17 ALTERATIONS OF HAEMATOLOGICAL FUNCTION ACROSS THE LIFE SPAN
INTRODUCTION
Under normal circumstances, changes occur to haematological function in early life; however, beyond childhood, little variation is seen in the healthy individual. Pathological alteration to function occurs either as a consequence of haematological disease or secondary to other processes. The incidence of some disorders affecting haematological function can vary greatly according to age. This chapter examines some of the important alterations to haematological function within the context of the life span.
Alterations to erythrocyte function involve either insufficient or excessive numbers of erythrocytes in the circulation or normal numbers of cells with abnormal components. Anaemia is a condition in which there are too few erythrocytes or an insufficient volume of erythrocytes in the blood. Polycythaemia is a condition in which erythrocyte numbers are excessive. Each condition has many causes and is a pathophysiological manifestation of a variety of disease states.
The primary role of haemostasis is to stop bleeding through the interaction of endothelium lining the vessels, platelets and clotting factors. A large number of disease states may be associated with a clinically significant increase or decrease in clotting, resulting from alterations in any of the three main components of the clotting process.
Alterations in leucocyte numbers may occur in response to infections or to proliferative disorders, such as leukaemia. Many haematological disorders are malignancies and many nonhaematological malignancies can metastasise to the bone marrow, affecting blood cell production.
In adults, extramedullary haematopoiesis — blood cell production in tissues other than bone marrow — is usually a sign of disease, occurring in pernicious anaemia, sickle cell anaemia, thalassaemia, haemolytic disease of the newborn (erythroblastosis fetalis), hereditary spherocytosis and certain leukaemias. Extramedullary haematopoiesis of apparently normal blood cells has been reported in the spleen, liver and, less frequently, lymph nodes, adrenal glands, cartilage, adipose tissue, intrathoracic areas and kidneys.
ALTERATIONS OF ERYTHROCYTE FUNCTION
Anaemia is the main cause of alteration in erythrocyte function. There are several different types of anaemia, which arise from insufficient red blood cells (or haemoglobin content) to meet the body’s needs and result in inadequate delivery of oxygen to tissues (hypoxia). The clinical manifestations of the types of anaemia are similar. Polycythaemia is a condition that arises from excessive levels of erythrocytes; it is much less common that anaemia. Both anaemia and polycythaemia result in altered levels of haematocrit, as seen by the proportion of erythrocytes in the blood (see Figure 17-1 and Table 17-1).
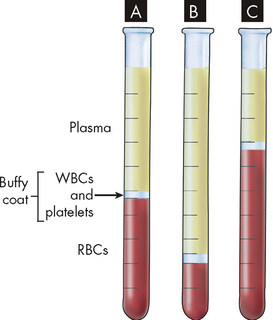
FIGURE 17-1 Haematocrit tubes showing normal blood, anaemia and polycythaemia.
Note the buffy coat located between the packed red blood cells (RBCs) and the plasma. A A normal percentage of red blood cells. B Anaemia (a low percentage of red blood cells). C Polycythaemia (a high percentage of red blood cells).
Source: Patton KT, Thibodeau GA. Anatomy & physiology. 7th edn. St Louis: Mosby; 2010.
Table 17-1 BLOOD TESTS FOR ERYTHROCYTE DISORDERS
| CELL TYPE AND TEST | PROPERTY EVALUATED BY TEST | POSSIBLE HAEMATOLOGICAL CAUSE OF ABNORMAL FINDINGS |
|---|---|---|
| Erythrocyte | ||
| Red cell count | Number (× 1012) of erythrocytes/litre of blood | Altered erythropoiesis, anaemias, haemorrhage, Hodgkin’s disease, leukaemia |
| Mean cell volume (MCV) | Size of erythrocytes | Anaemias, thalassaemias |
| Mean corpuscular haemoglobin (MCH) | Amount of haemoglobin in each erythrocyte (by weight) | Anaemias |
| Haemoglobin determination | Amount of haemoglobin (by weight)/litre of blood | Anaemias |
| Haematocrit determination | Proportion of a given volume of blood that is occupied by erythrocytes (expressed as L/L or %) | Haemorrhage, polycythaemia, erythrocytosis, anaemias, leukaemia |
| Reticulocyte count | Number of reticulocytes (× 109)/litre of blood | Hyperactive or hypoactive bone marrow function |
| Haemoglobin metabolism | ||
| Serum ferritin determination | Depletion of body iron (potential deficiency of haem synthesis) | Iron deficiency anaemia |
| Total iron-building capacity (TIBC) | Amount of iron in serum plus amount of transferrin available in serum | Haemorrhage, iron deficiency anaemia, haemochromatosis, iron overload, anaemias, thalassaemia |
| Transferrin saturation | Percentage of transferrin that is saturated with iron | Acute haemorrhage, haemochromatosis, iron deficiency anaemia, iron overload, thalassaemia |
Source: Based on Lewis SM et al. Dacie and Lewis practical hematology. Philadelphia: Elsevier; 2006; Key N et al. Practical hemostasis. 2nd edn. Chichester: Wiley-Blackwell; 2009.
Anaemia
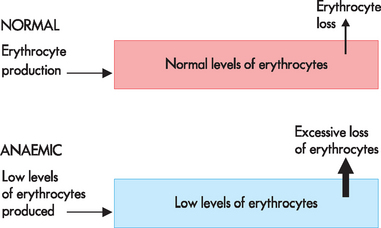
Anaemia is defined as a reduction in the haemoglobin concentration of the blood and this corresponds with a decrease in the total number of circulating erythrocytes. In broad terms, inadequate numbers of erythrocytes may result from either lack of red cell production (which is the more common type) or excessive destruction of red cells. Anaemia may also arise due to loss of blood volume in haemorrhage (see Figure 17-2).
Inherited defects can cause anaemia. With the notable exception of haemolytic disease of the newborn, acquired types of anaemia can occur at any stage of the life span. Iron deficiency, renal disease and chronic inflammation are common causes of anaemia. In the clinical setting, co-morbidities may contribute to the development of anaemia, such that no single cause may be identifiable.
Classification of anaemia
Different types of anaemia are classified by their causes or by the changes that affect the size, shape or substance of the erythrocyte. The most common classification is based on the changes that affect the cell’s size and haemoglobin content. The mean cell volume (MCV) and mean cell haemoglobin (MCH) are the laboratory measurements that are used respectively to determine red cell size and haemoglobin content (which gives the erythrocyte its red appearance). These measurements form part of a full blood count. Terms used to identify types of anaemia reflect these characteristics. Terms that end with cytic refer to cell size and those that end with chromic refer to haemoglobin content. Anaemia may be either microcytic (small), normocytic (normal) or macrocytic (large) in relation to red cell size and either hypochromic (pale) or normochromic in relation to red cell haemoglobin content. The term hyperchromic is not used, as red cells usually contain maximal amounts of haemoglobin.
A blood film examination can further assist in defining the type of anaemia and possible cause. Further laboratory tests (e.g. iron studies) may be required to complete the investigation.
General clinical manifestations of anaemia
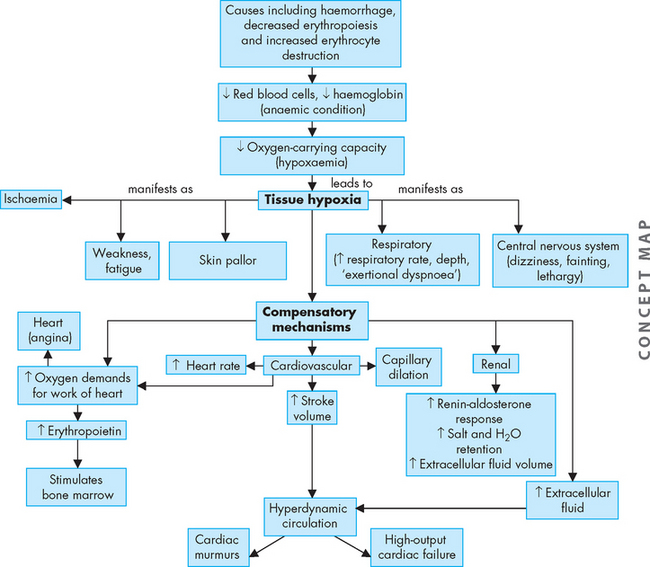
The fundamental alteration of anaemia is a reduced oxygen-carrying capacity of the blood resulting in tissue hypoxia (low oxygen content in the tissues). Symptoms of anaemia vary, depending on the body’s ability to compensate for the reduced oxygen-carrying capacity. The most common symptoms are fatigue, coldness and pallor, although mild cases are asymptomatic. Anaemia that is mild and starts gradually is usually easier to compensate for and may cause problems for the individual only during physical exertion. As red cell reduction continues, symptoms become more pronounced and alterations in specific organs and compensation effects are more apparent. Compensation generally involves the cardiovascular, respiratory and haematological systems (Figure 17-3).
A reduction in the number of blood cells in the blood causes a reduction in the consistency and volume of blood. Initial compensation for cellular loss is movement of interstitial fluid into the blood causing an increase in plasma volume (see Figure 17-4). This movement maintains an adequate blood volume, but the viscosity (thickness) of the blood decreases. The ‘thinner’ blood flows faster and more turbulently than normal blood, causing a hyperdynamic circulatory state. This hyperdynamic state creates cardiovascular changes — increased stroke volume and heart rate. These changes may lead to cardiac dilation and heart valve insufficiency if the underlying anaemic condition is not corrected.
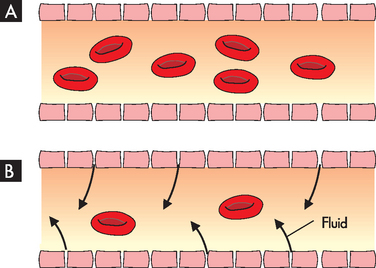
FIGURE 17-4 Fluid shift in anaemia.
A Normal proportion of erythrocytes and plasma in blood. B Fewer erythrocytes in fluid moving into plasma. Although blood volume is maintained, the blood is much less viscous (less thick).
Hypoxaemia, reduced oxygen level in the blood, further contributes to cardiovascular dysfunction by causing dilation of arterioles, capillaries and venules, thus increasing the volume of blood flow through them. Increased peripheral blood flow and venous return further contribute to an increase in heart rate and stroke volume in a continuing effort to meet normal oxygen demand and prevent cardiopulmonary congestion. These compensatory mechanisms may lead to heart failure (refer to Figure 17-3).
Tissue hypoxia creates additional demands and effects on the pulmonary and haematological systems. The rate and depth of breathing increases in an effort to increase oxygen availability and this is also accompanied by an increase in the release of oxygen from haemoglobin. All of these compensatory mechanisms may cause individuals to experience shortness of breath (dyspnoea), a rapid and pounding heartbeat, dizziness and fatigue. In mild chronic cases, these symptoms may be present only when there is an increased demand for oxygen (e.g. during physical exertion), but in severe cases symptoms may be experienced even at rest.
Manifestations of anaemia may be seen in other parts of the body. The skin, mucous membranes, lips, nail beds and conjunctivae become either pale because of reduced haemoglobin concentration or yellowish (jaundiced) because of the accumulation of the end products of red cell destruction (haemolysis) if that is the cause of the anaemia. Tissue hypoxia of the skin results in impaired healing and loss of elasticity, as well as thinning and early greying of the hair. Nervous system manifestations may occur where the cause of anaemia is a deficiency of vitamin B12. Myelin degeneration occurs, causing a loss of nerve fibres in the spinal cord, resulting in paraesthesias (numbness), gait disturbances, extreme weakness, spasticity and reflex abnormalities. Decreased oxygen supply to the gastrointestinal tract often produces abdominal pain, nausea, vomiting and anorexia. Low-grade fever occurs in some anaemic individuals and may result from the release of leucocyte pyrogens from ischaemic tissues (refer to Chapter 13).
When the anaemia is severe or acute in onset (e.g. due to haemorrhage), the initial compensatory mechanism is peripheral blood vessel constriction, diverting blood flow to essential vital organs (the brain, heart and lungs are the highest priority). Decreased blood flow detected by the kidneys activates the renin-angiotensin response, leading to salt and water retention in an attempt to increase blood volume. These situations are considered to be emergencies and require immediate intervention to correct the underlying problem that caused the acute blood loss; therefore, long-term compensatory mechanisms do not develop.
Therapeutic interventions for slowly developing anaemic conditions require treatment of the underlying condition and palliation of associated symptoms.1 Therapies include transfusion, dietary correction and administration of supplemental vitamins or iron.
Anaemia due to insufficient erythrocyte production
There are several different causes that may lead to inability to produce adequate levels of erythrocytes. Many of these causes include lack of availability of the necessary nutrients for the steps involved in red cell production.
Iron deficiency anaemia
Iron deficiency anaemia may result from inadequate iron intake or absorption, increased iron requirements (such as during growth) or excessive iron loss. The red cells are normocytic and normochromic, so otherwise normal but low in numbers. It is the most common type of anaemia throughout the world, occurring in both developing and developed countries.2 Females have a higher incidence than males for iron deficiency anaemia, with the peak incidence occurring in the reproductive years and decreasing at menopause. There are higher demands for iron during pregnancy, and, as fetal iron stores are accumulated in the third trimester (the final stages of pregnancy), premature infants have a greater risk of iron deficiency.
PATHOPHYSIOLOGY
Iron deficiency anaemia is the most common blood disorder of infancy and childhood, with the highest incidence occurring between the ages of 6 months and 2 years. Incidence is not related to gender or race, but socioeconomic factors are important because they affect nutrition. Iron deficiency anaemia is common in children because they need an extremely high amount of iron for normal growth to occur. During adolescence, iron deficiency anaemia is relatively common in menstruating females; menorrhagia (excessive menstrual bleeding) results in considerable iron loss with the menstrual fluid and causes iron deficiency anaemia.
Males and females may experience iron deficiency anaemia due to bleeding as a result of ulcers, hiatus hernia, oesophageal varices, cirrhosis, haemorrhoids, ulcerative colitis, drugs that cause gastrointestinal bleeding or cancer. Although iron is recycled in the body (see Chapter 16), blood loss disrupts this balance by creating a need for more iron, thus depleting the iron stores more rapidly to replace the iron lost from bleeding.
Insufficient dietary intake of iron leads to iron deficiency anaemia. In addition, surgical procedures that decrease the stomach acidity, decreased intestinal transit time and intestinal abnormality (such as coeliac disease) will limit the absorption of iron.
