
Bursae 1574
Bursitis 1574
Caplan syndrome 1600
Chondrogenic (cartilage-forming) tumor 1591
Chondroid 1591
Chondrosarcoma 1591
Chronic fatigue syndrome 1608
Closed (simple) fracture 1569
Collagenic (collagen-forming) tumor 1591
Comminuted fracture 1569
Compartment syndrome 1575
Complete fracture 1569
Contracture 1606
Delayed union 1571
Dermatomyositis 1611
Dislocation 1572
Disuse atrophy 1609
Enchondroma 1591
Endogenous (hematogenous) osteomyelitis 1587
Enthesis 1600
Epicondylitis 1574
Exogenous osteomyelitis 1587
Extracellular signal regulated kinases (ERKS) 1582
Fatigue fracture 1569
Fibromyalgia 1606
Fibrosarcoma 1591
Fracture 1568
Fragility fracture 1569
Giant cell tumor 1592
Glucocorticoid (e.g., cortisone)-induced osteoporosis 1582
Gout 1602
Gouty arthritis 1602
Greenstick fracture 1569
Hyperbaric oxygen therapy 1588
Hyperkalemic periodic paralysis 1609
Hypokalemic periodic paralysis 1609
Incomplete fracture 1569
Inflammatory joint disease (arthritis) 1596
Insufficiency fracture 1569
Involucrum 1588
Joint effusion 1595
Joint stiffness 1595
Lateral epicondylopathy (tennis elbow) 1574
Ligament 1573
Linear fracture 1569
Malunion 1571
McArdle disease 1610
Medial epicondylopathy (golfer’s elbow) 1574
Muscle strain 1675
Myelogenic tumor 1592
Myoadenylate deaminase deficiency (MDD) 1611
Myoglobin 1575
Myoglobinuria 1575
Myopathy 1612
Myositis 1611
Myositis ossificans 1575
Myotonia 1609
Noninflammatory joint disease 1592
Nonunion 1571
Oblique fracture 1569
Open (compound) fracture 1569
Osteoarthritis (OA) 1593
Osteogenic (bone-forming) tumor 1590
Osteomalacia 1584
Osteomyelitis 1587
Osteopenia 1577
Osteophyte 1593
Osteoporosis 1576
Osteoprotegerin (OPG) 1580
Osteosarcoma 1590
Paget disease (osteitis deformans) 1585
Pannus 1598
Pathologic fracture 1569
Peak bone mass 1577
Periodic paralysis 1609
Polymyositis 1611
Pompe disease 1610
Postmenopausal osteoporosis 1579
Primary gout 1603
Progressive relaxation training 1606
Pseudogout 1602
RANK 1580
Receptor activator of nuclear factor Kβ ligand (RANKL) 1580
Regional osteoporosis 1580
Rhabdomyolysis 1675
Rhabdomyosarcoma 1613
Rheumatoid arthritis (RA) 1596
Rheumatoid factor (RF) 1597
Rheumatoid nodule 1599
Secondary gout 1603
Secondary osteoporosis 1580
Sequestrum 1587
Spiral fracture 1569
Sprain 1573
Strain 1573
Stress fracture 1569
Subluxation 1572
Syndesmophyte 1601
Tendinitis 1574
Tendinosis 1574
Tendon 1573
Tophaceous gout 1605
Tophi (sing., tophus) 1602
Torus fracture 1569
Toxic myopathy 1612
Transchondral fracture 1569
Transverse fracture 1569
Volkmann ischemic contracture 1575
REFERENCES
1. Dennison, E., Mohamed, M.A., Cooper, C. Epidemiology of osteoporosis. Rheum Dis Clin North Am. 2006;32(4):617.
2. Bielby, R., Jones, E., McGonagle, D. The role of mesenchymal stem cells in maintenance and repair of bone. Injury. 2007;38(Suppl 1):S26.
3. Soma, I.L., Messer, T.M. Complications after treatment of flexor tendon injuries. J Am Acad Orthop Surg. 2006;14:387–396.
