Chapter 17 Drug therapy and poisoning
Drug therapy
The patient
The prerequisite of any form of therapeutic intervention is a reliable diagnosis or, at least, an assessment of clinical need. An accurate diagnosis ensures that a patient is not exposed, unnecessarily, to the hazards or costs of a particular intervention. Nevertheless, there are some circumstances when treatment is used in the absence of a clear diagnosis, for example:
 the symptomatic treatment of severe pain
the symptomatic treatment of severe pain
the initiation of ‘blind’ antimicrobial therapy where delay would expose a patient to hazard or discomfort (e.g. antimicrobial therapy for a patient with a suspected lower urinary tract infection).
In some instances a particular medicine is only effective in subgroups of patients who have a particular disorder. Trastuzumab, for example, is only of value in women with breast cancer whose malignant cells express the HER2 epidermal growth factor receptor. Tailoring treatment, depending on an individual’s specific genetic characteristics or gene expression, is increasingly used. This promising approach approach has become known as ‘personalized medicine’.
Medicines are also given to otherwise healthy individuals. In such circumstances there must be a very clear imperative to ensure that the benefits to the individual outweigh the harm. Examples include:
Immunization against serious microbial infections (e.g. influenza vaccination)
The reduction of individual risk factors to prevent later disease (e.g. the use of antihypertensive, or lipid-lowering, agents, to reduce the chances of ischaemic heart disease and stroke)
Oral contraceptives in sexually active women wishing to avoid pregnancy.
Co-morbidity may also significantly alter the way in which conditions are treated particularly in the elderly. Some examples are shown in Table 17.1.
Table 17.1 Examples of drugs to be avoided in people with co-morbidity
| Co-morbidity | Avoid | Effect |
|---|---|---|
Parkinson’s disease |
Neuroleptics |
Exacerbates Parkinsonian symptoms (including tremor) |
Hypertension |
Non-steroidal anti-inflammatory drugs |
Sodium retention |
Asthma |
Beta-blockers, adenosine |
Bronchospasm |
Respiratory failure |
Morphine, diamorphine |
Respiratory depression |
Renovascular disease |
ACE inhibitors/antagonists |
Reduction in glomerular filtration |
Chronic heart failure |
Trastuzumab |
Worsening of heart failure |
Chronic infections (e.g. tuberculosis, hepatitis C, histoplasmosis |
Cytokine modulators (e.g. etanercept) |
Increased risk of exacerbation |
Prescribing in neonates, infants, children and adolescents
The use of drugs in young people poses special problems. Extrapolating from adult dosage regimens, merely adjusting for weight, leads to excessive (and potentially toxic) doses because:
The rates of hepatic metabolism and renal excretion of drugs are reduced in neonates and infants.
Premature babies have approximately 1% of their body weight as fat (compared with 20% in adults), leading to a marked increase in plasma drug levels of fat-soluble drugs.
There are other difficulties in prescribing for children:
Many treatments have never been subject to formal trials in either children or adolescents and their benefits and risks have not, therefore, been appropriately assessed in these age groups. Efforts are being made, internationally, to redress this.
For many drugs, there are no paediatric preparations or formulations. Instead, adult products are used.
Precise oral dosing is often impossible in babies who spit out unpleasant-tasting products!
Adverse effect profiles of medicines may be different in children compared with adults (e.g. Reye’s syndrome in children given aspirin, suicidal ideas in depressed adolescents treated with selective serotonin reuptake inhibitors).
Prescribing for the elderly
The use of drugs in the elderly is often a problem:
Rates of hepatic drug metabolism and renal excretion decline with age. Extrapolation of drug dosages, from those appropriate in younger adults, may therefore lead to toxic plasma levels.
Changes in drug distribution due to changes in body composition, and the preferential distribution of the cardiac output to the brain, may also predispose to toxicity.
Co-morbidity, often associated with polypharmacy, leads to increased opportunities for disease–drug and drug–drug interactions.
Concordance with treatment regimens diminishes as the number of prescribed drugs increases, and is especially poor in the face of cognitive impairment.
Exaggerated pharmacodynamic effects of drugs acting on the central nervous, cardiovascular and gastrointestinal systems are common.
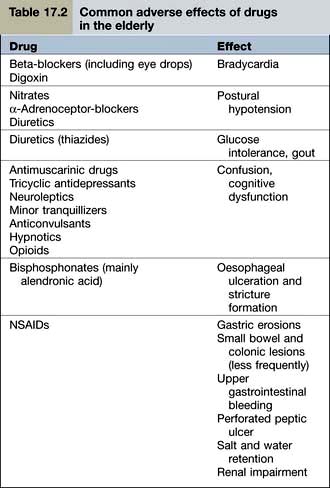
Examples of common problems encountered in the use of drugs amongst older people are shown in Table 17.2.
Drug use in pregnancy
Clinicians should be extremely cautious about prescribing drugs to pregnant women, and only essential treatments should be given. When a known teratogen is needed during pregnancy (e.g. an anticonvulsant drug or lithium), the potential adverse effects should be discussed with the parents, preferably before conception. If parents wish to go ahead with the pregnancy, they should be offered an appropriate ultrasound scan to assess whether there is any fetal damage. Some known human teratogens are shown in Table 17.3.
Table 17.3 Some human teratogens
| Drug | Effect |
|---|---|
ACE inhibitors/antagonists |
Oligohydramnios |
Retinoids, e.g. acitretin |
Multiple abnormalities |
Carbimazole |
Neonatal hypothyroidism |
|
Abnormalities of bone growth |
Antiepileptics |
|
Carbamazepine |
Cleft palate |
Lamotrigine |
|
Phenytoin |
|
Valproate |
Neural tube defects |
NSAIDs |
Delayed closure of the ductus arteriosus |
Cytotoxic drugs |
Most are presumed teratogens |
Lithium |
Ebstein’s anomaly |
Misoprostol |
Moebius’s syndrome |
Thalidomide (and possibly lenalidomide) |
Phocomelia |
Note: All drugs should be avoided in pregnancy unless benefit clearly outweighs the risk.
Breast-feeding
Although most drugs can be detected in breast milk, the quantity is generally small. This is because, for most drugs, the concentration in milk is in equilibrium with plasma water (i.e. the non-protein-bound fraction). A few drugs (e.g. aspirin, carbimazole) may, however, cause harm to the infant if ingested in breast milk. Relevant drug literature should be consulted when prescribing for nursing mothers.
The drug
Selecting the right drug involves three elements:
The drug’s clinical efficacy for the proposed use
The most common approach to assessing a drug’s efficacy is the randomized controlled trial (RCT), although other approaches (see p. 907) can be informative. The demonstration of absolute efficacy (against placebo) may, itself, be insufficient. Where there is more than one treatment for the same indication these should be compared with one another, taking account of the magnitude of their benefits, their individual adverse reaction profiles, and their costs.
Direct comparisons of one treatment versus another are particularly useful but are often unavailable. So-called indirect techniques, which involve comparing each drug against placebo and then imputing the comparison, are commonly used.
Patients’ own preferences should be discussed to enable them to be equal partners in decision-making about whether, and how, they wish to be treated. Moreover, a full understanding of the reasons for considering treatment, the likely benefits and the possible adverse reactions, has repeatedly been shown to improve ‘concordance’ with treatment regimens.
The dose
Appropriate drug dosages will have usually been determined from the results of so-called ‘dose-ranging’ studies during the original development programme. Such studies are generally conducted as RCTs covering a range of potential doses. Drug doses and dosage regimens may be fixed or adjusted.
Fixed dosage regimens
Drugs suitable (in adults) for prescribing at the same ‘fixed’ dose, for all patients, share common features. Efficacy is optimal in virtually all patients; and the risks of dose-related (type A) adverse reactions (see p. 904) are normally low. These drugs have a high ‘therapeutic ratio’ (i.e. the ratio between toxic and therapeutic doses). Examples of drugs prescribed at a fixed dose are shown in Table 17.4.
Table 17.4 Examples of fixed dose prescribing
| Drug | Indication |
|---|---|
Aspirin |
Secondary prevention of myocardial infarction |
Clopidogrel |
|
Bendroflumethiazide |
Hypertension |
Broad spectrum penicillins |
Lower urinary tract infection |
Cephalosporins |
|
Macrolides |
Upper and lower respiratory tract infection |
Levonorgestrel |
Emergency contraception |
Ulipristal |
|
Oestrogen antagonists |
Secondary prevention of breast cancer |
Aromatase inhibitors |
|
Vaccines |
e.g. Diphtheria, pertussis, mumps, measles, rubella, influenza, etc. |
Titrated dosage regimens
For many drugs, there are wide interindividual variations in response. As a consequence, whilst a particular dose may in one person lack any therapeutic effect, the same dose in another may cause serious toxicity. The reasons for such variability are partly due to pharmacokinetic factors (differences in the rates of drug absorption, distribution or metabolism) and partly due to pharmacodynamic factors (differences in the sensitivity of target organs).
Pharmacokinetics
Pharmacokinetics is the study of what the body does to a drug. The intensity of a drug’s action, immediately after parenteral administration, is largely a function of its volume of distribution. This, in turn, is predominantly governed by body composition and regional blood flow. Dosage adjustments, for body weight or surface area, are therefore common (e.g. in cancer chemotherapy) in order to optimize treatment.
The main determinants of a drug’s plasma concentration after oral administration are its bioavailability (the proportion of the unchanged drug that reaches the systemic circulation) and its rate of systemic clearance (by hepatic metabolism or renal excretion). A drug’s oral bioavailability depends on the extent to which it is:
destroyed in the gastrointestinal tract
able to cross the gastrointestinal epithelium
metabolized by the liver before reaching the systemic circulation (so-called presystemic or ‘first pass’ metabolism). First pass metabolism can be avoided by the intravascular (i.v.), intramuscular (i.m.) or sublingual routes.
Liver drug metabolism occurs in two stages:
Phase I is the modification of a drug, by oxidation, reduction or hydrolysis. Of these, oxidation is the most frequent route and is largely undertaken by a family of isoenzymes known as the cytochrome P450 system (see p. 902). Inhibition or induction of cytochrome P450 isoenzymes are major causes of drug interactions (Table 17.5).
Phase II involves conjugation with glucuronate, sulphate, acetate or other substances to render the drug more water soluble and therefore able to be excreted in the urine.
Table 17.5 Some inducers and inhibitors of cytochrome P450
Inducers |
Carbamazepine |
Hyperforina |
|
Nifedipine |
|
Non-nucleoside reverse transcriptase inhibitors (NNRTIs) |
|
Omeprazole |
|
Phenobarbital |
|
Phenytoin |
|
Rifampicin |
|
Ritonavir (see p. 180) |
|
Inhibitors |
Allopurinol |
Amiodarone |
|
Cimetidine |
|
Erythromycin, clarithromycin |
|
Fluoxetine, paroxetine |
|
Grapefruit juice (contains flavonoids) |
|
Imidazoles |
|
Quinolones |
|
Saquinavir |
|
Sulphonamides |
a Hyperforin is one of the ingredients of the herbal product known as St John’s wort used by herbalists to treat depression. Although it is marketed as a licensed medicine, it is a reminder that drug interactions can occur with alternative, as well as conventional, medicines.
Genetic causes of altered pharmacokinetics
Both presystemic hepatic metabolism, and the rate of systemic hepatic clearance, may vary markedly between healthy individuals.
Variability in the genes that encode drug-metabolizing enzymes (Table 17.6) is a major determinant of the inter-individual differences in the therapeutic and adverse responses to drug treatment. The most common involve polymorphisms of the cytochrome P450 family of enzymes, CYP. The first to be discovered was the polymorphism in the hydroxylation of the antihypertensive agent debrisoquin (CYP2D6). Defective catabolism was shown to be a monogenetically inherited trait, involving 5–10% of Caucasian populations, and leading to an exaggerated hypotensive response.
Table 17.6 Some genetic polymorphisms involving drug metabolism
| Enzyme | Drug |
|---|---|
P450 |
|
Cytochrome CYP1A2 |
Amitriptyline |
Clozapine |
|
Cytochrome CYP3A4 |
Amlodipine |
Ciclosporin |
|
Nifedipine |
|
Sildenafil |
|
Simvastatin |
|
Protease inhibitors |
|
Tacrolimus |
|
Cytochrome CYP2C9 |
Warfarin |
Glipizide |
|
Losartan |
|
Phenytoin |
|
Cytochrome CYP2D6 |
Beta-blockers |
Codeine |
|
Risperidone |
|
SSRIs |
|
Tramadol |
|
Venlafaxine |
|
Cytochrome CYP2C19 |
Clopidogrela |
Cyclophosphamide |
|
Diazepam |
|
Lansoprazole |
|
Omeprazole |
|
Plasma pseudocholinesterase |
Succinylcholine |
Mivacurium |
|
Thiopurine methyltransferase |
Azathioprine |
Mercaptopurine |
|
UDP-glucuronosyl transferase |
Irinotecan |
N-acetyl transferase |
Isoniazid |
CYP, cytochrome; SSRIs, Selective serotonin reuptake inhibitors.
a Clopidogrel is a prodrug and impaired metabolizers have a reduced response.
A substantial number of other drugs – estimated at 15–25% of all medicines in use – are substrates for CYP2D6. The frequencies of the variant alleles show racial variation and a small proportion of individuals may have two or more copies of the active gene. The phenotypic consequences of the defective CYP2D6 include the increased risk of toxicity with those antidepressants or antipsychotics that undergo metabolism by this pathway. Conversely, in individuals with multiple copies of the active gene, there are extremely rapid rates of metabolism and therapeutic failure at conventional doses.
Warfarin is predominantly metabolized by CYP2C9. In most populations, between 2% and 10% are homozygous for an allele that results in low enzyme activity. Such individuals will therefore metabolize warfarin more slowly leading to higher plasma levels, a greater risk of bleeding, and a requirement for lower doses if the international normalized ratio (INR) is to be maintained within the therapeutic range.
Cytochrome P450 is inhibited by the proton pump inhibitors but the consensus view is that co-prescribing with clopidogrel does not cause a significant increase in cardiovascular risk.
Individual differences in the activity of thiopurine methyltransferase (TPMT) determine the doses of mercaptopurine and azathioprine that are used. TMPT activity is therefore undertaken routinely in children undergoing treatment for acute lymphatic leukaemia and people with Crohn’s disease (see p. 233).
Many drugs undergo metabolism by more than one member of the cytochrome P450 family. Individuals deficient in one enzyme may have normal, or over-expressed, activities of others. Current knowledge (and cost) does not therefore permit predictions of an individual’s dosage requirements for the wide range of drugs for which polymorphisms in metabolism have been identified.
This may, however, become possible in the future, and would contribute – in part – to the prospect of ‘personalized prescribing’ (see p. 899).
Other causes of altered pharmacokinetics
Rates of hepatic drug clearance can also be influenced by environmental factors including diet, alcohol consumption and concomitant therapy with drugs capable of inducing or inhibiting (Table 17.5) drug metabolism. Hepatic drug clearance also decreases with age. By contrast, renal drug clearance does not show substantial variation between healthy individuals although it declines with age and in people with intrinsic renal disease.
Pharmacodynamics
Pharmacodynamics is the study of what the drug does to the body. Pharmacodynamic sources of variability in the intensity of drug action are at least partly due to drug receptor polymorphisms (Table 17.7). At present, the pharmacodynamic tests used in clinical practice to target therapy are largely confined to the expression of:
oestrogen and HER2 receptors in women with breast cancer (to determine, respectively, responsiveness to anti-oestrogens and trastuzumab)
epidermal growth factor receptor in lung cancer and glioblastomas (to predict responsiveness to gefitinib).
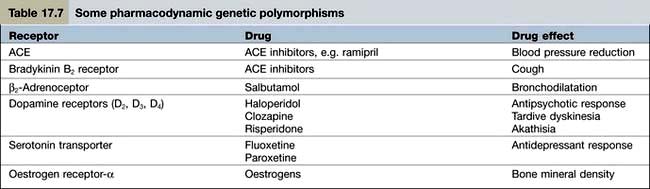
The prospect for ‘personalized prescribing’ will be enhanced further, when pharmacodynamic polymorphisms can be elicited by gene scanning. The interplay between pharmacokinetics and pharmacodynamics will then permit drug selection and dosing to become much more precise.
Monitoring the effects of treatment
The combination of pharmacokinetic and pharmacodynamic causes of variability makes monitoring of the effects of treatment essential. Three approaches are used.