CLINICAL MANIFESTATIONS
The onset of symptoms is gradual and individuals usually do not seek medical attention until haemoglobin levels drop to 70 or 80 g/L. Early symptoms are nonspecific and include fatigue, weakness, shortness of breath and pale ear lobes, palms and conjunctiva (see Figure 17-5).
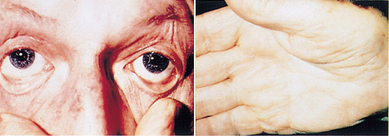
FIGURE 17-5 Pallor and iron deficiency.
Pallor of the skin, mucous membranes and palmar creases in an individual with haemoglobin of 90 g/L. The palmar creases become as pale as the surrounding skin when the haemoglobin level approaches 70 g/L.
Source: Hoffbrand AV, Pettit JE. Sandoz atlas of clinical hematology. London: Gower Medical; 1988.
As the condition progresses and becomes more severe, structural and functional changes occur in epithelial tissue. The fingernails become brittle and ‘spoon-shaped’ or concave (koilonychia) (see Figure 17-6). Tongue papillae atrophy and cause soreness along with redness and burning (see Figure 17-7). These changes can be reversed within 1 to 2 weeks of iron replacement. The corners of the mouth become dry and sore (angular stomatitis) and an individual may experience difficulty with swallowing because of a ‘web’ that develops from mucus and inflammatory cells at the opening of the oesophagus. These lesions have the potential to become cancerous.
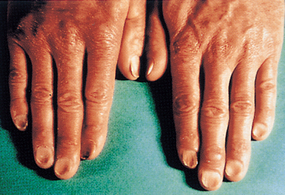
The nails are concave, ridged and brittle.
Source: Hoffbrand AV, Pettit JE. Sandoz atlas of clinical hematology. London: Gower Medical; 1988.
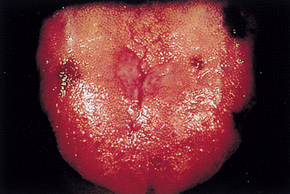
The tongue of an individual with iron deficiency anaemia has a bald, fissured appearance caused by loss of papillae and flattening.
Source: Hoffbrand AV, Pettit JE. Sandoz atlas of clinical hematology. London: Gower Medical; 1988.
Iron is a component of many enzymes in the body and lack of iron may alter other physiological processes and contribute to the clinical manifestations. Individuals with iron deficiency anaemia exhibit gastritis, neuromuscular changes, irritability, headache, numbness, tingling and vasomotor disturbances. Gait disturbances are rare. In the elderly, mental confusion, memory loss and disorientation may be wrongly perceived as normal events associated with ageing.
In children, parents generally do not note any change in the child’s behaviour or appearance until moderate anaemia has developed. General irritability, decreased activity tolerance, weakness and lack of interest in play are nonspecific indications of anaemia. When haemoglobin levels fall below 50 g/L, pallor, anorexia, tachycardia and systolic murmurs may occur. Other symptoms and signs include splenomegaly, widened skull sutures, decreased physical growth and developmental delays, pica (a behaviour in which non-food substances are eaten) and altered neurological and intellectual functions, especially those involving attention span, alertness and learning ability.
EVALUATION AND TREATMENT
Evaluation is based on clinical manifestations and laboratory tests. Iron stores are measured directly by bone marrow biopsy, or indirectly by tests that measure serum ferritin (circulating levels of iron in the blood), transferrin saturation (transferrin transports iron through the blood) or total iron-binding capacity.
The first step in the treatment of iron deficiency anaemia is to find and eliminate potential sources of blood loss. If this is not done, replacement therapy is ineffective. Iron replacement therapy is required and very effective. Initial doses are 150–200 mg/day and are continued until the serum ferritin level reaches 50 mg/L, indicating that adequate replacement has occurred. A rapid decrease in fatigue, lethargy and other associated symptoms is generally seen within the first month of therapy. Replacement therapy usually continues for 6–12 months after the bleeding has stopped but may continue for as long as 24 months. Menstruating females may need daily therapy (325 mg/day) until menopause.
Folate deficiency anaemia
Folate (folic acid) is an essential vitamin required for DNA synthesis within the developing erythrocyte. The erythrocytes are macrocytic (large) because of abnormal cell division, as well as normochromic. Humans are totally dependent on dietary intake to meet the daily requirement of 50–200 mg/day. Folate is absorbed from the upper small intestine and is then stored in the liver. Folate deficiency occurs more often than vitamin B12 deficiency (discussed next), particularly in alcoholics and individuals who are malnourished because of fad diets or diets low in vegetables. Increased amounts are required for lactating and pregnant females. Folate deficiency during pregnancy can result in the birth of infants with neural tube defect. This has led to the promotion of folic acid supplementation in women of childbearing age.3 Also, folate is now fortified in some foods in Australia and New Zealand (details in Chapter 9).
CLINICAL MANIFESTATIONS
Clinical manifestations are similar to the malnourished appearance of individuals with pernicious anaemia, except for the absence of neurological symptoms. Specific manifestations include cheilosis (scales and fissures of the mouth), stomatitis (inflammation of the mouth) and painful ulcerations of the buccal mucosa and tongue. Dysphagia, flatulence and watery diarrhoea may also be present, as well as histological changes in the gastrointestinal tract suggestive of coeliac disease (refer to Chapter 27). Neurological manifestations, if present, may be caused by thiamine deficiency, which often accompanies folate deficiency.
EVALUATION AND TREATMENT
Evaluation of folate deficiency is based on blood tests, measurement of serum folate levels and clinical manifestations. Treatment requires administration of oral folate preparations until adequate blood levels are obtained and manifestations are reduced or eliminated. Long-term therapy is not necessary except for maintaining an adequate daily intake of folate. Folate is essential for reducing blood levels of homocysteine, which has been recently recognised as a risk factor for the development of coronary artery disease.
Pernicious anaemia
Vitamin B12 deficiency can lead to pernicious anaemia and may be caused by inadequate intake, particularly seen with a strict vegan diet (as vitamin B12 is obtained from foods of animal source). Also, impaired absorption of vitamin B12 may result from abnormality in the stomach or intestines. Normal levels of stomach secretions are necessary for the secretion of intrinsic factor; this travels to the intestines, along with the vitamin B12, where intrinsic factor is necessary for vitamin absorption (refer to Figure 17-8). Pernicious anaemia may accompany chronic atrophic gastritis;4 complete or partial removal of the stomach (gastrectomy) also causes intrinsic factor deficiency and results in pernicious anaemia.
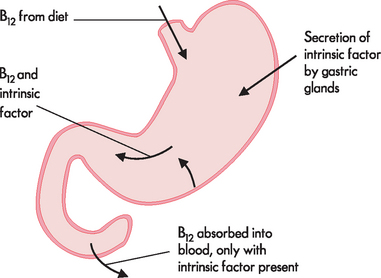
FIGURE 17-8 The absorption of vitamin B12.
Normal levels of intrinsic factor must be secreted in the stomach to facilitate the absorption of vitamin B12 in the small intestine.
Insufficient intrinsic factor affects normal red blood cell development, so that they are macrocytic (large cells), and these defective cells die early, resulting in insufficient numbers of erythrocytes. As for folate deficiency anaemia, they are also normochromic in pernicious anaemia. Pernicious means highly injurious or destructive and reflects the fact that this condition was once fatal. It typically develops in middle age or later (generally after 40 years of age) and is rare in children.
CLINICAL MANIFESTATIONS
Pernicious anaemia develops slowly (over 20 to 30 years), so that by the time an individual seeks treatment, it is usually severe. Early symptoms are often ignored because they are nonspecific and vague — they include infections, mood swings and gastrointestinal, cardiac or kidney ailments. When the haemoglobin has decreased to 70–80 g/L, the individual experiences the classic symptoms of anaemia: weakness, fatigue, paraesthesias of the feet and fingers, difficulty walking, loss of appetite, abdominal pain, weight loss and a sore tongue that is smooth and beefy red. The skin may become ‘lemon yellow’ (sallow), caused by a combination of pallor (being pale) and jaundice (yellow colouration due to build-up of bilirubin). Hepatomegaly, indicating right-sided heart failure, may be present in the elderly along with splenomegaly, which is nonpalpable. Neurological abnormalities may develop.
EVALUATION AND TREATMENT
Evaluation is based on blood tests, bone marrow aspiration, serological studies, gastric biopsy, clinical manifestations and the Schilling test. This test involves administering radioactive vitamin B12 to the patient and then measuring its excretion in the urine. Low urinary excretion is significant for pernicious anaemia. Serological studies reveal the presence of antibodies against gastric cells and gastric biopsy reveals achlorhydria, a total absence of hydrochloric acid.
Untreated pernicious anaemia is fatal, usually because of heart failure. With replacement therapy of vitamin B12, mortality has decreased significantly. Death from this disease is now rare and relapses are often the result of noncompliance with therapy. Initial replacement of vitamin B12 is accomplished by weekly injections until the deficiency is corrected. Monthly injections are then required for the remainder of an individual’s life. Although oral preparations were previously considered ineffective (as no intrinsic factor would prevent absorption of B12), recent practice has shown that oral administration of higher doses of B12 is beneficial.
There is an association between low folate and vitamin B12 levels and ageing populations. A study of almost 3000 Australians aged over 50 years found that 2.3% had low serum folate levels, whereas 22.9% had low serum vitamin B12 levels. These data suggest that vitamin B12 levels are of more concern than folate in the older Australian population.5
Aplastic anaemia
Aplastic anaemia is the condition of insufficient production of red blood cells due to cancer, chemotherapy or radiation therapy. Although low in numbers, the red cells are normocytic and normochromic. Bone marrow transplant may need to be considered, along with options for treating the underlying cancer. The effects of cancer on bone marrow and blood cell production are discussed further in Chapter 36.
Renal anaemia
Diseases affecting the kidneys can result in the impaired ability of the kidneys to detect hypoxaemia and respond by producing and secreting erythropoietin. Without adequate levels of erythropoietin, production of erythrocytes will be deficient. In this case, the anaemia is secondary to the kidney disease (refer to Chapter 30 for discussions on kidney disease).
Anaemia due to excessive erythrocyte loss
Loss or destruction of erythrocytes leads to anaemia. Loss of erythrocytes may occur following haemorrhage, and destruction of erythrocytes occurs with haemolytic disease of the newborn — this is now an extremely rare disease in Australia and New Zealand due to prophylactic treatment.
Post-haemorrhagic anaemia
Sudden blood loss occurs too quickly for production of replacement erythrocytes. This is referred to as post-haemorrhagic anaemia. Complications arising from haemorrhage are discussed in Chapter 23. Slower losses of blood such as due to gastric bleeds are able to be compensated.
Haemolytic disease of the newborn
Haemolytic disease of the newborn (erythroblastosis fetalis) is an acquired congenital haemolytic anaemia. It occurs when the fetal blood type differs from that of the mother. The pathophysiological effects of haemolytic disease of the newborn are more severe in Rh incompatibility than in ABO incompatibility. The risk occurs when a mother who is Rh-negative carries an Rh-positive fetus. The first Rh-incompatible pregnancy generally presents no difficulties because few fetal erythrocytes cross the placental barrier. When the placenta detaches at birth, however, a large number of fetal erythrocytes usually enter the mother’s bloodstream. This can cause the mother to develop anti-Rh antibodies (see Figure 17-9). During subsequent pregnancy, these antibodies can cross the placenta and bind to and destroy the fetal erythrocytes (haemolysis).
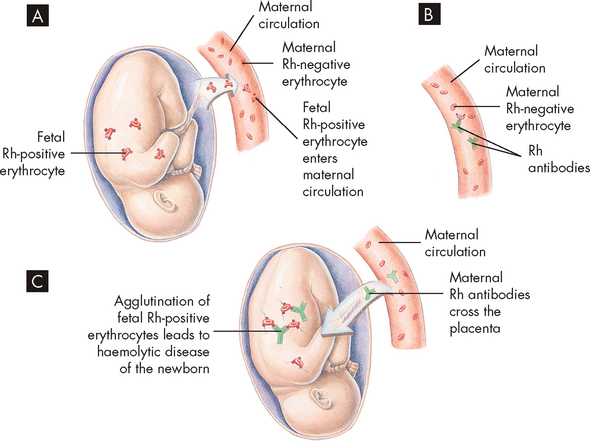
FIGURE 17-9 Haemolytic disease of the newborn.
A Before or during delivery, Rh-positive erythrocytes from the fetus enter the blood of the Rh-negative mother through a tear in the placenta. B The mother is sensitised to the Rh antigen and produces anti-Rh antibodies. Because this usually happens after delivery, there is no effect on the fetus in the first pregnancy. C During a subsequent pregnancy with an Rh-positive fetus, Rh-positive erythrocytes cross the placenta, enter the maternal circulation and stimulate the mother to produce antibodies against the Rh antigen. The anti-Rh antibodies from the mother cross the placenta, using agglutination and haemolysis of fetal erythrocytes, and haemolytic disease of the newborn develops.
Source: Seeley RR, Stephens TD, Tate P. Anatomy and physiology. 3rd edn. St Louis: Mosby; 1995.
CLINICAL MANIFESTATIONS
Neonates with mild haemolytic disease of the newborn may appear healthy or slightly pale, with slight enlargement of the liver or spleen. Pronounced pallor, splenomegaly and hepatomegaly indicate severe anaemia, which predisposes the neonate to cardiovascular failure and shock.
As haemolysis proceeds, the fetus becomes anaemic. Erythropoiesis accelerates, particularly in the liver and spleen, and immature erythroblasts are released into the bloodstream. Hyperbilirubinaemia occurs in the neonate after birth, which results in the accumulation of bilirubin products at a rate faster than the newborn’s liver is able to metabolise; bilirubin deposits in the brain (a condition termed kernicterus) may be fatal. Kernicterus produces cerebral damage and usually causes death (icterus gravis neonatorum); otherwise, infants may have mental retardation, cerebral palsy or high-frequency deafness. Rh incompatibility can cause severe or even life-threatening anaemia, death in utero or damage to the central nervous system.
EVALUATION AND TREATMENT
Routine evaluation of fetuses at risk for haemolytic disease of the newborn measures antibody in the mother’s circulation and indicates whether the fetus is at risk. Tests also include fetal blood sampling, amniotic fluid spectrophotometry and ultrasound fetal assessment.
