Nervous system infection and inflammation
Meningitis
Meningitis usually implies serious infection of the meninges (Box 22.25). Bacterial meningitis is fatal unless treated. Microorganisms reach the meninges either by direct extension from the ears, nasopharynx, cranial injury or congenital meningeal defect, or by bloodstream spread. Immunocompromised patients are at risk of infection by unusual organisms. Non-infective causes of meningeal inflammation include malignant meningitis, intrathecal drugs and blood following subarachnoid haemorrhage.
 Box 22.25
Box 22.25
Infective causes of meningitis in the UK
a These organisms account for 70% of acute bacterial meningitis outside the neonatal period. A wide variety of infective agents are responsible for the remaining 30% of cases. Haemophilus influenzae b (Hib) has been eliminated as a cause in many countries by immunization. Malaria often presents with cerebral symptoms and a fever.
Pathology
In acute bacterial meningitis, the pia-arachnoid is congested with polymorphs. A layer of pus forms. This may organize to form adhesions, causing cranial nerve palsies and hydrocephalus.
In chronic infection (e.g. TB), the brain is covered in a viscous grey-green exudate with numerous meningeal tubercles. Adhesions are invariable. Cerebral oedema occurs in any bacterial meningitis.
In viral meningitis there is a predominantly lymphocytic inflammatory CSF reaction without pus formation, polymorphs or adhesions; there is little or no cerebral oedema unless encephalitis develops.
Clinical features
The meningitic syndrome
This is a simple triad: headache, neck stiffness and fever. Photophobia and vomiting are often present. In acute bacterial infection there is usually intense malaise, fever, rigors, severe headache, photophobia and vomiting, developing within hours or minutes. The patient is irritable and often prefers to lie still. Neck stiffness and positive Kernig’s sign usually appear within hours.
In less severe cases (e.g. many viral meningitides), there are less prominent meningitic signs. However, bacterial infection may also be indolent, with a deceptively mild onset.
In uncomplicated meningitis, consciousness remains intact, although anyone with high fever may be delirious. Progressive drowsiness, lateralizing signs and cranial nerve lesions indicate complications such as venous sinus thrombosis (p. 1107), severe cerebral oedema, hydrocephalus, or an alternative diagnosis such as cerebral abscess (p. 1130) or encephalitis (p. 1128).
Specific varieties of meningitis
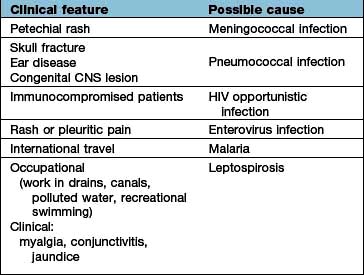
Clinical clues point to the diagnosis (Table 22.20). If there is access to the subarachnoid space via skull fracture (recent or old) or occult spina bifida, bacterial meningitis can be recurrent, and the infecting organism is usually pneumococcus.
Table 22.20 Clinical clues in meningitis
Acute bacterial meningitis
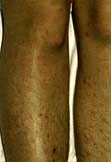
Onset is typically sudden, with rigors and high fever. Meningococcal meningitis is often heralded by a petechial or other rash, sometimes sparse (see Emergency Box 22.1). The meningitis may be part of a generalized meningococcal septicaemia (p. 127). Acute septicaemic shock may develop in any bacterial meningitis.
 Emergency Box 22.1 Meningococcal meningitis and meningococcaemia
Emergency Box 22.1 Meningococcal meningitis and meningococcaemia
emergency treatment
Suspicion of meningococcal infection is a medical emergency requiring treatment immediately.
Clinical features:
All these features may not be present – and meningococcal infection may sometimes begin like any apparently non-serious infection.
Immediate treatment for suspected meningococcal meningitis at first contact before transfer to hospital or investigation:
Viral meningitis
This is almost always a benign, self-limiting condition lasting 4–10 days. Headache may follow for some months. There are no serious sequelae, unless an encephalitis is present (p. 1128).
Differential diagnosis
It may be difficult to distinguish between the sudden headache of subarachnoid haemorrhage, migraine and acute meningitis. Meningitis should be considered seriously in anyone with headache and fever and in any sudden headache. Neck stiffness should be looked for – it may not be obvious. Chronic meningitis sometimes resembles an intracranial mass lesion, with headache, epilepsy and focal signs. Cerebral malaria can mimic bacterial meningitis.
