Stroke and cerebrovascular disease
Stroke is the third most common cause of death (11% of all deaths in the UK) and the leading cause of adult disability worldwide. Although data are difficult to obtain, approximately two-thirds of the global burden of strokes is in middle- and low-income countries. Stroke rates are higher in Asian and black African populations than in Caucasians. Stroke risk increases with age but one-quarter of all strokes occur before the age of 65. The death rate following stroke is 20–25%.
Definitions
 Stroke. To the public, stroke means weakness, usually permanent on one side, often with loss of speech. Stroke is defined as a syndrome of rapid onset of cerebral deficit (usually focal) lasting >24 h or leading to death, with no cause apparent other than a vascular one. Hemiplegia following middle cerebral arterial thromboembolism is the typical example.
Stroke. To the public, stroke means weakness, usually permanent on one side, often with loss of speech. Stroke is defined as a syndrome of rapid onset of cerebral deficit (usually focal) lasting >24 h or leading to death, with no cause apparent other than a vascular one. Hemiplegia following middle cerebral arterial thromboembolism is the typical example.
Transient ischaemic attack (TIA) means a brief episode of neurological dysfunction due to temporary focal cerebral or retinal ischaemia without infarction, e.g. a weak limb, aphasia or loss of vision, usually lasting seconds or minutes with complete recovery. TIAs may herald a stroke. The arbitrary time of <24 hours is no longer used.
Pathophysiology
The underlying pathology responsible for stroke is either infarction or haemorrhage. Stroke mechanism and pathophysiology depends on the population studied but is broadly as follows (Fig. 22.27):
Ischaemic stroke/infarction (80%)
Haemorrhagic stroke (17%) (see p. 1104)
Other (3%), e.g. arterial dissection, venous sinus thrombosis, vasculitis (see p. 1108).
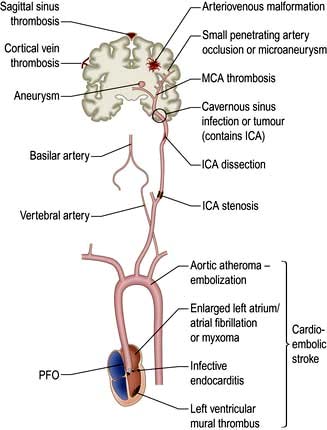
Figure 22.27 Pathophysiology of ischaemic stroke. ICA, internal carotid artery; MCA, middle cerebral artery; PFO, patent foramen ovale.
Ischaemic stroke
Arterial disease and atherosclerosis is the main pathological process causing stroke. Arterial branch points such as the origin of the great vessels arising from the aorta, the proximal internal carotid artery and its distal intracranial branches are particularly affected (Fig. 22.28). Non-Caucasian populations tend to have more intracranial narrowing and white populations more extracranial disease (which is strongly correlated with co-morbid coronary artery and peripheral vascular disease).
Thrombosis at the site of ruptured mural plaque leads to artery to artery embolism or vessel occlusion.
Large artery stenosis usually causes stroke by acting as an embolic source rather than by occlusion of the vessel (which may not in itself cause stroke if it occurs gradually and collateral circulation is adequate).
Small vessel disease. Small penetrating arterial branches supply the deep brain parenchyma and are affected by a different pathological process, an occlusive vasculopathy – lipohyalinosis – that is a consequence of hypertension. This leads to small infarcts called ‘lacunes’ and/or gradual accumulation of diffuse ischaemic change in deep white matter.
Cardio-embolic stroke. The heart is a common source of embolic material. Atrial fibrillation (and other arrhythmias) causing thrombosis in a dilated left atrium is the commonest cause. Cardiac valve disease, including congenital valve disorders, infective vegetations, rheumatic and degenerative calcific changes may cause embolization. Mural thrombosis may occur in a damaged or akinetic segment of the ventricle. A patent foramen ovale (PFO), which is a common variant, may occasionally allow passage of fragments of thrombus (e.g. from a lower limb DVT) from the right atrium to the left when Valsalva causes shunting of blood across the PFO. Pulmonary arteriovenous fistulas may also act as a conduit for paradoxical embolization. Rarer causes include fat emboli after long bone fracture, atrial myxomas and iatrogenic causes such as cardiac bypass and air embolism.
Simultaneous infarcts in different vascular territories are very suggestive of a proximal source of emboli in the heart or aorta.
Hypoperfusion. Severe hypotension, e.g. in cardiac arrest, may lead to borderzone infarction in the watershed areas between vascular territories, particularly if there is severe stenosis of proximal carotid vessels. The parieto-occipital area between the middle and posterior cerebral artery territories is particularly vulnerable.
Carotid and vertebral artery dissection
Dissection accounts for around 1 in 5 strokes below age 40 and is sometimes a sequel of trivial neck trauma or hyperextension, e.g. after whiplash, osteopathic manipulation, hairwashing in a salon, or exercise. It is now thought that subtle collagen disorders, e.g. partial forms of Marfan’s syndrome, may be a predisposing factor.
Most dissections are in large extracranial neck vessels. Blood penetrates the subintimal vessel wall forming a false lumen, but it is thrombosis within the true lumen due to tissue thromboplastin release that leads to embolization from the site of dissection and stroke, sometimes days after the initial event.
Pain in the neck or face is often the clue leading to diagnosis. Horner’s syndrome or lower cranial nerve palsies may occur with carotid dissection, as these structures are intimately associated with the carotid artery in the neck.
Venous stroke
Only 1% of strokes are venous. Thrombosis within intracranial venous sinuses, such as the superior sagittal sinus, or in cortical veins may occur in pregnancy, hypercoagulable states and thrombotic disorders or with dehydration or malignancy. Cortical infarction, seizures and raised intracranial pressure result.