Pre-treatment dose selection
In patients who have known, or suspected, impaired renal function, it is usually possible to predict dose requirements from their serum creatinine concentrations. If treatment needs to be started before the serum creatinine concentration is available, in patients who have very advanced renal impairment, or if renal function is fluctuating, then treatment can be started with conventional doses but the prescriber should be prepared to make adjustments within 24 hours.
Measuring plasma drug concentrations
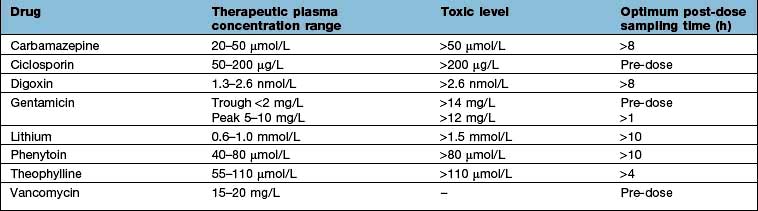
For a few drugs, dosages can be effectively monitored by reference to their plasma concentrations (Table 17.8). This technique is only useful, however, if both the following criteria are fulfilled:
Measuring drug effects
For many drugs, dosage adjustments are made in line with patients’ responses. Monitoring can involve dose titration against a therapeutic end-point or a toxic effect. Objective measures (such as monitoring antihypertensive therapy by measuring blood pressure, or cytotoxic therapy with serial white blood cell counts) are most helpful, but subjective ones are necessary in many instances (as with antipsychotic therapy in people with schizophrenia).
Affordability
The money available for healthcare varies widely across the world and there are marked differences (Fig. 17.1). All healthcare systems try to provide their populations with the highest standards of care within the resources they have at their disposal. The expenditure of large sums on a few people may deprive many of cost-effective remedies – a phenomenon known as the ‘opportunity cost’. The differences in healthcare expenditure shown in Figure 17.1 can be very largely accounted for by their differences in national wealth as reflected by their gross domestic products.
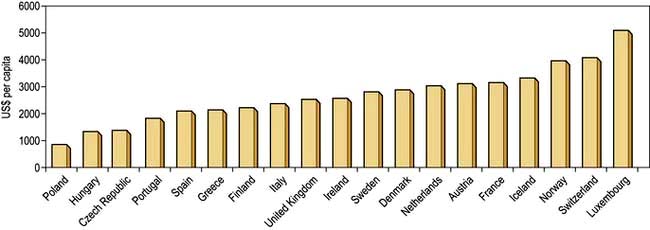
Figure 17.1 Annual expenditure on healthcare, as US$ per head of the population, in some developed countries.
(Source: OECD; http://www.oecd.org/document/4/0,3746,en_2649_37407_35101892_1_1_1_37407,00.html.)
In many countries cost-containment measures are encouraged (or mandated). For example, to reduce costs, all drugs should be prescribed by their generic (approved) names rather than their ‘brand’ ones because, once their patents have expired, generics products are cheaper. Despite occasional claims to the contrary, generic products are required to go through the same stringent regulatory processes as their branded counterparts.
Some countries, including Australia, Canada and Britain, assess the cost-effectiveness of new drugs (value for money) before they are available under their publicly funded healthcare systems.
Adverse drug reactions
Adverse drug reactions (ADRs), defined as ‘the unwanted effects of drugs occurring under normal conditions of use’, are a significant cause of morbidity and mortality. Around 5% of acute medical emergencies are admitted with ADRs, and around 10–20% of hospital inpatients suffer an ADR during their stay. Unwanted effects of drugs are five to six times more likely in the elderly than in young adults; and the risk of an ADR rises sharply with the number of drugs administered.
Classification
Two types of ADR are recognized.
Type A (augmented) reactions (Table 17.9) are:
qualitatively normal, but quantitatively abnormal, manifestations of a drug’s pharmacological or toxicological properties
predictable from its known pharmacological or toxicological actions
Table 17.9 Examples of adverse drug reactions
| Type of reaction and drug | Adverse reaction |
|---|---|
Type A (augmented) |
|
ACE inhibitors |
Hypotension |
ACE antagonists |
Hypotension |
Anticoagulants |
Gastrointestinal bleeding |
Antipsychotics |
Acute dystonia/dyskinesia |
Cytotoxic agents |
Bone marrow dyscrasias |
Erythromycin |
Nausea, vomiting |
Glucocorticosteroids |
Hypoadrenalism |
Insulin |
Hypoglycaemia |
Tricyclic antidepressants |
Dry mouth |
Type B (bizarre) |
|
Benzylpenicillin |
Anaphylaxis |
Broad-spectrum penicillins |
Maculopapular rash |
Carbamazepine |
Toxic epidermal necrolysis |
Carbamazepine |
Hepatotoxicity |
Isotretinoin |
Depression |
ACE, angiotensin-converting enzyme; SSRIs, selective serotonin reuptake inhibitors.
a In children and adolescents.
Whilst some such reactions as hypotension with ACE inhibitors may occur after a single dose, others may develop only after months (pulmonary fibrosis with amiodarone) or years (second malignancies with anti-cancer drugs).
Type B (idiosyncratic) reactions (Table 17.9) have no resemblance to the recognized pharmacological or toxicological effects of the drug. They are:
Diagnosis
All ADRs mimic some naturally occurring disease and the distinction between an iatrogenic aetiology, and an event unrelated to the drug, is often difficult. Although some effects are obviously iatrogenic (e.g. acute anaphylaxis occurring a few minutes after intravenous penicillin), many are less so. There are six characteristics that can help distinguish an adverse reaction from an event due to some other cause:
Appropriate time interval. The time interval between the administration of a drug and the suspected adverse reaction should be appropriate. Acute anaphylaxis usually occurs within a few minutes of administration, whilst aplastic anaemia will only become apparent after a few weeks (because of the life-span of erythrocytes). Drug-induced malignancy, however, will take years to develop.
Nature of the reaction. Some conditions (maculopapular rashes, angio-oedema, fixed drug eruptions, toxic epidermal necrolysis) are so typically iatrogenic that an adverse drug reaction is very likely.
Plausibility. Where an event is a manifestation of the known pharmacological property of the drug, its recognition as a type A adverse drug reaction can be made (e.g. hypotension with an antihypertensive agent, or hypoglycaemia with an antidiabetic drug). Unless there have been previous reports in the literature, the recognition of type B reactions may be very difficult. The first cases of depression with isotretinoin, for example, were difficult to recognize as an ADR even though a causal association is now acknowledged.
Exclusion of other causes. In some instances, particularly suspected hepatotoxicity, an iatrogenic diagnosis can only be made after the exclusion of other causes of disease.
Results of laboratory tests. In a few instances, the diagnosis of an adverse reaction can be inferred from the plasma concentration (Table 17.8). Occasionally, an ADR produces diagnostic histopathological features. Examples include putative reactions involving the skin and liver.
Results of dechallenge and rechallenge. Failure of remission when the drug is withdrawn (i.e. ‘dechallenge’) is unlikely to be an ADR. The diagnostic reliability of dechallenge, however, is not absolute: if the ADR has caused irreversible organ damage (e.g. malignancy) then dechallenge will result in a false-negative response. Rechallenge, involving re-institution of the suspected drug to see if the event recurs, is often regarded as an absolute diagnostic test. This is, in many instances, correct but there are two caveats. First, it is rarely justifiable to subject a patient to further hazard. Second, some adverse drug reactions develop because of particular circumstances which may not necessarily be replicated on rechallenge (e.g. hypoglycaemia with an antidiabetic agent).
Management
As a general rule, type A reactions can usually be managed by a reduction in dosage whilst type B reactions almost invariably require the drug to be withdrawn (and never re-instituted).
Specific therapy is sometimes required for ADRs such as bleeding with warfarin (vitamin K), acute dystonias (benztropine) or acute anaphylaxis (see Emergency Box 3.1, p. 69).
FURTHER READING
Pirmohamed M. The applications of pharmacogenetics to prescribing: what is currently known? Clin Med 2009; 9:493–495.
Relling M, Giacomini KM. Pharmacogenetics. In: Brunton LL, Lazo JS, Parker KL, eds. Gilman & Goodman’s The Pharmacological Basis of Therapeutics, 11th edn. New York: McGraw-Hill; 2006:93–115.
Wilkinson GR. Drug metabolism and variability among patients in drug response. N Engl J Med 2005; 352:2211–2221.
Woodcock J, Lesko LJ. Pharmacogenetics – tailoring treatment for outliers. N Engl J Med 2009; 360:811–813.
Evidence-based medicine
There is general acceptance that clinical practice should, as far as possible, be based on evidence of benefit rather than theoretical speculation, anecdote or pronouncement.
One of the main applications of ‘evidence-based medicine’ is in therapeutics. Treatments should be introduced into, and used in, routine clinical care only if they have been demonstrated to be effective in appropriate studies. Three approaches are used:
Randomized controlled trials
In this type of study, people with a particular condition are allocated to one of two (and sometimes more) treatments randomly. At the end of the study, the outcomes in the groups are compared. The purpose of the randomized controlled trial is to minimize bias and confounding. In order to minimize patient bias, the patients themselves are generally unaware of their treatment allocations (a ‘single-blind’ trial); and in order to reduce doctor bias, treatment allocations are also withheld from the investigators (a ‘double-blind’ trial). To recruit sufficient numbers of patients, and to examine the effects of treatment in different settings, it is often necessary to conduct the trial at several locations (a ‘multicentre’ trial).
Randomized controlled trials are designed to assess whether one treatment is better than another (a ‘superiority’ trial); or whether one treatment is similar to another (an ‘equivalence’ trial).
In a superiority trial the study treatment is usually compared to placebo or to current standard practice.
In an equivalence trial the treatment under study is compared to another treatment for the same condition.
Although RCTs were originally introduced to investigate the efficacy of drugs, the methodology can be used for surgical (and other) procedures and medical devices.
There are a number of variants of the conventional randomized controlled trial including cross-over trials, cluster randomized controlled trials, inferiority trials and futility trials (see Further Reading).
Assessing randomized controlled trials
In assessing the relevance and reliability of an RCT a number of features need to be taken into account.
Randomization
In any RCT the method of randomization should be robust. In particular, the investigator should be unaware of which treatment a patient entering a trial will receive. This avoids selection bias.
Maintaining blindness
Although, ideally, in RCTs neither the investigator nor the patient is aware of the treatment allocation until the end of the study, this is not always possible. Adverse drug reactions, for example, may make it obvious which treatment a patient has been given. Nevertheless maintaining ‘blindness’ is necessary where the outcome is subjective (e.g. relief of pain, alleviation of depression) if bias is to be avoided.
Were the treated and control groups comparable?
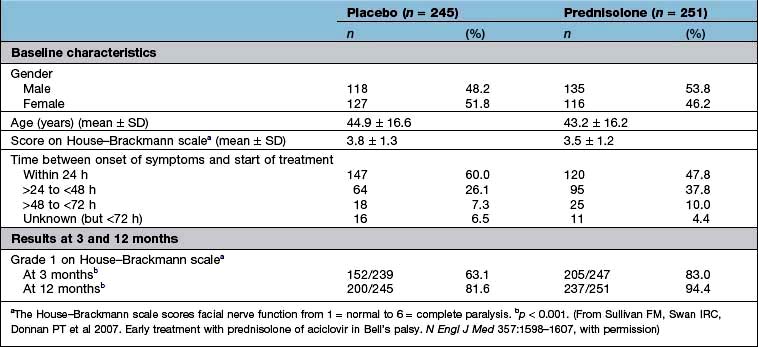
Were they similar in their ‘baseline’ characteristics? Were they, for example, of similar age, severity and duration of illness? If not, are the differences likely to influence the results? Has the statistical analysis (using analysis of covariance, or Cox’s proportional hazards model) (see below) tried to adjust for them? Table 17.10 shows some of the baseline characteristics of a trial comparing prednisolone with placebo in the treatment of Bell’s palsy (idiopathic facial paralysis).
Outcomes
There are two ways to look at the outcomes of an RCT.
Per protocol analysis: this includes only those who completed the study.
Intention-to-treat analysis: this includes all patients from the time of randomization.
Ideally, there should be no difference but in reality the results of a per protocol analysis are usually more advantageous to a treatment than an intention-to-treat analysis. The reason is that the intention-to-treat analysis will take account of patients who have withdrawn from the trial because of intolerance of the treatment or adverse drug reactions. It is therefore a much more robust approach. The results of the intention-to-treat analysis, in the trial of prednisolone in Bell’s palsy, are shown in Table 17.10. The trial results indicate, with a high probability, that treatment of Bell’s palsy with prednisolone will increase the chances of a full recovery of facial nerve function.
Are the results generalizable?
Were the patients enrolled into the study a reasonable reflection of those likely to be treated in routine clinical practice (a so-called pragmatic trial)? Or were they a selected population that excluded significant patient groups (such as the elderly)? If the latter, view the results with caution.
Analysis of a superiority trial
The analysis of a superiority trial is based on the premise – the ‘null hypothesis’ – that there is no difference between the treatments. The null hypothesis is rejected if the probability of the observed result occurring by chance, the p value, is less than 1 in 20 (i.e. p < 0.05). There are three caveats.
Any difference may still be due to chance; and it is often better to await the results of at least two independent studies before adopting a new treatment.
A trial may show no ‘statistically significant’ difference, when one in fact exists, because too few patients have been included, in other words the trial lacked sufficient ‘power’. The ‘power’ of a study (the number of patients needed in each treatment group to detect a predefined difference) should have been defined at the outset. If the study was underpowered, the results of the study should be interpreted with extreme care.
A statistically significant difference may not, necessarily, be clinically relevant.
Scrutiny of the magnitude of the effect, and its 95% confidence intervals (CI), is a far better guide than the p value.
Effect size. The results of the well-designed trial in Table 17.10 show, very convincingly, that the treatment of Bell’s palsy with prednisolone increases the chances of complete recovery of facial nerve function, at 12 months, from 81.6% to 94.4%. This is a far more convincing description of the benefits of treatment than the p value.
Another expression of the benefit of a treatment such as prednisolone can be derived from the number needed to treat (NNT). This is an estimate of the numbers of patients needed to be treated with a drug to achieve one positive result. In the study shown in Table 17.10, the NNT to enable one patient with Bell’s palsy to regain normal facial nerve function, after prednisolone treatment, is eight.
Analysis of an equivalence trial
The aim of an equivalence trial is to determine whether two (or possibly more) treatments produce similar benefits. During the design of such trials, it is necessary to decide what difference is unimportant and then to calculate the number of patients needed in order to have an 80% or 90% chance of showing this. In equivalence trials such power calculations show that the number of patients required is invariably greater than those needed for superiority trials. In such studies the comparator itself must, of course, already have been shown to be effective.
Meta-analysis
It is possible to summate all the controlled trials that have been performed in the treatment of a particular condition so as to refine the estimate of effectiveness. This technique minimizes random error in the assessment of the effect size of a treatment because more patients are included than could be accommodated in any single trial. A meta-analysis should be performed (and interpreted) carefully because of the heterogeneity of the individual studies used in it.
Controlled observational trials
Three types of observational study have been used to test the clinical effectiveness of therapeutic interventions:
Historical controlled trials
Despite the value of the prospective randomized controlled trial there are many treatments that have never been subjected to this technique, yet their efficacy is unquestioned. Examples include insulin in the treatment of diabetic ketoacidosis, thyroxine for hypothyroidism, vitamin B12 in pernicious anaemia and defibrillation for ventricular fibrillation. In a historical controlled trial the outcome in patients treated with the study drug is compared to that of previously untreated people with the same disease. Treatments can be accepted into routine use on the basis of favourable comparisons with historical controls when the following criteria are met:
There should be a biologically plausible basis for the observed benefits.
There should be no appropriate treatment that could be reasonably used as a control.
The condition should have a known and predictable natural history.
The treatment should not be expected to have adverse effects that would compromise its potential benefits.
There should be a reasonable expectation that the magnitude of the therapeutic effects of treatment will be large enough to make the interpretation of its benefits unambiguous.
Case–control studies
This type of study design compares people with a particular condition (the ‘cases’) with those without (the ‘controls’). The approach has predominantly been used to identify epidemiological ‘risk factors’ for specific conditions such as lung cancer (smoking) or sudden infant death syndrome (lying prone); or in the evaluation of potential adverse drug reactions (such as deep venous thrombosis with oral contraceptives).