4. Jaworski, C. Current understanding of tendinopathies and treatment options. Am Fam Physician. 2007;76(6):773.
5. Nirschl, R.P., Ashman, E.S. Elbow tendinopathy: tennis elbow. Clin Sports Med. 2003;22(4):813–836.
6. Shiri, R., et al. Prevalence and determinants of lateral and medial epicondylitis: a population study. Am J Epidemiol. 2006;164:1065.
7. Sirvanci, M., et al. Myositis ossificans of psoas muscle: magnetic resonance imaging findings. Acta Radiol. 2004;45(5):523–525.
8. Koya, S., Crenshaw, D., Agarwal, A. Rhabdomyolysis and acute renal failure after fire ant bites. J Gen Intern Med. 2007;22(1):145.
9. Shinzato, T., et al. Cowfish (Umisuzume, Lactoria diaphana) poisoning with rhabdomyolysis. Intern Med. 2008;47(9):853.
10. Yasue, H., et al. Severe hypokalemia, rhabdomyolysis, muscle paralysis, and respiratory impairment in a hypertensive patient taking herbal medicine containing licorice. Intern Med. 2007;46(9):575.
11. Kashani, A., et al. Risks associated with statin therapy: a systematic overview of randomized clinical trials. Circulation. 19(114), 2006.
12. Molokhia, M., et al. Statin induced myopathy and myalgia: time trend analysis and comparison of risk associated with statin class from 1991-2006. PLoS ONE. 2008;3(6):E2522.
13. Gourgiotis, S., et al. Acute limb compartment syndrome: a review. J Surg Educ. 2007;64(3):178.
14. E.Czerwiñski, et al. Current understanding of osteoporosis according to the position of the World Health Organization (WHO) and International Osteoporosis Foundation. Orthop Traumatol Rehabil. 2007;9(4):337.
15. Bone health and osteoporosis: a report of the surgeon general 2004. Available online at: www.surgeongeneral.gov/library/bonehealth/.
16. Poole, K.E., Compston, J.E. Osteoporosis and its management. BMJ. 333(7581), 2006.
17. Lewiecki, E.M., Laster, A.J. Clinical review: clinical applications of vertebral fracture assessment by dual-energy x-ray absorptiometry. J Clin Endocrinol Metab. 2006;91(11):4215–4222.
18. Ott S: Osteoporosis. Available online: http://courses.washington.edu/bonephys/oprisk.html.
19. Barrett-Connor, E., et al. Osteoporosis and fracture risk of women of different ethnic groups. J Bone Min Res. 2005;20:185.
20. Hochberg, M.C. Racial differences in bone strength. Trans Am Clin Climatol Assoc. 2007;118:305.
21. Ngyuen, T.V., et al. Bone loss, physical activity, and weight change in elderly women: the Dubbo Osteoporosis Epidemiology Study. J Bone Miner Res. 1998;13(9):1458–1467.
22. Brown, M. Skeletal muscle and bone: effect of sex steroids and aging. Adv Physiol Educ. 2008;32(2):120.
23. Fogle, R.H., et al. Ovarian androgen production in postmenopausal women. J Cin Endocrinol Metab. 2007;92(8):3040.
24. Kin, C.F., et al. Experience of famine and bone health in post-menopausal women. Int J Epidemiol. 2007;36(5):1143.
25. Moskowitz, R.W., Kelly, M.A., Lewallen, D.G. Understanding osteoarthritis of the knee—causes and effects. Am J Orthop. 2004;33(2 Suppl):5–9.
26. Gao, W., Dalton, J.T. Expanding the therapeutic use of androgens via selective androgen receptor modulators (SARMs). Drug Discov Today. 2007;12(5-6):241.
27. Korompilias, A.V., et al. Transient osteoporosis. J Am Acad Orthop Surg. 2008;16(8):480.
28. Niimi, R., et al. Changes on bone mineral density in transient osteoporosis of the hip. J Bone Joint Surg Br. 2006;88(11):1438.
29. Xyda, A., et al. Postpartum bilateral transient osteoporosis of the hip: MR imaging findings in three cases. Radiol Med. 2008;113(5):689.