For haemorrhagic stroke, see page 1104.
Transient ischaemic attacks
TIAs are usually the result of microemboli, but different mechanisms produce similar clinical events. For example, TIAs may be caused by a fall in cerebral perfusion (e.g. a cardiac dysrhythmia, postural hypotension or decreased flow through atheromatous arteries). Infarction is usually averted by autoregulation. Rarely, tumours and subdural haematomas cause episodes indistinguishable from thromboembolic TIAs. Principal sources of emboli to the brain are cardiac thrombus and atheromatous plaques/thrombus within the aortic arch, carotid and vertebral systems. Cardiac thrombus often results from atrial fibrillation or myocardial infarction. Cardiac valve disease may be a source of emboli, e.g. calcific material or vegetations in infective endocarditis. Polycythaemia is also a cause.
Risk factors for stroke
The principal risk factors and effects of altering these are shown in Table 22.12.
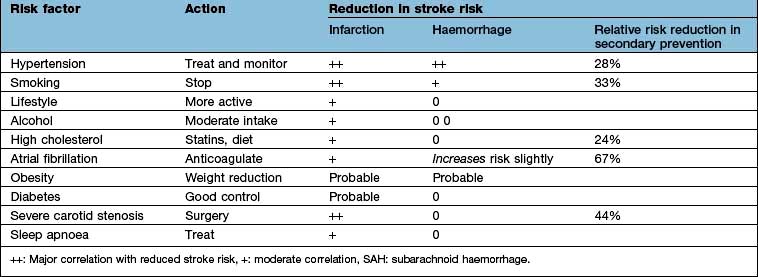
Hypertension is overall the most modifiable stroke risk factor; stroke is decreasing in the 40–60 age group partly because hypertension is now more effectively identified and treated. There is a linear relationship between blood pressure and stroke risk. Other risk factors are similar to vascular risk factors for cardiovascular disease, including smoking, dyslipidaemia, diabetes, obesity, inactivity and genetic/ethnic factors.
Other risk factors and rarer causes of stroke
Thrombocythaemia, polycythaemia and hyperviscosity states. Thrombophilia (e.g. protein C deficiency, factor V Leiden) is weakly associated with arterial stroke but predisposes to cerebral venous thrombosis.
Anti-cardiolipin and lupus anticoagulant antibodies (antiphospholipid syndrome, p. 538) predispose to arterial thrombotic strokes in young patients.
Low-dose oestrogen-containing oral contraceptives do not increase stroke risk significantly in healthy women but probably do so with other risk factors, e.g. uncontrolled hypertension or smoking.
Migraine is a rare cause of cerebral infarction.
Vasculitis (systemic lupus erythematosus (SLE), polyarteritis, giant cell arteritis, granulomatous CNS angiitis) is a rare cause of stroke.
Amyloidosis can present as recurrent cerebral haemorrhage (p. 1042).
Hyperhomocysteinaemia predisposes to thrombotic strokes. Folic acid therapy does not reduce the incidence.
Neurosyphilis, mitochondrial disease, Fabry’s disease (p. 1042).
Sympathomimetic drugs such as cocaine, and possibly over-the-counter cold remedies containing vasoconstrictors, neuroleptics in older patients.
CADASIL (cerebral dominant arteriopathy with subcortical infarcts and leucoencephalopathy) is a rare inherited cause of stroke/vascular dementia.
Vascular anatomy
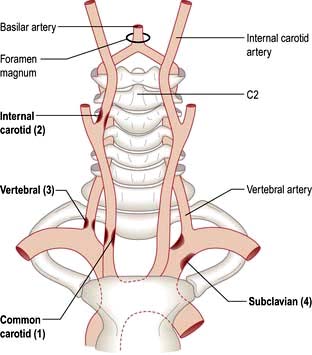
Knowledge of normal arterial anatomy and likely sites of atheromatous plaques and stenoses helps understanding of the main stroke syndromes.
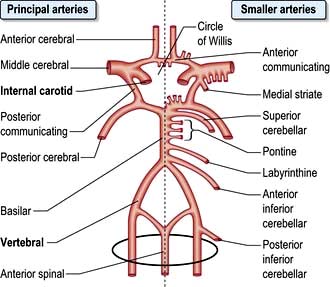
The circle of Willis is supplied by the two internal carotid arteries (the anterior cerebral circulation) and by the vertebro-basilar posterior cerebral circulation. The distribution of the anterior, middle and posterior cerebral arteries that supply the cerebrum is shown in Figures 22.28-22.30.
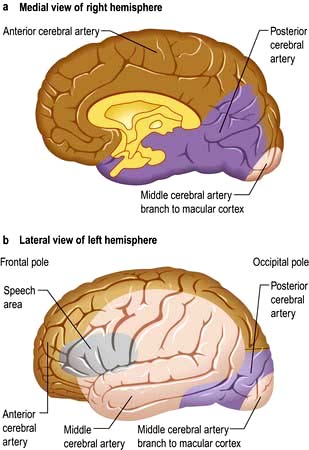
Clinical syndromes
Transient ischaemic attacks (TIAs)
Features
TIAs cause sudden loss of function, and usually last minutes or hours (by classical definition <24 h, but this is not used now). The site is often suggested by the type of attack. Clinical features of the principal forms of TIA are given in Box 22.12. Hemiparesis and aphasia are the commonest.
Amaurosis fugax
This is a sudden transient loss of vision in one eye. When due to the passage of emboli through the retinal arteries, arterial obstruction is sometimes visible through an ophthalmoscope during an attack. A TIA causing an episode of amaurosis fugax is often the first clinical evidence of internal carotid artery stenosis – and forerunner of a hemiparesis.