A case–control design allows an estimation of the odds ratio (OR), which is the ratio of the probability of an event occurring to the probability of the event not occurring (Box 17.1).
Box 17.1
Estimation of odds ratio
| Cases | Controls | |
|---|---|---|
Risk factor present |
a |
c |
Risk factor absent |
b |
d |
The odds ratio (OR) = (a ÷ b) / (c ÷ d) |
||
An OR that is significantly greater than unity indicates a statistical association that may be causal. The OR for deep venous thrombosis and current use of oral contraceptives equals 2–4 (depending on the preparation): this indicates that the risk of developing a deep venous thrombosis on oral contraceptives is between 2 and 4 times greater than the background rate.
In some studies, the OR for a particular observation has been found to be significantly less than unity, suggesting ‘protection’ from the condition under study. Some studies of women with myocardial infarction indicated protection in those using hormone-replacement therapies but it has been subsequently shown that the result was due to bias. On the other hand, case–control studies have consistently shown that aspirin and other non-steroidal anti-inflammatory drugs are associated with a reduced risk of colon cancer. This seems to be a causal effect.
Case–control studies claiming to demonstrate the efficacy of a drug need to be interpreted with great care: the possibility of bias and confounding is substantial as was seen in the studies of hormone-replacement therapy and myocardial infarction. Confirmation from one or more RCTs is usually essential.
Before-and-after studies
It has sometimes been inferred that observed improvements seen in patients before, and after, the application of a particular treatment is evidence of efficacy. Such an approach is fraught with difficulties: the combination of a placebo effect, as well as regression to the mean, is likely to negate most studies using this type of design. Nevertheless, there are some circumstances where genuine efficacy can be confidently observed with such designs: the consequences of hip replacement, and cataract surgery, are good examples. Such instances can be regarded as special examples of the use of implicit historical controls.
Uncontrolled observational studies
Uncontrolled case series cannot be considered as providing primary evidence of efficacy unless they are undertaken in circumstances that are virtually those of historical controlled trials. When used in this way to demonstrate clinical effectiveness their validity relies on the use of implicit historical trials. Case series can, however, sometimes be of value in demonstrating the generalizability of the results of RCTs.
Dangers
Drug trials are carried out in specific groups of selected patients under strict supervision. The results, particularly when dramatic, are often used outside the strict inclusion criteria for clinical trials. The dramatic effect of spironolactone in heart failure (30% reduction in all-cause mortality) has not always been replicated in routine clinical practice because the wrong patients have been treated, often with higher doses, leading to hyperkalaemia and death.
Evaluation of new drugs
New drugs are subjected to a vigorous programme of preclinical and clinical testing before they are licensed for general use (Table 17.11) and are also monitored for safety following licensing. Doctors are recommended to fill in yellow cards when they suspect an adverse reaction has taken place.
Table 17.11 Evaluation of new drugs
Phase I: Healthy human subjects (usually men) |
Phase II: First assessment in patients |
Phase III: Use in wider patient population |
Phase IV: Post-marketing surveillance |
Statistical analyses
The relevance of statistics is not confined to those who undertake research but also to anyone who wants to understand the relevance of research studies to their clinical practice.
The average
Clinical studies may describe, quantitatively, the value of a particular variable (e.g. height, weight, blood pressure, haemoglobin) in a sample of a defined population. The ‘average’ value (or ‘central tendency’ in statistical language) can be expressed as the mean, median or mode depending on the circumstances:

The mean is the average of a distribution of values that are grouped symmetrically around the central tendency.
The median is the middle value of a sample. It is used, particularly, where the values in a sample are asymmetrically distributed around the central tendency.
The mode is the interval, in a frequency distribution of values, that contains more values than any other.
In a symmetrically distributed population, the mean, median and mode are the same.
The average value of a sample, on its own, is of only modest interest. Of equal (and often greater) relevance is the confidence we can place on the sample average as truly reflecting the average value of the population from which it has been drawn. This is most often expressed as a confidence interval, which describes the probability of a sample mean being a certain distance from the population mean. If, for example, the mean systolic blood pressure of 100 undergraduates is 124 mmHg, with a 95% confidence interval of ± 15 mmHg, we can be confident that if we replicated the study 100 times the value of the mean would be within the range 109–139 mmHg on 95 occasions. It is intuitively obvious that the larger the sample the smaller will be the size of the confidence interval.
Correlation
In clinical studies two, or more, independent variables may be measured in the same individuals in a sample population (e.g. weight and blood pressure). The degree of correlation between the two can be investigated by calculating the correlation coefficient (often abbreviated to ‘r’). The correlation coefficient measures the degree of association between the two variables and may range from 1 to −1:
If r = 1, there is complete and direct concordance between the two variables
Statistical tables are available to inform investigators as to the probability that r is due to chance. As in other areas of statistics, if the probability is less than 1 in 20 (p < 0.05) then by custom and practice it is regarded as statistically significant. There are, however, two caveats:
The 1 in 20 rule is a convention and does not exclude the possibility that a presumed association is due to chance
The fact that there is an association between two variables does not necessarily mean that it is causal. For example, a correlation between blood pressure and weight, with r = 0.75 and p < 0.05, does not mean that weight has a direct effect on blood pressure (or vice versa).
Correlation analyses can become complicated. The simplest (least squares regression analysis) presumes a straight-line relationship between the two variables. More complicated techniques can be used to estimate r where a non-linear relationship is presumed (or assumed); where the distributions deviate from normal; where the scales of one or both variables are intervals or ranks; or where a correlation between three or more variables is sought.
Expressions of benefit and harm
There are three ways in which the outcomes, in clinical studies, are expressed:
Binary outcomes are often used in the design and analysis of RCTs. Such outcomes are dichotomous (such as alive or dead). The results are usually expressed as the relative risk (or risk ratio – RR). In a trial where the outcome is (say) mortality, the relative risk is the ratio of the proportion of treated patients dying to the proportion of control patients dying. Usually, RR of <1 is suggestive of benefit; an RR of >1 is suggestive of harm. RRs are almost invariably reported with their 95% confidence intervals. If the boundaries of the 95% confidence intervals do not cross unity the results are generally statistically significant (at least at the 5% level).
Survival analyses. In studies in which individuals are observed over a long(ish) period of time, and in which it is unreasonable (or erroneous) to assume that event rates are constant, the technique of survival analysis is used. This is most commonly reported as the hazard ratio (HR) and its 95% confidence interval. The HR is the probability that, if an event in question has not already occurred, it will happen in the next (short) time interval. It has, broadly, a comparable interpretation to the RR.
Continuous outcomes. Studies such as that in Table 17.10 may report outcomes using one or more continuous scales. In this study of the effects of prednisolone in the treatment of Bell’s palsy, the House–Brackmann measure of facial nerve function was used as the outcome measure. Conventional tests of statistical significance using Student’s t-test, for example, can be calculated to assess whether the null hypothesis should be rejected.
Number needed to treat (NNT). As discussed earlier, the NNT is an estimate of the number of patients that need to be treated for one to benefit compared to no treatment. If the probabilities of the end-points with the active drug and no treatment (i.e. placebo) are respectively pactive and pno treatment then the NNT can be calculated thus:
NNT = 1/ (pactive – pno treatment)
An analogous measure – the number needed to harm (NNH) – is the number of patients that need to be treated with a drug to cause one patient to be subject to a specific harm.
Other statistical techniques
Statisticians have developed a range of sophisticated methods to handle a wide variety of biomedical problems. Unless an investigator is supremely (and usually unwisely) confident it is wise to seek professional advice in analysing numerical data that look complicated. In doing so, it is invariably wiser to do so at the time the study is being designed rather than after the results have been generated!
Current information sources
Pharmacotherapy moves at a very rapid pace and it is impossible for anyone to keep up with contemporary advances. Details of current prescribing advice can be found in:
The Summary of Product Characteristics (SmPCs) produced by manufacturers but vetted by drug regulatory authorities (for the UK, these are the Medicines and Healthcare Products Regulatory Agency and the European Medicines Agency).
The relevant national formulary. Many countries have their own formularies, for example the UK has the British National Formulary (BNF), produced jointly by the British Medical Association and the Royal Pharmaceutical Society.
Guidance produced by the Technology Appraisals Guidance series from the UK’s National Institute for Health and Clinical Excellence (NICE).
Advice on the management of individual conditions is available in the form of clinical guidelines (systematically developed statements to assist practitioner and patient decisions about appropriate healthcare for specific clinical circumstances).
SIGNIFICANT WEBSITES
Sources of clinical guidelines:
National Institute for Clinical Excellence (NICE): http://www.nice-org.uk
Scottish Intercollegiate Guidelines Network (SIGN): http://www.sign.ac.uk/
National Guidelines Clearing House in the USA: http://www.guideline.gov
Poisoning
The nature of the problem
Exposure to a substance is often equated with poisoning. However, absorption is necessary for there to be a toxic effect and, even if this occurs, poisoning does not necessarily result, because the amount absorbed may be too small. In developed countries, poisoning causes approximately 10% of acute hospital medical presentations. In such cases poisoning is usually by self-administration of prescribed and over-the-counter medicines, or illicit drugs. Poisoning in children aged less than 6 months is most commonly iatrogenic and involves overtreatment with, e.g. paracetamol. Children between 8 months and 5 years of age also ingest poisons accidentally, or they may be administered deliberately to cause harm, or for financial or sexual gain. Occupational poisoning as a result of dermal or inhalational exposure to chemicals is a common occurrence in the developing world and still occurs in the developed world. Sometimes inappropriate treatment of a patient by a doctor is responsible for the development of poisoning, e.g. in the case of digoxin toxicity.
In adults, self-poisoning is commonly a ‘cry for help’. Those involved are most often females under the age of 35 who are in good physical health. They take an overdose in circumstances where they are likely to be found, or in the presence of others. In those older than 55 years of age, men predominate and the overdose is usually taken in the course of a depressive illness or because of poor physical health.
The type of agent taken in overdose is also heavily influenced by availability and culture. In the UK, paracetamol poisoning is responsible for approximately one-third of all admissions, whereas in Sri Lanka, for example, the agents ingested are more often pesticides or plants, such as oleander, and in South India, copper sulphate is a problem. In addition, ingestion of heating fuels (e.g. petroleum distillates), antimalarials, antituberculous drugs and traditional medicine is reported frequently in the developing world.
Self-poisoning can kill
All must be aware of the dangers of drugs and chemicals. Education on safe storage and careful handling of household and workplace chemicals is necessary on a continuing basis.
A third of patients admitted with an overdose in the UK state that they are unaware of the toxic effects of the substance involved; the majority take whatever drug is easily available at home (Box 17.2). Studies reveal that:
Acute overdoses often involve more than one agent; alcohol is the most commonly implicated second agent.
There is often a poor correlation between the drug history and the toxicological analytical findings. Therefore, a patient’s statement about the type and amount of drug ingested cannot always be relied upon.
Box 17.2
Prevention of self-poisoning
Patients usually take what is readily available at home.
Only small amounts of drugs should be bought
Foil-wrapped drugs are less likely to be taken in overdose
Keep drugs and liquids in their original containers
Child-resistant drug containers should be used
Doctors should be careful in prescribing all drugs
Prescriptions for any susceptible patient (e.g. the depressed) must be monitored carefully
Household products should be labelled and kept safely away from children
The majority of cases of self-poisoning do not require intensive medical management, but all patients require a sympathetic and caring approach, a psychiatric and social assessment and, sometimes, psychiatric treatment. However, as the majority of patients ingest relatively non-toxic agents, receive good supportive care and, when appropriate, the administration of specific antidotes, the in-hospital mortality in most developed countries is now less than 1%. Fatalities in the UK are due predominantly to carbon monoxide, antidepressants, paracetamol, analgesic combinations containing paracetamol and an opioid, heroin, methadone or cocaine. Deaths from poisoning in children are usually accidental and due to inappropriate storage of drugs such as digoxin and quinine and from drugs of abuse purchased or prescribed for a parent or carer.
The approach to the patient
History
More than 80% of adults are conscious on arrival at hospital and the diagnosis of self-poisoning can usually be made from the history (Table 17.12). In the unconscious patient a history from friends or relatives is helpful, and the diagnosis can often be inferred from the medicine containers or a ‘suicide note’ brought by the paramedics. In any patient with an altered level of consciousness, acute poisoning must always be considered in the differential diagnosis.
Table 17.12 Diagnostic process in acute poisoning
|
Obtain history, if possible, from the patient, relative, friend or paramedics Is there circumstantial evidence of an overdose? Are the circumstances in which the patient has been found suggestive? Are the symptoms suggestive of an overdose? Do the physical signs suggest an overdose? (Tables 17.13 and 17.14) |
Examination
On arrival at hospital, the patient must be assessed urgently (Airways, Breathing and Circulation). The following should be evaluated:
Level of consciousness: the Glasgow Coma Scale should be used (see p. 1092)
Ventilation: pulse oximetry can be used to measure oxygen saturation. The displayed reading may be inaccurate when the saturation is below 70%, there is poor peripheral perfusion and in the presence of carboxyhaemoglobin and methaemoglobin. Only measurement of arterial blood gases will indicate the presence both of hypercapnia and hypoxia
Pupil size and reaction to light
If the patient is unconscious, the following should also be checked:
Cough and gag reflex: present or absent.
Temperature: measured with a low-reading rectal thermometer if the ear temperature suggests it is low.
Some of the physical signs that may aid identification of the agents responsible for poisoning are shown in Table 17.13. The cluster of features on presentation may be distinctive and diagnostic. For example, sinus tachycardia, fixed dilated pupils, exaggerated tendon reflexes, extensor plantar responses and coma suggest tricyclic antidepressant poisoning (Table 17.13 and Table 17.14).
Table 17.13 Some physical signs of poisoning
| Features | Likely poisons |
|---|---|
Constricted pupils (miosis) |
Opioids, organophosphorus insecticides, nerve agents |
Dilated pupils (mydriasis) |
Tricyclic antidepressants, amfetamines, cocaine, antimuscarinic drugs |
Divergent strabismus |
Tricyclic antidepressants |
Nystagmus |
Carbamazepine, phenytoin |
Loss of vision |
Methanol, quinine |
Papilloedema |
Carbon monoxide, methanol |
Convulsions |
Tricyclic antidepressants, theophylline, opioids, mefenamic acid, isoniazid, amfetamines |
Dystonic reactions |
Metoclopramide, phenothiazines |
Delirium and hallucinations |
Amfetamines, antimuscarinic drugs, cannabis, recovery from tricyclic antidepressant poisoning |
Hypertonia and hyperreflexia |
Tricyclic antidepressants, antimuscarinic drugs |
Tinnitus and deafness |
Salicylates, quinine |
Hyperventilation |
Salicylates, phenoxyacetate herbicides, theophylline |
Hyperthermia |
Ecstasy (MDMA), salicylates |
Blisters |
Usually occur in comatose patients |
MDMA, 3,4-methylenedioxymetamfetamine.
Table 17.14 Common feature clusters in acute poisoning
| Feature clusters | Poisons |
|---|---|
Coma, hypertonia, hyperreflexia, extensor plantar responses, myoclonus, strabismus, mydriasis, sinus tachycardia |
Tricyclic antidepressants; less commonly antihistamines, orphenadrine, thioridazine |
Coma, hypotonia, hyporeflexia, plantar responses (flexor or non-elicitable), hypotension |
Barbiturates, benzodiazepine and alcohol combinations, tricyclic antidepressants |
Coma, miosis, reduced respiratory rate |
Opioid analgesics |
Nausea, vomiting, tinnitus, deafness, sweating, hyperventilation, vasodilatation, tachycardia |
Salicylates |
Hyperthermia, tachycardia, delirium, agitation, mydriasis |
Ecstasy (MDMA) or other amfetamine |
Miosis, hypersalivation, rhinorrhoea, bronchorrhoea |
Organophosphorus and carbamate insecticides, nerve agents |
Principles of management of poisoning (table 17.15)
Most people with self-poisoning require only general care and support of the vital systems. However, for a few drugs additional therapy is required.