30. Hofbauer, L.C., Schoppet, M. Clinical implications of the osteoprotegerin/RANKL/RANK System for bone and vascular diseases. JAMA. 2004;292(4):490–495.
31. Shoback, D. Update in osteoporosis and metabolic bone disorders. J Clin Endocrinol Metab. 2007;92(3):747.
32. Khosla, S., Melton, L.J., Riggs, B.L. Clinical review 144: estrogen and the male skeleton. J Clin Endocrinol Metab. 2002;87(4):1443–1450.
33. Plotkin, L.I., et al. Bisphosphonates and estrogens inhibit osteocyte apoptosis via distinct molecular mechanisms downstream of extracellular signal-regulated kinase activation. J Biol Chem. 2005;280(8):7317–7325.
34. Schwartz, E.M., Ritchlin, C.T. Clinical development of anti-RANKL therapy. Arthritis Res Ther. 2007;9(Suppl 1):S7.
35. ŽofkováI. Hormonal aspects of the muscle-bone unit. Physiol Res. 2008;57(Suppl 1):S159.
36. Seeman, E. Structural basis of growth-related gain and age-related loss of bone strength. Rheumtology (Oxford). 2008;47:iv2.
37. Genant, H.K., Engelke, K., Prevrhal, S. Advanced CT bone imaging in osteoporosis. Rheumatology (Oxford). 2008;47(Suppl 4):iv9.
38. Yago, M.D., et al. Intracellular magnesium: transport and regulation in epithelial secretory cells. Front Biosci. 2000;5:D602.
39. Khosla, S., Westendorf, J.J., Oursler, M.J. Building bone to reverse osteoporosis and repair fractures. Clin Invest. 2008;118(2):431.
40. Gallagher, J.C. Advances in bone biology and new treatments for bone loss. Maturitas. 2008;60(1):65.
41. Fukumoto, S. Physiological regulation and disorders of phosphate metabolism—pivotal role of fibroblast growth factor 23. Intern Med. 2008;47(5):337.
42. Emmett, M. What does serum fibroblast growth factor 23 do in hemodialysis patients? Kidney Inter. 2008;73:3.
43. Endo, I., et al. Clinical usefulness of measurement of fibroblast growth factor 23 (FGF 23) in hypophosphatemic patients: proposal of diagnostic criteria using FGF 23 measurement. Bone. 2008;42(6):1235.
44. Lewiecki, E.M., Urig, E.L., Jr., Williams, R.C., Jr. Tumor-induced osteomalacia: lessons learned. Arthritis Rheum. 2008;58(3):773.
45. Layfield, R. The molecular pathogenesis of Paget disease of bone. Expert Rev Mol Med. 2007;9(27):1.
46. Walsh, J.P. Paget’s disease of bone. Med J Aust. 2004;181(5):262.
47. Chen, W.C., et al. Spinal epidural abscess due to Staphylococcus aureus: clinical manifestations and outcomes. J Microbiol Immunol Infect. 2008;41(3):215.
48. Kapoor, A., et al. Magnetic resonance imaging for diagnosing foot osteomyelitis: a meta-analysis. Arch Intern Med. 2007;167(2):125.
49. Marina, N., et al. Biology and therapeutic advances for pediatric osteosarcoma. Oncologist. 2004;9(4):422–441.
50. Kivioja, A.H., et al. Cement is recommended in intralesional surgery of giant cell tumors: a Scandinavian Sarcoma Group study of 294 patients followed for a median time of 5 years. Acta Orthop. 2008;9(2):86.
51. Abramson, S.B. Inflammation in osteoarthritis. J Rheumatol (Suppl). 2004;70:70.
52. Alturfan, A.A., et al. Increased serum sialic acid levels in primary osteoarthritis and inactive rheumatoid arthritis. Tohoku J Exp Med. 2007;213(3):241.