Clinical findings in TIA
Diagnosis of TIA is often based solely upon its description. It is unusual to witness an attack as they are so brief. Consciousness is usually preserved in TIA. There may be clinical evidence of a source of embolus, e.g.:
Carotid arterial bruit (stenosis)
Atrial fibrillation or other dysrhythmia
An underlying condition may be evident:
Bradycardia or low cardiac output
Rarely, arteritis, polycythaemia, neurosyphilis, HIV
Antiphospholipid syndrome (p. 538).
Differential diagnosis
TIAs can be distinguished, usually on clinical grounds, from other transient episodes (p. 1116). Occasionally, events identical to TIAs are produced by mass lesions. Focal epilepsy is usually recognized by its positive features (e.g. limb jerking and loss of consciousness) and progression over minutes. In a TIA, involuntary limb movements do occur occasionally; deficit is usually instantaneous. A focal prodrome in migraine sometimes causes diagnostic difficulty. Headache, common but not invariable in migraine, is rare in TIA. Typical migrainous visual disturbances are not seen in TIA.
Prognosis
Prospective studies show that 5 years after a single thromboembolic TIA:
TIAs in the anterior cerebral circulation carry a more serious prognosis than those in the posterior circulation (see Table 22.12).
The ABCD2 score can help to stratify stroke risk in the first 2 days:
1 point |
|
|
|
1 point |
A score of <4 is associated with a minimal risk whereas >6 is high risk for a stroke within 7 days of a TIA.
If patients are considered to have had a high risk TIA, i.e. ABCD2 score >4, or have had two recent TIAs, especially within the same vascular territory, then the patient should ideally be admitted for investigation and commencement of secondary prevention. ALL patients should be referred to a TIA Clinic and ideally seen within 24 h. Investigation and treatment should be regarded as urgent and completed within 2 weeks.
Investigations should include Doppler of internal carotid arteries, cardiac echo, ECG and 24 h tape CT/MR brain including angiography. Treat with medical therapy (p. 1095) and surgical treatment if appropriate (p. 1104).
Cerebral infarction
Major thromboembolic cerebral infarction usually causes an obvious stroke. The clinical picture is thus very variable, depending on the infarct site and extent. While the general site can be deduced from physical patterns (e.g. cortex, internal capsule, brainstem), clinical estimations of precise vascular territories are often inaccurate, when compared to imaging.
Following vessel occlusion brain ischaemia occurs, followed by infarction. The infarcted region is surrounded by a swollen area which does not function but is structurally intact. This is the ischaemic penumbra, which is detected on MRI and can regain function with neurological recovery.
Within the ischaemic area, hypoxia leads to neuronal damage. There is a fall in ATP with release of glutamate, which opens calcium channels with release of free radicals. These alterations lead to inflammatory damage, necrosis and apoptotic cell death.
Clinical features
Stroke most typically seen is caused by infarction in the internal capsule following thromboembolism in a middle cerebral artery branch (Fig. 22.31). A similar picture is caused by internal carotid occlusion (see Fig. 22.28). Limb weakness on the opposite side to the infarct develops over seconds, minutes or hours (occasionally longer). There is a contralateral hemiplegia or hemiparesis with facial weakness. Aphasia is usual when the dominant hemisphere is affected. Weak limbs are at first flaccid and areflexic. Headache is unusual. Consciousness is usually preserved. Exceptionally, an epileptic seizure occurs at the onset. After a variable interval, usually several days, reflexes return, becoming exaggerated. An extensor plantar response appears. Weakness is maximal at first; recovery occurs gradually over days, weeks or many months.
Brainstem infarction
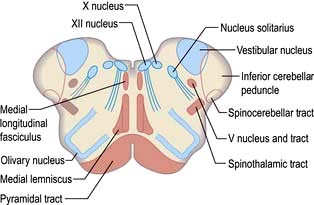
This causes complex signs depending on the relationship of the infarct to cranial nerve nuclei, long tracts and brainstem connections (Table 22.13).
The lateral medullary syndrome (posterior inferior cerebellar artery (PICA) thrombosis and Wallenberg’s syndrome) is a common example of brainstem infarction presenting as acute vertigo with cerebellar and other signs (Table 22.14 and Fig. 22.32). It follows thromboembolism in the PICA or its branches, vertebral artery thromboembolism or dissection. Features depend on the precise structures damaged.
Coma follows damage to the brainstem reticular activating system.
The locked-in syndrome is caused by upper brainstem infarction (p. 1095).
Pseudobulbar palsy (p. 1080) can follow lower brainstem infarction.