Table 17.15 Management strategy in acute poisoning
|
Is the use of an antidote appropriate? (Table 17.16) Is it appropriate to attempt to reduce poison absorption? Is it appropriate to perform toxicological investigations? Will non-toxicological investigations assist? (Table 17.17) Should urine alkalinization, multiple-dose activated charcoal, haemodialysis or haemodialfiltration be employed to increase poison elimination? |
Care of the unconscious patient (see also p. 1135)
In all cases the patient should be nursed in the lateral position with the lower leg straight and the upper leg flexed; in this position the risk of aspiration is reduced. A clear passage for air should be ensured by the removal of any obstructing object, vomit or dentures, and by backward pressure on the mandible. Nursing care of the mouth and pressure areas should be instituted. Immediate catheterization of the bladder in unconscious patients is usually unnecessary as it can be emptied by gentle suprapubic pressure. Insertion of a venous cannula is usual, but administration of intravenous fluids is unnecessary unless the patient has been unconscious for more than 12 hours or is hypotensive.
Ventilatory support
If respiratory depression is present, as determined by pulse oximetry or preferably by arterial blood gas analysis, an oropharyngeal airway should be inserted, and supplementaloxygen should be administered. Pulse oximetry alone will not detect hypercapnia. Loss of the cough or gag reflex is the prime indication for intubation. The gag reflex can be assessed by positioning the patient on one side and making him or her gag using a suction tube. In many severely poisoned patients, the reflexes are depressed sufficiently to allow intubation without the use of sedatives or relaxants. The complications of endotracheal tubes are discussed on page 885 in Chapter 16. If ventilation remains inadequate after intubation, as shown by hypoxaemia and hypercapnia, intermittent positive-pressure ventilation (IPPV) should be instituted.
Cardiovascular support
Although hypotension (systolic blood pressure below 80 mmHg) is a recognized feature of acute poisoning, the classic features of shock: tachycardia and pale cold skin, are observed only rarely.
Hypotension and shock may be caused by:
A direct cardio-depressant action of the poison (e.g. beta-blockers, calcium channel blockers, tricyclic antidepressants)
Vasodilation and venous pooling in the lower limbs (e.g. ACE inhibitors, phenothiazines)
A decrease in circulating blood volume because of gastrointestinal losses (e.g. profuse vomiting in theophylline poisoning), increased insensible losses (e.g. salicylate poisoning), increased renal losses (e.g. poisoning due to diuretics) and increased capillary permeability.
Hypotension may be exacerbated by co-existing hypoxia, acidosis and dysrhythmias. In people with marked hypotension, volume expansion with crystalloids should be used, guided by monitoring of central venous pressure (CVP). Urine output (aiming for 35–50 mL/h) is also a useful guide to the adequacy of the circulation. If a patient fails to respond to the above measures, more intensive therapy is required. In such patients, it is helpful to undertake invasive haemodynamic monitoring to confirm that adequate volume replacement has been administered. Volume replacement and the use of inotropes are discussed on page 874. All patients with cardiogenic shock should have ECG monitoring.
Systemic hypertension can be caused by a few drugs when taken in overdose. If this is mild and associated with agitation, a benzodiazepine may suffice. In more severe cases, for example those due to a monoamine oxidase inhibitor, there may be a risk of arterial rupture, particularly intracranially. To prevent this, an α-adrenergic blocking agent such as phentolamine, 2–5 mg i.v. every 10–15 min, or intravenous isosorbide dinitrate 2–10 mg/h up to 20 mg/h if necessary, or sodium nitroprusside 0.5–1.5 µg/kg per min by intravenous infusion, should be administered until the blood pressure is controlled.
Arrhythmias can occur, e.g. tachyarrhythmias following ingestion of a tricyclic antidepressant or theophylline; bradyarrhythmias with digoxin poisoning. Known arrhythmogenic factors such as hypoxia, acidosis and hypokalaemia should be corrected.
Other problems
A rectal temperature below 35°C is a recognized complication of poisoning, especially in older patients or those who are comatose. The patient should be covered with a ‘space blanket’ and, if necessary, given intravenous and intragastric fluids at normal body temperature. The administration of heated (37°C), humidified oxygen delivered by face mask is also useful.
Rarely, body temperature may increase to potentially fatal levels after poisoning with central nervous system stimulants such as cocaine, amfetamines including ecstasy (MDMA), monoamine oxidase inhibitors or theophylline. Muscle tone is often increased and convulsions and rhabdomyolysis are common. Cooling measures, sedation with diazepam and, in severe cases, i.v. dantrolene 1 mg/kg body weight should be given.
Skin blisters may be found in poisoned patients who are, or have been, unconscious. Such lesions are not diagnostic of specific poisons, but are sufficiently common in poisoned patients (and sufficiently uncommon in patients unconscious from other causes) to be of diagnostic value.
Rhabdomyolysis can occur from pressure necrosis in drug-induced coma, or it may complicate, e.g. ecstasy (MDMA) abuse in the absence of coma. People with rhabdomyolysis are at risk of developing, firstly, acute kidney injury from myoglobinaemia, particularly if they are hypovolaemic and have an acidosis and, secondly, wrist or ankle drop from the development of a compartment syndrome (see p. 509).
These may occur, e.g. in poisoning due to tricyclic antidepressants, mefenamic acid or opioids. Usually the seizures are short-lived but, if they are prolonged, diazepam 10–20 mg i.v. or lorazepam 4 mg i.v. should be administered. Persistent fits must be controlled rapidly to prevent severe hypoxia, brain damage and laryngeal trauma. If diazepam or lorazepam in repeated dose is ineffective, the patient should also receive a loading dose of phenytoin (20 mg/kg) administered intravenously at not more than 50 mg/min, with ECG monitoring.
Stress ulceration and bleeding
Measures to prevent stress ulceration of the stomach should be started on admission in all patients who are unconscious and require intensive care. A proton pump inhibitor should be administered intravenously.
Body ‘packers’ and body ‘stuffers’
Body ‘packers’ (sometimes called ‘mules’ or ‘swallowers’) are those who swallow a substantial number of packages containing illicit drugs for the purpose of smuggling. Heroin used to be the drug of choice but this has been superseded by cocaine. Although each package contains a potentially lethal amount of drug, packets are now usually machine manufactured using a material which usually does not leak. Body packers may ingest up to 100–200 packages.
Body ‘stuffers’ are those who swallow a small number of packages containing an illicit drug, usually heroin, cocaine, cannabis or an amfetamine, in an unplanned attempt to conceal evidence when on the verge of being arrested. These drugs are usually either unpackaged or poorly packaged and as a consequence leakage may occur over the ensuing 3–6 hours and cause significant symptoms. Some also hide illicit drug packages in their rectum or vagina with the same intent (these are sometimes known as body ‘pushers’).
The role of imaging is confined to body packers; imaging has little role in the care of body stuffers or pushers. Ultrasound is of similar accuracy to abdominal X-ray in locating packages and less accurate than CT. A urine screen for drugs of misuse should be performed. A screen that is positive for one or more drugs of misuse suggests that either the patient has used the drug in the previous few days, or at least one packet is leaking. A negative screen strongly suggests that no packet is leaking. Screens should be repeated daily, or immediately if the patient develops features of intoxication, to confirm the diagnosis.
Packages can be removed most expeditiously in body stuffers by employing whole bowel irrigation (see p. 913). In the past early surgery was advocated in body packers. However, with the development of improved packaging, a more conservative approach (the use of lactulose or whole bowel irrigation) can now be adopted with which there is a complication rate of <5%. Immediate surgery is indicated if acute intestinal obstruction develops, or when packets can be seen radiologically and there is clinical or analytical evidence to suggest leakage, particularly if the drug involved is cocaine.
Specific management
Antidotes
Specific antidotes are available for only a small number of poisons (Table 17.16).
Table 17.16 Antidotes of value in poisoning
| Poison | Antidotes |
|---|---|
Aluminium (aluminum) |
Desferrioxamine (deferoxamine) |
Arsenic |
DMSA, dimercaprol |
Benzodiazepines |
Flumazenil |
β-adrenoceptor blocking drugs |
Atropine, glucagon |
Calcium channel blockers |
Atropine |
Carbamate insecticides |
Atropine |
Carbon monoxide |
Oxygen |
Copper |
D-penicillamine, DMPS |
Cyanide |
Oxygen, dicobalt edetate, hydroxocobalamin, sodium nitrite, sodium thiosulphate |
Diethylene glycol |
Fomepizole, ethanol, |
Digoxin and digitoxin |
Digoxin-specific antibody fragments |
Ethylene glycol |
Fomepizole, ethanol |
Hydrogen sulphide |
Oxygen |
Iron salts |
Desferrioxamine |
Lead (inorganic) |
DMSA (succimer), sodium calcium edentate |
Methaemoglobinaemia |
Methylthioninium chloride (methylene blue) |
Methanol |
Fomepizole, ethanol |
Mercury (inorganic) |
Unithiol (DMPS) |
Nerve agents |
Atropine, HI-6, obidoxime, pralidoxime |
Oleander |
Digoxin-specific antibody fragments |
Opioids |
Naloxone |
Organophosphorus insecticides |
Atropine, HI-6, obidoxime, pralidoxime |
Paracetamol |
Acetylcysteine |
Thallium |
Berlin (Prussian) blue |
Warfarin and similar anticoagulants |
Phytomenadione (vitamin K) |
DMSA, dimercaptosuccinic acid; DMPS, dimercaptopropanesulphonate.
Antidotes may exert a beneficial effect by:
Forming an inert complex with the poison (e.g. desferrioxamine (deferoxamine), D-penicillamine, dicobalt edetate, digoxin-specific antibody fragments, dimercaprol, HI-6, hydroxocobalamin, obidoxime, pralidoxime, protamine, Prussian (Berlin) blue, sodium calcium edetate, succimer (DMSA), unithiol (DMPS))
Accelerating detoxification of the poison (e.g. acetylcysteine, sodium thiosulphate)
Reducing the rate of conversion of the poison to a more toxic compound (e.g. ethanol, fomepizole)
Competing with the poison for essential receptor sites (e.g. oxygen, naloxone, phytomenadione)
Blocking essential receptors through which the toxic effects are mediated (e.g. atropine)
By-passing the effect of the poison (e.g. oxygen, glucagon).
Reducing poison absorption
To reduce poison absorption through the lungs, remove the casualty from the toxic atmosphere, making sure that rescuers themselves are not put at risk. Contaminated clothing should be removed to reduce dermal absorption and contaminated skin washed thoroughly with soap and water.
Gut decontamination. While it appears logical to assume that removal of unabsorbed drug from the gastrointestinal tract will be beneficial (gut decontamination), the efficacy of gastric lavage and syrup of ipecacuanha remains unproven and efforts to remove small amounts of non-toxic drugs are clinically not worthwhile or appropriate.
Gastric lavage should only be performed if a patient has ingested a potentially life-threatening amount of a poison, e.g. iron, and the procedure can be undertaken within 60 minutes of ingestion. Intubation is required if airway protective reflexes are lost. Lavage is also contraindicated if a hydrocarbon with high aspiration potential or a corrosive substance has been ingested.
Syrup of ipecacuanha should not be used as the amount of drug recovered is highly variable, diminishes with time and there is no evidence that it improves the outcome of poisoned patients.
Single-dose activated charcoal. Activated charcoal is able to adsorb a wide variety of compounds. Exceptions are strong acids and alkalis, ethanol, ethylene glycol, iron, lithium, mercury and methanol.
In studies in volunteers given 50 g activated charcoal, the mean reduction in absorption was 40%, 16% and 21%, at 60 min, 120 min and 180 min, respectively after ingestion. Based on these studies, activated charcoal should be given in those who have ingested a potentially toxic amount of a poison (known to be adsorbed by charcoal). There are insufficient data to support or exclude its use after 1 hour. There is no evidence that administration of activated charcoal improves the clinical outcome.
Cathartics have no role in the management of the poisoned patient.
Whole bowel irrigation requires the insertion of a nasogastric tube into the stomach and the introduction of polyethylene glycol electrolyte solution 1500–2000 mL/h in an adult, which is continued until the rectal effluent is clear. Whole bowel irrigation may be used for potentially toxic ingestions of sustained-release or enteric-coated drugs or to remove illicit drug packets.
FURTHER READING
Barceloux D, McGuigan M, Hartigan-Go K et al. Position paper: cathartics. Clin Toxicol 2004; 42:243–253.
Chyka PA, Seger D, Krenzelok EP et al. Position paper: single-dose activated charcoal. Clin Toxicol 2005; 43:61–87.
Krenzelok EP, McGuigan M, Lheureux P et al. Position paper: ipecac syrup. Clin Toxicol 2004; 42:133–143.
Kulig K, Vale JA. Position paper: gastric lavage. Clin Toxicol 2004; 42:933–943.
Tenenbein M, Lheureux P. Position paper: whole bowel irrigation. Clin Toxicol 2004; 42:843–854.
Increasing poison elimination
Multiple-dose activated charcoal (MDAC) involves the repeated administration of oral activated charcoal to increase the elimination of a drug that has already been absorbed into the body. Drugs are secreted in the bile and re-enter the gut by passive diffusion if the concentration in the gut is lower than that in the blood. The rate of passive diffusion depends on the concentration gradient and the intestinal surface area, permeability and blood flow. Activated charcoal will bind any drug that is in the gut lumen.
Elimination of drugs with a small volume of distribution (<1 L/kg), low pKa (which maximizes transport across membranes), low binding affinity and prolonged elimination half-life following overdose is particularly likely to be enhanced by MDAC. MDAC also improves total body clearance of the drug when endogenous processes are compromised by liver and/or renal failure.
Although MDAC has been shown to significantly increase drug elimination, it has not reduced morbidity and mortality in controlled studies. At present, MDAC should only be used in patients who have ingested a life-threatening amount of carbamazepine, dapsone, phenobarbital, quinine or theophylline.
Dosage. In adults, charcoal should be administered in an initial dose of 50–100 g and then at a rate of not less than 12.5 g/h, preferably via a nasogastric tube. If the patient has ingested a drug that induces protracted vomiting (e.g. theophylline), intravenous ondansetron 4–8 mg is effective as an antiemetic and thus enables administration of MDAC.
Urine alkalinization. Increasing the urine pH enhances elimination of salicylate, phenobarbital, chlorpropamide and chlorophenoxy herbicides (e.g. 2,4-dichlorophenoxyacetic acid) by mechanisms which are not clearly understood. Urine alkalinization is not recommended as first-line therapy for poisoning with phenobarbital as MDAC is superior, and supportive care is invariably adequate for chlorpropamide. A substantial diuresis is required in addition to urine alkalinization to achieve clinically relevant elimination of chlorophenoxy herbicides.
Urine alkalinization is a metabolically invasive procedure requiring frequent biochemical monitoring and medical and nursing expertise. Before commencing urine alkalinization, correct plasma volume depletion, electrolytes (administration of sodium bicarbonate exacerbates pre-existing hypokalaemia) and metabolic abnormalities. Sufficient bicarbonate is administered to ensure that the pH of the urine, which is measured by narrow range indicator paper or a pH meter, is more than 7.5 and preferably close to 8.5. In one study, sodium bicarbonate 225 mmol was the mean amount required initially. This is most conveniently administered as 225 mL of an 8.4% solution (1 mmol bicarbonate/mL) i.v. over 1 hour.
Haemodialysis and haemodialfiltration. Haemodialysis and haemodialfiltration are of little value in patients poisoned with drugs with large volumes of distribution (e.g. tricyclic antidepressants), because the plasma contains only a small proportion of the total amount of drug in the body. These methods are indicated in people with severe clinical features and high plasma concentrations of ethanol, ethylene glycol, isopropanol, lithium, methanol and salicylate.
Toxicological investigations
On admission, or at an appropriate time post overdose, a timed blood sample should be taken if it is suspected that aspirin, digoxin, ethylene glycol, iron, lithium, methanol, paracetamol, paraquat, quinine or theophylline has been ingested. The determination of the concentrations of these drugs will be valuable in management. Drug screens on blood and urine are occasionally indicated in severely poisoned patients in whom the cause of coma is unknown. A poison information service will advise.
Non-toxicological investigations (Table 17.17)
Some routine investigations are of value in the differential diagnosis of coma or the detection of poison-induced hypokalaemia, hyperkalaemia, hypoglycaemia, hyperglycaemia, hepatic or renal failure or acid–base disturbances (Table 17.18). Measurement of carboxyhaemoglobin, methaemoglobin and cholinesterase activities are of assistance in the diagnosis and management of cases of poisoning due to carbon monoxide, methaemoglobin-inducing agents such as nitrites and organophosphorus insecticides, respectively.