Management (Emergency Box 22.1)
Recognition and immediate treatment of acute bacterial meningitis is vital. Minutes save lives. Bacterial meningitis is lethal. Even with optimal care, mortality is around 15%. The following applies to adult patients; management is similar in children.
When meningococcal meningitis is diagnosed clinically by the petechial rash, immediate i.v. antibiotics should be given and blood cultures taken; lumbar puncture is unnecessary. In other causes of meningitis, a lumbar puncture is performed if there is no clinical suspicion of a mass lesion (p. 1091). If the latter is suspected an immediate CT scan must be performed because coning of the cerebellar tonsils may follow LP. Typical CSF changes are shown in Table 22.21. CSF pressure is characteristically elevated. If a presumptive diagnosis of the organism can be made (e.g. pneumococcus is likely with skull fracture or sinus infection), targeted treatment should be started immediately. Immediate antibiotic treatment in acute bacterial meningitis is shown in Table 22.22.

Table 22.22 Antibiotics in acute bacterial meningitis
| Organism | Antibiotic | Alternative (e.g. allergy) |
|---|---|---|
Unknown pyogenic |
Cefotaxime |
Benzylpenicillin and chloramphenicol |
Meningococcus |
Benzylpenicillin |
Cefotaxime |
Pneumococcus |
Cefotaxime |
Penicillin |
Haemophilus |
Cefotaxime |
Chloramphenicol |
Blood should be taken for cultures, glucose and routine tests. Chest and skull films should be obtained if appropriate.
CSF stains demonstrate organisms (e.g. Gram-positive intracellular diplococci – pneumococcus; Gram-negative cocci-meningococcus). Ziehl–Neelsen stain demonstrates acid-fast bacilli (TB), though TB organisms are rarely numerous. Indian ink stains fungi.
Meticulous attention should focus on microbiological studies in suspected CNS infection with close liaison between clinician and microbiologist. Specific techniques (e.g. polymerase chain reaction for meningococci and other bacteria) are invaluable. Syphilitic serology should always be carried out.
The clinical picture and CSF examination should thus yield a presumptive cause for acute meningitis within hours. Antibiotics, however, must be started before the actual organism is identified.
If bacterial meningitis is diagnosed, further discussion with the microbiologist should include antibiotics, drug resistance, recent infections in the locality, barrier nursing and prophylaxis.
In adults with pneumococcal meningitis, dexamethasone should be given first with the initial antibiotics.
Intrathecal antibiotics are no longer used.
Local infection (e.g. paranasal sinus) should be treated surgically if necessary. Repair of a depressed skull fracture or meningeal tear may be required.
Prophylaxis
Meningococcal infection should be notified to public health authorities, and advice sought about immunization and prophylaxis of contacts, e.g. with rifampicin or ciprofloxacin. MenC, a meningococcal C conjugate vaccine, is part of many countries’ immunization programme and often given to case contacts. A combined A and C meningococcal vaccine is sometimes used prior to travel from the UK to endemic regions, e.g. Africa, Asia; and a quadrivalent ACWY vaccine for specific events, e.g. the Hajj and Umrah in Mecca. There is no vaccine for Group B.
A polyvalent pneumococcal vaccine is used after recurrent meningitis, e.g. after a CSF leak following skull fracture.
Hib (Haemophilus influenzae) vaccine is given routinely in childhood in the UK and other countries, e.g. Gambia, virtually eliminating a common cause of fatal meningitis.
Chronic meningitis
Tuberculous meningitis (TBM) and cryptococcal meningitis commence typically with vague headache, lassitude, anorexia and vomiting. Acute meningitis can occur but is unusual. Meningitic signs often take some weeks to develop. Drowsiness, focal signs (e.g. diplopia, papilloedema, hemiparesis) and seizures are common. Syphilis, sarcoidosis and Behçet’s also cause chronic meningitis. In some cases of chronic meningitis, an organism is never identified.