The key to treatment of haemolytic disease of the newborn resulting from Rh incompatibility lies in prevention (immunoprophylaxis). One of the success stories of immunology has been the result obtained with Rh immune globulin, a preparation of IgG antibody against Rh antigen (Rhogam in Australia). If an Rh-negative woman is given Rh immune globulin within 72 hours of exposure to Rh-positive erythrocytes, she will not produce an antibody against the D antigen and the next Rh-positive baby she conceives will therefore be protected.
If antigenic incompatibility of the mother’s erythrocytes is not discovered in time to administer prophylactic immune globulin and a child is born with this disease, treatment consists of exchange transfusions in which the neonate’s blood is replaced with new Rh-negative blood that is not contaminated with anti-Rh antibodies. Phototherapy is also used to reduce the toxic effects of bilirubin accumulation.
Myeloproliferative red cell disorders
Haematological dysfunction results from an overproduction of cells, as well as a deficiency. One or more marrow elements may be produced in excess, responding to processes arising from within the body, such as due to a physiological compensatory response or an immune disorder. Also, external factors such as radiation and drugs can lead to haematological dysfunction. Excessive red cell production is classified as polycythaemia. Polycythaemia exists in two forms: relative and absolute.
 Relative polycythaemia results from haemoconcentration of the blood associated with dehydration. It is of minor consequence and resolves with fluid administration or treatment of underlying conditions. Absolute polycythaemia consists of two forms: primary and secondary. Secondary polycythaemia, the more common of the two, is a physiological response resulting from erythropoietin secretion caused by hypoxia. This hypoxia is noted in individuals living at higher altitudes (> 3000 metres), smokers with increased blood levels of carbon monoxide and individuals with chronic obstructive pulmonary disease or coronary heart failure, or both. Abnormal types of haemoglobin, which have a greater affinity for oxygen, also cause secondary polycythaemia, as does inappropriate secretion of erythropoietin by certain tumours (some renal, hepatic or brain tumours). The absolute primary form of polycythaemia is referred to as polycythaemia vera and is due to excessive production of red blood cells despite low levels of erythropoietin; this is a rare condition.
Relative polycythaemia results from haemoconcentration of the blood associated with dehydration. It is of minor consequence and resolves with fluid administration or treatment of underlying conditions. Absolute polycythaemia consists of two forms: primary and secondary. Secondary polycythaemia, the more common of the two, is a physiological response resulting from erythropoietin secretion caused by hypoxia. This hypoxia is noted in individuals living at higher altitudes (> 3000 metres), smokers with increased blood levels of carbon monoxide and individuals with chronic obstructive pulmonary disease or coronary heart failure, or both. Abnormal types of haemoglobin, which have a greater affinity for oxygen, also cause secondary polycythaemia, as does inappropriate secretion of erythropoietin by certain tumours (some renal, hepatic or brain tumours). The absolute primary form of polycythaemia is referred to as polycythaemia vera and is due to excessive production of red blood cells despite low levels of erythropoietin; this is a rare condition.ALTERATIONS OF PLATELETS AND COAGULATION
In this section, we consider abnormalities that relate to excessive or insufficient levels of haemostasis. These may arise due to alterations in either platelet formation (which normally forms a platelet plug) or the coagulation process (which leads to the formation of the fibrin mesh). Common laboratory tests to assess platelet and clotting factor function are listed in Table 17-2.
Table 17-2 LABORATORY TESTS OF HAEMOSTASIS
| CELL TYPE AND TEST | PROPERTY EVALUATED BY TEST | POSSIBLE HAEMATOLOGICAL CAUSE OF ABNORMAL FINDINGS |
|---|---|---|
| Platelets and clotting factors | ||
| Platelet count | Number of circulating platelets (× 109)/litre of blood | Anaemias, multiple myeloma, myelofibrosis, polycythaemia vera, leukaemia, disseminated intravascular coagulation (DIC), haemolytic disease of the newborn, transfusion reaction, lymphoproliferative disorders |
| Platelet function analysis | Ability of platelets to adhere and aggregate under standardised conditions simulating small blood vessel injury | Inherited and acquired platelet function disorders, storage pool disorder, von Willebrand’s disease |
| Platelet aggregation tests | Ability of platelets to adhere to one another (aggregate) in response to different activators | Inherited and acquired platelet function disorders, storage pool disorder, von Willebrand’s disease |
| Activated partial thromboplastin time (aPTT) | Effectiveness of plasma clotting factors of the intrinsic and common pathways of the coagulation cascade, as measured by the time taken for a clot to form in a test tube (in seconds) | Presence of circulating anticoagulants, DIC, clotting factor deficiencies, excessive fibrinolysis, haemorrhagic disease of the newborn, hypofibrinogenaemia, dysfibrinogenaemia and afibrinogenaemia, prothrombin deficiency, von Willebrand’s disease, acute haemorrhage |
| Prothrombin time | Effectiveness of plasma clotting factors of the extrinsic and common pathways of coagulation cascade, as measured by the time taken for a clot to form in a test tube (in seconds) | Hypofibrinogenaemia, dysfibrinogenaemia, and afibrinogenaemia; presence of circulating anticoagulants; DIC; clotting factor deficiency; presence of fibrin degradation products, increased fibrinolytic activity, haemolytic jaundice, haemorrhagic disease of the newborn; acute leukaemia, polycythaemia vera, multiple myeloma |
| Thrombin time | Quantity and activity of fibrinogen as measured in a test tube (in seconds) | Hypofibrinogenaemia, dysfibrinogenaemia, and afibrinogenaemia; presence of circulating anticoagulants; haemorrhagic disease of the newborn, polycythaemia vera; increase in fibrinogen-fibrin degradation products; increased fibrinolytic activity |
| Fibrinogen assay | Amount of fibrinogen available for fibrin formation | Acute leukaemia, congenital hypofibrinogenaemia or afibrinogenaemia, DIC, increased fibrinolytic activity, severe haemorrhage |
| Fibrin-fibrinogen degradation products (fibrin-fibrinogen split products) | Fibrinogenic activity as measured by levels of fibrin-fibrinogen degradation products (in mg/L of blood) | Transfusion reactions, DIC, internal haemorrhage in the newborn, deep vein thrombosis, pulmonary embolism |
Source: Lewis SM et al. Dacie and Lewis practical hematology. Philadelphia: Elsevier; 2006; Key N et al. Practical hemostasis. 2nd edn. Chichester: Wiley-Blackwell; 2009.
Platelet disorders
Alterations in the number or functions of platelets can interrupt normal blood coagulation and prevent haemostasis. The main quantitative abnormality (abnormal number of platelets) is thrombocytopenia, a decrease in the number of circulating platelets. Qualitative disorders affect the structure or function of individual platelets and can coexist with quantitative disorders. Qualitative disorders usually result in the prevention of platelet adherence and aggregation, thereby preventing formation of a platelet plug.
Thrombocytopenia
Thrombocytopenia is defined as a platelet count below 150 × 109/L of blood, although most individuals do not consider the decrease significant unless it falls below 100 × 109/L. The risk for haemorrhage associated with minor trauma is not substantial until the count falls below 50 × 109/L. Spontaneous bleeding without trauma can occur with counts ranging from 10 to 15 × 109/L. When this happens, skin manifestations (petechiae, ecchymoses and larger purpuric spots) are observed or frank bleeding from mucous membranes occurs. Severe bleeding results if the count falls below 10 × 109/L and can be fatal if it occurs in the gastrointestinal, respiratory or central nervous systems. In the bone marrow transplant or oncology setting, a platelet count as low as 10 × 109/L can be tolerated before platelet transfusions are required.
Before thrombocytopenia is diagnosed, careful attention needs to be made to the procedures used in obtaining the blood sample. A traumatic venepuncture or incorrect mixing of blood samples after blood collection can result in platelet activation or clot formation (thus platelet aggregation) in the blood collection tube. Also, any abnormalities in the laboratory testing of the blood need to be ruled out. In approximately 1 in 1000 to 10,000 blood samples, the platelets may form an aggregate (platelet plug) after the blood sample is obtained; platelets that are incorporated into the aggregate are not available to be counted by an automated cell counter. Finally, other physiological conditions such as hypothermia (< 25°C) can predispose to a thrombocytopenic state, which is reversed when temperatures return to normal, suggesting that the platelets are being sequestered by the spleen for later release back to the blood.
PATHOPHYSIOLOGY
Thrombocytopenia results from decreased platelet production or increased consumption, or both. The condition may be either congenital or acquired and either primary or secondary to other conditions.5,6 Thrombocytopenia secondary to congenital conditions occurs in a large number of different diseases, although each is relatively rare.7 Acquired thrombocytopenia is more common and may occur in relationship with acute viral infections (Epstein-Barr virus, rubella and HIV), drug reactions, autoimmune diseases, nutritional deficiencies, aplastic anaemia or cancer. Thrombocytopenia that results from decreased platelet production is usually the result of nutritional deficiencies (vitamin B12 or folic acid), drugs (e.g. chemotherapeutic agents, alcohol), radiation therapy or bone marrow infiltration by some cancers.
Most common forms of thrombocytopenia are the result of increased platelet consumption. The main examples are heparin-induced thrombocytopenia and disseminated intravascular coagulation (discussed in the section on disorders of coagulation).
Heparin-induced thrombocytopenia syndrome
Heparin is used clinically prior to surgery to inactivate clotting factor X and thrombin, thereby preventing coagulation during surgery. Specifically, it can reduce the risk of venous thromboembolism (see Chapter 23) and pulmonary embolism (see Chapter 25). Heparin is also used in the days after surgery to continue to prevent coagulation, particularly until the patient is mobilised, when the risk of unwanted clotting is lower. Approximately 4% of individuals treated with heparin develop heparin-induced thrombocytopenia syndrome. The incidence is lower (about 0.1%) with the use of low-molecular-weight heparin, a structurally different form of heparin that may be more effective in some types of surgery. Heparin-induced thrombocytopenia syndrome is an immune-mediated adverse drug reaction, as IgG antibodies bind to platelet receptors and activate platelet aggregation; this ultimately results in decreased numbers of free platelets.
CLINICAL MANIFESTATIONS
The hallmark of heparin-induced thrombocytopenia syndrome is the actual thrombocytopenia. However, 30% or more individuals are also at risk for thrombosis. If the syndrome is not recognised and treated, intravascular aggregation of platelets causes rapid development of arterial and venous thrombosis. Venous thrombosis is more common and results in deep venous thrombosis and pulmonary emboli. Arterial thrombosis affects the lower extremities causing limb ischaemia (impaired oxygen delivery). Cardiovascular accidents and myocardial infarctions also may be experienced (refer to Chapter 23).
EVALUATION AND TREATMENT
Diagnosis is based primarily on clinical observations. The individual presents with dropping platelet counts after 5 days or longer of heparin treatment. On average, platelet counts may reach 60 × 109/L. The onset of symptoms, including thrombosis, may be delayed until after release from the hospital. Most people are diagnosed postsurgery, therefore other possible causes of thrombocytopenia (e.g. infection, other drugs) must be considered. A number of laboratory tests are available to measure the antiplatelet antibodies and the amount of platelet aggregation.
Treatment is the withdrawal of heparin and use of alternative anticoagulants. Warfarin (which blocks the action of vitamin K and hence prevents the liver from producing clotting factors) should not be used until the symptoms of heparin-induced thrombocytopenia syndrome have resolved because of an increased risk of initiating skin necrosis. The thrombocytopenia should then progressively resolve. The risk of blood clots can be diminished by using a thrombin inhibitor (such as lepirudin).
Alterations of platelet function
Alterations in platelet function have similar clinical effects to thrombocytopenia, irrespective of the platelet count. The diagnosis can be made on the basis of abnormal laboratory tests. As these tests require a threshold number of platelets to be present for the results to be meaningful, diagnosing platelet function abnormalities in the presence of thrombocytopenia can be difficult, if not impossible. Associated clinical manifestations include spontaneous petechiae and purpura, bleeding from the gastrointestinal tract, genitourinary tract, pulmonary mucosa and gums.
Acquired disorders of platelet function may result from the use of drugs, with aspirin being the most common. It irreversibly inhibits cyclo-oxygenase function for several days after administration. Non-steroidal anti-inflammatory drugs also affect cyclo-oxygenase, although in a reversible fashion; this means that if the drug is not taken, the platelet function can return to normal.
Other disorders of platelet function include some systemic disorders, such as chronic renal disease, liver disease, cardiopulmonary bypass surgery and severe deficiencies of iron or folate. Haematological disorders associated with platelet dysfunction include chronic myeloproliferative disorders, multiple myeloma, leukaemias and myelodysplastic syndromes.
Disorders of coagulation
Disorders of coagulation are usually caused by defects or deficiencies in one or more of the clotting factors. (Normal function of the clotting factors is described in Chapter 16.) Qualitative or quantitative abnormalities interfere with or prevent the enzymatic reactions that transform clotting factors, circulating as plasma proteins, into a stable fibrin clot (see Figure 16-11).
Some clotting factor defects are inherited and involve one single factor, such as haemophilia (see Table 17-3). Other defects are acquired and tend to result from deficient synthesis of clotting factors by the liver. Causes include liver disease and dietary deficiency of vitamin K.
Table 17-3 COAGULATION FACTORS AND ASSOCIATED DISORDERS
| COAGULATION FACTOR | ASSOCIATED DISORDER |
|---|---|
| I | Fibrinogen deficiency |
| II | Hypoprothrombinaemia |
| V | Factor V deficiency |
| VII | Factor VII deficiency |
| VIII | Factor VIII deficiency (haemophilia A); von Willebrand’s disease |
| IX | Factor IX deficiency (haemophilia B) |
| X | Factor X deficiency |
| XI | Factor XI deficiency |
| XII | Hageman trait |
| XIII | Factor XIII deficiency (fibrin stabilising factor deficiency) |
Some coagulation disorders are attributed to pathological conditions that trigger coagulation inappropriately, engaging the clotting factors and causing detrimental clotting within blood vessels. For example, any cardiovascular abnormality that alters normal blood flow by speeding it up, slowing it down or obstructing it can create conditions in which coagulation proceeds within the vessels. An example of this is thromboembolic disease, in which blood clots obstruct blood vessels.