Table 17.17 Relevant non-toxicological investigations
|
Serum sodium (e.g. hyponatraemia in MDMA poisoning) and potassium (e.g. hypokalaemia in theophylline poisoning and hyperkalaemia in digoxin poisoning) concentrations Plasma creatinine concentration (e.g. eGFR in acute kidney injury in ethylene and diethylene glycol poisoning) Acid–base disturbances, including metabolic acidosis (Table 17.18) Blood sugar concentration (e.g. hypoglycaemia in insulin poisoning or hyperglycaemia in salicylate poisoning) Serum calcium concentration (e.g. hypocalcaemia in ethylene glycol poisoning) Liver function (e.g. in paracetamol poisoning) Carboxyhaemoglobin concentration (in carbon monoxide poisoning) Methaemoglobinaemia (e.g. in nitrite poisoning) Cholinesterase activities (e.g. organophosphorus insecticide and nerve agent poisoning) ECG (e.g. wide QRS in tricyclic antidepressant poisoning) – see page 915 |
Table 17.18 Some poisons inducing metabolic acidosis
Calcium channel blockers |
Iron |
Carbon monoxide |
Metformin |
Cocaine |
Methanol |
Cyanide |
Paracetamol |
Diethylene glycol |
Topiramate |
Ethanol |
Tricyclic antidepressants |
Ethylene glycol |
|
Routine ECG is of limited diagnostic value, but continuous ECG monitoring should be undertaken in those ingesting potentially cardiotoxic drugs; for example, sinus tachycardia with prolongation of the PR and QRS intervals in an unconscious patient suggests tricyclic antidepressant overdose. Q–T interval prolongation is an adverse effect of several drugs (e.g. quetiapine and quinine).
Routine radiology is of little diagnostic value. It can confirm ingestion of metallic objects (e.g. coins, button batteries) or injection of globules of metallic mercury. Rarely, hydrocarbon solvents (e.g. carbon tetrachloride) may be seen as a slightly opaque layer floating on the top of the gastric contents with the patient upright, or outlining the small bowel. Some enteric-coated or sustained-release drug formulations may be seen on plain abdominal radiographs, but, with the exception of iron salts, ordinary formulations are seldom seen. Ingested packets of illicit substances can sometimes been seen on CT (see p. 912). Radiology can confirm complications of poisoning, e.g. aspiration pneumonia, non-cardiogenic pulmonary oedema (salicylates), acute respiratory distress syndrome (ARDS).
Specific poisons: drugs
In this section, only specific treatment regimens will be discussed. The general principles of management of self-poisoning will always be required.
Amfetamines including ecstasy (MDMA)
The medicinal product is usually the dextro-isomer, dexamfetamine. The N-methylated derivative, metamfetamine (the crystalline form of this salt is known as ‘crystal meth’ or ‘ice’), and 3,4-methylenedioxymetamfetamine (MDMA), commonly known as ecstasy, are used worldwide.
Amfetamines are CNS and cardiovascular stimulants. These effects are mediated by increasing synaptic concentrations of adrenaline (epinephrine) and dopamine.
Clinical features
Amfetamines cause euphoria, extrovert behaviour, a lack of desire to eat or sleep, tremor, dilated pupils, tachycardia and hypertension. More severe intoxication is associated with agitation, paranoid delusions, hallucinations and violent behaviour. Convulsions, rhabdomyolysis, hyperthermia and cardiac arrhythmias may develop in severe poisoning. Rarely, intracerebral and subarachnoid haemorrhage occur and can be fatal.
MDMA poisoning is characterized by agitation, tachycardia, hypertension, widely dilated pupils, trismus and sweating. In more severe cases, hyperthermia, disseminated intravascular coagulation, rhabdomyolysis, acute kidney injury and hyponatraemia (secondary to inappropriate antidiuretic hormone secretion) predominate.
Anticonvulsants
Clinical features
The clinical features of poisoning with anticonvulsant drugs are summarized in Table 17.19.
Table 17.19 Clinical features of poisoning with anticonvulsant drugs
| Anticonvulsant drug | Clinical features of poisoning |
|---|---|
Carbamazepine |
Dry mouth, coma, convulsions, ataxia, incoordination, hallucinations (particularly in the recovery phase) |
Ocular: nystagmus dilated pupils (common), divergent strabismus, complete external ophthalmoplegia (rare) |
|
Phenytoin |
Nausea, vomiting, headache, tremor, cerebellar ataxia, nystagmus, loss of consciousness (rare) |
Sodium valproate |
Most frequent: drowsiness, impairment of consciousness, respiratory depression |
Uncommon complications: liver damage, hyperammonaemia, metabolic acidosis |
|
Very severe poisoning: myoclonic jerks and seizures; cerebral oedema has been reported |
|
Gabapentin and pregabalin |
Lethargy, ataxia, slurred speech and gastrointestinal symptoms |
Lamotrigine |
Lethargy, coma, ataxia, nystagmus, seizures, cardiac conduction abnormalities |
Levetiracetam |
Lethargy, coma, respiratory depression |
Tiagabine |
Lethargy, facial grimacing, nystagmus, posturing, agitation, coma, hallucinations, seizures |
Topiramate |
Lethargy, ataxia, nystagmus, myoclonus, coma, seizures, non-anion gap metabolic acidosis |
Metabolic acidosis can appear within hours of ingestion and persist for days |
Treatment
Multiple-dose activated charcoal has been shown to significantly increase elimination of carbamazepine. Early intravenous supplementation with L-carnitine should be used in severe valproate poisoning if hepatotoxicity and encephalopathy are present. Haemodialysis should also be instituted if severe hyperammonaemia and electrolyte and acid–base disturbances occur.
Antidepressants: MAOIs
Monoamine oxidase inhibitors are now used less frequently in the treatment of depression because of the dangers of dietary and drug interactions.
Clinical features
Features after overdose may be delayed for 12–24 hours and include excitement, restlessness, hyperpyrexia, hyper-reflexia, convulsions, opisthotonos, rhabdomyolysis and coma. Sinus tachycardia and either hypo- or hypertension have also been observed.
Treatment
Treatment is supportive with control of convulsions and marked excitement; diazepam 10–20 mg i.v. should be given as necessary and repeated. Dantrolene 1 mg/kg i.v. should be administered if hyperpyrexia develops. Hypotension should be treated with plasma expansion and hypertension by the administration of an α-adrenoceptor blocker such as chlorpromazine.
Antidepressants: tricyclics and SSRIs
Tricyclic antidepressants block the reuptake of noradrenaline (norepinephrine) into peripheral and intracerebral neurones, thereby increasing the concentration of monoamines in these areas, and also have antimuscarinic actions and class 1 antiarrhythmic (quinidine-like) activity. Citalopram, fluoxetine, fluvoxamine, paroxetine and sertraline are selective serotonin reuptake inhibitors (SSRIs) and lack the antimuscarinic actions of tricyclic antidepressants.
Clinical features
Even large overdoses of SSRIs appear to be relatively safe unless potentiated by ethanol. Most patients will show no signs of toxicity but drowsiness, nausea, diarrhoea and sinus tachycardia have been reported. Rarely, junctional bradycardia, seizures and hypertension have been encountered and influenza-like symptoms may develop. In contrast, drowsiness, sinus tachycardia, dry mouth, dilated pupils, urinary retention, increased reflexes and extensor plantar responses are the most common features of mild tricyclic antidepressant poisoning. Severe intoxication leads to coma, often with divergent strabismus and convulsions. Plantar, oculocephalic and oculovestibular reflexes may be abolished temporarily. An ECG will often show a wide QRS interval and there is a reasonable correlation between the width of the QRS complex and the severity of poisoning. Metabolic acidosis and cardiorespiratory depression are observed in severe cases.
Treatment
The majority of patients recover with supportive therapy alone (adequate oxygenation, control of convulsions and correction of acidosis), although a small percentage of patients who ingest a tricyclic will require assisted ventilation for 24–48 hours. The onset of supraventricular tachycardia and ventricular tachycardia should be treated with sodium bicarbonate (8.4%) 50 mmol intravenously over 20 min, even if there is no acidosis present. If ventricular tachycardia is compromising cardiac output, amiodarone 300 mg i.v. over 20–60 min should be administered.
FURTHER READING
Bradberry SM, Thanacoody HK, Watt BE et al. Management of the cardiovascular complications of tricyclic antidepressant poisoning: role of sodium bicarbonate. Toxicol Rev 2005; 24:195–204.
Isbister GK, Bowe SJ, Dawson A et al. Relative toxicity of selective serotonin reuptake inhibitors (SSRIs) in overdose. Clin Toxicol 2004; 42:277–285.
Thanacoody HK, Thomas SH. Tricyclic antidepressant poisoning: cardiovascular toxicity. Toxicol Rev 2005; 24:205–214.
Antidiabetic drugs
Insulin (if injected but not if ingested) and sulfonylureas cause hypoglycaemia, not seen with metformin, since its mode of action is to increase glucose utilization, but lactic acidosis is a potentially serious complication of metformin poisoning.
Clinical features
Features of severe hypoglycaemia include drowsiness, coma, convulsions, depressed limb reflexes, extensor plantar responses and cerebral oedema. Hypokalaemia may be associated. Neurogenic diabetes insipidus and persistent vegetative states are possible long-term complications if hypoglycaemia is prolonged. Cholestatic jaundice has been described as a late complication of chlorpropamide poisoning.
Treatment
The blood or plasma glucose concentration should be measured urgently and intravenous glucose given, if necessary. Glucagon produces only a slight rise in blood glucose, although it can reduce the amount of glucose required (see p. 1001).
Severe insulin poisoning. A continuous infusion of 10–20% glucose (with K+ 10–20 mmol/L) together with carbohydrate-rich meals are required, though there may be difficulty in maintaining normoglycaemia.
Sulfonylurea poisoning. The administration of glucose increases already high circulating insulin concentrations. Octreotide (50 µg i.v.), which inhibits insulin release, should be given as well as glucose.
Antimalarials
Clinical features
Chloroquine. Hypotension is often the first clinical manifestation of chloroquine poisoning. It may progress to acute heart failure, pulmonary oedema and cardiac arrest. Agitation and acute psychosis, convulsions and coma may ensue. Hypokalaemia is common and is due to chloroquine-induced potassium channel blockade. Bradyarrhythmias and tachyarrhythmias are common and ECG conduction abnormalities are similar to those seen in quinine poisoning.
Quinine. Cinchonism (tinnitus, deafness, vertigo, nausea, headache and diarrhoea) is common. In more severe poisoning, convulsions, hypotension, pulmonary oedema and cardiorespiratory arrest is seen (due to ventricular arrhythmias which are often preceded by ECG conduction abnormalities, particularly QT prolongation). Quinine cardiotoxicity is due to sodium channel blockade. Patients may also develop ocular features, including blindness, which can be permanent.
Primaquine. The main concern regarding primaquine is its propensity to cause methaemoglobinaemia and haemolytic anaemia.
Treatment
Multiple-dose oral activated charcoal increases quinine and probably chloroquine clearance. Hypokalaemia should be corrected. Sodium bicarbonate 50–100 mmol i.v. is given if the ECG shows intraventricular block but will exacerbate hypokalaemia, which should be corrected first. Mechanical ventilation, the administration of an inotrope and high doses of diazepam (1 mg/kg as a loading dose and 0.25–0.4 mg/kg per hour maintenance) may reduce the mortality in severe chloroquine poisoning. Overdrive pacing may be required if torsades de pointes (p. 710) occurs in quinine poisoning and does not respond to magnesium sulphate infusion. If clinically significant methaemoglobinaemia (generally above 30%) develops in primaquine poisoning, methylthioninium (methylene blue) 1–2 mg/kg body weight should be administered.
β-Adrenoceptor blocking drugs
Clinical features
In mild poisoning, sinus bradycardia is the only feature, but if a substantial amount has been ingested, coma, convulsions and hypotension develop. Less commonly, delirium, hallucinations and cardiac arrest supervene.
Treatment
Glucagon 50–150 µg/kg (typically 5–10 mg in an adult) followed by an infusion of 1–5 mg/h is the most effective agent. It acts by by-passing the blocked beta-receptor thus activating adenyl cyclase and promoting formation of cyclic AMP from ATP; cyclic AMP in turn exerts a direct beta-stimulant effect on the heart. Atropine 0.6–1.2 mg i.v. can be used to treat bradycardia but is usually less effective.
Benzodiazepines
Benzodiazepines are commonly taken in overdose but rarely produce severe poisoning except in the elderly or those with chronic respiratory disease.
Clinical features
Benzodiazepines produce drowsiness, ataxia, dysarthria and nystagmus. Coma and respiratory depression develop in severe intoxication.
Treatment
If respiratory depression is present in patients who have severe benzodiazepine poisoning, flumazenil 0.5–1.0 mg i.v. is given in an adult and this dose often needs repeating. Flumazenil use often avoids ventilation. It is contraindicated in mixed tricyclic antidepressant (TCA)/benzodiazepine poisoning and in those with a history of epilepsy because it may cause convulsions.
Calcium channel blockers
Calcium channel blockers all act by blocking voltage-gated calcium channels. Dihydropyridines (e.g. amlodipine, felodipine, nifedipine) are predominantly peripheral vasodilators while verapamil and, to a lesser extent, diltiazem also have significant cardiac effects. Poisoning, particularly with verapamil and diltiazem, causes heart block and hypotension and there is a substantial fatality rate.
Clinical features
Hypotension occurs due to peripheral vasodilatation, myocardial depression and conduction block. The electrocardiogram may progress from sinus bradycardia through first, then higher, degrees of block, to asystole. Cardiac and non-cardiac pulmonary oedema may ensue in severely poisoned patients. Other features include nausea, vomiting, seizures and a lactic acidosis. When a sustained-release preparation has been ingested the onset of severe features is delayed, sometimes for more than 12 hours. Overdose with even small amounts can have profound effects.
Treatment
Intravenous atropine 0.6–1.2 mg, repeated as required, should be given for bradycardia and heart block. The initial dose can be repeated every 3–5 min but if there is no response in pulse rate or blood pressure after three such doses it is unlikely that further boluses will be helpful. The response to atropine is sometimes improved following intravenous 10% calcium chloride, 5–10 mL (at 1–2 mL/min). If there is an initial response to calcium, a continuous infusion is warranted; this is given as 10% calcium chloride, 1–10 mL/h.
Cardiac pacing has a role if there is evidence of AV conduction delay but failure to capture occurs.
Treat hypotension initially with intravenous crystalloid. If significant hypotension persists despite volume replacement, administer glucagon (see p. 409) as it activates myosin kinase independent of calcium. Give i.v. glucagon 10 mg (150 µg/kg) as a slow bolus and repeat. If there is a favourable response in blood pressure, an infusion of 5–10 mg/h can be commenced; if there is no response after the initial boluses, discontinue.
Insulin–glucose euglycaemia has been shown to improve myocardial contractility and systemic perfusion. If hypotension persists despite the above measures, insulin is given as a bolus dose of 1 U/kg, followed by an infusion of 1–10 U/kg per hour with 10% glucose and frequent monitoring of blood glucose and potassium.
Acidosis impairs L-type channel function (see p. 708) and is corrected by the administration of sodium bicarbonate, which has been shown experimentally to improve myocardial contractility and cardiac output.
Cannabis (marijuana)
Cannabis is usually smoked but may be ingested as a ‘cake’, made into a tea or injected intravenously. Apart from alcohol, it is the drug most widely used in developed countries. The major psychoactive constituent is delta-11-tetrahydrocannabinol (THC). THC possesses activity at the benzodiazepine, opioid and cannabinoid receptors. Street names include pot, grass, ganga, reefs and spliff. It is prepared as marijuana (ganga) from the female flowers; hashish or charas – a concentrated resin of glandular trichomes; kief – chopped female plants; and bhang – a drink prepared from cannabis leaves boiled in milk with spices.
Clinical features
Initially there is euphoria, followed by distorted and heightened images, colours and sounds, altered tactile sensations and sinus tachycardia. Visual and auditory hallucinations and acute psychosis are particularly likely to occur after substantial ingestion in naive cannabis users. Intravenous injection leads to watery diarrhoea, tachycardia, hypotension and arthralgia.
Heavy users suffer impairment of memory and attention and poor academic performance. There is an increased risk of anxiety and depression. Regular users are at risk of dependence. Cannabis use results in an overall increase in the relative risk for later schizophrenia and psychotic episodes (see p. 1185). Cannabis smoke is probably carcinogenic.