Table 22.13 Features of brainstem infarction
| Clinical feature | Structure involved |
|---|---|
Hemiparesis or tetraparesis |
Corticospinal tracts |
Sensory loss |
Medial lemniscus and spinothalamic tracts |
Diplopia |
Oculomotor system |
Facial numbness |
Vth nerve nuclei |
Facial weakness |
VIIth nerve nucleus |
Nystagmus, vertigo |
Vestibular connections |
Dysphagia, dysarthria |
IXth and Xth nerve nuclei |
Dysarthria, ataxia, hiccups, vomiting |
Brainstem and cerebellar connections |
Horner’s syndrome |
Sympathetic fibres |
Coma, altered consciousness |
Reticular formation |
Table 22.14 Clinical deficits associated with problems in vascular supply
| Vascular supply | Neurological deficits |
|---|---|
Left middle cerebral artery |
Right-sided weakness involving face and arm > leg with dysphasia |
Right middle cerebral artery |
Left-sided weakness involving face and arm > leg, visual and/or sensory neglect, denial of disability |
Lateral medulla (posterior inferior cerebral artery and/or parent vertebral artery) |
Ipsilateral Horner’s syndrome, Xth nerve palsy, facial sensory loss, limb ataxia with contralateral spinothalamic sensory loss. Vertiginous and unable to eat due to failing laryngeal closure and ineffective coughing. Cervical radiculopathies if involvement of radicular branches of the vertebral artery |
Posterior cerebral artery |
Homonymous hemianopia with varied deficits due to parietal and/or temporal lobe |
Internal capsule |
Motor, sensory or sensorimotor loss, face = arm = leg. Possible profound dysarthria from involvement of corticobulbar fibres but not dysphasia or other cortical deficits |
Bilateral paramedian thalamus |
Coma or disturbed vigilance, ophthalmoplegia (internal and/or external), ataxia and memory impairment. Some require ventilation |
Carotid artery dissection |
Ipsilateral Horner’s syndrome from compression of sympathetic plexus around the carotid artery, can also affect lower cranial nerves (Xth and XIIth most clinically obvious). If ipsilateral cerebral infarction follows, clinical picture can mimic brain stem event |
Other patterns of infarction
Lacunar infarction
Lacunes are small (<1.5 cm3) infarcts seen on MRI or at autopsy. Hypertension is commonly present. Minor strokes (e.g. pure motor stroke, pure sensory stroke, sudden unilateral ataxia and sudden dysarthria with a clumsy hand) are syndromes caused typically by single lacunar infarcts. Lacunar infarction is often symptomless.
Hypertensive encephalopathy (p. 778)
This is due to cerebral oedema, causing severe headaches, nausea and vomiting. Agitation, confusion, fits and coma occur if the hypertension is not treated. Papilloedema develops, either due to ischaemic optic neuropathy or following the brain swelling due to multiple acute infarcts. MRI shows oedematous white matter in the parieto-occipital regions.
Multi-infarct dementia (vascular dementia)
Multiple lacunes or larger infarcts cause generalized intellectual loss seen with advanced cerebrovascular disease. In the late stages, there is dementia, pseudobulbar palsy and a shuffling gait – the marche à petits pas (small steps), sometimes called atherosclerotic Parkinsonism. Binswanger’s disease is a term for widespread low attenuation in cerebral white matter, usually with dementia, TIAs and stroke episodes in hypertensive patients (the changes being seen on imaging/autopsy).
Visual cortex infarction
Posterior cerebral artery infarction or infarction of the middle cerebral artery macular branch causes combinations of hemianopic visual loss and cortical blindness (Anton’s syndrome, Fig. 22.30 and p. 1073).
Weber’s syndrome
Ipsilateral IIIrd nerve palsy with contralateral hemiplegia is due to a unilateral infarct in the midbrain. Paralysis of upward gaze is usually present.
Watershed (borderzone) infarction
Cortical infarcts, often multiple, follow prolonged periods of low perfusion (e.g. hypotension after massive myocardial infarction or cardiac bypass surgery). Infarcts occur in the borderzones, between areas supplied by the anterior, middle and posterior cerebral arteries. Cortical visual loss, memory loss and intellectual impairment are typical. In some cases, a vegetative state or minimal conscious state follows (p. 1096).
Acute stroke: immediate care and thrombolysis
(See also Box 22.14)
 Box 22.14 Stroke
Box 22.14 Stroke
immediate management
1. Admit to multidisciplinary stroke unit
3. Is thrombolysis appropriate?
CT should always be available. This will indicate haemorrhage, other pathology and sometimes infarctionParamedics and members of the public are encouraged to make the diagnosis of stroke on a simple history and examination – FAST:
Face – sudden weakness of the face
Arm – sudden weakness of one or both arms
Dedicated units with multidisciplinary, organized teams deliver higher standards of care than a general hospital ward, reducing stroke mortality and long-term disability. Evidence-based guidelines have contributed to clear protocols. Admission to hospital should proceed without delay, for imaging, care and investigation.
Following a stroke, immediate, continued and meticulous attention to the airway and to swallowing is essential. Management of unconscious or stuporose patients is outlined on page 1096.
In cerebral infarction, the issue is thrombolysis. Practical Box 22.4 outlines the current proposals for stroke thrombolysis. Alteplase is commonly used although tenecteplase is also effective. The benefit of thrombolysis is shown on CT perfusion scans (Fig. 22.33) and decreases with time, even within the time window of 4.5 h. Every minute counts. If thrombolysis is not given, aspirin 300 mg daily should be given as soon as a diagnosis of ischaemic stroke or thromboembolic TIA is confirmed, reducing to 75 mg after several days. Following thrombolysis aspirin should not be started until 24–48 h later.
 Practical Box 22.4
Practical Box 22.4
Thrombolysis in acute ischaemic stroke
Eligibility
Exclusion criteria
Rapidly improving stroke syndrome
Minor and isolated neurological signs
Seizure at the onset of stroke if the residual impairments are due to postictal phenomena
Symptoms suggestive of subarachnoid haemorrhage, even if the CT is normal
Acute MI or post-MI pericarditis
Persistent systolic BP >185, diastolic BP >110 mmHg, or requiring aggressive therapy to control BP
Adapted from Adams HP Jr, del Zoppo G, Alberts MJ et al. Stroke 2007; 38(5):1655–1711.
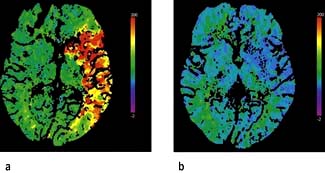
Figure 22.33 CT perfusion scans: (a) pre-thrombolysis; (b) post-thrombolysis showing reperfusion of the ischaemic site.
(Courtesy of Professor Adrian Dixon, Cambridge Radiology Department, UK.)