53. Krasnokutsky, S., Samuels, J., Abramson, S.B. Osteoarthritis in 2007. Bull NYU Hosp Jt Dis. 2007;65(3):222.
54. Rutsch, F., Terkeltaub, R. Deficiencies of physiologic calcification inhibitors and low grade inflammation in arterial calcification: lessons for cartilage calcification. Joint Bone Spine. 2005;72(2):110–118.
55. Hashimoto, S., et al. Chondrocyte-derived apoptotic bodies and calcification of articular cartilage. Proc Natl Acad Sci. 1998;95(6):3094–3099.
56. Fan, Z., et al. Activation of interleukin-1 signaling cascades in normal and osteoarthritic articular cartilage. Am J Pathol. 2007;171(3):938.
57. Barron, M.C., Rubin, B.R. Managing osteoarthritic knee pain. J Am Osteopath Assoc. 2007;107(10 Suppl 6):ES21.
58. Otero, M., Goldring, M.B. Cells of the synovium in rheumatoid arthritis. Chondrocytes. Arthritis Res Ther. 2007;9(5):220.
59. Mauri, C., Ehrenstein, M.R. Cells of the synovium in rheumatoid arthritis. B cells. Arthritis Res Ther. 2007;9(2):205.
60. Sweeney, S.E., Firestein, G.S. Rheumatoid arthritis: regulation of synovial inflammation. Int J Biochem Cell Biol. 2004;36(3):372–378.
61. Abeles, A.M., Pillinger, M.H. The role of the synovial fibroblast in rheumatoid arthritis: cartilage destruction and the regulation of matrix metalloproteinases. Bull NYU Hosp Jt Dis. 2006;64(1-2):20.
62. Alonso-Ruiz, A., et al. Tumor necrosis factor alpha drugs in rheumatoid arthritis: systematic review and meta-analysis of efficacy and safety. BMC Musculoskelet Disord. 2008;9:52.
63. Davis, J.C., et al. Definition of disease duration in ankylosing spondylitis: reassessing the concept. Ann Rheum Dis. 2006;65(11):1518.
64. Tuite, M.J. Sacroiliac joint imaging. Semin Musculoskelet Radiol. 2008;12(1):72.
65. Zochling, J., et al. Magnetic resonance imaging in ankylosing spondylitis. Curr Opin Rheumatol. 2007;19(4):346.
66. Ikbal, M., et al. Association of chromatid exchange frequencies in patients with ankylosing spondylitis with and without HLA-B27. Ann Rheum Dis. 2003;62(8):775.
67. Samji, M.F., Bafaquh, M., Tsai, E. The pathogenesis of ankylosing spondylitis. Neurosurg Focus. 2008;24(1):E3.
68. Katana, R.K., Brent, L.J. Spondyloarthropathies. Am Fam Physican. 2004;60(12):3853.
69. Inman, R.D. Mechanisms of disease: infection and spondyloarthritis. Nat Clin Pract Rheumatol. 2006;2(3):163.
70. Ogrendik, M. Treatment of ankylosing spondylitis with moxifloxacin. South Med J. 2007;100(4):366.
71. Pillinger, M.H., Rosenthal, P., Abeles, A.M. Hyperuricemia and gout: new insights into pathogenesis and treatment. Bull NYU Hosp Jt Dis. 2007;65(3):215.
72. Nyhan, W.L. Lesch-Nyhan disease. J Hist Neurosci. 2005;14(1):1–10.
73. So, A., et al. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res Ther. 2007;9(2):R28.
74. Leistad, R.B., et al. Similarities in stress physiology among patients with chronic pain and headache disorders: evidence for a common pathophysiological mechanism? J Headache Pain. 2008;9(3):165.
75. Nilsen, K.B., et al. Autonomic and muscular responses and recovery to one-hour laboratory mental stress in healthy subjects. BMC Musculoskelet Disord. 2007;8:81.
76. Meeus, M., Nijs, J. Central sensitization: a biopsychosocial explanation for chronic widespread pain in patients with fibromyalgia and chronic fatigue syndrome. Clin Rheumatol. 2007;26(4):46.