Cocaine
Cocaine hydrochloride (‘street’ cocaine, ‘coke’) is a water-soluble powder or granule that can be taken orally, intravenously or intranasally. ‘Freebase’ or ‘crack’ cocaine comprises crystals of relatively pure cocaine without the hydrochloride moiety and is obtained in rocks (150 mg of cocaine). It is more suitable for smoking in a pipe or mixed with tobacco and can also be heated on foil and the vapour inhaled (approximately 35 mg of drug per ‘line’ or a ‘rail’). The ‘effects’ of cocaine are experienced almost immediately with i.v. or smoking routes, about 10 min in the intranasal route and 45–90 min when taken orally. The effects start resolving in about 20 min and last up to 90 min. In severe poisoning, death occurs in minutes but survival beyond 3 hours is not usually fatal.
Cocaine blocks the reuptake of biogenic amines. Inhibition of dopamine reuptake is responsible for the psychomotor agitation which commonly accompanies cocaine use. Blockade of noradrenaline (norepinephrine), reuptake produces tachycardia, and inhibition of serotonin reuptake induces hallucinations. Cocaine also enhances CNS arousal by potentiating the effects of excitatory amino acids. Cocaine is also a powerful local anaesthetic and vasoconstrictor.
Clinical features
After initial euphoria, cocaine produces agitation, tachycardia, hypertension, sweating, hallucinations, convulsions, metabolic acidosis, hyperthermia, rhabdomyolysis and ventricular arrhythmias. Dissection of the aorta, myocarditis, myocardial infarction, dilated cardiomyopathy, subarachnoid haemorrhage, and cerebral haemorrhage and infarction also occur. If a young person presents with a stroke or myocardial infarction, cocaine poisoning, because of its vasoconstrictor effect, is a possible diagnosis.
Treatment
Diazepam 10–20 mg i.v. is used to control agitation and convulsions. Active external cooling should be used for hyperthermia. Hypertension and tachycardia usually respond to sedation and cooling. If hypertension persists, give i.v. nitrates such as glyceryl trinitrate starting at 1–2 mg/h and gradually increase the dose (maximum 12 mg/h) until BP is controlled. Calcium channel blockers such as nifedipine, verapamil or diltiazem are an alternative as second-line therapy. The use of beta-blockers is controversial. Early use of a benzodiazepine is often effective in relieving cocaine-associated non-cardiac chest pain. Aspirin and nitrates should be given to all people with chest pain suspected of being cardiac in origin. Treat myocardial ischaemia/infarction conventionally.
Digoxin
Toxicity occurring during chronic administration is common, though acute poisoning is infrequent.
Clinical features
These include nausea, vomiting, dizziness, anorexia and drowsiness. Rarely, confusion, visual disturbances and hallucinations occur. Sinus bradycardia is often marked and may be followed by supraventricular arrhythmias with or without heart block, ventricular premature beats and ventricular tachycardia. Hyperkalaemia occurs due to the inhibition of the sodium-potassium activated ATPase pump.
Treatment
Sinus bradycardia, atrioventricular block and sinoatrial standstill are often reduced or even abolished by atropine 1.2–2.4 mg i.v. If cardiac output is compromised, however, digoxin-specific antibody fragments (digoxin-Fab) should be administered. In both acute and chronic poisoning, only half the estimated dose (calculated from amount of drug taken or serum digoxin concentration) required for full neutralization need be given initially; a further dose is given if clinically indicated.
Gamma-hydroxybutyric acid (GHB)
Gamma-hydroxybutyric acid occurs naturally in mammalian brain where it is derived metabolically from gamma-aminobutyric acid (GABA). GHB has emerged as a major recreational drug for body building, weight loss and for producing a ‘high’. Street names include cherry meth and liquid X. It is taken as a colourless liquid dissolved in water.
Clinical features
Poisoning with GHB is characterized by aggressive behaviour, ataxia, amnesia, vomiting, drowsiness, bradycardia, respiratory depression and apnoea, seizures and characteristically coma, which is short-lived.
Management
In a patient who is breathing spontaneously, the management of GHB poisoning is primarily supportive with oxygen supplementation and the administration of atropine for persistent bradycardia, as necessary. Those who are severely poisoned will require mechanical ventilation, although recovery is usually complete within 6–8 hours.
Iron
Unless more than 60 mg of elemental iron per kg of body weight is ingested (a ferrous sulphate tablet contains 60 mg of iron), features are unlikely to develop. As a result poisoning is seldom severe but deaths still occur. Iron salts have a direct corrosive effect on the upper gastrointestinal tract.
Clinical features
The initial features are characterized by nausea, vomiting (the vomit may be grey or black in colour), abdominal pain and diarrhoea. Severely poisoned patients develop haematemesis, hypotension, coma and shock at an early stage. Usually, however, most patients only suffer mild gastrointestinal symptoms. A small minority deteriorate 12–48 hours after ingestion and develop shock, metabolic acidosis, acute tubular necrosis and hepatocellular necrosis. Rarely, up to 6 weeks after ingestion, intestinal strictures due to corrosive damage occur. The serum iron concentration should be measured some 4 hours after ingestion and if the concentration exceeds the predicted normal iron binding capacity (usually >5 mg/L; 90 µmol/L), free iron is circulating and treatment with desferrioxamine is required.
Treatment
The majority of patients ingesting iron do not require desferrioxamine therapy. If a patient develops coma or shock, desferrioxamine should be given without delay in a dose of 15 mg/kg per hour i.v. (total amount of infusion usually not to exceed 80 mg/kg in 24 hours). If the recommended rate of administration is continued for several days, adverse effects including pulmonary oedema and ARDS have been reported.
Lithium
Lithium toxicity is usually the result of therapeutic overdosage (chronic toxicity) rather than deliberate self-poisoning (acute toxicity). However, single large doses are occasionally ingested by individuals on long-term treatment with the drug (acute on therapeutic toxicity).
Clinical features
Features of intoxication include thirst, polyuria, diarrhoea and vomiting and in more serious cases impairment of consciousness, hypertonia and convulsions; irreversible neurological damage occurs. Measurement of the serum lithium concentration confirms the diagnosis. Acute massive overdose may produce concentrations of 5 mmol/L (34.7 mg/L) without causing toxic features, whereas chronic toxicity is associated with neurological features at concentrations >1.5 mmol/L (6.94 mg/L).
Treatment
Forced diuresis with sodium chloride 0.9% is effective in increasing elimination of lithium, though haemodialysis is far superior and should be undertaken particularly if neurological features are present, if renal function is impaired and if chronic toxicity or acute on chronic toxicity are the modes of presentation.
Neuroleptics and atypical neuroleptics
Neuroleptic (antipsychotic) drugs are thought to act predominantly by blockade of the dopamine D2 receptors. Older neuroleptics include the phenothiazines, the butyrophenones (benperidol, haloperidol) and the substituted benzamides (sulpiride). More selective ‘atypical’ antipsychotics include amisulpride, aripiprazole, clozapine, olanzapine, quetiapine and risperidone.
Clinical features
These include impaired consciousness, hypotension, respiratory depression, hypothermia or hyperthermia, antimuscarinic effects such as tachycardia, dry mouth and blurred vision, occasionally seizures, rhabdomyolysis, cardiac arrhythmias (both atrial and ventricular) and acute respiratory distress syndrome. Extrapyramidal effects, including acute dystonic reactions, occur but are not dose related. Most ‘atypical’ antipsychotics have less profound sedative actions than the older neuroleptics. Q–T interval prolongation and subsequent ventricular arrhythmias (including torsades de pointes) have occurred following overdose with the atypical neuroleptics. Unpredictable fluctuations in conscious level, with variations between agitation and marked somnolence, have been particularly associated with olanzapine overdose.
Treatment
Procyclidine 5–10 mg i.v. in an adult is occasionally required for the treatment of dyskinesia and oculogyric crisis. If hypotension is severe and does not respond to intravenous fluids, a sympathomimetic amine such as noradrenaline (norepinephrine) is used. After correcting acidosis with sodium bicarbonate, the preferred treatment for arrhythmias caused by antipsychotic drugs (usually torsades de pointes) is intravenous magnesium or cardiac pacing. Amiodarone is used if multifocal ventricular arrhythmias occur.
Non-steroidal anti-inflammatory agents (NSAIDs)
Self-poisoning with NSAIDs has increased, particularly now that ibuprofen is available without prescription in many countries.
Clinical features and treatment
In most cases minor gastrointestinal disturbance is the only feature but, in more severe cases, coma, convulsions and acute kidney injury have occurred. Transient renal impairment is common after ibuprofen overdose. Poisoning with mefenamic acid commonly results in convulsions though these are usually short-lived.
Opiates and opioids
Clinical features
Cardinal signs of opiate poisoning are pinpoint pupils, reduced respiratory rate and coma. Hypothermia, hypoglycaemia and convulsions are occasionally observed in severe cases. In severe heroin overdose, non-cardiogenic pulmonary oedema has been reported.
Treatment
Naloxone 1.2–2.0 mg i.v. will reverse at least partially severe respiratory depression and coma. In severe poisoning larger initial doses or repeat doses will be required. The duration of action of naloxone is often less than the drug taken in overdose, e.g. methadone, which has a very long half-life. For this reason an infusion of naloxone is often required. Non-cardiogenic pulmonary oedema should be treated with mechanical ventilation.
Paracetamol (acetaminophen)
Paracetamol is the most common form of poisoning encountered in the UK. In therapeutic dose, paracetamol is conjugated with glucuronide and sulphate. A small amount of paracetamol is metabolized by mixed function oxidase enzymes to form a highly reactive compound (N-acetyl-p-benzoquinoneimine, NAPQI), which is then immediately conjugated with glutathione and subsequently excreted as cysteine and mercapturic conjugates. In overdose, large amounts of paracetamol are metabolized by oxidation because of saturation of the sulphate conjugation pathway. Liver glutathione stores become depleted so that the liver is unable to deactivate the toxic metabolite. Paracetamol-induced renal damage probably results from a mechanism similar to that which is responsible for hepatotoxicity.
The severity of paracetamol poisoning is dose related. There is, however, some variation in individual susceptibility to paracetamol-induced hepatotoxicity. People with pre-existing liver disease, those suffering from acute or chronic starvation (patients not eating for a few days for example due to a recent febrile illness or dental pain), those suffering from anorexia nervosa and other eating disorders, those receiving enzyme-inducing drugs, and those with HIV infection should be considered to be at greater risk and given treatment at plasma paracetamol concentrations lower than those normally used for interpretation (Fig. 17.2).
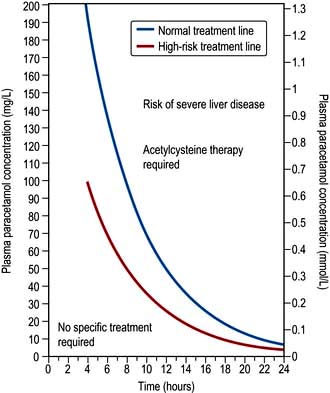
Clinical features
Following the ingestion of an overdose of paracetamol, patients usually remain asymptomatic for the first 24 hours or at the most develop anorexia, nausea and vomiting. Liver damage is not usually detectable by routine liver function tests until at least 18 hours after ingestion of the drug. Liver damage usually reaches a peak, as assessed by measurement of alanine transferase (ALT) activity and prothrombin time (INR), at 72–96 hours after ingestion. Without treatment, a small percentage of patients will develop fulminant hepatic failure. Acute kidney injury due to acute tubular necrosis occurs in 25% of patients who have severe hepatic damage and in a few without evidence of serious disturbance of liver function.
Treatment
The treatment protocol is dependent on the time of presentation and this is summarized in Table 17.20. Acetylcysteine has emerged as an effective protective agent provided that it is administered within 8–10 hours of ingestion of the overdose. It acts by replenishing cellular glutathione stores, though it may also repair oxidation damage caused by NAPQI. The treatment regimen is shown in Table 17.21. If a staggered overdose has been taken (multiple ingestions over several hours), acetylcysteine should be given when the paracetamol dose exceeds 150 mg/kg body weight in any one 24-hour period or 75 mg/kg body weight in those at high risk (see above).
Table 17.20 Management of people with paracetamol poisoning
≤8 h after ingestion |
|
|
8–15 h after ingestion |
|
|
15–24 h after ingestion |
|
|
Table 17.21 Regimen for acetylcysteine
Up to 15% of patients treated with intravenous acetylcysteine (20.25-h regimen) develop rash, angio-oedema, hypotension and bronchospasm. These reactions, which are related to the initial bolus, are seldom serious and discontinuing the infusion is usually all that is required. In more severe cases, chlorphenamine 10–20 mg i.v. in an adult should be given.
If liver or renal failure ensues, this should be treated conventionally though there is evidence that a continuing infusion of acetylcysteine (continue 16-h infusion until recovery) will improve the morbidity and mortality. Liver transplantation has been performed successfully in patients who have paracetamol-induced fulminant hepatic failure (see p. 316).
Salicylates
Aspirin is metabolized to salicylic acid (salicylate) by esterases present in many tissues, especially the liver, and subsequently to salicyluric acid and salicyl phenolic glucuronide (Fig. 17.3); these two pathways become saturated with the consequence that the renal excretion of salicylic acid increases after overdose; this excretion pathway is extremely sensitive to changes in urinary pH.
Clinical features
Salicylates stimulate the respiratory centre, increase the depth and rate of respiration, and induce a respiratory alkalosis. Compensatory mechanisms, including renal excretion of bicarbonate and potassium, result in a metabolic acidosis. Salicylates also interfere with carbohydrate, fat and protein metabolism, and disrupt oxidative phosphorylation, producing increased concentrations of lactate, pyruvate and ketone bodies, all of which contribute to the acidosis.
Thus, tachypnoea, sweating, vomiting, epigastric pain, tinnitus and deafness develop. Respiratory alkalosis and metabolic acidosis supervene and a mixed acid–base disturbance is observed commonly. Rarely, in severe poisoning, non-cardiogenic pulmonary oedema, coma and convulsions ensue.
Treatment
Fluid and electrolyte replacement is required and special attention should be paid to potassium supplementation. Severe metabolic acidosis requires at least partial correction with the administration of sodium bicarbonate intravenously. Mild cases of salicylate poisoning are managed with parenteral fluid and electrolyte replacement only. Patients whose plasma salicylate concentrations are in excess of 500 mg/L (3.62 mmol/L) should receive urine alkalinization (see p. 913). Haemodialysis is the treatment of choice for severely poisoned patients (plasma salicylate concentration >700 mg/L; >5.07 mmol/L), particularly those with coma and metabolic acidosis.
Theophylline
Poisoning may complicate therapeutic use as well as being the result of deliberate self-poisoning. If a slow-release preparation is involved, peak plasma concentrations are not attained until 6–12 hours after overdosage and the onset of toxic features is correspondingly delayed.
Clinical features
Nausea, vomiting, hyperventilation, haematemesis, abdominal pain, diarrhoea, sinus tachycardia, supraventricular and ventricular arrhythmias, hypotension, restlessness, irritability, headache, hyperreflexia, tremors and convulsions have been observed. Hypokalaemia probably results from activation of Na+/K+ ATPase. A mixed acid–base disturbance is common. Most symptomatic patients have plasma theophylline concentrations in excess of 25 mg/L (430 µmol/L). Convulsions are seen more commonly when concentrations are >50 mg/L (>860 µmol/L). Plasma potassium concentrations of <2.6 mmol/L, metabolic acidosis, hypotension, seizures and arrhythmias are indications of severe poisoning.
Treatment
There is good evidence that multiple-dose (12.5 g/h) activated charcoal enhances the elimination of theophylline. However, protracted theophylline-induced vomiting may mitigate the benefit of this therapy, unless vomiting is suppressed by a 5HT3 antagonist such as ondansetron 4–8 mg i.v. Correction of hypokalaemia to prevent or treat tachyarrhythmias is of great importance. A non-selective β-adrenoceptor blocking drug, such as propranolol, is also useful in the treatment of tachyarrhythmias secondary to hypokalaemia, but should not be given if the patient has severe airways disease. Convulsions should be treated with diazepam 10–20 mg i.v. in an adult.
Specific poisons: chemicals
Carbamate insecticides
Carbamate insecticides inhibit acetylcholinesterase, as do organophosphorus insecticides, but the duration of this inhibition is comparatively short-lived in comparison since the carbamate–enzyme complex tends to dissociate spontaneously. The clinical features and treatment are similar except that an oxime such as pralidoxime is not usually required because the enzyme complex dissociates spontaneously; recovery invariably occurs within 24 hours (see p. 923).