Investigations
The purpose of investigations in stroke is:
to confirm the clinical diagnosis and distinguish between haemorrhage and thromboembolic infarction;
Sources of embolus should be sought (e.g. carotid bruit, atrial fibrillation, valve lesion, evidence of endocarditis, previous emboli or TIA) and hypertension/postural hypotension assessed. Brachial BP should be measured on each side; >20 mmHg difference is suggestive of subclavian artery stenosis.
Routine investigations in thromboembolic stroke and TIA are listed in Box 22.13.
Box 22.13 Stroke
further investigation and management
Further management
Drugs for hypertension, heart disease, diabetes, other medical conditions
Antiplatelet agents, e.g. aspirin and elopidogrel
Question of anticoagulation – Table 22.15
Speech therapy, dysphagia care, physiotherapy, occupational therapy
Imaging in acute stroke
Non-contrast CT will demonstrate haemorrhage immediately but cerebral infarction is often not detected or only subtle changes are seen initially (Fig. 22.34a).
MRI shows changes early in infarction (Fig. 22.35a) and a later MRI shows the full extent of the damaged area or penumbra (Fig. 22.35b).
Diffusion-weighted MRI (DWI) can detect cerebral infarction immediately (see Fig. 22.34b) but is as accurate as CT for the detection of haemorrhage.
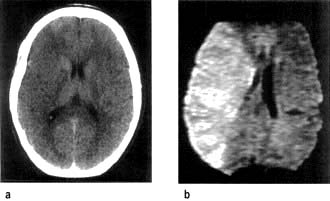
Figure 22.34 Middle cerebral artery infarction. (a) CT performed initially shows only very subtle low density changes in right MCA territory. (b) Diffusion-weighted MRI done at the same time shows full extent of the area of ischaemia.
(Courtesy of Dr Paul Jarman.)
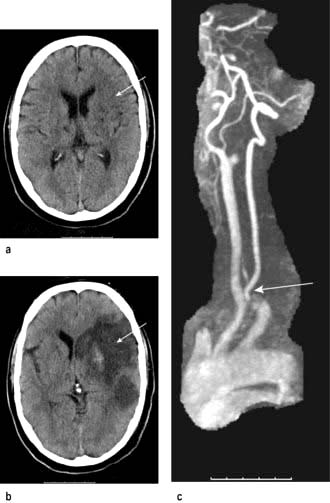
Figure 22.35 MRI of cerebral infarction and subsequent MRA performed in the same patient. (a) Early changes. (b) Changes after 5 days. (c) MR angiography showing internal carotid artery occlusion.
(Courtesy of Dr Paul Jarman.)
CT is still more widely available than MRI and should be performed if MRI is unavailable so that there is no delay in giving thrombolysis for cerebral infarction.
More detailed studies involving perfusion-weighted images and diffusion-weighted MRI will differentiate the infarct core and the penumbral area which is potentially recoverable.
FURTHER READING
Chalela JA, Kidwell CS, Nentwich LM et al. Magnetic resonance imaging and computed tomography in emergency assessment of patients with suspected acute stroke: a prospective comparison. Lancet 2007; 369:293–298.
Donnan GA, Fisher M, Macleod M et al. Stroke. Lancet 2008; 371:1612–1623.
Hacke W, Kaste M, Bluhmki E et al. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med 2008; 359:1319–1329.
Khaja AM, Grotta JC. Established treatments for acute ischaemic stroke. Lancet 2007; 369:319–330.
Royal College of Physicians’ Intercollegiate Stroke Working Party. National Clinical Guidelines for Stroke, 2nd edn. London: Royal College of Physicians; 2008.
Strong K, Mather C, Bonita R. Preventing stroke; saving lives around the world. Lancet Neurol 2007; 6:182–187 + Series.
Treatment of acute stroke
This is shown in Box 22.14. Thrombolysis has been shown to improve outcome and should be used immediately if there are no contraindications. In a massive middle cerebral artery infarct, hemispheric swelling occurs with oedema (Fig. 22.36). Decompressive hemicraniectomy reduces the intracranial pressure and the mortality but extensive neurological deficits remain.
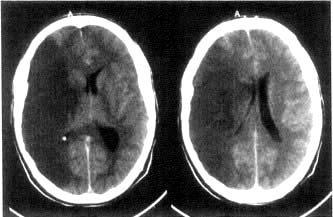
Figure 22.36 CT scan of massive infarction in the middle carotid artery territory. There is hemisphere swelling with brain shift.
(Courtesy of Dr Paul Jarman.)
Later investigations
MR angiography (MRA) or CT angiography is valuable in anterior circulation TIAs to confirm surgically accessible arterial stenoses, mainly internal carotid stenosis (Fig. 22.35c). If ultrasound suggests carotid stenosis, normotensive patients with TIA or stroke in the anterior circulation should have vascular imaging.
Carotid Doppler and duplex scanning. These screen for carotid (and vertebral) stenosis and occlusion: in skilled hands they demonstrate accurately the degree of internal carotid stenosis.
Long-term management
Medical therapy
Risk factors (Table 22.12) should be identified and addressed.
Antihypertensive therapy
Recognition and good control of high blood pressure is the major factor in primary and secondary stroke prevention. Transient hypertension, often seen following stroke, usually does not require treatment provided diastolic pressure does not rise >100 mmHg. Sustained severe hypertension needs treatment (p. 781); BP should be lowered slowly to avoid any sudden fall in perfusion.