Management of tuberculous meningitis
TBM is a common and serious disease worldwide. Brain imaging, usually with MRI, may show meningeal enhancement, hydrocephalus and tuberculomas (in this chapter), although it may remain normal (see Table 22.21 for CSF changes). In many cases the sparse TB organisms cannot be seen on staining and PCR testing should be performed, although results may be negative. Repeated CSF examination is often necessary and it will be some weeks before cultures are confirmatory. Treatment with antituberculosis drugs (p. 842) – rifampicin, isoniazid and pyrazinamide – must commence on a presumptive basis and continue for at least 9 months. Ethambutol should be avoided because of its eye complications. Adjuvant corticosteroids, e.g. prednisolone 60 mg for 3 weeks, are now recommended (often tapered off). Relapses and complications (e.g. seizures, hydrocephalus) are common in TBM. The mortality remains over 60% even with early treatment.
Malignant meningitis
Malignant cells can cause a subacute or chronic non-infective meningitic process. A meningitic syndrome, cranial nerve lesions, paraparesis and root lesions are seen, often in confusing and fluctuating patterns. The CSF cell count is raised, with high protein and low glucose. Treatment with intrathecal cytotoxic agents is rarely helpful.
Cells in a sterile CSF (pleocytosis)
A raised CSF cell count is present without an evident infecting organism. CSF pleocytosis, i.e. a mixture of lymphocytes and polymorphs, is the usual situation (Box 22.26).
Box 22.26
Causes of sterile CSF pleocytosis
Encephalitis
Encephalitis means acute inflammation of brain parenchyma, usually viral. In viral encephalitis fever (90%) and meningism are usual, but in contrast to meningitis the clinical picture is dominated by brain parenchyma inflammation. Personality and behavioural change is a common early manifestation which progresses to a reduced level of consciousness and even coma. Seizures (focal and generalized) are very common and focal neurological deficits, e.g. speech disturbance, often occur (especially in herpes simplex encephalitis).
Viral encephalitis
The viruses isolated from adult UK cases are usually herpes simplex, VZV and other herpes group viruses, HHV-6, 7, enteroviruses and adenovirus. HSV encephalitis (HSVE) typically affects the temporal lobes initially, often asymmetric. Often, the virus is never identified. Outside the UK in endemic regions different pathogens cause encephalitis including Flaviviruses (Japanese encephalitis, West Nile virus, tick- borne encephalitis) and rabies.
Local epidemics can occur. For example, in New York in the 1990s, West Nile virus caused an epidemic and Venezuelan equine virus was isolated from encephalitis cases in South America.
Investigations
 MR imaging shows areas of inflammation and swelling, generally in the temporal lobes in HSV encephalitis. Raised intracranial pressure and midline shift may occur leading to coning.
MR imaging shows areas of inflammation and swelling, generally in the temporal lobes in HSV encephalitis. Raised intracranial pressure and midline shift may occur leading to coning.
EEG shows periodic sharp and slow wave complexes.
CSF shows an elevated lymphocyte count (95%).
Viral detection by CSF PCR is highly sensitive for several viruses such as HSV and VZV. However, a false negative result may occur within the first 48 h after symptom onset. Serology (blood and CSF) is also helpful.
Brain biopsy is rarely required since the advent of MRI and PCR.
Treatment
Suspected HSV and VZV encephalitis is treated immediately with i.v. aciclovir (10 mg/kg 3 times a day for 14–21 days), even before investigation results are available. Early treatment significantly reduces both mortality and long-term neurological damage in survivors. Seizures are treated with anticonvulsants. Occasionally decompressive craniectomy is required to prevent coning but coma confers a poor prognosis.
Long-term complications are common including memory impairment, personality change and epilepsy.
Post-infectious encephalomyelitis
Acute disseminated encephalomyelitis (ADEM) follows many infections (e.g. measles, mycoplasma, mumps and rubella) and rarely follows immunization after 1–2 weeks. There is a monophasic illness with multifocal brain, brainstem and often spinal cord inflammatory lesions in white matter, with demyelination. ADEM is caused by an immune mediated host response to infection and occurs principally in children and young adults. Mild cases recover completely. Survivors often have permanent brain damage. Treatment is supportive, with steroids and anticonvulsants.