Carbon monoxide
The commonest source of carbon monoxide is an improperly maintained and poor, ventilated heating system. In addition, inhalation of methylene chloride (found in paint strippers) may also lead to carbon monoxide poisoning as methylene chloride is metabolized in vivo to carbon monoxide. The affinity of haemoglobin for carbon monoxide is some 240 times greater than that for oxygen. Carbon monoxide combines with haemoglobin to form carboxyhaemoglobin, thereby reducing the total oxygen carrying capacity of the blood and increasing the affinity of the remaining haem groups for oxygen. This results in tissue hypoxia. In addition, carbon monoxide also inhibits cytochrome oxidase a3.
Clinical features
Symptoms of mild to moderate exposure to carbon monoxide may be mistaken for a viral illness. A peak carboxyhaemoglobin (COHb) concentration of less than 10% is not normally associated with symptoms and peak COHb concentrations of 10–30% usually result only in headache and mild exertional dyspnoea. Higher concentrations of COHb are associated with coma, convulsions and cardiorespiratory arrest. Metabolic acidosis, myocardial ischaemia, hypertonia, extensor plantar responses, retinal haemorrhages and papilloedema also occur. Neuropsychiatric features may develop after apparent recovery from carbon monoxide exposure.
Treatment
In addition to removing the patient from carbon monoxide exposure, high flow oxygen should be administered using a tightly fitting face mask. Endotracheal intubation and mechanical ventilation is required in those who are unconscious. Several controlled studies of hyperbaric oxygen have been published but none have shown long-term clinical benefit.
Cyanide
Cyanide and its derivatives are used widely in industry. Hydrogen cyanide is also released during the thermal decomposition of polyurethane foams. Cyanide reversibly inhibits cytochrome oxidase a3 so that cellular respiration ceases.
Clinical features
Inhalation of hydrogen cyanide produces symptoms within seconds and death within minutes. By contrast, the ingestion of a cyanide salt may not produce features for 1 hour. After exposure initial symptoms are non-specific and include a feeling of constriction in the chest and dyspnoea. Coma, convulsions and metabolic acidosis may then supervene.
Treatment
Oxygen should be administered and, if available, dicobalt edetate 300 mg should be administered intravenously; the dose is repeated in severe cases. Dicobalt edetate (and the free cobalt contained in the preparation) complexes free cyanide. An alternative but expensive antidote is hydroxocobalamin, which enhances endogenous cyanide detoxification mechanisms: 5 g i.v. is administered, and a second dose may be required in severe cases. If these two antidotes are not available sodium thiosulphate 12.5 g i.v., which acts by enhancing endogenous detoxification, and sodium nitrite 300 mg i.v. should be administered. Sodium nitrite produces methaemoglobinaemia; methaemoglobin combines with cyanide to form cyanmethaemoglobin.
Ethanol
Ethanol is commonly ingested in beverages and deliberately with other substances in overdose. It is also present in many cosmetic and antiseptic preparations. Following absorption, ethanol is oxidized to acetaldehyde and then to acetate. Ethanol is a CNS depressant and the features of ethanol intoxication are generally related to blood concentrations (Table 17.22).
Table 17.22 Clinical features of ethanol poisoning
| Blood (ethanol) | Clinical features | |
|---|---|---|
| mg/L | mmol/L | |
500–1500 |
11.0–32.5 |
|
1500–3000 |
32.5–65.0 |
|
3000–5000 |
65.0–108.5 |
|
>5000 |
>108.5 |
|
Clinical features
In children in particular severe hypoglycaemia may accompany alcohol intoxication due to inhibition of gluconeogenesis. Hypoglycaemia is also observed in those who are malnourished or who have fasted in the previous 24 hours. In severe cases of intoxication, coma and hypothermia are often present and lactic acidosis, ketoacidosis and acute kidney injury have been reported.
Treatment
As ethanol-induced hypoglycaemia is not responsive to glucagon, i.v. glucose 10–20% should be infused at a rate determined by the blood sugar. Haemodialysis is used if the blood ethanol concentration exceeds 7500 mg/L and if a severe metabolic acidosis is present, which has not been corrected by i.v. fluids and bicarbonate.
Ethylene and diethylene glycol
Ethylene and diethylene glycol are found in a variety of common household products including antifreeze, windshield washer fluid, brake fluid and lubricants. The features observed are due to metabolites predominantly, not the parent chemical. Ethylene glycol (Fig. 17.4) is metabolized to glycolate, the cause of the acidosis. A small proportion of glyoxylate is metabolized to oxalate. Calcium ions chelate oxalate to form insoluble calcium oxalate, which is responsible for renal toxicity. Diethylene glycol is metabolized to 2-hydroxyethoxyacetate (Fig. 17.5), which is the cause of metabolic acidosis, and diglycolic acid (the cause of renal failure).

Figure 17.4 The metabolism of ethylene glycol. ADH, alcohol dehydrogenase; ALDH, aldehyde dehydrogenase; AO, aldehyde oxidase; GO, glycolate oxidase; LDH, lactate dehydrogenase.
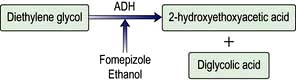
Clinical features
Initially, the features of ethylene glycol poisoning are similar to ethanol intoxication (though there is no ethanol on the breath). Coma and convulsions follow and a variety of neurological abnormalities, including nystagmus and ophthalmoplegias, are seen. Severe metabolic acidosis, hypocalcaemia and acute kidney disease are well-recognized complications. In diethylene glycol poisoning nausea and vomiting, headache, abdominal pain, coma, seizures, metabolic acidosis and acute kidney injury commonly occur. Pancreatitis and hepatitis, together with cranial neuropathies and demyelinating peripheral neuropathy, are also seen.
Treatment
If the patient presents early after ingestion, the first priority is to inhibit metabolism using either intravenous fomepizole or ethanol; the former does not require monitoring. Fomepizole 15 mg/kg body weight should be administered followed by four 12-hourly doses of 10 mg/kg, then 15 mg/kg every 12 hours until glycol concentrations are not detectable. Following a substantial ingestion, haemodialysis or haemodialfiltration should be employed to remove the glycol and metabolites. If dialysis is employed the frequency of fomepizole dosing should be increased to 4-hourly because fomepizole is dialysable. Alternatively, a loading dose of ethanol 50 g should be administered followed by an i.v. infusion of ethanol 10–12 g/h to produce blood ethanol concentrations of 500–1000 mg/L (11–22 mmol/L). The infusion is continued until the glycol is no longer detectable in the blood. If haemodialysis is employed, the rate of ethanol administration will need to be increased to 17–22 g/h as ethanol is dialysable. Supportive measures to combat shock, hypocalcaemia and metabolic acidosis should be instituted.
Household products
The agents most commonly involved are bleach, cosmetics, toiletries, detergents, disinfectants and petroleum distillates such as paraffin and white spirit. Ingestion of household products is usually accidental and is most common among children less than 5 years of age.
Clinical features
If the ingestion is accidental, features very rarely occur except in the case of petroleum distillates where aspiration is a recognized complication because of their low surface tension. Powder detergents, sterilizing tablets, denture cleaning tablets and industrial bleaches (which contain high concentrations of sodium hypochlorite) are corrosive to the mouth and pharynx if ingested. Nail polish and nail polish remover contain acetone that may produce coma if ingested in substantial quantities. Inhalation by small children of substantial quantities of talcum powder has occasionally given rise to severe pulmonary oedema and death.
Lead
Exposure to lead occurs occupationally, children may eat lead-painted items in their homes (pica) and the use of lead-containing cosmetics or ‘drugs’ has also resulted in lead poisoning.
Clinical features
Mild intoxication may result in no more than lethargy and occasional abdominal discomfort, though abdominal pain, vomiting, constipation and encephalopathy (seizures, delirium, coma) may develop in more severe cases. Encephalopathy is more common in children than in adults but is now rare in the developed world. Typically, though very rarely, lead poisoning results in foot drop attributable to peripheral motor neuropathy. A bluish discoloration of the gum margins due to the deposition of lead sulphide is observed occasionally.
The characteristic haematological features include:
Normochromic normocytic anaemia, due to inhibition by lead of several enzymes involved in haem synthesis, including ALA synthetase
Haemolysis, which is usually mild, resulting from damage to the red cell membrane
Punctate basophilia (or basophilic stippling: the blood film shows red cells with small, round, blue particles), due to aggregates of RNA in immature red cells owing to inhibition by lead of pyrimidine-5-nucleotidase, which normally disperses residual RNA to produce a diffuse blue staining seen in reticulocytes on blood films (polychromasia).
Treatment
The social and occupational dimensions of lead poisoning must be recognized. Simply giving the patient chelation therapy and then returning them to a contaminated environment is of no value.
The decision to use chelation therapy is based not only on the blood lead concentration but on the presence of symptoms. Parenteral sodium calcium edetate 75 mg/kg per day has been the chelating agent of choice for over 50 years, but there is now evidence to suggest that oral DMSA 30 mg/kg per day is of similar efficacy. At least 5 days’ treatment is usually required.
Mercury
Mercury is the only metal that is liquid at room temperature. It exists in three oxidation states (elemental/metallic Hg0, mercurous Hg22+ and mercuric Hg2+) and can form inorganic (e.g. mercuric chloride) and organic (e.g. methylmercury) compounds. Metallic mercury is very volatile and when spilled, has a large surface area so that high atmospheric concentrations may be produced in enclosed spaces, particularly when environmental temperatures are high. Thus, great care should be taken in clearing up a spillage. If ingested, metallic mercury will usually be eliminated per rectum, though small amounts may be found in the appendix. Mercury salts are well absorbed following ingestion as are organometallic compounds where mercury is covalently bound to carbon.
Clinical features
Inhalation of acute mercury vapour causes headache, nausea, cough, chest pain, bronchitis and occasionally pneumonia. Proteinuria and nephrotic syndrome are observed rarely. In addition a fine tremor and neurobehavioural impairment occurs and peripheral nerve involvement has also been observed. Ingestion of inorganic and organic mercury compounds causes an irritant gastroenteritis with corrosive ulceration, bloody diarrhoea and abdominal cramps and may lead to circulatory collapse and shock. Mercurous compounds are less corrosive and toxic than mercuric salts.
Methanol
Methanol is used widely as a solvent and is found in antifreeze solutions. Methanol is metabolized to formaldehyde and formate (Fig. 17.6). The concentration of formate increases greatly and is accompanied by accumulation of hydrogen ions causing metabolic acidosis.
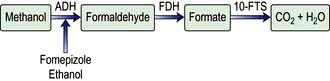
Figure 17.6 The metabolism of methanol. ADH, alcohol dehydrogenase; FDH, formaldehyde dehydrogenase; 10-FTS, 10-formyl tetrahydrofolate synthetase.
Clinical features
Methanol causes inebriation and drowsiness. After a latent period coma supervenes. Blurred vision and diminished visual acuity occur due to formate accumulation. The presence of dilated pupils that are unreactive to light suggests that permanent blindness is likely to ensue. A severe metabolic acidosis may develop and be accompanied by hyperglycaemia and a raised serum amylase activity. A blood methanol concentration of 500 mg/L (15.63 mmol/L) confirms severe poisoning. The mortality correlates well with the severity and duration of metabolic acidosis. Survivors may show permanent neurological sequelae including parkinsonian-like signs as well as blindness.
Treatment
Treatment is similar to that of ethylene glycol poisoning (see p. 921) with the addition of folinic acid 30 mg i.v. 6-hourly, which accelerates formate metabolism thereby reducing ocular toxicity.
Nerve agents
Nerve agents are related chemically to organophosphorus insecticides (see below) and have a similar mechanism of toxicity, but a much higher mammalian acute toxicity, particularly via the dermal route. In addition to inhibition of acetylcholinesterase, a chemical reaction known as ‘ageing’ also occurs rapidly and more completely than in the case of insecticides. This makes the enzyme resistant to spontaneous reactivation or by treatment with oximes (pralidoxime, obidoxime or HI-6).
Two classes of nerve agent are recognized: G agents (named for Gerhardt Schrader who synthesized the first agents) and V agents (V allegedly stands for venomous). G agents include tabun, sarin, soman and cyclosarin. The V agents were introduced later, e.g. VX. The G agents are both dermal and respiratory hazards, whereas the V agents, unless aerosolized, are contact poisons.
Agents used in bioterrorism are described on page 935.
Clinical features
Systemic features include increased salivation, rhinorrhoea, bronchorrhoea, miosis and eye pain, abdominal pain, nausea, vomiting and diarrhoea, involuntary micturition and defecation, muscle weakness and fasciculation, tremor, restlessness, ataxia and convulsions. Bradycardia, tachycardia and hypotension occur, dependent on whether muscarinic or nicotinic effects predominate. Death occurs from respiratory failure within minutes but mild or moderately exposed individuals usually recover completely. Diagnosis is confirmed by measuring the erythrocyte cholinesterase activity
Treatment
The administration of atropine 2 mg i.v. repeated every 3–5 min as necessary to patients presenting with rhinorrhoea and bronchorrhoea may be life-saving. In addition, an oxime should be given to all patients requiring atropine as soon as possible after exposure before ‘ageing’ has occurred. For example, pralidoxime chloride 30 mg/kg i.v., followed by an infusion of pralidoxime chloride 8–10 mg/kg per hour; alternatively boluses of pralidoxime chloride 30 mg/kg may be given 4- to 6-hourly. Intravenous diazepam 10–20 mg, repeated as required, is useful in controlling apprehension, agitation, fasciculation and convulsions.
Organophosphorus insecticides
Organophosphorus (OP) insecticides are used widely throughout the world and are a common cause of poisoning, causing thousands of deaths annually, in the developing world. Intoxication may follow ingestion, inhalation or dermal absorption. Organophosphorus insecticides inhibit acetylcholinesterase causing accumulation of acetylcholine at central and peripheral cholinergic nerve endings, including neuromuscular junctions. Many OP insecticides require biotransformation before becoming active and so the features of intoxication may be delayed.
Clinical features
Poisoning is characterized by anxiety, restlessness, tiredness, headache, and muscarinic (cholinergic) features such as nausea, vomiting, abdominal colic, diarrhoea, tenesmus, sweating, hypersalivation and chest tightness. Miosis may be present. Nicotinic effects include muscle fasciculation and flaccid paresis of limb muscles, respiratory muscles, and, occasionally of extraocular muscles. Respiratory failure will ensue in severe cases and is exacerbated by the development of bronchorrhoea and pulmonary oedema. Coma and convulsions occur in severe poisoning. Diagnosis is confirmed by measuring the erythrocyte cholinesterase activity; plasma cholinesterase activity is less specific but may also be depressed. Delayed polyneuropathy is a rare complication of acute exposure to some OP insecticides not marketed in most countries. It is initiated by phosphorylation and subsequent ageing of at least 70% of neuropathy target esterase (NTE) in peripheral nerves.
The intermediate syndrome usually becomes established 2–4 days after exposure when the symptoms and signs of the acute cholinergic syndrome are no longer obvious. The characteristic features of the syndrome are weakness of the muscles of respiration (diaphragm, intercostal muscles and accessory muscles including neck muscles) and of proximal limb muscles. Accompanying features often include weakness of muscles innervated by some cranial nerves.
Treatment
Mild cases require no specific treatment other than the removal of soiled clothing. Atropine 2 mg i.v. should be given every 3–5 min if necessary to reduce increased secretions, rhinorrhoea and bronchorrhoea. Symptomatic patients should also be given an oxime (pralidoxime, obidoxime or HI-6) to reactivate inhibited acetylcholinesterase: for example, pralidoxime chloride 30 mg/kg by slow i.v. injection followed by an infusion of pralidoxime mesylate 8–10 mg/kg per hour.
Phosphides
Aluminium and zinc phosphides are used as rodenticides and insecticides. They react with moisture in the air (and the gastrointestinal tract) to produce phosphine, the active pesticide. Acute poisoning with these compounds may be direct, due to ingestion of the salts, or indirect from accidental inhalation of phosphine generated during their approved use.
Clinical features
Ingestion causes vomiting, epigastric pain, peripheral circulatory failure, severe metabolic acidosis, acute kidney injury and disseminated intravascular coagulation, in addition to the features induced by phosphine. Exposure to phosphine causes lacrimation, rhinorrhoea, productive cough, breathlessness, chest tightness, dizziness, headache, nausea and drowsiness. Acute pulmonary oedema, hypertension, cardiac arrhythmias, convulsions and jaundice have been described in severe cases. Ataxia, intention tremor and diplopia are found on examination.