Haemophilia
Awareness of a serious bleeding disorder in males was documented nearly 2000 years ago in the Babylonian Talmud, which exempted from the rite of circumcision those boys having male relatives prone to excessive bleeding. In 1803 the first description of this disorder appeared in the medical literature, where it was noted to be X chromosome-linked in nature and associated with joint bleeding and crippling. This disease, haemophilia, is caused by genetic abnormalities that are linked to deficiencies in the production of clotting factors. Being X-linked, this disorder affects predominantly males. Males have XY sex chromosomes, whereas females have XX. If a male inherits the defective factor VIII gene on the X chromosome, the disease will be expressed. If a female inherits a defective gene on an X chromosome and there is a fully functional gene on the other X chromosome, the reduction in factor VIII levels will usually not be sufficient to cause bleeding. The female in this case will, however, be a carrier of the disease. A female may be affected by haemophilia in the extremely rare instance where defective genes are inherited on both X chromosomes. Many boys with haemophilia have undergone circumcision without excessive bleeding — we now know that normal haemostasis is achieved in these infants because clotting is activated through the extrinsic coagulation cascade. Haemophilia A is a deficiency in factor VIII, while haemophilia B is a deficiency in factor IX. Haemophilia A is the more common, but affects only approximately 1 in 4000 to 1 in 10,000 males.
CLINICAL MANIFESTATIONS
Prolonged bleeding will often become apparent in the first few years of the child’s life. Easy bruising and haemarthrosis (bleeding into joints) may occur and minor cuts take a long time to clot. Haemorrhage into the elbows, knees and ankles causes pain, limits joint movement and predisposes the child to degenerative joint changes. The extent of haemorrhagic disease and the age at which symptoms manifest are related to the severity of the deficiency. Recurrent bleeding, both spontaneous and after minor trauma, is a lifelong problem. Many affected individuals experience phases or cycles of spontaneous bleeding episodes. Mechanisms that cause this phenomenon are unknown. Intracranial haemorrhage and bleeding into the tissues of the neck or abdomen constitute life-threatening emergencies.
EVALUATION AND TREATMENT
Treatment options have been largely focused on transfusion of blood products, to allow the coagulation factor from the donor to enter the patient’s blood, thereby temporarily correcting the deficiency. However, in the early 1980s unfortunately many haemophilia patients became infected with blood-borne viruses such as HIV and hepatitis C. By the end of the 1980s, substantially improved screening and treatment of donated blood products led to them being safe, with minimal risk of transmitting these infections. Thus individuals can now be treated with plasma clotting factor concentrates much more safely. Haemophilia treatment may also include recombinant clotting factors, which are produced in laboratories rather than from blood products. These are safer still as there is no risk of viral contamination; however, they are more expensive. As a result of improved treatment options, it is likely that the prevalence of this disease may increase in the future, as patient survival is increasing.
Impaired haemostasis
Impaired haemostasis, or the inability to promote coagulation and the development of a stable fibrin clot, is commonly associated with liver dysfunction, as the liver is responsible for producing the clotting factors. This process is dependent on vitamin K and hence deficiency in this vitamin will interfere with clotting factor production. Other liver abnormalities may also contribute to impaired production of the clotting factors.
Vitamin K deficiency
Vitamin K is a fat-soluble vitamin that is required for the production of many of the clotting factors. Parenteral administration of vitamin K is the treatment of choice and usually results in correction of the deficiency. Fresh frozen plasma also may be administered but is usually reserved for individuals with life-threatening haemorrhages or those who require emergency surgery.
Liver disease
Individuals who have liver disease present with a broad range of haemostatic derangements that may be characterised by defects in the clotting or fibrinolytic system and by platelet function. The usual sequence of events is an initial reduction in clotting factors, which parallels the degree of liver cell damage or destruction. Factor VII is the first to decline because of its rapid turnover, followed by declines in factors II and X. Factor IX levels are less affected and do not decline until liver destruction is well advanced. Protein C (an anticoagulant) levels decline early, similar to levels of factor VII, and protein S (also an anticoagulant) levels decline in the later stages of liver disease. Declines of factor V are of special importance because factor V plasma levels appear to be a direct reflection of liver cell damage.
Other alterations of haemostasis in liver disease include an increase in fibrinolytic activity that is either primary in origin or is a manifestation that is secondary to disseminated intravascular coagulation (see next section).
Thrombocytopenia and thrombocytopathies are manifestations of liver disease. Thrombocytopenia is caused by splenomegaly, which often accompanies liver disease. Splenic pooling of platelets is the major cause of thrombocytopenia. Thrombocytopathies are associated with elevated levels of fibrin fragments, alcohol or drugs.
Treatment of alterations to haemostasis in liver disease must be comprehensive to cover all aspects of dysfunction. Fresh frozen plasma administration is the treatment of choice; however, not all individuals tolerate the volume needed to adequately replace all deficient factors. Platelet concentrates may also be transfused depending on the degree of thrombocytopenia.
Thromboembolic disorders
Certain conditions within the blood vessels predispose an individual to develop clots spontaneously. A clot attached to the vessel wall is called a thrombus (see Figure 17-10); this may form as unwanted clotting that contributes to the formation of an atherosclerotic plaque (refer to Chapter 23). A thrombus is composed of fibrin and blood cells and can develop in either the arterial system or the venous system. Arterial clots form under conditions of high blood flow and are composed mostly of platelet aggregates held together by fibrin strands. Venous clots form in conditions of low flow and are composed mostly of red cells with larger amounts of fibrin and few platelets.
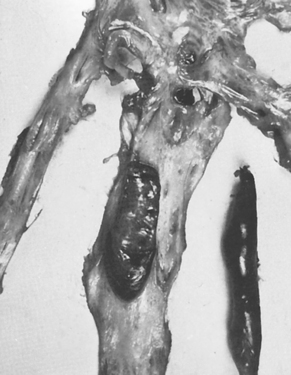
A thrombus arising in a valve pocket at the upper end of the superficial femoral vein. A postmortem clot on the right is shown for comparison.
Source: McLachlin J, Paterson JC. Some basic observations on venous thrombosis and pulmonary embolism. Surg Gynecol Obstet 1951; 93(1):1–8.
A thrombus eventually reduces or obstructs blood flow to tissues or organs, such as the heart, brain or lungs, depriving them of essential nutrients critical to survival. A thrombus also has the potential of detaching from the vessel wall and circulating within the bloodstream (referred to as an embolus). The embolus may become lodged in smaller blood vessels, blocking blood flow into the local tissue or organ and leading to ischaemia. Whether episodes of thromboembolism are life-threatening depends on the site of vessel occlusion. Thromboembolic disorders are mainly found in the adult population and the risk increases with age.
Therapy consists of removal or breakdown of the clot and supportive measures. Anticoagulant therapy is effective in treating or preventing venous thrombosis; it is not as useful in treating or preventing arterial thrombosis. Parenteral heparin is the major anticoagulant used to treat thromboembolism. Oral coumarin drugs also are widely used, particularly for individuals not hospitalised. More aggressive therapy may be indicated for such conditions as pulmonary embolism, coronary thrombosis or thrombophlebitis. Streptokinase and urokinase activate the fibrinolytic system and are administered to accelerate the lysis of known thrombi. Thrombolytic therapy has limited uses and is prescribed with a high degree of caution because it can cause haemorrhagic complications.
The risk for developing spontaneous thrombi is related to several factors, referred to as the Virchow triad:
Endothelial injury to blood vessels can result from atherosclerosis (plaque deposits on arterial walls; see Chapter 23). Atherosclerosis initiates platelet adhesion and aggregation, promoting the development of atherosclerotic plaques that enlarge, causing further damage and occlusion. Other causes of vessel endothelial injury may be related to haemodynamic alterations associated with hypertension and turbulent blood flow. Injury also is caused by radiation injury, exogenous chemical agents (toxins from cigarette smoke), endogenous agents (cholesterol), bacterial toxins or endotoxins, or immunological mechanisms. Whatever the precipitating cause of endothelial injury, it is a potent thrombogenic agent.
Sites of turbulent blood flow in the arteries and stasis of blood flow in the veins are at risk for thrombus formation. In areas of turbulence, platelets and endothelial cells may be activated, leading to thrombosis. In sites of stasis, platelets may remain in contact with the endothelium for prolonged lengths of time and clotting factors that would normally be diluted with fresh flowing blood are not diluted and may become activated. The most common clinical conditions that predispose to venous stasis and subsequent thromboembolic phenomena are major surgery (e.g. orthopaedic surgery), acute myocardial infarction, congestive heart failure, limb paralysis, spinal injury, malignancy, advanced age, the postpartum period and bed rest longer than 1 week. Turbulence and stasis occur with ruptured atherosclerotic plaques (myocardial infarction), hyperviscosity (polycythaemia) and conditions with deformed red cells (sickle cell anaemia).
The events leading to thrombus formation at sites of atherosclerotic plaque rupture are not fully understood. However, recent research shows that the effects of the mechanical forces associated with flow alteration lead to an accumulation of platelets tethered together on the downstream side of the blockage. This leads to growth of the blockage, which in turn creates a vortex or backflow immediately downstream of the platelet clump. Soluble activators released by the tethered platelets (see Chapter 16) can then accumulate in the backflow to cause platelet activation and further increasing the blockage. In blood vessels where there is no blockage the mechanism for platelet aggregation is different. In this case platelet aggregation is solely dependent on release of soluble platelet activators such ADP, thrombin and thromboxane A2.8
Hypercoagulability is the condition in which an individual is at risk for thrombosis, but by itself it is a rare cause of thrombosis. An individual may be in a hypercoagulable state if they are deficient in anticoagulation proteins, such that there is an increased tendency towards coagulation. Hypercoagulability (thrombophilia) is differentiated according to whether it results from primary (hereditary) or secondary (acquired) causes.
Disseminated intravascular coagulation
Disseminated intravascular coagulation (DIC) is an acquired clinical syndrome characterised by widespread activation of coagulation resulting in the formation of fibrin clots in medium and small vessels throughout the body.9 Widespread clotting may lead to blockage of blood flow to organs, resulting in multiple organ failure. The magnitude of clotting may result in the consumption of platelets and clotting factors, which can result in severe bleeding.
This is a complex systemic disorder that arises as a result of a major physiological event or trauma, such as sepsis, malignancy, complications of pregnancy and severe trauma. The clinical course of this condition is largely determined by the individual circumstances. It is characterised by subacute haemorrhage and diffuse microcirculatory thrombosis.
PATHOPHYSIOLOGY
Coagulation is designed to function at local areas of vascular damage, resulting in cessation of bleeding and activation of repair to the vessels. DIC results from abnormally widespread and ongoing activation of clotting.
Excessive exposure of tissue factor appears to be the trigger for activating coagulation (see Figure 17-11). Not only is the clotting system extensively activated in DIC, but the predominant natural anticoagulants (such as tissue factor pathway inhibitor, antithrombin III) are also greatly diminished. Tissue factor pathway inhibitor (TFPI) in association with factor Xa inactivates the TF-VIIa complex, preventing further activation of clotting. Antithrombin III is the principal inhibitor of thrombin. The rate of fibrinolysis is also diminished in DIC, as the activity of plasmin is diminished.
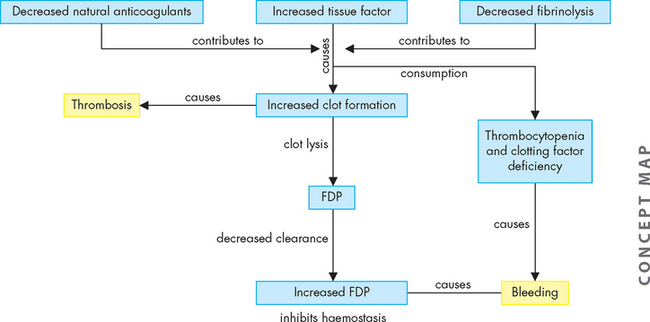
FIGURE 17-11 The pathophysiology of disseminated intravascular coagulation (DIC).
DIC is initiated by exposure of tissue factor causing clot formation. This is enhanced by a decrease in the natural anticoagulants (tissue factor pathway inhibitor, antithrombin-III and protein C). There is also a reduction in fibrinolysis by plasmin. The combined effect is to cause thrombosis. The thrombotic activity consumes coagulation factors and platelets. Slow degradation of the fibrin clots produces fibrin degradation products (FDP). FDP have inhibitory effects upon thrombin and platelets. The inhibition of coagulation, combined with the depletion of factors and platelets, then creates a bleeding tendency. DIC is a thrombohaemorrhagic disorder.
Although thrombosis is generalised and widespread, individuals with DIC are paradoxically at risk for haemorrhage. Haemorrhage is secondary to the abnormally high consumption of clotting factors and platelets, as well as the anticoagulant properties of fibrin degradation products. Thrombin causes platelet activation and aggregation — an event that occurs early in the development of DIC — which facilitates microcirculatory coagulation and obstruction in the initial phase. However, platelet consumption exceeds production, resulting in a thrombocytopenia that increases bleeding.
The deposition of fibrin clots in the circulation interferes with blood flow, causing widespread organ hypoperfusion. This condition may lead to ischaemia, infarction and necrosis, further potentiating and complicating the existing DIC process by causing further release of tissue factor and eventually organ failure. Whatever initiates the process of DIC, the cycle of thrombosis and haemorrhage persists until the underlying cause of the DIC is removed or appropriate therapeutic interventions are used.
CLINICAL MANIFESTATIONS
Clinical signs and symptoms of DIC present a wide spectrum of possibilities, depending on the underlying disease process that initiates DIC and whether the DIC is acute or chronic in nature. Most symptoms are the results of either bleeding or thrombosis. Acute DIC presents with rapid development of haemorrhaging (oozing) from venipuncture sites, arterial lines or surgical wounds or development of ecchymotic lesions (purpura, petechiae) and haematomas. Other sites of bleeding include the eyes (sclera, conjunctiva), nose and gums. Shock may also be observed.
Manifestations of thrombosis are not always as evident, even though it is often the first pathological alteration to occur. Several organ systems are susceptible to microvascular thrombosis associated with dysfunction: cardiovascular, pulmonary, central nervous, renal and hepatic systems. Acute and accurate clinical interpretations are critical to preventing progression of DIC that may lead to multisystem organ dysfunction and failure. Indicators of multisystem dysfunction include changes in the level of consciousness, behaviour and confusion, seizure activity, oliguria (low volume urine), haematuria (blood in the urine), hypoxia, hypotension, haemoptysis (coughing blood), chest pain and tachycardia. Symmetric cyanosis of fingers and toes (blue finger/toe syndrome), nose and breast may be observed and indicates macrovascular thrombosis. This may lead to infarction and gangrene that may require amputation.