77. Bennett, R. Myofacial pain syndromes and their evaluation. Best Pract Res Clin Rheumatol. 2007;21(3):427.
78. Dannecker, E.A., Knoll, V., Robinson, M.E. Sex differences in muscle pain: self-care behaviors and effects on daily activities. J Pain. 2008;9(3):200.
79. Martinez-Lavin, M. Biology and therapy of fibromyalgia. Stress, the stress response system, and fibromyalgia. Arthritis Res Ther. 2007;9(4):216.
80. Verbunt, J.A., Pernot, D.H., Smeets, R.J. Disability and quality of life in patients with fibromyalgia. Health Qual Life Outcomes. 2008;6:8.
81. Buckwalter, J.A., Lappin, D.R., The disproportionate impact of chronic arthralgia and arthritis among women. Clin Orthop. 2000(372):159.
82. Mease, P.J., et al. A randomized, double-blind, placebo-controlled, phase III trial of pregabalin in the treatment of patients with fibromyalgia. J Rheumatol. 2008;35(3):502.
83. Centers for Disease Control: www.cdc.gov/cfs/draft_5yr_research_plan.htm. Accessed 6/2/2009.
84. Centers for Disease Control:http://www.cdcgov/cfs/cfsatrisk.htm. Accessed 6/2/2009.
85. Meeus, M., Nijs, J. Central sensitization: a biopsychosocial explanation for chronic widespread pain in patients with fibromyalgia and chronic fatigue syndrome. Clin Rheumatol. 2007;26(4):465.
86. Gur, A., Oktayoglu, P. Central nervous abnormalities in fibromyalgia nad chronic fatigue syndrome: new concepts in treatment. Curr Pharm Des. 2008;14(13):1274.
87. Lorusso, L. Immunological aspects of chronic fatigue syndrome. Autoimmun Rev. 2009;8(4):287.
88. Fulle, S. Specific correlations between muscle oxidative stress and chronic fatigue syndrome: a working hypothesis. J Muscle Res Cell Motil. 2007;28(6):355.
89. Centers for Disease Control: Toolkit: Fact sheets for healthcare professionals: basic CFS overview. www.cdc.gov/cfs/toolkit.htm, Accessed 06/02/09.
90. Powers, S.K., Kavazis, A.N., McClung, J.M. Oxidative stress and disuse muscle atrophy. J Appl Physiol. 2007;102(6):2389.
91. Koeberl, D.D., Kishnani, P.S., Chen, Y.T. Glycogen storage disease type I and II: treatment updates. J Inherit Metab Dis. 2007;320:159.
92. Pereira, S.J., Berditchevisky, C.R., Suely, K.N.M. Report of the first Brazilian infantile Pompe disease patient to be treated with recombinant human acid alpha-glucosidase. J Pediatr (Rio J). 2008;84(3):272.
93. Dorph, C., et al. Signs of inflammation in both symptomatic and asymptomatic muscles from patients with polymyositis and dermatomyositis. Ann Rheum Dis. 2006;65(12):1565.
94. Koenig, M., et al. Heterogeneity of autoantibodies in 100 patients with autoimmune myositis: insights into clinical features and outcomes. Arthritis Res Ther. 2007;9(4):R78.
95. Walsh, R.J., et al. Type 1 interferon-inducible gene expression in blood is present and reflects disease activity in dermatomyositis and polymyositis. Arthritis Rheum. 2007;56(11):3784.
96. Bradshaw, E.M., et al. A local antigen-driven humoral response is present in the inflammatory myopathies. J Immunol. 2007;178(1):547.
97. Chung, Y.L., et al. Creatine supplements in patients with idiopathic inflammatory myopathies who are clinically weak after conventional pharmacologic treatment: six-month, double-blind, randomized, placebo-controlled trial. Arthritis Rheum. 2007;57(4):694.
98. Scola, R.H., et al. Toxic myopathies: muscle biopsy features. Arg Neuropsiquiatr. 2007;65(1):82.
99. Urbano-Marquez, A., Fernandez-Sola, J. Effects of alcohol on skeletal and cardiac muscle. Muscle Nerve. 2004;30(6):689–707.