Antiplatelet therapy (see also p. 425)
Long-term soluble aspirin (75 mg daily) reduces substantially the incidence of further infarction following thromboembolic TIA or stroke. Aspirin inhibits cyclo-oxygenase, which converts arachidonic acid to prostaglandins and thromboxanes; predominant therapeutic effects are reduction of platelet aggregation. Clopidogrel and dipyridamole are also used (p. 425). Combined aspirin 75 mg daily and clopidogrel 75 mg daily provide optimal prophylaxis against further thromboembolic stroke or TIA. Dipyridamole 200 mg twice daily is used if clopidogrel is contraindicated.
Anticoagulants
Heparin and warfarin should be given when there is atrial fibrillation, other paroxysmal dysrhythmias or when there are cardiac valve lesions (uninfected) or cardiomyopathies. Brain haemorrhage must be excluded by CT/MRI. Patients must be aware of the small risk of cerebral (and other) haemorrhage. Anticoagulants are potentially dangerous in the two weeks following infarction because of the risk of provoking cerebral haemorrhage; there are wide differences in clinical practice. Antithrombins are now being used. Table 22.15 outlines the issues in secondary stroke prevention.
Table 22.15 Anticoagulants and stroke prevention
| Indication | Comment |
|---|---|
Valvular heart disease |
Heparin/warfarin of benefit in chronic rheumatic heart disease, particularly mitral stenosis |
Recent MI |
|
Intracardiac thrombus |
Heparin/warfarin if there is evidence of intracardiac thrombus |
Atrial fibrillation |
Anticoagulants long term reduce stroke incidence in atrial fibrillation |
Acute internal carotid artery thrombus |
Anticoagulants reserved for imaging-confirmed cases of arterial thrombosis or dissection. They have not been shown to be beneficial in stroke prevention after thromboembolism from carotid or vertebrobasilar sources |
Acute basilar artery thrombus |
|
Internal carotid artery dissection |
|
Extracranial vertebral artery dissection |
|
Prothrombic states, e.g. protein C deficiency |
Anticoagulation, in consultation with haematologist |
Recurrent TIAs or stroke on full antiplatelet therapy |
If no remediable cause, a trial of anticoagulants may be justified |
Cerebral venous thrombosis including sinus thrombosis |
Benefits of anticoagulation outweigh risks of haemorrhage |
Adapted from Brown M. Medicine 2000; 28(7):64.
Other measures
Polycythaemia and any clotting abnormalities should be treated (p. 402). Statin therapy should be given for all.
Surgical approaches
Internal carotid endarterectomy
Surgery is usually recommended in TIA or stroke patients with internal carotid artery stenosis >70% (see ABCD2, above). Successful surgery reduces the risk of further TIA/stroke by around 75%. Endarterectomy has a mortality around 3%, and a similar risk of stroke. Percutaneous transluminal angioplasty (stenting) is an alternative. The value of surgery for asymptomatic carotid stenosis is debatable.
Stroke in the elderly
While in the elderly the yield of investigation in stroke diminishes, age is no barrier to recovery; elderly patients benefit the most from good rehabilitation. Consider social isolation, pre-existing cognitive impairment, nutrition, skin and sphincter care, and reassess swallowing. Carotid endarterectomy over 75 years carries little more risk than in younger cases. Fibrinolysis is not contraindicated.
Rehabilitation: multidisciplinary approach
Physiotherapy has particular value in the first few weeks after stroke to relieve spasticity, prevent contractures and teach patients to use walking aids. The benefits of physiotherapy for longer-term outcome are still inadequately researched. Baclofen and/or botulinum toxin are sometimes helpful in the management of severe spasticity.
Speech and language therapists have a vital understanding of aphasic patients’ problems and frustration. Return of speech is hastened by conversation generally. If swallowing is unsafe because of the risk of aspiration, either nasogastric feeding or percutaneous gastrostomy will be needed. Video-fluoroscopy while attempting to swallow is helpful.
Physiotherapy, occupational and speech therapy have a vital role in assessing and facilitating the future care pathway. Stroke is frequently devastating and, particularly during working life, alters radically the patient’s remaining years. Many become unemployable, lose independence and are financially embarrassed. Loss of self-esteem makes depression common.
At home, aids and alterations may be needed: stair and bath rails, portable lavatories, hoists, sliding boards, wheelchairs, tripods, stair lifts, electric blinds and modified sleeping arrangements, kitchen, steps, flooring and doorways. Liaison between hospital-based and community care teams, and primary care physician is essential.
Prognosis
About 25% of patients die within 2 years of a stroke, nearly 10% within the 1st month. This early mortality is higher following intracranial haemorrhage than thromboembolic infarction. Poor outcome is likely when there is coma, a defect in conjugate gaze and hemiplegia. Many complications, particularly in the elderly, are preventable, e.g. aspiration. Coordinated care reduces deaths.
Recurrent strokes are, however, common (10% in the 1st year) and many patients die subsequently from myocardial infarction. Of initial stroke survivors, some 30–40% remain alive at 3 years.
Gradual improvement usually follows stroke, although late residual deficits are typically substantial. One-third of survivors return to independent mobility and one-third have disability requiring institutional care.
The outlook for recovery of language varies: in general, if language is intelligible at all at 3 weeks, prognosis for fluent speech is good, but many are left with word-finding difficulties.
Intracranial haemorrhage
Intracerebral haemorrhage
Aetiology
Intracerebral haemorrhage causes approximately 15% of strokes. Rupture of microaneurysms (Charcot–Bouchard aneurysms, 0.8–1.0 mm diameter) and degeneration of small deep penetrating arteries are the principal pathology. Such haemorrhage is usually massive, often fatal, and occurs in chronic hypertension and at well-defined sites – basal ganglia, pons, cerebellum and subcortical white matter.