EVALUATION AND TREATMENT
No single laboratory test can be used to effectively diagnosis DIC. Diagnosis is based primarily on clinical symptoms and confirmed by a combination of laboratory tests. The individual must present with a clinical condition that is known to be associated with DIC. The most commonly used combination of laboratory tests usually confirm thrombocytopenia or a rapidly decreasing platelet count on repeated testing, prolongation of clotting times, the presence of fibrin degradation products, reduced fibrinogen levels and decreased levels of coagulation inhibitors.
Detection of fibrin degradation products is more specific for DIC. Detection of D-dimers is a widely used test for DIC. D-dimer is a molecule produced by plasmin degradation of cross-linked fibrin in clots. D-dimers in the blood can be quantified by a number of different methods, some of which have been modified for use on automated analysers, greatly improving the turnaround time for the laboratory results. It is important to note that D-dimer levels may also be raised in conditions where DIC is not present (e.g. deep vein thrombosis).
Treatment of DIC is directed towards:
Eliminating the underlying pathology is the initial intervention in the treatment phase in order to eliminate the trigger for activation of clotting. Once the stimulus is gone, production of coagulation factors in the liver leads to restoration of normal plasma levels within 24–48 hours.
Controlling ongoing thrombosis is more difficult to attain. Heparin has been used for this; however, its use is controversial because its mechanism of action is binding to and activating antithrombin III, which is deficient in many types of DIC. The anticoagulant action of heparin may also create a bleeding risk if the clotting factors are depleted. Currently, heparin is indicated only in certain types of situations related to DIC. Replacement of deficient coagulation factors, platelets and other coagulation elements is gaining recognition as an effective treatment modality. Their use is not without controversy, however, because a major concern with replacement therapy is the possible risk of adding components that will increase the rate of thrombosis. Clinical judgement is the key factor in determining whether replacement is to be used as a treatment modality.
Maintaining organ function is achieved by fluid replacement to sustain adequate circulating blood volume and maintain optimal tissue and organ perfusion. Fluids may be required to restore blood pressure, cardiac output and urine output to normal parameters.
Haemostasis therapy
For many years, the only agents used to inhibit coagulation were heparin and warfarin. These drugs are still widely used to treat or prevent thrombosis. Heparin is a fast-acting anticoagulant that is administered intravenously or subcutaneously. It combines with antithrombin III to exert an anticoagulant effect. Warfarin has an advantage over heparin in that it is taken orally; however, the anticoagulant effect develops slowly over several days. Warfarin is an antagonist of vitamin K, which is required for the production of clotting factors II, VII, IX and X in the liver. An individual taking warfarin will not be able to produce functional versions of these clotting factors and so the ability of their blood to clot will be reduced.
The dose of anticoagulant given to a patient needs to be sufficient to reduce the risk of thrombosis, but the anticoagulant effect should not be so great as to create a high risk of bleeding. For this reason, therapeutic ranges have been established and the degree of anticoagulation can be monitored by laboratory tests (see Table 17-2). Unfractionated heparin therapy can be monitored by the activated partial thromboplastin time (APTT). This is a clotting test that assesses the intrinsic and common pathways of the coagulation cascade and is sensitive to the anticoagulant effect of unfractionated heparin. The APTT test is not sensitive to the effect of low-molecular-weight heparin, which is sometimes used in clinical practice.
Dark chocolate, wine and platelet-inhibitory functions
An increasing number of foods have been reported to have platelet-inhibitory functions. Recent studies showed flavanol-rich cocoa inhibited several measures of platelet activity. Dark chocolate contains much more cocoa than does light chocolate. Additional cardioprotective effects may include antioxidant properties and activation of nitric oxide (NO). Low to moderate consumption of red wine reportedly has a greater benefit than other alcoholic beverages on cardioprotective mechanisms. Emerging are the effects of the polyphenol resveratrol known to be abundant in red wine. Investigators documented that the polyphenolic antioxidants, resveratrol and proanthocyanidins provide cardioprotection by their function in vivo as antioxidants.
Source: Pearson DA et al. Flavonols and platelet reactivity. Clin Dev Immunol 2005; 12(1):1–9; Holt RR et al. Chocolate consumption and platelet function. JAMA 2002; 287(17):2212–2213; Engler MB. Flavonoid-rich dark chocolate improves endothelial function and increase plasma epicatechin concentrations in healthy adults. J Am Coll Nutr 2004; 23(3):197–204; Sato M, Maulik N, Das DK. Cardioprotection with alcohol and polyphenolic antioxidants. Annals NY Acad Sci 2002; 957:122–135.
Warfarin therapy is monitored by the prothrombin time (PT). This clotting test assesses the extrinsic and common pathways and is sensitive to the effect of warfarin. A calculation is applied by the laboratory to the PT test result, such that the PT test result divided by the average time obtained for a normal control plasma produces the prothrombin ratio (PR). The INR (international normalised ratio) calculation allows test results from different laboratories and different batches to be comparable — otherwise, prothrombin time results for a patient could only be reliably compared with other prothrombin time tests prepared at a similar time, which is not achievable. By using the INR, patient results can be compared over time and from one location to another to allow effective monitoring of warfarin therapy. Because of this, the INR is one of the most frequently requested laboratory tests.
New oral anticoagulants targeting activated factor X or thrombin are in advanced stages of development.10 These new drugs may not need the same level of monitoring as warfarin.
Aspirin has been used successfully to inhibit platelets for many years. In more recent times other specific inhibitors of platelet activation, mostly targeting platelet glycoprotein receptors, have emerged.11
The management of individuals with bleeding disorders has been advanced by the development of recombinant factor agents, the prolonged half-life of agents or agents with reduced activation of immune responses. First introduced for haemophiliacs, recombinant factor VIIa (rFVIIa) is now used for several inherited and acquired bleeding disorders.
ALTERATIONS OF LEUCOCYTE FUNCTION
Leucocyte function is affected if too many or too few white cells are present in the blood or if the cells that are present are structurally or functionally defective (see Table 17-4). Phagocytic cells (granulocytes, monocytes, macrophages) may lose their ability to act as effective phagocytes and lymphocytes may lose their ability to respond to antigens. Other leucocyte alterations include infectious mononucleosis and cancers of the blood — leukaemia and multiple myeloma.
Table 17-4 WHITE BLOOD CELL COUNTS
| CELL TYPE AND TEST | PROPERTY EVALUATED BY TEST | POSSIBLE HAEMATOLOGICAL CAUSE OF ABNORMAL FINDINGS |
|---|---|---|
| Leucocytes: differential white cell count (absolute number of a type of leucocyte/litre of blood) | See below | See below |
| Neutrophil count | Neutrophils (× 109)/L | Myeloproliferative disorders, haematopoietic disorders, haemolysis, infection |
| Lymphocyte count | Lymphocytes (× 109)/L | Infectious lymphocytosis, infectious mononucleosis, haematopoietic disorders, anaemias, leukaemia, lymphosarcoma, Hodgkin’s disease |
| Monocyte count | Monocytes (× 109)/L | Hodgkin’s disease, infectious mononucleosis, monocytic leukaemia, non-Hodgkin’s lymphoma, polycythaemia vera |
| Eosinophil count | Eosinophils (× 109)/L | Haematopoietic disorders |
| Basophil count | Basophils (× 109)/L | Chronic myeloid leukaemia, haemolytic anaemias, Hodgkin’s disease, polycythaemia vera |
Source: Lewis SM et al. Dacie and Lewis practical hematology. Philadelphia: Elsevier; 2006; Key N et al. Practical hemostasis. 2nd edn. Chichester: Wiley-Blackwell; 2009.
Quantitative alterations of leucocytes
Quantitative alterations are increases or decreases in the number of leucocytes in the blood. Leucocytosis is used to describe a white cell count that is higher than normal, whereas leucopenia refers to when the count is lower than normal. Leucocytosis and leucopenia may affect a specific type of white blood cell and may result from a variety of physiological conditions and alterations.
Leucocytosis occurs as a normal protective response to physiological stressors, such as invading microorganisms, strenuous exercise, emotional changes, temperature changes, anaesthesia, surgery, pregnancy and some drugs, hormones and toxins. It is also caused by pathological conditions, such as malignancies and haematological disorders.
If the leucocyte count falls to less than 1 × 109/L, the risk of infection increases drastically. With counts below 0.5 × 109/L, the possibility for life-threatening infections is high. Leucopenia may be caused by radiation, anaphylactic shock, autoimmune disease (e.g. systemic lupus erythematosus), immune deficiencies and certain chemotherapeutic agents.
Granulocyte and monocyte alterations
Granulocytosis — an increase in granulocytes (neutrophils, eosinophils or basophils) — begins when stored blood cells are released. Neutrophilia is another term that may be used to describe granulocytosis because neutrophils are the most numerous of the granulocytes. Neutrophilia is seen in the early stages of infection or inflammation and is established when the absolute count exceeds 7.5 × 109/L. Release and depletion of stored neutrophils stimulates granulopoiesis to replenish neutrophil reserves.
When the demand for circulating mature neutrophils exceeds the supply, immature neutrophils (and other leucocytes) are released from the bone marrow. The immature cells can be observed by microscopic examination of a blood smear.
Neutropenia is a condition associated with a reduction in circulating neutrophils and exists clinically when the neutrophil count is less than 2 × 109/L. Reduction in neutrophils occurs in severe prolonged infections when production of granulocytes cannot keep up with demand.12
Other causes of neutropenia, in the absence of overwhelming infection, may be:
Haematological disorders that cause ineffective or decreased production include hypoplastic or aplastic anaemia, megaloblastic anaemia, leukaemia or drug/toxin-induced neutropenia. Neutropenia is also seen in starvation and anorexia nervosa because of an inadequate supply of protein. Decreased neutrophil survival is seen in autoimmune disorders (e.g. systemic lupus erythematosus, rheumatoid arthritis). Abnormal neutrophil distribution and sequestration are associated with hypersplenism and pseudoneutropenia, which in the presence of rheumatoid arthritis constitute Felty’s syndrome. Viral infections (HIV, Epstein-Barr virus) also may cause neutropenia, as do chemotherapy and other toxic drugs received for cancer treatment and transplantation.
If neutrophils are drastically reduced (< 0.5 × 109/L) and the entire granulocyte count is extremely low, granulocytopenia or agranulocytosis results. Usually, when this occurs, haematopoiesis is arrested in the bone marrow or cell destruction increases in the circulation. Chemotherapeutic agents used to treat haematological and other malignancies cause bone marrow suppression. Several other drugs cause agranulocytosis, which occurs rarely but carries a high mortality rate of 10–48%. Clinical manifestations of agranulocytosis include infection (particularly of the respiratory system), general malaise, septicaemia, fever, tachycardia and ulcers in the mouth and colon. If untreated, sepsis results in death within 3–6 days.
Infectious mononucleosis
Infectious mononucleosis is an acute infection of B lymphocytes (B cells) with Epstein-Barr virus.13 Infections with Epstein-Barr virus are common in children, particularly those from low socioeconomic environments. Approximately 50–85% of these children are infected with the virus by age 4 and more than 90% of adults have indications of previous exposure. These early infections are usually asymptomatic and provide immunity to Epstein-Barr virus; thus children with an early infection rarely develop infectious mononucleosis.
The incidence of infectious mononucleosis is approximately 45 in 10,000 individuals and is most commonly seen in young adults between 15 and 35 years of age, with the peak incidence being between 15 and 19 years of age. Transmission of the virus is usually through saliva from close personal contact (e.g. kissing, hence the name kissing disease). The virus also may be secreted in other mucosal secretions of the genital, rectal and respiratory tract, as well as blood. The virus initially infects the oropharynx, nasopharynx and salivary epithelial cells with later extension into lymphoid tissues and B cells.
Unaffected B cells produce antibodies (IgG, IgA, IgM) against the virus. Cytotoxic T lymphocytes are activated and multiply to assist the B cells in attacking the virus and virus-infected cells directly (see Chapter 12). The production of B and T cells and the process of removing dead and damaged leucocytes are largely responsible for lymphoid tissue swelling (lymph nodes, spleen, tonsils and, occasionally, liver). Sore throat and fever, two initial manifestations of infectious mononucleosis, are caused by inflammation and infection in the mouth and throat, where the virus initially entered the body.
CLINICAL MANIFESTATIONS
The incubation period for infectious mononucleosis is quite long: 30–50 days. Early flu-like symptoms, such as headache, malaise, joint pain and fatigue, may appear during the first 3–5 days, although some individuals are without symptoms. At the time of diagnosis, the individual commonly presents with the classic group of symptoms: fever, sore throat, cervical (neck) lymph node enlargement and fatigue. As the condition progresses, generalised lymph node enlargement may also develop, as well as enlargement of the spleen and liver (25–75% of individuals). Splenic rupture is rare and can occur spontaneously or as a result of mild trauma, occurring primarily in males (90%) between day 4 and day 21 after symptom onset, and may be fatal. Other causes of the rare fatalities associated with infectious mononucleosis are hepatic failure, extensive bacterial infection or viral myocarditis. Other organ systems are rarely involved, but such involvement may be present with characteristic manifestations, such as fulminant hepatitis with jaundice and anaemia, encephalitis, meningitis and Guillain-Barré inflammation syndrome. Eye manifestations may include eyelid and periorbital oedema, dry eyes, inflammation and conjunctivitis. Pulmonary involvement is rare, although incidences of pneumonia and respiratory failure have been documented in immunocompromised individuals.
Infectious mononucleosis is usually self-limiting and recovery occurs in a few weeks; severe clinical complications are rare (5%). Fatigue may last for 1–2 months after resolution of other symptoms.
EVALUATION AND TREATMENT
The blood of affected individuals typically demonstrates a moderate leucocytosis due to lymphocytosis, and numerous atypical lymphocytes are observed in the peripheral blood film. Serological tests are necessary to diagnose Epstein-Barr virus infection.14
Treatment is supportive and consists of rest and alleviation of symptoms with analgesics and antipyretics. Streptococcal pharyngitis, which occurs in 20–30% of cases, is treated with penicillin or erythromycin, not ampicillin — ampicillin is known to cause a rash. Bed rest with avoidance of strenuous activity and contact sports is indicated. Steroids are used when severe complications, such as impending airway obstruction or other organ involvement (central nervous system manifestations, thrombocytopenic purpura, myocarditis, pericarditis), is evident. Aciclovir has been used in immunocompromised individuals but is not considered standard therapy.
Qualitative alterations of leucocytes
Leukaemia
Leukaemia is a clonal malignant disorder of the blood and blood-forming organs.15 The common pathological feature of all forms of leukaemia is an uncontrolled proliferation of malignant leucocytes, causing an overcrowding of bone marrow and decreased production and function of normal hematopoietic cells.