Autoimmune encephalitis
This group of disorders have been described in recent years and are increasingly recognized. Autoantibodies directed against neuronal epitopes cause a subacute encephalitic illness – limbic encephalitis or panencephalitis. Limbic encephalitis presents over weeks or months with memory impairment, confusion, psychiatric disturbance, and seizures – usually temporal lobe seizures reflecting involvement of the hippocampus and mesial temporal lobes.
Paraneoplastic limbic encephalitis (PLE). Seen particularly with small cell lung cancer and testicular tumours and associated with a variety of antibodies including anti-Hu and anti-Ma2. Antibodies can be detected in 60% of cases. MRI usually shows a hippocampal high signal. PLE precedes the diagnosis of cancer in most cases and should prompt investigation to identify the tumour.
Voltage gated potassium channel (VGKC) limbic encephalitis. VGKC antibodies (which can be tested for) produce a variety of disorders including limbic encephalitis, characteristic faciobrachial dystonic seizures, neuromyotonia and peripheral nerve hyperexcitability syndromes. This usually occurs in patients older than 50 but is rarely associated with cancer (thymoma).
Anti NMDA receptor antibody panencephalitis. Presents as limbic encephalitis followed by coma and often status epilepticus. Orofacial dyskinesias are characteristic. Usually younger patients, some have ovarian teratomas.
Patients may respond to immunotherapy – i.v. immunoglobulin or plasma exchange initially followed by steroids, rituximab or cyclophosphamide. PLE responds less well to treatment.
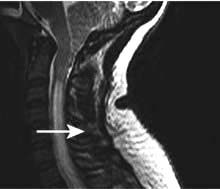
Herpes zoster (shingles)
This is caused by reactivation of varicella zoster virus (VZV), usually within dorsal root ganglia. Primary infection with VZV causes chickenpox following which the virus remains latent in sensory ganglia. Development of shingles may indicate a decline in cell mediated immunity, e.g. due to age or malignancy.
Clinical patterns and complications
Dermatomal shingles. Thoracic dermatomes are most commonly affected. Tingling or painful dysaesthesias precede the vesicular rash by a few days (p. 81). Motor radiculopathy can occur (usually lumbar or cervical, as thoracic involvement is often clinically silent). It rarely occurs without the rash – zoster sin herpete. Antiviral drugs such as aciclovir, famciclovir or valaciclovir reduce the incidence of postherpetic neuralgia.
Postherpetic neuralgia. Defined as pain lasting more than 4 months after developing shingles; occurs in 10% of patients (often elderly). Burning, intractable pain responds poorly to analgesics. Response to treatment is unsatisfactory but there is a trend towards gradual recovery over 2 years. Amitriptyline or gabapentin are commonly used and topical lidocaine patches may help.
Cranial nerve involvement. Only cranial nerves with sensory fibres are affected, particularly the trigeminal and facial nerves. Ophthalmic herpes is due to involvement of V1. This can lead to corneal scarring and secondary panophthalmitis. Involvement of the geniculate ganglion of the facial nerve is also called Ramsay Hunt syndrome (p. 1098).
Myelitis may occur in the context of shingles when the inflammatory process spreads from the dorsal root ganglion to the adjacent spinal cord.
Immunization. The Centers for Disease Control and Prevention (CDC) in America have suggested that all adults over 60 years old should be vaccinated against herpes zoster (even those who have had shingles previously), as it reduces the incidence of shingles by about 50%.
Neurosyphilis
Many neurological symptoms occur, sometimes mixed (see also syphilis, p. 166).
Tabes dorsalis
Demyelination in dorsal roots causes a complex deafferentation syndrome. The elements of tabes:
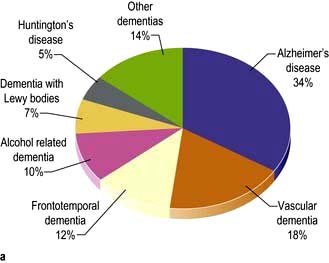
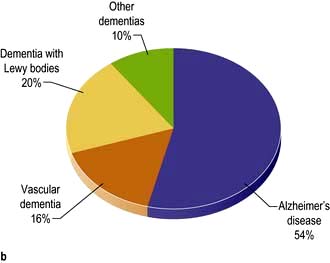
General paralysis of the insane (GPI)
The grandiose title describes dementia and weakness. GPI dementia is typically similar to Alzheimer’s (p. 1138). Progressive cognitive decline, seizures, brisk reflexes, extensor plantar reflexes and tremor develop. Death follows within 3 years. Argyll Robertson pupils are usual. GPI and tabes are rarities in the UK.