In normotensive patients, particularly over 60 years, lobar intracerebral haemorrhage occurs in the frontal, temporal, parietal or occipital cortex. Cerebral amyloid angiopathy (rare) is the cause in some of these haemorrhages, and the tendency to rebleed is associated with particular apolipoprotein E genotypes.
Recognition
At the bedside, there is no entirely reliable way of distinguishing between haemorrhage and thromboembolic infarction. Both produce stroke. Intracerebral haemorrhage tends to be dramatic with severe headache. It is more likely to lead to coma than thromboembolism.
Brain haemorrhage is seen on CT imaging immediately (cf. infarction, p. 1091) as intraparenchymal, intraventricular or subarachnoid blood. Routine MRI may not identify an acute small haemorrhage correctly in the first few hours but MRI diffusion-weighted (MRI-DW) is as good as CT.
Management: haemorrhagic stroke
The principles are those for cerebral infarction. The immediate prognosis is less good. Antiplatelet drugs and, of course, anticoagulants are contraindicated. Control of hypertension is vital. Urgent neurosurgical clot evacuation is occasionally necessary when there is deepening coma and coning (particularly in cerebellar haemorrhage).
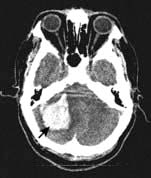
Cerebellar haemorrhage (Fig. 22.37)
There is headache, often followed by stupor/coma and signs of cerebellar/brainstem origin (e.g. nystagmus, ocular palsies). Gaze deviates towards the haemorrhage. Skew deviation (p. 1094) may develop. Cerebellar haemorrhage sometimes causes acute hydrocephalus, a potential surgical emergency.
Subarachnoid haemorrhage (SAH)
SAH means spontaneous arterial bleeding into the subarachnoid space, and is usually clearly recognizable clinically from its dramatic onset. SAH accounts for some 5% of strokes and has an annual incidence of 6 per 100 000.
Causes
The causes of SAH are shown in Table 22.16; it is unusual to find any contributing disease.
Table 22.16 Underlying causes of subarachnoid haemorrhage
| Cause of aneurysm | (%) |
|---|---|
Saccular (berry) aneurysms |
70% |
Arteriovenous malformation (AVM) |
10% |
No arterial lesion found |
15% |
Rare associations |
(<5%) |
Bleeding disorders Mycotic aneurysms – endocarditis Acute bacterial meningitis Tumours, e.g. metastatic melanoma, oligodendroglioma Arteritis (e.g. SLE) Spinal AVM → spinal SAH Coarctation of the aorta Marfan’s, Ehlers–Danlos syndrome Polycystic kidneys |
|
Saccular (berry) aneurysms (Fig. 22.38)
Saccular aneurysms develop within the circle of Willis and adjacent arteries. Common sites are at arterial junctions:
Between posterior communicating and internal carotid artery – posterior communicating artery aneurysm
Between anterior communicating and anterior cerebral artery – anterior communicating and anterior cerebral artery aneurysm
At the trifurcation or a bifurcation of the middle cerebral artery – middle cerebral artery aneurysm.
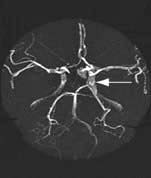
Other aneurysm sites are on the basilar, posterior inferior cerebellar, intracavernous internal carotid and ophthalmic arteries. Saccular aneurysms are an incidental finding in 1% of autopsies and can be multiple.
Aneurysms cause symptoms either by spontaneous rupture, when there is usually no preceding history, or by direct pressure on surrounding structures; for example, an enlarging unruptured posterior communicating artery aneurysm is the commonest cause of a painful IIIrd nerve palsy (p. 1081).
Arteriovenous malformation (AVM)
AVMs are vascular developmental malformations, often with a fistula between arterial and venous systems causing high flow through the AVM and high pressure arterialization of draining veins. An AVM may also cause epilepsy, often focal. The risk of a first haemorrhage (20% fatal and 30% resulting in permanent disability) is approximately 2–3% per year. Once an AVM has caused a haemorrhage, the risk of rebleeds is increased– to approximately 10% per year. AVMs may be ablated with endovascular treatment (catheter injection of glue into the nidus usually), surgery or stereotactic radiotherapy. A multidisciplinary team approach with neurologist, interventional neuro-radiologist and neurosurgeon is required in deciding on treatment options.
Cavernous haemangiomas (cavernomas) are common (0.1–0.5% prevalence) and consist of a tangle of low pressure dilated vessels without a major feeding artery; they are frequently symptomless (Fig. 22.39) and seen incidentally on imaging. Multiple cavernomas often have a genetic basis, with linkage to two loci on chromosome 7q. Cavernomas may cause seizures. Small haemorrhages may occur but are usually low pressure bleeds and rarely cause severe deficits. Surgical resection is rarely needed except where the cavernoma is gradually enlarging or causing significant neurological symptoms.
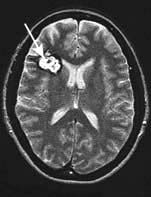
Clinical features of subarachnoid haemorrhage
There is a sudden very severe headache, often occipital (mean time to peak headache 3 min). Headache is usually followed by vomiting and often by coma and death. Survivors of SAH may remain comatose or drowsy for hours, days, or longer. SAH is a possible diagnosis in any sudden headache.
Following major SAH there is neck stiffness and a positive Kernig’s sign. Papilloedema is sometimes present, with retinal and/or subhyaloid haemorrhage (tracking beneath the retinal hyaloid membrane). Minor bleeds cause few signs, but almost invariably headache (approximately 17% of patients have small ‘sentinel bleeds’ in the weeks before presenting with SAH).