The classification of leukaemia is based on the predominant cell of origin; myeloid cells are those that develop and mature within the bone marrow (erythrocytes, platelets, all leucocytes except for the lymphocytes and natural killer cells), whereas lymphoid cells undergo some development outside of the bone marrow (lymphocytes and natural killer cells). Also, leukaemia is classified according to the degree of differentiation that took place before the cell became malignant: acute refers to the rapid growth of immature blood cells; chronic refers to the slow growth of more differentiated cells. Thus, there are four types of leukaemia (see Figure 17-12):
FIGURE 17-12 Leukaemia can arise from different cell types.
The lymphoid stem cells are the precursors to the lymphocytes and natural killer cells, while the myeloid stem cells ultimately develop into all other blood cell types (erythrocytes, platelet, all other leucocytes). Types of acute leukaemia occur in less matured cell types, while types of chronic leukaemia occur in cells that have developed further.
Further classification of types of acute leukaemia is based on characteristics that may provide significant therapeutic prognostic information, such as structure, number of cells, genetics, identification of surface markers and histochemical staining.
Acute leukaemia is characterised by a proliferation of undifferentiated or immature cells, usually blast cells. Blast cells are very immature blood cells normally found only in bone marrow and not the peripheral blood. In the case of many types of leukaemia, the proliferation of the malignant cells in the marrow ‘spills over’ into the blood, due to a blockage in cell maturation and accelerated cell division. This can lead to very high white cell counts in many patients, although the leucocytes are immature and therefore not functional. In the case of acute leukaemia, a high white cell count in the peripheral blood is associated with the presence of blast cells. Laboratory tests can determine whether the blast cells are myeloid or lymphoid.
The correct classification of leukaemia is important for determining the treatment protocol. Classification is mostly based on a range of laboratory tests that examine cell structure and appearance, expression of surface markers (molecules), gene mutations and biochemical make-up of the malignant cells. In most cases of leukaemia, a bone marrow sample is required in addition to a peripheral blood sample. An aspirate (fluid) and a trephine (core) sample are collected from the marrow, usually from the iliac crest. The bone marrow samples supply test material from the source of the disease and also provide useful information regarding the stage of progression and likely prognosis. Patient outcome is highly variable and depends on the type of leukaemia and numerous biological factors. Age is a significant determinant, as there is a high cure rate among children, but older individuals may not tolerate aggressive therapies well.
The onset of acute leukaemia is abrupt and rapid. Disease progression results in a short survival time. In chronic leukaemia, the predominant cell is more mature but still does not function normally. The onset of the disease is gradual and the prolonged clinical course results in a relatively longer survival time; however, chronic leukaemia can be difficult to cure.
Leukaemia occurs with varying frequencies at different ages and is more common in adults than in children. Table 17-5 shows the incidence of the four major types of leukaemia in Australia in 2005. The most common type was CLL, closely followed by AML, then ALL and CML. The most common form of childhood cancer is leukaemia and most children have ALL.18
Table 17-5 THE INCIDENCE OF MAJOR HAEMATOLOGICAL CANCERS IN AUSTRALIA, 2005
| TYPE | NUMBER OF NEW CASES | PERCENTAGE OF ALL CANCERS (%) |
|---|---|---|
| Acute myeloid leukaemia (AML) | 865 | 0.8 |
| Chronic myeloid leukaemia (CML) | 242 | 0.3 |
| Acute lymphoblastic leukaemia (ALL) | 304 | 0.3 |
| Chronic lymphocytic leukaemia (CLL) | 969 | 0.8 |
| Multiple myeloma | 1196 | 1.2 |
| Hodgkin’s lymphoma | 527 | 0.5 |
| Non-Hodgkin’s lymphoma | 3903 | 3.7 |
Source: Australia Institute of Health and Welfare. Cancer in Australia: an overview. Cancer series no. 46. Cat no. CAN 42. Canberra: AIHW; 2008.
Over recent decades, remission and survival rates for most forms of leukaemia have increased. Current 5-year survival rates (that is, the percentage of people alive 5 years after being diagnosed) are approximately 48%; the rate is particularly promising for children, with a 5-year survival over 80%.19 This progress is the result of more effective chemotherapeutic agents, improved blood product and antimicrobial support and specialised nursing care. Chemotherapy and bone marrow transplants (stem cell transplants) have significantly increased the survival time for individuals with acute leukaemia.
PATHOPHYSIOLOGY
Although the exact cause of leukaemia is unknown, several risk factors and related genetic aberrations are associated with the onset of malignancy. There is a significant tendency for leukaemia to reappear in families. There is also an increased incidence of leukaemia in association with other hereditary abnormalities (such as Down syndrome and trisomy 13) and some immune deficiencies. One of the chromosomal (genetic) abnormalities results in an error with mitosis, which occurs in 95% of those with CML and 30% of adults with ALL.
Risk factors for the onset of leukaemia include environmental factors as well as other diseases. Increased risk has been linked to cigarette smoke and exposure to benzene. Large doses of ionising radiation particularly result in an increased incidence of myeloid leukaemia. Infections with HIV or hepatitis C virus increase the risk for leukaemia. Drugs that cause bone marrow depression can also predispose an individual to leukaemia. AML is the most frequently reported secondary cancer after high doses of chemotherapy for treatments of Hodgkin’s lymphoma, non-Hodgkin’s lymphoma, multiple myeloma, ovarian cancer and breast cancer. Acute leukaemia also may develop secondary to certain acquired disorders, including CML, CLL, polycythaemia vera and ovarian cancer.
The leukaemia blasts (immature cells) literally ‘crowd out’ the marrow and cause cellular proliferation of the other cell lines to cease (known as bone marrow failure). Normal granulocytic-monocytic, lymphocytic, erythrocytic and megakaryocytic progenitor cells cease to function, eventually resulting in pancytopenia (a reduction in all cellular components of the blood).
Acute leukaemia
Acute leukaemia is characterised by a proliferation of blast cells. It can be defined as the presence of more than 20% of blast cells in the blood or bone marrow at presentation. Acute leukaemia can be either myeloid (AML) or lymphoid (ALL). AML is found in all age groups; however, it is most common in adults and the incidence increases with age. In contrast, ALL is the most common childhood leukaemia.
About 85% of ALL cases arise from the B cell line and about 15% arise from T cell lineage. Acute leukaemia is seen in both genders and in all ages, with the incidence increasing dramatically in individuals older than 50 years.
CLINICAL MANIFESTATIONS
The clinical manifestations of all varieties of acute leukaemia are generally similar. Mechanisms associated with common manifestations are summarised in Table 17-6. Signs and symptoms related to bone marrow depression include fatigue caused by anaemia, bleeding resulting from thrombocytopenia and fever caused by infection. Bleeding may occur in the skin, gums, mucous membranes and gastrointestinal tract. Visible signs include petechiae and ecchymosis, as well as discolouration of the skin, gingival bleeding, haematuria and midcycle or heavy menstrual bleeding.
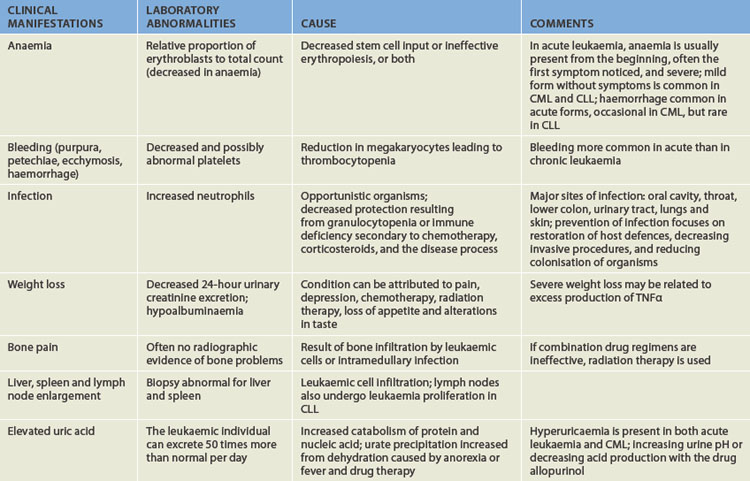
Infection sites include the mouth, throat, respiratory tract, lower colon, urinary tract and skin and may be caused by gram-negative bacilli (Escherichia coli), Pseudomonas and Klebsiella. Fever is an early sign often accompanied by chills.
Anorexia is accompanied by weight loss, diminished sensitivity to sour and sweet tastes, wasting of muscle and difficulty swallowing. Liver, spleen and lymph node enlargement occurs more commonly in ALL than in CML. Liver and spleen enlargement commonly occur together. The leukaemic individual often experiences abdominal pain and tenderness and also breast tenderness.
Neurological manifestations are common and may be caused by either leukaemic infiltration or cerebral bleeding. Headache, vomiting, papilloedema (swelling of the optic disc), facial palsy, blurred vision, auditory disturbances and meningeal irritation can occur if leukaemic cells infiltrate the cerebral or spinal meninges. Because chemotherapeutic agents do not penetrate the blood–brain barrier, leukaemia cells can grow easily in these locations.
EVALUATION AND TREATMENT
Because leukaemia often is confused with other conditions, early detection is difficult. Persistent symptoms need intensive medical investigation. The diagnosis is made through blood tests and examination of bone marrow.
Chemotherapy, used in various combinations, is the treatment of choice for leukaemia. Supportive measures include blood transfusions, antibiotics, antifungals and antivirals. Allopurinol is used to prevent uric acid production and elevation that occurs because of cellular death caused by treatment. Bone marrow transplantation as a treatment has increased since the 1980s. Survival rates have dramatically increased because of improvements in donor matching, transfusion support, conditioning regimens and antibiotics.
Factors influencing the increased survival rate seen in recent years include the use of combined and multimodality treatment methods, improved supportive services such as blood banking and nutritional support, and antimicrobial treatment.
Stimulation of blood cell growth and development with haematopoietic drugs has increased neutrophil recovery during chemotherapy and bone marrow transplant. Blood granulocyte numbers (e.g. neutrophils, eosinophils, basophils/mast cells) are normally in the range of 4–6 × 109/L and susceptibility to infection develops below 1 × 109/L. During a natural response to a bacterial infection, granulocytes usually rise in number to 10–20 × 109/L. The administration of specific colony-stimulating factors to target the deficient cells in a leucopenic patient can raise white cell numbers and afford protection from infections. Leukaemia itself, as well as the chemotherapeutic agents used to treat the disease, can result in dramatic decreases in circulating granulocytes.
Chronic leukaemia
The two main types of chronic leukaemia are chronic myeloid leukaemia and chronic lymphocytic leukaemia. Several forms of CML can occur, depending on the lineage of the malignant cells (for example, whether neutrophils or eosinophils are affected). Unlike cells in acute leukaemia, chronic leukaemic cells are well differentiated (refer to Figure 17-12) and can be readily identified. Individuals with chronic leukaemia have a longer life expectancy, usually extending several years from the time of diagnosis.
Chronic leukaemia accounts for the majority of cases in adults. The incidences of CLL and CML increase significantly in individuals over 40 years of age, with prevalence in the sixth through to the eighth decades. CML is included in a group of diseases called myeloproliferative disorders, which also include polycythaemia vera and myelofibrosis (invasion of bone marrow by fibrous tissue).
PATHOPHYSIOLOGY AND CLINICAL MANIFESTATIONS
Chronic leukaemia advances slowly and insidiously. Individuals are generally unaware of the condition until symptoms appear and sometimes the diagnosis is made as a consequence of a routine blood test. When symptoms do appear, they present as splenomegaly, extreme fatigue, weight loss, night sweats and low-grade fever. Individuals with CML may progress through three phases of the disease: a chronic phase lasting 2–5 years during which symptoms may not be apparent; an accelerated phase of 6–18 months during which the primary symptoms develop; and a terminal blast phase with a survival of only 3–6 months. The accelerated phase is characterised by excessive proliferation and accumulation of malignant cells. Splenomegaly is prominent and more painful, but lymphadenopathy generally is not present. Liver enlargement also occurs, but liver function is rarely altered. Hyperuricaemia is common and produces gouty arthritis. Infections, fever and weight loss are seen often. The terminal blast phase is characterised by rapid and progressive leucocytosis with an increase in basophils. In the later stages of the terminal phase, which then resembles AML, blast cells or promyelocytes predominate and the individual experiences a ‘blast crisis’.
Chromosomal abnormalities (Philadelphia chromosome) are useful in diagnosing CML and are observed in 95% of these individuals. The median age for persons with CML is 40–45 years. The chromosomal abnormality actually occurs in red cells, white cells and platelets, but appears to affect only white cell production and function. There is no known specific cause for CML except exposure to ionising radiation.
CLL involves predominantly malignant transformation of B cells; rarely (< 5%) are T cells involved. The malignant transformation is thought to be caused by failure of the normal mechanisms of programmed cell destruction (apoptosis), allowing these cells to have an extended life; hence the chronic nature of the disease. These cells fail to develop into antibody-producing cells and fail to respond to stimulation by helper T cells.
Suppression of normal antibody production is the most significant effect in CLL. Individuals are thus at risk for recurrent bacterial and other infections that are commonly sensitive to antibodies. Anaemia, thrombocytopenia and neutropenia are typically present with overt CLL. Invasion of most organs by leukaemic cells is uncommon, but infiltration of lymph nodes, liver, spleen and salivary glands is observed.
EVALUATION AND TREATMENT
Therapeutic approaches include bone marrow transplantation, biological response modifiers and combination chemotherapy. Alone, state-of-the-art chemotherapy for CML does not cure the disease, prevent blastic transformation or prolong the average survival time. New drugs, including imatinib (which is highly specific for CML), have been used successfully; however, relapse with drug-resistant disease has been a problem. Bone marrow transplantation, when compared with biological response modifiers and combination chemotherapy, appears to increase the survival time more significantly. Allogenic bone marrow transplants involve the patient receiving stem cells from a matched donor, often a family member. With these transplants, survival rates increase 20–30% with concurrent high-dose radiation, chemotherapy and interferon therapy. Because of the physiological stress associated with the procedure, bone marrow transplantation is usually considered a viable option only in patients younger than 55 years of age.
When to begin treatment for CLL is difficult to determine and is related to the degree of symptoms. In low-grade disease, early treatment can result in a poorer outcome. Treatment consists of alkylating agent or purine analogue chemotherapy. Steroids and, later, splenectomy also may be used to control leucocytosis and cytopenias. Radiation therapy may be used to alleviate lymphadenopathy. Late stages of the disease require combination chemotherapy. Regardless of the approach, cure rates for CLL are poor.