Other forms of neurosyphilis
In congenital neurosyphilis (acquired in utero), features of combined tabes and GPI develop in childhood – taboparesis.
Secondary syphilis can be symptomless or cause a self-limiting subacute meningitis.
Treatment
Benzylpenicillin 1 g daily i.m. for 10 days in primary infection eliminates any risk of neurosyphilis. Allergic (Jarisch–Herxheimer) reactions can occur; steroid cover is usually given with penicillin (p. 1129). Established neurological disease is arrested but not reversed by penicillin.
Neurocysticercosis
The pork tapeworm, Taenia solium, is endemic in Latin America, Africa, India and much of South-east Asia (p. 159). Epilepsy is the commonest clinical manifestation of neurocysticercosis and one of the commonest causes of epilepsy in endemic countries. Most infected people remain asymptomatic.
Brain CT and MRI show ring-enhancing lesions with surrounding oedema when the cyst dies and later calcification. Multiple cysts are often seen in both brain and skeletal muscle. Serological tests indicate infection but not activity. Biopsy of a lesion is rarely necessary. Management is primarily the control of seizures and the anthelminthic agent albendazole is often also given (usually with steroid cover).
HIV and neurology
HIV-infected individuals frequently present with or develop neurological conditions. The HIV virus itself is directly neuroinvasive and neurovirulent. Immunosuppression leads to indolent, atypical clinical patterns (p. 176). HIV patients also have a high incidence of stroke. The pattern of disease is changing where antiretroviral (ARV) therapy is available.
CNS and peripheral nerve disease in HIV
HIV seroconversion can cause meningitis, encephalitis, Guillain–Barré syndrome and Bell’s palsy (the commonest cause of Bell’s palsy in South Africa).
Chronic meningitis occurs with fungi (e.g. Cryptococcus neoformans or Aspergillus), TB, listeria, coliforms or other organisms. Raised CSF pressure is common in cryptococcal meningitis.
AIDS-dementia complex (ADC). A progressive, HIV-related dementia, sometimes with cerebellar signs, is still seen where antiretroviral therapy is unavailable.
Encephalitis and brain abscess. Toxoplasma, cytomegalovirus, herpes simplex and other organisms cause severe encephalitis. Multiple brain abscesses develop in HIV infection, usually due to toxoplasmosis.
CNS lymphoma. this is typically fatal (Chapter 9).
Progressive multi-focal leucoencephalopathy (PML) is due to JC virus and occurs with very low CD4 counts (p. 191).
Spinal vacuolar myelopathy occurs in advanced disease.
Peripheral nerve disease. HIV related peripheral neuropathy is common (70%) and can be difficult to distinguish from the effects of ARV treatment which is also toxic to peripheral nerves.
Other infections
Many other infections involve the CNS and are discussed in Chapter 4, e.g. rabies, tetanus, botulism, Lyme disease and leprosy.
Other inflammatory conditions
Subacute sclerosing panencephalitis (SSPE)
Persistence of measles antigen in the CNS is associated with this rare late sequel of measles. Progressive mental deterioration, fits, myoclonus and pyramidal signs develop, typically in a child. Diagnosis is made by high measles antibody titre in blood and CSF. Measles immunization protects against SSPE, which has now been almost eliminated in the UK.
Progressive rubella encephalitis
Some 10 years after primary rubella infection, this causes progressive cognitive impairment, fits, optic atrophy, cerebellar and pyramidal signs. Antibody to rubella viral antigen is produced locally within the CNS. It is even rarer than SSPE.
Mollaret’s meningitis
This is recurrent self-limiting episodes of aseptic meningitis (i.e. no bacterial cause is found) over many years. Viral (possibly herpes simplex) infection is postulated.