Specific poisons: marine animals
Amnesic shellfish (domoic acid) poisoning
The syndrome should be known more accurately as domoic acid poisoning because amnesia is not always present. In one outbreak, the first symptoms were experienced between 15 minutes and 38 hours after mussel consumption.
Diarrhoeic shellfish (okadaic) poisoning
Okadaic acid is produced by dinoflagellates belonging to the genera Dinophysis spp. Okadaic acid inhibits the activity of the protein phosphatases 1 and 2a.
Neurotoxic shellfish (brevetoxin) poisoning
Neurotoxic shellfish poisoning is caused by brevetoxins produced by the dinoflagellate Gymnodinium breve. Brevetoxins open voltage-gated sodium ion channels in cell walls and enhance the inward flow of sodium ions into the cell.
Clinical features and treatment
The symptoms of neurotoxic shellfish poisoning occur within 30 minutes to 3 hours, last a few days and include nausea, vomiting, diarrhoea, chills, sweats, reversal of temperature, hypotension, arrhythmias, numbness, tingling, paraesthesiae of the lips, face and extremities, cramps, bronchoconstriction, paralysis, seizures and coma. Treatment is symptomatic and supportive.
Paralytic shellfish (saxitoxin) poisoning
This is caused by bivalve molluscs being contaminated with neurotoxins, including saxitoxin, produced by toxic dinoflagellates on which the molluscs graze. Saxitoxin blocks voltage-gated sodium channels in nerve and muscle cell membranes, thereby blocking nerve signal transmission.
Clinical features and treatment
Symptoms develop within 30 minutes. The illness is characterized by paraesthesiae of the mouth, lips, face and extremities and is often accompanied by nausea, vomiting and diarrhoea. In more severe cases, dystonia, dysphagia, muscle weakness, paralysis, ataxia and respiratory depression occur. In one outbreak involving 187 cases, there were 26 deaths. Treatment is symptomatic and supportive.
Ciguatera fish poisoning
Over 400 fish species have been reported as ciguatoxic (Cigua is Spanish for poisonous snail), though barracuda, red snapper, amberjack and grouper are most commonly implicated. Ciguatera fish contain ciguatoxin, maitotoxin and scaritoxin, which are lipid soluble, heat stable compounds that are derived from dinoflagellates such as Gambierdiscus toxicus. Ciguatoxin opens voltage-sensitive sodium channels at the neuromuscular junction and maitotoxin opens calcium channels of the cell plasma membrane.
Clinical features and treatment
The onset of symptoms occurs from a few minutes to 30 hours after ingestion of toxic fish. Typically features appear between one and six hours and include abdominal cramps, nausea, vomiting and watery diarrhoea. In some cases, numbness and paraesthesiae of the lips, tongue and throat occur. Other features described include malaise, dry mouth, metallic taste, myalgia, arthralgia, blurred vision, photophobia and transient blindness. In more severe cases, hypotension, cranial nerve palsies and respiratory paralysis have been reported. Treatment is symptomatic and supportive. Recovery takes from 48 hours to 1 week in the mild form and from 1 to several weeks in the severe form. The mortality in severe cases may be as high as 12%.
Scombroid fish poisoning
This is due to the action of bacteria such as Proteus morgani and Klebsiella pneumoniae in decomposing flesh of fish such as tuna, mackerel, mahi-mahi, bonito and skipjack if the fish are stored at insufficiently low temperatures. The spoiled fish can contain excessively high concentrations of histamine (muscle histidine is broken down by the bacteria to histamine), though the precise role of histamine in the pathogenesis of the clinical syndrome is uncertain.
Clinical features and treatment
Clinically, the mean incubation period is 30 min. The illness is characterized by flushing, headache, sweating, dizziness, burning of the mouth and throat, abdominal cramps, nausea, vomiting and diarrhoea and is usually short-lived; the mean duration is 4 hours. Treatment is symptomatic and supportive. Antihistamines may alleviate the symptoms.
Stings from marine animals
Several species of fish have venomous spines in their fins. These include the weaver fish, short-spine cottus, spiny dogfish and the stingray. Bathers and fishermen may be stung if they tread on or handle these species. The immediate result of a sting is intense local pain, swelling, bruising, blistering, necrosis and, if the poisoned spine is not removed, chronic sepsis (although this is uncommon). Occasionally systemic symptoms including, vomiting, diarrhoea, hypotension and tachycardia occur. Treatment by immersing the affected part in hot water may relieve local symptoms as this denatures the thermolabile toxin.
Jellyfish
Most of the jellyfish found in North European coastal waters are non-toxic as their stings cannot penetrate human skin. A notable exception is the ‘Portuguese man-o’-war’ (Physalia physalis) whose sting contains a toxic peptide, phospholipase A, and a histamine-liberating factor. Toxic jellyfish are found more frequently in Australia and some cause the Irukandji syndrome.
Clinical features and treatment
Local pain occurs followed by myalgia, nausea, griping abdominal pain, dyspnoea and even death. The cluster of severe systemic symptoms that constitute the Irukandji syndrome occur some 30 min after the jellyfish sting. The symptoms include severe low back pain, excruciating muscle cramps in all four limbs, the abdomen and chest, sweating, anxiety, restlessness, nausea, vomiting, headache, palpitations, life-threatening hypertension, pulmonary oedema and toxic global heart dilatation.
Adhesive tape may be used to remove any tentacles still adherent to the bather. Local application of 5% acetic acid is said to prevent stinging cells adherent to the skin discharging. Local analgesia and antihistamine creams provide symptomatic relief. Other features should be treated symptomatically and supportively.
Specific poisons: venomous animals
Insect stings and bites
Insect stings from wasps and bees, and bites from ants, produce pain and swelling at the puncture site. Following the sting or bite, patients should be observed for 2 hours for any signs of evolving urticaria, pruritus, bronchospasm or oropharyngeal oedema. The onset of anaphylaxis requires urgent treatment (see p. 69).
Scorpions
Scorpion stings are a serious problem in North Africa, the Middle East and the Americas. Scorpion venoms stimulate the release of acetylcholine and catecholamines causing both cholinergic and adrenergic symptoms.
Clinical features and treatment
Severe pain occurs immediately at the site of puncture, followed by swelling. Signs of systemic involvement, which may be delayed for 24 hours, include vomiting, sweating, piloerection, abdominal colic, diarrhoea. In some cases, depending on the species, shock, respiratory depression and pulmonary oedema may develop.
Local infiltration with anaesthetic or a ring block will usually alleviate local pain, though systemic analgesia may be required. Specific antivenom, if available, should be administered as soon as possible.
Spiders
The black widow spider (Latrodectus mactans) is found in North America and the tropics and occasionally in Mediterranean countries.
Venomous snakes
Approximately 15% of the 3000 species of snake found worldwide are considered to be dangerous to humans. Snake bite is common in some tropical countries (Table 17.23).
Table 17.23 Examples of snake bite incidence andmortality
Sri Lanka |
6 bites per 100 000 population and 900 deaths per year |
Nigeria |
500 bites per 100 000 population with a 12% mortality |
Myanmar |
15 deaths per 100 000 population |
USA |
45 000 bites per year (in a population of 301 million), 8000 by venomous species, with 6 deaths annually |
UK |
Approximately 100 people admitted to hospital annually (population 60 million) but only one death since 1970 |
Australia |
2 or 3 deaths annually (population 20 million) |
There are three main groups of venomous snakes, representing some 200 species, which have in their upper jaws a pair of enlarged teeth (fangs) that inject venom into the tissues of their victim. These are:
Viperidae (with two subgroups: Viperinae: European adders and Russell’s vipers; and Crotalinae: American rattlesnakes, moccasins, lance-headed vipers and Asian pit vipers)
Elapidae (cobras, kraits, mambas, coral snakes, Australian venomous snakes)
In addition, some members of the family Colubridae are mildly venomous (mongoose snake).
Clinical features
Viperidae (Viperinae and Crotalinae)
Russell’s viper causes most of the snake-bite mortality in India, Pakistan and Myanmar. There is local swelling at the site of the bite (Fig. 17.7) which may become massive. Local tissue necrosis may occur. Evidence of systemic involvement (envenomation) occurs within 30 minutes, including vomiting, evidence of shock and hypotension. Haemorrhage due to incoagulable blood can be fatal.
There is not usually any swelling at the site of the bite, except with Asian cobras and African spitting cobras – here the bite is painful and is followed by local tissue necrosis. Vomiting occurs first followed by shock and then neurological symptoms and muscle weakness, with paralysis of the respiratory muscles in severe cases. Cardiac muscle can be involved.
Treatment
As a first aid measure, a firm pressure bandage should be placed over the bite and the limb immobilized. This may delay the spread of the venom. Arterial tourniquets should not be used, and incision or excision of the bite area should not be performed. Local wounds often require little treatment. If necrosis is present, antibiotics should be given. Skin grafting may be required later. Antitetanus prophylaxis must be given. The type of snake should be identified if possible.
In about 50% of cases, no venom has been injected by the bite. Nevertheless, careful observation for 12–24 hours is necessary in case envenomation develops. General supportive measures should be given, as necessary. These include intravenous fluids with volume expanders for hypotension and diazepam for anxiety. Treatment of acute respiratory, cardiac and kidney injury is instituted as necessary.
Antivenoms are not generally indicated unless envenomation is present, as they can cause severe allergic reactions. Antivenoms can rapidly neutralize venom, but only if an amount in excess of the amount of venom is given. Large quantities of antivenom may be required. As antivenoms cannot reverse the effects of the venom, they must be given early to minimize some of the local effects and may prevent necrosis at the site of the bite. Antivenoms should be administered intravenously by slow infusion, the same dose being given to children and adults.
Allergic reactions are frequent, and adrenaline (epinephrine) 1 in 1000 solution should be available. In severe cases, the antivenom infusion should be continued even if an allergic reaction occurs, with subcutaneous injections of adrenaline being given as necessary. Some forms of neurotoxicity, such as those induced by the death adder, respond to anticholinesterase therapy with neostigmine and atropine.
Specific poisons: plants
Life-threatening poisoning from plant ingestion is rare though many plants contain potentially toxic substances. These include antimuscarinic agents, calcium oxalate crystals, cardiogenic glycosides, pro-convulsants, cyanogenic compounds, mitotic inhibitors, nicotine-like alkaloids, alkylating agent precursors, sodium channel activators and toxic proteins (toxalbumins). While many plants contain gastrointestinal toxins, these rarely give rise to life-threatening sequelae. In contrast, other botanical poisons may cause specific organ damage and death may occur from only small ingestions of yew (genus: Taxus), oleander (Thevetia peruviana and Nerium oleander) and cowbane (genus: Cicuta).
Atropa belladonna
Atropa belladonna (deadly nightshade) contains hyoscyamine and atropine and causes antimuscarinic effects – a dry mouth, nausea and vomiting – leading to blurred vision, hallucinations, confusion and hyperpyrexia.
Cicuta species
Cicuta spp. (water hemlock) and the related genus Oenanthe contain cicutoxin, a potent central nervous system (CNS) stimulant that produces violent seizure activity. The CNS effects of cicutoxin are similar to those of picrotoxin, a known inhibitor of GABA. Severe gastrointestinal symptoms, diaphoresis, salivation and skeletal muscle stimulation may precede the seizure activity.
Conium maculatum
Conium maculatum (poison hemlock) contains a variety of volatile pyridine alkaloids, including coniine, N-methylconiine and gammaconiceine. Coniceine is significantly more toxic than coniine and is thought to be the precursor to coniine. The toxic activity of the alkaloids is similar to that of nicotine. Large doses produce non-polarizing neuromuscular blockade which may result in respiratory depression and death.
Datura stramonium
Datura stramonium (jimsonweed) and other Datura species contain L-hyoscyamine and atropine. These alkaloids are potent antagonists of acetylcholine at muscarinic receptors and produce the anticholinergic syndrome. While morbidity is significant, fatalities are rare and are the consequence of hyperthermia, seizures and/or arrhythmias.
Digitalis purpurea, Nerium oleander, Thevetia peruviana
Ingestion of Digitalis purpurea, or the common (Nerium oleander) or yellow (Thevetia peruviana) oleander can produce a syndrome similar to digoxin poisoning (see p. 917). A randomized controlled trial has shown that digoxin-specific antibody fragments rapidly and safely reverse yellow oleander-induced arrhythmias, restore sinus rhythm, and rapidly reverse bradycardia and hyperkalaemia. The administration of multiple doses of activated charcoal is used, but the effect on survival is debated.
Specific poisons: mushrooms
Poisoning due to mushrooms is usually accidental, though ingestion of hallucinogenic (‘magic’) mushrooms is invariably intentional.
Cytotoxic mushrooms
Cytotoxic mushroom poisoning is caused by amatoxins and orellanin. Amatoxins are found in Amanita phalloides, A. virosa and A. verna, and in some Galerina and Lepiota spp. Amatoxins inhibit transcription from DNA to mRNA by the blockade of nuclear RNA polymerase II; this results in impaired protein synthesis and cell death.
Clinical features and treatment
Intense watery diarrhoea starts 8–24 hours after ingestion and persists for 24 hours or longer. Patients often become severely dehydrated. Signs of liver damage appear during the 2nd day and hepatic failure may ensue. Impaired kidney function is often seen both because of fluid loss and as a result of direct kidney injury. In all patients, fluid, electrolyte and acid–base disturbances should be corrected and renal and hepatic function supported. The value of silibinin and benzylpenicillin is not proven. Occasionally, liver transplantation is necessary.
Gyromitrin poisoning
Gyromitrin is found in Gyromitra spp., including in particular the false morel (Gyromitra esculenta) and Cudonia circinans. Gyromitrin decomposes in the stomach, to form hydrazines that reduce pyridoxine in the CNS and, hence, GABA synthesis, causing glutathione depletion in red blood cells and may form free oxygen radicals that bind to hepatic macromolecules.
Clinical features and treatment
Vapours from the mushrooms are irritating to the eyes and respiratory tract. Gastrointestinal symptoms appear 5–8 hours after exposure. Vertigo, sweating, diplopia, headache, dysarthria, incoordination and ataxia may follow. Symptomatic and supportive care are required. Pyridoxine 25 mg/kg as an infusion over 30 min, should be given if severe CNS toxicity develops; repeat doses may be required.
Hallucinogenic mushroom poisoning
Psilocybin produces pharmacological effects similar to those of LSD and is found in Psilocybe and Panaeolus spp.
Clinical features and treatment
Symptoms occur within 20–60 min. Effects include altered time and space sense, depersonalization, hallucinations, derealization and euphoria. Symptoms are usually maximal within 2 hours and disappear within 4–6 hours, though ‘flashbacks’ may recur after weeks or months. Anxiety and agitation should be treated with diazepam, 10–20 mg i.v., repeated if necessary.
Isoxazole poisoning
Isoxazoles (e.g. ibotenic acid, muscimol, muscazone) occur in Amanita muscaria and A. pantherina and act as γ-aminobutyric acid agonists.
Clinical features and treatment
Nausea, vomiting, inebriation, euphoria, confusion, anxiety, visual disturbances and hallucinations occur often within 30 minutes. Severe agitation and violent behaviour are occasionally seen. Other features include myoclonic jerks, muscle fasciculation, seizures and coma. Symptomatic and supportive care should be given as necessary. Diazepam, 10–20 mg, repeated as required, should be administered for anxiety, agitation and seizures.
Neurotoxic mushroom poisoning
Muscarine is found in, for example, Inocybe spp., Clitocybe spp. and Mycena pura. Muscarine stimulates cholinergic receptors in the autonomic nervous system.
Orellanin poisoning
Orellanin is a potent nephrotoxin found in, for example, Cortinarius orellanus and C. speciosissimus. A metabolite of orellanin inhibits protein synthesis in the kidneys.
Clinical features and treatment
Symptoms are typically delayed for 2–4 days. Some patients suffer a mild gastrointestinal disturbance before developing signs of renal impairment, headache, fatigue, intense thirst, chills, muscular discomfort, and abdominal, lumbar and flank pain. Transient polyuria with proteinuria, haematuria and, characteristically, leucocyturia is followed by oliguria and then anuria. Renal function may recover only partially; chronic kidney disease is reported in about 10–40% of cases. Management involves careful monitoring and haemodialysis/haemofiltration if renal failure supervenes. Renal transplantation may be required.
Toxbase – Database of UK National Poisons Information Service
Database of the New Zealand Poisons Centre
National Library of Medicine’s Toxnet
www.who.int/ipcs/poisons/centre/directory/en