ALTERATIONS OF LYMPHOID FUNCTION
Lymphadenopathy
Lymphadenopathy is characterised by enlarged lymph nodes (see Figure 17-13). Lymph node enlargement is caused by an increase in the size and number of its germinal centres as a result of proliferation of lymphocytes and monocytes (immature phagocytes) or invasion by malignant cells. Normally, lymph nodes are not palpable or are barely palpable. Enlarged lymph nodes are characterised by being palpable and often also may be tender or painful to touch, although not in all situations.
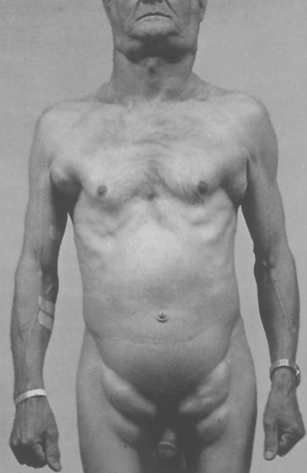
An individual with lymphocyte leukaemia with extreme but symmetric lymphadenopathy.
Source: Courtesy Dr AR Kagan, Los Angeles. From del Regato JA, Spjut HJ, Cox JD. Cancer: diagnosis, treatment, and prognosis. 6th edn. St Louis: Mosby; 1985.
Localised lymphadenopathy (reactive lymph nodes) usually indicates drainage of an area associated with an inflammatory or infectious lesion. Generalised lymphadenopathy, associated with infection, occurs less often and is generally seen in the presence of malignant or non-malignant disease. Lymphadenopathy is of more significance in adult disease than in children. The location and size of the enlarged nodes are important factors in diagnosing the cause of the lymphadenopathy, as are the individual’s age and sex. Generalised lymphadenopathy occurs with non-Hodgkin’s lymphomas, CLL and disorders that produce lymphocytosis. In general, lymphadenopathy results from four types of conditions:
Diseases of unknown cause, including autoimmune diseases and reactions to drugs, also may lead to generalised lymphadenopathy.
Malignant lymphomas
Lymphomas consist of a diverse group of neoplasms that develop from the proliferation of malignant lymphocytes in the lymphatic system. The most recent classification of lymphomas was published by the World Health Organization (WHO) and is derived from the Revised European-American Lymphoma (REAL) Classification. This classification is based on the cell type from which the lymphoma probably originated. The groups include Hodgkin’s lymphoma and two that were previously classified as non-Hodgkin’s lymphoma (B-cell neoplasms, and T-cell and natural killer-cell neoplasms). With the new classification, multiple myeloma, which was previously classified independently, is included as a B-cell lymphoma.
Currently in Australia, about 4300 individuals per year are diagnosed with lymphoma, which is the fifth most common cancer in Australia.20 By far the majority of these people have non-Hodgkin’s lymphoma (about 89%). Since the early 1970s, the incidence of non-Hodgkin’s lymphoma has risen substantially. The exact reason for this increase remains a mystery; however, a modest portion of the increase has been attributed to lymphomas developing in association with immune deficiencies, including AIDS and organ transplants. Conversely, the incidence of Hodgkin’s lymphoma has declined over the same time period, especially among the elderly.
Non-Hodgkin’s lymphomas
The previously used generic classification of non-Hodgkin’s lymphoma (NHL) has been reclassified into B-cell neoplasms (which include a variety of lymphomas including myelomas that originate from B cells at various stages of differentiation) and T-cell and natural killer-cell (NK-cell) neoplasms. Because malignant changes can occur at various stages of B-cell, T-cell or NK-cell development, these cancers present with a variety of clinical states. In the following section, the types of tumours previously classified as non-Hodgkin’s lymphoma are considered together, and myeloma is described separately.
PATHOPHYSIOLOGY
As with all cancers, lymphomas most likely originate from mutations in cellular genes (many of which are environmentally induced) in a single cell that lead to loss of control of proliferation and other aspects of cell growth. The most common type of chromosomal alteration is translocation, which disrupts the genes encoded at the breakpoints. Risk factors include a family history, exposure to a variety of mutagenic chemicals, irradiation, infection with certain cancer-related viruses (e.g. Epstein-Barr virus, human herpes virus-8, HIV, hepatitis C) and immune suppression related to organ transplantation. Gastric infection with Helicobacter pylori increases the risk for gastric lymphomas. NHL is a disease of middle age, usually found in those over 50 years old. The reported incidence of NHL in Australia in 2005 was 18.4 per 100,000 persons (see Table 17-7).
Table 17-7 CLINICAL DIFFERENCES BETWEEN NON-HODGKIN’S LYMPHOMA AND HODGKIN’S LYMPHOMA
| CHARACTERISTICS | NON-HODGKIN’S LYMPHOMA | HODGKIN’S LYMPHOMA |
|---|---|---|
| Nodal involvement | Multiple peripheral nodes | Localised to single axial group of nodes (i.e. cervical, mediastinal, para-aortic) |
| Mesenteric nodes and Waldeyer’s ring commonly involved | Mesenteric nodes and Waldeyer’s ring rarely involved | |
| Spread | Noncontiguous | Orderly spread by contiguity |
| B symptoms* | Uncommon | Common |
| Extranodal involvement | Common | Rare |
| Extent of disease | Rarely localised | Often localised |
CLINICAL MANIFESTATIONS
Clinical manifestations of NHL usually start out as localised or generalised lymphadenopathy, similar to Hodgkin’s lymphoma. Differences in clinical features are noted in Table 17-7. The cervical, axillary, inguinal and femoral chains are the most commonly affected sites. Generally, the swelling is painless and the nodes have enlarged and transformed over a period of months or years. Other sites of involvement are the nasopharynx, gastrointestinal tract, bone, thyroid, testes and soft tissue. Some individuals have retroperitoneal and abdominal masses with symptoms of abdominal fullness, back pain, ascites (fluid in the peritoneal cavity) and leg swelling.
EVALUATION AND TREATMENT
Individuals with NHL can survive for extended periods. Survival with nodular lymphoma ranges up to 15 years. Individuals with diffuse disease generally do not survive as long. Overall, the survival rates for NHL are less than those for Hodgkin’s lymphoma. For NHL, the survival rates are: 1-year, 77%; 5-year, 59%; and 10-year, 42%. Many investigators think that more aggressive treatment increases the cure rate. High-grade NHL is seen with increasing frequency in those with AIDS and has an extremely poor prognosis.
Success of treatment is dependent on several parameters, including the type of lymphoma, stage of disease, cell type, involvement of organs outside the lymph nodes, age of the person and the severity of the body’s reaction to the disease (e.g. fever, night sweats, weight loss).21 Treatment with chemotherapy alone may be adequate in many cases, although radiation therapy is frequently included. Chemotherapy has been followed by autologous stem cell transplantation (where one’s own marrow is removed, stored and re-infused later) in some NHLs or for recurrent disease. Treatment of B-cell lymphomas with rituximab has proven effective. Rituximab is a commercial monoclonal antibody against antigen CD20, which is expressed on the surface of all B cells, including malignant ones. Administration of rituximab depletes most B cells and allows the replenishment of normal B cells from the lymphoid stem cell pool. It has also proven useful in a variety of autoimmune diseases, including immune thrombocytopenia purpura, autoimmune anaemia, systemic lupus erythematosus and rheumatoid arthritis.
PATHOPHYSIOLOGY
Hodgkin’s lymphoma is characterised by its progression from one group of lymph nodes to another, the development of systemic symptoms (see Figure 17-14) and the presence of Reed-Sternberg (RS) cells.22 It is widely accepted that RS cells represent the malignant transformation of lymph cells.23 RS cells are often large and binucleate, with occasional mononuclear variants. RS cells are necessary for the diagnosis of Hodgkin’s lymphoma; however, they are not specific to it. In rare instances, cells resembling them can be found in benign illnesses, as well as in other forms of cancer, including non-Hodgkin’s lymphomas, solid tissue cancers and infectious mononucleosis.

FIGURE 17-14 Hodgkin’s lymphoma and enlarged cervical lymph node.
Typical enlarged cervical lymph node in the neck of a woman with Hodgkin’s lymphoma.
Source: del Regato JA, Spjut HJ, Cox JD. Cancer: diagnosis, treatment, and prognosis. 6th edn. St Louis: Mosby; 1985.
The incidence of Hodgkin’s lymphoma in Australia is low (refer to Table 17-5). The disease peaks at two different times over the life span — during the second and third decades, and later during the sixth and seventh decades.
The triggering mechanism for the malignant transformation of cells remains unknown. Classical Hodgkin’s lymphoma appears to be derived from precursor B cells that have altered during development (see Chapter 21). Survival of this cell may be linked to infection with Epstein-Barr virus. Laboratory and epidemiological studies have linked Hodgkin’s lymphoma with Epstein-Barr virus infections, and Epstein-Barr virus DNA, RNA and proteins are frequently observed in HL cells.24 RS cells secrete and release cytokines (e.g. IL-10, TGF-β) that result in the accumulation of inflammatory cells that produce the local and systemic effects.
CLINICAL MANIFESTATIONS
Many clinical features of Hodgkin’s lymphoma can be explained by the complex action of cytokines and other growth factors that are secreted and released by the malignant cells. These substances induce infiltration and proliferation of inflammatory cells, resulting in an enlarged, painless lymph node in the neck (often the first sign of Hodgkin’s lymphoma; see Figure 17-14). The discovery of an asymptomatic mediastinal mass on routine chest X-ray is not uncommon. The cervical, axillary, inguinal and retroperitoneal lymph nodes are commonly affected (see Figure 17-15). Local symptoms caused by pressure and obstruction of the lymph nodes are the result of the lymphadenopathy.
About one-third of individuals will have some degree of systemic symptoms. Intermittent fever, without other symptoms of infection, drenching night sweats, itchy skin (pruritus) and fatigue are relatively common. These constitutional symptoms accompanied by weight loss are associated with a poor prognosis. The Cotswold staging classification system used for Hodgkin’s lymphoma (also known as the Ann Arbor staging system) is able to establish a correlation between the anatomic extent of the disease and prognosis (see Table 17-8). This classification system is based on the individual’s medical history, examination (presence of symptoms and palpable lymph nodes) and other radiological and haematological results. Prognostic indicators include clinical stage, histological type, tumour cell concentration and tumour burden, constitutional symptoms and age.
Table 17-8 MODIFIED COTSWOLD STAGING CLASSIFICATION SYSTEM
| STAGE | CRITERIA |
|---|---|
| I | Involvement of a single lymph node region or single extranodal organ or site |
| II | Involvement of two or more lymph node regions on the same side of the diaphragm or a single extranodal organ or site and its regional lymph nodes |
| III | Involvement of lymph node regions or structures on both sides of the diaphragm |
| IV | Disseminated involvement of one or more extralymphatic organs or an isolated extralymphatic organ with distant nodal involvement |
Modifying characteristics for all four stages
A: no B symptoms
B: unexplained fever of > 38°C, drenching night sweats, unexplained loss of > 10% of body weight in the 6 months preceding diagnosis
E: large mediastinal mass with direct extension into extranodal sites
Source: Lister TA, Crowther D. Staging for Hodgkin’s disease. Semin Oncol 1990; 17:696.
Although Hodgkin’s lymphoma rarely arises in the lung, adenopathy of nodes in the mediastinum and hilus can cause secondary involvement of the trachea, bronchi, pleura or lungs. Retroperitoneal nodes can involve vertebral bodies and nerves and can also cause displacement of the ureters. Spinal cord involvement is more common in the dorsal and lumbar regions than in the cervical region. Skin lesions, although uncommon, include psoriasis and eczematoid lesions, causing itching and scratching.
As a result of direct invasion from mediastinal lymph nodes, pericardial involvement can cause pericardial friction rub, pericardial effusion and engorgement of neck veins. The gastrointestinal tract and urinary tract are rarely involved. Anaemia is often found in individuals with Hodgkin’s lymphoma accompanied by a low serum iron and iron-binding capacity. Other laboratory findings include elevated erythrocyte sedimentation rate (ESR), leucocytosis and eosinophilia. Leucopenia occurs in advanced stages of Hodgkin’s lymphoma.
Splenic involvement in Hodgkin’s lymphoma depends on histological type. In mixed cellularity and lymphocytic deletion types of Hodgkin’s lymphoma, the spleen is involved in 60% of cases. With lymphocyte and nodular sclerosis types, 34% of cases involve the spleen.
EVALUATION AND TREATMENT
Because of the variability in symptoms, early definitive detection may be difficult. Asymptomatic lymphadenopathy can progress undetected for several years. Careful evaluation, including chest X-ray films, positron emission tomography (PET) scans and biopsy, should be carried out for individuals with fever of unknown origin and peripheral lymphadenopathy. A lymph node biopsy with scattered RS cells and a cellular infiltrate is highly indicative of Hodgkin’s lymphoma. The effectiveness of treatment is related to the age of the individual and the extent of the disease. Approximately 75% of individuals diagnosed with Hodgkin’s lymphoma can be cured, largely because of successful treatment with irradiation and chemotherapy.
Those with stage III or IV disease, bulky disease (> 10 cm mass or mediastinal disease with a transverse diameter exceeding 33% of the transthoracic diameter) or presence of B symptoms require combined chemotherapy with or without additional radiation treatment. Those with stage I or II disease are candidates for chemotherapy, combined or radiation therapy alone. The survival rate depends on many factors, including the age of the individual, the stage of the disease, gender and other variables. The 5-year survival rate with no additional factors is about 85%, but drops precipitously with each additional factor to about 42% with five or more factors.
Multiple myeloma
Multiple myeloma is a B-cell cancer characterised by the proliferation of malignant plasma cells that infiltrate the bone marrow and aggregate into tumour masses throughout the skeletal system.25 The reported incidence of multiple myeloma has doubled in the past 2 decades, possibly as a result of more sensitive testing used for diagnosis. In Australia, multiple myeloma accounts for 1.2% of all cancers (see Table 17-5). It rarely occurs before the age of 40 years — the peak age of incidence is about 65 years. It is slightly more common in men than women.
Neoplastic cells of multiple myeloma reside in the bone marrow and are usually not found in the peripheral blood. Occasionally, however, it may spread to other tissues, especially in very advanced disease. The basic defect is genetic, which may result from chronic stimulation of B cells with bacterial or viral antigens.