Whipple’s disease
CNS Whipple’s disease, due to Tropheryma whipplei infection is characterized by myoclonus, dementia and supranuclear ophthalmoplegia (p. 268). Diagnosis of CNS involvement is made by CSF PCR (only 50% sensitivity) or brain biopsy.
Neurosarcoidosis
Neurosarcoid with or without systemic sarcoid causes chronic meningoencephalitis, cord lesions, cranial nerve palsies, particularly bilateral VIIth nerve lesions, polyneuropathy and myopathy (p. 845).
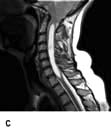
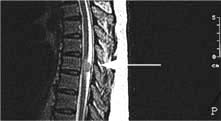
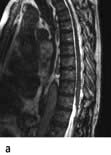
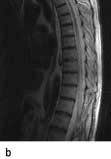
Brain and spinal abscesses
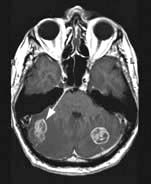
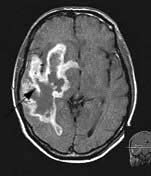
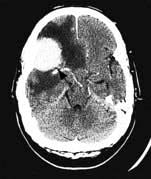
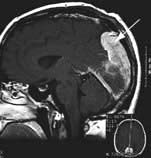
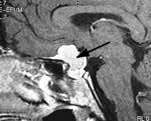
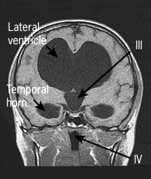
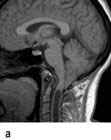
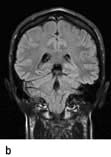
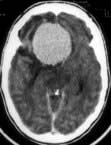
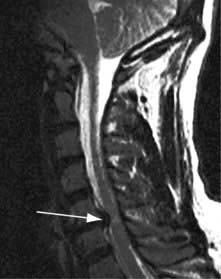
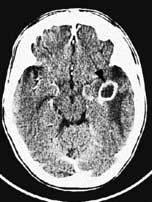
Brain abscess (Fig. 22.46)
Focal bacterial infection behaves as any expanding mass. Typical bacteria found are Streptococcus anginosus and Bacteroides species (paranasal sinuses and teeth) and staphylococci (penetrating trauma). Mixed infections are common. Multiple abscesses develop, particularly in HIV infection. Fungi also cause brain abscesses. A parameningeal infective focus (e.g. ear, nose, paranasal sinus, skull fracture) or a distant source of infection (e.g. lung, heart, abdomen) may be present. Frequently no underlying cause is found. An abscess is more than 10 times rarer than a brain tumour in the UK.
Clinical features and management
Headache, focal signs (e.g. hemiparesis, aphasia, hemianopia), epilepsy and raised intracranial pressure develop. Fever, leucocytosis and raised ESR are usual although not invariable.
Urgent imaging is essential. MRI shows a ring-enhancing mass, usually with considerable surrounding oedema. The search for a focus of infection should include a detailed examination of the skull, ears, paranasal sinuses and teeth, and distant sites such as heart and abdomen. Lumbar puncture is dangerous and should not be performed. Neurosurgical aspiration with stereotactic guidance allows the infective organism to be identified. Treatment is with high-dose antibiotics and sometimes surgical resection/decompression. Despite treatment, mortality remains high at approximately 25%. Epilepsy is common in survivors.
Brain tuberculoma
TB causes chronic caseating intracranial granulomatous masses – tuberculomas. These are the commonest intracranial masses in countries where TB is common, e.g. India. Brain tuberculomas either present as mass lesions de novo or develop during tuberculous meningitis; they are also found as symptomless intracranial calcification on imaging. Spinal cord tuberculomas also occur. Treatment is described on page 842.
Subdural empyema and intracranial epidural abscess
Intracranial subdural empyema is a collection of subdural pus, usually secondary to local skull or middle ear infection. Features are similar to those of a cerebral abscess. Imaging is diagnostic.
In intracranial epidural abscess a layer of pus, 1–3 mm thick, tracks along the epidural space causing sequential cranial nerve palsies. There is usually local infection, e.g. in the middle ear. MRI shows the collection; CT is typically normal. Drainage is required, and antibiotics.