Investigations
CT imaging is the immediate investigation (Fig. 22.40). Subarachnoid and/or intraventricular blood is usually seen (sensitivity of CT to detect subarachnoid blood is 95% within 24 h of onset but much lower over subsequent days). Lumbar puncture is not necessary if SAH is confirmed by CT, but should be performed if doubt remains. CSF becomes yellow (xanthochromic) within 12 h of SAH and remains detectable for 2 weeks. Visual inspection of supernatant CSF is usually sufficiently reliable for diagnosis. Spectrophotometry to estimate bilirubin in the CSF released from lysed cells is used to define SAH with certainty. CT angiography or catheter angiography to identify the aneurysm or other source of bleeding is performed in patients potentially fit for surgery. In some, no aneurysm or source of bleeding is found, despite a definite SAH.
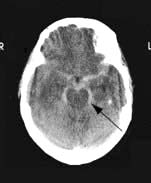
Differential diagnosis
SAH must be differentiated from migraine. This is sometimes difficult – a short time to maximal headache intensity and the presence of neck stiffness usually indicate SAH. Thunderclap headache is used (confusingly) to describe either SAH or a sudden (benign) headache for which no cause is ever found. The syndrome of reversible cerebral vasoconstriction (Call–Fleming syndrome) presents with thunderclap headache. Acute bacterial meningitis occasionally causes a very abrupt headache, when a meningeal microabscess ruptures; SAH also occasionally occurs at the onset of acute bacterial meningitis. Cervical arterial dissection can present with a sudden headache.
Complications
Blood in the subarachnoid space can lead to obstructive hydrocephalus, seen on CT. Hydrocephalus can be asymptomatic but may cause deteriorating consciousness following SAH. Shunting may be necessary. Arterial spasm (visible on angiography and a cause of coma or hemiparesis) is a serious complication of SAH and a poor prognostic feature.
Management
Immediate treatment of SAH is bed rest and supportive measures. Hypertension should be controlled. Nimodipine, a calcium-channel blocker given for 3 weeks, reduces mortality.
All SAH cases should be discussed urgently with a neurosurgical centre. Nearly half of SAH cases are either dead or moribund before reaching hospital. Of the remainder, a further 10–20% rebleed and die within weeks. Failure to diagnose SAH, e.g. mistaking SAH for migraine, contributes to this mortality.
Where angiography demonstrates an aneurysm (the cause of the vast majority of SAH), endovascular treatment by placing platinum coils via a catheter in the aneurysm sac to promote thrombosis and ablation of the aneurysm, is now the first line treatment. Endovascular coiling has a lower complication rate than surgery but direct surgical clipping of the aneurysm neck is still required in some selected cases. For asymptomatic (unruptured) aneurysms over 8 mm in diameter the risk of treatment is less than the risk of haemorrhage if not treated. Patients who remain comatose or who have persistent severe deficits after SAH have a poor outlook.
Subdural and extradural bleeding
These conditions can cause death following head injuries unless treated promptly.
Subdural haematoma (SDH)
SDH means accumulation of blood in the subdural space following rupture of a vein. This usually follows a head injury, sometimes trivial. The interval between injury and symptoms can be days, or extend to weeks or months. Chronic, apparently spontaneous SDH is common in the elderly, and also occurs with anticoagulants.
Headache, drowsiness and confusion are common; symptoms are indolent and can fluctuate. Focal deficits, e.g. hemiparesis or sensory loss, develop. Epilepsy occasionally occurs. Stupor, coma and coning may follow.
Extradural haemorrhage (EDH)
EDH typically follows a linear skull vault fracture tearing a branch of the middle meningeal artery. Extradural blood accumulates rapidly over minutes or hours. A characteristic picture is of a head injury with a brief duration of unconsciousness, followed by improvement (the lucid interval). The patient then becomes stuporose, with an ipsilateral dilated pupil and contralateral hemiparesis, with rapid transtentorial coning. Bilateral fixed dilated pupils, tetraplegia and respiratory arrest follow. An acute progressive SDH presents similarly.
Management
Possible extradural or subdural bleeding needs immediate imaging. CT (Fig. 22.41a) is the most widely used investigation because of its immediate availability. MRI is more sensitive for the detection of small haematomas. T1-weighted MRI (Fig. 22.41b) shows bright images due to the presence of methaemoglobin.
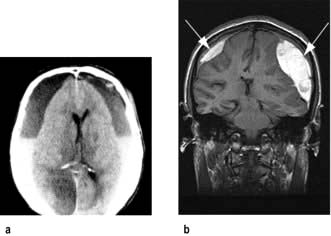
EDHs require urgent neurosurgery: if it is performed early, the outlook is excellent. When far from a neurosurgeon, e.g. in wartime or at sea, drainage through skull burr-holes has been lifesaving when an EDH has been diagnosed clinically.
Subdural bleeding usually needs less immediate attention but close neurosurgical liaison is necessary. Even large collections can resolve spontaneously without drainage. Serial imaging is needed to assess progress.
Cortical venous thrombosis and dural venous sinus thrombosis
Intracranial venous thromboses are usually (>50%) associated with a pro-thrombotic risk factor, e.g. oral contraceptives, pregnancy, genetic or acquired pro-thrombotic states and dehydration. Head injury is also a cause. Infection, e.g. from a paranasal sinus, may be present. Venous thromboses can also arise spontaneously.
Cortical venous thrombosis
The venous infarct leads to headache, focal signs (e.g. hemiparesis) and/or epilepsy, often with fever.
Dural venous sinus thromboses
Cavernous sinus thrombosis causes ocular pain, fever, proptosis and chemosis. External and internal ophthalmoplegia with papilloedema develops.
Sagittal and lateral sinus thrombosis cause raised intra-cranial pressure with headache, fever, papilloedema and often epilepsy.