PATHOPHYSIOLOGY
Most, if not all, multiple myelomas involve chromosomal translocations (break points), which recur in many individuals. In about half of all cases, one of the chromosomal partners is 14 (the site of genes for the immunoglobulin heavy chain), which recombines with a number of other chromosomal sites of oncogenes, most commonly located on chromosomes 11, 4, 16, 20 and 6, resulting in probable dysregulation of the oncogenes (refer to Chapter 37). Deletions in chromosome 13 are observed in about 50% of cases. The molecular pathogenesis of multiple myeloma also involves proto-oncogene mutations and, more rarely, inactivation of tumour-suppressor genes. The precise timing and reason for the genetic alteration and accumulation is unknown.
Malignant plasma cells arise from one clone of B cells that produce abnormally large amounts of one class of immunoglobulin (usually IgG, occasionally IgA and rarely IgM, IgD or IgE). The malignant transformation may begin early in B cell development, possibly before encountering antigen in the secondary lymphoid organs. The myeloma cells return to either the bone marrow or other soft tissue sites. Their return is aided by cell adhesion molecules that help them to target favourable sites that promote continued expansion and maturation. Cytokines, particularly interleukin-6 (IL-6), have been identified as essential factors that promote the growth and survival of multiple myeloma cells. (Lymphocytes and cytokines are described in Chapter 12.)
Myeloma cells in the bone marrow produce several cytokines themselves (e.g. IL-6, IL-1, TNFα). IL-6 in particular acts as an osteoclast-activating factor and stimulates osteoclasts to resorb (break down and dissolve) bone, which releases calcium in the blood. This process results in bone lesions and hypercalcaemia (high calcium levels in the blood).
The antibody produced by the transformed plasma cell is frequently defective, containing truncations, deletions and other abnormalities, and is frequently referred to as a paraprotein (abnormal protein in the blood). Because of the large number of malignant plasma cells, the abnormal antibody, called the M protein, becomes the most prominent protein in the blood and is readily detected by laboratory tests. Suppression of normal plasma cells by the myeloma results in diminished or absent normal antibodies. The excessive amount of M protein may also contribute to many of the clinical manifestations of the disease. If the myeloma produces IgM, the excessive amount of large-molecular-weight proteins (about 900,000 daltons) can lead to abnormally high blood viscosity (hyperviscosity syndrome). Frequently, the myeloma produces free immunoglobulin light chain (Bence Jones protein) that is present in the blood and urine and contributes to damage of renal tubular cells.
CLINICAL MANIFESTATIONS
The common presentation of multiple myeloma is characterised by elevated levels of calcium in the blood (hypercalcaemia), renal failure, anaemia and bone lesions. The hypercalcaemia and bone lesions result from infiltration of the bone by malignant plasma cells and stimulation of osteoclasts to reabsorb bone. This process results in the release of calcium (hypercalcaemia) and development of ‘lytic lesions’ (round, ‘punched out’ regions of bone). Destruction of bone tissue causes pain, the most common presenting symptom, and pathological fractures. The bones most commonly involved, in decreasing order of frequency, are the vertebrae, ribs, skull, pelvis, femur, clavicle and scapula. Spinal cord compression, because of the weakened vertebrae, occurs in about 10% of individuals.
Proteinuria (protein present in the urine) is observed in 90% of individuals. Renal failure may be either acute or chronic and is usually secondary to the hypercalcaemia. Bence Jones protein is present in about 80% of individuals and may also lead to damage of the proximal tubules. Anaemia is usually normocytic and normochromic and results from inhibited erythropoiesis caused by tumour cell infiltration of the bone marrow.
Suppression of the humoral (antibody-mediated) immune response results in repeated infections, primarily pneumonias and pyelonephritis. The most commonly involved organisms are encapsulated bacteria that are particularly sensitive to the effects of antibody; pneumonia caused by S. pneumoniae, S. aureus or K. pneumoniae, or pyelonephritis caused by E. coli or other gram-negative organisms. Cell-mediated (T cell) function is relatively normal. Overwhelming infection is the leading cause of death from multiple myeloma.
EVALUATION AND TREATMENT
Diagnosis of multiple myeloma is made by symptoms, radiographic and laboratory studies and a bone marrow biopsy. Quantitative measurements of immunoglobulins (IgG, IgM, IgA) are usually performed. Typically, one class of immunoglobulin (the M protein produced by the myeloma cell) is greatly increased, while the others are suppressed. Laboratory analysis of serum reveals increased levels of M protein, and Bence Jones protein can be detected in the urine or serum. Usually M protein coexists with Bence Jones protein. However, variants of multiple myeloma exist where only parts of the immunoglobulin molecule are present (either light chain or heavy chain). Measurement of another protein, free β2-microglobulin, is used as an indicator of prognosis or effectiveness of therapy.
Although chemotherapy, radiation therapy and marrow transplant have been used for treatment, the prognosis for those with multiple myeloma remains poor. Autologous peripheral blood stem cell transplantation is preferred to bone marrow transplantation. Individuals with multiple bone lesions, if untreated, rarely survive more than 6–12 months. Individuals with inactive (indolent) myeloma, however, can survive for many years. With chemotherapy and aggressive management of complications, the prognosis can improve significantly, with a median survival of 24–30 months and a 10-year survival rate of 3%. The median survival for all states of multiple myeloma is 3 years.
A recent addition to treatment of multiple myeloma in individuals who have a relapse after conventional chemotherapy is the drug thalidomide. The use of thalidomide is based on its suppression of TNFα and its anti-angiogenesis ability.
Alterations of erythrocyte function
The most common classification of anaemia is based on changes in the cell size (MCV) and changes in the cell’s haemoglobin content (MCH). Clinical manifestations of anaemia can be found in all organs and tissues throughout the body. Decreased oxygen delivery to tissues causes fatigue, dyspnoea, syncope, angina, compensatory tachycardia and organ dysfunction. Macrocytic (megaloblastic) anaemia is caused by deficiency of vitamin B12 or folate. Pernicious anaemia can be fatal unless vitamin B12 replacement is given. Microcytic-hypochromic anaemia is characterised by abnormally small red cells with insufficient haemoglobin content. The most common cause is iron deficiency. Iron deficiency anaemia usually develops slowly, with a gradual insidious onset of symptoms, including fatigue, weakness, dyspnoea, alteration of various epithelial tissues and vague neuromuscular complaints. Iron deficiency anaemia is the most common blood disorder of infancy and childhood; the highest incidence occurs between 6 months and 2 years of age. Iron deficiency anaemia is usually a result of a chronic blood loss or decreased iron intake. Once the source of blood loss is identified and corrected, iron replacement therapy can be initiated. Normocytic-normochromic anaemia is characterised by insufficient numbers of normal erythrocytes. Included in this category are aplastic, post-haemorrhagic and haemolytic anaemia and anaemia of chronic inflammation. Haemolytic disease of the newborn (HDN) results from incompatibility between the maternal and fetal blood, which may involve differences in Rh antigens or ABO blood type. Maternal antibodies enter the fetal circulation and cause haemolysis of fetal erythrocytes. Because the immature liver of the newborn is unable to conjugate and excrete the excess bilirubin that results from the haemolysis, icterus neonatorum or kernicterus, or both, can develop. Kernicterus causes brain damage and can be fatal. Polycythaemia vera is characterised by excessive proliferation of erythrocyte precursors in the bone marrow. Signs and symptoms result directly from increased blood volume and viscosity. Therapeutic phlebotomy to remove excessive blood volume and use of radioactive phosphorus have been helpful in decreasing the excessive red cell pool.Alterations of platelets and coagulation
Thrombocytopenia is characterised by a platelet count below 100 × 109/L of blood; a count below 50 × 109/L increases the potential for haemorrhage associated with minor trauma. Thrombocytopenia exists in primary or secondary forms and is commonly associated with autoimmune diseases and viral infections; bacterial sepsis with disseminated intravascular coagulation also results in thrombocytopenia. Qualitative alterations in normal platelet adherence or aggregation prevent platelet plug formation and may result in prolonged bleeding times. Disorders of coagulation are usually caused by defects or deficiencies of one or more clotting factors. Haemophilia A is the most common of the inherited coagulation factor deficiencies. Because it is an X-linked disorder it mainly affects males; females are carriers. Coagulation is impaired when there is a deficiency of vitamin K because of insufficient production of prothrombin and synthesis of clotting factors II, VII, IX and X, often associated with liver diseases. Disseminated intravascular coagulation (DIC) is a complex syndrome resulting from a variety of clinical conditions that release tissue factor, causing an increase in fibrin and thrombin activity in the blood producing augmented clot formation and accelerated fibrinolysis. Sepsis is a condition that is often associated with DIC. DIC is characterised by a cycle of intravascular clotting followed by active bleeding caused by the initial consumption of coagulation factors and platelets and diffuse fibrinolysis. Diagnosis of DIC is based on measurement in the blood of end products characteristic of dysfunctional coagulation activity. Treatment is complex and nonstandardised and focused on removing the primary cause, restoring haemostasis and preventing further organ damage. Thromboembolic disease results from a fixed (thrombus) or moving (embolus) clot that blocks flow within a vessel, denying nutrients to tissues distal to the occlusion; death can result when clots obstruct blood flow to the heart, brain or lungs.Alterations of leucocyte function
Quantitative alterations of leucocytes (too many or too few) can be caused by bone marrow dysfunction or premature destruction of cells in the circulation. Many quantitative changes in leucocytes occur in response to invasion by microorganisms. Leucocytosis is a condition in which the leucocyte count is higher than normal and is usually a response to stress and invasion of microorganisms. Leucopenia is a condition in which the leucocyte count is lower than normal and is caused by pathological conditions, such as malignancies, and haematological disorders. Granulocytosis (particularly as a result of an increase in neutrophils) occurs in response to infection. The marrow releases immature cells when responding to an infection that has created a demand for neutrophils that exceeds the supply in the circulation. Eosinophilia results most commonly from parasitic invasion and ingestion or inhalation of toxic foreign particles. Basophilia is seen in hypersensitivity reactions because of the high content of histamine and subsequent release. Monocytosis occurs during the late or recuperative phase of infection when macrophages (mature monocytes) phagocytose surviving microorganisms and debris. Granulocytopenia, a significant decrease in neutrophils, can be a life-threatening condition if sepsis occurs; it is often caused by chemotherapeutic agents, severe infection and radiation. Infectious mononucleosis is an acute infection of B lymphocytes most commonly associated with the Epstein-Barr virus, a type of herpes virus. Transmission of Epstein-Barr virus is through close personal contact, commonly through saliva — thus its nickname, the kissing disease. Two of the earliest manifestations of infectious mononucleosis are sore throat and fever caused by inflammation at the primary site of viral entry. Most causes of Epstein-Barr virus infectious mononucleosis include fever lasting 7–10 days, sore throat and enlargement and tenderness of the cervical lymph nodes. It is self-limiting and treatment consists of rest and symptomatic treatment. The common pathological feature of all forms of leukaemia is an uncontrolled proliferation of leucocytes, overcrowding the bone marrow and resulting in decreased production and function of the other blood cell lines. Leukaemia is classified by the cell type involved — lymphoid or myeloid — and is differentiated by onset — acute or chronic. Thus, there are four major types of leukaemia: acute lymphocytic leukaemia (ALL), chronic lymphocytic leukaemia (CLL), acute myeloid leukaemia (AML) and chronic myeloid leukaemia (CML). Although the exact cause of leukaemia is unknown, it is considered a clonal disorder. A high incidence of acute leukaemia and CLL is reported in certain families, suggesting a genetic predisposition. The major clinical manifestation of leukaemia includes fatigue caused by anaemia, bleeding caused by thrombocytopenia, fever secondary to infection, anorexia and weight loss. Chemotherapy is the treatment of choice for leukaemia. Acute leukaemia is associated with an increasing survival rate of 80–90%, with long-term survival of 30–40%. Chronic leukaemia is associated with a longer life expectancy than acute leukaemia.Alterations of lymphoid function
The number of lymphocytes is decreased (lymphocytopenia) in most acute infections and in some immunodeficiency syndromes. Lymphocytosis occurs in viral infections (infectious mononucleosis and infectious hepatitis, in particular), leukaemia, lymphomas and some chronic infections. Lymphomas are tumours of primary lymphoid tissue (thymus, bone marrow) or secondary lymphoid tissue (lymph nodes, spleen, tonsils, intestinal lymphoid tissue). The two major types of malignant lymphomas are Hodgkin’s lymphoma and non-Hodgkin’s lymphoma. Distinctive abnormal chromosomes are present in multiple cells of the lymph nodes of an individual with Hodgkin’s lymphoma. The abnormal cell is called the Reed-Sternberg cell. A virus might be involved in the pathogenesis of Hodgkin’s lymphoma. Some familial clustering suggests an unknown genetic mechanism. An enlarged, painless mass or swelling, most commonly in the neck, is an initial sign of Hodgkin’s lymphoma. Local symptoms are produced by lymphadenopathy, usually caused by pressure or obstruction. Treatment of Hodgkin’s lymphoma includes radiation therapy and chemotherapy. A cure is possible regardless of the stage of Hodgkin’s lymphoma; however, individuals treated with chemotherapy who relapse in less than 2 years have a poor prognosis. The cause of lymph node enlargement and cancerous transformation in non-Hodgkin’s lymphoma is unknown. Immunosuppressed persons have a higher incidence of non-Hodgkin’s lymphoma, suggesting an immune mechanism. Generally, with non-Hodgkin’s lymphoma, the swelling of lymph nodes is painless and the nodes enlarge and transform over a period of months or years. Individuals with non-Hodgkin’s lymphoma can survive for long periods. The treatment used is chemotherapy. Multiple myeloma is a neoplasm of B cells (immature plasma cells) and mature plasma cells. It is characterised by multiple malignant tumour masses of plasma cells scattered throughout the skeletal system and sometimes found in soft tissue. The exact cause of multiple myeloma is unknown, but genetic factors and chronic stimulation of the mononuclear phagocyte system by bacteria, viral agents and chemicals have been suggested.Matthew is a 28-year-old chemical engineer. He has been feeling unwell for the past two weeks and has gone to see his doctor. He presents with fatigue, fever, night sweats and bruising. Matthew’s doctor decides to request a full blood count. The blood sample is sent to a haematology laboratory — the laboratory report that comes back shows that Matthew has normocytic-normochromic anaemia, marked leucocytosis and marked thrombocytopenia. A blood film was made from Matthew’s sample and examined microscopically. The report states that numerous blast cells were present in the blood film (50% of the white cells in Matthew’s blood film are blast cells).